

Abstract
Herpes simplex virus-type 1 (HSV-1) infection of sensory neurons may lead to a significant reduction in the expression of voltage-activated Na+ and Ca2+ channels, which can disrupt the transmission of pain information. Viral infection also results in the secretion of various pro-inflammatory cytokines, including interleukin (IL)-6. In this work, we tested whether IL-6 regulates the expression of Na+ and Ca2+ channels post HSV-1 infection in ND7/23 sensory-like neurons. Our results demonstrate that HSV-1 infection causes a significant decrease in the protein expression of the Cav3.2 T-type Ca2+ channel subunit, despite increasing Cav3.2 mRNA synthesis. Neither Cav3.2 mRNA nor total protein content was affected by IL-6 treatment post HSV-1 infection. In ND7/23 cells, HSV-1 infection caused a significant reduction in the expression of Na+ and T-type Ca2+ channels within 48 h. Exposure of ND7/23 cells to IL-6 for 24 h post infection reverses the effect of HSV-1, resulting in a significant increase in T-type Ca2+ current density. However, Na+ currents were not restored by 24 h-treatment with IL-6 post HSV-1 infection of ND7/23 cells. The ability of IL-6 to increase the functional expression of T-type Ca2+ channels on the membrane was blocked by inhibition of protein trafficking with brefeldin-A and ERK1/2 activation. These results indicate that IL-6 release following HSV-1 infection regulates the expression of T-type Ca2+ channels, which may alter the transmission of pain information.
Keywords: sensory neuron, calcium channel, pain, electrical excitability, interleukin-6
Grapical Abstract
In this Issue
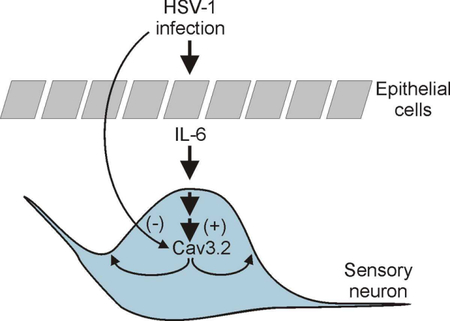
Regulation of T-type Ca2+ channel expression by interleukin-6 in sensory-like ND7/23 cells post herpes simplex virus (HSV-1) infection.
In this work, we tested the hypothesis that IL-6 upregulates the expression of T-type Ca2+ channel expression in ND7/23 sensory-like neurons post-HSV-1 infection. We propose that herpes simplex virus-type 1 (HSV-1) infection of sensory neurons negatively regulates the functional expression of T-type Ca2+ channels. This effect can be overcome by HSV-1 evoked release of interleukin-6 (IL-6) from neighboring epithelial cells. The paracrine effect of IL-6 release from epithelial cells promotes the trafficking of T-type Ca2+ channel to the membrane in infected neurons via an ERK dependent mechanisms. Thus, IL-6 release following HSV-1 infection regulates the expression of T-type Ca2+ channels, which can potentially alter the transmission of pain information in sensory neurons.
1.1. INTRODUCTION
Changes in ion channel expression regulate a variety of neuronal functions, including electrical excitability and the transmission of sensory information. Herpes simplex virus-type 1 (HSV-1) infection of sensory neurons alters the expression of voltage-gated ion channels, disrupting transmission of pain stimuli (Storey et al., 2002; Zhang et at., 2017). HSV-1 infection of dorsal root ganglion (DRG) neurons downregulates the expression of voltage-gated Na+ channels (Oakes et at., 1981; Fukuda et at., 1983; Storey et al., 2002). Changes in Na+ channel expression during early HSV-1 infection disrupt the electrical excitability of sensory neurons, which impair the transmission of pain stimuli (Mayer, 1986a). Similarly, HSV-1 infection of rat DRG neurons causes a significant reduction in inward rectifying currents while sparing outward rectifying K+ channels (Mayer, 1986b). We have previously demonstrated that HSV-1 infection of ND7/23 sensory-like neurons caused a significant reduction in the expression of T-type Ca2+ channels (Zhang et al., 2017). T-type Ca2+ channels are activated at negative membrane potentials and generate transient, fast inactivating currents (Perez-Reyes, 2003). Activation of T-type Ca2+ channels at membrane potentials close to rest can lower the threshold for the generation of action potentials, increasing electrical excitability (Chemin et al., 2002; Todorovic & Jevtovic-Todorovic, 2011).
Primary and secondary HSV-1 infections often disrupt pain sensory transmission resulting in either diminished or enhanced pain signaling. Diminished pain transmission can result from decreased functional expression of voltage-gated ion channels, whereas increased pain signaling may be generated by increased expression of ion channels in the membrane. HSV-1 outbreaks can evoke pain sensations, formication, paresthesia, or even numbness or tingling around initial infection area, which have been reported to be associated with decreased or enhanced neuronal excitability (Andoh et al., 1995). Diminished sensory transmission immediately after HSV-1 infection contrasts with increased painful signals generated in infected patients suffering from cold sores generated by HSV-1 reactivation. Previous studies have demonstrated that HSV-1 infection often evokes significant changes in the expression of pro-inflammatory cytokines and chemokines (Wuest & Carr, 2008). For example, HSV-1 infection of trigeminal neurons in vitro triggers the expression of the transcripts of several cytokines, including IL-6, IFN-γ, TNF-α, (Halford et al., 1996). HSV-1 infection of epithelial corneal cells also results in a significant release of IL-6 and other cytokines 2 h post-infection (Li et al., 2006). It is unclear whether these factors have the potential to alter the expression of voltage-activated channels in pain-transmitting neurons post HSV-1 infection and its implication for the development of post-herpetic neuralgia.
In this work, we tested the hypothesis that IL-6 upregulates the expression of T-type Ca2+ channel expression in ND7/23 sensory-like neurons post-HSV-1 infection. Our choice of IL-6 is based on previous findings showing a significant secretion of IL-6 following HSV-1 infection of epithelial tissue (Li et al., 2006), and the well characterized effect of cytokines in regulating the expression of T-type Ca2+ channel expression during neuronal differentiation (Trimarchi et al., 2009; Dey et al., 2011). Changes in T-type Ca2+ channel expression may underlie the sensory abnormalities in patients following HSV-1 infection. Those changes could be triggered not only by the direct effect of the virus on pain transmitting neurons but also by the secretion of pro-inflammatory cytokines.
1.2. METHODS
1.2.1. Cell culture, differentiation and infection of ND7/23 cells:
ND7/23 cells were obtained from Sigma-Aldrich (RRID:CVCL_4259). ND7/23 cells were generated by the fusion of mouse neuroblastoma and rat dorsal root ganglion cells, generating a more homogeneous cell population with sensory neuron-like properties (Wood et al., 1990). Culture and differentiation of ND7/23 cells was performed as previously described by Zhang et al. (2017). Briefly, differentiation of ND7/23 cells was evoked by treatment with DMEM/F12 culture media (Millipore, Cat.#DF-041-B), supplemented with 0.5% fetal bovine serum (Invitrogen, Cat.#10437010), db-cAMP (1 mM, Sigma-Aldrich, Cat.#D0627), and NGF (50 ng/mL, Sigma-Aldrich, Ca.#N2513) as previously described (Wood et al., 1990). The differentiation culture media was also supplemented with uridine (20 µM, Sigma-Aldrich, Cat.#U3003) and fluorodeoxyuridine (20 µM, Sigma-Aldrich, Cat.#F0503) post plating to remove any proliferating cells. After induction of differentiation for 4 d, cell were maintained in differentiation media without uridine and fluorodeoxyuridine. Human corneal epithelial cells (HCEC) were purchased from Millipore (Cat.#SCCE016, purchased Apr. 2018) and cultured in EpiGro human ocular epithelia complete media (Millipore, Cat.#SCMC001) according to the manufacturer’s recommendations. Cells were grown in an incubator at 37°C in the presence of 5% CO2/95% air humidified atmosphere. Cells passaged less than 20 times were used in this work. ND7/23 cells were maintained in differentiation media for ≥4 days. Cells were grown either in poly-d-lysine-coated 6-well plates or on glass coverslips (for whole cell recordings). None of the cell lines used in this work has been misidentified according to the International Cell Line Authentication Committee (ICLAC). Cell line authentication was performed by the providers (Sigma-Aldrich or Millipore) using short-tandem repeat (STR) analysis.
