

Summary
Herpes simplex virus-2 infection is characterized by frequent episodic shedding in the genital tract. Expansion in HSV-2 viral load early during episodes is extremely rapid. However, the virus invariably peaks within 18 hours and is eliminated nearly as quickly. A critical feature of HSV-2 shedding episodes is their heterogeneity. Some episodes peak at 108 HSV DNA copies, last for weeks due to frequent viral re-expansion, and lead to painful ulcers, while others only reach 103 HSV DNA copies and are eliminated within hours and without symptoms. Within single micro-environments of infection, tissue-resident CD8+ T cells (TRM) appear to contain infection within a few days. Here, we review components of TRM biology relevant to immune-surveillance between HSV-2 shedding episodes and containment of infection upon detection of HSV-2 cognate antigen. We then describe the use of mathematical models to correlate large spatial gradients in TRM density with the heterogeneity of observed shedding within a single person. We describe how models have been leveraged for clinical trial simulation, as well as future plans to model the interactions of multiple cellular subtypes within mucosa, predict the mechanism of action of therapeutic vaccines, and describe the dynamics of three-dimensional infection environment during the natural evolution of an HSV-2 lesion.
Keywords: Tissue-resident T cells, HSV-2, Mathematical modeling, Viral dynamics, mucosal immunology
1. Introduction
Herpes simplex virus-2 (HSV-2) is an important human pathogen that is the leading cause of recurrent genital ulcers, a risk factor for enhanced HIV acquisition, and the occasional cause of severe disseminated disease (1). Partially effective antiviral therapies licensed over two decades ago limit the extent of recurrent ulcers, lower the clinical recurrence rate and genital shedding rate when given prophylactically (2–4), and diminish the risk of sexual transmission between infected persons when taken on a daily basis (5). However, the quest for licensure of an effective prophylactic vaccine has been expensive and thus far unsuccessful (6–9). Recent development of therapeutic vaccines that appear to be nearly as effective as licensed antiviral therapies has energized the field (10, 11). Moreover, new technologies applied in conjunction with creative study protocols have shed considerable light on the nature of the tissue-resident T cell response to reactivations during chronic HSV-2 (12–14). Nevertheless, it is clear that optimizing the development and dosing of future vaccines will require a deeper understanding of the tissue-specific immune response that underlies containment of HSV-2.
HSV-2 replicates in highly accessible mucosal sites of infection. This fact has allowed completion of perhaps the most comprehensive studies to date of interactions between the virus and tissue-resident host immune cells for a human viral pathogen. Our group has used the kinetics of viral shedding as a probe to better understand the timing and intensity of the endogenous immune response to HSV-2 (2, 15–25). We developed protocols employing highly frequent and spatially granular sampling for local viral load. These studies demonstrated that the local immune response against HSV-2 exerts significant pressure within 6-18 hours of viral detection (26, 27). However, HSV-2 reactivation within a single host follows a unique pattern, in which some shedding episodes are extremely rapidly contained within hours whereas others persist for more than a week. This observed variability in episode duration suggests that immunologic success fluctuates spatially and temporally within an infected individual (16, 19).
Because genital lesions and healing tissue can be safely and repeatedly biopsied, HSV-2 has proven to be an ideal platform for assessing the potential role of specific immunological factors in dictating episode heterogeneity. Detailed, serial analysis of infected human tissue has revealed a role for tissue resident CD8+ T cells (TRM) in limiting the duration of HSV-2 episodes (12, 13, 28). TRM are a subset of T-cells which remain lodged within solid tissues without recirculating in blood and provide in situ immunosurveillance at prior sites of infection (29–33). In human tissues, TRM can be differentiated from circulating effector memory T cells (TEM) by their cell surface markers, transcriptional profile (34), and micro-localization within tissue (12–14, 35).
Based on its replication in peripheral tissues and broad cellular and animal tropism, HSV-2 has emerged as a crucial tool for immunologists who develop murine systems to characterize the precise functions of TRM (33, 36–38). Recent evidence from experiments in mice highlights a critical role of TRM in determining the extent of pathogen spread and resultant tissue damage prior to recognition and clearance (39–41). TRM persist in tissue following pathogen clearance (42–44), actively patrol between target cells (45, 46), express dendritic arms to interface with a large number of possibly-infected cells (45, 46), proliferate locally (40, 47), express a rapid alarm signal upon contact with cognate antigen (44, 48), and can rapidly and cooperatively kill infected cells via T cell receptor mediated granzyme release (49). In human studies, TRM express gene signatures of T-cell activation months after pathogen elimination (14, 35). The multi-faceted immune-surveillance functionality of TRM raises the exciting possibility that these cells may be harnessed to provide rapid protection of barrier and non-lymphoid tissues following vaccination or immunotherapies.
Many questions regarding TRM activities during and after elimination of virally infected cells can only be answered with elegant transgenic murine systems in which tissue resident T cells can be generated, labelled, expanded and observed longitudinally in situ (40). While many of these mechanisms are likely to be fundamental to HSV-2 control in humans, it is currently extremely challenging to directly confirm their clinical relevance. A major priority in the field is to identify links between murine and human experimental systems, and to ultimately establish mechanistic correlates of vaccine protection following therapeutic vaccination (50).
Mathematical models are a useful technique for filling in some of these persistent gaps. Models allow the synthesis of available viral kinetics data with multiple qualitative features of the TRM response observed in humans. More detailed quantitative observations of TRM proliferation, killing and movement observed in mice can be imported into model equations to assess whether they improve or damage model fit to observed human viral load data. Differential equations are in fact essential to properly account for the non-linearity of pathogen-host interactions, as well as the stochastic factors inherent to early reactivation. When applied optimally, models are employed in an iterative fashion in close conjunction with experimental work. They are designed to help explain observed experimental data, but also to generate hypotheses which can be tested with subsequent experimental protocols.
In this review, we describe our research group’s efforts to use mathematical models as a method to infer the dynamics underling TRM control of HSV-2. Our modeling approach has generated numerous predictions, many of which have been experimentally verified in recent animal model and human studies. These predictions include the concepts that 1) immunologic control within the dorsal root ganglia is leaky, which allows persistent drip of infectious HSV-2 from neuronal cell bodies ganglia into the genital tract via neuronal connections (17); 2) TRM density at the precise site of reactivation dictates the extent of infection spread prior to local HSV-2 elimination (16, 20); 3) rapid local expansion of the TRM response leads to contraction in viral loads within 12-24 hours of mucosal reactivation (15, 19); 4) heterogeneous but highly organized TRM clustering is a feature of chronic infection; and 5) TRM pressure must be considered in order to accurately simulate clinical trials of small molecular antiviral agents (20, 51).
Our modeling approach also highlights key knowledge gaps in the field as well as challenges in studying mechanisms of viral pathogen containment in tissue. Because elimination of virally infected cells is never directly observed in real time in humans, the relative contributions of different cellular subsets including CD4+ T-cells, antigen presenting cells and natural killer cells are currently impossible to discern. This fundamental problem is exacerbated by the extremely rapid timeframes and microscopic distances over which infected cells are eliminated. While animal models allow a more detailed mechanistic understanding of TRM behavior, pathogen containment is still rarely observed directly. Moreover, for various reasons, many experimental models do not accurately recapitulate all aspects of human infection conditions. Mathematical models are essential to capture the non-linear spatiotemporal features of TRM-pathogen interface. Yet even the most complex models dramatically oversimplify the infection micro-environment and invariably do not include key features of the host response. For these reasons, we suggest that continued progress in the field requires tight synergy between human studies, animal models and theoretical modeling approaches.
2. HSV-2 pathogenesis
HSV-2 is a sexually transmitted infection. Primary infection is initiated during a sexual act when the transmitting partner is shedding virus, often in an asymptomatic fashion (52). Virus replication initiates in genital keratinocytes and may spread to thousands of cells. While not all transmissions are symptomatic, development of painful persistent lesions requiring medical attention is the sine qua non, of symptomatic primary infection. Virus invariably enters neuron endings and takes a remarkable journey via single neurons to the dorsal root ganglia where lifelong latency is established (53).
Virus periodically reactivates within the ganglia and travels back down the neuron leading to either asymptomatic, low titer shedding or less commonly recurrent ulcers which are typically associated with prolonged high titer shedding (2, 22, 23, 25). The genital tract shedding rate and lesion rate are highest during the first year after primary infection but generally stabilize thereafter (54), signaling a détente of sorts between the virus and host immune system. Transmission to new partners is efficient based on the high frequency of asymptomatic and therefore unnoticed shedding, the lifelong nature of infection and the high shedding rate (5, 55).
Cellular immunity is vital for containing infection. Patients with severe T-cell immunodeficiency including those with AIDS or who have received immunosuppression for stem cell transplantation or solid organ transplantation are at risk of severe, persistent localized genital disease, development of drug resistance and systemic spread (56, 57). The critical aspects of the cellular immune response are measured in genital mucosal tissue and sites of latency (dorsal root ganglia) rather than blood (12–14, 28). While generation of neutralizing antibodies is believed to be a critical component of any prophylactic vaccine, humoral immune deficiency does not correlate with more severe disease. Mouse models of genital HSV-2 infection further suggest that T cell immunity is more important than humoral immunity for viral control (58, 59).
2.1. Genital shedding as a probe of virus-host interactions
Over the last 20 years, over a thousand study participants have participated in research studies at the University of Washington which have allowed detailed characterization of genital HSV-2 shedding kinetics. Typically study participants perform longitudinal self-genital swabs at home and return these samples to the clinic. This method is as reliable, and more efficient, than clinician sampling (60). Swabs are subsequently processed for quantitative HSV-2 PCR which provides a measure of the burden of viral replication.
Individual studies vary according to study population, intervention, sampling location and sampling frequencies. Collectively, these studies have allowed us to precisely define the kinetic patterns of HSV-2 shedding in the genital tract. Genital shedding rate (percentage of swabs with detectable HSV-2 DNA) and lesion rate are the most relevant outcomes for clinical trials and vary across infected persons (61, 62). To better understand the basic pathogenesis of infection, we developed more detailed quantitative methods to characterize viral expansion and clearance (18, 19, 21). Our approach provides a rough estimation of the timing of the immune response against the virus over various temporal and spatial scales.
2.2. Episodic genital HSV-2 shedding implies local immunologic clearance of episodes.
Our most comprehensive analysis of HSV-2 shedding kinetics to date was from 531 immunocompetent study participants who donated 30-60 daily swabs for analysis. Several critical patterns from prior studies were confirmed. First, HSV-2 shedding is episodic with an annualized episode rate of 17.2, signifying a high frequency of HSV-2 reactivation from the ganglia leading to replication in the genital mucosa (21). Second, in a majority of immunocompetent persons, each episode terminates, which suggests that despite the high frequency of recurrent battles in the genital mucosa, the immune system ultimately wins each battle in a majority of immunocompetent persons. However, it can be argued that the virus wins the war, given the high overall rate of shedding (~12-18% of swabs positive for HSV-2 depending on the study) that facilitates efficient transmission and development of multiple recurrences per year in many infected people (2, 3, 21–25, 27, 62, 63).
2.3. Variable effectiveness of immunologic clearance of HSV-2 within single infected persons over narrow time intervals.
A critical feature of HSV-2 shedding episodes is their heterogeneity (Fig 1a), which we discovered by developing quantitative tools to characterize and compare multiple features of HSV-2 shedding episodes. Specifically, we used frequency histograms to capture variability in episode characteristics such as peak HSV DNA viral load and duration. Episode peak viral load varied from 102-109 HSV DNA copies with the highest-copy, most severe episodes typically associated with prolonged duration of 5-14 days and development of ulcers (Fig 1b,c) (21). A higher proportion of episodes (~40%) were moderate in severity, peaking between 10,000 and 1,000,000 HSV DNA copies, often lasting 1-3 days. We consider the immune response during these moderate episodes to be partially effective: virus is usually eliminated prior to reaching a high enough burden of killed epithelial cells to cause symptomatic lesions. On the other hand, the high overall frequency, relatively prolonged shedding phase, and higher viral loads of these episodes likely contribute to highly efficient transmissibility of the virus between sexual partners (55).
Figure 1. Herpes simplex virus-2 genital shedding episodes are notable for extraordinary heterogeneity in terms of peak viral load and duration.
