


Abstract
The postreplication repair gene, HLTF, is often amplified and overexpressed in cancer. Here we model HLTF dysregulation through the functionally conserved Saccharomyces cerevisiae ortholog, RAD5. Genetic interaction profiling and landscape enrichment analysis of RAD5 overexpression (RAD5OE) reveals requirements for genes involved in recombination, crossover resolution, and DNA replication. While RAD5OE and rad5Δ both cause cisplatin sensitivity and share many genetic interactions, RAD5OE specifically requires crossover resolving genes and drives recombination in a region of repetitive DNA. Remarkably, RAD5OE induced recombination does not require other post-replication repair pathway members, or the PCNA modification sites involved in regulation of this pathway. Instead, the RAD5OE phenotype depends on a conserved domain necessary for binding 3′ DNA ends. Analysis of DNA replication intermediates supports a model in which dysregulated Rad5 causes aberrant template switching at replication forks. The direct effect of Rad5 on replication forks in vivo, increased recombination, and cisplatin sensitivity predicts similar consequences for dysregulated HLTF in cancer.
INTRODUCTION
Post-replication repair (PRR) is essential for damage tolerance during DNA replication. In the absence of this pathway, cells are acutely sensitive to intrastrand DNA crosslinking agents such as cisplatin—one of several platinum-based drugs used in an estimated 50% of all chemotherapeutic regimens (1–4). PRR enables damage tolerance during replication either through recruitment of specialized polymerases that can synthesize across damaged bases in a process called translesion synthesis, or through bypassing lesions by continuing synthesis on undamaged sister chromatid DNA in a process called template switching (5).
Translesion synthesis and template switching are sometimes referred to as error-prone and error-free PRR respectively owing to high rates of nucleotide misincorporation by translesion synthesis polymerases across from damaged bases (6). However, template switching can cause errors through ectopic recombination, which can drive loss or gain of information in repetitive DNA (7,8). Moreover, fork stalling and template switching resulting in microhomology-mediated break-induced replication (BIR) is the proposed driving mechanism behind disease-associated human genome rearrangements (9,10). In the absence of PRR, replication forks encountering a DNA lesion are more prone to fork collapse which may also result in a recombinogenic double-stranded DNA break and chromosomal rearrangements (11). Entry into PRR is mediated by monoubiquitination of proliferating cell nuclear antigen (PCNA), performed in Saccharomyces cerevisiae by the Rad6–Rad18 complex (human UBE2A/UBE2B-RAD18) (12). This initial modification of PCNA is thought to recruit translesion synthesis polymerases to a stalled replication fork. Subsequent regulation of translesion synthesis/template switching pathway choice is then thought to be mediated by the Mms2–Ubc13–Rad5 complex (human UBE2N-UBE2V2-HLTF), which can polyubiquitinate PCNA and perform replication fork remodeling to initiate template switching (13,14).
Given the importance of PRR in responding to lesions caused by commonly used chemotherapeutic drugs, we sought to determine whether this pathway incurs any frequent genetic alterations in cancer genomes. In this study, we perform an analysis of primary tumor samples from The Cancer Genome Atlas (TCGA), which reveals HLTF to be commonly amplified and overexpressed in esophageal, uterine and several types of squamous cell carcinoma (15). This observation corroborates previous work showing overexpression of HLTF at the RNA level in several cancer cell lines and at the protein level in hypopharyngeal squamous cell carcinomas, as well as during early stages of carcinogenesis in a kidney tumor model (16–18). Paradoxically, HLTF is also found to be frequently silenced by promoter methylation in colorectal cancers (19), leading to conflicting descriptions of HLTF as both a tumor suppressor and driver. Ultimately, the role of variable HLTF expression in carcinogenesis remains unclear. Our goal is to understand whether dysregulation of this gene could have functional consequences for cells, especially with respect to cellular fitness, response to chemotherapy and maintenance of genome stability.
Human HLTF and its budding yeast homolog, Rad5, are remarkably conserved in domain structure and biochemical function. HLTF and Rad5 share the eponymous HIP116, Rad5p N-terminal (HIRAN) domain — an ancient region of amino acid sequence homology that has been shown to form an OB-fold structure which can bind to the 3′ end of single-stranded DNA (ssDNA) (20–23). Downstream of the HIRAN domain, both HLTF and Rad5 have SWI2/SNF2 helicase sequence homology interrupted by a RING finger domain (20,24). Biochemically, both proteins have adenosine triphosphate (ATP)-dependent DNA translocase activity, and are capable of reversing model replication forks in vitro (25,26). Additionally, both proteins physically interact with a Rad6-Rad18 counterpart and cooperate with an Mms2-Ubc13 E2 ubiquitin ligase heterodimer to mediate Lys-63-linked PCNA polyubiquitination (12,27,28). Genetically, HLTF is capable of partially complementing rad5Δ in ultraviolet radiation (UV)-treated yeast, albeit in a translesion synthesis deficient background (28).
The RAD5 gene was initially discovered on the basis of UV sensitivity, and reduced forward mutation rates in forward genetic screens performed in the late 1960s and early 1970s and much progress has been made since in elucidating its roles in PRR (29–32). While it is clear that Rad5 contributes to regulating PRR through PCNA modification, the mechanism of PCNA polyubiquitination in promoting template switching is unclear. Furthermore, while many studies have considered the effect of RAD5 loss of function alleles, little is known about the consequences of RAD5 overexpression (RAD5OE) – a genetic alteration frequently observed for HLTF in human cancers. In this study, we reveal the genomic repercussions of RAD5 dysregulation using synthetic dosage interaction (SDI) profiling with the yeast gene disruption and temperature-sensitive mutant collections. We present a genetic landscape enrichment analysis indicating that efficient DNA replication, recombination, and crossover resolution are essential in the context of RAD5OE. We demonstrate that rad5Δ and RAD5OE are both sensitive to cisplatin treatment. However, RAD5OE, but not rad5Δ, requires crossover resolution and drives recombination in the rDNA tandem repeat array. Remarkably, we observe no requirement for upstream PRR signaling events on PCNA, and a previously characterized Rad5 RING domain mutant, known to disrupt polyubiquitination activity (33), has no effect on the RAD5OE phenotype. We find that a Rad5 ATPase mutant, defective in translocase activity (25), only partially relieves the consequences of RAD5OE, whereas a Rad5 HIRAN point mutation, identified using a homology model of the HLTF ssDNA binding HIRAN domain, nearly eliminates the RAD5OE phenotype. By quantitative physical analysis of DNA replication intermediates, we demonstrate that RAD5OE promotes formation of crossover products near an early firing replication origin. These products persist in the absence of Sgs1, a RecQ helicase involved in crossover resolution and depend on a functional Rad5 HIRAN domain. Our data support a model whereby RAD5OE bypasses PRR regulatory events to promote aberrant template switching via HIRAN domain-mediated replication fork remodeling. The genomic ramifications of RAD5OE presented here inform the mechanistic consequences of HLTF misexpression and its potential effect on carcinogenesis, genome instability, and response to platinum-based chemotherapy.
MATERIALS AND METHODS
Strains and plasmids
All strains are RAD5 derivatives of W303 (34), unless otherwise stated. Library strains used in our genetic screens are derivatives of S288C (35). Strains and plasmids constructed for this study, or used in validation experiments, were verified by Sanger sequencing. All genotypes for strains and plasmids used in this study and primers used for construction and validation are listed in Supplementary Tables S7 and 8.
Cancer genomics
Copy number and mRNA expression data for primary tumor samples originate from The Cancer Genome Atlas (https://cancergenome.nih.gov). These data were accessed via the Memorial Sloan Kettering computational biology portal R package (http://www.cbioportal.org) which provides access to TCGA data processed using the Broad institute GDAC firehose pipeline (https://gdac.broadinstitute.org) (15,36,37). Gene level threshold GISTIC scores were used to classify the predicted ploidy of genes in a given tumor sample with a score of 2 representing gene copy number amplification. Median expression Z-scores were used to assess increases in mRNA transcript abundance for tumors with a given gene amplified (GISTIC = 2) as compared to diploid tumors (GISTIC = 0). An expression Z-score of 0 indicates no change in transcript abundance relative to diploid tumors, whereas an expression Z-score of 2 indicates two standard deviation higher mRNA transcript abundance. Summaries for each PRR gene considered in this study are provided in Supplementary Table S1.
Genome-wide identification of SDL interactions
Selective ploidy ablation was used as described by (38) to transfer galactose-inducible overexpression plasmids into a collection of (1) 4999 MATa KanMX gene disruption strains representing individual deletions of 4899 genes (35), and (2) a collection of 1920 MATa temperature-sensitive strains representing 476 essential genes (39). Plates were scanned on a Microtek ScanMaker 9800XL plus flatbed scanner with transparency cover. Background subtracted colony densities were measured using the screenmill software package available on GitHub (https://github.com/ericedwardbryant/screenmill). Plates were manually reviewed to exclude regions of pinning failure resulting in 4876 and 363 genes from the non-essential gene deletion and essential temperature-sensitive collections, respectively. For the MATa screen, colony measurements were normalized to the plate median. For the temperature-sensitive screen, colony measurements were normalized to the mean of healthy strains on each plate at a given temperature. Normalized colony measurements were then compared via growth ratio to an empty vector control screen performed in parallel to each RAD5OE screen. Scores for all strains considered in these screens are provided in Supplementary Tables S2–4.
Landscape enrichment analysis
Landscape enrichment analysis was performed using genetic interaction profile correlations from The Cell Map (40,41). These correlations were filtered to include only genes that were considered in our SDL screens. An additional filter was applied to remove genes with more than 30% missing values. Remaining missing values were imputed as 0 (i.e. no correlation assumed). Dimension reduction was then performed using t-SNE at the default perplexity of 30 (42). The resulting two-dimensional matrix was then used to visualize the genetic landscape of S. cerevisiae where local clustering represents groups of genes with similar patterns of genetic profile correlations. MATa gene disruption SDL hits (Z-score < −2) or the top 50 temperature-sensitive SDL hits were then visualized on the landscape to identify clusters. Clusters were assessed for significance by permutation testing. This test involved repeatedly sampling the same number of SDL and TS strains from the landscape to determine the empirical probability of observing a given local density. To encourage further exploration of the landscape, all coordinates are included in Supplementary Table S5.