Viral infections were performed with a GFP-expressing HSV-1 strain 17Syn+-GFP virus (A1 strain) (Foster et al.; 1998). The recombinant viral construct was engineered from the HSV-1 wild-type strain 17syn+, expressing enhanced GFP under the control of a cytomegalovirus (CMV) promoter (Foster et al.; 1998). Viral particle were propagated in African green monkey kidney (Vero) cells (ATCC, RRID:CVCL_0059) were cultured in MEM media (ThermoFisher, Cat.# 41090–036), supplemented with 10% fetal bovine serum. GFP expression was used to facilitate the identification of infected cells. Cell cultures were exposed to HSV-1 for 1 h in a cell culture incubator, as previously described (Bedadala et al.; 2014). For electrophysiological recordings, cells were infected with HSV-1 at a MOI of 0.5; whereas for western blotting, cells were infected at a MOI of 0.2, to insure we can get enough proteins after 48 h incubation. After this time period, unbound viral particles were washed out and fresh differentiation media supplemented with different drug combinations was applied. Custom-made materials, including viral constructs, will be shared upon reasonable request.
1.2.2. Western blot analysis:
Immunoblot analysis of the Cav3.2 T-type Ca2+ channel subunit was conducted as previously described by Zhang et al. (2017), using a rabbit anti-Cav3.2 primary antibody (1:500, Santa Cruz, RRID:AB_2259537). Detection of ERK1/2 activation was performed using a rabbit anti-phospho-ERK1/2 or anti-ERK1/2 antibodies (1:1000, Cell Signaling, RRID:AB_331775 and RRID:AB_330744, respectively). Briefly, cell lysates were combined with Bolt LDS sample buffer (ThermoFisher, Cat.#B0007), supplemented with reducing agent (ThermoFisher, Cat.#B0004) and boiled for 10 min at 80°C. Proteins were separated on pre-cast SDS-PAGE 8% gels (Bolt Bis-Tris Plus Gels, ThermoFisher, Cat.# NW00080BOX). Proteins were transferred to nitrocellulose membranes using the Invitrogen iBlot system, followed by incubation in SuperBlock blocking buffer for 45 min (ThermoFisher, Cat.#37535) before overnight incubation with the primary antibodies. BupH Tris buffered saline (ThermoFisher, Cat.#28379) was used to wash the membranes following incubation with the antibodies. Blots were analyzed using a horseradish peroxidase-conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch Lab, RRID:AB_2339150) and a chemiluminescent substrate (SuperSignal West Pico, ThermoFisher, Cat.#34580). To control for equal loading of protein in each sample, membranes were stripped using Restore Plus stripping buffer (ThermoFisher, Cat.#46430) for 30 min at room temperature (22–24ºC) and reprobed with a tubulin-specific antibody (1:2000 dilution, Millipore, RRID:AB_310035) followed by incubation with a peroxidase-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch Lab, RRID:AB_2307392) and immunodetection. Signals were captured using a ChemiDoc™ XRS+ documentation system (Bio-Rad, Hercules). Changes in Cav3.2 protein expression was determined by densitometry analysis using Image Lab software (Bio-Rad). The Cav3.2/tubulin intensity ratio was normalized to the control values (non-stimulated cells).
1.2.3. Biotinylation of cell surface proteins:
Labeling of cell surface proteins was performed as previously described by Hall et al. (2018). Briefly, ND7/23 cells were cultured in 6-well plates at 60% confluency. Surface proteins were biotinylated using Pierce™ Premium Grade Sulfo NHS-SS-Biotin (1mg/mL; ThermoFisher, Cat.#PG82077). A 5 min-incubation period of the mixture in PBS supplemented with 100 mM glycine terminated the biotinylation reaction. After washing with PBS, proteins were extracted with a RIPA cell lysis buffer, collected as the supernatant fraction post centrifugation (6000 x g for 10 min at 4°C), and quantitatively assessed using the Pierce BCA protein assay kit (ThermoFisher, Cat.#23227). Pull-down of the biotinylated proteins was accomplished by overnight incubation of equal amounts of protein samples with Pierce Streptavidin Agarose Resin (ThermoFisher, Cat.#20347). Agarose beads were collected by centrifugation (6000 x g for 5 min) and after three washes with cell lysis buffer the biotinylated proteins were eluted with Bolt sample buffer supplemented with reducing agent. The samples were subjected to immune-detection to assess surface expression of Cav3.2 proteins using an anti-Cav3.2 antibody (1:500, Santa Cruz, RRID:AB_2259537). To control for equal loading of protein in each sample, biotinylated membrane fractions were treated with stripping buffer (Restore Plus, ThermoFisher, Cat.#46430) for 30 min at room temperature and reprobed with an ATPase-specific antibody (1:1000 dilution, Sigma, RRID:AB_2061127) followed by incubation with the corresponding secondary antibody (Jackson ImmunoResearch Lab, RRID:AB_2307392) and immuno-detection.
1.2.4. Electrophysiology:
A Nikon Eclipse Ti inverted microscope equipped with Hoffman optics and epifluorescence filters was used to visualize the ND7/23 cells during recordings. Infected cells were identified by the expression of GFP. Recordings were performed at room temperature (22–24ºC) using glass electrodes made from thin wall borosilicate glass (3–4 MΩ). The pipette solution consisted of (in mM) CsCl (120), MgCl2 (2), HEPES-KOH (10), EGTA (10), ATP (1), and GTP (0.1), pH 7.4 with CsOH. The composition to of the normal external saline used for measurements of Ca2+ currents was (in mM) tetraethylammonium chloride (TEACl, 145), CaCl2 (10), MgCl2 (1), and HEPES (10), pH 7.4 adjusted with CsOH. Ca2+ currents were generated by applying a 400 ms-depolarizing step to various potentials from a holding potential of −110 mV. The external solution used to measure Na+ currents contained (in mM) NaCl (145), KCl (5.3), CaCl2 (5.4), MgCl2 (0.8), and HEPES (13), pH 7.4 adjusted with KOH. Na+ currents were generated by applying a 20 ms-depolarizing step to various potentials from a holding potential of −100 mV. A MultiClamp 700A amplifier and Pclamp software (Axon Instruments) was used to deliver voltage commands and to perform data acquisition and analysis. Pipette offset, whole cell capacitance, and series resistance were compensated automatically with the MultiClamp 700B Commander. Only cells with stable seals and series resistance (≤10 MΩ) were analyzed. Sampling rates were between 5 and 10 kHz. For quantitative analysis, cell size was normalized by dividing current amplitudes by cell capacitance, determined by integration of the transient current evoked by a 10-mV voltage step from a holding potential of −60 mV (Pachuau & Martin-Caraballo, 2007a,b).