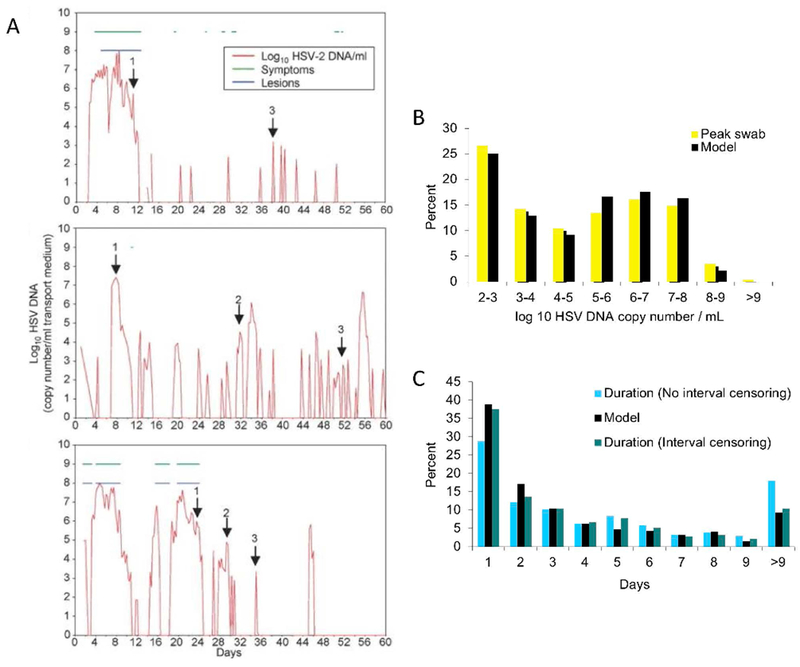
(A) Individual shedding trajectories from 3 study participants who self-sampled every 6 hours over 60 days. Arrows indicate high (1), medium (2) and low (3) copy episodes. Data is from reference 27. (B & C) Data from 1020 shedding episodes in 531 study participants (figure adapted from references 19 and 21) who performed daily self-sampling of the anogenital region for 30-60 days. Colored bars are actual data and black bars are from optimized simulations of the mathematical model in Figure 5. (B) Frequency histogram of observed episode peak viral load. (C) Frequency histogram of observed episode duration. Interval censoring means that episodes which were not observed completely (i.e. initiated before sampling began or ended after sampling terminated) are excluded. It is notable that the cohort described in B and C is likely overestimating median peak viral load and duration because brief episodes lasting less than 12 hours which compromise ~50% of observed episodes are missed with daily sampling.
The critical role of the local immune response in episode heterogeneity was further confirmed in studies of immunocompetent participants who performed self-sampling every 6 hours for HSV-2 DNA. In these studies, over 50% of episodes peaked at only 100 to 10,000 copies, lasted fewer than 12 hours, and were asymptomatic (27). These results demonstrate that local immune cells are often capable of extremely rapid recognition and elimination of virally infected cells.
The most extraordinary feature of the extreme variability in HSV-2 shedding episode severity is that this variability is observed across study participants, but also within the same study participants over the short time window of a couple weeks (Fig 1a). It is a signature feature of HSV-2 infection that instances of relative immunologic success and failure occur commonly within the same person over time. For nearly all human pathogens, including HSV-2, a major focus of clinical and immunologic research is to develop a better understanding of what drives differences in disease severity or manifestations between persons by identifying viral and host-related correlates of protection from infection or disease (64). Indeed, HSV-2 infection is characterized by “low” and “high” shedders, the latter of whom is most likely to experience frequent and bothersome disease manifestations (23, 63). In the case of HSV-2, similarly important comparisons must also be made within a single infected person.
2.4. Early potent activity of the local anti-HSV-2 immune response regardless of episode severity.
We used our frequency histograms to describe variability in the first detected value of HSV DNA genomic copies per episode and identified a wide range of viral loads (102-108 HSV DNA genomic copies) (19, 21). This heterogeneity reflects in part the age of the episode at detection (which might vary from 0-24 hours), but also the variability in severity of the episode itself. The fact that HSV-2 viral loads can reach 108 genomic copies in the first 24 hours of an episode reflects extraordinarily high rates of replication and spread and is a signal that the tissue resident immune response must be poised, spatially and functionally, to achieve immediate efficacy upon re-exposure to HSV-2.
Remarkably nearly all of the episodes sampled every 6 hours in our database peak and enter a viral clearance phase within 18 hours of viral detection, implying very quick recognition of, and response to, reactivating infection (27). Accordingly, local and systemic sequelae related to HSV-2 reactivation during chronic infection are less severe relative to those occurring during primary HSV-2 infection (1). The rapid kinetic pattern of HSV-2 reactivations demonstrates that the classical T cell memory recall response, which takes several days to evolve in lymphatic tissue and become detectable in blood (65), is unlikely to be relevant for containment of infection within tissue. If HSV-2 spread was left completely unchecked for several days in genital mucosa, the likely result would be massive disfiguring ulcers and systemic spread of virus.
The last detected viral load during episodes also varies over wide ranges reflecting the rapidity of viral elimination. The last detected viral load is on average lower than the first detected value, meaning that the late stage of viral clearance is slightly slower than the early phase of expansion. Nevertheless, a 3-4 log decrease in viral load over 24 hours is commonly observed as episodes terminate, reflecting the intensity of the local immune response (19, 21).
Individual shedding episodes are notable for complex HSV-2 dynamics, which likely reflect viral host interactions. We designed protocols in which participants collected samples every two hours during symptomatic genital HSV-2 lesions (19). A surprising finding from these studies was that shedding episodes lasting for more than two days nearly always consisted of multiple viral load peaks (Fig 2a). Specifically, episodes typically exhibit a very rapid early expansion phase: the first peak typically occurs within 6 to 18-hours of viral detection. This is followed by a period of non-monotonic viral contraction, and then another period of expansion to a second peak, usually at a lower viral load. To ensure that this observation is biologically valid and not due to variability in sampling or laboratory methods, we performed a study in which participants self-sampled every 5 minutes for 4 hours: extremely stable viral loads were noted implying that fluctuating levels over 2-hour intervals reflect the true biology of the disease (19). Thus, the observed rebound in viral load during shedding episodes must ultimately be explained on immunologic grounds.
Figure 2. Prolonged HSV-2 shedding episodes display temporal and spatial complexity.
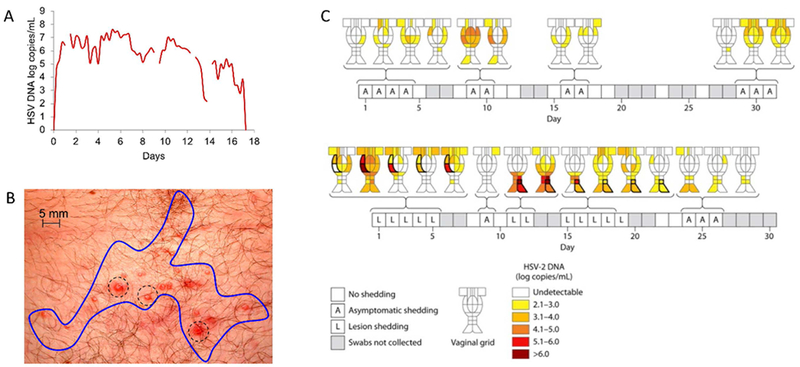
(A) An example of a 17-day HSV-2 shedding episodes with multiple peaks (adapted from reference 19). In this study, participants sampled every 6 hours. A typical early peak and transition to viral clearance is noted, as are multiple peaks and re-expansion phases. Missing lines indicate missed samples. (B) Spatial dispersion of individual vesicles (dotted circles) typically observed during an HSV-2 recurrence. Each vesicle represents a single viral-immune battlefield of infection. The blue line circumscribes the whole lesion which is typically sampled in its entirety in shedding protocols. (C) Spatial heterogeneity in viral load noted in a participant who had 23 regions of the genital tract sampled separately on a daily basis. This result highlights that HSV-2 reactivations may consist of multiple concurrent viral immune battles with differing outcomes in terms of peak regional viral load. Certain reactivations occur in single regions, are asymptomatic, and achieve low viral loads. Other reactivations are associated with lesions, achieve higher viral loads and involve multiple regions. Data is adapted from reference 67.
2.5. Shedding characteristics are shared across immunocompromised hosts.
It is widely recognized that persons with impaired T cell function shed virus more frequently and at higher levels, and have greater risk of severe persistent localized disease, systemic spread and development of drug resistance (56, 66). We applied the detailed quantitative tools described above to HIV-infected cohorts and noted a dose response of shedding severity according to CD4+ T-cell depletion. HIV infected persons with CD4+ T cell counts greater than 500/uL of blood shed at the same rate and quantity as persons without HIV. HIV infected persons with CD4+ T cell counts <200/uL shed at higher rates and with higher quantities (66). Surprisingly, the sole driver of this effect was a higher frequency of shedding episodes. Specific episode characteristics including duration, peak viral load, expansion rate and clearance rate were equivalent between HIV infected and uninfected persons. This unexpected finding may imply that certain features of virus specific immunity are preserved while others are lost during prolonged untreated HIV infection.
2.6. Spatial heterogeneity in mucosal HSV-2 viral load across spatial gradients.
HSV-2 genital lesions consist of single or multiple individual skin erosions which form and heal sequentially during clinical recurrences (Fig 2b). To account for the fact that shedding may also be similarly spatially dispersed, a landmark study was performed at the University of Washington in which several study participants were sequentially sampled at 23 separate micro-environments on a daily basis for one month (Fig 2c) (67). A key result was that during high copy shedding episodes, the virus is often shed concurrently at multiple locations across the genital tract, with heterogeneous levels noted across time and space. Brief, more rapidly controlled episodes were often contained at lower viral loads within single mucosal locations but appeared to initiate in multiple non-adjacent regions over longer periods of sampling. The critical message for considering immunity is that there is also likely to be extraordinary heterogeneity across genital mucosa and skin in terms of the intensity and effectiveness of the cellular immune response. Moreover, concurrent viral-host interactions are likely to be occurring at dozens of micro-anatomic sites during large reactivations. When developing mathematical models of this system, it is critical to specify whether a single battlefield, or the entire, war, are being described.
3. Tissue-resident T cells during HSV-2 infection.
Tissue resident T cells are a subset of T cells that are defined by their permanent establishment in tissues throughout the body, without recirculation in blood (29, 33). This concept of permanent tissue lodgement has been defined using parabiosis models in mice (68). While the study of human TRM does not benefit from such elegant experimental systems, phenotypic markers as well as defined tissue localization has allowed for parallel study of human TRM in various mucosal tissues. Specifically, human TRM can be defined with varying degrees of specificity according to cell-surface markers, transcriptional profiling, and lack of observed recirculation (32, 34, 69–71).
It is increasingly clear that tissue-resident T-cells are established in nearly every organ in the body (70, 72). Their importance is particularly crucial at mucosal portals of entry such as the gastro-intestinal tract, oral mucosa and reproductive tract. Tissue-resident T cells are also predictably present in tissues where chronic reactivating viruses — such as cytomegalovirus, Epstein-Barr virus and HSV — reside (13, 73, 74). It is believed that the number of tissue-resident T cells within a barrier tissue reflects whether exposure to a known pathogen has previously occurred. Excess or aberrant tissue resident T cells may lead to autoimmune diseases such as psoriasis (75), necessitating mechanisms of restraint or regulation of these TRM responses, perhaps in the form of tissue regulatory T cells (Treg) (76, 77). For each of these infections or conditions, it is clear that measuring attributes of infection response in circulating T cells is a poor surrogate for directly assessing tissue-resident T cells (78).
Recent estimates suggest that the total proportion of T cells within murine tissue that are tissue resident exceed 80% in all organs except lymph nodes, the liver and the spleen, with over 95% of T cells displaying markers of tissue residence in female genital tract organs (72). Similar proportions are noted from human organ donor studies in which 97% of CD8+ T cells in the colon and 73% in the lung were either resident effector memory or resident central memory phenotype (69). Distributions of T cell subsets and homeostatic proliferation patterns of tissue resident cells appears to be organ specific (70). The proportion of these cells is likely to be lower in infants who have yet to undergo exposure to numerous commensals and pathogens.
For the study of chronic HSV-2 infection, we are most focused on the immunosurveillance and pathogen clearance capabilities of tissue-resident T cells. Immunosurveillance functions include local trafficking within small micro-environments of infection (45, 46), rapid activation in the case of antigen re-exposure (44, 48), and a prolonged lifespan in tissue (12, 13). During clearance of infected cells, tissue-resident T cells acts as sentinels which detect the nearby presence of infected cells, proliferate locally (40, 47), release alarm signals to rapidly induce regional resistance to spread of infection (44, 48), and directly kill infected cells expressing the appropriate cognate antigen (33).
3.1. Characterization of tissue residence in T-cells.
Many subsets of T cells achieve tissue residence, including both CD4+ T cells and CD8+ T cells. Tissue resident CD4+ T cells may be further sub-classified according to subsets including T-regulatory cells (77, 79, 80). For the purpose of this review, we focus on tissue-resident CD8+ T cells (TRM) because these cells localize precisely to the site of HSV-2 replication and have known potent antiviral effects (13, 14, 28). This is not meant to imply that tissue-resident CD4+ T-cells do not play a vital role in elimination of infected cells, but rather to reflect that quantitative data describing the interactions between CD4+ and CD8+ tissue-resident T-cells during a local immune response is currently too limited for inclusion in mathematical models.
