

Abstract
A drug−drug cocrystal of two anticonvulsants, lamotrigine and phenobarbital, is presented. In the crystal structure, molecules form heterodimers via N−H···O and N−H···N hydrogen bonding. The intrinsic dissolution rate (IDR) and solubility of the cocrystal were measured in phosphate buffer (pH 7.2) and simulated gastric fluid (without pepsin), and compared to pure APIs. Dissolution experiments found suppressed IDR of the cocrystal with rates in the order pure PB > pure LTG > cocrystal. The solubility measurements were consistent with the dissolution behavior. The presence of strong heterodimers in the cocrystal compared to weaker homodimers in the parent drugs is implicated for the reduced solubility and dissolution rate.
Graphical Abstract
Multidrug cocrystals (MDCs) have been the subject of growing interest in the pharmaceutical industry, as they potentially offer many advantages over single component parent drugs, such as enhanced dissolution rate and bioavailability, and tandem therapeutic e ffects.1–3 Additionally, MDCs fulfill all the criteria for patent applications such as novelty, utility, and nonobviousness; recently, the US FDA approved the first MDC−Entresto comprising Sacubitril (a neprilysin inhibitor) and valsartan (an angiotensin II receptor blocker)−for chronic heart failure.4 Yet, most MDCs reported in the literature are only characterized structurally and are not studied for physiochemical properties.5–8 A recent survey suggested that of 36 examples of drug−drug combination,9 many fail to contribute increased solubility/dissolution to a parent drug. For example, the dissolution rate of the theophylline-phenobarbital cocrystal was found to be less than that of either single component drug,9 and a diflunisal-theophylline cocrystal displayed a similar dissolution profile as each parent drug.10 Although design strategies for cocrystals are well-defined,4,11–13 these approaches cannot always be extended directly to the design of MDCs. Crystal engineering of cocrystals14,15 often relies on strong heterodimer synthons to ensure interaction between API and coformer. In the case of cocrystals containing only a single API, improved solubility is imparted to a material by incorporation of a highly soluble coformer.16–19 Even when the cocrystal displays formidable solid-state stability, it nonetheless undergoes fast dissolution of API mediated by dissolution of coformer.20 Yet, for MDCs, the same strategies are no longer applicable, as the solubility difference between a Biopharmaceutics Classification System (BCS) Class I/III drug and a BCS Class II/IV drug can be small.
Lamotrigine (originally marketed as Lamictal, hereafter LTG) is considered a first-line drug for primary generalized tonic-clonic seizures and classified as a class II drug under the BCS with poor aqueous solubility (0.17 mg/mL at 25 °C).21 The presence of multifunctional moieties with both hydrogen bond donors and acceptors (see Scheme 1 for molecular structure) on the periphery of the weak base (pKa 5.7) make the drug desirable for forming both cocrystals and salts.22–24 A screen for cocrystals produced three novel salts of LTG with oxalic acid (OA), malonic acid (MA), and sebacic acid (SA)−however, their dissolution behavior was not further investigated in this work (crystal structure description is provided in Supporting Information, Section SI 3). The following work presents an analysis of the drug−drug cocrystal formed between two anticonvulsants, lamotrigine (LTG) and phenobarbital (PB). Patients not responding to a single medication alone are frequently treated with combination anticonvulsant drugs,25 and the therapeutic benefits of drug combinations have been demonstrated in the literature.26 It is found that these combinations offer a significant reduction of seizures and/or favorable adverse-effect profiles. Phenobarbital, (PB, 5-ethyl-5-phenylpyrimidine-2,4,6(1H,3H,5H)-trione), is a BCS class I drug (aqueous solubility 1 mg/mL) and is included in the list of essential medicines by the World Health Organization (see Scheme 1 for molecular structure). PB is used widely for treating epilepsy and neonatal seizures. The relative low cost of PB compared to other antiepileptic drugs (in current use), makes it affordable and suitable for use in low income countries.27 It has several polymorphic forms28–30−most recently, the crystal structure of form V was reported by our group using polymer-induced heteronucleation.31 Lamotrigine and phenobarbital combination in the dose ratios of 1:3 and 3:1 are reported to offer favorable effects in terms of increased anticonvulsant effcacy and reduced neurotoxicity in maximal electroshock induced seizures.25 Further, given that the aqueous solubility of PB is roughly 10-fold that of LTG and that PB is a weak acid and LTG is a weak base, it was hypothesized that a MDC between the two pharmaceuticals would impart increased solubility and dissolution to LTG. Therefore, in the light of the enhanced therapeutic benefits of these drug combinations, cocrystallization of LTG and PB was carried out and the multidrug cocrystal between lamotrigine and phenobarbital was structurally characterized by single-crystal and powder X-ray diffraction, Raman spectroscopy, and thermal analysis (DSC/TGA), and its dissolution behavior quantified in phosphate buffer saline (PBS, pH 7.2) and simulated gastric fluid (SGF, without pepsin). The influence of pH on cocrystal solubility was examined and it was found to be consistent with the dissolution behavior. The reduced dissolution rate and solubility of the LTG-PB cocrystal relative to its constituents is ascribed to the presence of strong heterodimers compared to weaker homodimer interactions in the single component crystals (LTG and PB).
Scheme 1.

Molecular Structure of (a) Lamotrigine (LTG) and (b) Phenobarbital (PB)
Recrystallization of a ground 1:1 LTG−PB mixture from methanol/acetonitrile resulted in blocky crystals. Powder X-ray diffraction and Raman spectroscopy reveal significant differences compared to LTG and PB in the PXRD pattern and Raman spectra suggesting cocrystal formation (Figures S1−S3; Supporting Information). Single crystal X-ray diffraction reveals monoclinic crystals in the noncentrosymmetric space group (Cc) consisting of one molecule of LTG and one molecule of PB in the asymmetric unit (Figure S4; Supporting Information). Intermolecular hydrogen bonds are facilitated by amino groups and the triazine ring on LTG interacting with carbonyl groups and N−H on PB. Via N−H···O and N−H···N hydrogen bonding, the cocrystal is stabilized by a 3-point interaction (heterodimer, shown in blue highlight in Figure 1). Another heterodimer formed by N−H···O and N−H···N hydrogen bonding leads to the interaction highlighted in red (Figure 1). These interactions propagate further forming a 2-dimensional sheet.
Figure 1.

Hydrogen bonding features in LTG-PB cocrystal.
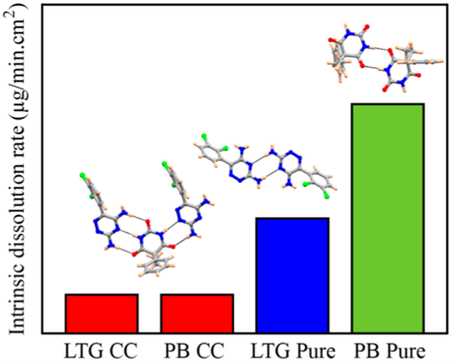
The intrinsic dissolution rate (IDR) and solubility of LTGPB cocrystal were measured in phosphate buffer saline (pH 7.2) and simulated gastric fluid (without pepsin), and compared with the IDR and solubility of LTG and PB. Dissolution behavior of each component of the cocrystal was described by the concentration change of LTG and PB in the dissolution media,20 which was quantified based on UV−vis spectra, applied to monitor the dissolution process (details in Supporting Information). The dissolution rate was calculated by evaluating the slope of concentration−time profiles in the initial few minutes (LTG-PB cocrystal dissolution profile in PBS is shown in Figure S11, Supporting Information). There was no improvement in the IDR of the cocrystal compared to parent drugs in both dissolution media (PBS and SGF); IDR was greatest for the pure PB drug, followed by pure LTG, and slowest for the cocrystal (see Figure 2). Dissolution profiles of LTG, PB, and cocrystal in simulated gastric fluid and their IDR are shown in Figures S12−13; Supporting Information. No solid transformation was found after dissolution (Figure S14; Supporting Information).