1.2.5. Plaque assay:
The plaque assay was performed as previously described (Zhang et al., 2017). Briefly, Vero cells were plated in 12-well plates and seeded at a concentration of 1.6×105 cells/mL overnight. The supernatant of infected cells was collected and serially diluted before being added to cultured cells. After 48 hpi, the culture media was removed, and the cells fixed with methanol and stained with crystal violet prior to counting plaque formation. Data was collected from triplicates.
1.2.6. Quantitative PCR:
Quantification of thymidine kinase (TK) expression was performed as previously described (Zhang et al., 2017). Briefly, genomic DNA was isolated using the GeneElute™ Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich, Cat.#G1N70, purchased in 2017). RNA was isolated using the iScript sample preparation reagent (BioRad, Cat.#170–8898, purchased in 2018). Synthesis of cDNA by RT reaction was performed with iScript RT Supermix (Bio-Rad, Cat.#17008841, purchased in 2017). The SsoAdvanced Universal SYBR green supermix (Bio-Rad, Cat.#1725271, purchased in 2018) was used to perform the qPCR reactions on triplicate samples. The set of primers used to amplify the TK gene expression was: forward, 5’-ATG GCT TCG TAC CCC TGC CAT-3’, reverse, 5’-GGT ATC GCG CGG CCG GGT A-3’. qPCR reactions were carried out at 98°C for 3 min, 98°C for 10 s, followed by 58°C for 30 s (39 cycles), 65°C for 5 s. TK expression was normalized to that of peptidylpropyl isomerase A (PPIA, forward, 5’-GGT GGC AAG TCC ATC TAC GG-3’, reverse, 5’-CTT GCC ATC CAG CCA CTC A-3’). Quantification of Cav3.2 mRNA expression was performed as previously described (Hall et al., 2018). The set of primers used to assess Cav3.2 gene expression was: forward, 5’-GTT CGT GCT GGT CAA TGT GG-3’, reverse, 5’-GGC TTT CCT GTG CTG TAG GT-3’. qPCR reactions were carried out at 95°C for 30 s, 95°C for 10 s, followed by 56°C for 30 s (39 cycles), 65°C for 5 s. Cav3.2 mRNA expression was normalized to that of peptidylpropyl isomerase A (PPIA).
1.2.7. ELISA:
IL-6 secretion following infection of HCEC with HSV-1 was performed with the human IL-6 ELISA kit according to the manufacturer’s instructions (ThermoFisher, Cat.# KHC0061, purchased in Apr. 2018). HCEC were infected with HSV-1 for 1 h. After washing to remove viral particles, cells were returned to the incubator for 6–24 h with fresh media. Detection of IL-6 release was performed on the harvested cultured media. The total IL-6 secretion was normalized to the total protein content in the cell culture. After harvesting the culture media, cells were lysed with RIPA buffer (ThermoFisher, Cat.#89901). Following centrifugation of cell lysates, the supernatant was collected and protein concentration was determined using the Pierce BCA protein assay kit (ThermoFisher, Cat.#23227, purchased in Nov. 2017). Secreted IL-6 (in pg) was normalized to the protein content of the cell lysate (in mg).
1.2.8. Reagents:
IL-6 (Invitrogen, Cat.#RP-8619), brefeldin A (Sigma-Aldrich, Cat.#B7651), and U0126 (Sigma-Aldrich, Cat.#19–147).
1.2.9. Data Analysis:
All electrophysiological data are presented as box plots, showing the median value, first and third quartile, with the whiskers showing the maximum and minimum values. Data from immunoblot analysis, PCR, plaque assay, and ELISA are presented as scatter plots. Statistical analyses consisted of Student’s unpaired t-test when single comparisons were made, and one-way ANOVA followed by post hoc analysis using Tukey’s honest significant difference test for unequal n for the more typical experimental designs that entailed comparisons between multiple groups (Statistica software, version 11). Throughout, p ≤ 0.05 was regarded as significant. No statistical analysis was performed to predetermine sample sizes or to test outlier values. The normal distribution of the sample values was assessed by the Shapiro-Wilk test using the Statistica software.
1.2.10. Institutional Approval:
All performed experiment using HSV-1 infection received institutional approval from the Institutional Biosafety Committee (#20110913–02).
1.3. RESULTS
Our previous findings demonstrate that both undifferentiated and differentiated ND7/23 cells express a variety of voltage-gated ion channels (Zhang et al., 2017). However, differentiated ND7/23 cells express larger transient currents generated by T-type Ca2+ channels. There is significant expression of Cav3.2 T-type Ca2+ channel subunits in differentiated ND7/23 cells, resulting in Ca2+ currents that are sensitive to inhibition by low concentrations of nicker ions and NNC55–0396 (Zhang et al., 2017).
ND7/23 cells were cultured in differentiation media, supplemented with NGF+db-cAMP for ~4 days. Following 1 h exposure of differentiated ND7/23 cells to HSV-1, cells were cultured overnight prior to treatment with IL-6 (20 ng/mL) for 24 h. As previously reported (Zhang et al., 2017), HSV-1 infection of differentiated ND7/23 cells for 24–48 h causes a significant reduction in the amplitude of the transient Ca2+ currents generated by a voltage step to −20 mV (Fig. 1A-B). Treatment of HSV-1 infected cells with IL-6 for 24 h restore the expression of T-type Ca2+ currents (Fig. 1C). IL-6 treatment of ND7/23 cells post HSV-1 infection did not alter the current-voltage relationship of T-type Ca2+ channels with peak current at −20 mV (Fig. 1D). Current amplitudes (generated by a voltage step to −20 mV) were normalized to cell size by dividing current values by cell capacitance (Pachuau & Martin-Caraballo, 2007a,b). The resulting current densities were used to assess changes in the functional expression of T-type Ca2+ channels in the membrane under different culture conditions. Infection of differentiated ND7/23 cells for 48 h caused a significant reduction in the T-type Ca2+ current densities without any significant change in cell capacitance (Fig. 1E-F). However, there was an increase in cell capacitance following treatment of ND7/23 cells with IL-6 compared with controls without any changes in current density, indicating an increase in cell size following IL-6 stimulation (Fig. 1E-F). IL-6 treatment of ND7/23 cells did not alter the cell capacitance post HSV-1 infection but evoked a considerable increase in current density in HSV-1 infected cells (Fig. 1E-F). As represented in Fig. 2, HSV-1 infection with or without IL-6 treatment causes significant changes in the distribution of current densities. HSV-1 infection of ND7/23 cells caused a leftward shift in the current density plots compared with control cells (Fig. 2A vs. 2C). In HSV-1 infected ND7/23 cells, IL-6 evoked a rightward shift in the current density plots (Fig. 2C vs. 2D). The density plots for non-infected cell with or without IL-6 treatment was very similar with peak current densities between 5 and 9.9 pA/pF (Fig. 2A vs. 2B).
Figure 1.