The most fundamental feature of TRM is their sessile phenotype. The rarity of TRM recirculation was established in parabiosis studies, in which the circulatory systems of two mice are joined to allow even mixing of circulating T cells: tellingly, T cells in peripheral tissues equilibrate to a much lesser extent due to their lack of recirculation (68). Labelling experiments demonstrate that persistence of this population does not depend on contribution from circulating T cells (40, 47). In humans, donor tissue-resident T cells with tissue-resident markers were noted in a biopsy following a facial transplantation (71); these results replicated similar results in murine tissue-graft experiments (39, 81). Moreover, in patients with cutaneous T cell lymphomas who received an anti CD52 antibody, there was subsequent depletion of all T cells in blood but tissue-resident T cells were spared, presumably due to their lack of recirculation (32).
Identification of cell-surface markers allow a less-labor intensive approach to establish the presence of TRM, particularly in human immunology studies. Fortunately, markers such as CD69, CD103 and CD49a are consistent across mice and humans, and can be ascertained by flow cytometry (69, 70, 74). Less auspiciously, the presence of these markers is not entirely sensitive or specific for tissue-residence, and their expression within TRM may vary from organ to organ (72). Notably, these cell surface markers are linked to functional attributes of the cells such as CD103 tethering to epithelial cells, as well as responsiveness to the local signaling milieu, including TGF-β, which promotes CD103 upregulation (42). Particularly in genital tissue, cells with these markers often cluster in the presence of antigen presenting cells (12). Transcriptional signatures can also be used as a surrogate to identify TRM cells across tissues and species (34). Detailed methods for identification of TRM have been comprehensively reviewed elsewhere by Gebhardt et al (31).
3.2. Mechanisms of rapid pathogen clearance mediated by tissue resident T-cells in genital tissue
By virtue of its natural replication in external anatomic sites such as skin and mucosa, HSV-2 has proven to be an ideal model pathogen for interrogating the potent and varied antiviral mechanisms of TRM (39, 40, 82, 83). Mouse models have been essential for delineating these mechanisms, many of which can only be measured indirectly in humans. Mathematical models can assist by linking mechanisms observed in mice with the available human data, thereby forming the third, in silico arm of the trifecta of approaches used to study mechanisms of immune control of HSV-2.
The most important HSV-2 clearance mechanisms studied to date include in situ proliferation, rapid and cooperative TCR-mediated killing, and generation of a tissue wide alarm response that in many ways resembles an innate response. Two recent papers, one using HSV challenge, utilized elegant labelling and intravital imaging techniques to demonstrate that the massive local surge in TRM during the first three days of pathogen elimination is almost entirely due to local TRM proliferation (Fig 3a), rather than off site generation of new cells capable of migrating to tissue sites (40, 47). This timeframe is consistent with the observed interval to rapid viral disappearance from local sites of infection during chronic human HSV-2. Trafficking of effector memory cells to the inflamed site trails local TRM proliferation and occurs at least four days after antigen presentation (40, 47).
Figure 3. Pathogen containment features of TRM include local proliferation, rapid cooperative killing of infected cells, and generation of a tissue-wide antiviral state.
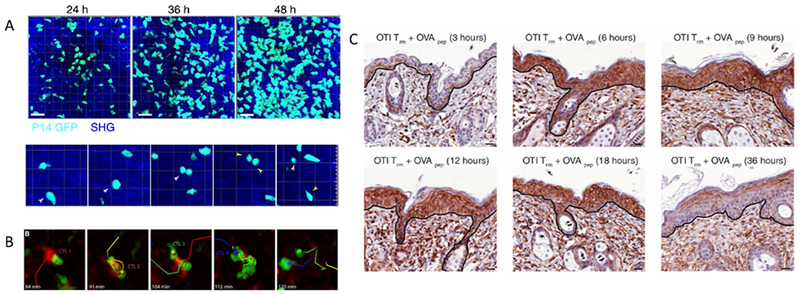
(A) Intravital image evidence of in situ proliferation of TRM in the murine cervix following challenge with gp33 peptide (reference 47). The top panels show accumulation of TRM (cyan) over 48 hours while the bottom shows an actual cell division event: a single cell denoted with a yellow arrow divides into two daughter cells (yellow arrows). Local proliferation was also confirmed with BrdU and Ki67+ labelling. (B) Intravital image evidence of rapid, cooperative killing of a single murine-cytomegalovirus infected cell (red) by multiple cytotoxic lymphocytes (green). In these experiments, killing occurred in a median time of 40 minutes and was more rapid in the presence of multiple CTL (reference 49) (C) Immunohistochemical evidence of a tissue-wide antiviral cytokine response. TRM specific for the OVA peptide were generated in skin followed by OVA challenge. Surrounding epidermal cells intensely express interferon-induced transmembrane protein 3 (brown staining), an antiviral protein, within 6 hours, indicating a tissue-wide state of protection against equivalent or differing virus infection (reference 48).
The in vivo killing rate of HSV-2 infected cells by TRM is unknown in humans because the relative contribution of other features of the local immune response such as antibody-dependent cytotoxicity, neutralizing antibodies, natural killer cells and other innate cells cannot be directly measured. Moreover, the indirect killing rate afforded by bystander TRM is also likely to be important but cannot be directly quantified (84). Nevertheless, experiments which show killing of infected cells within 20 minutes of antigen presentation, as well as cooperative killing of a single cell by two adjoining TRM (Fig 3b), are vital for developing model assumptions (49).
The sense and alarm response utilized by TRM is an astonishing feature of these cell populations. A TRM with the appropriate TCR to recognize a viral peptide is capable of initiating the local immune response (Fig 3c), possibly prior to recognition of the viral pathogen by innate sensors. This cell promotes a rapidly diffusing cytokine response that lowers infectivity towards surrounding uninfected cells, limits replication in infected cells, and activates local innate cells and bystander TRM (44, 48), which in effect function like innate cells by amplifying the cytokine response despite lack of specificity of the TCR for an HSV-2 epitope. These mechanisms are likely to underlie the rapidity of the TRM response and may explain how a rapidly expanding virus like HSV-2 can be contained despite a low initial precursor frequency of pathogen-specific TRM at the site of reactivation and shedding (13, 16, 19, 26, 27).
When developing mathematical models of chronic, established HSV-2 infection, certain features of TRM behavior, such as their initial establishment during primary infection, are not explicitly captured. Although mechanistic details remain to be elucidated, the general developmental pathway of TRM formation is well established and includes antigen stimulation of naïve T cells in lymph nodes, activation and clonal expansion of these cells, infiltration of infected tissue sites, downregulation of functional markers of tissue egress and establishment of a TRM phenotype (42, 85). These steps would need to be formally included in any viral dynamic model of primary HSV-2infection: unfortunately, there is no available temporo-spatial data describing viral or TRM dynamics during primary HSV-2 infection.
3.3. Immunosurveillance mechanisms of tissue resident T-cells in genital tissue
The vast majority of a TRM’s lifespan will be spent in waiting, rather than in the heat of battle. However, the transcriptional profiles of TRM do not suggest a fully quiescent state. In human genital biopsies obtained many weeks after viral elimination and tissue healing, perforin granules are present in TRM (Fig 4a) and the cellular transcriptional profile is notable for upregulation of activation, cytolysis and inflammation pathways (14, 35) (Fig 4b).
Figure 4. Immunosurveillance features of TRM include prolonged tissue residence, secretion of cytotoxic molecules, an activated transcriptional profile, intra-epithelial trafficking and self-organizational behavior.
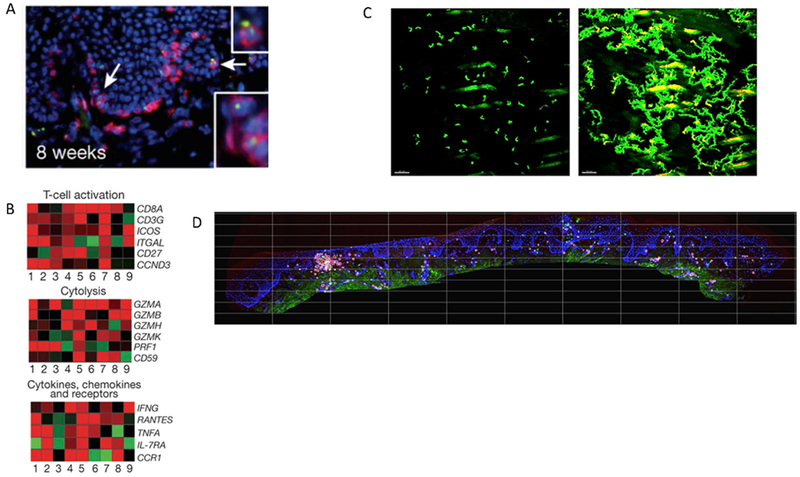
(A) Immunohistochemical evidence of perforin granules (green) within multiple tissue-resident CD8+ T cells from human genital skin biopsies at a timepoint 8 weeks post lesion healing when HSV-2 antigen is not detected in tissue (reference 14) (B) Transcriptional evidence of an activated state in CD8+ T cells from human genital skin biopsies at a timepoint 8 weeks post healing when HSV-2 antigen is not detected in tissue. Gene expression is compared between CD8+ T cells detected at the dermal-epidermal junction (presumed TRM) versus control cells from the same individual with red and green indicating up and down regulated genes respectively (reference 14) (C) Intravital imaging evidence that TRM express dendritic arms and patrol extensively within but not beyond local micro-environment (reference 46). The left panel is an initial frame of a time-lapse movie in which the branching nature of TRM in murine flank skin is evident. The right panel is superimposed frames taken every 3 minutes over 7.5 hours showing that a majority of the local tissue region is patrolled. However, over more than a year, cells rarely migrate more than 5 mm from their starting location. (D) Immunohistochemistry evidence that TRM demonstrate self-organizational behavior in human genital skin with clustering of CD8+ T cells at the dermal epidermal junction. We determined that the ratio of CD8+ T cells to epithelial cells within the square regions follows an exponential slope on a rank order distribution, indicating a strict slope to CD8+ T cell structure in tissue.
TRM also remain clustered at the dermal-epidermal junction, which suggests that these cells manifest a self-organizational capacity. Elegant murine investigations in concert with theoretical approaches demonstrate that TRM are in constant motion, patrolling between epithelial cells while projecting dendritic arms to contact as many cells as possible (45, 46) (Fig 4c). Despite this surveilling phenotype, TRM remain within a 1-2 mm neighborhood for months rather than diffusing evenly across at-risk tissue and cease trafficking upon antigen recognition (40, 45–47). This observation in mice helps explain observed aggregation patterns of TRM in human biopsies: certain areas are notable for high density clusters of cells while nearby regions may only have tissue resident T cells at a ratio of 1 to 1000 relative to epithelial target cells (Fig 4d). If TRM were moving about freely without any signaling mechanism for retention in their original site, then it is mathematically projected that a more homogeneous dispersion of TRM would result. Given the presence of other interacting immune cells such as dendritic cells and CD4+ T helper cells within or close to these clusters of cells (33, 83), it is possible that these clusters represent quasi-organelles akin to gastro-intestinal-associated lymphoid tissues, or GALT.
Ultimately the number and spacing of TRM in tissue is a key parameter of protection upon HSV re-exposure. In mouse model experiments, there is an inverse relationship between total genital tract TRM density and subsequent peak viral titers upon challenge with HSV-2 (39, 40). Another study identified that a parenteral prime and topical chemokine pull strategy can draw circulating effector memory T cells permanently into tissue-residence within the genital mucosa. Interestingly, this strategy is lifesaving for the mice prior to HSV-2 challenge despite similar viral dynamics in the control and experimental groups (83). In humans, the role of tissue resident T cells in HSV-2 containment is circumstantial but strongly suggestive (12, 13, 16).
3.4. Tissue resident CD4+ and CD8+ T-cells in HSV latently infected ganglia
The idea of true and complete HSV-2 latency within dorsal root ganglia has been largely rejected. Rather, the site of latent HSV-2 infection appears to be a highly dynamic viral immune environment. The traditional criticism of murine HSV-2 models is that the virus does not reactivate spontaneously in these animals. Nevertheless, studies demonstrate that a high proportion of infected neuronal cell bodies in mice contain transcriptionally active HSV (86, 87). Sessile CD8+ T cells are retained in murine ganglia; interestingly, these cells produce granzyme, which inhibits HSV-2 reactivation without leading to cell body damage (88). A higher number of these T cells has been shown to decrease the frequency of reactivation in these tissues (89, 90).
Human studies are more limited in their ability to identify mechanisms of T cell immunity in ganglia but are consistent with an active role for these cells. In trigeminal ganglia, HSV-1 specific CD4+ and CD8+ T cells cluster around HSV-2 infected neurons (73, 91). While similar studies have not been performed for HSV-2 in dorsal root ganglia, T-cell deficiency is associated with a higher episode rate of HSV-2, suggesting that T cells are critical to immune control within the dorsal root ganglia (66).