Figure 2.

Intrinsic dissolution rate of LTG and PB from single component molecules and a newly discovered cocrystal at 37 ± 0.1 °C in phosphate bu ffer saline.
Solubility behavior of the cocrystal is consistent with the dissolution measurements as shown in Figure 3. These results demonstrated that the LTG-PB cocrystal is thermodynamically stable between pH values of 2.6 and 9.0. These are pHmax values with PB and LTG, respectively. The cocrystal solubility curve intersects with PB at pH 2.6 and LTG at pH 9.0, which proves that the cocrystal is less soluble than its components in this pH range. The cocrystal is shown to alter the solubility-pH dependence of its components, resulting in a U-shaped curve with a solubility minimum between the pKa of LTG and the first pKa of PB (pKa 7.5).17,32,33 The equations for calculating the pH dependent solubility of LTG, PB, and LTG-PB cocrystal are provided in the Supporting Information (Section SI 7).
Figure 3.

Solubility-pH dependence of the LTG-PB cocrystal, basic drug LTG, and diprotic acidic drug PB. Diamond and square symbols represent solubilities determined from solutions saturated with LTG or PB, respectively. Triangular and circular symbols represent solubilities determined from eutectic solutions saturated with both LTG and LTG-PB. pH values correspond to equilibrium pH. The pH values at the intersections of PB and LTG-PB curves and of LTG and LTG-PB curves correspond to pHmax. LTG-PB thermodynamic stability and solubility are determined by the solution pH as indicated by the pHmax values of 2.6 and 9.0.
In general, solubility is the main factor which determines dissolution rate, and thermodynamic solubility is governed by the relative energies of both the solid-state and solvated form of a material. Solubility is a function of both solid and solution interactions or homogeneous and heterogeneous equilibria. Solution thus increases the dissolution rate of an otherwise sparingly soluble API.19,34,35 By using highly soluble coformers, any relative increase in the solid-state stability of a cocrystal is negligible relative to the dramatic improvement in solubility. For the formation of MDCs, the relative heightened solubility of BCS Class I or III drugs is often insignificant compared to a poorly soluble API, and thus the solid-state stability of such materials dictates their rate of dissolution. If a multidrug cocrystal is thermodynamically stable relative to its single components, it is likely that this material will not show the solubility improvements often expected in the formation of cocrystals, when ionization of cocrystal constituents is negligible. This failure is most apparent when MDCs are designed by a common methodology applied for cocrystal formation−the formation of heterodimer interactions in a cocrystal as a combination of homodimer interactions found in each single component species. The LTG-PB cocrystal, as described above, is stabilized by a heterodimer comprising discrete homodimers found in each parent drug. The lamotrigine crystal is held by N−H···N amine/pyridine interactions36 and the phenobarbital crystal is held by N−H···O amine/carbonyl intermolecular bonds;27 the cocrystal contains a heterodimer with both interactions (Figure 5). Given that the pKa of PB is 7.5; its ionization under the conditions studied is negligible. The relative increase in stability of this cocrystal is seen not only in its lower dissolution rate but also in its higher melting point; the cocrystal melts at 262 °C (Figure 4) whereas LTG melts at 217 °C, and PB at 175 °C (Figure S15, Supporting Information).
Figure 5.

Crystal structure analysis of LTG, PB, and LTG-PB reveal the homodimer interactions in the LTG and PB single component crystals (a and b) are combined to form a heterodimer (c) in the LTG-PB cocrystal.
Figure 4.

DSC and TGA plot of the LTG-PB cocrystal.