Changes in whole cell Ca2+ currents in differentiated ND7/23 cells following HSV-1 infection and treatment with IL-6. A-C) Example of whole cell T-type Ca2+ currents generated in a differentiated ND7/23 cell following HSV-1 infection and treatment with IL-6. Differentiated ND7/23 cells were infected overnight with HSV-1. After a 24 h infection period, cells were treated with IL-6 (20 ng/mL) for another 24 h. Recordings from control (non-treated) and treatment groups were performed at the end of the 48 h period. In this and subsequent figures, the voltage step protocol is shown below the current trace. Note that the transient component (arrow) generated by voltage step to −20 mV from a holding potential of −110 mV was eliminated following HSV-1 infection (A,B). Stimulation with IL-6 reverses the inhibitory effect of HSV-1 infection on T-type Ca2+ currents (B,C). Scale bars in A are the same for B and C. D). Normalized current-voltage (I-V) relationship generated by the activation of T-type Ca2+ channels in differentiated ND7/23 cells (non-infected vs. HSV+IL-6 treated cells). Current amplitudes at different voltages were normalized to that generated by a voltage step to −20 mV from a holding potential of −110 mV (I/I −20). E) Comparison of cell capacitance in ND7/23 cells following HSV-1 infection and treatment with IL-6. The number of cells recorded under each condition is presented in parenthesis in panel F. F) Mean T-type Ca2+ current densities generated in ND7/23 cells following HSV-1 infection and treatment with IL-6. T-type Ca2+ current density was calculated from the peak current amplitude generated by a voltage step to –20 mV from a holding potential of −110 mV. The number of cells recorded under each condition is presented in parenthesis from at least 3 different cell cultures. * denotes p ≤ 0.05 vs. control (non-treated) cells; ** denotes p ≤ 0.05 vs. HSV-1 infected cells.
Figure 2.

Density plot of T-type Ca2+ currents generated in differentiated ND7/23 cells following HSV-1 infection and treatment with IL-6. A, B, C) HSV-1 infection causes a leftward shift in current densities. D) IL-6 treatment for 24 h reverses the HSV-1 evoked shift in current densities. The number of cells analyzed under each condition was: control (n=22), +IL-6 (n=15), +HSV (n=22), and HSV+Il-6 (n=25) from at least 3 different cell cultures.
We also assessed whether IL-6 had the potential to restore the expression of Na+ channels post HSV-1 infection in ND7/23 cells. As represented in Fig. 3A-B, infection of ND7/23 cells with HSV-1 evoked a significant reduction in the functional expression of Na+ channels. However, IL-6 treatment of ND7/23 cells post HSV-1 infection did not reverse the effect of viral infection on Na+ channels (Fig. 3C-D).
Figure 3.

Effect of HSV-1 infection on Na+ currents generated in differentiated ND7/23 cells following treatment with IL-6. A-B) Examples of whole cell Na+ currents generated in a differentiated ND7/23 cell following HSV-1 infection. Note that the inward Na+ current generated by voltage step to +20 mV from a holding potential of −100 mV was eliminated following HSV-1 infection. C) Comparison of cell capacitance in ND7/23 cells following HSV-1 infection and treatment with IL-6. The number of cells recorded under each condition is presented in parenthesis in panel D. D) Infection of differentiated ND7/23 cells with HSV-1 causes a significant reduction in the density of Na+ currents under all conditions tested (* denotes p < 0.05 vs. HSV-1 infected cells). IL-6 treatment does not reverse the inhibitory effect of HSV-1 infection on Na+ current densities. The number of cells recorded under each condition is presented in parenthesis from at least 3 different cell cultures.
Next, we investigated the cellular mechanisms responsible for the effect of IL-6 post HSV-1 infection on T-type Ca2+ channel expression. First, we assessed whether HSV-1 infection followed by IL-6 stimulation evokes changes in Cav3.2 mRNA expression by quantitative PCR. As represented in Fig. 4A, HSV-1 infection of ND7/23 cells caused a significant upregulation in Cav3.2 mRNA expression. IL-6 does not have any effect on the level of Cav3.2 transcript expression in non-infected or HSV-1 infected ND7/23 cells. Although, our results indicate that HSV-1 infection causes a significant increase in Cav3.2 mRNA levels, electrophysiological data demonstrates a reduction in the functional expression of T-type Ca2+ channels. To investigate whether HSV-1 infection alters the expression of Cav3.2 channel proteins, we performed immunoblot analysis (Fig. 4B). HSV-1 infection evokes a significant reduction in the amount of Cav3.2 proteins in whole cell lysates as presented in Fig. 4B-C for ND7/23 cells (Fig. 4B). However, stimulation of ND7/23 cells with IL-6 post HSV-1 infection did not increase Cav3.2 protein levels in whole cell lysates (Fig. 4B). These findings indicate that although HSV-1 infection evokes a considerable increase in Cav3.2 mRNA expression, there is a concomitant reduction in Cav3.2 protein expression.
Figure 4.

Effect of HSV-1 infection and treatment with IL-6 on the expression of the Cav3.2 transcripts and channel proteins. A) HSV-1 infection of differentiated ND7/23 cells evokes a considerable increase in the expression of Cav3.2 mRNA as assessed by real time PCR analysis. IL-6 does not alter the expression of Cav3.2 mRNA in control or HSV-1 infected cells. * denotes p ≤ 0.05 vs. control (non-treated) cells; ns denotes no significant difference (p > 0.05); n=4 (number of independent cell cultures). B) Western blot analysis showing changers in the expression of Cav3.2 channel protein following HSV-1 infection and treatment with IL-6. Note that HSV-1 infection caused a significant reduction in Cav3.2 protein expression. IL-6 did not reverse the reduction in Cav3.2 protein expression. * denotes p ≤ 0.05 vs. control (non-treated) cells; ns denotes no significant difference (p > 0.05); n=4 (number of independent cell cultures).
Thus, the question arises on how IL-6 can promote the functional expression of T-type Ca2+ channels following a reduction in the total amount of Cav3.2 proteins. We tested the hypothesis that IL-6 promotes the trafficking of channel proteins to the membrane. First, we assessed whether IL-6 promotes the insertion of new channel proteins in the membrane by measuring the level of biotinylated Ca3.2 subunits. Labeling of membrane-bound proteins with EZ-link sulfo-NHS-SS biotin was followed by immunodetection of Cav3.2 protein expression. As represented in Fig. 5A, infection of ND7/23 cells with HSV-1 caused a significant reduction in the amount of biotinylated Cav3.2 proteins. However, stimulation of ND7/23 cells with IL-6 post HSV-1 infection caused an increase in the level of Cav3.2 biotinylated membrane proteins. This finding suggests that IL-6 stimulation of ND7/23 cells post HSV-1 infection causes an increase in the trafficking of Cav3.2 channel subunit to the membrane. To further confirm this finding, we tested the effect of brefeldin-A (BFA, 1 μg/mL) on the functional expression of T-type Ca2+ channels in IL-6 treated ND7/23 cells. Brefeldin-A disrupts protein trafficking by altering the integrity of the Golgi apparatus. As represented in Fig. 5B, brefeldin-A evoked a considerable reduction in the cell capacitance of non-infected or HSV-1 infected ND7/23 cells, as would be expected from blocking vesicular trafficking (Pachuau & Martin-Caraballo, 2007b). However, in non-infected cells, brefeldin-A treatment had no effect on T-type Ca2+ channels already present in the membrane (Fig. 5C). In HSV-1 infected cells treated with IL-6, brefeldin-A caused a significant reduction in T-type Ca2+ current density, indicating that blockage of trans-Golgi trafficking prevents the functional expression of T-type Ca2+ channels (Fig. 5C). Changes in the range of current densities observed in ND7/23 cells following brefeldin-A treatment under different conditions are represented in Fig. 5D-G. In non-infected ND7/23 cells, brefeldin-A had no significant effect in the distribution of current densities (Fig. 5D vs. 5E). However, in HSV-1 infected cells treated with IL-6, brefeldin-A caused a left-ward shift in the distribution of current densities (Fig. 5F vs. 5G).