Colony growth and fitness measurements
Overnight liquid cultures were grown to saturation in appropriate selective media. Saturated cultures were equilibrated to an OD600 of 1.0, arrayed in a 96-well microtiter plate, and pinned three times at 384 colony density (i.e. four technical replicates for each position of the microtiter plate) using a Singer RoToR robot. Strains were grown on synthetic complete (SC) solid agar media with 2% galactose and appropriate nutrient drop-out to maintain plasmid selection. Solubilized drugs (e.g. cisplatin in 0.9% NaCl), were added immediately before pouring plates. Plates with pinned colonies were incubated at 30°C for 65 h. For growth curves, colonies were scanned hourly. Colony density was measured using screenmill. Expected fitness of a double mutant, 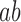 (
(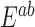 ) was calculated as the multiplicative fitness contributions of each single mutant (
) was calculated as the multiplicative fitness contributions of each single mutant (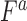 , and
, and 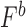 ) scaled to fitness of wild-type (WT) (
) scaled to fitness of wild-type (WT) (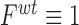 ).
).
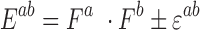 |
Error in expected fitness (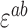 ) was computed by propagating error from estimates of
) was computed by propagating error from estimates of 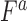 , and
, and 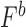 using the equation below. Care was taken to minimize systematic bias in experiments (e.g. by distributing strains evenly throughout the 96-well plate to minimize position and neighboring strain effects).
using the equation below. Care was taken to minimize systematic bias in experiments (e.g. by distributing strains evenly throughout the 96-well plate to minimize position and neighboring strain effects).
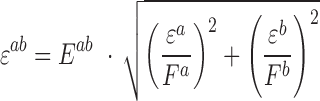 |
Dose response models for cisplatin experiments were fit to a logistic curve using nonlinear least squares regression.
Statistics
All error bars are 95% confidence intervals based on a 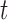 -distribution with
-distribution with 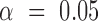 and
and 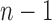 degrees of freedom where
degrees of freedom where 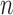 is the number of independent biological replicates with a sample standard deviation
is the number of independent biological replicates with a sample standard deviation 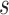 .
.
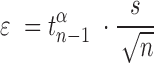 |
A significant difference between samples at 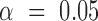 is determined when neither interval embraces the other sample's point estimate (i.e. P < 0.05 for a two-sided
is determined when neither interval embraces the other sample's point estimate (i.e. P < 0.05 for a two-sided 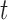 -test for independent samples (43)). For 2D gel quantification, measurements were paired within a batch of experiments by comparing X-spike and Y-arc signal to that of a WT strain harboring an empty vector. In this case, a significant change in signal at
-test for independent samples (43)). For 2D gel quantification, measurements were paired within a batch of experiments by comparing X-spike and Y-arc signal to that of a WT strain harboring an empty vector. In this case, a significant change in signal at 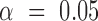 is determined when the confidence interval does not embrace zero (i.e. P < 0.05 for a two-sided paired-sample
is determined when the confidence interval does not embrace zero (i.e. P < 0.05 for a two-sided paired-sample 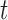 -test (43)).
-test (43)).
Ribosomal DNA recombination
Ribosomal DNA (rDNA) recombination assays were performed by inoculating 3 ml SC -Leu -Ade with a single colony of an rDNA recombination assay strain (rDNA::ADE2-CAN1) (44) transformed with a LEU2-containing plasmid. The following morning, 30 μl were inoculated into 3 ml SC raffinose -Leu -Ade and grown overnight to saturation. The following morning, ∼30 μl were inoculated into 3 ml SC Galactose -Leu to a final OD600 of 0.015. Cells were grown for 24 h then concentrated to an OD600 = 5 followed by plating 100 μl of a 10−5 dilution onto fresh SC plates, and 250 μl of a 10−4 dilution onto fresh SC -Arg +Canavanine plates to detect total cell numbers and recombinants, respectively. Cells were spread on plates using glass beads. Colonies were counted after 3 to 4 days growth. Individual cultures, representing biological replicates, were plated as three technical replicates with the average colony count from SC plates being used to estimate the number of viable cells present in the culture, and the average colony count on SC -Arg +Canavanine plates being used to estimate the rDNA recombination frequency. Nearly all colonies turned pink indicating simultaneous loss of ADE2 and CAN1 in the rDNA array. All incubation steps took place at 30°C.
Galactose induction
Galactose inductions for protein blots and two-dimensional gel analysis was performed by growing cells in SC 3% glycerol 2% lactate media with appropriate nutrient drop-out to maintain selection of a plasmid when present. Cells in log-phase growth were induced by adding galactose to a final concentration of 2%.
Immunoblots
Pelleted yeast cultures were resuspended in 5% trichloroacetic acid to precipitate proteins. Protein pellets were then washed in acetone and allowed to dry. Dry pellets were solubilized by bead-beating for 40 s in 100 μl TE buffer (50 mM Tris–HCl, 1 mM ethylenediaminetetraacetic acid, pH 7.5) followed by addition of 50 μl 3× sodium dodecyl sulphate (SDS) sample buffer (9% SDS, 0.75 mM Bromophenol blue, 187.5 mM Tris–HCl [pH 6.8], 30% glycerol and 810 mM β-mercaptoethanol). Samples were boiled for 5 min and micro-centrifuged at max speed for 5 min before sodium dodecyl sulphate-polyacrylamide gelelectrophoresis (SDS-PAGE) separation using 4–15% gradient mini-PROTEAN® TGX™ gels (BioRad) and transferred to nitrocellulose membranes with a Trans-Blot® Turbo transfer system (BioRad). Membranes were incubated overnight at 4°C with either goat α-Rad5 polyclonal antibody (SC15548 Santa Cruz Biotechnology), mouse α-FLAG® M2 monoclonal antibody (F3165 Sigma), mouse α-V5 monoclonal antibody (R96025 Invitrogen) or mouse α-Pgk1 monoclonal antibody (22C5D8 Thermo-Fisher). Protein was then visualized using secondary antibodies for α-mouse-IgG or α-goat-IgG conjugated to horseradish peroxidase with light emission catalyzed by SuperSignal® West Pico Chemiluminescent Substrate (Thermo). Light emission was captured using an Amersham™ Imager 600 (GE Healthcare).
Immunoprecipitation
Pelleted yeast cultures were prepared as described for protein blots. A total of 1350 μl of denaturing IP buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 2mM MgCl2, 1% NP-40, 10% glycerol, 2% bovine serum albumin) was added to 150 μl of denatured protein sample and then clarified by centrifugation at 15 000 RCF for 15 min at 4°C. A total of 20 μl of equilibrated α-V5-coupled agarose (Invitrogen) was added and the samples were incubated with rotation for 2 h at 4°C. Beads were collected by centrifugation and washed 4× in denaturing IP buffer. Bound factors were released by boiling in 30 μl 1× SDS sample buffer for 5 min. A total of 5 μl of sample per well were separated by SDS-PAGE using 10% gels and blotted as described above using a 1:2000 dilution of α-V5 (R96025 Invitrogen), or a 1:4000 dilution of α-SUMO (gift from X. Zhao lab). Horseradish peroxidase-conjugated α-mouse-IgG (GE Healthcare) was used at a 1:10 000 dilution as a secondary antibody.
Structural homology and sequence alignment
Structural homology modeling was performed using SWISS-MODEL (https://swissmodel.expasy.org) (45–47). HIRAN domain sequences were accessed from Pfam (https://pfam.xfam.org) and sequence alignment was performed using Clustal Omega (48,49).
Analysis of replication intermediates
All strains used for analysis of replication intermediates are bar1Δ RAD5 derivatives of W303 containing an empty vector control plasmid or RAD5 alleles under the control of the galactose-inducible GAL1 promoter (see Supplementary Table S1). Cells were pre-grown overnight in SC medium lacking leucine using 3% glycerol and 2% lactate as carbon sources to relieve glucose repression of the GAL1 promoter. RAD5 expression was then induced in log phase cultures by addition of 2% galactose. At the same time alpha-mating pheromone (Zymo Research, Irvine, CA) was added to 1 μM final concentration. Cultures were incubated with shaking at 30°C for 3.5 h and monitored for G1 cell-cycle arrest by microscope. Alpha pheromone was removed and cells were released into S phase by pelleting the culture, resuspending in sterile water, re-pelleting and suspending in the original volume of SC-leucine medium containing 2% galactose. Streptomyces griseus protease (P8811; Millipore-Sigma) was added to a final concentration of 70 μg/ml to inactivate residual alpha pheromone and hydroxyurea (HU) (H8627; Millipore-Sigma) was added to 30 mM final concentration. Cultures were incubated with shaking at 30°C and samples were collected and arrested by addition of sodium azide to 5mM final concentration then stored on ice or at 4°C until further processing.
Cell cycle progression of samples collected for replication analysis was monitored by staining DNA with SYBR Green I (Thermo Fisher) as previously described (50) and analyzed using an LSRII analyzer (Becton Dickinson) at the Columbia University Medical Center Department of Microbiology & Immunology Flow Cytometry Core facility. FACS data was analyzed using the flowCore package for R (http://bioconductor.org/packages/flowCore/). Stabilization and isolation of replicating DNA molecules in the presence of CTAB was performed as described (51). Approximately 2 μg of each DNA sample was digested with NcoI before electrophoresis.
Neutral-neutral two-dimensional agarose gel electrophoresis was adapted from the procedure of (52). The first dimension was 0.4% agarose and was run for 16 h at 1 V/cm in TBE buffer (89 mM Tris base, 89 mM boric acid, 2 mM ethylenediaminetetraacetic acid [pH 8.0]). The second dimension was 0.8% agarose run for 6 h at 5 V/cm in TBE at 4°C with 0.4 μg/ml ethidium bromide. DNA was transferred to Hybond(TM)-XL nylon membranes (GE Healthcare) using an alkaline buffer transfer procedure (53). Eight PCR products of 300–500 bp were used as hybridization probes to cover the 5.1 Kb NcoI fragment containing ARS305. The amplified probes were pooled in equimolar concentrations and labeled with 32P-CTP via random priming. Primers for probe PCRs are listed in Supplementary Table S7. Hybridized blots were exposed to storage phosphor screens (GE Healthcare) and imaged on a Typhoon 7100 phosphorimager (Amersham). Measurements of RI signal intensity were performed by selecting specific RI features using ImageJ 2.0 software (54). A mean background signal was subtracted and each measurement was normalized to the 1N spot. All measurements were performed using images in which the 1N spot signal was below saturation of the phosphor screen. RI feature signal intensity differences between empty vector and RAD5OE were calculated for samples collected within the same experimental set.
