

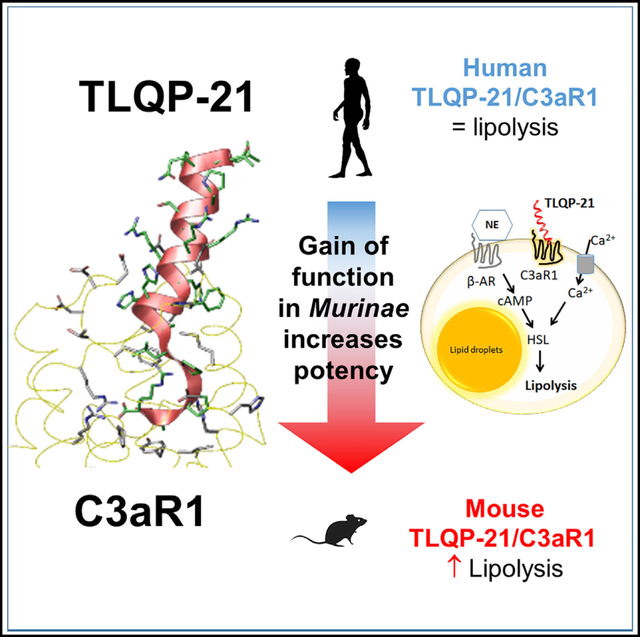
SUMMARY
Structural and functional diversity of peptides and GPCR result from long evolutionary processes. Even small changes in sequence can alter receptor activation, affecting therapeutic efficacy. We conducted a structure-function relationship study on the neuropeptide TLQP-21, a promising target for obesity, and its complement 3a receptor (C3aR1). After having characterized the TLQP-21/C3aR1 lipolytic mechanism, a homology modeling and molecular dynamics simulation identified the TLQP-21 binding motif and C3aR1 binding site for the human (h) and mouse (m) molecules. mTLQP-21 showed enhanced binding affinity and potency for hC3aR1 compared with hTLQP-21. Consistently, mTLQP-21, but not hTLQP-21, potentiates lipolysis in human adipocytes. These findings led us to uncover five mutations in the C3aR1 binding pocket of the rodent Murinae subfamily that are causal for enhanced calculated affinity and measured potency of TLQP-21. Identifying functionally relevant peptide/receptor co-evolution mechanisms can facilitate the development of innovative pharmacotherapies for obesity and other diseases implicating GPCRs.
In Brief
GPCRs and neuropeptide ligands are under intense evolutionary pressure and are major pharmacological targets. Sahu et al. identify a cluster of mutations within the C3aR1 receptor and the TLQP-21 peptide in the Murinae subfamily of rodents, resulting in enhanced binding affinity and potency, leading to potentiation of adrenergic-receptor-induced lipolysis.
Graphical Abstract
INTRODUCTION
G protein-coupled receptors (GPCR) are one of the largest membrane protein families in eukaryotes, with over 800 different GPCRs found in the human proteome (Shimada et al., 2019). Importantly, GPCRs are associated with a large number of human diseases (Tschöp et al., 2016; Wacker et al., 2017), a finding that has stimulated, over the last several decades, increased interest in identifying their endogenous ligands and mechanism of activation as well as the development of pharmacological agonists and antagonists with improved therapeutic efficacy (Hauser et al., 2017). In this respect, obesity and obesity-associated diseases, such as type 2 diabetes, hypertension, etc., are a therapeutic area in which the study of the biological mechanisms regulated by peptides/GPCRs and the development of drugs based on these mechanisms have flourished (Rodgers et al., 2012). For example, insulin and glucagon-like peptide 1 analogs are currently in the market or in advanced stages of clinical trials for several common diseases (Clemmensen et al., 2019; Müller et al., 2018). Pharmacotherapy for obesity aimed at increasing lipolysis and free fatty acid utilization would be ideal because it would directly target the excess in fat stores. Lipolysis is a complex and finely regulated multistep biological process in which norepinephrine (NE), released by the postganglionic sympathetic neurons innervating adipose fat depots, activates the GPCR β-adrenergic receptors (β-ARs) on adipocyte membranes and the downstream signaling cascade, including cyclic AMP (cAMP)-dependent activation of protein kinase A (PKA), and phosphorylation and translocation of hormone-sensitive lipase (HSL) to lipid droplets (Arner, 1999; Granneman and Moore, 2008; Lafontan and Langin, 2009; Lönnqvist et al., 1992; Reilly and Saltiel, 2017). As an end result of lipolysis, free fatty acids and glycerol are released from adipocytes and delivered to other metabolic organs and undergo β-oxidation. Obesity perturbs the physiological regulation of lipolysis, leading to lipolytic catecholamine resistance, characterized by, among other molecular changes, downregulation of β-ARs and HSL (Arner, 1999; Lafontan and Langin, 2009; Lönnqvist et al., 1992). Identifying new mechanisms that can overcome lipolytic catecholamine resistance and safely enhance lipolysis would be extremely helpful for obese patients because it would directly target excessive fat mass (Cero et al., 2016; Reilly and Saltiel, 2017). However, previous attempts largely failed because most potent lipolytic sympathomimetic drugs (such as β-AR agonists) induced adverse cardiovascular effects or aggravated metabolic syndrome (Clapham and Arch, 2011; James et al., 2010; Rodgers et al., 2012). Several neurotransmitters and neuropeptides, often co-secreted with NE by sympathetic nerves, endocrine glands, or macrophages/adipocytes and targeting GPCRs on adipocyte membranes play a key role as sensitizers of adrenergic-induced lipolysis (Bandyopadhyay et al., 2012; Nguyen et al., 2011; Pellegrinelli et al., 2018; Pirzgalska et al., 2017; Villarroya and Vidal-Puig, 2013; Whittle et al., 2012). These sensitizer mechanisms have the potential to serve as targets alternative to or synergistic with β-ARs agonists for pharmacotherapy of obesity, directly targeting excess adiposity, but so far, this approach remains poorly understood and investigated. In the present study, we focused the complement 3a receptor (C3aR1) (Hollmann et al., 1998), a receptor expressed in adipocytes (Mamane et al., 2009; Quell et al., 2017), and its ligand, the VGF-derived neuropeptide TLQP-21, a sensitizer of β-AR-induced lipolysis shown previously to oppose obesity and normalize molecular markers of catecholamine resistance without causing cardiovascular side effects, a chronic increase in circulating fatty acids, or ectopic fat deposition in mice, overall suggesting a concomitant increase in tissue β-oxidation (Bartolomucci et al., 2006; Cero et al., 2014, 2016; Fargali et al., 2014; Guo et al., 2018; Jethwa et al., 2007; Possenti et al., 2012). Here, combining pharmacology, homology modeling, and evolutionary analysis, we first characterized the mechanism of TLQP-21 potentiation of β-AR-induced lipolysis in adipocytes and subsequently identified a unique peptide/receptor co-evolution in the Murinae subfamily of rodents, which explains its pharmacological efficacy by virtue of enhanced hydrophobicity representing a gain of function for a neuropeptide that becomes a potent agonist at C3aR1. Because we also demonstrated that changes in gene expression of critical nodes in TLQP-21/C3aR1-mediated lipolysis are conserved in obesity in mice and humans, and because the rodent peptide retains its biological activity in humans (potentiation of adrenergic-induced lipolysis), our discovery can pave the way for investigation of this molecular target to develop innovative pharmacotherapies for obesity and associated diseases.
RESULTS
An Essential Role of C3aR1 in TLQP-21-Induced Potentiation of Adrenergic-Induced Lipolysis
To test the hypothesis that C3aR1 is required for TLQP-21-mediated lipolysis and to understand the molecular mechanism of action of TLQP-21, we generated C3aR1 knockdown (KD) 3T3-L1 cell lines. Cells were infected with 3 different lentiviruses harboring short hairpin RNA (shRNA) against C3aR1, or a non-targeting shRNA was used as a control. qPCR showed strong depletion of C3aR1 in KD cells (Figure 1A). We selected one of the KD lines for the experiments and replicated critical findings in a second cell line (see below). Adipogenesis and expression of key genes involved in adipogenesis and lipolysis, such as PPAR-γ, leptin, βARs, HSL, etc., were normal, except for a modest increase in β3AR (Figure 1A; Figure S1). C3aR1 KD and shRNA control cells were differentiated in adipocytes and incubated with TLQP-21 (using previously validated doses; Cero et al., 2016) in the presence or absence of the nonselective β-AR agonist isoproterenol (ISO), essentially as established previously (Cero et al., 2016) and as described in the STAR Methods. As expected (Cero et al., 2016; Possenti et al., 2012), TLQP-21 enhanced ISO-induced lipolysis in control cells without being pro-lipolytic per se (Figure 1B). Conversely, TLQP-21 failed to potentiate ISO-induced lipolysis in C3aR1 KD cells (Figure 1B). Essentially the same phenotype was obtained in a second independent cell line harboring a different shRNA targeting C3aR1 (Figure S1F), ruling out nonspecific targeting of the shRNA. Next we carried out immunoblots and densitometry analysis for phosphorylated hormone sensitive lipase (pHSL) and phosphorylated extracellular signal–regulated kinase (pERK), the key signaling nodes in the TLQP-21 pro-lipolytic pathway (Cero et al., 2016; Possenti et al., 2012). Consistent with previous findings, incubating 3T3-L1 adipocytes with TLQP-21 enhanced pERK and potentiated ISO-induced phosphorylation of ERK and HSL (Figure 1C). Importantly, experiments conducted in C3aR1 KD cells established that C3aR1 expression is necessary for TLQP-21-induced phosphorylation of ERK and potentiation of ISO-induced pERK and pHSL (Figure 1C). Overall, our data demonstrate that C3aR1 is necessary for TLQP-21-mediated lipolysis via ERK activation of HSL in murine adipocytes. However, the upstream signals remained unknown. To answer this question, we focused on the mechanism of peptide-mediated calcium uptake.
Figure 1. C3aR1 Is Required for TLQP-21-Induced Lipolysis.

(A) Generation and characterization of stable C3aR1 KD cell lines. Shown is qPCR analysis of C3aR1 mRNA in 3 independent shRNA lines in comparison with the scramble shRNA C3aR1 KD cell line expressing normal leptin and PPAR-γ levels and normal differentiation using oil red O staining.
(B) Free glycerol release in C3aR1 KD and control cells. C3aR1 KD prevents TLQP-21 (100 nM) potentiation of isoproterenol (ISO, 50 nM)-induced lipolysis (cell line x treatment F(3,58) = 5.07, p < 0.005, N = 4–14).
(C) Western blot analysis of pERK and pHSL in C3aR1 KD and control adipocytes incubated for 5 min with ISO (50 nM) or TLQP-21 (10 μM). C3aR1 KD prevented TLQP-21-induced phosphorylation of HSL (cell line × treatment F(3,24) = 4.7, p < 0.01, N = 4) and ERK (cell line × treatment F(3,24) = 26.1, p < 0.00001, N = 4).
Tukey’s post hoc tests; NS, not significant; *p < 0.05, **p < 0.01, ***p < 0.001. Data are expressed as average and SEM.
See also Figure S1.