Figure 5.

IL-6 increases the expression of Cav3.2 channel proteins on the membrane following HSV-1 infection of differentiated ND7/23 cells (A). The effect of IL-6 post HSV-1 infection was quantified on biotinylated proteins to assess changes in Cav3.2 channel proteins on the membrane. * denotes p ≤ 0.05 vs. control (non-treated) cells; n=4 (number of independent cell cultures). B-C) Inhibition of protein trafficking with brefeldin-A (BFA, 1 μg/mL) reduces T-type Ca2+ channel functional expression as assessed by whole cell recordings. Effect of brefeldin-A on cell capacitance (B) and T-type Ca2+ current density (C) on differentiated ND7/23 cells. Note that brefeldin-A evoked a significant reduction in the cell capacitance of IL-6 or HSV-1/IL-6 treated cells. T-type Ca2+ current density was significantly reduced following HSV-1/IL-6 treatment of ND7/23 cells. The number of cells recorded under each condition is presented in parenthesis in panel C from 2 different cell cultures. * denotes p ≤ 0.05 vs. control (non-treated) cells; ** denotes p ≤ 0.05 vs. HSV-1 infected cell cultures treated with IL-6. D-G) Density plot of T-type Ca2+ currents generated in differentiated ND7/23 cells following treatment with brefeldin-A. Treatment of ND7/23 cells with brefeldin-A caused a leftward shift in current densities.
As previously reported, IL-6 can activate the JAK/STAT signaling pathway resulting in transcriptional changes in gene expression, which does not seem to explain the changes evoked by IL-6 stimulation in differentiated ND7/23 cells post HSV-1 infection. Since IL-6 can also activate ERK1/2 signaling, we tested the effect of this signaling pathway in promoting the functional expression of T-type Ca2+ channels in differentiated ND7/23 cells (Trimarchi et al., 2009; Dey et al., 2011). Our data indicate that there is significant increase in phosphorylated ERK (p-ERK) in differentiated ND7/23 cells compared to cells cultured in the presence of growth media (GM) alone as assessed by the presence of two bands with a molecular weight between 40–50 kDa (Fig. 6A, insert). Infection of differentiated ND7/23 cells with HSV-1 caused a decrease in p-ERK (Fig. 6A, insert). Overall changes in the p-ERK levels normalized to t-ERK are presented in Fig. 6A. Stimulation of differentiated ND7/23 cells with IL-6 (30 min-3 h) did not result in significant changes in p-ERK (Fig. 6B, insert). However, pre-treatment of differentiated ND7/23 cells with the ERK inhibitor U0126 prevented ERK activation as indicated by the lack of the two p-ERK bands (Fig. 6B insert). Infection of ND7/23 cells with HSV-1 downregulates ERK phosphorylation, which can be increased by IL-6 stimulation (Fig. 6C, insert). In HSV-1 infected ND7/23 cells, stimulation with IL-6 (for 30 min to 3 h) evoked a considerable increase in the phosphorylated levels of p-ERK (Fig. 6C). Thus, these results suggest that IL-6 stimulation of differentiated ND7/23 cells pre- or post-HSV-1 infection results in different patterns of ERK1/2 activation. ERK1/2 activation plays a role in the functional expression of T-type Ca2+ channels in differentiated ND7/23 cells (Fig. 6D). Thus, 1 h inhibition of ERK activation with U0126 did not have any effect on T-type Ca2+ channels already present in the membrane, suggesting that blockade of ERK activity does not alter channel expression by an allosteric mechanism such as phosphorylation (Fig. 6D). On the other hand, overnight treatment of differentiated ND7/23 cells with the ERK inhibitor U0126 causes a ~50% reduction in the T-type Ca2+ current density in non-infected cells (Fig. 6D). In HSV-1 infected ND7/23 cells treated with IL-6, we detected significant expression of functional T-type Ca2+ channels as assessed by whole cell recordings. This effect was eliminated by treatment with U0126. These findings suggest that the stimulatory effect of IL-6 on T-type Ca2+ channel expression post HSV-1 infection can be eliminated by inhibition of ERK activation.
Figure 6.

Effect of ERK1/2 activation on the functional expression of T-type Ca2+ channels in differentiated ND7/23 cells. A) Effect of cell differentiation and HSV-1 infection on ERK activation as assessed by changes in phosphorylated ERK (p-ERK). Note that differentiated cells (following 4 d culture in differentiation media-DM) expressed higher levels of p-ERK compared with ND7/23 cells cultured in growth media (GM). Infection of differentiated ND7/23 cells with HSV-1 evoked a significant reduction in p-ERK. Overall changes in p-ERK was normalized to total ERK (t-ERK) expression under each culture condition (number of independent cell cultures n=3). B) Time course of ERK activation following stimulation of differentiated ND7/23 cells with IL-6 (20 ng/mL). Immunoblot analysis was used to determine changes in ERK activation by assessing the levels of phosphorylated and total ERK. ERK activation in differentiated ND7/23 cells can be inhibited by pre-treatment with the ERK blocker U0126 (10 μM). In these experiments, cultures were pre-treated with U0126 for 1 h prior to stimulation with IL-6 (number of independent cell cultures n=4). C) In HSV-1 infected cells, IL-6 evokes an increased in ERK activation (number of independent cell cultures n=4). D) The ERK inhibitor U0126 blocked the stimulatory effect of IL-6 on T-type Ca2+ channel expression. Note that 1 h treatment with U0126 did not alter the functional expression of T-type Ca2+ channels, suggesting a lack of an allosteric effect on channels already present in the membrane. Overnight incubation with U0126 prevents the normal expression of T-type Ca2+ channels on the membrane. In HSV-1 infected cells treated with IL-6, inhibition of ERK activity with U0126 caused a completed disruption of channel expression. The number of cells recorded under each condition is presented in parenthesis from 2 independent cell cultures. * denotes p ≤ 0.05 vs. control (non-infected cultures); ** denotes p ≤ 0.05 vs. HSV-1 infected cell cultures treated with IL-6.
The effect of HSV-1 infection (strain 17Syn+-GFP virus) on IL-6 secretion has not been reported. Therefore, we assessed the amount of IL-6 secretion in the culture media following HSV-1 infection of HCEC. As represented in Fig. 7A, there is very little secretion of IL-6 in HCEC between 6 and 12 h in culture. However, infection of human epithelial corneal cells with the 17Syn+-GFP virus strain evoked a considerable increase in IL-6 secretion after 6 h and continued for 12 h. Finally, we assessed the ability of IL-6 to alter viral replication and release (Zhang et al., 2017). Viral replication was assessed by qPCR analysis to measure viral genome copy number using primer pairs against the thymidine kinase (TK) gene. Plaque assay analysis was used to assess viral release. As shown in Fig. 7B-C, treatment of differentiated ND7/23 cells with IL-6 post HSV-1 infection caused a significant increase in TK gene expression without any changes in plaque formation.
Figure 7.