3.5. Host-pathogen equilibrium during chronic HSV-2 infection
Given the explosive interactions between infected cells and tissue resident T cells during mucosal reactivations and the high variability in shedding episode severity, perhaps the most remarkable feature of chronic infection is its stability over longer time scales. HSV-2 shedding rates equilibrate to a steady state after the first year of infection and remain quite stable over subsequent decades (54). HSV-2 shedding continues at levels and frequencies sufficient for high per coital transmission rates (55, 92). While a severe nuisance for many people, chronic HSV-2 rarely impacts human lifespan and is most often shed asymptomatically. There is also little evidence that an over exuberant tissue resident T cell response to HSV-2 contributes to immunopathogenesis effects. Tissue-resident T cell responses appear to be precisely regulated, perhaps by local Tregs (76), to ensure HSV-2 elimination during every single reactivation well before there is risk of systemic spread, while not inducing excess tissue damage.
4. Mathematical modeling of viral dynamics
Viral dynamic modeling is a discipline within applied mathematics which was created out of necessity to account for non-linearities inherent to viral load and immunologic data during infections. These models also differ fundamentally from statistical modeling approaches in that model equations have mechanistic assumptions. Often, models with competing hypotheses are compared in silico for fit to the quantitative data and for general consistency with qualitative data. Under ideal circumstances, theoretical modeling is paired tightly with experimental work to allow iterative hypothesis development and testing.
4.1. Challenges and limitations of viral dynamic modeling.
The “art” of viral dynamic modeling involves the design of sets of equations that are just complex enough to answer the proposed scientific question given a certain set of available data, but no more so. Mathematically, models that contain too many equations and free parameters lack analytic solutions. Even in models that use simulation techniques, issues of model and parameter identifiability invariably arise (93). The most complex models may contain thousands of equations and require weeks to simulate despite parallel computing: interpretation of such models is extraordinarily complicated and lack of reproducibility may be an issue. On the other hand, an overly simplistic model is not biologically informative. Even the most useful viral dynamic models predictably underestimate the extraordinary complexity underlying the virus-host interactions which determine the outcomes of human infections.
The utility of mathematical modeling is ultimately predicated on the availability of serial, quantitative data to confirm, alter or altogether reject model developed hypotheses. It is impossible to gather every piece of relevant data to completely inform model assumptions. Ideally, for every modeling problem, there would be available molecular, cellular and host-level data measured across sufficiently granular temporal and spatial scales. In reality, this data is often only partially available. Moreover, very subjective decisions need to be made regarding whether observations or parameter values from in vitro or animal models can be applied to human models.
The nature of available data therefore dictates the types of questions that can be answered, as well as the precision with which a mathematical model can make mechanistically verifiable predictions. On one end of the spectrum, carefully designed in vitro experiments can provide experimental data which can often be modelled with high accuracy. As an example, dose response curves between a drug and its target (or between effector T cells and their targets) allow models which provide a nearly complete mechanistic interpretation of the experiment: this approach has been used to directly classify the potency of nearly all HIV antiviral agents (94).
Animal models of infection and immune response allow control over key experimental variables. For instance, well-defined murine systems permit immune cell types of interest to be followed at reasonably narrow intervals within multiple organ systems. Many papers have used these systems to estimate key parameters such as CD8+ T-cell proliferation rates, elimination rates of infected cells and within-tissue trafficking rates (95–97). Nevertheless, there are limits to the degree of control that can be exerted within animal systems. Heterogeneity of infection (even within inbred colonies), limitations in sampling, and most critically, unmeasured critical driver variables, can limit a model’s ability to make predictions. For instance, no standard model seems to reliably describe early SIV dynamics in non-human primates (98).
At the other extreme is the most relevant data, which is obtained directly from human infection. However, the challenges related to this data are substantial. Often, available data for modeling is derived from purely observational studies which were not originally performed with modeling in mind. In this case, the availability of limited samples may severely impede the ability of a model to derive mechanistic interpretations. Even if there is strategically gathered data from a clinical trial in which sampling is adequate and key variables are controlled, the issue of critical missing data remains. This is particularly germane to measurements of the immune response, which is mediated by multiple coordinating cell types and complex interactions in tissues which are not easily assessed longitudinally in infected persons.
These limitations are most evident in the richest area of viral dynamic modeling, HIV infection. Despite a rich 25-year history of HIV modeling which has contributed enormously to the survival of infected patients and to our bedrock of knowledge regarding HIV pathogenesis (described below), the equations typically used to model cytolytic anti-HIV immunity remain strikingly coarse, describing cytolytic immune pressure in the broadest sense only. Due to lack of relevant data, most existing models appropriately do not consider the anatomic location or cellular subtypes involved in the HIV response. Rather, models are usually limited to one or two equations describing the expansion, contraction and killing rates of the bulk cell-mediated response (99). Even more sophisticated models that consider concepts such as neutralizing antibody generation and T-cell exhaustion (100), do so in a speculative and phenomenological fashion, owing to the lack of specific, relevant data for model fitting to these variables. In general, available models are able to compare the global assessment of the total cytolytic response across patients. The nature of existing data for HSV-2 infection is slowly allowing us to develop more sophisticated models to characterize the antiviral immune response.
4.2. Highlights of viral dynamic and immune modeling for infections other than HSV-2.
Despite these limitations, modeling has resulted in foundational conclusions regarding pathogenesis of chronic viral diseases, particularly HIV, and has had a major impact on the development of the most important clinical intervention to date, antiretroviral therapy. The original viral dynamic model was applied to bi-phasic HIV clearance data in two seminal papers during antiretroviral therapy (101, 102). This model is notable for its extreme simplicity and also for its obviously false assumption of no underlying immunologic response. This approach allowed linearization of the model and provided the fundamental insight that during viral load steady state, infected cells are replaced on a nearly daily basis while free viruses are replaced in the serum on a time scale of hours (103, 104). Given that the viral population size is enormous during untreated infection (~1011 viruses generated per day) and the error rate of the HIV reverse transcriptase is extremely high, this rapid rate of infected cell turnover guarantees the appearance of, and selection for, drug resistance to monotherapy. Strikingly, this extremely simple model was central to the concept of multi-drug therapy to avoid drug resistance.
Subsequent models have provided innumerable insights into very fundamental aspects of infection. Examination of HIV-specific CD8+ T-cell clone kinetics, as well as patient-specific viral dynamics (105), has allowed precise specification of the impact of the immune response versus target cell limitation on determining HIV peak viral load and set point. An entire discipline is dedicated to phylodynamic approaches to estimate the average rate of HIV diversification and immune escape (106). Mathematical models have helped define the complex dose response curves of all available HIV agents, and in turn can predict combinations of drugs which are most likely to completely eliminate infection of new cells without allowing emergence of drug resistant strains (107). More recent models focused on HIV cure established the rate and mechanisms underlying the establishment and maintenance of a population of persistent latently infected cells despite decades of antiretroviral therapy (108). In the near future, possible curative or prevention approaches will extend to include immunotherapies such as therapeutic vaccines, neutralizing antibodies, synthetic antiviral peptides, and CAR T cells, as well as gene therapies such as vector-expressed DNA cleavage enzymes and stem cell transplant with gene modified HIV resistant cells. Novel pharmacokinetic and pharmacodynamic models will be vital for dose optimization of each of these new strategies (109). While the most mature versions of these models currently exist only for HIV infection, they will undoubtedly become highly relevant for HSV-2 infection in the years to come.
4.3. Rationale for mathematical modeling of HSV-2.
In 2008, our group elected to develop mathematical models of HSV-2 for two broad reasons. First, the necessary conditions for developing reasonable models for HSV-2 shedding were in place. As described above, detailed clinical evaluations of HSV-2 shedding kinetics in humans had been performed, consisting of serial quantitative viral load measurements. Viral kinetics were clearly non-linear in nearly all study participants, suggesting the need for coupled differential equations to fully explain our empirical observations (21). Moreover, a qualitative understanding of the tissue-resident immune response was also beginning to emerge.
Second, the field was teeming with unanswered, extremely basic questions, some of which we are still attempting to address today. Why is HSV-2 shedding episodic? Why are episodes so heterogeneous in viral load and duration? What explains the different shedding patterns observed between infected persons? Why are current antiviral therapies, which appear so potent in vitro, only partially suppressive in the clinic despite lack of apparent drug resistance in most treated persons? Why are TRM generated during repeated exposure to the virus not fully protective against all reactivations? What accounts for the wide spatial distribution of TRM densities across small distances in the genital tract? If HSV-2 initiated replication in a region with very few T cells, then how can the virus still be eliminated? Is local T cell amplification during infection due to local proliferation of TRM or trafficking of effector memory cells from blood? How long will TRM remain in place following natural infection and vaccination, and can they be expected to traffic to other adjacent mucosal regions to allow a broader field of protection? How are mouse models of infection relevant, and how are they different from infection in humans? How might different subsets of tissue resident T cells and antigen presenting cells coordinate a response to infection? Is it necessary for all T cells in tissue to be specific for the virus, or are bystander T cells helpful for clearance of virus?
These questions share a common thread in that they all are directly or indirectly related to viral and TRM dynamics. Accordingly, modeling can be useful, if not mandatory, to focus these questions and develop hypotheses which can be tested in murine and human experiments.
4.4. Unique challenges in developing useful models of HSV-2 infection.
There are several features of HSV-2 pathogenesis which present a unique set of problems for modeling HSV-2 relative to other chronic viral infections. First, individual HSV-2 shedding data is not amenable to traditional deterministic mathematical model data fitting. Viruses such as HIV, hepatitis B and hepatitis C follow relatively simple bi-exponential decay trajectories during therapy (104, 110, 111), as well as exponential expansion and achievement of a steady-state during initial infection (112, 113). HSV-2 kinetics are far more complex. The timing of episode takeoff is completely unpredictable (114). Moreover, as described above, our study protocols that employed sampling every two-hours during shedding episodes revealed very frequent and erratic re-expansion events. Many of the most clinically relevant episodes have multiple peaks of various height and width. This complex kinetics profile precludes recapitulating the data with a simplified set of deterministic differential equations, without making numerous unproven assumptions and ultimately over-fitting to the data. In our hands, it is not currently possible to recapitulate shedding data within individuals through modeling. As an alternative, we have employed stochastic differential equation models which allow for random episode initiation as well as highly heterogeneous episodes containing multiple peaks (17). The result is a population-level model which is useful for describing broadly relevant trends in HSV-2 biology, but not in differentiating between individuals, or predicting shedding within a single person.
A second challenge for HSV-2 modeling is the relative paucity of detailed quantitative data on the longitudinal kinetics and functions of specific components of the tissue-based immune response in humans, throughout all phases of local infection including pre-infection, viral expansion, viral elimination and immunosurveillance. Therefore, despite having the most detailed available datasets of infection site human viral shedding at our disposal, our models can often only provide a very accurate assessment of timing and the overall intensity of the mucosal response rather than experimentally verifiable estimates of particular immune effectors (such as in vivo tissue-resident T-cell expansion and killing rates).
As with HIV models, our approximations of the local immune response are oversimplifications. We assume all of the local immune response is due to proliferation and killing by local tissue-resident CD8+ T cells (TRM). We select these cells as a model variable because they reside at the terminus of nerve endings which are at the precise site of HSV-2 replication in the genital epithelium (13), and because they demonstrate effector functions in the absence of antigen (14). We do not as of yet explicitly model or account for tissue-resident CD4+ T cells which reside deeper in dermis but likely provide key helper, and direct cytolytic, functions (33, 58, 115, 116), or regulatory CD4+ T cells which could restrain or limit CD8+ TRM activity (76, 77). The models ignore any potential dynamic effects of antibodies or innate like cells simply because data is lacking on whether these components of local immunity expand and contract in the presence of viral antigen to the same degree as local CD8+ T cells: it is possible that their antiviral activity is vital but expressed in a constitutive fashion; in this sense, basic model parameters of viral infectivity and replication rate may capture the role of innate immunity.
We also cannot directly model the role of antigen presenting cells (117), such as dendritic cells based on lack of sufficient data, though these cells are known to contribute to the innate protection (115, 118). Instead, the models tend to make the very basic assumption that a certain density of infected cells is necessary to allow local T cell proliferation. This delay in the local proliferative response is necessary for fitting data (19), and does suggest that given the right data, antigen presentation could be captured with a model. In summary, our models can only generalize the totality of the local immune response at the whole tissue scale due to the lack of adequate contributory data. As described below, our long-term research agenda is to gradually address these limitations.
5. Applied models to HSV-2 shedding data
Our mathematical model development has proceeded in an iterative fashion with the goal of answering specific questions regarding HSV-2 infection. We first developed a simple stochastic differential equation model to explain the high frequency of detectable HSV-2 shedding episodes in the human genital tract. The model had separate equations for susceptible cells, infected cells (defined according to their rate of viral production and more rapid death rate relative to uninfected cells), virus, and cytolytic effector cells. This model has since expanded to accommodate spatial features of infection and to recapitulate our comprehensive dataset of HSV-2 shedding.