Software and data analysis
Data analysis was performed using the R project for statistical computing version 3.4.4 (https://www.r-project.org) and RStudio version 1.1.456 (https://www.rstudio.com), making extensive use of packages from Bioconductor version 3.5 (http://bioconductor.org) and a snapshot of CRAN from 2018–03-01 (https://cran.microsoft.com/snapshot/2018–03-01). Specific R packages and versions used for analysis are described in the Supplementary Materials.
RESULTS
The RAD5 ortholog, HLTF, is overexpressed in specific cancer types
Deletions of PRR regulating genes in S. cerevisiae (i.e. rad5Δ, mms2Δ, ubc13Δ, rad18Δ and rad6Δ) cause acute cisplatin sensitivity. Given the importance of PRR in responding to this common chemotherapeutic, we sought to determine whether the corresponding PRR regulating human genes (i.e. HLTF, UBE2V2, UBE2N, RAD18 and UBE2A/B) are genetically altered in cancer genomes. Using somatic copy number and mRNA expression data from The Cancer Genome Atlas (TCGA), we assessed the frequency of amplification and deletion events in all available cancer cohorts. This analysis reveals UBE2V2 (8q11.21), an ortholog of yeast MMS2, to have a mild increase in amplification events (>5% of samples) in eight different cohorts. Additionally, RAD18 (3p25.3) is amplified in some bladder cancers (9%), and deep deletions of RAD18 are present in kidney renal clear cell carcinomas (11%) (Supplementary Figure S1A and Table S1). Most strikingly, we find HLTF (3q24), a human ortholog of yeast RAD5, to be commonly amplified in lung (26%), head and neck (14%), and cervical squamous cell carcinomas (12%), as well as ovarian serous cystadenocarcinoma (14%), and esophageal carcinomas (15%) (Figure 1A). Transcript abundance correlates well with gene copy number for all PRR genes considered in this analysis. HLTF overexpression in the TCGA database corroborates the HLTF protein overexpression previously observed by immunohistochemistry in an independent study of hypopharyngeal squamous cell carcinomas (18). Since little is known about the consequences of overexpressing either human HLTF, or the functionally conserved RAD5, we pursue a systematic genetic study of RAD5 overexpression (RAD5OE)—an approach previously used to identify cancer-specific pathway dependencies, and to reveal novel pathway relationships (55,56).
Figure 1.

RAD5 overexpression (RAD5OE) causes a requirement for DNA replication and HDR genes. (A) The RAD5 human ortholog, HLTF, is overexpressed in squamous cell carcinomas. The percent of TCGA tumor samples with amplified HLTF copy number are shown (see ‘Materials and Methods’ section). Bar color corresponds to the median of mRNA expression Z-scores within each cohort. Annotated on each bar are the number of samples with HLTF amplified and the total number of samples for each cohort. (B) Schematic for SDI profiling via Selective Ploidy Ablation (SPA) (38). An engineered donor strain harboring a galactose-inducible RAD5 overexpression vector (RAD5OE) is mated to an array of mutant strains. Donor chromosomes are then destabilized and counter selected. SDIs are scored by comparing colony growth of RAD5OE to an empty vector control (see ‘Materials and Methods’ section). (C) Genetic interaction landscape enrichment for RAD5OE highlights a requirement for DNA replication and HDR genes. On the left, genes represented in the MATa non-essential gene disruption and temperature-sensitive strain collections were clustered with t-SNE in two dimensions using genetic interaction profile correlation data from The Cell Map (see ‘Materials and Methods’ section) (40,41). Dark red spots represent high-density clusters of multiple genes with similar genetic interaction profiles. The locations of RAD5OE SDIs in the landscape are shown in the middle. Two significantly enriched clusters were identified using a permutation test (P < 0.001). On the right, the region of the landscape representing the primary enriched cluster is shown. RAD5OE SDIs are labeled and colored by function. Genes in this region of the landscape that were not identified as having a RAD5OE genetic interaction are labeled background and shown as gray dots. (D) Using a quantitative spot assay, we observe HDR mutants to be the most sensitive to RAD5OE. On the left, growth curves used to quantify the rad51Δ RAD5OE SDI are shown with representative images of colony growth over time shown as an inset. Shaded regions represent the 95% confidence interval (CI) of at least four independent experiments. Expected growth of rad51Δ RAD5OE was modeled by multiplying the independent contributions of rad51Δ and RAD5OE to colony fitness (see ‘Materials and Methods’ section). On the right, differences in observed from expected colony fitness at 65 h of growth are summarized using strains from the MATα gene disruption collection for genes selected from the clusters identified by landscape enrichment analysis.
RAD5OE causes a requirement for DNA replication and recombination genes
To study the consequences of RAD5OE, we constructed a galactose-inducible RAD5OE system on a CEN plasmid. In WT yeast, Rad5 is a low abundance protein—around 100 Rad5 molecules per cell (57). When expressed from the GAL1 promoter (GAL1p), immunoblotting indicates Rad5 protein levels increase ∼100- to 500-fold over endogenous expression levels (Supplementary Figure S1B). This observation is also supported by fluorescence intensity comparing an endogenously tagged Turquoise2-Rad5 (TQ2-Rad5) with galactose-induced overexpression of TQ2-Rad5 (Supplementary Figure S1C). Both endogenous and overexpressed Rad5 localize almost exclusively to the nucleus.
We next performed two reverse genetic screens for RAD5OE synthetic lethality. For this experiment, we used selective ploidy ablation to pair a RAD5OE plasmid with 4876 non-essential yeast gene disruption mutants, and 525 temperature-sensitive mutants (38)—see experimental diagram in Figure 1B. Fitness effects were scored by comparing growth of arrayed colonies on solid agar for RAD5OE compared to an empty vector control. Reduced colony density in the RAD5OE condition relative to the vector control for a given mutant strain is indicative of a RAD5OE SDI, with extreme examples indicative of synthetic dosage lethality (SDL) (58).
To summarize the genetic interactions of RAD5OE, we performed a landscape enrichment analysis. This approach to gene-set visualization takes advantage of the yeast genetic interactome provided by The Cell Map, to cluster genes in a 2D landscape using the t-SNE dimension reduction algorithm (40–42). Clusters in this landscape represent major genetic features and biological processes included in our genetic screens (Figure 1C, left). Using this landscape, we then visualized where RAD5OE SDIs cluster. By permutation test, there are two significant clusters of RAD5OE SDI hits (P < 0.001) (Figure 1C, middle). The primary cluster highlights DNA replication, DNA damage checkpoint, homology-directed repair (HDR), and crossover resolution as sub-clusters that are essential when RAD5 is overexpressed (Figure 1C right). RAD5OE SDI screen hits found elsewhere on the landscape are shown in Supplementary Figure S1D. Landscape coordinates and interaction scores for all strains are provided in Supplementary Tables S2–5.
To validate our screen results, we developed a quantitative colony growth assay to determine the magnitude of each genetic interaction (see ‘Materials and Methods’ section, Figure 1D, left). From the quantitative measurements, RAD5OE strains grow at ∼80% fitness relative to an empty vector control strain (compare WT to RAD5OE in Figure 1D, left). Expected growth values for each mutant strain were calculated by multiplying each single mutant growth effect (without RAD5OE) to the RAD5OE effect in WT (yellow bars in Figure 1D). Observed growth for each mutant with RAD5OE and difference from expected growth is shown (red bars and blue columns in Figure 1D, right). Mutants in the 9–1-1 replication checkpoint complexes (rad17Δ/mec3Δ/rad24Δ) and a mutant involved in fork protection (csm3Δ) show only small but reproducible RAD5OE SDIs that are difficult to discern in a standard serial-dilution drop assay. Notably, the strongest interactions occur with HDR and crossover resolution mutants. Specifically, HDR machinery (e.g. rad51Δ, rad52Δ and the MRX complex mre11Δ/rad50Δ/xrs2Δ), as well as two crossover resolution complexes (sgs1Δ/top3Δ and mms4Δ/mus81Δ) are critically important in RAD5OE cells (Figure 1D and Supplementary Figure S1E).
RAD5OE and rad5Δ have similar genetic interaction profiles
When analyzing the landscape of RAD5OE SDIs, we noticed an overlap with previously published genetic interactions for rad5Δ (59). Indeed, there are 17 shared genetic interactions for RAD5OE and rad5Δ, all of which are present in the two significant landscape enrichment clusters. This overlap includes essentially all of the HDR members of the primary cluster, as well as the replication checkpoint and some DNA replication associated mutants (Figure 2A, green). There are, however, some revealing differences in genetic interaction profiles. The major differences being that rad5Δ uniquely requires nucleotide excision repair (NER) proteins (i.e. Rad4, Rad23, Rad1 and Rad14), Ctf18–RFC complex proteins (i.e. Ctf18, Ctf8 and Dcc1), and translesion synthesis proteins (i.e. Rev3 and Rev7) (Supplementary Figure S2A and Table S6). Conversely, RAD5OE uniquely requires crossover resolution proteins (e.g. Sgs1, Top3, Rmi1, Mms4 and Mus81) (Figure 2A). To confirm the RAD5OE specific requirement for crossover resolution, we measured colony fitness to quantify an interaction with sgs1Δ in a rad5Δ and RAD5OE background. As expected, there is no enhanced requirement for SGS1 in the rad5Δ background, and a strongly enhanced requirement for SGS1 in the RAD5OE background (Figure 2B). We also confirmed that rad5Δ does not exhibit negative genetic interactions with MMS4, MUS81 and TOP3 (Figure 1D and Supplementary Figure S2A). This distinct pattern of genetic requirements indicates that RAD5OE does not simply phenocopy rad5Δ and that RAD5OE produces functional protein. Based on these interaction profiles, we hypothesize that RAD5OE likely drives the formation of crossovers, possibly by promoting replication fork collapse, or aberrant fork regression and template switching.
Figure 2.