Effect of HSV-1 infection on IL-6 secretion, and its contribution to viral replication and release. A) IL-6 release was assessed from the supernatant of HSV-1 infected HCEC as assessed by ELISA (* denotes p < 0.05 vs. HSV-1 infected cells for 6 h; number of independent cell cultures n=3). B) IL-6 evokes a considerable increase in the expression of the HSV-1 TK following infection of differentiated ND7/23 cells (* denotes p < 0.05 vs. HSV-1 infected cells; number of independent cell cultures n=4). C) IL-6 had no effect on plaque formation following HSV-1 infection of differentiated ND7/23 cells (p > 0.05 vs. HSV-1 infected cells; number of independent cell cultures n=4).
1.4. DISCUSSION
In this study we examined the effect of IL-6 on sensory-like neurons post HSV-1 infection. Several conclusions can be drawn from these experiments. First, HSV-1 infection causes IL-6 release from epithelial cells, which can potentially regulate the properties of neurons in a paracrine manner. Second, HSV-1 infection causes a significant reduction in the expression of both voltage-gated Na+ and T-type Ca2+ channels in differentiated ND7/23 cells. Third, IL-6 specifically restore the functional expression of T-type Ca2+ channels on the membrane, which can potentially regulate the electrical excitability of pain-transmitting neurons post-HSV-1 infection. Four, IL-6 evoked upregulation of T-type Ca2+ channel expression post HSV-1 infection does not involve inhibition of viral replication or viral protein synthesis, but increased Cav3.2 protein trafficking to the membrane via an ERK1/2-dependent signaling mechanism.
Our present results demonstrate that IL-6 treatment promotes the functional expression of T-type Ca2+ channels in the membrane of HSV-1 post-infected neurons. As our previous work has shown, infection of differentiated ND7/23 cells with HSV-1 causes over 80% reduction of T-type Ca2+ functional channels 24–48 h post infection (Zhang et al., 2017). Our present findings demonstrate that 24 h-treatment with IL-6 post HSV-1 infection restores the functional expression of T-type Ca2+ channels to the membrane. We should note that IL-6 treatment does not have any effect on the functional expression of T-type Ca2+ channels on non-infected ND7/23 cells, which indicates that IL-6 has no functional activity on T-type Ca2+ channels already present on the membrane. Western blot analysis reveals that HSV-1 infection causes a significant reduction in the protein expression of the Cav3.2 T-type Ca2+ channel subunit (both the cytosolic and biotinylated fractions), whereas IL-6 treatment does not prevent the downregulation of Cav3.2 protein expression caused by HSV-1 infection.
IL-6 evoked upregulation of T-type Ca2+ channel functional expression on the membrane does not involved increased Cav3.2 mRNA expression nor de novo total protein synthesis in HSV-1 infected ND7/23 cells. Surprisingly, our present results demonstrate that Cav3.2 mRNA expression is upregulated by HSV-1 infection alone. Thus, it appears that virion-associated host shutoff (vhs) does not trigger rapid degradation of Cav3.2 mRNA in the host cells (Matis & Kúdelová, 2001). This effect could be a compensatory response to reverse the downregulation of Cav3.2 protein expression in HSV-1 infected cells. Alternatively, the 24 h-treatment with IL-6 post HSV-1 infection may be not long enough for the translation of Cav3.2 mRNA. Although HSV-1 infection of ND7/23 cells evokes a considerable increase in Cav3.2 mRNA synthesis, there is a marked downregulation in the expression of the Cav3.2 channel subunit. This process could be the result of increased protein degradation by an ubiquitin-proteasome pathway (Everett, 2000).
IL-6 stimulation of differentiated ND7/23 cells post HSV-1 infection promotes the trafficking of Cav3.2 channel subunits to the membrane. Thus, our biotinylation experiments reveal a significant increase in the amount of membrane-localized Cav3.2 channel subunits after IL-6 stimulation in post HSV-1 infected cells. Furthermore, inhibition of trans-Golgi trafficking with brefeldin-A evokes a considerable reduction in the functional expression of T-type Ca2+ currents generated by IL-6 stimulation of infected cells. Thus, these results indicate that trans-Golgi trafficking play an important role in the cytokine-evoked trafficking of T-type Ca2+ channels both in HSV-1 infected neurons and during neuronal differentiation (Dey et al., 2011). It appears that ERK1/2 activation was required to promote the functional expression of T-type Ca2+ channels on the membrane. Thus, inhibition of ERK1/2 activation with U0126 prevents the stimulatory effect of IL-6 in promoting the functional expression of T-type Ca2+ channels on the membrane. We have previously reported that ciliary neurotrophic factor (CNTF) and leukemia inhibitory factor (LIF), which belong to the IL-6 family of trophic factors, evoke ERK1/2 activation, resulting in increased T-type Ca2+ channel trafficking to the membrane during neuronal differentiation (Trimarchi et al., 2009; Dey et al., 2011).
HSV-1 infection or viral reactivation could trigger the release of cytokines, such as IL-6 from both immune cells and non-immune cells, which in turn help the elimination of infectious virus from the host (Paludan, 2001; Azher et al., 2017). Specifically, it was reported that HSV-1 infection with the HSV-1 KOS strain could cause the release of considerable amount of IL-6 from HCEC, which are widely used as a model for testing cytokine released into the media due to the use of serum-free media culture conditions (Terasaka et al., 2010; Li et al., 2006). In the present study we also tested the ability of the HSV-1 17Syn+-GFP strain to evoke IL-6 secretion from infected epithelial cells. Indeed, infection of HCEC with the HSV-1 17Syn+-GFP strain evokes a significant increase in IL-6 secretion. These findings indicate that HSV-1 infection of targeted tissue such as epithelial cells triggers the release of IL-6, which then can act on neighboring cells in a paracrine manner. Thus, it is plausible that secretion of IL-6 following HSV-1 infection alters the functional properties of neighboring cells, in particular T-type Ca2+ channel expression. HSV-1 evoked IL-6 release may also regulate various aspects of viral function including latency establishment and reactivation (Kriesel a et al., 1997). Under our experimental conditions, IL-6 promotes viral replication without increasing viral release.
Functional expression of T-type Ca2+ channels on the membrane is regulated by a variety of factors, including neurotransmitters and hormones through the activation of various signaling mechanisms (Chemin et al., 2006; Iftinca & Zamponi, 2009). Our previous work has revealed that cytokine signaling specifically targets the functional expression of T-type Ca2+ channels during neuronal differentiation (Pachuau & Martin-Caraballo, 2007b). Our present results demonstrate that the stimulatory effect of IL-6 on the functional expression of T-type Ca2+ channels post HSV-1 infection was specific. Stimulation of HSV-1 infected ND7/23 cells with IL-6 had no effect on the functional expression of voltage-gated Na+ channels. This finding is consistent with our previous work demonstrating that cytokines regulates the functional expression of T-type Ca2+ channels but no other conductances, including other voltage gated Ca2+ channels (Pachuau & Martin-Caraballo, 2007a). We have previously demonstrated that CNTF and LIF, selectively increase T-type Ca2+ channel functional expression in developing sensory neurons (Trimarchi et al., 2009; Dey et al., 2011). IL-6 induced neuroendocrine differentiation of prostate cancer cells also results in increased functional expression of T-type Ca2+ channels through a post-transcriptional mechanism without having any effect on other voltage-gated Na+ channels or Ca2+ conductances (Lee et al., 2007; Weaver et al., 2015). Thus, the question arises about what factors may regulate the functional expression of voltage-gated Na+ channels post HSV-1 infection. As previously demonstrated, HSV-1 infection evoked the secretion of a variety of cytokines and chemokines (Li et al., 2006; Paludan, 2001; Azher et al., 2017). We are currently investigating the possibility that other factors may alter the expression of voltage-gated Na+ channels specifically.