5.1. Immunologic control of HSV-2 within latently infected ganglia is leaky.
The key unknown parameter in our initial model was the release rate of virus from the total body mass of infected dorsal root ganglia, to the genital tract, that would allow approximately bi-weekly detection of shedding episodes. We inferred that most plausible mechanism to explain the high frequency of genital shedding episodes is a nearly constant drip of small numbers of HSV-2 virions (50-100 per day) from the dorsal root ganglia towards the genital tract (17). Small weekly pulses of virus could also explain the observed episode rate. This prediction does not overturn the latency paradigm of chronic HSV-2 infection, as it still assumes that the vast majority of infected neuronal cell bodies maintain latent HSV-2 at any given point in time. Reactivation from a single neuron is likely adequate to explain our model prediction, which incidentally has remained essentially unchanged with sequentially more complex iterations of the model (19, 20, 119).
Recent work provides supporting evidence to verify the “drip” hypothesis of HSV-2 latency. Unfortunately, it is impossible to longitudinally sample dorsal root ganglia in humans due to inaccessibility of these tissues. However, studies of human trigeminal ganglia following surgical removal revealed varying HSV-1 viral loads on a cell-to-cell basis, suggesting that oral HSV-1 reactivates disproportionately within a tiny fraction of neurons relative to others (120, 121). Murine models of the ganglia also demonstrate various stages of viral reactivation, with transcripts from all stages of the replication cycle noted (86). Overall, the state of latency across the entire ganglia seems to be far more spatially and temporally dynamic and heterogeneous than previously predicted.
Our model predictions also suggest that the tissue-resident immune response within human ganglia may be important in regulating latency versus reactivation. Several studies demonstrated that CD8+ T-cells with markers of tissue residence and different HSV-1 antigen specificities preferentially surround HSV-1 infected cell bodies within the human trigeminal ganglia (73). Murine models revealed non-cytolytic mechanisms by which these cells may limit reactivation (88, 122). Further modeling by our group demonstrates that the increased shedding rate and episode rate noted in many patients with late stage HIV is likely related to more frequent release of HSV-2 from latency: a highly plausible underlying mechanism is poor T cell function within ganglia.
Nevertheless, latency is an extremely complex process. Viral and epigenetic features are likely to be critical (123), and may vary from person to person. Our model emphasizes a major unmet research need: the role of tissue resident immunity within latently affected ganglia must be more carefully defined. Enhancing this response in latent and lytic anatomic sites of infection may be critical for future vaccines.
5.2. Heterogeneity in the density of mucosal tissue resident CD8+ T cells explains the high variability in observed shedding episode severity.
We next attempted to understand the mechanism responsible for the extraordinary heterogeneity observed among shedding episodes within infected persons over time. Using a similar model, we developed the prediction that density of TRM at the precise site and time of mucosal HSV-2 reactivation was the prime determinant of peak viral load and shedding duration within that single microenvironment (16). This prediction is highly relevant because infected genital skin and mucosa is notable for high variability in the density of CD8+ T cells: densely packed clusters of these cells occupy nearly the entire dermal-epidermal region in certain regions following high titer viral replication. In other areas, which may be less than 0.1 millimeters away, the ratio of CD8 cells epithelial target cells is much lower, often less than 1:1000 (Fig 4d).
After nearly a decade, this model prognostication was formally proven in a murine model system (39, 40). Using an intravital imaging system, the authors also verified our model’s other key prediction that motility cessation and local proliferation of TRM, rather than trafficking of new T cells from blood or lymph, occurs in conjunction with rapid early extinguishment of infection (40, 47). This result is crucial because in human studies it is daunting task to identify a relevant 1-millimeter microenvironment in which an HSV-2 reactivation is certain to occur within the timeframe of a clinical study. Indeed, we have formally shown, using a principal components approach, that the timing and location of subsequent HSV-2 shedding episodes is impossible to predict based on prior knowledge of shedding or clinical characteristics, and that HSV-2 shedding data shows features compatible with chaos (114).
While the model used up to this point was simple and helped develop two fundamental, testable hypotheses, it had important shortcomings. First, we derived most of the parameters relating to viral replication rate and infectivity, as well as tissue-resident T-cell expansion by fitting a deterministic model to single episodes and then carrying these values over to the stochastic model (17). This approach required strategic selection of appropriate episodes for analysis: only episodes with monotonic expansion and contraction phases, rather than multiple viral re-expansions shown in Fig 2a, could be modeled. We were therefore ignoring a majority of our observed episodes and fundamentally biasing our parameter estimates. Second, while the model accurately reproduced key quantitative features of the shedding data, it was not trained to provide precise recapitulations of more detailed shedding-episode kinetic features, as described above. Qualitatively, the model was unable to generate prolonged episodes, some of which last longer than two weeks, and never generated episodes with multiple erratic peaks of various height. Third, the model did not account for the fact that viral-immune battles are clearly occurring in dozens of concurrent spatially discrete locations during more severe HSV-2 reactivations (67). To achieve a more comprehensive understanding of HSV-2 infection, and in particular to simulate therapy, it was necessary to address these shortcomings.
5.3. Modeling multi-focal reactivations suggests that elimination of HSV-2 replication within single micro-environments occurs extremely rapidly.
The goal of our next model was to more accurately recapitulate the entirety of our HSV-2 shedding data set in 531 immunocompetent persons (Fig 1b, c). We developed a hive model in which several hundred regions of the genital tract were modeled concurrently over time (Fig 5a). These regions were arrayed in an interlocking hexagon formation, with each region representing a 2 mm diameter area of possible HSV-2 replication and spread (15, 19). Model regions were assumed to be immunologically independent such that T-cells do not traffic between regions or kill across regions. However, free virus from an ulcer in region was allowed to seed adjacent regions. We assumed random neuronal seeding of each region at rates sufficient to produce empirically observed episode rates.
Figure 5. Mathematical model simulation of a prolonged HSV-2 shedding episode.
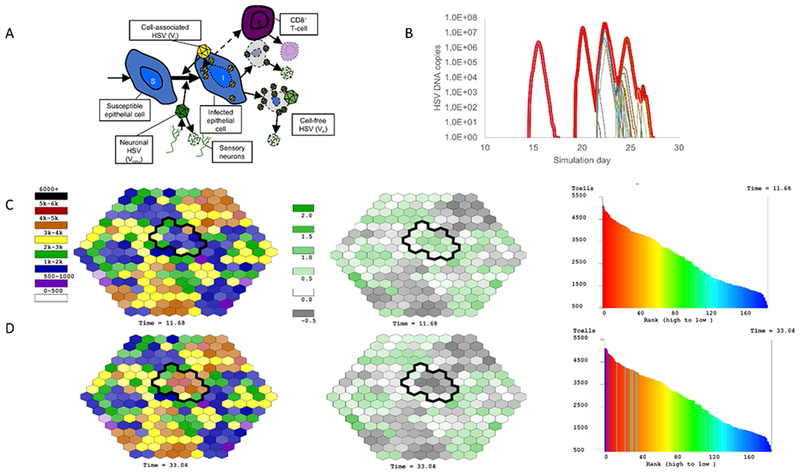
(A) Cartoon of mathematical model including susceptible cells, infected cells, cell-associated virus, cell-free virus and CD8+ T cells (TRM). These equations are simulated stochastically in 200 regions concurrently as shown in (C) & (D). (B) Simulated HSV-2 shedding episode notable for multiple peaks. The thick red line represents total viral load that would be detected with a clinical swab, while the thin lines represent viral load generated within single-microregions of infection. Simulated TRM structure and degree of protection (C) before and (D) following clearance of the shedding episode in (B). The left column represents the density of TRM within each micro-region before and after the episode. The circumscribed black regions had viral replication during the episode (thin lines in (B)). Regions which generated high viral loads transitioned from low to high TRM density as a large amount of local TRM proliferation was required to contain infection. The middle column demonstrates the log10 effective reproductive number (Re) of each region at the two timepoints. This number is inversely correlated with TRM density and denotes the potential for persistent cell-to-cell spread of virus prior to elimination. Green scale regions have Re>1 (log10 Re>0) indicating the need for TRM proliferation prior to elimination of all infected cells. Grey scale regions have Re<1 indicating no need for TRM proliferation prior to very rapid elimination of only a few infected cells. The right column plots the rank of each region according to TRM regional density. The shape of the distribution is stable before and after the episode, in keeping with the stable long-term détente established between HSV-2 and its host, despite dynamic reshuffling of individual region’s rank due to localized TRM proliferation events.
We were ultimately able to fit the updated model to 1020 heterogeneous shedding episodes: when the equations are run over 100 years, the model generates thousands of episodes which match observed variability in episode duration, first observed HSV DNA copy number, peak observed HSV DNA copy number, last observed HSV DNA copy number, and re-expansion frequency (19, 21). Model fitting was achieved by exploring thousands of potential parameter ranges with Latin Hypercube sampling and then identifying individual parameter values associated with best fit to the frequency histograms noted above. Several models with competing assumptions such as T-cell infusion from separate sites of infection rather than local proliferation, selective release of HSV-2 into only certain regions only, lack of inter-regional viral seeding, and the addition of inter-regional T-cell trafficking, worsened data fitting despite added complexity (19).
Our updated model was now finely tuned to observed population-level data from 531 study participants and as such generated new hypotheses relevant to tissue-resident immune responses. First, elimination of infected cells within a single micro region was predicted to occur much more rapidly than in our prior models. Even when local viral loads exceeded 100 million HSV DNA copies, all infected cells in the region were usually eliminated within 24 hours. This observation points to the incredible pace of HSV-2 infection but also the extraordinary effectiveness of the endogenous tissue-resident immune response. It also emphasizes that substantial hurdles that must be overcome for a novel therapeutic vaccine to induce an even more effective and rapid system of immunologic defense.
Model simulations also predicted that prolonged episodes occurred entirely due to seeding of cell-free virus from active ulcers into surrounding regions (Fig 5b,c). The serpiginous spread of HSV-2 ulcers and multi-peak episodes were also explained by this phenomenon. Spread of virus to multiple regions resulted in high TRM densities in these regions following viral elimination (Fig 5d).
5.4. Inter-patient variability of HSV-2 shedding is attributable to differences in specific immune parameters in the model.
While our model is an accurate simulator of patient shedding data in a cohort of hundreds of study participants, an insurmountable limitation is its inability to predict future, or even reproduce an individual’s past, shedding data. The model’s stochastic nature means that in keeping with human HSV-2 shedding data, the timing and trajectories of individual episodes in simulated data are, like the weather, also unpredictable over timeframes exceeding several days (114).
With the aim of providing general predictions about different observed categories of shedders, we performed large multi-parameter sensitivity analyses. These analyses suggest certain value ranges for a small number of model parameters that correlate with higher shedding rates, including high neuronal drip rate of HSV-2, shorter lifespan of TRM in genital mucosa and low clearance rate of cell-free virus. Globally, these predictions suggest that deficiencies in ganglionic immunity, shortened duration of cytolytic immune memory within the genital mucosa, and lack of mucosal non-neutralizing antibody activity may contribute to more severe disease (15).
We next separately fit the model to cohorts of HIV infected persons within different strata of CD4+ T cell counts. A first important observation is that HSV-2 shedding was not strictly defined according to strata. Several high shedders were noted in the least immunocompromised hosts (HIV negative and HIV positive with CD4>500/uL), while low shedders with clinical AIDS (CD4<200/uL) were also observed (66). Nevertheless, the population averaged shedding rate was considerably higher in the cohort with AIDS. The model achieving best fit to this cohort data assumed a higher neuronal drip rate of HSV-2, shorter lifespan of tissue-resident T-cells in genital mucosa and lower clearance rate of free-virus relative to the other cohorts (66), all in keeping with our original sensitivity analysis.
5.5. Tissue-resident T-cells form a highly structured and temporally resilient spatial geographic structure.
A recent analysis of emergent model TRM density data from our model suggests that a highly organized spatial structure of TRM is induced by chronic HSV-2 infection (manuscript under review). Specifically, when we rank model regions according to abundance of TRM, we observe an exponential relationship between this rank and local TRM abundance. The morphology and slope of this relationship is nearly constant over timeframes of years despite highly dynamic and unpredictable within-region fluctuations in TRM density during simulated lesions and accordingly, frequent shuffling of individual region ranks (Fig 5e,f). This finding may explain why HSV-2 shedding is highly turbulent and chaotic over weeks and months, but much more stable over years and decades (114). In effect, our model provides a platform for explaining the climate but not for forecasting the weather.
To validate the observation of a fixed spatial structure of TRM density in whole infected tissue, we analyzed density of CD8+ T-cells in 8 post-healing, lesional biopsies and identified a very similar spatial structure, albeit across more narrow diameters of tissue and with a steeper exponential slope: the feature driving this finding in the biopsies were clusters of TRM of various size and density scattered across infected tissue (Fig 4d).