RAD5OE causes a specific requirement for crossover resolution, sensitivity to replication inhibitors and increased recombination. (A) Panel shows the primary cluster from Figure 1C highlighting genetic interactions specific to RAD5OE (red circles), rad5Δ (blue squares) or shared between RAD5OE and rad5Δ (green Xs). Interaction data for rad5Δ was acquired from BioGRID (59). (B) Quantified colony fitness confirms the absence of an sgs1Δ rad5Δ genetic interaction, and presence of an sgs1Δ RAD5OE interaction. Horizontal and vertical bars represent the mean and 95% CI of at least four independent experiments for observed (red) and expected (yellow) fitness. Turquoise and yellow rectangles represent negative and positive interaction magnitudes respectively. (C) Both RAD5OE and rad5Δ cause cisplatin sensitivity. Cisplatin dosage response curves for WT, RAD5OE, and rad5Δ are shown. Colony fitness was scored relative to untreated WT. Points and vertical bars represent the mean and 95% CI of four independent experiments. Shaded regions represent the 95% confidence band of the modeled dose response curves. (D) RAD5OE, but not rad5Δ, promotes tandem repeat recombination in ribosomal DNA (rDNA). rDNA recombination frequency was measured by loss of ADE2 and CAN1 markers incorporated within the rDNA tandem repeat array. Frequencies are normalized to the median recombination frequency observed for a WT assay strain with an empty vector. Horizontal and vertical bars indicate the median and 95% CI for at least 6 independent experiments shown as points.
RAD5OE and rad5Δ cause sensitivity to DNA replication fork blocking drugs
RAD5 deletion causes dramatic cisplatin sensitivity, and given the considerable overlap of RAD5OE and rad5Δ genetic interactions, we reason that RAD5OE might also sensitize cells to cisplatin treatment. Using the same quantitative spot assay used to measure genetic interactions, RAD5OE and rad5Δ both show cisplatin sensitivity (50% inhibitory concentrations (IC50) of 75 and 40 μM, respectively) (Figure 2C). RAD5OE can suppress rad5Δ cisplatin sensitivity, conferring the same IC50 observed when overexpressing RAD5 in a WT background (Supplementary Figure S2B). This result again shows that RAD5OE produces functional Rad5 protein, but still causes a limited capacity to resist DNA crosslinks. RAD5OE also causes sensitivity to two other replication inhibitors, HU and 4-nitroquinoline 1-oxide (4NQO) (Supplementary Figure S2C). However, RAD5OE is not sensitive to DNA double-strand break-inducing ionizing radiation, at a dose sufficient to kill rad51Δ cells (Supplementary Figure S2D). We conclude that cells overexpressing RAD5 can recover from radiation-induced breaks, but struggle to tolerate chronic replication stress, indicating RAD5OE likely impacts DNA replication thereby causing a differential response to two distinct classes of cancer therapeutics: gamma radiation, and DNA replication inhibitors.
RAD5OE promotes recombination
While rad5Δ and RAD5OE are both sensitive to DNA replication inhibitors, and share requirements for HDR, RAD5OE uniquely requires crossover resolution proteins, suggesting that it actively promotes recombination. To test this notion, we measured direct-repeat recombination within the ribosomal DNA (rDNA) array (44)—see locus diagram in Supplementary Figure S2E. RAD5OE causes an approximate 9-fold increase in marker loss, which is not observed in rad5Δ cells (Figure 2D). Interestingly, the RAD5OE-induced increase in rDNA recombination is not dependent on Fob1, the programmed DNA replication fork blocking protein known to stimulate rDNA recombination (60). While fob1Δ cells do show a 7-fold decrease in rDNA recombination as expected, overexpressing RAD5 in these cells causes an 18-fold increase in rDNA recombination to levels ∼3-fold higher than WT (Supplementary Figure S2F). Thus, RAD5OE stimulates rDNA recombination independently of the programmed rDNA replication fork block.
Titrating expression quantitatively impacts the RAD5OE phenotype
To determine the level of overexpression required to produce the RAD5OE phenotype, we cloned RAD5 under the control of three different galactose inducible promoters that each achieve different levels of Rad5 protein production. Using the GAL10, GALL and HXK1 promoters allows titration of overexpression to 40, 10, and 4% of GAL1 promoter driven expression, respectively (Supplementary Figure S2G). Lowering Rad5 protein levels has a remarkably linear effect on rDNA recombination with GAL1, GAL10 and GALL having 10.6-, 4.3- and 2.5-fold increases in recombination frequency, respectively, (Supplementary Figure S2H). A similar effect of varying RAD5 expression occurs with cisplatin sensitivity at concentrations below 75 μM where sensitivity falls off with lower expression (Supplementary Figure S2I). However, at higher doses of cisplatin, RAD5 expression from all three of the alternative promoters shows considerable cisplatin sensitivity. Indeed, at a concentration of cisplatin that is only 25% inhibitory to WT cells, HXK1-driven RAD5OE is sufficient to cause near complete lethality (Supplementary Figure S2I). Genetic requirements for RAD51 and SGS1 are also reduced when RAD5 is expressed from the GAL10 promoter, and no requirement for RAD51, or SGS1 is observed when RAD5 is expressed from the GALL, or HXK1 promoters. However, both GALL- and HXK1-driven RAD5OE enhance the requirement for RAD51 and SGS1 when cells are challenged with a low dose of cisplatin (Supplementary Figure S2J). Altogether, titrating RAD5 expression has a quantitative impact on recombination, genetic interactions, and cisplatin sensitivity that correlates with the amount of Rad5 protein production. Importantly, a 10- to 50-fold increase in Rad5 protein levels is sufficient to cause genome instability, and a 4- to 20-fold increase in Rad5 protein levels is sufficient to cause sensitivity to cisplatin mediated replication stress.
RAD18OE and MMS2OE do not phenocopy RAD5OE
Of the PRR genes considered for overexpression in cancer genomes, HLTF (yeast RAD5) is the most commonly amplified and overexpressed gene. However, RAD18 (yeast RAD18) shows some evidence for overexpression in bladder cancers, and UBE2V2 (yeast MMS2) shows evidence for overexpression in uterine carcinosarcoma (Supplementary Figure S1A). Given their shared role in PRR, we were interested in determining whether RAD18OE, or MMS2OE might have an effect similar to RAD5OE on cisplatin sensitivity and genome instability. To test this notion, we cloned RAD18 and MMS2 coding sequence into our inducible GAL1 promoter overexpression plasmid and assayed colony growth on cisplatin, and rDNA recombination frequency. RAD18OE has essentially no phenotype in either assay, and MMS2OE only has a mild impact on cisplatin sensitivity (IC50 of 120 μM), and 1.6-fold increased rDNA recombination frequency (Supplementary Figure S3A and B). We therefore conclude that RAD18OE and MMS2OE are insufficient to phenocopy RAD5OE, and that the effects we observe are not simply generalizable to overexpression of any PRR gene.
PRR genes are not essential for the RAD5OE phenotype
Rad5 is an E3 ubiquitin ligase whose RING domain is essential for establishing a physical relationship with its E2 partner—the Mms2-Ubc13 heterodimer (12). Together, Mms2-Ubc13 and Rad5 can polyubiquitinate Pol30 (PCNA) at lysine residues 127 and 164 (13). This polyubiquitination occurs during PRR downstream of initiating monoubiquitination events mediated by the Rad6 and Rad18 E2/E3 ubiquitin ligase complex. Given the well-studied role of Rad5 in regulating PRR pathway choice, we suspected that the phenotype of RAD5OE could depend on upstream PCNA monoubiquitination and its Mms2-Ubc13 E2 partner. First, we examined cisplatin sensitivity of RAD5OE in cells that cannot ubiquitinate Pol30 at the critical lysine residues 127 and 164 using the pol30K127R,K164R (pol30RR) and rad18Δ mutant backgrounds. The pol30RR mutant only partially suppresses the cisplatin sensitivity induced by RAD5OE, and RAD5OE mildly suppresses the cisplatin sensitivity of a rad18Δ strain. Thus, neither mutation shows a simple epistatic relationship with RAD5OE (Supplementary Figure S3C). Additionally, in pol30RR and rad18Δ backgrounds, the effect of RAD5OE on rDNA recombination is reduced but not eliminated. While pol30RR has a decreased, and rad18Δ has an increased frequency of rDNA recombination, RAD5OE still causes a 3-fold increase in recombination events in both of these backgrounds (Supplementary Figure S3D). Importantly, neither pol30RR nor rad18Δ alleviated the RAD5OE requirement for the crossover-resolution gene, SGS1 (Figure 3A). Next, we tested the requirement for the Rad5 E2 partner, Mms2-Ubc13, by examining the RAD5OE phenotype in an mms2Δ ubc13Δ background. Strikingly, absence of its E2 partner does not suppress the requirement for SGS1, cisplatin sensitivity, or increased rDNA recombination caused by RAD5OE (Figure 3). These results indicate that RAD5OE is capable of promoting recombination in the absence of upstream PRR signaling events, and that genome instability caused by RAD5OE is separable from the role of Rad5 as a ubiquitin ligase.
Figure 3.

Other PRR members are not essential for RAD5OE driven recombination. (A) Quantified colony fitness reveals RAD5OE requires SGS1 even in the absence of PRR members RAD18, MMS2, UBC13 and in the absence of canonical PRR PCNA ubiquitination sites (i.e. pol30RR). Horizontal and vertical bars represent the mean and 95% CI of at least four independent experiments for observed (red) and expected (yellow) fitness. Turquoise and yellow rectangles represent negative and positive interaction magnitudes respectively. (B) Quantified colony growth reveals cisplatin sensitivity of RAD5OE does not depend on the Rad5 E2 partner—Mms2-UBC13. Colony fitness was scored relative to untreated WT. Points and vertical bars represent the mean and 95% CI of four independent experiments. Shaded regions represent the 95% confidence band of the modeled dose response curves. (C) RAD5OE drives rDNA recombination even in the absence of Mms2-Ubc13. Frequencies are normalized to the median recombination frequency observed for a WT assay strain with an empty vector. Horizontal and vertical bars indicate the median and 95% CI for at least 6 independent experiments shown as points.