TLQP-21 Mediates Lipolysis via Calcium Influx from the Extracellular Compartment
TLQP-21-mediated lipolysis can be prevented by pretreatment with the calcium chelant EGTA, suggesting calcium influx from the extracellular compartment (Cero et al., 2016). However, the mechanism is not well understood. We carried out PCR-based microarray experiments and found a number of calcium channels expressed in both 3T3-L1 fibroblasts and adipocytes (Table S2), including transient receptor potential channel 1 (TRPC1), TRPC5, and TRPC6, channels that have been identified previously in this cell type (Sukumar et al., 2012). Based on the TLQP-21 pharmacological profile, we tested the hypothesis that C3aR1 is functionally coupled to a putative TRPC channel upon TLQP-21 activation, leading to calcium influx from the extracellular compartment. To test this hypothesis, we used two methods, the non-ratiometric Fluo-4 Ca2+ indicator and radiometric Ca452+ assays. Fluo-4, unlike ratiometric indicators such as Fura-2-acetoxymethyl ester (FURA-2AM), is less prone to quenching of the signal by lipid droplets in adipocytes and more sensitive to transient calcium influx, which would be undetectable by FURA-2AM (Cero et al., 2016; Sukumar et al., 2012). Using the Fluo-4 assay, we established that TLQP-21 rapidly and concentration-dependently increased intracellular calcium ([Ca2+]i) in 3T3-L1 cells (Figures S2A and S2B). This effect is specific to TLQP-21/C3aR1 activation, as demonstrated by the following: (1) the TLQP-21 mutant R21A (Cero et al., 2014) did not increase [Ca2+]i (Figure S2A); (2) the C3aR1 antagonist SB290157 antagonized TLQP-21-mediated calcium influx (Figure 2A); (3) C3aR1 KD prevented the TLQP-21-mediated increase in [Ca2+]i (Figure 2B; Figure S2C). Additionally, multiple pharmacological experiments allowed us to exclude a role of the C5a receptor in TLQP-21-induced increases in [Ca2+]i (Figures S3A and S3B). Next, to unambiguously establish that TLQP-21 causes an increase in calcium from the extracellular compartment, we used a high-sensitivity radiometric Ca452+ assay. TLQP-21 caused significant Ca452+ uptake compared with control treatment, whereas the R21A mutant consistently failed to elicit calcium influx (Figure 2C; Figure S4A). Additionally, ISO per se did not increase Ca452+ uptake under our experimental conditions and did not potentiate TLQP-21-mediated effects (Figure 2D), further supporting the hypothesis that the first messengers downstream of NE- and TLQP-21-mediated signaling pathways are independent in our cellular model.
Figure 2. TLQP-21 Mediates Extracellular Calcium Influx in 3T3-L1 Cells.

(A) TLQP-21 (10 μM) increases [Ca2+]i (as measured by Fluo-4 fluorescence) during a 60-s incubation, and this effect is antagonized by the C3aR1 antagonist SB290157 (F(4,178) = 18.9, p < 0.00001, N = 25–60 cells from 2 independent experiments).
(B) TLQP-21 (10 μM) increases [Ca2+]i in non-targeting (NT) shRNA controls but not C3aR1 KD cells (F(1,102) = 17.05, p < 0.00001, N = 20–29 cells from 2 independent experiments). Additional data can be found in Figure S2C.
(C) TLQP-21 but not the R21A mutant increases uptake of Ca452+ from the medium (F(1,14) = 17.3, p < 0.001, N = 3–9 from 3 independent experiments.
(D) ISO does not modify TLQP-21-induced Ca452+ uptake from the medium (F(1,8) = 17.3, p < 0.005, N = 3).
(E) TLQP-21-induced potentiation of ISO-induced lipolysis is blocked by SKF-96365 (F(1,35) = 65.8, p < 0.0001; N = 5–6; ISO = 50 nM, TLQP-21 = 100 nM, SKF-96365 = 20 μM).
(F) TLQP-21-induced increase in [Ca2+]i, measured by Fluo-4 fluorescence, is blocked by SKF-96365 (F(1,126) = 6.8, p < 0.0001; N = 8–50 cells from 2 different experiments; ISO = 50 nM, TLQP-21 = 10 μM, SKF-96365 = 20 μM, UTP =10 μM) (Figure S4 presents the dose response curves).
Tukey’s post hoc tests; *p < 0.05, **p < 0.01, ***p < 0.001. Data are expressed as average and SEM.
See also Figures S2 and S4 and Table S2.
Next we directly tested the hypothesis that TLQP-21 mediates lipolysis via activation of a TRPC channel. We first measured TLQP-21-mediated calcium influx in 3T3-L1 cells in the presence or absence of the pan-specific TRPC inhibitor SKF-96365 (Singh et al., 2010). SKF-96365 blocked the TLQP-21-induced increase in [Ca2+]i, whereas the response to the purinergic agonist uridine triphosphate (UTP) was maintained even in the presence of a mild SKF-96365-induced elevation of basal fluorescence (Figure 2E; Figure S4B). Next we incubated 3T3-L1 adipocytes with TLQP-21 in the presence or absence of ISO and in the presence or absence of SKF-96365. SKF-96365 concentration-dependently blocked TLQP-21-mediated enhancement of ISO-induced lipolysis without affecting basal or ISO-induced lipolysis (Figure 2F; Figures S4C and S4D). Because SKF-96365 can potentially inhibit store-operated calcium release (Sabourin et al., 2016), we also carried out a second set of lipolysis experiments, incubating adipocytes with AncoA4, a selective inhibitor of store-operated calcium release (Sadaghiani et al., 2014). Unlike SKF-96365, AncoA4 did not prevent TLQP-21-mediated lipolysis (Figure S4E), further strengthening the conclusion that TLQP-21 activates calcium influx from the extracellular compartment (Figure 2). Finally, Ca2+/calmodulin-dependent protein kinase II (CaMKII) is activated by increased [Ca2+]i and causes phosphorylation of the ERK/HSL pathways (Rapold et al., 2013), which are critical nodes in TLQP-21 signaling (Cero et al., 2016). Consistently, the CaMKII inhibitor KN-62 prevented TLQP-21-induced potentiation of ISO-induced lipolysis without affecting basal or ISO-induced lipolysis per se (Figure S4F). Overall, our results demonstrate that the TLQP-21 lipolytic mechanism is mediated by C3aR1 activation of a TRPC channel that is responsible for calcium uptake from the extracellular compartment and activation of the CaMKII/ERK signaling cascade, resulting in potentiation of adrenergic-induced HSL phosphorylation.
Having identified the TLQP-21 mechanism of action in vitro, we aimed to establish whether the identified signaling pathway is conserved in mouse and human obesity.
Obesity Results in Low βAR, HSL, and TRPC1 Expression and High C3aR1 Expression in White Adipose Tissue of Mice and Humans
The current knowledge of TLQP-21/C3aR1-mediated biological effects is limited to rodent models. Therefore, we first aimed to determine whether the critical molecular nodes in the TLQP-21/C3aR1 lipolytic pathway identified in this study and elsewhere are conserved in adipose tissue in humans (Figure 3A). We quantified the expression of several genes in 208 well-characterized and non-diabetic humans (see STAR Methods for a description of the study population). We observed a negative correlation between BMI and/or percent fat mass with β2ARs, TRPC1, and HSL and a positive association of the same biometrical parameters with C3aR1 (Figure 3B). Consistently, 9 weeks of a high-fat diet (HFD) induced very similar molecular changes in obese mice, with a significant increase in C3aR1 expression and downregulation of βARs (particularly β3AR), HSL, and TRPC1 (Figures 3C and 3D). Conversely, the expression of C5aR1 and C5aR2, receptors closely related to C3aR1, is not significantly associated with obesity in humans or in mice (Figure S3C). These results suggest that the molecular pathway critical for TLQP-21-mediated lipolysis is conserved in obese mice and humans. Specifically, although C3aR1 is upregulated in adipose tissue in obesity in both species, the critical nodes downstream of C3aR1 (i.e., TRPC1 and HSL) are downregulated in obesity. This finding is in line with and adds to the existing literature on molecular changes observed in lipolytic catecholamine resistance (Arner, 1999; Cero et al., 2016; Reilly and Saltiel, 2017), suggesting that dysregulation of this lipolytic pathway can be causal for fat accumulation.
Figure 3. Expression of Key Signaling Nodes in the TLQP-21/C3aR1-Mediated Pathway Is Conserved in Obese Mice and Humans.

(A) Simplified model of key nodes in the TLQP-21/C3aR1 pathway.
(B) Association of normalized gene expression in human adipose tissue biopsies with BMI (coefficient of determination (R2):C3aR1 = 9.4%, ADRB2 = 2.2%, ADRB3 = 4.2%, TRPC1 = 2.7%, LIPE = 14%) and percent fat mass (R2: C3aR1 = 6.4%, ADRB2 = 5.3%, ADRB3 = 2.0%, TRPC1 = 7.0%, LIPE = 5.5%).
(C) Body weight and percent fat mass in mice fed a standard diet (STD) or high-fat diet (HFD) for 9 weeks starting at 9 weeks of age.
(D) Difference in gene expression in subcutaneous WAT in mice fed a STD and HFD (N = 6).
The silhouettes of Homo sapiens and Mus musculus are from http://www.phylopic.org. *p < 0.05, **p < 0.01. Data are expressed as average and SEM.
See also Figure S3.
Modeling and Simulation of TLQP-21 Binding to C3aR1
Our data identified TLQP-21/C3aR1 as a promising translational target for obesity. To shed light on how the mouse and human peptides activate C3aR1, we conducted homology modeling and targeted pharmacological experiments. The activity of human TLQP-21 (hTLQP-21) has been shown to be lower at the human, murine, and hamster C3aR1 compared with mouse TLQP-21 (mTLQP-21) (Cero et al., 2014; Hannedouche et al., 2013). To establish the molecular basis of peptide/receptor binding and activity, we conducted modeling and simulation of hTLQP-21 and mTLQP-21 bound to human C3aR1 (hC3aR1) or mouse C3aR1 (mC3aR1), respectively. Homology modeling of hC3aR1 and mC3aR1 was carried out based on our previous structural data (Cero et al., 2014) and the recently solved X-ray crystallographic structure of C5aR1 (Liu et al., 2018), a much more closely evolved receptor to C3aR1 compared with the human chemokine receptor CXCR4 used previously to model C3aR1 (Reid et al., 2013). This X-ray structure by Liu et al. (2018) was strategically chosen over a related structure (Robertson et al., 2018) because of its resolved orthosteric site with bound PMX53, a cyclic peptide. Notably, the carbon α root-mean-square deviation (CαRMSD) between the two structures (PDB: 5O9H and 6C1Q) was 0.87 Å, which does not provide a significant advantage for structural refinement (Figure S5A). hTLQP-21 and mTLQP-21 bound to C3aR1 were modeled as an ideal α helix, with their C terminus mimicking the observed mode of binding of the cyclic peptide PMX53 using Maestro’s builder (STAR Methods). Consistent with previous modeling of C3aR1 using smaller fragments of C3a and small molecules (Reid et al., 2013), our final homology model lacks the extracellular region consisting of amino acids 178–324, known to bind the 77-amino-acid, full-length C3a that can be removed without affecting C3a binding/activity (Chao et al., 1999).
Our model revealed that mTLQP-21 retained its secondary helical structure in the presence of mC3aR1 with a very low average CαRMSD of 1.7 Å (Figure S5B; Video S3). Importantly, although hTLQP-21 had a much higher average CαRMSD of 4.0 Å in the presence of hC3aR1, indicating partial unfolding, mTLQP-21 retained a lower average CαRMSD of 2.1 Å and a relative α-helicity of 0.7 Å in the presence of hC3aR1, indicating a more stable helical structure relative to its unbound form (Figures 4A–4C; Figures S5C and S5D), even when extending the simulation to up to 500 ns (Figures S5C and S5D). As expected (Cero et al., 2014; Chakraborty et al., 2015), both hTLQP-21 and mTLQP-21 spontaneously unfolded to completion in water, with an average CαRMSD of 7.9 and 8.6 Å, respectively (Figure 4C; Figure S5B; Videos S1 and S2). The CαRMSD and carbon α root-mean-square fluctuation (CαRMSF) of mC3aR1 and hC3aR1 for all three bound states remained less than 3.0 Å throughout the molecular dynamic (MD) simulations, indicative of a stable GPCR membrane-bound structure (Figures S5C and S5D; Videos S3, S4, and S5). These data complement previous binding results showing a half-maximal effective concentration (EC50) for mTLQP-21 to be approximately seven times lower (10.3 μM) than that of hTLQP-21 (68.8 μM) in human cells expressing hC3aR1 (Cero et al., 2014). Most importantly, the final conformation of mTLQP-21 bound to hC3aR1 showed specific salt bridge interactions between R21 of the peptide with surrounding D417, R161, and R340 in C3aR1, which validates their importance for receptor binding and activation (Figure 4B; Figure S6A). Amidation of the TLQP-21 peptide C terminus prevents TLQP-21 biological activity (Cero et al., 2014), which could result in loss of salt bridge interactions between its terminal carboxylate group to D417, R161, and R340 (Figure 4B). Furthermore, it could further explain the previous site-directed mutagenesis study (Sun et al., 1999), which showed that the R161A, R340A, and D417A mutations result in complete loss of C3a binding affinity to C3aR1. The presence of two consecutive prolines (P18 and P19), near the C terminus of TLQP-21, is a partial disruptor of the α helix structure motif that was observed both in our earlier NMR study (Cero et al., 2014) and in our simulation. Interestingly, the bound TLQP-21 within the C3aR1 retained a high level of α-helical structure in our simulation in the absence of the 178–324 extracellular region, confirming that this extracellular loop is not required for binding of small peptides. As shown in Figure 4B, a significant number of acidic residues are located at the opening of the hC3aR1 binding site that form salt bridge interactions with the conserved R9RRHXHH15 motif of both human and mouse TLQP-21. These include D167, D325, D326, and D327, located at the truncated extracellular loop 2 (ECL2) region between transmembrane helix 4 and 5 (TM4 and TM5), as well as E406, located on ECL3 adjacent to TM6. Among them, D167, D326, and E406 in hC3aR1 were found to be conserved as aspartate or glutamate in both EL2 and EL3 among other mammalian C3aR1s (Table S3). Overall, based on our modeling and simulation study, the C-terminal motif (PPAR in mTLQP-21 and PPSR in hTLQP-21) of TLQP-21 was identified as the substrate recognition motif required for binding and activation of C3aR1. Both C-terminal motifs contain R21, which is essential for C3aR1 activation and is also found in the C terminus LAR motif of C3a (Reid et al., 2013).