This amazingly fixed TRM structure implies an underlying dynamic mechanism that is also predicted by the model. The first principle is that the TRM structure is intrinsically linked with the pattern of HSV-2 release from the ganglia during latency. For data fitting purposes, our model must assume that the distribution of HSV-2 release is random across the spatial regions (19). The stochastic nature of this release process creates a natural distribution of time between episodes within a single region ranging from 0-10 years (median 3 years). Accordingly, the time between local replenishment of CD8+ T-cells in single regions (which only occurs when HSV-2 infected cells are present at a sufficient level) is also normally distributed with a wide variance (range: 2.5 -10 years, median 4.5 years). Because TRM are assumed to very slowly decay when infected cells are not present, micro-regions which have not had recent infection have low TRM densities at the time of viral seeding: the downstream events are a high number of infected cells (>10,000) and high viral load (>10,000,000 HSV DNA copies), as well as a higher likelihood of a painful, visible ulcer. A higher degree of TRM local proliferation is then required to achieve elimination of virally infected cells. This region will therefore be protected for several years going forward, owing to the high density of local TRM. It will also undergo a shift from very low to high rank in our rank abundance curves due to massive local TRM proliferation (Fig 5e).
As a comparison, if viral takeoff occurs in a region with slightly higher TRM density, the subsequent peak viral load is only moderate, a lesion may not form locally, and a less substantial amount of TRM proliferation is required to achieve local pathogen elimination. This region will undergo only a slight increase in rank in our rank abundance curves. Episodes which take off in regions of high TRM density require no local proliferation to achieve viral clearance and these regions do not shift position on the rank abundance curve. The net effect over time is that the curve does not change its shape, and there is structured heterogeneous TRM density across previously infected tissue (Fig 5e).
Functionally, at any point in time a moderate proportion of the genital tract (20-60%) is fully protected against extensive replication. These episodes are defined mathematically as having an effective reproductive number less than one: due to a threshold density of TRM, virus cannot spread beyond more than a few cells before elimination (19). This explains why over 50% of observed shedding episodes last fewer than 12 hours and peak at extremely low viral loads. Given the explosiveness of HSV-2 replication and the high sensitivity of our assays, we suspect that many of these episodes reflect HSV-2 replication in a single cell (16, 17). A lower proportion of regions, roughly 5-10%, have low enough TRM density to allow a high local effective reproductive number (>5) which in turn puts the region at risk for a high viral load episode with enough killed cells for visual lesion development.
Surprisingly, when we simulate the model with prolonged, potent antiviral treatment, the TRM spatial structure is maintained. In accordance with data from treated patients, the spatial imprint of TRM as a result of chronic infection appears to be extremely resilient (12). If similar trends are assumed to occur with HSV-2 specific CD4+ T cells (124), then this may explain why treatment of HSV-2 did not decrease risk of HIV-1 acquisition in two clinical trials (125, 126). Numbers of CD4+ T cells, targets for HIV infection, are increased in the genital tract during chronic HSV-2; this is the proposed mechanism by which chronic HSV-2 infection enhances HIV risk (12). Our simulations suggest that antiviral therapy would need to be taken for years to diminish this inflammatory footprint of HSV-2. One caveat to this conclusion is that measured clearance rates of CD4+ T cells were obtained in genital skin and it is unknown if similar rates are observed in mucosal tissues (12).
5.6. Consideration of heterogeneous, spatial tissue-resident CD8+ T-cell density allows accurate and predictive clinical trial simulations.
A useful byproduct of developing a model of shedding in the absence of antiviral treatment was the ability to use this model as a baseline for simulated trials. We first estimated the impact of the currently licensed HSV-directed agents, acyclovir and famciclovir, by simulating both the pharmacokinetics (drug concentration curves over time) and pharmacodynamics (dose response curves) of these agents. We then assumed drug concentration-dependent inhibition of HSV-2 replication. These models closely reproduced detailed shedding kinetic data from studies in which participants served as their own controls. We identified that these drugs only eliminate shedding by 50 to 70% because of their short half-life (3.5 hours) (18, 20). Even narrow windows of subtherapeutic drug concentrations allowed breakthrough shedding. From an immunological perspective, this approach demonstrated that immune cells remain the key drivers of viral clearance during antiviral treatment (Fig 6); furthermore, density of TRM at the site of viral reactivation is still likely to be the key driver of episode severity.
Figure 6. The pressure exerted by TRM during antiviral therapy remains vital for containment of HSV-2 replication.
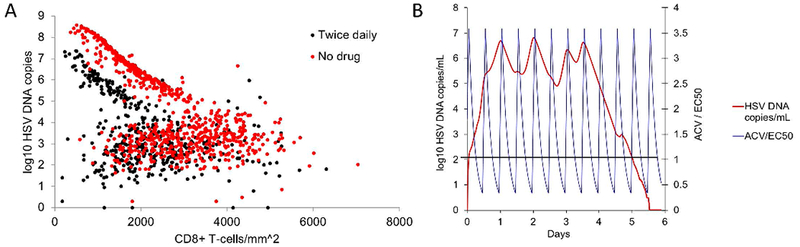
(A) Simulated episodes on doses of 400 mg acyclovir twice daily (red dots) and placebo (black dots). These simulations recapitulated clinical trial data accurately. For both study arms, a clear inverse relationship between pre-episode TRM density and peak episode viral load is noted. Antiviral therapy blunts the peak viral load at each given TRM density. (B) A single simulated episode shows that HSV-2 expansion (red line, left y-axis) decelerates when drug levels are high (blue line, right y-axis is ratio of drug concentration to EC50 at which inhibition of replication is 50%). Nevertheless, when virus is being cleared, the activity of the drug does not accelerate clearance, owing to the intensity of the cytolytic response (Adapted from reference 20).
We next simulated the phase 2 multi-dose trial of a promising HSV helicase inhibitor that benefits from a much longer half-life (70 hours) relative to acyclovir (51). The model was again able to fully recapitulate shedding episode kinetics across multiple doses. We made the important practical conclusion that drug potency measures using the IC50 (concentration at which replication is inhibited by 50%) are overestimated within in vitro systems relative to the human body. This study again confirmed the finding that accurate clinical trial simulations of HSV-2 cannot be achieved without considering the spatial heterogeneity of the HSV-2 tissue resident immune response.
6. Future directions
6.1. Precise modeling of the infection micro-environment.
HSV-2 infections occur within the three-dimensional genital tissue micro-environment. To fully capture the spatial features of virus spread and the full spectrum of tissue-resident T-cell activity, agent-based mathematical models can be employed to recapitulate the dynamic histopathology of herpetic genital ulcers. Agent-based mathematical models follow the activity of every single target cell, T-cell, virus and cytokine at each time step (127). As a result, these models are extremely computationally intensive and often require parallel computing on a cluster for efficient simulation.
Agent-based models of HSV-2 infection have obvious advantages relative to our prior methods. First, they provide attractive in silico representations of in situ infection that are accessible to biologists who lack a mathematical background. Agent-based models also capture components of infection that are missed with differential equation approaches. For instance, agent-based models more accurately capture the effects that targets cell limitation have on viral spread to new cells: as infection spreads a higher proportion of cells in contact a newly infected cell may already be infected; this will limit the rate of viral growth even in complete absence of an immune response and will dictate the peak viral load within a given micro-environment. Several papers have also shown the importance of a lattice-based approach for capturing the inter-epithelial trafficking patterns of tissue resident T cells (45, 46). Finally, agent-based models allow a new set of research questions to be addressed. While our past models were scaled to address the dynamics of the long-term war between HSV-2 and host across many cm2 of tissue and over many years, agent-based approaches will be tuned to address viral elimination within 2 mm2 areas of tissues over several days.
Past agent-based models, while elegant, were highly theoretical based on the challenges of obtaining sufficiently detailed spatial data for accurate model parameterization and data fitting (127). However, this is no longer the case for HSV-2. As we have described, detailed studies of TRM proliferation, trafficking, and killing from mouse experiments are now available to inform models, as are experiments tracking HSV-2 cell-to-cell spread (40, 44–48). Our group is therefore actively developing complex, but extremely well parameterized, models of the genital ulcer micro-environment. An area of particular interest is the vexing question of how pathogen control is achieved in the 40% of genital tract regions with very low TRM density.
6.2. HSV-2 intra-host phylodynamics.
Another area of enormous potential for modeling is intra-host HSV-2 evolution driven by mutation and positive selection. RNA viruses such as HIV and hepatitis C accrue mutations in a predictable and linear fashion, within an infected person who is not being treated (106, 113, 128). These mutations occur due to the high error rate of viral replication enzymes, as well as occasional recombination events. Certain mutations become fixed in the population as a result of ongoing positive selection pressure from the host T cell response. Remarkable population sweeps occur during untreated HIV infection: the dominant strain is replaced approximately once per month (129).
It is currently unknown whether a comparable process exists for HSV-2. The error rate per base pair of HSV DNA polymerase is extremely low relative to HIV reverse transcriptase (though high relative to human DNA polymerase) (130). Moreover, in a majority of infected patients, a continual stream of viral replication is not established as with HIV. Rather, the virus replicates periodically in the genital tract, but is eliminated on each occasion. Finally, the total effective viral population size is many orders of magnitude lower for HSV-2 relative to systemic infections such as HIV (19, 103), offering fewer opportunities for new mutations to accrue and achieve fixation.
However, HSV-2 has a much larger genome than HIV-1; if measured on a whole genome basis, the error rate of the viral DNA polymerase is only 5-10 times lower than HIV (130). In addition, it is quite clear that virus-host interactions are intense and nearly constant in both the peripheral mucosal tissues and the dorsal root ganglia (13, 14, 73). The latent ganglia serve as a persistent reservoir of infection. Therefore, if the ganglia are re-seeded during large peripheral reactivations during which new selected mutations might accrue, then the possibility of intra-host evolution exists. Our preliminary data suggests fixation of new mutations over several days in chronically infected persons. However, there are caveats to interpreting this data, because the ganglia may archive all past large peripheral replication events with new mutations. It is possible that reactivations may occur with past archived strains, rather than the strain with the greatest distance from the common ancestral strain established during primary infection. We hypothesize that HSV-2 phylogenies will demonstrate multiple evolving branches suggestive of weak positive selective pressure, though much more sequencing data is required to test this hypothesis. Perhaps the most relevant data might emerge from deep sequencing of viruses within cadaveric dorsal root ganglia, in order to generate comprehensive phylodynamic structure of infection that has been historically archived over several decades.
Once such data becomes available, modeling will be crucial to identify the frequency of new mutations and population sweeps during the lifetime of an infected person. This information will be highly relevant for vaccine development, to facilitate selection of epitopes that undergo the least variation over time.
6.3. Mathematical modeling of therapeutic vaccines.
Mathematical modeling may also be useful in the development of therapeutic vaccines. Designed to lower shedding rate, lesion incidence, and transmissibility in already infected persons, therapeutic vaccines are being pursued more aggressively than prophylactic vaccines at present. Studies for therapeutic vaccines are far less expensive and time consuming than for prophylactic vaccines. Modern study designs allow each participant to serve as their own control, with pre-vaccine shedding rate used a comparator to post-vaccine time points (131, 132). Licensure of a therapeutic vaccine would likely promote testing of equivalent or similar products in a prophylactic context (133, 134).
Several vaccine products appear promising, based on data from recent trials. The most powerful reduction in shedding was noted for GEN–003, which generated a ~50% reduction in shedding at 3, 6 and 12 months after vaccination (11). Interestingly, this vaccine generates an increase in HSV-2 specific neutralizing antibodies, non-neutralizing antibodies, and HSV-2 specific T-cells (10). However, each of these potential surrogates of immunologic protection was measured in blood. Studies of vaccine-induced immunity within relevant sites of lytic viral replication or neuronal latency have not yet been performed. It is impossible to infer the vaccine’s mechanisms of activity based on the tantalizing existing data.
From a modeling perspective, it may be possible to use virologic data following therapeutic vaccination to infer the predominant mechanism of action. Effective neutralizing antibodies, non-neutralizing antibodies and cytotoxic T cells would likely have different effects on specific features of HSV-2 shedding, including episode rate, expansion rate, and clearance rate (15). Mathematical modeling may be able to identify these differences within the existing data. Data from prophylactic vaccine trials would be much more difficult to model, as breakthrough infections would represent vaccine failure.
6.4. Expanding model of tissue resident immunity beyond CD8+ T cells.
There are several features of the tissue-resident immune response that are likely to be vital for pathogen containment but are not currently amenable to modeling in human infection. In particular, the role of CD4+ T-cell help, direct CD4+ antiviral activity, and broader immunosurveillance remains to be determined, though there is clear evidence that these cells can exert potent antiviral effects (83, 115). Similarly, it is unknown whether local antigen presenting cells are dispensable for achieving early elimination of virally infected cells. As murine knock-out and transgenic systems develop to allow specific focus on the role of each of these subsets, we anticipate constructing accompanying models to capture the intricacies of tripartite interactions between local CD4+ T-cells, CD8+ T-cells and antigen presenting cells. Finally, human Tregs are capable of dampening anti-HSV T cell responses (135). High numbers of Tregs infiltrate the site of virus reactivation in genital skin biopsies of lesions and persist proximal to conventional T cells beyond the lesion healing period (76), suggesting that the balance between Tregs and effector T cells influences human HSV-2 disease. Tregs could be a critical player to include in future models.