PCNA is sumoylated in response to RAD5OE
While RAD5OE is still capable of causing recombination in the pol30RR mutant, the frequency of recombination is considerably lower than RAD5OE in a WT background. This observation raises the possibility that Pol30 (PCNA) may be modified in response to RAD5OE. To test this possibility, we investigated PCNA modification in cells overexpressing either RAD5, or the polyubiquitination defective rad5I916A (RING) mutant (27), and compared both to an empty vector control. MMS treatment was used as a positive control to induce PCNA ubiquitination. In this assay, RAD5OE does not have a detectable effect on PCNA ubiquitination. Bands at the expected size of mono- and di-ubiquitinated PCNA increase in abundance in response to MMS treatment, but are unaffected by both RAD5 and RING mutant overexpression (Supplementary Figure S3E—PCNA-Ub and PCNA-2xUb; expected sizes are derived from (61)). However, RAD5OE and RINGOE cause a significant accumulation of higher molecular weight PCNA bands the size of which correspond to sumoylated PCNA (61). Using a SUMO-specific antibody, we determined that these higher weight species were indeed SUMO-modified PCNA (Supplementary Figure S3E and F). Thus, rather than driving PRR associated PCNA ubiquitination, RAD5OE instead induces PCNA sumoylation, an event that occurs primarily during S phase. SUMO modification of PCNA occurs at lysine residues 127 and 164, and is necessary for template switching, but also recruits the Srs2 anti-recombinase to limit homologous recombination (61–63).
The conserved HIRAN domain is essential for RAD5OE genetic interactions
To gain mechanistic insight into the consequences of RAD5 overexpression, we sought to determine which domains within Rad5 were necessary to convey the RAD5OE phenotype. The RAD5 gene encodes a protein containing three primary domains, (i) a SNF2 helicase domain that is essential for Rad5 to reverse model replication forks in vitro, (ii) a RING domain that mediates a physical interaction with Ubc13 and is essential for PCNA polyubiquitination and (iv) a HIRAN domain, which is a conserved N-terminal region that, in HLTF, forms an OB-fold which can bind the 3′-end of DNA (Figure 4A). The helicase and RING domains each have well characterized separation of function alleles. Two such alleles are the ATPase defective rad5DE681,682AA (AA) mutation in the SNF2 Walker B motif, and the ubiquitination defective rad5I916A (RING) mutation (25,27,33,64).
Figure 4.

The HIRAN domain is essential for the RAD5OE phenotype. (A) RAD5 domain map with N-terminal truncations (N164Δ and N293Δ) and HIRAN domain deletion (173–281Δ) variants indicated below. (B) Predicted secondary and tertiary structures from the RAD5 HIRAN domain HLTF homology model. The corresponding putative 3′OH ssDNA binding pocket, described in HLTF by (21), is indicated with a dotted line. Three highly conserved positively charged residues are annotated. Colors indicate vector cluster (red), intermediate cluster (green) and RAD5 cluster (blue) for variants described in panel (C). Red shaded regions highlight loops critical for RAD5OE genetic interactions. (C) Unrooted dendrogram visualizing the SDI profile distance between 39 overexpressed RAD5 variants scored for interaction against 25 RAD5OE SDIs using selective ploidy ablation (38). Variants having no effect on the interaction profile fall in the RAD5 cluster (blue). Variants having a partial effect fall in the intermediate cluster (green). Variants that nearly eliminate RAD5OE genetic interactions fall in the vector cluster (red). Bold RAD5 variants are discussed in the text. (D–F) Experiments, as described for previous figures, comparing sgs1Δ interaction, cisplatin sensitivity and rDNA recombination for RAD5, rad5R187E (R187E), rad5DE681,682AA (AA) and rad5I916A (RING) variants (see also ‘Materials and Methods’ section ). (E) Endogenous refers to variants expressed from the native RAD5 promoter in the chromosome. Overexpressed refers to variants expressed from the plasmid-born GAL1 promoter in the corresponding RAD5 variant background.
To dissect the relative contributions of RAD5 domains to the RAD5OE phenotype, we generated overexpression constructs for the AA and RING mutations, as well as three N-terminal deletions designed to separate the contribution of a poorly conserved N-terminal region from the more conserved HIRAN domain (N164Δ, 173–281Δ and N293Δ—see Figure 4A). We crossed each overexpression construct into a mini-array of genetic backgrounds representing 25 of the strongest RAD5OE SDI mutant strains. The resulting genetic interaction scores were used to calculate SDI profile distances that are visualized in the clustering dendrogram of Figure 4C. As expected from our experiments in the mms2Δ ubc13Δ background, overexpressing the RING domain mutant results in an interaction profile that is nearly indistinguishable from RAD5OE (Figure 4C—RAD5 cluster). This result further strengthens our conclusion that direct PCNA ubiquitination by Rad5 is not the source of the genomic instability we observe in cells overexpressing RAD5. The AA ATPase mutant partially alleviates many of the RAD5OE genetic requirements (e.g. reduced requirement for SGS1, Figure 4D), and cluster along with several RAD5 mutant alleles that have a weak effect on function (Figure 4C—Intermediate cluster). Among this group is the rad5G535R—a mutation adjacent to the SNF2 Walker A motif that is present in the original W303 laboratory strain (65,66). Strikingly, when we truncate the RAD5 N-terminus up to the HIRAN domain (N164Δ) we observe no effect on the interaction profile, but when we further truncate beyond the HIRAN domain (N293Δ), or delete just the HIRAN domain (173–281Δ), we see near complete elimination of all 25 RAD5OE genetic interactions (Figure 4C—Vector cluster). We conclude that the HIRAN domain is necessary for the RAD5OE interactions.
The conserved HIRAN residue, R187, is integral to the RAD5OE phenotype
While the HIRAN domain has been characterized for human HLTF, less is known about the role of this domain in yeast. To predict structural features of the Rad5 HIRAN domain, we used SWISS-MODEL to perform an unsupervised homology search using the Rad5 HIRAN domain amino-acid sequence (45–47). The top hits included three independent crystal structures of the HLTF HIRAN domain, which were then used to construct a structural homology model of the Rad5 HIRAN domain (Figure 4B) (PDB IDs: 5BNH, 4XZF and 4S0N from (21,22,67)). A visual comparison of an HLTF HIRAN domain structure and the Rad5 HIRAN homology model is shown in Supplementary Figure S4A. The predicted yeast structure has a much larger loop between the predicted β2 and β3 strands, which forms part of the putative 3′OH ssDNA binding pocket. A sequence alignment among yeast species shows considerable variability in sequence and length for this region, hence we refer to it as the ‘divergent loop’ (Figure 4B; Supplementary Figure S4A and B). As expected, given the low sequence conservation and variable length of this region, no alanine substitution tested in the divergent loop has any effect on the RAD5OE interaction profile, and deleting the portion of the loop that does not face the binding pocket (204–214Δ) also has no effect. However, deleting the portion of the loop facing the binding pocket (215–225Δ) has an intermediate effect, and deleting the whole loop (204–225Δ) has a strong effect, eliminating most of the RAD5OE genetic interactions (Figure 4C). Therefore, like HLTF, this loop is important for HIRAN domain function.
Examination of the HIRAN domain sequence alignment reveals four highly conserved positively charged residues, three of which face the putative ssDNA binding pocket (R187, R229 and R241)—see Figure 4B; Supplementary Figure S4A and B. Remarkably, every alanine and/or acidic substitution tested for these three residues in SDI profiling has at least an intermediate effect (R187A, R229A, R229E and R241E) and the R187E substitution alone has a strong effect (Figure 4C). R187E resembles HLTFR71E in the homology model, which is the HIRAN mutation that had the strongest biochemical effect on ssDNA binding and in vitro fork regression observed by (21). Importantly, the R187E substitution does not cause major changes in protein expression (Supplementary Figure S4C).
We next compared R187E, AA, and RING mutants to determine their impact on other important features of the RAD5OE phenotype. Consistent with the interaction profile data, RINGOE phenocopies RAD5OE levels of sumoylation, while AAOE causes only a mild increase, and overexpression of the R187E mutant eliminates the increased PCNA sumoylation (Supplementary Figure S4D). With respect to the SGS1 requirement during RAD5OE, mutation of R187E completely removes the colony fitness defect, supporting a role for the HIRAN domain in promoting recombination (Figure 4D). Next, we integrated the R187E, AA, and RING alleles at the endogenous locus to compare cisplatin sensitivity at normal expression levels. AA and R187E show equivalent cisplatin sensitivity (Figure 4E, left), demonstrating that the HIRAN domain is important for damage tolerance at endogenous expression levels. However, cisplatin sensitivity of R187E can be dosage suppressed by R187EOE (Figure 4E, right), consistent with the idea that this mutant causes a binding defect in the HIRAN domain that can be overcome by its increased expression (21,23). Lastly, when we measure rDNA recombination rates, we find a pattern similar to that observed from genetic interaction profiling. RAD5OE and RINGOE have identical rDNA recombination frequencies, whereas AAOE partially, and R187EOE strongly suppresses the increase in rDNA recombination (Figure 4F). Taken together, these results clearly indicate that the conserved HIRAN residue R187, plays a major role in RAD5OE genetic interactions, cisplatin tolerance, PCNA sumoylation and recombination. We propose that this effect is due to a reduced affinity of Rad5 to DNA 3′-ends, as was recently demonstrated for the nearby Rad5 K194E substitution (23).
RAD5OE promotes fork stalling and X-shaped DNA accumulation during replication
The above genetic observations, and the known requirement for Rad5 in PRR template switching, support a mechanism in which excess Rad5 is capable of driving aberrant recombination during DNA replication, resulting in intermediates that are resolved by SGS1. Additionally, the analysis of Rad5 separation of function mutants predicts that this mechanism would depend on the 3′-end ssDNA binding HIRAN domain. Given this model, we used 2D gel electrophoresis to physically analyze the recombination intermediates that occur in cells misexpressing RAD5 during DNA replication. WT or sgs1Δ mutant yeast strains containing an empty vector control or RAD5OE vectors were cell cycle synchronized by arrest in G1 phase followed by release into S phase. Expression was induced during the course of alpha pheromone treatment so that Rad5 would accumulate before synchronous release (Supplementary Figure S5A). HU was added to cultures at the time of release to deplete cellular dNTP pools and slow DNA replication resulting in cells with less than a G2 DNA content even at 120 min after release into S phase (Supplementary Figure S5B and C). Digested DNA was separated on a 2D gel and probed to detect a 5.1 Kbp DNA fragment containing the early firing replication origin ARS305 in the middle of the fragment (Supplementary Figure S5D). The 2D gel electrophoresis separates DNA by mass in the first dimension and by shape in the second, resulting in characteristic migration patterns depending on the orientation of replication through the probed DNA fragment (Figure 5A). The centrally located ARS305 initiates bidirectional replication resulting in a bubble arc signal in WT cells containing the vector control at 30 and 60 min timepoints which dissipates at later times as replication proceeds beyond the limits of the DNA fragment (Figure 5B). Slowing replication by release into HU induces template switch recombination in PRR resulting in X-shaped molecules in the replicating DNA observed as diagonal spikes extending from the 2N spot (X-spike)—see Figure 5A and B. WT cells with the RAD5OE vector show an increase in the X-spike signal at 60 min compared to the vector control. The X-shaped DNA structures are largely resolved by the 90-min time point and are not present at 120 min. Mutant sgs1Δ strains have a reduced ability to resolve X-shaped replication intermediates (68). We find that sgs1Δ cells with RAD5OE accumulate X-shaped DNA to higher levels than WT and fail to resolve these structures even 120 min after release from arrest (Figure 5C). Quantification of X-spike signal difference between RAD5OE and vector control shows that RAD5OE induces formation of X-shaped DNA and that this structure accumulates in the absence of Sgs1 (Figure 5E, top).