Figure 4. Homology Modeling and Biological Significance of TLQP-21/C3aR1 in Humans.

(A) Homology modeling of hC3aR1 (yellow ribbon) with bound mTLQP-21 (red ribbon).
(B) Binding site of hC3aR1 (labeled in black) with bound mTLQP-21 (labeled in green) after 50 ns simulation showed that TLQP-21 R21 forms multiple salt bridges with nearby R161, R340, and D417. mTLQP-21 containing the conserved RRRH motif was found to be surrounded by D167, D325, D326, D327, and E406, which could stabilize the α helix portion of TLQP-21 upon C3aR1 binding.
(C) Molecular dynamics simulation of TLQP-21 (mouse [m], human [h]) in water and in complex with hC3aR1. The CαRMSDs are shown for unbound mTLQP-21 (salmon) and unbound hC3aR1 (light blue), hC3aR1 (gray and black), mTLQP-21-bound hC3aR1 (red), and hTLQP-21-bound hC3aR1 (blue). The simulation showed that mTLQP-21 binding to hC3aR1 retained its secondary helical structure compared with the partially unfolded hTLQP-21 bound to hC3aR1 and the completely unfolded m/hTLQP-21 in water.
(D) mTLQP-21 potentiates ISO (10 nM)-induced lipolysis in human adipocytes (F(9,23) = 59.4, p < 0.0001). **p < 0.00.
(E) Phylogenetic analysis of the combined C3a and C3aR1 sequence of the 87 species for which the three proteins (VGF, C3, and C3aR1) are present in NCBI. The color highlights the sequence at the C terminus of TLQP-21. Red, PAR; black (blue for humans), PSR; purple, other sequences (full details are presented in Table S3). The specific Murinae and Cricetinae subfamilies have the exclusive invariant PAR motif in TLQP-21. The phylogenetic tree was built using the Itol software (https://itol.embl.de).
(F) Diagrams of critical motifs in humans and mice. The silhouettes of Homo sapiens and Mus musculus are from http://www.phylopic.org. Data are expressed as average and SEM.
See also Figures S5–S7 and Table S3.
Mouse TLQP-21 Enhances Adrenergic-Induced Lipolysis in Human Adipocytes
To determine the functional significance of our modeling and receptor binding data (Cero et al., 2014), we used human mesenchymal stem cells differentiated into adipocytes as a model system (Figure S7). Human adipocytes were incubated with different doses of either hTLQP-21 or mTLQP-21 in the presence or absence of ISO using a protocol similar to what we described previously for murine adipocytes. The results demonstrate that mTLQP-21 dose-dependently potentiates ISO-induced lipolysis without affecting basal lipolysis and that the mouse peptide is more potent compared with hTLQP-21 in human cells (Figure 4D). This result matches the results obtained in rodent adipocytes and highlights a conserved functional role of C3aR1 in mediating lipolysis in rodent and humans systems. Importantly, the lipolysis data provide pharmacological validation of the modeling data, which demonstrate that the average CαRMSD is much higher for hTLQP-21 compared with mTLQP-21 in the presence of hC3aR1 (Figure 4C). Furthermore, the CαRMSD of mTLQP-21 is lower in the presence of mC3aR1 compared with hC3aR1, a finding that supports the observation that mTLQP-21 potentiate ISO-induced lipolysis in rodent cells with doses in the nanomolar range (Figure 1; Cero et al., 2016), whereas micromolar-range doses are required in human cells. Overall, these results demonstrate that mTLQP-21 potentiates adrenergic-induced lipolysis in human adipocytes.
Co-evolution of Critical Amino Acids Explains the Biological Activity of TLQP-21/C3aR1
To explore the basis of the surprising outcome of our modeling and pharmacological experiments and to understand the sequence specificity required for activation of hC3aR1 (which would be relevant for future drug discovery endeavors), we examined all full-chain sequences of mammalian TLQP-21 and C3aR1 available from the NCBI and compared them with C3a (Table S3). Multiple sequence alignment was carried out for each protein to quantify the occurrence of established binding motif and to identify other potential candidates. Of the 71 non-redundant mammalian TLQP-21 sequences found in the VGF pro-peptide, the C-terminal PSR motif was found to be the most predominant (56%), followed by PSH (14%) (Table 1). Together with other observed motifs, 96% of all available mammalian sequences require a positively charged arginine or histidine at the C terminus of TLQP-21, which strongly supports our identified salt bridge interaction with D417 as an essential and structurally conserved interaction for hC3aR1 activation (Figure 4B). In all 15 primate species analyzed, including humans, the TLQP-21 C-terminal motif was invariably PSR (Figures 4E and 4F; Table 1). In comparison, only 52% of the rodent species analyzed had the C-terminal PSR motif. However, all species included in the Murinae (including mice and rats) or the Cricetinae (including Cricetulus griseus, used to generate Chinese hamster ovary [CHO] cells, see Molteni et al., 2017; and closely related to Phodopus sungorus, see Jethwa et al., 2007) subfamilies of rodents invariably expressed an alanine at the 20th position of TLQP-21 (Figures 4E and 4F; Table 1). Based on the present data, the PSR motif, commonly observed across other mammalian species, can be considered the ancestral sequence in mammals, whereas the PAR sequence represents an evolved sequence in a subgroup of rodents (Figure S6).
Table 1.
Sequences of Critical Amino Acid Motifs in TLQP-21, C3a, and C3aR1
| TLQP-21 | |||||
| Motif | All Species | Primate | Rodent | ||
| All | Murinae | Cricetinae | |||
| PSR | 40 | 15 | 10 | 0 | 0 |
| PAR | 9 | 0 | 9 | 4 | 4 |
| PSH | 10 | 0 | 0 | 0 | 0 |
| Others | 12 | 0 | 0 | 0 | 0 |
| Total | 71 | 15 | 19 | 4 | 4 |
| C3a | |||||
| Motif | All Species | Primate | Rodent | ||
| All | Murinae | Cricetinae | |||
| LAR | 58 | 9 | 18 | 4 | 4 |
| LGT | 22 | 7 | 0 | 0 | 0 |
| Others | 2 | 0 | 0 | 0 | 0 |
| Total | 82 | 16 | 18 | 4 | 4 |
| C3aR1 | |||||
| Motif | All Species | Primate | Rodent | ||
| All | Murinae | Cricetinae | |||
| DHVS | 49 | 8 | 2 | 0 | 0 |
| DHVF | 12 | 12 | 0 | 0 | 0 |
| DHVC | 8 | 4 | 0 | 0 | 0 |
| DHMS | 5 | 0 | 5 | 4 | 1 |
| Others | 36 | 2 | 13 | 0 | 3 |
| Total | 110 | 26 | 20 | 4 | 4 |
To compare the evolutionary history of the C-terminal XR motif required for activation of C3aR1 (Cero et al., 2014; Doolen et al., 2017; Reid et al., 2013), we further examined the available mammalian full-length sequences of C3a (Table 1). Of the 82 species examined, 56 species possess the C-terminal LAR motif (71%), followed by 22 species with an LGT motif (27%). Interestingly, all rodents exclusively possess the terminal LAR motif in C3a, whereas in primates, 56% (including humans) express the LAR sequence and 44% express the LGT sequence, which have never been explored for their C3aR1 activity, to the best of our knowledge.
Subsequently, we examined the specific sequence motif within the identified C3aR1 binding site. D417, crucial for the salt bridge interaction of the TLQP-21 C-terminal arginine 21 side chain, was conserved in all 110 mammalian species analyzed (Table S3A). Furthermore, when exploring the contiguous amino acids, we found that the most common motif across taxa was the DHV* (67%), with 92% of all primates expressing a DHVS/C/F motif (Table 1; Table S3A). Conversely, there was larger variability in rodents, with the predominant C3aR1 motif being DHMS (25%). However, all species belonging to the Murinae subfamily of rodents (including mice and rats) invariantly expressed the DHMS motif (Figure 4F).
Finally, to explain the increased binding affinity of mTLQP-21 at both mC3aR1 and hC3aR1 compared with hTLQP-21 (Figure 4C; Figure S3B), we identified the amino acid residues within 4 Å of the TLQP-21 C-terminal PPAR/PPSR motif in the h/mC3aR1 substrate binding site. Of the 47 mammals available in NCBI, consisting of all three reported TLQP-21, C3a, and C3aR1 sequences, 9 of 17 amino acid residues were highly conserved among all mammals (from 96% to 100% identity across all species; Table S3B), including the critical R161, R340, and D417A. Importantly, the only sequence variability differentiating the human and the mouse receptor corresponded to V103, S400, L413, V419, and C420 in hC3aR1 compared with the corresponding I, L, V, M, and S residues in mC3aR1 (Table 2; Table S3C). Although these mutations are still permissive for binding of the PPAR or PPSR motifs, the increase in hydrophobic residue makeup going from the hC3aR1 (VSLVC, 53% hydrophobicity) to the mC3aR1 (ILVMS, 59% hydrophobicity) binding sites is consistent with (1) the higher binding affinity and relative higher potency of mTLQP-21 (PPAR motif, 75% hydrophobicity) to mC3aR1 than to hC3aR1 (Figure 4C; Figures S5C and S5D) and (2) the higher binding affinity and higher potency of mTLQP-21 at hC3aR1 compared with hTLQP-21 (PPSR motif, 50% hydrophobicity) (Figures 4C and 4D).
Table 2.
Amino Acid Residues within 4 Å of the TLQP-21 C-Terminal PPAR/PPSR Motif in the C3aR1 Binding Site
| VGF_Motif | C3aR_Accession | Residue (Numbers According to the Human Receptor) | Species | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 81 | 82 | 88 | 99 | 102 | 103 | 161 | 340 | 393 | 400 | 413 | 414 | 417 | 418 | 419 | 420 | 421 | |||
| PPSR | NP_001313406 | H | L | W | P | I | V | R | R | Y | S | L | M | D | H | V | C | I | Homo sapiens sapiens |
| PPAR | NP_033909 | H | L | W | P | I | I | R | R | Y | L | V | M | D | H | M | S | I | Mus musculus domesticus |
To directly test the hypothesis that the five residues in the C3aR1 receptor binding pocket that are mutated in mice (V103I, S400L, L413V, V419M, and C420S; Figure 5A) can explain the receptor activation potency of TLQP-21, we conducted in silico and in vitro experiments in wild-type (WT) and murinized hC3aR1 (m*C3aR1, generated by mutating the 5 aforementioned amino acid residues). First we conducted a mutagenesis thermodynamic cycle to evaluate the binding affinity (ΔGbind) by using the molecular mechanics generalized born solvent-accessible model (see STAR Methods for details). Murinizing hTLQP-21 at the 5 critical residues led to a decrease in ΔGbind in the presence of both hTLQP-21 and the mTLQP-21 (Figure 5B), indicating increased binding affinity. Consistent with the result of the lipolysis assay (Figure 4D), the most negative ΔGbind was obtained for m*C3aR1 in the presence of mTLQP-21 (Figure 5B). Next we conducted C3aR1 β-arrestin recruitment assays as described previously (Cero et al., 2014; Kroeze et al., 2015) in HTLA cells transfected with hC3aR1 or m*C3aR1. Supporting our hypothesis, and consistent with the results from the mutagenesis thermodynamic cycle, murinizing the human receptor at the five residues led to a 42-fold increase in potency for mTLQP-21 (hC3aR1, EC50 = 2.3 μM; m*C3aR1, EC50 = 0.055 μM) and a more modest 28-fold increase in potency for hTLQP-21 (hC3aR1, EC50 = 16.9 μM; m*C3aR1, EC50 = 0.59 μM) (Figures 5C, 5D, and 5F). Interestingly, the potency of the C3aR1 agonist C3a63–77 was only increased by 8.5-fold at m*C3aR1 (hC3aR1, EC50 = 3.6 μM; m*C3aR1, EC50 = 0.4 μM; Figures 5E and 5F). These results directly validate the outcome of our evolutionary analysis and demonstrate that these 5 mutations in the C3aR1 binding pocket are important for the enhanced functional potency of TLQP-21.