A greater insight into structure−property relation is provided by a detailed analysis of intermolecular interactions in LTG, PB, and LTG-PB cocrystal. It is postulated that much of the increased stability in the cocrystal form of LTG-PB is derived from the increased strength in a heterodimer interaction between the molecules compared to the homodimer interactions connecting the single component forms. The strength of such interactions can be correlated to their intermolecular bond distances. In LTG, the aminopyridine homodimer governs the crystal packing with N−H···N hydrogen bonds distances of 3.178 Å.33 In the cocrystal, this same interaction is shortened to intermolecular bond length of 2.904 and 2.824 Å. In PB (ref code: PHBARB07, Form I, most stable form), N− H···O homodimers30 show intermolecular bond lengths of 2.926, 2.868, and 2.902 Å, whereas in the cocrystal the same interaction has bond distances of 2.895, 2.979, and 2.922 Å (Figure 5).
A survey of the Cambridge Structural Database (CSD) computed the average intermolecular bond length between homodimer and heterodimer N−H···O and N−H···N inter-actions in crystalline materials with such functionalities. As shown in Figure 6, the average bond distance of N−H···O and N−H···N in homodimers is longer than that in the heterodimer. This difference implies that a cocrystal built by this heterodimer will be more stable than either single component comprising the homodimer interaction. In the case of MDCs, where the solubility of an included BCS I/III pharmaceutical may be relatively small, this novel form will not show an increase in dissolution rate and therefore few of the solubility advantages commonly discussed in the crystal engineering of cocrystal pharmaceuticals.
Figure 6.

Average intermolecular bond lengths in pyridine/amino homodimer, amino/carbonyl homodimer, and the heterodimer formed by both functionalities in CSD. Error bars show standard deviation of the mean.
In conclusion, three novel salts of lamotrigine and a drug− drug cocrystal of lamotrigine-phenobarbital combination have been synthesized and characterized structurally. The LTG-PB cocrystal is sustained by heterodimers via N−H···O and N− H···N hydrogen bonding and displays a higher melting point and slower dissolution compared to parent APIs. This decrease in dissolution rate and solubility is rationalized by the increased strength of heterodimer interactions in the cocrystal compared to homodimer interactions in each single component form, and the generality of this finding is supported by a survey of such interactions in the CSD; MDCs containing these functionalities will often fail to impart solubility to a weakly soluble API. On the other hand, this suppression of solubility may be useful to achieve controlled release from MDCs for applications where sustained release of low concentrations is advantageous.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of Health Grant Number RO1 GM106180. We thank Dr. Jeff W. Kampf for single crystal X-ray analysis. We would also like to thank Mr. Derek Frank for helpful discussions.
Footnotes
Accession Codes
CCDC 1549452−1549455 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Sekhon BS Daru, J. Pharm. Sci 2012, 20, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Aitipamula S; Chow PS; Tan RB H. CrystEngComm 2009, 11, 1823. [Google Scholar]
- (3).Simon F Nat. Rev. Drug Discovery 2006, 5, 881. [DOI] [PubMed] [Google Scholar]
- (4).Duggirala NK; Perry ML; Almarsson O; Zaworotko MJ Chem. Commun 2016, 52, 640. [DOI] [PubMed] [Google Scholar]
- (5).Bhatt PM; Azim Y; Thakur TS; Desiraju GR Cryst. Growth Des 2009, 9, 951. [Google Scholar]