Understanding how HSV-1 mediated secretion of IL-6 and other factors alter the electrical properties of sensory neurons may have important implications in the treatment of post-herpetic neuralgia. HSV-1 infections often cause significant disruption in pain sensory transmission, resulting in both diminished and enhanced pain signaling (Andoh et al., 1995). Reduced functional expression of ion channels can diminish the transmission of pain information through sensory neurons, whereas increased ion channel expression may have the opposite effect on pain transmission. Of significant clinical importance is the development of post-herpetic neuralgia, which maybe the result of changes in the expression of voltage-gated Na+ and Ca2+ channels (Kennedy et al., 2013). Although expression of voltage-gated Na+ channels is required for the transmission of pain information, increased expression of T-type Ca2+ channels can alter the electrical excitability of neuronal cells, resulting in increased transmission of pain signals to the CNS (Todorovic & Jevtovic-Todorovic, 2011). Upregulation of IL-6 and its receptor in DRG sensory neuronal cells have been reported in various pain models. Thus, IL-6 enhances neuropathic pain in wild-type mice, whereas in the IL-6 knockout mice, development of spinal nerve lesion-induced mechanical allodynia was delayed and became attenuated (Ramer et al., 1998). Similarly, the IL-6 knock-out mice is less sensitive to pain in the chronic constriction injury (CCI) model (Murphy et al., 1999). Furthermore, anti-IL-6 or anti-IL-6R neutralizing antibodies can effectively reverse these pain-related issues (Ramer et al., 1998; Murakami et al., 2013; Brunner et al., 2015; Nishimoto et al., 2000). Thus, the restoration of T-type Ca2+ channels on the membrane by IL-6 secretion post HSV-1 infection could have several consequences. First, activation of T-type Ca2+ channels at membrane potentials closed to rest can increase neuronal excitability and promote the activation of voltage-gated Na+ channels involved in the generation of action potential. Pain information transmission is highly dependent on the generation of action potential which requires activation of voltage-gated Na+ channels. Because of their hyperpolarized voltage activation range, T-type Ca2+ channels serve as preamplifier in action potential generation and regulator of neuronal excitability (Bourinet et al., 2016). Second, increased Ca2+ influx into neuronal cells can regulate viral replication and release. Intracellular Ca2+ concentration is critical for viral entry, replication, and release (Cheshenko et al., 2003; Zhou et al., 2009; Arimoto et al., 2006). Previous studies have demonstrated that HSV-1 infection promotes Ca2+ release from IP3-sensitive ER stores, resulting in increased intracellular Ca2+ concentrations, which stimulate viral entry. Voltage-gated Ca2+ channels have a slight effect on rising the intracellular Ca2+ and it is less important for the virus entry process (Cheshenko et al., 2003; Zhou et al., 2009; Cheshenko et al., 2007). However, increased T-type Ca2+ channel functional expression by IL-6 in HSV-1 post-infected neurons may play a role in facilitating viral replication and release.
Acknowledgement
This work was supported by funds provided by the UMES School of Pharmacy and grant R01NS081109 to SCH from NINDS/NIH. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NINDS/NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This work was presented at the 2018 Colorado Alphaherpesvirus Latency Society Symposium as a poster presentation. The abstract presented at the symposium was published in the J. Neurobiol (Baird et al., 2018).
List of Abbreviations:
- CNTF
Ciliary neurotrophic factor
- ERK1/2
Extracellular signal-regulated kinases 1/2
- GFP
Green fluorescence protein
- HCEC
Human corneal epithelial cells
- HSV-1
Herpes simplex virus type 1
- IL-6
Interleukin-6
- IFN-γ
Interferon-γ
- LIF
Leukemia inhibitory factor
- TNF-α
Tumor necrosis factor-α
Footnotes
Declaration of Conflicting Interests
The authors declare no potential conflicts of interest regarding the research, authorship, and/or publication of this article.
Open Science Badges
This article has received a badge for *Open Materials* because it provided all relevant information to reproduce the study in the manuscript. The complete Open Science Disclosure form for this article can be found at the end of the article. More information about the Open Practices badges can be found at https://cos.io/our-services/open-science-badges/.
REFERENCES
- Andoh T, Shiraki K, Kurokawa M (1995) Paresthesia induced by cutaneous infection with herpes simplex virus in rats. Neurosci. Lett 190(2), 101–104. [DOI] [PubMed] [Google Scholar]
- Arimoto E, Iwai S, Sumi T, Ogawa Y, Yura Y (2006) Involvement of intracellular free Ca2+ in enhanced release of herpes simplex virus by hydrogen peroxide. Virol. J 3, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azher T, Yin X, Stuart P (2017) Understanding the Role of Chemokines and Cytokines in Experimental Models of Herpes Simplex Keratitis. J. Immunol. Res 2017, 7261980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedadala G, Chen F, Figliozzi R, Balish M, Hsia V (2014) Construction and characterization of recombinant HSV-1 expressing early growth response-1. ISRN Virol 2014 pii: 629641. [DOI] [PMC free article] [PubMed]
- Baird NL, Depledge DP, Cohrs RJ (2018) 2018 Colorado Alphaherpesvirus Latency Society Symposium. J Neurovirol 24(6), 797–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Francois A, Laffray S (2016) T-type calcium channels in neuropathic pain. Pain 157, S15–S22. [DOI] [PubMed] [Google Scholar]
- Brunner HI, Ruperto N, Zuber Z, Keane C, Harari O, Kenwright A, Lu P, Cuttica R, Keltsev V, Xavier RM (2015) Efficacy and safety of tocilizumab in patients with polyarticular-course juvenile idiopathic arthritis: results from a phase 3, randomised, double-blind withdrawal trial. Ann. Rheum. Dis 74, 1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Perez-Reyes E, Bourinet E, Nargeot J, Lory P (2002) Specific contribution of human T-type calcium channel isotypes (alpha(1G), alpha(1H) and alpha(1I)) to neuronal excitability. J Physiol 540(Pt 1), 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Traboulsie A, Lory P (2006) Molecular pathways underlying the modulation of T-type calcium channels by neurotransmitters and hormones. Cell Calcium 40(2), 121–134. [DOI] [PubMed] [Google Scholar]
- Cheshenko N, Del Rosario B, Woda C, Marcellino D, Satlin L, Herold BC (2003) Herpes simplex virus triggers activation of calcium-signaling pathways. J. Cell Biol 163(2), 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshenko N, Liu W, Satlin LM, Herold BC (2007) Multiple Receptor Interactions Trigger Release of Membrane and Intracellular Calcium Stores Critical for Herpes Simplex Virus Entry. Mol. Biol. Cell 18(8), 3119–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey D, Shepherd A, Pachuau J, Martin-Caraballo M (2011) Leukemia inhibitory factor (LIF) regulates the trafficking of T-type Ca2+ channels. Am. J. Physiol.-Cell Physiol 300, C576–C587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD (2000) ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 22(8), 761–770. [DOI] [PubMed] [Google Scholar]