Figure 5. Murinizing the hC3aR1 by Site-Directed Mutagenesis Enhances TLQP-21 Binding Affinity and Potency.

(A) Murinized hC3aR1 (m*C3aR1) from the homology model, highlighting conserved amino acids (purple circles) and amino acids affected by variants in rodents (V103I, S400L, L413V, V419M, and C420S, green circles).
(B) Change in binding affinity (ΔGbind) of mouse and human TLQP-21 to hC3aR1 and m*C3aR1 was calculated based on the mutagenesis thermodynamic cycle.
(C-E) β-Arrestin recruitment Tango assay transfected with either hC3aR1 or m*C3aR1 and incubated with a range of concentrations with either mTLQP-21 (hC3aR1, EC50 = 2.3 μM; m*C3aR1, EC50 = 0.055 μM) (C), hTLQP-21 (hC3aR1, EC50 = 16.9 μM; m*C3aR1, EC50 = 0.59 μM) (D), or C3a63–77 (hC3aR1, EC50 = 3.6 μM; m*C3aR1, EC50 = 0.4 μM) (E). Data represent mean and SE of triplicates from three independent experiments.
(F) Quantitative comparison of β-arrestin recruitment functional activity as measured by log(Emax/EC50) from three independent experiments in (C)–(E) (ANOVA F(5,12) = 2.5, p = 0.032). *p < 0.05, **p < 0.01 comparing m*C3aR1 with hC3aR1. Emax, maximal effect at high drug concentrations when all the receptors are occupied by the drug. Data are expressed as average and SEM.
Overall, we identified unique peptide/receptor co-evolution in the Murinae subfamily of rodents (which includes mice and rats), which explains the increased estimated binding affinity and functional potency of the mTLQP-21 peptide at the mouse and hC3aR1 compared with the ancestral hTLQP-21 sequence. Importantly, in spite of this evolved enhanced function, the potent mouse peptide retains its ability to bind and activate the human receptor and can thus be considered a relevant template for drugs designed to target C3aR1.
DISCUSSION
GPCRs and their substrate ligands are major molecular targets for pharmacotherapies (Shimada et al., 2019; Wacker et al., 2017). However, one constraint when developing effective and safe biologic therapies is the high specificity of the amino acid motif required for optimal peptide/receptor binding and receptor activation. Subtle species-specific differences in key amino acids in peptide pharmacophore and receptor binding pockets may compromise the ability to translate promising therapeutic efficacy from animal models to humans (Lahti et al., 2012; Lau and Dunn, 2018; Shimada et al., 2019). Here, combining phylogenetic analysis, molecular dynamics, and molecular pharmacology, we established that one amino acid substitution in the TLQP-21 peptide (S20A) and 5 amino acid substitutions in the C3aR1 receptor binding site (Table 2), mutations that, based on our evolutionary analysis, likely occurred in a common ancestor of mice and rats (and other species in the Murinae and Cricetinae subfamilies), enabled the TLQP-21 neuropeptide to become a potent agonist at C3aR1. Importantly, despite humans carrying the ancestral sequence of TLQP-21 and C3aR1, the rodent peptide retains its ability to bind hC3aR1 and potentiate adrenergic-induced lipolysis in human adipocytes. Because TLQP-21 exerts anti-obesity effects in the absence of cardiac side effects while improving hypertension in rodents (Cero et al., 2016; Fargali et al., 2014; Possenti et al., 2012), our finding might pave the way to target an alternative mechanism for pharmacotherapy of obesity.
The Mechanism of TLQP-21/C3aR1-Induced Lipolysis
TLQP-21 recently emerged as the second endogenous ligand of C3aR1 (Cero et al., 2014; Coulthard and Woodruff, 2015; Doolen et al., 2017; Hannedouche et al., 2013). C3aR1, a member of the GPCR superfamily, is characterized by seven transmembrane domains and a large second extracellular loop (Klos et al., 2013; Roglic et al., 1996). Although its role in the innate immune response is well established, C3aR1 is now emerging with a much broader pattern of expression in different tissues, including adipose tissue and the CNS, and functions involving cell metabolism (Coulthard and Woodruff, 2015; Lo et al., 2014; Mamane et al., 2009). In obesity, a significant increase in adipose tissue-specific C3aR1 expression has been found (Cero et al., 2016; Mamane et al., 2009), with a concomitant increase in TLQP-21 membrane binding affinity (Possenti et al., 2012). TLQP-21 is present in a subpopulation of sympathetic nerve fibers innervating white adipose tissue (WAT) but not in adipocytes. Chronic peripheral TLQP-21 treatment in rodents increases tyrosine hydroxylase (TH) enzymatic activity and NE in WAT, decreases body weight and fat mass (Cero et al., 2016; Possenti et al., 2012), and improves glucose tolerance (Stephens et al., 2012) and obesity-induced hypertension (Fargali et al., 2014) without reported adverse effects and in the presence of normal plasma lipids, suggesting enhanced tissue β-oxidation. All of these peripheral effects appear to be independent of its central effect and to match the tissue-specific expression of C3aR1 (Guo et al., 2018). In the present study, we showed that pharmacological antagonism or KD of C3aR1 compromises TLQP-21-mediated biological activity, including calcium uptake and pERK and blunted potentiation of adrenergic-induced activation of HSL and lipolysis. Conversely, our results appear to exclude a role of C5aR in TLQP-21-mediated biological activity. Additionally, using a pharmacological approach, we established that TLQP-21/C3aR1 induces calcium uptake from the extracellular compartment via a TRPC channel. We deliberately chose a pharmacological approach rather than genetic manipulation of TRPC because (1) TRPC channels exist as dimers (Liao et al., 2014; Vazquez et al., 2004), and, often, depleting one subtype by KD or knockout (KO) results in compensatory upregulation of other subtypes (Krout et al., 2017; Sexton et al., 2016), and (2) genetic manipulation of the TRPC1 channel results in altered ERK protein levels and adipogenesis (Krout et al., 2017). Specifically, the pan-specific TRPC inhibitor SKF96365 blocked TLQP-21 mediated lipolysis. Because there is some evidence that this compound can also block store-operated Ca2+ channels and that sustained exposure to compounds that inhibit store-operated Ca2+ channels disrupts Ca2+ homeostasis at the level of the endoplasmic reticulum (ER) and can significantly affect resting Ca2+ concentrations within the cytoplasm (Liu et al., 2012), we used AncoA4, a specific inhibitor of store-operated calcium release mediated by Orai I. As opposed to SKF96365, the AncoA4 inhibitor did not affect TLQP-21-mediated lipolysis. Overall, our data support the conclusion that TLQP-21 potentiates adrenergic-induced lipolysis by a mechanism requiring C3aR1-mediated influx of calcium from the extracellular compartment (via a TRPC channel) and phosphorylation of CaMKII, ERK, and HSL but not PKA or protein kinase C (PKC) (Cero et al., 2016; Petrocchi Passeri et al., 2013; Possenti et al., 2012).
Therapeutic Relevance of the TLQP-21/C3aR1 Pathway in Humans
We investigated the role of TLQP-21/C3aR1 in human metabolic functions. We discovered that the pattern of gene expression changes due to obesity in key signaling nodes of the TLQP-21/C3aR1 and adrenergic pathways in adipose tissue is conserved in humans and mice. The observed downregulation of β-ARs and HSL in adipose tissue in obesity is consistent with previous data describing a key signature of lipolytic catecholamine resistance (Lönnqvist et al., 1992; Mowers et al., 2013). Furthermore, present and previous data (Cero et al., 2016) suggest that a concomitant increase in C3aR1 and decrease in TRPC1 expression, which is conserved in mice and humans, can be included in the molecular signature of this clinically relevant condition. Remarkably, our in vitro study demonstrates that mouse TLQP-21 can potentiate adrenergic-induced lipolysis in human adipocytes without exerting a pro-lipolytic effect per se. This result matches the lipolytic effect of mTLQP-21 in rodent adipocytes, whereas it contrasts with the inability of hTLQP-21 to exert lipolysis in human adipocytes (at least at the micromolar doses used in the present study) and is consistent with previous data demonstrating that hTLQP-21 is a very weak agonist at hC3aR1 (Cero et al., 2014). To examine the molecular basis for TLQP-21/C3aR1 activation, we conducted homology modeling of the rodent and human sequences. As expected (Cero et al., 2014; Chakraborty et al., 2015), both hTLQP-21 and mTLQP-21 spontaneously unfolded to completion in solution, confirming the intrinsically disordered nature of the native peptide. Importantly, although hTLQP-21 had a partially unfolded structure in the presence of hC3aR1, mTLQP-21 retained a strong helical structure in the presence of both mC3aR1 as well as hC3aR1. Additionally, our analysis identified D417, R161, and R340 in the C3aR1 binding motif (conserved in both human and mouse peptide/receptor) as the critical residues for recognition of the C-terminal arginine of TLQP-21. Our analysis supports previous findings indicating that mutagenesis of the same residues prevents C3a binding and receptor activation (Reid et al., 2013; Sun et al., 1999). Amidation, mutation of arginine to alanine, or cleavage of the C-terminal arginine in both TLQP-21 and C3a prevents their biological activity (Cero et al., 2016; Sun et al., 1999). This is consistent with the notion that changes to the C-terminal arginine carboxylate group would result in loss of salt bridge interactions with the D417, R161, and R340 in the C3aR1.
Our analysis also identified a key role of the amino acid in position 20 of the TLQP-21 sequence, S in human and A in rodents, respectively. S20A is the only amino acid substitution at the C-terminal tetrapeptide of TLQP-21 interacting with the 17 amino acids included in the C3aR1 binding pocket. Serine is a polar amino acid, whereas alanine is a hydrophobic amino acid. Although our model revealed that both the -PPAR (mouse) and -PPSR (human) motifs can bind to hC3aR1, the relative increase in hydrophobicity caused by the S20A substitution in the rodent peptide can explain its enhanced binding affinity and potency in the hydrophobic C3aR1 binding pocket. Additionally, increased hydrophobicity of mTLQP-21 because of the S20A substitution is consistent with the higher potency at the relatively more hydrophobic mC3aR1 compared with the hC3aR1 biding pocket. Our data on the C terminus of TLQP-21 are consistent with evidence showing that increasing the hydrophobicity markedly enhanced the potency of synthetic C3a analogs (Ember et al., 1991; Gerardy-Schahn et al., 1988).
Finally, our homology modeling identified a significant number of conserved acidic residues (D167, D325, D326, and D327) located at the opening of the C3aR1 binding site (conserved in mice and humans) that form salt bridge interactions with the conserved R9RRHXHH15 motif of TLQP-21. This result explains why the nonapeptide truncated after R12 (HFHHALPPAR) only retained minimal biological activity (Cero et al., 2014), whereas the RRRHFHHALPPAR fragment retains full biological activity (Rivolta et al., 2017), and identifies this stretch of charged amino acids in the median portion of TLQP-21 as critical for peptide/receptor interaction.
Peptide/Receptor Co-evolution Explains the Pharmacology and Function of TLQP-21/C3aR1
Our evolutionary analysis identified one amino acid substitution in the TLQP-21 peptide (S20A) and 5 amino acid substitutions in the C3aR1 receptor in mice compared with humans (Table 2) as critical to explain TLQP-21/C3aR1 biological activation. We identified a consensus sequence motif within the C3aR1 active site and TLQP-21 in the Murinae subfamily of rodents, suggesting peptide/receptor co-evolution behind the enhanced metabolic function observed in these species. Conversely, humans and most other primates express the ancestral sequence for both TLQP-21 and C3aR1. The evolutionary analysis was supported by modeling and experimental data demonstrating that “murinizing” hC3aR1 at those five residues in the receptor binding pocket is sufficient to enhance the calculated binding affinity and measured pharmacological potency of both hTLQP-21 and mTLQP-21.