- (6).Grobelny P; Mukherjee A; Desiraju GR CrystEngComm 2011, 13, 4358. [Google Scholar]
- (7).Caira MR J. Crystallogr. Spectrosc. Res 1992, 22, 193. [Google Scholar]
- (8).Vishweshwar P; McMahon JA; Peterson ML; Hickey MB; Shattock TR; Zaworotko MJ Chem. Commun 2005, 4601. [DOI] [PubMed]
- (9).Thipparaboina R; Kumar D; Chavan RB; Shastri NR Drug Discovery Today 2016, 21, 481. [DOI] [PubMed] [Google Scholar]
- (10).Surov AO; Voronin AP; Manin AN; Manin NG; Kuzmina LG; Churakov AV; Perlovich GL Mol. Pharmaceutics 2014, 11, 3707. [DOI] [PubMed] [Google Scholar]
- (11).Kuminek G; Cao F; Bahia de Oliveira da Rocha A; Goncalveş Cardoso S; Rodríguez-Hornedo N Adv. Drug Delivery Rev 2016, 101, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Vishweshwar P; McMahon JA; Bis JA; Zaworotko MJ J. Pharm. Sci 2006, 95, 499. [DOI] [PubMed] [Google Scholar]
- (13).Shan N; Zaworotko MJ Drug Discovery Today 2008, 13, 440. [DOI] [PubMed] [Google Scholar]
- (14).Mukherjee A Cryst. Growth Des 2015, 15, 3076. [Google Scholar]
- (15).Shattock TR; Arora KK; Vishweshwar P; Zaworotko MJ Cryst. Growth Des 2008, 8, 4533. [Google Scholar]
- (16).Good DJ; Rodríguez-Hornedo N Cryst. Growth Des 2009, 9, 2252. [Google Scholar]
- (17).Bethune SJ; Huang N; Jayasankar A; Rodríguez-Hornedo N Cryst. Growth Des 2009, 9, 3976. [Google Scholar]
- (18).Good DJ; Rodríguez-Hornedo N Cryst. Growth Des 2010, 10, 1028. [Google Scholar]
- (19).Babu NJ; Nangia A Cryst. Growth Des 2011, 11, 2662. [Google Scholar]
- (20).For a recently exception see: Li Z; Matzger A J. Mol. Pharmaceutics 2016, 13, 990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Miller AA; Wheatley P; Sawyer DA; Baxter MG; Roth B Epilepsia 1986, 27, 483. [DOI] [PubMed] [Google Scholar]
- (22).Galcera J; Molins E Cryst. Growth Des 2009, 9, 327. [Google Scholar]
- (23).Thipparaboina R; Kumar D; Mittapalli S; Balasubramanian S; Nangia A; Shastri NR Cryst. Growth Des 2015, 15, 5816. [Google Scholar]
- (24).Chadha R; Saini A; Arora P; Jain DS; Dasgupta A; Guru Row TN CrystEngComm 2011, 13, 6271. [Google Scholar]
- (25).Luszczki JJ; Czuczwar M; Kis J; Krysa J; Pasztelan I; Swiader M; Czuczwar SJ Epilepsia 2003, 44, 1003. [DOI] [PubMed] [Google Scholar]
- (26).Perucca E Br. J. Clin. Pharmacol 2006, 61, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ilangaratne NB; Mannakkara NN; Bell GS; Sander JW Bull. World Health Organ 2012, 90, 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Platteau C; Lefebvre J; Hemon S; Baehtz C; Danede F; Prevost D Acta Crystallogr., Sect. B: Struct. Sci 2005, 61, 80. [DOI] [PubMed] [Google Scholar]
- (29).Zencirci N; Gelbrich T; Apperley DC; Harris RK; Kahlenberg V; Griesser UJ Cryst. Growth Des 2010, 10, 302. [Google Scholar]
- (30).Williams P Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem 1974, 30, 12. [Google Scholar]
- (31).Roy S; Goud NR; Matzger AJ Chem. Commun 2016, 52, 4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kuminek G; Rodriguez-Hornedo N; Siedler S; Rocha HVA; Cuffini SL; Cardoso SG Chem. Commun 2016, 52, 5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Maheshwari C; Andre V; Reddy S; Roy L; Duarte T; Rodriguez-Hornedo N CrystEngComm 2012, 14, 4801. [Google Scholar]
- (34).Thakuria R; Delori A; Jones W; Lipert MP; Roy L; Rodríguez-Hornedo N Int. J. Pharm 2013, 453, 101. [DOI] [PubMed] [Google Scholar]
- (35).Serajuddin ATM Adv. Drug Delivery Rev 2007, 59, 603. [DOI] [PubMed] [Google Scholar]
- (36).Sridhar B; Ravikumar K Acta Crystallogr., Sect. C: Cryst. Struct. Commun 2009, 65, o460. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.