Figure 5.

RAD5OE promotes aberrant template switching during DNA replication. (A) Representation of replication intermediate shapes detectable by 2D gel analysis. Replicating DNA samples are separated by electrophoresis in one dimension based on mass (top arrow on leftmost diagram) and in a second dimension by shape (left arrow). Each panel depicts a mode of replication through a restriction fragment. A representative DNA molecule shape (blue) in the inset and their migration are represented by the black arc in the diagram. Non-replicating linear molecules migrate to a single spot (1N) and the dashed diagonal line represents the line of linear DNA molecules. Thin dashed arcs representing Y-arcs are included in other panels for reference. (B) The 2D gel electrophoresis analysis of WT cells. Replicating DNA was isolated after arresting cells in G1 and releasing them into medium containing 30 mM HU with either a control plasmid (vector) or a RAD5 expression plasmid (RAD5OE). Expression was induced during arrest by addition of galactose. Samples were collected at the indicated times after release and replicating DNA was visualized by probing a 5.1 Kbp restriction fragment from chromosome III containing an origin of replication at its midpoint. (C) Replicating DNA from an sgs1Δ mutant strain was collected and analyzed as in B. (D) The 2D gel analysis of an sgs1Δ strain with vector control, RAD5OE or the mutant allele R187EOE. Samples shown are from the same experiment 60 min after release. Vector and RAD5OE samples are the same as those shown in panel C. (E) Hybridization signal intensities for the X-spike and the Y-arc were measured in multiple experiments and normalized to the 1N spot signal. Normalized measurements from vector control samples were subtracted from the measurements in RAD5OE or R187EOE cells to obtain the change in signal from vector (points) for each experiment. Vertical bars indicate the 95% CI. Stars indicate increased signal relative to an empty vector control (P < 0.05 two-sided paired-sample 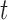 -test).
-test).
In addition to an increase in X-shaped DNA, we note an accumulation of Y-arc DNA in samples with RAD5OE (Figure 5B and C). The Y-arc DNA is increased to a similar extent in both WT and sgs1Δ mutant cells at the 30 and 60 min time points indicating that the effect is not dependent on SGS1. We also note that the excess Y-arc DNA is largely absent in later samples. An increased Y-arc signal suggests that replication from ARS305 becomes asymmetric in cells with RAD5OE giving a transition from a bubble to a Y-arc signal and is evidence for random pausing of replication forks in the RAD5OE samples. Quantification of Y-arc signal difference from multiple experiments is shown in Figure 5E, bottom.
We next tested the effect of the R187E HIRAN domain mutation on replicating DNA. This mutant was overexpressed in sgs1Δ cells and compared to the vector control and RAD5OE (Figure 5C). sgs1Δ samples from 60-min post release show the largest accumulation of X-shaped DNA with RAD5OE but do not show this increase with R187EOE (Figure 5D; Supplementary Figure S5E and F). Likewise, Y-arc signal in the sgs1Δ cells is maximal at 60 min, but no significant accumulation is observed in the R187EOE samples. Comparison of X-shape and Y-arc signals to vector control samples is shown in Figure 5E, right. The R187E mutation suppresses the effect of RAD5OE on replicating DNA. Thus, the conserved position of the R187E mutation in the HIRAN domain indicates that 3′ DNA end binding is required for in vivo replication fork stalling and remodeling.
DISCUSSION
PRR is an essential pathway for DNA damage tolerance during replication in mitotic cells. As such, dysregulation of this pathway may have important implications for actively dividing tumor cells challenged with DNA replication-inhibiting chemotherapies. Here we highlight a frequent copy number amplification event in ovarian, esophageal, and squamous cell carcinoma that results in overexpression of HLTF—a large multifunctional protein implicated in multiple aspects of PRR (Figure 1A). HLTF is a well-conserved gene that shares many common features with S. cerevisiae RAD5, including PCNA polyubiquitination activity, ATP-dependent DNA translocase activity, physical interactions with a Rad18 counterpart, and an ancient region of conserved sequence known as the HIRAN domain, important for binding ssDNA 3′ ends. Given this striking conservation of function, we have modeled the potential genomic repercussions of HLTF overexpression through a systematic genetic characterization of RAD5OE in budding yeast.
We estimate a 2- to 10-fold increase in HLTF transcript abundance in amplified tumors relative to diploid tumors based on RNAseq data from TCGA (15,36,37). Of course, fold-change of mRNA expression in tumor biopsies that possibly contain non-cancerous infiltrate may underestimate fold-change mRNA expression. On the other hand, increased transcript abundance might not result in increased HLTF protein. However, small scale studies using immunohistochemistry to determine relative overexpression of HLTF protein in cancer cells have placed the physiologically relevant level of overexpression in the range of 10- to 20-fold (17). Most RAD5 overexpression experiments in this study use more than 100-fold overexpression, which yields easily measured effects and allows for genetic dissection of the RAD5OE phenotypes. The magnitude of these phenotypes depends upon the level of RAD5 overexpression, but they are detectable at lower levels of expression. For example, a 4- to 20-fold Rad5 overexpression is sufficient to cause cisplatin sensitivity, which is the most clinically compelling aspect of the RAD5OE phenotype. While due caution needs to be exercised when comparing such distantly related organisms, our results in Saccharomyces point to an important set of genome stability phenotypes relevant to tumor cells.
Genetic interaction profiling and landscape enrichment analysis of RAD5OE revealed a clear requirement for DNA replication, homology directed repair, and crossover resolution factors (Figure 1B–D). Many of these genetic requirements are shared between RAD5OE and rad5Δ (Figure 2A). Accordingly, both RAD5OE and rad5Δ cause cisplatin sensitivity, underscoring the importance of Rad5 regulation in maintaining DNA damage tolerance (Figure 2C). From the comparison of RAD5OE and rad5Δ genetic interactions, we identified the necessity for crossover resolution genes as specific to RAD5OE (Figure 2A, B). In line with this observation, RAD5OE, but not rad5Δ, results in nearly an order of magnitude increase in rDNA recombination demonstrating that excess Rad5 promotes genome instability (Figure 2D). This genome instability is specific to RAD5OE as overexpression of other PRR members is insufficient to cause high levels of recombination (Supplementary Figure S3B).
The RAD5OE genetic interaction and damaging agent sensitivity profiles support a model where RAD5OE acts during replication—an observation consistent with the known role of Rad5 in PRR. While we expected the RAD5OE phenotype to require other members of PRR, this requirement was not observed. For example, the Rad5 E2 partner (Mms2-Ubc13) is completely dispensable for the SGS1 genetic requirement, cisplatin sensitivity and rDNA recombination (Figure 3). Similarly, a well-characterized RING domain mutation in Rad5 fails to suppress the RAD5OE phenotype (Figure 4). Additionally, in rad18Δ, and pol30RR backgrounds, which lack canonical PRR associated PCNA modification, RAD5OE still causes a requirement for SGS1, and increases rDNA recombination (Figure 3A and Supplementary Figure S3D). Rather than causing PRR associated PCNA ubiquitination, RAD5OE predominantly causes PCNA sumoylation—a modification that is also associated with template switch recombination during DNA replication (Supplementary Figure S3E and F) (61–63). Thus, we reason that RAD5 overexpression bypasses PRR signaling events that are usually required to mediate template switching at DNA replication forks.
We next investigated other functional aspects of the Rad5 protein and identified R187E as a single residue substitution that nearly eliminates the RAD5OE phenotype (Figure 4 and Supplementary Figure S4D). Notably, based on primary sequence alignment and structural homology modeling, this mutation closely resembles HLTFR71E, which has a strong effect on 3′OH dependent ssDNA affinity and in vitro fork regression activity (Supplementary Figure S4A and B) (21). Analysis of replication intermediates by 2D gel electrophoresis confirmed that RAD5OE drives X-shaped DNA formation and an accumulation of a Y-arc signal during replication near an early firing origin (Figure 5). In addition, the X-shaped structures accumulate in an sgs1Δ background (Figure 5). Importantly, X-shaped DNA accumulation during replication is severely reduced, if not eliminated, when overexpressing the R187E mutation, emphasizing the importance of the 3′-end binding HIRAN domain for generating crossovers during replication (Figure 5D and E).
Model of Rad5 overexpression
Any model for the genomic outcomes of RAD5OE must account for increased recombination at a replication fork. One possible model is that Rad5 binding at a replication fork induces collapse leading to dsDNA breaks (DSBs) followed by repair via BIR (Figure 6 model 1). BIR involves a single-ended DNA break invading homologous sequence to establish a replication fork. This process has been implicated in alternative maintenance of telomeres and may drive complex genome rearrangements (69,70). However, we do not see evidence for DSBs with RAD5OE. In Figure 5, we observe replication proceeding as a replication bubble with some conversion to Y-arc by RAD5OE. Breakage occurring in a replication bubble should produce smaller asymmetric Y-arcs that would resolve below the full-length Y-arc signal, and these are not observed (71). Furthermore, the genetic requirements for BIR do not match the genetic interaction profile of RAD5OE. BIR that is induced from single-ended DSBs outside of the context of DNA replication requires the Pif1 helicase and the DNA polymerase delta subunit Pol32 (72,73). In contrast, BIR induced at a replication fork does not require Pif1 or Pol32 (74). RAD5OE requires Pol32, but not Pif1 (Supplementary Figure S6A), which does not fit either model of BIR. We therefore think that fork breakage is unlikely to account for effects that we see with RAD5OE.