Our analysis reveals that the evolution of the C3a system (C3a+C3aR1) was likely ancestral and independent of TLQP-21 in mammals. Conversely, TLQP-21 showed a divergent branch in two rodent subfamilies. All analyzed species in the Murinae and Cricetinae subfamilies express the DHMS motif in C3aR1 as well as the unique PAR motif in TLQP-21. This sequence difference can be explained by a single point mutation in TLQP-21 (S20 to A20, encoded by tcg→gcg), resulting in enhanced function of this VGF-derived peptide. Furthermore, we can speculate that multiple mutation events led to a five-amino-acid-residue difference in the C3aR1 binding pocket and were favored for their increased hydrophobicity, favoring enhanced peptide folding, binding, and pharmacological efficacy. Based on our current knowledge of TLQP-21 physiological function, we can speculate that this co-evolution was positively selected in the ancestral rodent because it conferred adaptive advantages in stimulating lipolysis in a small mammal. However, other selective pressures cannot be excluded at the moment because of incomplete knowledge of TLQP-21 biological activity (Bartolomucci et al., 2011).
Our analysis also sheds light on the biology of C3a/C3aR1. Indeed, C3a has long been regarded as an anaphylaxotoxin. However, several inconsistencies in the literature, including an anti-inflammatory role (Coulthard and Woodruff, 2015) and our findings in TLQP-21-treated mice, suggest that the role of C3aR1 might be more complex than originally thought. The best-established model of anaphylactic response is the guinea pig, whereas rats and mice are notoriously suboptimal models for C3a-induced anaphylactic response (Hugli and Müller-Eberhard, 1978; Regal and Klos, 2000; Regal et al., 1993). Our modeling and evolutionary analysis sheds light on this issue. Indeed, the critical motifs for C3a, TLQP-21, and C3aR1 in Cavia porcellus (the guinea pig) are the ancestral LAR, PSR, and DHVS, respectively (Table S3) which are shared with humans but not with rodents. Based on our findings, we postulate that the differential pharmacology of C3a in different species can be explained by these variations in the receptor sequence. With the guinea pig sequence of the C3aR1 binding site conserved in humans, care should be taken when designing C3aR1 agonists based on the TLQP-21/C3a mechanism of receptor activation. However, recent data obtained by our laboratory suggest that TLQP-21 has a lower potency compared with the C3aR1 agonist C3a70–77 in guinea pigs (unpublished data), suggesting that TLQP-21 can be a safer template to target C3aR1 functions in multiple cell types, including adipocytes, while minimizing side effects. Additionally, our study revealed that murinizing hC3aR1 at five residues in the receptor binding pocket is sufficient to enhance the β-arrestin potency of mTLQP-21 (48-fold) or hTLQP-21 (28-fold) to a greater extent than C3a (8.5-fold), suggesting the existence of differences in agonist-mediated activation, possibly implying a biased agonism mechanism (Lotta et al., 2019; McCorvy et al., 2018; Pandey et al., 2019), which will be explored in future studies. A therapeutically beneficial biological activity of TLQP-21 has been established for energy balance, lipolysis, sexual behavior, blood pressure regulation, and glucose homeostasis, whereas the peptide can cause pain behavior (Bartolomucci et al., 2011; Cero et al., 2016; Doolen et al., 2017; Pinilla et al., 2011). To the best of our knowledge, the role of TLQP-21 in innate immunity has yet to be investigated; thus, further studies are required to address the complex biological role of this evolutionary adaptation in rodents as well as to fully understand the therapeutic potential of C3aR1 agonists/antagonists based on the TLQP-21 versus the C3a mechanism of receptor activation.
Conclusion
Our multidisciplinary study identified peptide/receptor co-evolution that allowed the TLQP-21 neuropeptide to become a potent agonist at an ancient receptor, C3aR1, and, based on its mechanism of action, arguably conferred a selective advantage to mobilize energy substrates in small mammals. Because our data demonstrate that the TLQP-21/C3aR1 pathway is conserved in humans, and because mTLQP-21 is a functional agonist at hC3aR1, our discovery can pave the way for investigation of this molecular target to develop innovative pharmacotherapies directed toward adipocytes and other organs in which C3aR1 is expressed.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources, reagents, and data should be directed to and will be fulfilled by the Lead Contact, Alessandro Bartolomucci (abartolo@umn.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
3T3-L1 cells were obtained from ATCC (ATCC, CL-173) and Minnesota Obesity Centre. Commercially available human mesenchymal stem cells (hMSC) (ATCC, PCS-500–011). 3T3-L1 are from male mice (Shah et al., 2014), while hMSC are from a female donor.
3T3-L1 cells were plated on 6-well plates and maintained in DMEM supplemented with 10% fetal calf serum (FCS) (Lonza) and with 100 units/ml of penicillin/streptomycin (Invitrogen, Carlsbad, CA) in a humidified atmosphere of 5% CO2 at 37°C. Media was changed alternate days (~48 hours) till confluent. Once confluent, differentiation into adipocytes was initiated by using a differentiation cocktail containing 10% fetal bovine serum (FBS) (Atlas) 0.5 mM methylisobutylxanthine (Sigma Aldrich, Saint Louis, MO), 10 μg/ml insulin (Sigma Aldrich, Saint Louis, MO), and 0.25 μM dexamethasone (Sigma Aldrich, Saint Louis, MO). After 48 h, the media was refreshed with Insulin only FBS medium with 10 μg/ml insulin, which was removed after 2 days. The differentiated cells were maintained in DMEM with 10% FBS and media changed every other day until used in experiments 8–9 days after induction.
hMSC were differentiated using adipocyte differentiation tool kit available from ATCC (ATCC® PCS-500–050). We validated the differentiation by Oil red O staining and by QPCR analysis of FABP4 and PPARγ expression levels.
Human subjects
208 non-diabetic volunteers of American Indian ethnicity (63% male, mean age = 30 years, mean BMI = 34 kg/m2) were admitted to the Clinical Research Unit, NIDDK, Phoenix, AZ, USA, where they consumed a weight-maintaining diet (containing 50% of calories as carbohydrate, 30% as fat and 20% as protein) for at least 3 days prior to clinical testing. BMI was calculated as weight (in kg) divided by the square of height (in m2). Body composition was measured by dual-energy X-ray absorptiometry (DXA) using a total body scanner (DPX-L; Lunar Radiation, Madison, WI, USA). Before participation, volunteers were fully informed of the nature and purpose of the studies, and written informed consent was obtained. All protocols were approved by the Institutional Review Board of the National Institute of Diabetes and Digestive and Kidney Diseases.
Following an overnight fast, subjects underwent a subcutaneous abdominal fat needle biopsy under local anesthesia with 1% lidocaine. The adipose tissue was then digested with collagenase and the floating adipocyte fraction was collected. Total RNA was extracted from adipose tissue and isolated mature adipocytes using an RNeasy Lipid Mini Kit and an RNeasy Mini Kit from QIAGEN (Valencia, CA, USA), respectively. During the extraction, RNA was treated with DNase using the RNase-free DNase Set (QIAGEN) according to the manufacturer’s instructions. First-strand cDNA was synthesized from the adipocyte total RNA using a BD Advantage RT-for-PCR Kit (BD Bioscience/Clontech) following the manufacturer’s instructions. The cDNA was analyzed on Human Exon 1.0 ST Array microarray chips (Affymetrix, Santa Clara, CA, USA) as previously described (Mason et al., 2011).
ANIMALS
Wild-type male mice on a mixed C57BL6/J X 129Sv background were housed with same sex littermates and maintained in a 12:12h light-dark cycle at 22 ± 2°C. The mice were ad libitum fed either a 10% kcal% fat control diet (STD, D12450B, Research Diets, New Brunswick, NJ) or a 60% kcal% from fat high fat diet (HFD, D12492, Research Diets, New Brunswick, NJ) for 3 months. Body composition was determined by quantitative nuclear magnetic resonance using EchoMRI-100 (QNMR Systems, Houston, TX). All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Minnesota.
METHOD DETAILS
Peptides and drugs
mTLQP-21, R21A mutant and the hTLQP-21 peptides were synthesized as previously described (Cero et al., 2014). SKF-96365 and KN-62 were purchased from Tocris while AnC0A4 was purchased from EMD-millipore.
Lipolysis
For lipolysis experiments,3T3-L1 adipocytes were serum-starved in Krebs-Ringer buffer containing HEPES (KRH buffer) (NaCl at 120 mM; KCl at 4.7 mM; CaCl2 at 2.2 mM; HEPES at 10 mM; KH2PO4 at 1.2 mM; MgSO4 at 1.2 mM; glucose at 5.4 mM) supplemented with 1% bovine serum albumin (BSA) fatty acid free (Roche) for 3 h. Following starvation, the cells were incubated with KRH buffer containing 4% BSA with various treatments. ISO 50 nM, TLQP-21 100nM, were used in all the experiments according to previous results (Cero et al., 2016). For inhibitory lipolysis experiments with SKF-96365, AnC0A4 and KN-62, cells were pretreated for 1h with doses close to the reported IC-50/EC-50, and then exposed to ISO and TLQP-21. In case of SKF-96365 a series of doses were used while for AnC0A4 and KN-62 (1μM) doses were used in the experiments.
In all experiments, lipolysis was measured as the rate of glycerol release into the induction media. Following the incubation period, the media was collected, placed on ice for 10 min, and then placed in a water bath at 60°C for 20 min to inactivate any residual enzymatic activity. The induction media was then stored at −20°C until the glycerol assay was performed. Glycerol concentration in the conditioned media was measured using the Free Glycerol Determination kit (Sigma) in a flat-bottom 96-well plate following the manufacturer’s instructions. All samples incubated for 15 min at room temperature prior to measuring the absorbance at 540 OD on a plate reader (Synergy H1, BioTEK). Glycerol content was normalized to total cellular protein content determined by Bradford Assay (Thermo Scientific). The data were normalized to the control response detected in the same experiment and expressed as fold change over controls.
Lipolysis was conducted in hMSC on 18th day, by using both Isoproterenol, human and mouse TLQP-21 peptides essentially as detailed above for 3T3-L1.
mRNA extraction, reverse transcription and quantitative real-time PCR
RNA was obtained by homogenizing around 50–100 mg of frozen tissue in 500 μL of TRI REAGENT (Molecular Research Center, Inc., Cincinnati, Ohio) on ice following manufacturer’s instructions. For cells 500μl of tri reagent was directly added to the wells and processed further as per manufacturer’s instructions. Total RNA was digested with Dnase I using DNA-free water (Ambion, Austin, TX) and tested for the presence of DNA contamination using PCR. Total RNA concentration and purity was then determined by spectrophotometer at 260 nm (NanoDrop 2000 UV–Vis Spectrophotometer, Thermo Scientific). 500 ng of RNA was converted into cDNA using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA) and relative quantification of mRNAs was performed with 3.5 μL of cDNA used in each 11.5 μL real-time-RT-PCR reaction using C1000 thermal cycler (Bio-Rad Laboratories, Hercules, CA). The PCR reactions were carried out using IQ Syber Green Supermix (BIO-RAD). Primers for the target genes are presented in Table S1. Thermal cycling parameters were as follows: an initial denaturing step (95°C for 10 min), followed by 40 cycles of denaturing, annealing, and extending (95°C for 45 s, 58°C for 45 sand then 60°C for 1 min, respectively) in a 96-well BioRad plate. The results were calculated by the comparative Ct method using β-actin as an endogenous reference gene, according to the Applied Biosystems ABI PRISM 7700 User Bulletin #2. The expression relative to β-actin was determined by calculating 2−ΔCt.