- Foster TP, Rybachuk GV, Kousoulas KG (1998) Expression of the enhanced green fluorescent protein by herpes simplex virus type 1 (HSV-1) as an in vitro or in vivo marker for virus entry and replication. J Virol. Methods 75(2), 151–160. [DOI] [PubMed] [Google Scholar]
- Fukuda J, Kurata T, Yamaguchi K (1983) Specific reduction in Na currents after infection with herpes simplex virus in cultured mammalian nerve cells. Brain Res 268(2), 367–371. [DOI] [PubMed] [Google Scholar]
- Halford WP, Gebhardt BM, Carr DJ (1996) Persistent cytokine expression in trigeminal ganglion latently infected with herpes simplex virus type 1. J Immunol 157(8), 3542–3549. [PubMed] [Google Scholar]
- Hall M, Todd B, Allen ED, Nga N, Kwon YJ, Nguyen V, Hearne JL, Martin-Caraballo M (2018) Androgen receptor signaling regulates T-type Ca2+ channel expression and neuroendocrine differentiation in prostate cancer cells. Amer. J. Cancer Res 8(4), 732–747. [PMC free article] [PubMed] [Google Scholar]
- Iftinca MC, Zamponi GW (2009) Regulation of neuronal T-type calcium channels. Trends Pharmacol. Sci 30(1), 32–40. [DOI] [PubMed] [Google Scholar]
- Kennedy PG, Montague P, Scott F, Grinfeld E, Ashrafi GH, Breuer J, Rowan EG (2013) Varicella-zoster viruses associated with post-herpetic neuralgia induce sodium current density increases in the ND-23 Nav1.8 neuroblastoma cell line. PLoS One 8, e51570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriesel JD, Ricigliano J, Spruance SL, Garza HH, Hill JM (1997) Neuronal reactivation of herpes simplex virus may involve interleukin-6. J Neurovirol 3(6), 441–448. [DOI] [PubMed] [Google Scholar]
- Lee SO, Chun JY, Nadiminty N, Lou W, Gao AC (2007) Interleukin-6 undergoes transition from growth inhibitor associated with neuroendocrine differentiation to stimulator accompanied by androgen receptor activation during LNCaP prostate cancer cell progression. Prostate 67(7), 764–773. [DOI] [PubMed] [Google Scholar]
- Li H, Zhang J, Kumar A, Zheng M, Atherton SS, Yu FS (2006) Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 117(2), 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matis J, Kúdelová M (2001) Early shutoff of host protein synthesis in cells infected with herpes simplex viruses. Acta Virol 45(5–6), 269–277. [PubMed] [Google Scholar]
- Mayer ML, James MH, Russell RJ, Kelly JS, Pasternak CA (1986a) Changes in excitability induced by herpes simplex viruses in rat dorsal root ganglion neurons. J. Neurosci 6(2), 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML (1986b) Selective block of inward but not outward rectification in rat sensory neurons infected with herpes simplex virus. J. Physiol 375, 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PG, Ramer MS, Borthwick L, Gauldie J, Richardson PM, Bisby MA (1999) Endogenous interleukin-6 contributes to hypersensitivity to cutaneous stimuli and changes in neuropeptides associated with chronic nerve constriction in mice. Eur. J. Neurosci 11, 2243–2253. [DOI] [PubMed] [Google Scholar]
- Murakami T, Kanchiku T, Suzuki H, Imajo Y, Yoshida Y, Nomura H (2013) Anti-interleukin-6 receptor antibody reduces neuropathic pain following spinal cord injury in mice. Exp. Therap. Med 6(5), 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto N, Sasai M, Shima Y, Nakagawa M, Matsumoto T, Shirai T, Kishimoto T, Yoshizaki K (2000) Improvement in Castleman’s disease by humanized anti-interleukin-6 receptor antibody therapy. Blood 95, 56–61. [PubMed] [Google Scholar]
- Oakes SG, Petry RW, Ziegler RJ, Pozos RS (1981) Electrophysiological changes of HSV-1-infected dorsal root ganglia neurons in culture. J. Neuropathol. Exp. Neurol 40(4), 380–389. [DOI] [PubMed] [Google Scholar]
- Pachuau J, Martin-Caraballo M (2007a) Expression pattern of T-type Ca2+ channels in embryonic chick nodose ganglion neurons. Dev. Neurobiol 67(14), 1901–1914. [DOI] [PubMed] [Google Scholar]
- Pachuau J, Martin-Caraballo M (2007b) Extrinsic regulation of T-type Ca(2+) channel expression in chick nodose ganglion neurons. Dev Neurobiol 67(14), 1915–1931. [DOI] [PubMed] [Google Scholar]
- Paludan S (2001) Requirements for the Induction of Interleukin-6 by Herpes Simplex Virus-Infected Leukocytes. J. Virol 75(17), 8008–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E (2003) Molecular physiology of low-voltage-activated T-type calcium channels. Physiol. Rev 83, 117–161. [DOI] [PubMed] [Google Scholar]
- Ramer MS, Murphy PG, Richardson PM, Bisby MA (1998) Spinal nerve lesioninduced mechanoallodynia and adrenergic sprouting in sensory ganglia are attenuated in interleukin-6 knockout mice. Pain 78, 115–121. [DOI] [PubMed] [Google Scholar]
- Storey N, Latchman D, Bevan S (2002) Selective internalization of sodium channels in rat dorsal root ganglion neurons infected with herpes simplex virus-1. J. Cell Biol 158(7), 1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaka Y, Miyazaki D, Yakura K, Haruki T, Inoue Y (2010) Induction of IL-6 in Transcriptional Networks in Corneal Epithelial Cells after Herpes Simplex Virus Type 1 Infection. Investigative Opthalmology & Visual Science 51(5), 2441. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic-Todorovic V (2011) T-type voltage-gated calcium channels as target for the development of novel pain therapies. Br. J. Pharmacol 163(3), 484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi T, Pachuau J, Shepherd A, Dey D, Martin-Caraballo M (2009) CNTF-evoked activation of JAK and ERK mediates the functional expression of T-type Ca2+ channels in chicken nodose neurons. J. Neurochem 108(1), 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver E, Zamora F, Hearne J, Martin-Caraballo M (2015) Posttranscriptional regulation of T-type Ca 2+ channel expression by interleukin-6 in prostate cancer cells. Cytokine 76(2), 309–320. [DOI] [PubMed] [Google Scholar]
- Wolfe JT, Wang H, Perez-Reyes E, Barrett PQ (2002) Stimulation of recombinant Ca(v)3.2, T-type, Ca(2+) channel currents by CaMKIIgamma (C). J Physiol 538(Pt 2), 343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JN, Bevan SJ, Coote PR, Dunn PM, Harmar A, Hogan P, Latchman DS, Morrison C, Rougon G, Theveniau M (1990) Novel cell lines display properties of nociceptive sensory neurons. Proc. Biol Sci 241(1302), 187–194. [DOI] [PubMed] [Google Scholar]
- Wuest TR, Carr DJ (2008) The role of chemokines during herpes simplex virus-1 infection. Front Biosci 13, 4862–4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Frey TK, Yang JJ (2009) Viral calciomics: Interplays between Ca2+ and virus. Cell Calcium 46(1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QJ, Hsia SC, Martin-Caraballo M (2017) Regulation of T-type Ca2+ channel expression by herpes simplex virus-1 infection in sensory-like ND7 cells. J. Neurovirol 23(5), 657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]