7. Conclusions.
Viral dynamics are central to understanding HSV-2 intra-host dynamics, and mathematical models are a necessary tool to interrogate these dynamics. We have developed accurate simulations that describe the leaky nature of ganglionic latency and the vital role of mucosal TRM in early recognition and containment of infection after HSV-2 reactivation. These models have progressed and now can accurately simulate modern clinical trials of HSV-2 antiviral agents. Future work should address the activity of TRM density within specific infection micro-environments, identify the modes of action of therapeutic vaccines, and establish the role of viral evolution in propagating infection.
Acknowledgements:
We thank Ashley Sherrid for her helpful review and editing of the article.
Funding: National Institute of Allergy and Infectious Diseases. Grant Numbers: P01 AI030371-24, R01 AI121129-01.
References
- 1.Corey L, Holmes KK. Genital herpes simplex virus infections: current concepts in diagnosis, therapy, and prevention. Ann Intern Med.1983;98:973–983. [DOI] [PubMed] [Google Scholar]
- 2.Wald A, Corey L, Cone R, Hobson A, Davis G, Zeh J. Frequent genital herpes simplex virus 2 shedding in immunocompetent women. Effect of acyclovir treatment. J Clin Invest.1997;99:1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wald A, Zeh J, Barnum G, Davis L, Corey L. Suppression of subclinical shedding of herpes simplex virus type 2 with acyclovir. Ann Intern Med.1996;124:8–15. [DOI] [PubMed] [Google Scholar]
- 4.Johnston C, Saracino M, Kuntz S, et al. Standard-dose and high-dose daily antiviral therapy for short episodes of genital HSV-2 reactivation: three randomised, open-label, cross-over trials. Lancet.2012;379:641–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corey L, Wald A, Patel R, et al. Once-daily valacyclovir to reduce the risk of transmission of genital herpes. N Engl J Med.2004;350:11–20. [DOI] [PubMed] [Google Scholar]
- 6.Belshe RB, Leone PA, Bernstein DI, et al. Efficacy results of a trial of a herpes simplex vaccine. N Engl J Med.2012;366:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnston C, Koelle DM, Wald A. HSV-2: in pursuit of a vaccine. J Clin Invest.2011;121:4600–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langenberg AG, Corey L, Ashley RL, Leong WP, Straus SE. A prospective study of new infections with herpes simplex virus type 1 and type 2. Chiron HSV Vaccine Study Group. N Engl J Med.1999;341:1432–1438. [DOI] [PubMed] [Google Scholar]
- 9.Straus SE, Corey L, Burke RL, et al. Placebo-controlled trial of vaccination with recombinant glycoprotein D of herpes simplex virus type 2 for immunotherapy of genital herpes. Lancet.1994;343:1460–1463. [DOI] [PubMed] [Google Scholar]
- 10.Flechtner JB, Long D, Larson S, et al. Immune responses elicited by the GEN-003 candidate HSV-2 therapeutic vaccine in a randomized controlled dose-ranging phase ½a trial. Vaccine.2016;34:5314–5320. [DOI] [PubMed] [Google Scholar]
- 11.Bernstein DI, Wald A, Warren T, et al. Therapeutic Vaccine for Genital Herpes Simplex Virus-2 Infection: Findings From a Randomized Trial. The Journal of infectious diseases.2017;215:856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu J, Hladik F, Woodward A, et al. Persistence of HIV-1 receptor-positive cells after HSV-2 reactivation is a potential mechanism for increased HIV-1 acquisition. Nature medicine.2009;15:886–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu J, Koelle D, Cao J, et al. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J Exp Med.2007;204:595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu J, Peng T, Johnston C, et al. Immune surveillance by CD8alphaalpha+ skin-resident T cells in human herpes virus infection. Nature.2013;497:494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schiffer JT. Mucosal HSV-2 Specific CD8+ T-Cells Represent Containment of Prior Viral Shedding Rather than a Correlate of Future Protection. Frontiers in immunology.2013;4:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiffer JT, Abu-Raddad L, Mark KE, et al. Mucosal host immune response predicts the severity and duration of herpes simplex virus-2 genital tract shedding episodes. Proceedings of the National Academy of Sciences of the United States of America.2010;107:18973–18978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schiffer JT, Abu-Raddad L, Mark KE, et al. Frequent release of low amounts of herpes simplex virus from neurons: results of a mathematical model. Science translational medicine.2009;1:7ra16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schiffer JT, Magaret A, Selke S, Corey L, Wald A. Detailed analysis of mucosal herpes simplex virus-2 replication kinetics with and without antiviral therapy. The Journal of antimicrobial chemotherapy.2011;66:2593–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schiffer JT, Swan D, Al Sallaq R, et al. Rapid localized spread and immunologic containment define Herpes simplex virus-2 reactivation in the human genital tract. eLife.2013;2:e00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schiffer JT, Swan DA, Corey L, Wald A. Rapid viral expansion and short drug half-life explain the incomplete effectiveness of current herpes simplex virus 2-directed antiviral agents. Antimicrobial agents and chemotherapy.2013;57:5820–5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schiffer JT, Wald A, Selke S, Corey L, Magaret A. The kinetics of mucosal herpes simplex virus-2 infection in humans: evidence for rapid viral-host interactions. The Journal of infectious diseases.2011;204:554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wald A, Zeh J, Selke S, Warren T, Ashley R, Corey L. Genital Shedding of Herpes Simplex Virus among Men. The Journal of infectious diseases.2002;186 Suppl 1:S34–39. [DOI] [PubMed] [Google Scholar]
- 23.Wald A, Zeh J, Selke S, et al. Reactivation of genital herpes simplex virus type 2 infection in asymptomatic seropositive persons. N Engl J Med.2000;342:844–850. [DOI] [PubMed] [Google Scholar]
- 24.Wald A, Huang ML, Carrell D, Selke S, Corey L. Polymerase chain reaction for detection of herpes simplex virus (HSV) DNA on mucosal surfaces: comparison with HSV isolation in cell culture. The Journal of infectious diseases.2003;188:1345–1351. [DOI] [PubMed] [Google Scholar]
- 25.Wald A, Zeh J, Selke S, Ashley RL, Corey L. Virologic characteristics of subclinical and symptomatic genital herpes infections. N Engl J Med.1995;333:770–775. [DOI] [PubMed] [Google Scholar]
- 26.Mark KE, Wald A, Magaret AS, et al. Rapidly cleared episodes of oral and anogenital herpes simplex virus shedding in HIV-infected adults. J Acquir Immune Defic Syndr.2010;54:482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mark KE, Wald A, Magaret AS, et al. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. The Journal of infectious diseases.2008;198:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Posavad CM, Zhao L, Dong L, et al. Enrichment of herpes simplex virus type 2 (HSV-2) reactive mucosal T cells in the human female genital tract. Mucosal Immunol.2017;10:1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science.2001;291:2413–2417. [DOI] [PubMed] [Google Scholar]
- 30.Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity.2014;41:886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gebhardt T, Palendira U, Tscharke DC, Bedoui S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev.2018;283:54–76. [DOI] [PubMed] [Google Scholar]
- 32.Clark RA, Watanabe R, Teague JE, et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Science translational medicine.2012;4:117ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wakim LM, Waithman J, van Rooijen N, Heath WR, Carbone FR. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science.2008;319:198–202. [DOI] [PubMed] [Google Scholar]
- 34.Kumar BV, Ma W, Miron M, et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep.2017;20:2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng T, Zhu J, Phasouk K, Koelle DM, Wald A, Corey L. An effector phenotype of CD8+ T cells at the junction epithelium during clinical quiescence of herpes simplex virus 2 infection. J Virol.2012;86:10587–10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linehan MM, Richman S, Krummenacher C, Eisenberg RJ, Cohen GH, Iwasaki A. In vivo role of nectin-1 in entry of herpes simplex virus type 1 (HSV-1) and HSV-2 through the vaginal mucosa. J Virol.2004;78:2530–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDermott MR, Smiley JR, Leslie P, Brais J, Rudzroga HE, Bienenstock J. Immunity in the female genital tract after intravaginal vaccination of mice with an attenuated strain of herpes simplex virus type 2. J Virol.1984;51:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parr MB, Kepple L, McDermott MR, Drew MD, Bozzola JJ, Parr EL. A mouse model for studies of mucosal immunity to vaginal infection by herpes simplex virus type 2. Lab Invest.1994;70:369–380. [PubMed] [Google Scholar]
- 39.Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol.2009;10:524–530. [DOI] [PubMed] [Google Scholar]
- 40.Park SL, Zaid A, Hor JL, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol.2018;19:183–191. [DOI] [PubMed] [Google Scholar]
- 41.Davies B, Prier JE, Jones CM, Gebhardt T, Carbone FR, Mackay LK. Cutting Edge: Tissue-Resident Memory T Cells Generated by Multiple Immunizations or Localized Deposition Provide Enhanced Immunity. J Immunol.2017;198:2233–2237. [DOI] [PubMed] [Google Scholar]
- 42.Mackay LK, Rahimpour A, Ma JZ, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol.2013;14:1294–1301. [DOI] [PubMed] [Google Scholar]
- 43.Mackay LK, Stock AT, Ma JZ, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proceedings of the National Academy of Sciences of the United States of America.2012;109:7037–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science.2014;346:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ariotti S, Beltman JB, Chodaczek G, et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proceedings of the National Academy of Sciences of the United States of America.2012;109:19739–19744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaid A, Mackay LK, Rahimpour A, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proceedings of the National Academy of Sciences of the United States of America.2014;111:5307–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beura LK, Mitchell JS, Thompson EA, et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol.2018;19:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ariotti S, Hogenbirk MA, Dijkgraaf FE, et al. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science.2014;346:101–105. [DOI] [PubMed] [Google Scholar]
- 49.Halle S, Keyser KA, Stahl FR, et al. In Vivo Killing Capacity of Cytotoxic T Cells Is Limited and Involves Dynamic Interactions and T Cell Cooperativity. Immunity.2016;44:233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Belshe RB, Heineman TC, Bernstein DI, et al. Correlate of immune protection against HSV-1 genital disease in vaccinated women. The Journal of infectious diseases.2014;209:828–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schiffer JT, Swan DA, Magaret A, et al. Mathematical modeling of herpes simplex virus-2 suppression with pritelivir predicts trial outcomes. Science translational medicine.2016;8:324ra315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mertz GJ, Schmidt O, Jourden JL, et al. Frequency of acquisition of first-episode genital infection with herpes simplex virus from symptomatic and asymptomatic source contacts. Sex Transm Dis.1985;12:33–39. [DOI] [PubMed] [Google Scholar]
- 53.Stevens JG, Cook ML. Latent herpes simplex virus in spinal ganglia of mice. Science.1971;173:843–845. [DOI] [PubMed] [Google Scholar]
- 54.Phipps W, Saracino M, Magaret A, et al. Persistent genital herpes simplex virus-2 shedding years following the first clinical episode. The Journal of infectious diseases.2011;203:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schiffer JT, Mayer BT, Fong Y, Swan DA, Wald A. Herpes simplex virus-2 transmission probability estimates based on quantity of viral shedding. J R Soc Interface.2014;11:20140160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Erard V, Wald A, Corey L, Leisenring WM, Boeckh M. Use of long-term suppressive acyclovir after hematopoietic stem-cell transplantation: impact on herpes simplex virus (HSV) disease and drug-resistant HSV disease. The Journal of infectious diseases.2007;196:266–270. [DOI] [PubMed] [Google Scholar]
- 57.Posavad CM, Koelle DM, Shaughnessy MF, Corey L. Severe genital herpes infections in HIV-infected individuals with impaired herpes simplex virus-specific CD8+ cytotoxic T lymphocyte responses. Proceedings of the National Academy of Sciences of the United States of America.1997;94:10289–10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harandi AM, Svennerholm B, Holmgren J, Eriksson K. Differential roles of B cells and IFN-gamma-secreting CD4(+) T cells in innate and adaptive immune control of genital herpes simplex virus type 2 infection in mice. J Gen Virol.2001;82:845–853. [DOI] [PubMed] [Google Scholar]
- 59.Parr EL, Parr MB. Immunoglobulin G is the main protective antibody in mouse vaginal secretions after vaginal immunization with attenuated herpes simplex virus type 2. J Virol.1997;71:8109–8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hobbs MM, van der Pol B, Totten P, et al. From the NIH: proceedings of a workshop on the importance of self-obtained vaginal specimens for detection of sexually transmitted infections. Sex Transm Dis.2008;35:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burrel S, Rouard C, Boutolleau D. Helicase-primase inhibitor pritelivir for HSV-2 infection. N Engl J Med.2014;370:1663–1664. [DOI] [PubMed] [Google Scholar]
- 62.Wald A, Timmler B, Magaret A, et al. Effect of Pritelivir Compared With Valacyclovir on Genital HSV-2 Shedding in Patients With Frequent Recurrences: A Randomized Clinical Trial. JAMA.2016;316:2495–2503. [DOI] [PubMed] [Google Scholar]
- 63.Tronstein E, Johnston C, Huang ML, et al. Genital shedding of herpes simplex virus among symptomatic and asymptomatic persons with HSV-2 infection. JAMA.2011;305:1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Magaret A, Dong L, John M, et al. HLA Class I and II alleles, heterozygosity and HLA-KIR interactions are associated with rates of genital HSV shedding and lesions. Genes Immun.2016;17:412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wherry EJ, Teichgraber V, Becker TC, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol.2003;4:225–234. [DOI] [PubMed] [Google Scholar]