Western blot
Cells were rinsed 3X with cold PBS and harvested in RIPA buffer containing protease (Complete Mini, Roche) and phosphates inhibitors (Thermo Scientific). Cells were incubated for 10 min (13) with ISO (50 nM), TLQP-21 (10 μM) or a combination of the two treatments. Lysates were then sonicated and centrifuged at 12,000 rpm for 10 min to remove nuclei. Protein was determined by BCA assay (Thermo Scientific) and an equivalent concentration of cell lysates were prepared in sodium dodecyl sulfate (SDS) sample buffer and boiled for 5 min at 95°C. Proteins were resolved by a (4%–20%) SDS-polyacrylamide gel electrophoresis (Bio-Rad) and transferred to a PVDF membrane (Bio-Rad) by using a turbo blot system from Bio-Rad. Individual proteins were detected with the specific primary antibodies by overnight incubation at 4°C (tubulin #2146), pHSL (Ser660; #4126), pERK1/2 (Thr202/Tyr204, #9101) all from Cell Signaling at 1:1000 dilution. The HRP conjugated anti-rabbit antibody was used as a secondary antibody at 1:7000 dilutions and the blot was exposed to luminol enhancer solution by using ECL prime reagent (GE health care), further imaged using a chemidoc documentation system from Bio-Rad laboratories.
Generation of stable C3aR1 knockdown cells
C3aR1 ShRNA plasmid (Sigma, Cat# SHCLNG-NM_009779, and control ShRNA plasmid (Sigma,SHC002) were purchased and the lentivirus were packed by Minnesota Obesity Center. 3T3-L1 cells (from ATCC) were infected by C3aR1 shRNA lentivirus with 8ug/ml polybrene. Two days after infection, cells were selected with 2.5 μg/ml puromycin. One week after puromycin selection, the knockdown efficiency was detected by qPCR using the primers mentioned in S1 table. Knockdown cells and controls cells were also differentiated to adipocytes for lipolysis assay.
Ca452+ Uptake assay
Ca452+ uptake assay was conducted as described previously (Mahata et al., 1997). Differentiated 3T3-L1 adipocytes were washed with buffer (150 mmol/L NaCl, 5 mmol/L KCl, 2 mmol/L CaCl2, 10 mmol/L HEPES buffer, pH 7.4) every 15 minutes two times at 37°C and cells were then incubated for 3 minutes with 1 mL of Calcium-free release buffer that contained 2 μCi of Ca452+ (12.40 mCi/mg) plus the peptides at different concentrations and Isoproterenol at 100nM. Uptake was terminated by the addition of 2 mL of ice-cold Calcium-free buffer that contained 2mmol/L EGTA and 1 mmol/L of LaCl3, with further washing 3 times with 2 mL of the same buffer. One milliliter of cell lysis buffer (buffer containing 0.1% Triton X-100) was added to each well and collected for scintillation counting. The data were expressed as cpm/well.
Fluo-4
3T3-L1 cells were plated at 60% confluency on μ-Slide 8 Well ibiTreat dishes previously coated with collagen coating solution (Cell applications Inc.). On the second day, FCS media was replaced with Hanks’ Balanced Salt Solution 1X (GIBCO) and treated with 2.5 uM Fluo-4 AM (Invitrogen) for 30 minutes. Cells were then washed with HBSS for 30 minutes, and then replaced with clean HBSS. The dishes were placed on a live cell environmental chamber in a Nikon Ti-E Deconvolution Microscope System with a New Lambda coat anti-reflective coated 20X objective lens. Elements software was used for the measurement and analysis of the assays. Images of individual wells were taken every second for a total of three minutes. After 30 s, the wells were treated with one of the following TLQP-21 concentrations: 100 nM, 1 μM, and 10 μM, the mutant version of TLQP-21 (R21A) 10 μM. HBSS was used as a negative control, and lonomycin and UTP (Uridine-5′-triphosphate) as a positive controls. For each cell, the area under the curve of the first 60 s was calculated by first removing the background noise, and then normalizing each cell to their respective basal intensity. Each experiment was then normalized to their control and then analyzed by transforming the values to a 0–1 range.
Homology Modeling
All molecular modeling was carried out using the Schrodinger modeling package (Schrödinger LLC NY, NY. 2013). Homology modeling of the Hs. C3aR1 (NP_004045.1) and Mm. C3aR1 (NP_033909.1) were based on the structural conserved regions identified from multiple sequence alignment of C3aR1 and C5aR1 across various mammalian species. The approach was based on our earlier approach (Zhang et al., 2005) that has been widely adopted in other GPCR studies (Ahmed et al., 2014; Falls and Zhang, 2018). The sequence alignment used for the homology modeling is shown in the Figure S8. The sequence homology between Hs C3aR1 and Hs C5aR1 was 57%. The large variation is due to residues 178 – 324 extracellular region that is uniquely found in C3aR1 which is excluded in our final c3aR1 homology model. The X-ray crystallographic structure of the C5aR1 in complex with an orthosteric cyclic peptide PMX53 and an allosteric antagonist NDT9513727 (PDB code: 6C1Q) (Liu et al., 2018) was used as the structural template. All missing sidechains, sequence gaps, and hydrogen atoms of the template structure were first added with standard protein preparation protocols at physiological pH, followed by energy minimization using OPLS-AA 2005 force field (Jorgensen et al., 1996) with Generalized Born implicit solvent model (Still et al., 1990) to optimize all hydrogen-bonding networks. Modeling of C3aR1 bound TLQP-21 was based on an ideal alpha helix with its C terminus mimicking the observed mode of binding of cyclic peptide PMX53. Modeling of TLQP-21 in solution was based on an ideal alpha helix.
Molecular dynamics simulation
Molecular dynamics (MD) simulations were carried out to examine the specific conformation of TLQP-21 and its interactions with C3aR1. Each of the C3aR1 –TLQP21 bound complexes was embedded in a POPC lipid membrane. Both the TLQP-21 in solution and in membrane bound C3aR1 complex were solvated by a buffer region of 15 A of TIP3P explicit water (Jorgensen et al., 1983) from its farthest edge with at 0.1 M NaCl salt concentration. Each simulation was carried out using Desmond (D. E. Shaw Research NY, NY. 2018) with default initialization protocol, followed by 50-ns simulation under constant area isothermal isobaric (NPAT) conditions at 300 K and of 1 atm pressure using the OPLS-AA 2005 force field. For comparison, simulation of TLQP-21 was carried out to identified its overall stability in solution as compared to its bound state in C3aR1. The root mean square deviation (CαRMSD) relative to its starting structure was evaluated to determine the overall structural stability and changes during the course of simulation. Our final homology model and MD simulations of C3aR1–TLQP-21 bound complex were extended for an additional 500 ns in triplicates using different random seed numbers and compared with unbound TLQP-21. CαRMSD of the receptor and TLQP-21 in complex with C3aR1 and in water was averaged over all triplicate runs and reported with SEM.
Helicity of TLQP-21
Relative alpha helicity was evaluated to compare the relative stability of TLQP-21 in solution and in bound complex with C3aR1 over the course of MD simulation. The secondary structure propensity of each residues of TLQP-21 was evaluated using the Timeline module within VMD 1.9.3. The alpha helicity of TLQP-21 was normalized relative to the starting conformation. The average relative alpha helicity of the three replicate runs for free and bound TLQP-21 is plotted over time with reported standard error bars.
TLQP-21 Binding Affinity
The change in the binding affinity of the TLQP-21 to the human and murinized-human C3aR1 was evaluated based on the thermodynamic cycle shown in Figure 5. The peptide affinity, ΔGbind, was evaluated using the molecular mechanics generalized born solvent accessible (MMGBSA) model (Beard et al., 2013) implemented within the Bioluminate module of the Schrodinger modeling software suite. The hTLQP-21-bound hC3aR1 complex was used as the starting point of our in-silico mutagenesis study. Either the peptide or the receptor was mutated to determine the relative change in the binding affinity in each step of the thermodynamic cycle. The murinized-human C3aR1 model was obtained based on V400L, S400L, L413V, V419M, C420S mutations of the human C3aR1. The mTLQP-21 was obtained based on the S6A, A7S, L8S, Y13F, S20A mutations of the hTLQP-21.
Tango Arrestin Recruitment Assay
The C3aR β-arrestin recruitment assays were performed as previously described (Cero et al., 2014; Kroeze et al., 2015) with indicated modifications. Briefly, HTLA cells (a kind gift from Julius Axelrod) expressing the TEV fused-β-arrestin2 and tTA-driven luciferase were cultured and transfected with 6 μg of C3aR wild-type or murinized (at five resides: V103I, S400L, L413V, V419M, C420S) C3aR1 DNA per 10-cm dish using calcium phosphate precipitation. After approximately 16–20 hours post-transfection, cells were detached using trypsin, centrifuged, counted and plated into white 384-well plates at a density of 10,000 cells per well in 40 μL of 1% dialyzed FBS DMEM. After approximately 6 hour incubation at 37° and 5% CO2, plates were stimulated with drugs ranging in concentration from 100 μM to 10 pM diluted in drug buffer (20 mM HEPES, 1X HBSS, 0.1% BSA, 0.01% ascorbic acid, pH 7.4) at a 5 × concentration, using a FLIPR Tetra (Molecular Devices). After approximately 20 hours of incubation at 37°C and 5% CO2, media was decanted, plates blotted for residual media, and 20 μL of BriteGlo (Promega, after 1:20 dilution) was immediately added per well. After 20 min incubation, plates were read on a MicroBeta Trilux (Perkin Elmer) at 1 s per well. Luminescence counts per second were plotted as a function of drug concentration and analyzed using log (agonist) versus response (variable slope) using Graphpad Prism 5.0. Data were normalized to percent C3a 66–77 response, which was present on every plate for every experiment.
Phylogenetic Study
Analysis was carried for mammalian species whose protein sequence for VGF, C3aR1 and C3a are known within the NCBI protein database (Table S3). Constraints-based multiple sequence alignment and phylogenetic analysis were carried out using Cobalt (Papadopoulos and Agarwala, 2007) available through NCBI-webserver with unadjusted default parameters.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed with ANOVA followed by Tuckey’s HSD or Bonferroni post hoc test or by unpaired t test where appropriate using Statistica 13. Bayesian phylogenetic analysis was conducted with Mr. Bayes software. Gene expression levels in the human study were log-transformed and then batch- and sex- standardized prior to analyses. Values of BMI and %Fat Mass were also log-transformed to approximate a normal distribution. Linear regression analyses were performed after adjustment for age, sex and the first genetic principal component obtained from a genome-wide association study, with significance level correct for multiple comparisons. Unless otherwise noted data are presented as average + standard error of the mean. Significance was set at 0.05 unless otherwise noted.