Figure 6.

Models for excess Rad5 at a replication fork. A replication fork is shown with blue and red solid lines representing the parental strands. Nascent DNA is represented as dashed lines, and arrowheads indicate the direction of DNA polymerization. Rad5 (green oval) is shown binding to the 3′ end of the leading strand via the HIRAN domain (cleft in the oval). See text of Discussion section for description of the models.
A second possible model, which involves template switching is shown in Figure 6 (model 2). In this model, Rad5 binding at a stalled replication fork results in reprimed leading strand synthesis leaving a single-strand gap behind the replication fork (75). The ssDNA gap would then be processed by HDR proteins to generate crossovers. This model accounts for the accumulation of X-spikes in replicating DNA and the genetic requirement for HDR and crossover proteins in RAD5OE cells. However, this model does not account for the accumulation of Y-arc DNA as repriming would allow continuation of the replication fork. In addition, this model does not involve Rad5 translocase activity, which is responsible for at least half of the increased recombination we observe in rDNA. Thus, recombination in ssDNA gaps cannot fully account for our observations.
Therefore, we favor the fork capture and reversal model as it accounts for every aspect of the RAD5OE phenotype (Figure 6, model 3). In this model, the Rad5 HIRAN domain captures a replication fork. The DNA is reorganized such that the two nascent strands can anneal (fork capture) and, when coupled with a translocase activity, leads to extrusion to form a 4-way junction (fork reversal). Both Rad5 and HLTF exhibit this activity in vitro on fork-like oligonucleotide substrates, and both ATPase activity and the ssDNA-binding HIRAN domain are important for this function (21,23,25). HIRAN domain-mediated capture of a 3′ DNA end has been shown to form an initial 3-way junction (23,76), and stabilization of this junction would explain the accumulation of Y-arc DNA in 2D gels (Figure 5). In addition, this Y-arc DNA accumulation is absent when overexpressing the R187E HIRAN mutant, which should not interact with a 3′ DNA end. The Rad5-stabilized 3-way junction would be converted to a 4-way junction aided by Rad5 translocase activity. We propose that the partial requirement for Rad5 translocase activity in our results could indicate that other mechanisms of fork reversal or branch migration can catalyze fork remodeling once the HIRAN domain has engaged DNA at the fork. Candidates for accessory translocases include the yeast FANCM helicase Mph1, or Rad54 (77,78). Alternative branch migration mechanisms could explain the partial requirement for Rad5 translocase activity. This reversed fork structure is likely transient and is quickly resolved by HDR and crossover resolution to re-establish the replication fork, explaining the requirement for HDR and crossover resolution genes.
While we favor the fork capture and reversal model (Figure 6 model 3), it is possible that multiple pathways are invoked upon Rad5 interaction with a replication fork. For instance, the Mus81–Mms4 structure-specific nuclease complex contributes to cell viability in the context of RAD5OE and a simple model is that it functions redundantly with Sgs1. Alternatively, Mus81–Mms4 could be acting early after the formation of a 3-way junction (Figure 6 model 3) to catalyze conversion to a DNA break (Figure 6 model 1). Consequently, the three models described may not be mutually exclusive. In fact, another explanation for the partial dependence of RAD5OE phenotypes on ATPase function is that all three pathways could be initiated by HIRAN-dependent DNA binding, but only pathway 3 would depend on translocase activity. In the ATPase mutant, DNA breakage or leading strand repriming may become more likely. Nevertheless, our observations demonstrate that the HIRAN domain is the predominant driver of RAD5OE-induced genome instability.
In conclusion, RAD5 overexpression causes hyperrecombination during DNA replication, which can result in genome instability. This finding implies that human cells with amplified and overexpressed HLTF may be prone to similar genome instability—a hallmark of carcinogenesis. Importantly, we also show that RAD5 dysregulation results in impaired cisplatin tolerance. Our studies suggest that key recombination and repair pathways may become important in cancers harboring HLTF copy number alterations. In view of the widespread use of platinum-based chemotherapy in the treatment of cancer, our observations encourage further study into the effect of dysregulated HLTF on DNA damage tolerance.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Xiaolan Zhao for the gift of the anti SUMO antibody. We would also like to thank Alberto Ciccia, Chao Lu and Lorraine Symington for critical reading of the manuscript. E.E.B., I.S., R.R. and R.J.D.R. conceived the project and designed the experiments. E.E.B., I.S. and R.J.D.R. performed the experiments. L.E.B. conceived and performed the analysis of PCNA modifications. E.E.B. conceived and performed the landscape enrichment analysis. E.E.B. and R.J.D.R. developed the quantitative colony fitness assay. Scanner automation was developed by R.J.D.R. The screenmill R package developed by E.E.B., R.J.D.R performed the 2D gel analysis assisted by I.S., R.R. supervised E.E.B., E.E.B. and R.J.D.R. wrote the manuscript. E.E.B., L.E.B., I.S., R.R. and R.J.D.R. edited and approved the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [T32CA009503, T32GM008798, TL1TR001875 to E.E.B.; U54 CA209997, R35GM118180 to R.R.; R35GM124633 to L.E.B.] and P30CA013696 for use of the HICCC shared radiation resource. Funding for open access charge: National Institutes of Health [R35GM118180].
Conflict of interest statement. None declared.
REFERENCES
- 1. Galanski M., Jakupec M.A., Keppler B.K.. Update of the preclinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches. Curr. Med. Chem. 2005; 12:2075–2094. [DOI] [PubMed] [Google Scholar]
- 2. Simon J.A., Szankasi P., Nguyen D.K., Ludlow C., Dunstan H.M., Roberts C.J., Jensen E.L., Hartwell L.H., Friend S.H.. Differential toxicities of anticancer agents among DNA repair and checkpoint mutants of Saccharomyces cerevisiae. Cancer Res. 2000; 60:328–333. [PubMed] [Google Scholar]
- 3. Wu H.I., Brown J.A., Dorie M.J., Lazzeroni L., Brown J.M.. Genome-wide identification of genes conferring resistance to the anticancer agents cisplatin, oxaliplatin, and mitomycin C. Cancer Res. 2004; 64:3940–3948. [DOI] [PubMed] [Google Scholar]
- 4. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer. 2007; 7:573–584. [DOI] [PubMed] [Google Scholar]
- 5. Boiteux S., Jinks-Robertson S.. DNA repair mechanisms and the bypass of DNA damage in Saccharomyces cerevisiae. Genetics. 2013; 193:1025–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiao W., Chow B.L., Broomfield S., Hanna M.. The Saccharomyces cerevisiae RAD6 group is composed of an error-prone and two error-free postreplication repair pathways. Genetics. 2000; 155:1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin J.-R., Zeman M.K., Chen J.-Y., Yee M.-C., Cimprich K.A.. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol. Cell. 2011; 42:237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang M.-E., Rio A.-G., Galibert M.-D., Galibert F.. Pol32, a subunit of Saccharomyces cerevisiae DNA polymerase delta, suppresses genomic deletions and is involved in the mutagenic bypass pathway. Genetics. 2002; 160:1409–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee J.A., Carvalho C.M.B., Lupski J.R.. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007; 131:1235–1247. [DOI] [PubMed] [Google Scholar]
- 10. Zhang F., Khajavi M., Connolly A.M., Towne C.F., Batish S.D., Lupski J.R.. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009; 41:849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Motegi A., Liaw H.-J., Lee K.-Y., Roest H.P., Maas A., Wu X., Moinova H., Markowitz S.D., Ding H., Hoeijmakers J.H.J. et al.. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:12411–12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ulrich H.D., Jentsch S.. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000; 19:3388–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoege C., Pfander B., Moldovan G.-L., Pyrowolakis G., Jentsch S.. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002; 419:135–141. [DOI] [PubMed] [Google Scholar]
- 14. Branzei D., Seki M., Enomoto T.. Rad18/Rad5/Mms2-mediated polyubiquitination of PCNA is implicated in replication completion during replication stress. Genes Cells. 2004; 9:1031–1042. [DOI] [PubMed] [Google Scholar]
- 15. Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gong X., Kaushal S., Ceccarelli E., Bogdanova N., Neville C., Nguyen T., Clark H., Khatib Z.A., Valentine M., Look A.T. et al.. Developmental regulation of Zbu1, a DNA-binding member of the SWI2/SNF2 family. Dev. Biol. 1997; 183:166–182. [DOI] [PubMed] [Google Scholar]
- 17. Debauve G., Nonclercq D., Ribaucour F., Wiedig M., Gerbaux C., Leo O., Laurent G., Journé F., Belayew A., Toubeau G.. Early expression of the helicase-like transcription factor (HLTF/SMARCA3) in an experimental model of estrogen-induced renal carcinogenesis. Mol. Cancer. 2006; 5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Capouillez A., Decaestecker C., Filleul O., Chevalier D., Coppée F., Leroy X., Belayew A., Saussez S.. Helicase-like transcription factor exhibits increased expression and altered intracellular distribution during tumor progression in hypopharyngeal and laryngeal squamous cell carcinomas. Virchows Arch. 2008; 453:491–499. [DOI] [PubMed] [Google Scholar]
- 19. Moinova H.R., Chen W.-D., Shen L., Smiraglia D., Olechnowicz J., Ravi L., Kasturi L., Myeroff L., Plass C., Parsons R. et al.. HLTF gene silencing in human colon cancer. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:4562–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iyer L.M., Babu M.M., Aravind L.. The HIRAN domain and recruitment of chromatin remodeling and repair activities to damaged DNA. Cell Cycle. 2006; 5:775–782. [DOI] [PubMed] [Google Scholar]
- 21. Kile A.C., Chavez D.A., Bacal J., Eldirany S., Korzhnev D.M., Bezsonova I., Eichman B.F., Cimprich K.A.. HLTF’s ancient HIRAN domain binds 3′ DNA ends to drive replication fork reversal. Mol. Cell. 2015; 58:1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hishiki A., Hara K., Ikegaya Y., Yokoyama H., Shimizu T., Sato M., Hashimoto H.. Structure of a novel DNA-binding domain of helicase-like transcription factor (HLTF) and its functional implication in DNA damage tolerance. J. Biol. Chem. 2015; 290:13215–13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shin S., Hyun K., Kim J., Hohng S.. ATP binding to Rad5 initiates replication fork reversal by inducing the unwinding of the leading arm and the formation of the Holliday junction. Cell Rep. 2018; 23:1831–1839. [DOI] [PubMed] [Google Scholar]