- 66.Schiffer JT, Swan DA, Magaret A, Schacker TW, Wald A, Corey L. Mathematical Modeling Predicts that Increased HSV-2 Shedding in HIV-1 Infected Persons Is Due to Poor Immunologic Control in Ganglia and Genital Mucosa. PLoS One.2016;11:e0155124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnston C, Zhu J, Jing L, et al. Virologic and immunologic evidence of multifocal genital herpes simplex virus 2 infection. J Virol.2014;88:4921–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klonowski KD, Williams KJ, Marzo AL, Blair DA, Lingenheld EG, Lefrancois L. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity.2004;20:551–562. [DOI] [PubMed] [Google Scholar]
- 69.Sathaliyawala T, Kubota M, Yudanin N, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity.2013;38:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thome JJ, Yudanin N, Ohmura Y, et al. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell.2014;159:814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lian CG, Bueno EM, Granter SR, et al. Biomarker evaluation of face transplant rejection: association of donor T cells with target cell injury. Mod Pathol.2014;27:788–799. [DOI] [PubMed] [Google Scholar]
- 72.Steinert EM, Schenkel JM, Fraser KA, et al. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell.2015;161:737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Velzen M, Jing L, Osterhaus AD, Sette A, Koelle DM, Verjans GM. Local CD4 and CD8 T-cell reactivity to HSV-1 antigens documents broad viral protein expression and immune competence in latently infected human trigeminal ganglia. PLoS Pathog.2013;9:e1003547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Woon HG, Braun A, Li J, et al. Compartmentalization of Total and Virus-Specific Tissue-Resident Memory CD8+ T Cells in Human Lymphoid Organs. PLoS Pathog.2016;12:e1005799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martini E, Wiken M, Cheuk S, et al. Dynamic Changes in Resident and Infiltrating Epidermal Dendritic Cells in Active and Resolved Psoriasis. J Invest Dermatol.2017;137:865–873. [DOI] [PubMed] [Google Scholar]
- 76.Milman N, Zhu J, Johnston C, et al. In Situ Detection of Regulatory T Cells in Human Genital Herpes Simplex Virus Type 2 (HSV-2) Reactivation and Their Influence on Spontaneous HSV-2 Reactivation. The Journal of infectious diseases.2016;214:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Graham JB, Da Costa A, Lund JM. Regulatory T cells shape the resident memory T cell response to virus infection in the tissues. J Immunol.2014;192:683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moss NJ, Magaret A, Laing KJ, et al. Peripheral blood CD4 T-cell and plasmacytoid dendritic cell (pDC) reactivity to herpes simplex virus 2 and pDC number do not correlate with the clinical or virologic severity of recurrent genital herpes. J Virol.2012;86:9952–9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanchez Rodriguez R, Pauli ML, Neuhaus IM, et al. Memory regulatory T cells reside in human skin. J Clin Invest.2014;124:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ugur M, Schulz O, Menon MB, Krueger A, Pabst O. Resident CD4+ T cells accumulate in lymphoid organs after prolonged antigen exposure. Nat Commun.2014;5:4821. [DOI] [PubMed] [Google Scholar]
- 81.Masopust D, Choo D, Vezys V, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med.2010;207:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mackay LK, Wakim L, van Vliet CJ, et al. Maintenance of T cell function in the face of chronic antigen stimulation and repeated reactivation for a latent virus infection. J Immunol.2012;188:2173–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature.2012;491:463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chu T, Tyznik AJ, Roepke S, et al. Bystander-activated memory CD8 T cells control early pathogen load in an innate-like, NKG2D-dependent manner. Cell Rep.2013;3:701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mackay LK, Wynne-Jones E, Freestone D, et al. T-box Transcription Factors Combine with the Cytokines TGF-beta and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity.2015;43:1101–1111. [DOI] [PubMed] [Google Scholar]
- 86.Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC. Lytic gene expression is frequent in HSV-1 latent infection and correlates with the engagement of a cell-intrinsic transcriptional response. PLoS Pathog.2014;10:e1004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proceedings of the National Academy of Sciences of the United States of America.2002;99:978–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.St Leger AJ, Hendricks RL. CD8+ T cells patrol HSV-1-infected trigeminal ganglia and prevent viral reactivation. J Neurovirol.2011;17:528–534. [DOI] [PubMed] [Google Scholar]
- 89.Hoshino Y, Pesnicak L, Cohen JI, Straus SE. Rates of reactivation of latent herpes simplex virus from mouse trigeminal ganglia ex vivo correlate directly with viral load and inversely with number of infiltrating CD8+ T cells. J Virol.2007;81:8157–8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoshino Y, Pesnicak L, Straus SE, Cohen JI. Impairment in reactivation of a latency associated transcript (LAT)-deficient HSV-2 is not solely dependent on the latent viral load or the number of CD8(+) T cells infiltrating the ganglia. Virology.2009;387:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Verjans GM, Hintzen RQ, van Dun JM, et al. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proceedings of the National Academy of Sciences of the United States of America.2007;104:3496–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wald A, Krantz E, Selke S, Lairson E, Morrow RA, Zeh J. Knowledge of partners’ genital herpes protects against herpes simplex virus type 2 acquisition. The Journal of infectious diseases.2006;194:42–52. [DOI] [PubMed] [Google Scholar]
- 93.Wu H, Zhu H, Miao H, Perelson AS. Parameter identifiability and estimation of HIV/AIDS dynamic models. Bull Math Biol.2008;70:785–799. [DOI] [PubMed] [Google Scholar]
- 94.Shen L, Peterson S, Sedaghat AR, et al. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nature medicine.2008;14:762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Antia R, Pilyugin SS, Ahmed R. Models of immune memory: on the role of cross-reactive stimulation, competition, and homeostasis in maintaining immune memory. Proceedings of the National Academy of Sciences of the United States of America.1998;95:14926–14931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De Boer RJ, Oprea M, Antia R, Murali-Krishna K, Ahmed R, Perelson AS. Recruitment times, proliferation, and apoptosis rates during the CD8(+) T-cell response to lymphocytic choriomeningitis virus. J Virol.2001;75:10663–10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Regoes RR, Barber DL, Ahmed R, Antia R. Estimation of the rate of killing by cytotoxic T lymphocytes in vivo. Proceedings of the National Academy of Sciences of the United States of America.2007;104:1599–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Noecker C, Schaefer K, Zaccheo K, Yang Y, Day J, Ganusov VV. Simple mathematical models do not accurately predict early SIV dynamics. Viruses.2015;7:1189–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gadhamsetty S, Coorens T, de Boer RJ. Notwithstanding Circumstantial Alibis, Cytotoxic T Cells Can Be Major Killers of HIV-1-Infected Cells. J Virol.2016;90:7066–7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Conway JM, Perelson AS. Post-treatment control of HIV infection. Proceedings of the National Academy of Sciences of the United States of America.2015;112:5467–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature.1995;373:123–126. [DOI] [PubMed] [Google Scholar]
- 102.Wei X, Ghosh SK, Taylor ME, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature.1995;373:117–122. [DOI] [PubMed] [Google Scholar]
- 103.Perelson AS, Essunger P, Cao Y, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature.1997;387:188–191. [DOI] [PubMed] [Google Scholar]
- 104.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science.1996;271:1582–1586. [DOI] [PubMed] [Google Scholar]
- 105.Graw F, Regoes RR. Predicting the impact of CD8+ T cell polyfunctionality on HIV disease progression. J Virol.2014;88:10134–10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zanini F, Brodin J, Thebo L, et al. Population genomics of intrapatient HIV-1 evolution. eLife.2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rosenbloom DI, Hill AL, Rabi SA, Siliciano RF, Nowak MA. Antiretroviral dynamics determines HIV evolution and predicts therapy outcome. Nature medicine.2012;18:1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Reeves DB, Duke ER, Hughes SM, Prlic M, Hladik F, Schiffer JT. Anti-proliferative therapy for HIV cure: a compound interest approach. Sci Rep.2017;7:4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Roychoudhury P, De Silva Feelixge HS, Pietz HL, Stone D, Jerome KR, Schiffer JT. Pharmacodynamics of anti-HIV gene therapy using viral vectors and targeted endonucleases. The Journal of antimicrobial chemotherapy.2016;71:2089–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Neumann AU, Lam NP, Dahari H, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science.1998;282:103–107. [DOI] [PubMed] [Google Scholar]
- 111.Ribeiro RM, Lo A, Perelson AS. Dynamics of hepatitis B virus infection. Microbes Infect.2002;4:829–835. [DOI] [PubMed] [Google Scholar]
- 112.Ribeiro RM, Qin L, Chavez LL, Li D, Self SG, Perelson AS. Estimation of the initial viral growth rate and basic reproductive number during acute HIV-1 infection. J Virol.2010;84:6096–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ribeiro RM, Li H, Wang S, et al. Quantifying the diversification of hepatitis C virus (HCV) during primary infection: estimates of the in vivo mutation rate. PLoS Pathog.2012;8:e1002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dhankani V, Kutz JN, Schiffer JT. Herpes simplex virus-2 genital tract shedding is not predictable over months or years in infected persons. PLoS Comput Biol.2014;10:e1003922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Iijima N, Iwasaki A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science.2014;346:93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Milligan GN, Bernstein DI. Interferon-gamma enhances resolution of herpes simplex virus type 2 infection of the murine genital tract. Virology.1997;229:259–268. [DOI] [PubMed] [Google Scholar]
- 117.Kim M, Truong NR, James V, et al. Relay of herpes simplex virus between Langerhans cells and dermal dendritic cells in human skin. PLoS Pathog.2015;11:e1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lund JM, Linehan MM, Iijima N, Iwasaki A. Cutting Edge: Plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J Immunol.2006;177:7510–7514. [DOI] [PubMed] [Google Scholar]
- 119.Schiffer JT, Corey L. Rapid host immune response and viral dynamics in herpes simplex virus-2 infection. Nature medicine.2013;19:280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang K, Lau TY, Morales M, Mont EK, Straus SE. Laser-capture microdissection: refining estimates of the quantity and distribution of latent herpes simplex virus 1 and varicella-zoster virus DNA in human trigeminal Ganglia at the single-cell level. J Virol.2005;79:14079–14087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang K, Mahalingam G, Hoover SE, et al. Diverse herpes simplex virus type 1 thymidine kinase mutants in individual human neurons and Ganglia. J Virol.2007;81:6817–6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science.2008;322:268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Knipe DM. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology.2015;479-480:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Byrne CM, Gantt S, Coombs D. Effects of spatiotemporal HSV-2 lesion dynamics and antiviral treatment on the risk of HIV-1 acquisition. PLoS Comput Biol.2018;14:e1006129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Celum C, Wald A, Hughes J, et al. Effect of aciclovir on HIV-1 acquisition in herpes simplex virus 2 seropositive women and men who have sex with men: a randomised, double-blind, placebo-controlled trial. Lancet.2008;371:2109–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Watson-Jones D, Weiss HA, Rusizoka M, et al. Effect of herpes simplex suppression on incidence of HIV among women in Tanzania. N Engl J Med.2008;358:1560–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Graw F, Perelson AS. Modeling Viral Spread. Annu Rev Virol.2016;3:555–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gray RR, Parker J, Lemey P, Salemi M, Katzourakis A, Pybus OG. The mode and tempo of hepatitis C virus evolution within and among hosts. BMC Evol Biol.2011;11:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lemey P, Rambaut A, Pybus OG. HIV evolutionary dynamics within and among hosts. AIDS Rev.2006;8:125–140. [PubMed] [Google Scholar]
- 130.Sanjuan R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci.2016;73:4433–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bender Ignacio RA, Perti T, Magaret AS, et al. Oral and Vaginal Tenofovir for Genital Herpes Simplex Virus Type 2 Shedding in Immunocompetent Women: A Double-Blind, Randomized, Cross-over Trial. The Journal of infectious diseases.2015;212:1949–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Perti T, Saracino M, Baeten JM, et al. High-dose valacyclovir decreases plasma HIV-1 RNA more than standard-dose acyclovir in persons coinfected with HIV-1 and HSV-2: a randomized crossover trial. J Acquir Immune Defic Syndr.2013;63:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schiffer JT, Gottlieb SL. Biologic interactions between HSV-2 and HIV-1 and possible implications for HSV vaccine development. Vaccine.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Spicknall IH, Looker KJ, Gottlieb SL, et al. Review of mathematical models of HSV-2 vaccination: Implications for vaccine development. Vaccine.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Diaz GA, Koelle DM. Human CD4+ CD25 high cells suppress proliferative memory lymphocyte responses to herpes simplex virus type 2. Journal of virology.2006;80:8271–8273. [DOI] [PMC free article] [PubMed] [Google Scholar]