DATA AND CODE AVAILABILITY
This study did not generate custom code. All original data are available from the lead contact upon request.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti β-Tubulin | Cell signaling technology | Cat. No. 2146; RRID: AB_2210545 |
| Phospho-HSL (Ser660) | Cell signaling technology | Cat. No. 4126; RRID: AB_490997 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Antibody | Cell signaling technology | Cat. No. 9101; RRID: AB_331646 |
| goat anti-rabbit IgG-HRP | SantaCruz | Cat. No. sc-2004; RRID: AB_631746 |
| Biological Samples | ||
| 208 non-diabetic volunteers of American Indian ethnicity (63% male, mean age = 30 years, mean BMI 34 kg/m2). | NIDDK | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SKF 96365 | TOCRIS | Cat. No. 1147 |
| KN-62 | TOCRIS | Cat. No. 1277 |
| AnCoA4 | EMD Millipore | Cat. No. 532999 |
| Uridine 5′-triphosphate trisodium salt hydrate | SIGMA | Cat. No. U6625 |
| Isoproterenol hydrochloride | SIGMA | Cat. No. I6504 |
| Ionomycin calcium salt | SIGMA | Cat. No. I3909 |
| Fluo-4, AM, cell permeant | SIGMA | Cat. No. F14201 |
| 3-Isobutyl-1-methylxanthine | SIGMA | Cat. No. I5879 |
| Dexamethasone | SIGMA | Cat. No. D1756 |
| Insulin from bovine pancreas | SIGMA | Cat. No. I6634 |
| Halt Phosphatase Inhibitor Cocktail | Thermo Scientific | Cat. No. 78420 |
| Halt Protease Inhibitor Cocktail, EDTA-free (100X) | Thermo Scientific | Cat. No. 78425 |
| Calcium-45 Radionuclide | Perkin Elmer | Cat. No. NEZ013001MC |
| Puromycin | TAKARA | Cat. No. 631306 |
| Polybrene Infection | SIGMA | Cat. No. TR-1003 |
| Collagen coating solutions | Cell Applications, Inc. | Cat. No. 125–50 |
| TLQP-21, human, mouse and R21A | Cero et al.,2014 | N/A |
| SB290157 | SIGMA | Cat. No. 1192 |
| C5a Receptor Agonist FKP - (D - Cha) - Cha - r | Anaspec | Cat. No. AS-65121 |
| W54011 | Tocris | Cat. No. 5455 |
| Primestar | Takara/Fisher | Cat. No. R045A |
| DpnI | New England Biolabs | Cat. No. R0176L |
| Blasticydin | Invivogen | Cat. No. ant-bl-10p |
| Hygromycin B | KSE Scientific | Cat. No. 15140–122 |
| BrightGlo | Promega | Cat. No. E2620 |
| DMEM | VWR | Cat. No. 45000–306 |
| Fetal Bovine Serum (FBS) | VWR | Cat. No. 97068–085 |
| 10xHBSS | Invitrogen | Cat. No. 14065–056 |
| Fatty acid free bovine serum albumin | Sigma Aldrich | Cat. No. A7030–10G |
| L-Ascorbic acid | Sigma Aldrich | Cat. No. A92902–25G |
| Critical Commercial Assays | ||
| Pierce BCA Protein Assay Kit | Thermo Scientific | Cat. No. 23225 |
| Free Glycerol Reagent | SIGMA | Cat. No. F6428 |
| RNeasy lipid tissue mini kit | QIAGEN | Cat. No. 74804 |
| RNase-Free DNase Set | QIAGEN | Cat. No. 79254 |
| iScript cDNA Synthesis Kit | BIORAD | Cat. No. 1708890 |
| iQ SYBR® Green Supermix | BIORAD | Cat. No. 1708880 |
| Amersham ECL Prime Western Blotting Detection Reagent | GE health care | RPN2236 |
| C3aR1 ShRNA plasmid | SIGMA | Cat. No. SHCLNG-NM_009779 |
| control ShRNA plasmid | SIGMA | SHC002 |
| Adipocyte differentiation tool kit | ATCC | ATCC® PCS-500–050 |
| RNeasy Micro Kit | QIAGEN | Cat. No. 74004 |
| Human Exon 1.0 ST Array microarray chip | ThermoFisher | Cat. No. 900651 |
| GeneChip Whole Transcript Sense Target Labeling Assay kit | ThermoFisher | Cat. No. 900652 |
| GeneChip Fluidics Station 450 | ThermoFisher | Cat. No. 00–0079 |
| GeneChip Scanner 3000 7G | ThermoFisher | Cat. No. 00–00213 |
| Experimental Models: Cell Lines | ||
| 3T3-L1 cells | ATCC | ATCC® CL-173 |
| 3T3-L1 cells | Minnesota Obesity Center | N/A |
| human mesenchymal stem cells | ATCC | ATCC® PCS-500–011 |
| HTLA | A gift from Dr. Richard Axel, Columbia University | N/A |
| Experimental Models: Organisms/Strains | ||
| 3 months old male WT mice on a mixed C57BL6/J and 129Sv (N = 6/group). | Produced in house | N/A |
| Recombinant DNA | ||
| C3aR1 Tango | Kroeze et al., 2015 | Addgene, Cat. No. 66231 |
| Software and Algorithms | ||
| Image lab | BIORAD | N/A |
| Molecular Modeling | Small-Molecule Drug Discovery Suite 2018–4, Schrödinger, LLC, New York, NY, 2018 | N/A |
| MD Simulation | Desmond 2018–4, D. E. Shaw Research, New York, NY, 2018 | N/A |
| MD Simulation Analysis | Small-Molecule Drug Discovery Suite 2018–4, Schrödinger, LLC, New York, NY, 2018. | N/A |
| Secondary structure analysis | VMD 1.9.3, Theoretical and Computational Biophysics Group, NIH Center for Macromolecular Modeling and Bioinformatics, University of Illinois - Urbana-Champaign | N/A |
| Evolutionary sequence alignment | Constraint-based Multiple Alignment Tool (COBALT), National Center for Biotechnology Information | N/A |
| Evolutionary analysis | MrBayes, version 3.2 http://nbisweden.github.io/MrBayes/ | N/A |
| Phylogenetic tree design | Itol software. https://itol.embl.de | N/A |
| NIS-Elements | Nikon | N/A |
| Statistica 13.3 | Tibco software Inc. | N/A |
| Correlations were assessed using the statistical analysis system of the SAS institute | Sas Institute, Cary, NC | N/A |
| Prism | GraphPad Software Inc. | N/A |
Highlights.
TLQP-21 binds to C3aR1 and potentiates lipolysis via intracellular calcium
A cluster of mutations was identified in C3aR1 and TLQP-21 in Murinae
These mutations result in enhanced binding affinity and pharmacological potency
Mouse TLQP-21 enhances adrenergic-receptor-induced lipolysis in human adipocytes
ACKNOWLEDGMENTS
This study was supported by NIH/NIDDK DK102496 and DK117504 (to A.B.), a JCSTF-180217 travelling research fellowship from the Company of Biologists (to B.S.S.), AI128729 and AES0016096 (to S.O.), and Department of Veterans Affairs I01BX000323 (to S.K.M.). This study was also supported by the Intramural Program of the NIDDK, NIH (to L.B.), and BT-/RLF/Re-entry/38/2016, Department of Biotechnology, Government of India (to B.S.S.). The mouse study was conducted at the IBP Phenotyping Core (University of Minnesota).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.101.
REFERENCES
- Ahmed MH, Kellogg GE, Selley DE, Safo MK, and Zhang Y (2014). Predicting the molecular interactions of CRIP1a-cannabinoid 1 receptor with integrated molecular modeling approaches. Bioorg. Med. Chem. Lett 24, 1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner P (1999). Catecholamine-induced lipolysis in obesity. Int. J. Obes. Relat. Metab. Disord 23 (Suppl 1), 10–13. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay GK, Vu CU, Gentile S, Lee H, Biswas N, Chi NW, O’Connor DT, and Mahata SK (2012). Catestatin (chromogranin A(352–372)) and novel effects on mobilization of fat from adipose tissue through regulation of adrenergic and leptin signaling. J. Biol. Chem 287, 23141–23151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomucci A, La Corte G, Possenti R, Locatelli V, Rigamonti AE, Torsello A, Bresciani E, Bulgarelli I, Rizzi R, Pavone F, et al. (2006). TLQP-21, a VGF-derived peptide, increases energy expenditure and prevents the early phase of diet-induced obesity. Proc. Natl. Acad. Sci. USA 103, 14584–14589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomucci A, Possenti R, Mahata SK, Fischer-Colbrie R, Loh YP, and Salton SRJ (2011). The extended granin family: structure, function, and biomedical implications. Endocr. Rev 32, 755–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard H, Cholleti A, Pearlman D, Sherman W, and Loving KA (2013). Applying physics-based scoring to calculate free energies of binding for single amino acid mutations in protein-protein complexes. PLoS ONE 8, e82849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cero C, Vostrikov VV, Verardi R, Severini C, Gopinath T, Braun PD, Sassano MF, Gurney A, Roth BL, Vulchanova L, et al. (2014). The TLQP-21 peptide activates the G-protein-coupled receptor C3aR1 via a folding-upon-binding mechanism. Structure 22, 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cero C, Razzoli M, Han R, Sahu BS, Patricelli J, Guo Z, Zaidman NA, Miles JM, O’Grady SM, and Bartolomucci A (2016). The neuropeptide TLQP-21 opposes obesity via C3aR1-mediated enhancement of adrenergic-induced lipolysis. Mol. Metab 6, 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Akhter S, Requena JR, and Basu S (2015). Probing the Conformational Dynamics of the Bioactive PeptideTLQP-21 in Solution: A Molecular Dynamics Study. Chem. Biol. Drug Des 86, 938–944. [DOI] [PubMed] [Google Scholar]
- Chao TH, Ember JA, Wang M, Bayon Y, Hugli TE, and Ye RD (1999). Role of the second extracellular loop of human C3a receptor in agonist binding and receptor function. J. Biol. Chem 274, 9721–9728. [DOI] [PubMed] [Google Scholar]
- Clapham JC, and Arch JRS (2011). Targeting thermogenesis and related pathways in anti-obesity drug discovery. Pharmacol. Ther 131, 295–308. [DOI] [PubMed] [Google Scholar]
- Clemmensen C, Finan B, Müller TD, DiMarchi RD, Tschöp MH, and Hofmann SM (2019). Emerging hormonal-based combination pharmacotherapies for the treatment of metabolic diseases. Nat. Rev. Endocrinol 15, 90–104. [DOI] [PubMed] [Google Scholar]
- Coulthard LG, and Woodruff TM (2015). Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J. Immunol 194, 3542–3548. [DOI] [PubMed] [Google Scholar]
- Doolen S, Cook J, Riedl M, Kitto K, Kohsaka S, Honda CN, Fairbanks CA, Taylor BK, and Vulchanova L (2017). Complement 3a receptor in dorsal horn microglia mediates pronociceptive neuropeptide signaling. Glia 65, 1976–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ember JA, Johansen NL, and Hugli TE (1991). Designing synthetic superagonists of C3a anaphylatoxin. Biochemistry 30, 3603–3612. [DOI] [PubMed] [Google Scholar]
- Falls BA, and Zhang Y (2018). Insights into the Allosteric Mechanism of Setmelanotide (RM-493) as a Potent and First-in-Class Melanocortin-4 Receptor (MC4R) Agonist To Treat Rare Genetic Disorders of Obesity through an in Silico Approach. ACS Chem. Neurosci 10, 1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fargali S, Garcia AL, Sadahiro M, Jiang C, Janssen WG, Lin WJ, Cogliani V, Elste A, Mortillo S, Cero C, et al. (2014). The granin VGF promotes genesis of secretory vesicles, and regulates circulating catecholamine levels and blood pressure. FASEB J. 28, 2120–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerardy-Schahn R, Ambrosius D, Casaretto M, Grötzinger J, Saunders D, Wollmer A, Brandenburg D, and Bitter-Suermann D (1988). Design and biological activity of a new generation of synthetic C3a analogues by combination of peptidic and non-peptidic elements. Biochem. J 255, 209–216. [PMC free article] [PubMed] [Google Scholar]
- Granneman JG, and Moore HPH (2008). Location, location: protein trafficking and lipolysis in adipocytes. Trends Endocrinol. Metab 19, 3–9. [DOI] [PubMed] [Google Scholar]
- Guo Z, Sahu BS, He R, Finan B, Cero C, Verardi R, Razzoli M, Veglia G, Di Marchi RD, Miles JM, and Bartolomucci A (2018). Clearance kinetics of the VGF-derived neuropeptide TLQP-21. Neuropeptides 71, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannedouche S, Beck V, Leighton-Davies J, Beibel M, Roma G, Oakeley EJ, Lannoy V, Bernard J, Hamon J, Barbieri S, et al. (2013). Identification of the C3a receptor (C3AR1) as the target of the VGF-derived peptide TLQP-21 in rodent cells. J. Biol. Chem 288, 27434–27443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, and Gloriam DE (2017). Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann TJ, Haviland DL, Kildsgaard J, Watts K, and Wetsel RA (1998). Cloning, expression, sequence determination, and chromosome localization of the mouse complement C3a anaphylatoxin receptor gene. Mol. Immunol 35, 137–148. [DOI] [PubMed] [Google Scholar]
- Hugli TE, and Müller-Eberhard HJ (1978). Anaphylatoxins: C3a and C5a. Adv. Immunol 26, 1–53. [DOI] [PubMed] [Google Scholar]
- James WPT, Caterson ID, Coutinho W, Finer N, Van Gaal LF, Maggioni AP, Torp-Pedersen C, Sharma AM, Shepherd GM, Rode RA, and Renz CL; SCOUT Investigators (2010). Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N. Engl. J. Med 363, 905–917. [DOI] [PubMed] [Google Scholar]
- Jethwa PH, Warner A, Nilaweera KN, Brameld JM, Keyte JW, Carter WG, Bolton N, Bruggraber M, Morgan PJ, Barrett P, and Ebling FJ (2007). VGF-derived peptide, TLQP-21, regulates food intake and body weight in Siberian hamsters. Endocrinology 148, 4044–4055. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935. [Google Scholar]
- Jorgensen WL, Maxwell DS, and Tirado-Rives J (1996). Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc 118, 11225–11236. [Google Scholar]