- 24. Johnson R.E., Henderson S.T., Petes T.D., Prakash S., Bankmann M., Prakash L.. Saccharomyces cerevisiae RAD5-encoded DNA repair protein contains DNA helicase and zinc-binding sequence motifs and affects the stability of simple repetitive sequences in the genome. Mol. Cell Biol. 1992; 12:3807–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blastyák A., Pintér L., Unk I., Prakash L., Prakash S., Haracska L.. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell. 2007; 28:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blastyák A., ú I.H., Unk I., Haracska L.. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol. Cell Biol. 2010; 30:684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ulrich H.D. Protein-protein interactions within an E2-RING finger complex. implications for ubiquitin-dependent DNA damage repair. J. Biol. Chem. 2003; 278:7051–7058. [DOI] [PubMed] [Google Scholar]
- 28. Unk I., ú I.H., Fátyol K., Hurwitz J., Yoon J.-H., Prakash L., Prakash S., Haracska L.. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:3768–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Snow R. Mutants of yeast sensitive to ultraviolet light. J. Bacteriol. 1967; 94:571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cox B.S., Parry J.M.. The isolation, genetics and survival characteristics of ultraviolet light-sensitive mutants in yeast. Mutat. Res. 1968; 6:37–55. [DOI] [PubMed] [Google Scholar]
- 31. Lemontt J.F. Mutants of yeast defective in mutation induced by ultraviolet light. Genetics. 1971; 68:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Game J.C., Mortimer R.K.. A genetic study of x-ray sensitive mutants in yeast. Mutat. Res. 1974; 24:281–292. [DOI] [PubMed] [Google Scholar]
- 33. Chen S., Davies A.A., Sagan D., Ulrich H.D.. The RING finger ATPase Rad5p of Saccharomyces cerevisiae contributes to DNA double-strand break repair in a ubiquitin-independent manner. Nucleic Acids Res. 2005; 33:5878–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thomas B.J., Rothstein R.. The genetic control of direct-repeat recombination in Saccharomyces: The effect of Rad52 and Rad1 on mitotic recombination at GAL10, a transcriptionally regulated gene. Genetics. 1989; 123:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H. et al.. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999; 285:901–906. [DOI] [PubMed] [Google Scholar]
- 36. Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. et al.. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013; 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reid R.J.D., González-Barrera S., Sunjevaric I., Alvaro D., Ciccone S., Wagner M., Rothstein R.. Selective ploidy ablation, a high-throughput plasmid transfer protocol, identifies new genes affecting topoisomerase I-induced DNA damage. Genome Res. 2011; 21:477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Z., Vizeacoumar F.J., Bahr S., Li J., Warringer J., Vizeacoumar F.S., Min R., Vandersluis B., Bellay J., Devit M. et al.. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat. Biotechnol. 2011; 29:361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Usaj M., Tan Y., Wang W., VanderSluis B., Zou A., Myers C.L., Costanzo M., Andrews B., Boone C.. TheCellMap.org: A web-accessible database for visualizing and mining the global yeast genetic interaction network. G3. 2017; 7:1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Costanzo M., VanderSluis B., Koch E.N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S.D. et al.. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016; 353:aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van der Maaten L., Hinton G.. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008; 9:2579–2605. [Google Scholar]
- 43. Pfister R., Janczyk M.. Confidence intervals for two sample means: calculation, interpretation, and a few simple rules. Adv. in Cogn. Psychol. 2013; 9:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roy N., Runge K.W.. Two paralogs involved in transcriptional silencing that antagonistically control yeast life span. Curr. Biol. 2000; 10:111–114. [DOI] [PubMed] [Google Scholar]
- 45. Guex N., Peitsch M.C., Schwede T.. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis. 2009; 30:S162–S173. [DOI] [PubMed] [Google Scholar]
- 46. Bienert S., Waterhouse A., de Beer T.A.P., Tauriello G., Studer G., Bordoli L., Schwede T.. The SWISS-MODEL repository-new features and functionality. Nucleic Acids Res. 2017; 45:D313–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L. et al.. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018; 46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sievers F., Higgins D.G.. Clustal omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014; 1079:105–116. [DOI] [PubMed] [Google Scholar]
- 49. Finn R.D., Bateman A., Clements J., Coggill P., Eberhardt R.Y., Eddy S.R., Heger A., Hetherington K., Holm L., Mistry J. et al.. Pfam: the protein families database. Nucleic Acids Res. 2014; 42:D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fortuna M., JoãoSousa M., Côrte-Real M., Leão C., Salvador A., Sansonetty F.. Cell cycle analysis of yeasts. Curr. Protoc. Cytom. 2001; 13:11.13.1–11.13.9. [DOI] [PubMed] [Google Scholar]
- 51. Neelsen K.J., Chaudhuri A.R., Follonier C., Herrador R., Lopes M.. Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol. Biol. 2014; 1094:177–208. [DOI] [PubMed] [Google Scholar]
- 52. Brewer B.J., Fangman W.L.. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell. 1987; 51:463–471. [DOI] [PubMed] [Google Scholar]
- 53. Brown T. Southern Blotting. Curr. Protoc. Mol. Biol. 1993; 21:2.9.1–2.9.20. [DOI] [PubMed] [Google Scholar]
- 54. Rueden C.T., Schindelin J., Hiner M.C., DeZonia B.E., Walter A.E., Arena E.T., Eliceiri K.W.. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics. 2017; 18:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reid R.J.D., Du X., Sunjevaric I., Rayannavar V., Dittmar J., Bryant E., Maurer M., Rothstein R.. A synthetic dosage lethal genetic interaction between CKS1B and PLK1 is conserved in yeast and human cancer cells. Genetics. 2016; 204:807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Šuštić T., van Wageningen S., Bosdriesz E., Reid R.J.D., Dittmar J., Lieftink C., Beijersbergen R.L., Wessels L.F.A., Rothstein R., Bernards R.. A role for the unfolded protein response stress sensor ERN1 in regulating the response to MEK inhibitors in KRAS mutant colon cancers. Genome Med. 2018; 10:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kulak N.A., Pichler G., Paron I., Nagaraj N., Mann M.. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods. 2014; 11:319–324. [DOI] [PubMed] [Google Scholar]
- 58. Kroll E.S., Hyland K.M., Hieter P., Li J.J.. Establishing genetic interactions by a synthetic dosage lethality phenotype. Genetics. 1996; 143:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stark C., Breitkreutz B.-J., Reguly T., Boucher L., Breitkreutz A., Tyers M.. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006; 34:D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kobayashi T., Horiuchi T.. A yeast gene product, Fob1 protein, required for both replication fork blocking and recombinational hotspot activities. Genes Cells. 1996; 1:465–474. [DOI] [PubMed] [Google Scholar]
- 61. Papouli E., Chen S., Davies A.A., Huttner D., Krejci L., Sung P., Ulrich H.D.. Crosstalk between sumo and ubiquitin on pcna is mediated by recruitment of the helicase srs2p. Mol. Cell. 2005; 19:123–133. [DOI] [PubMed] [Google Scholar]
- 62. Choe K.N., Moldovan G.-L.. Forging ahead through darkness: PCNA, still the principal conductor at the replication fork. Mol. Cell. 2017; 65:380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Branzei D., Vanoli F., Foiani M.. SUMOylation regulates Rad18-mediated template switch. Nature. 2008; 456:915–920. [DOI] [PubMed] [Google Scholar]
- 64. Gangavarapu V., Haracska L., Unk I., Johnson R.E., Prakash S., Prakash L.. Mms2-Ubc13-dependent and -independent roles of Rad5 ubiquitin ligase in postreplication repair and translesion DNA synthesis in Saccharomyces cerevisiae. Mol. Cell Biol. 2006; 26:7783–7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fan H.Y., Cheng K.K., Klein H.L.. Mutations in the RNA polymerase II transcription machinery suppress the hyperrecombination mutant hpr1 delta of Saccharomyces cerevisiae. Genetics. 1996; 142:749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Elserafy M., Abugable A.A., Atteya R., El-Khamisy S.F.. Rad5, HLTF, and SHPRH: A fresh view of an old story. Trends Genet. 2018; 34:574–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Neculai D., Walker J.R., Weigelt J., Bountra C., Edwards A.M., Arrowsmith C.H., Dhe-Paganon S.. Co-crystal structure of the HLTF HIRAN domain with a ssDNA fragment. 2015; doi:10.2210/pdb5bnh/pdb.
- 68. Liberi G., Maffioletti G., Lucca C., Chiolo I., Baryshnikova A., Cotta-Ramusino C., Lopes M., Pellicioli A., Haber J.E., Foiani M.. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005; 19:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Llorente B., Smith C.E., Symington L.S.. Break-induced replication: What is it and what is it for. Cell Cycle. 2008; 7:859–864. [DOI] [PubMed] [Google Scholar]
- 70. Sakofsky C.J., Malkova A.. Break induced replication in eukaryotes: mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017; 52:395–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Noguchi E., Noguchi C., Du L.-L., Russell P.. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol. Cell Biol. 2003; 23:7861–7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Saini N., Ramakrishnan S., Elango R., Ayyar S., Zhang Y., Deem A., Ira G., Haber J.E., Lobachev K.S., Malkova A.. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013; 502:389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wilson M.A., Kwon Y., Xu Y., Chung W.-H., Chi P., Niu H., Mayle R., Chen X., Malkova A., Sung P. et al.. Pif1 helicase and polδ promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013; 502:393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mayle R., Campbell I.M., Beck C.R., Yu Y., Wilson M., Shaw C.A., Bjergbaek L., Lupski J.R., Ira G.. DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science. 2015; 349:742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Marians K.J. Lesion bypass and the reactivation of stalled replication forks. Annu. Rev. Biochem. 2018; 87:217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chavez D.A., Greer B.H., Eichman B.F.. The HIRAN domain of helicase-like transcription factor positions the DNA translocase motor to drive efficient DNA fork regression. J. Biol. Chem. 2018; 293:8484–8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zheng X.-F., Prakash R., Saro D., Longerich S., Niu H., Sung P.. Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae Mph1 protein. DNA Repair (Amst.). 2011; 10:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Symington L.S., Rothstein R., Lisby M.. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics. 2014; 198:795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.