- Klos A,Wende E,Wareham KJ, and Monk PN (2013). International Union of Basic and Clinical Pharmacology. [corrected]. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol. Rev 65, 500–543. [DOI] [PubMed] [Google Scholar]
- Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguère PM, Sciaky N, and Roth BL (2015). PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol 22, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krout D, Schaar A, Sun Y, Sukumaran P, Roemmich JN, Singh BB, and Claycombe-Larson KJ (2017). The TRPC1 Ca2+-permeable channel inhibits exercise-induced protection against high-fat diet-induced obesity and type II diabetes. J. Biol. Chem 292, 20799–20807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontan M, and Langin D (2009). Lipolysis and lipid mobilization in human adipose tissue. Prog. Lipid Res 48, 275–297. [DOI] [PubMed] [Google Scholar]
- Lahti JL, Tang GW, Capriotti E, Liu T, and Altman RB (2012). Bioinformatics and variability in drug response: a protein structural perspective. J. R. Soc. Interface 9, 1409–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau JL, and Dunn MK (2018). Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem 26, 2700–2707. [DOI] [PubMed] [Google Scholar]
- Liao Y, Abramowitz J, and Birnbaumer L (2014). The TRPC Family of TRP Channels: Roles Inferred (Mostly) from Knockout Mice and Relationship to ORAI Proteins. Handb. Exp. Pharmacol 223, 1055–1075. [DOI] [PubMed] [Google Scholar]
- Liu XR, Zhang MF, Yang N, Liu Q, Wang RX, Cao YN, Yang XR, Sham JSK, and Lin MJ (2012). Enhanced store-operated Ca2+ entry and TRPC channel expression in pulmonary arteries of monocrotaline-induced pulmonary hypertensive rats. Am. J. Physiol. Cell Physiol 302, C77–C87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Kim HR, Deepak RNVK, Wang L, Chung KY, Fan H, Wei Z, and Zhang C (2018). Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol 25, 472–481. [DOI] [PubMed] [Google Scholar]
- Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, Kelly ME, Chatterjee Bhowmick D, Murano I, Cohen P, et al. (2014). Adipsin is an adipokine that improves β cell function in diabetes. Cell 158, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lönnqvist F, Wahrenberg H, Hellström L, Reynisdottir S, and Arner P (1992). Lipolytic catecholamine resistance due to decreased beta 2-adrenoceptor expression in fat cells. J. Clin. Invest 90, 2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotta LA, Mokrosiński J, Mendes de Oliveira E, Li C, Sharp SJ, Luan J, Brouwers B, Ayinampudi V, Bowker N, Kerrison N, et al. (2019). Human Gain-of-Function MC4R Variants Show Signaling Bias and Protect against Obesity. Cell 177, 597–607.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahata SK, O’Connor DT, Mahata M, Yoo SH, Taupenot L, Wu H, Gill BM, and Parmer RJ (1997). Novel autocrine feedback control of catecholamine release. A discrete chromogranin a fragment is a noncompetitive nicotinic cholinergic antagonist. J. Clin. Invest 100, 1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamane Y, Chung Chan C, Lavallee G, Morin N, Xu LJ, Huang J, Gordon R, Thomas W, Lamb J, Schadt EE, et al. (2009). The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes 58, 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason CC, Hanson RL, Ossowski V, Bian L, Baier LJ, Krakoff J, and Bogardus C (2011). Bimodal distribution of RNA expression levels in human skeletal muscle tissue. BMC Genomics 12, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy JD, Wacker D, Wang S, Agegnehu B, Liu J, Lansu K, Tribo AR, Olsen RHJ, Che T, Jin J, and Roth BL (2018). Structural determinants of 5-HT2B receptor activation and biased agonism. Nat. Struct. Mol. Biol 25, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molteni L, Rizzi L, Bresciani E, Possenti R, Petrocchi Passeri P, Ghè C, Muccioli G, Fehrentz JA, Verdié P, Martinez J, et al. (2017). Pharmacological and Biochemical Characterization of TLQP-21 Activation of a Binding Site on CHO Cells. Front. Pharmacol 8, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowers J, Uhm M, Reilly SM, Simon J, Leto D, Chiang SH, Chang L, and Saltiel AR (2013). Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKε and TBK1. eLife 2, e01119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller TD, Clemmensen C, Finan B, DiMarchi RD, and Tschöp MH (2018). Anti-Obesity Therapy: from Rainbow Pills to Polyagonists. Pharmacol. Rev 70, 712–746. [DOI] [PubMed] [Google Scholar]
- Nguyen KD, Qiu Y, Cui X, Goh YPS, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, and Chawla A (2011). Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Li XX, Srivastava A, Baidya M, Kumari P, Dwivedi H, Chaturvedi M, Ghosh E, Woodruff TM, and Shukla AK (2019). Partial ligand-receptor engagement yields functional bias at the human complement receptor, C5aR1. J. Biol. Chem 294, 9416–9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos JS, and Agarwala R (2007). COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23, 1073–1079. [DOI] [PubMed] [Google Scholar]
- Pellegrinelli V, Peirce VJ, Howard L, Virtue S, Türei D, Senzacqua M, Frontini A, Dalley JW, Horton AR, Bidault G, et al. (2018). Adipocyte-secreted BMP8b mediates adrenergic-induced remodeling of the neurovascular network in adipose tissue. Nat. Commun 9, 4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrocchi Passeri P, Biondini L, Mongiardi MP, Mordini N, Quaresima S, Frank C, Baratta M, Bartolomucci A, Levi A, Severini C, and Possenti R (2013). Neuropeptide TLQP-21, a VGF internal fragment, modulates hormonal gene expression and secretion in GH3 cell line. Neuroendocrinology 97, 212–224. [DOI] [PubMed] [Google Scholar]
- Pinilla L, Pineda R, Gaytán F, Romero M, García-Galiano D, Sánchez-Garrido MA, Ruiz-Pino F, Tena-Sempere M, and Aguilar E (2011). Characterization of the reproductive effects of the anorexigenic VGF-derived peptide TLQP-21: in vivo and in vitro studies in male rats. Am. J. Physiol. Endocrinol. Metab 300, E837–E847. [DOI] [PubMed] [Google Scholar]
- Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sánchez NM, Mahú I, Mendes R, Gres V, Kubasova N, Morris I, et al. (2017). Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat. Med 23, 1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possenti R, Muccioli G, Petrocchi P, Cero C, Cabassi A, Vulchanova L, Riedl MS, Manieri M, Frontini A, Giordano A, et al. (2012). Characterization of a novel peripheral pro-lipolytic mechanism in mice: role of VGF-derived peptide TLQP-21. Biochem. J 441, 511–522. [DOI] [PubMed] [Google Scholar]
- Quell KM, Karsten CM, Kordowski A, Almeida LN, Briukhovetska D, Wiese AV, Sun J, Ender F, Antoniou K, Schröder T, et al. (2017). Monitoring C3aR Expression Using a Floxed tdTomato-C3aR Reporter Knock-in Mouse. J. Immunol 199, 688–706. [DOI] [PubMed] [Google Scholar]
- Rapold RA, Wueest S, Knoepfel A, Schoenle EJ, and Konrad D (2013). Fas activates lipolysis in a Ca2+-CaMKII-dependent manner in 3T3-L1 adipocytes. J. Lipid Res 54, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regal JF, and Klos A (2000). Minor role of the C3a receptor in systemic anaphylaxis in the guinea pig. Immunopharmacology 46, 15–28. [DOI] [PubMed] [Google Scholar]
- Regal JF, Fraser DG, and Toth CA (1993). Role of the complement system in antigen-induced bronchoconstriction and changes in blood pressure in the guinea pig. J. Pharmacol. Exp. Ther 267, 979–988. [PubMed] [Google Scholar]
- Reid RC, Yau MK, Singh R, Hamidon JK, Reed AN, Chu P, Suen JY, Stoermer MJ, Blakeney JS, Lim J, et al. (2013). Downsizing a human inflammatory protein to a small molecule with equal potency and functionality. Nat. Commun 4, 2802. [DOI] [PubMed] [Google Scholar]
- Reilly SM, and Saltiel AR (2017). Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol 13, 633–643. [DOI] [PubMed] [Google Scholar]
- Rivolta I, Binda A, Molteni L, Rizzi L, Bresciani E, Possenti R, Fehrentz JA, Verdié P, Martinez J, Omeljaniuk RJ, et al. (2017). JMV5656, A Novel Derivative of TLQP-21, Triggers the Activation of a Calcium-Dependent Potassium Outward Current in Microglial Cells. Front. Cell. Neurosci 11, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson N, Rappas M, Doré AS, Brown J, Bottegoni G, Koglin M, Cansfield J, Jazayeri A, Cooke RM, and Marshall FH (2018). Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 553, 111–114. [DOI] [PubMed] [Google Scholar]
- Rodgers RJ, Tschöp MH, and Wilding JPH (2012). Anti-obesity drugs: past, present and future. Dis. Model. Mech 5, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roglic A, Prossnitz ER, Cavanagh SL, Pan Z, Zou A, and Ye RD (1996). cDNA cloning of a novel G protein-coupled receptor with a large extracellular loop structure. Biochim. Biophys. Acta 1305, 39–43. [DOI] [PubMed] [Google Scholar]
- Sabourin J, Bartoli F, Antigny F, Gomez AM, and Benitah JP (2016). Transient Receptor Potential Canonical (TRPC)/Orai1-dependent Store-operated Ca2+ Channels: NEW TARGETS OF ALDOSTERONE IN CARDIOMYOCYTES. J. Biol. Chem 291, 13394–13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaghiani AM, Lee SM, Odegaard JI, Leveson-Gower DB, McPherson OM, Novick P, Kim MR, Koehler AN, Negrin R, Dolmetsch RE, and Park CY (2014). Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem. Biol 21, 1278–1292. [DOI] [PubMed] [Google Scholar]
- Sexton JE, Desmonds T, Quick K, Taylor R, Abramowitz J, Forge A, Kros CJ, Birnbaumer L, and Wood JN (2016). The contribution of TRPC1, TRPC3, TRPC5 and TRPC6 to touch and hearing. Neurosci. Lett 610, 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, McCormack CE, and Bradbury NA (2014). Do you know the sex of your cells? Am. J. Physiol. Cell Physiol 306, C3–C18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada I, Ueda T, Kofuku Y, Eddy MT, and Wüthrich K (2019). GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov 18, 59–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Hildebrand ME, Garcia E, and Snutch TP (2010). The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br. J. Pharmacol 160, 1464–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens SB, Schisler JC, Hohmeier HE, An J, Sun AY, Pitt GS, and Newgard CB (2012). A VGF-derived peptide attenuates development of type 2 diabetes via enhancement of islet β-cell survival and function. Cell Metab. 16, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Still WC, Tempczyk A, Hawley RC, and Hendrickson T (1990). Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc 112, 6127–6129. [Google Scholar]
- Sukumar P, Sedo A, Li J, Wilson LA, O’Regan D, Lippiat JD, Porter KE, Kearney MT, Ainscough JFX, and Beech DJ (2012). Constitutively active TRPC channels of adipocytes confer a mechanism for sensing dietary fatty acids and regulating adiponectin. Circ. Res 111, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ember JA, Chao TH, Fukuoka Y, Ye RD, and Hugli TE (1999). Identification of ligand effector binding sites in transmembrane regions of the human G protein-coupled C3a receptor. Protein Sci. 8, 2304–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschöp MH, Finan B, Clemmensen C, Gelfanov V, Perez-Tilve D, Müller TD, and DiMarchi RD (2016). Unimolecular Polypharmacy for Treatment of Diabetes and Obesity. Cell Metab. 24, 51–62. [DOI] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Aziz O, Trebak M, and Putney JW Jr. (2004). The mammalian TRPC cation channels. Biochim. Biophys. Acta 1742, 21–36. [DOI] [PubMed] [Google Scholar]
- Villarroya F, and Vidal-Puig A (2013). Beyond the sympathetic tone: the new brown fat activators. Cell Metab. 17, 638–643. [DOI] [PubMed] [Google Scholar]
- Wacker D, Stevens RC, and Roth BL (2017). How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle AJ, Carobbio S, Martins L, Slawik M, Hondares E, Vázquez MJ, Morgan D, Csikasz RI, Gallego R, Rodriguez-Cuenca S, et al. (2012). BMP8B increases brown adipose tissue thermogenesis through both central and peripheral actions. Cell 149, 871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sham YY, Rajamani R, Gao J, and Portoghese PS (2005). Homology modeling and molecular dynamics simulations of the mu opioid receptor in a membrane-aqueous system. ChemBioChem 6, 853–859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate custom code. All original data are available from the lead contact upon request.