


Abstract
Transcription and maintenance of genome integrity are fundamental cellular functions. Deregulation of transcription and defects in DNA repair lead to serious pathologies. The Mediator complex links RNA polymerase (Pol) II transcription and nucleotide excision repair via Rad2/XPG endonuclease. However, the functional interplay between Rad2/XPG, Mediator and Pol II remains to be determined. In this study, we investigated their functional dynamics using genomic and genetic approaches. In a mutant affected in Pol II phosphorylation leading to Mediator stabilization on core promoters, Rad2 genome-wide occupancy shifts towards core promoters following that of Mediator, but decreases on transcribed regions together with Pol II. Specific Mediator mutations increase UV sensitivity, reduce Rad2 recruitment to transcribed regions, lead to uncoupling of Rad2, Mediator and Pol II and to colethality with deletion of Rpb9 Pol II subunit involved in transcription-coupled repair. We provide new insights into the functional interplay between Rad2, Mediator and Pol II and propose that dynamic interactions with Mediator and Pol II are involved in Rad2 loading to the chromatin. Our work contributes to the understanding of the complex link between transcription and DNA repair machineries, dysfunction of which leads to severe diseases.
INTRODUCTION
Maintenance of genome integrity is essential for the normal cell function. A number of mechanisms have evolved to repair DNA damages induced by genotoxic agents or cellular metabolism. Nucleotide excision DNA repair (NER) is a unique evolutionarily conserved pathway that specifically removes bulky and/or helix-distorting DNA lesions including photoproducts induced by UV light (1,2). These DNA lesions can interfere with replication and transcription, emphasizing the importance of NER for cellular physiology. Given an essential role of NER, inherited defects in this DNA repair pathway lead to severe diseases including xeroderma pigmentosum (XP) and Cockayne syndrome (CS). NER factors recognize the DNA lesions, incise and excise the damaged DNA fragment from the genomic DNA allowing repair synthesis. Two subpathways have been proposed for NER mechanisms. Global genome repair (GGR) removes DNA lesions in the genome overall, and transcription-coupled repair (TCR) removes DNA lesions interfering with RNA polymerase II (Pol II) progression through actively-transcribed regions (3–7). TCR-specific Rad26/CSB protein was shown to bind the stalled Pol II to initiate the TCR complex assembly, but the existence of Rad26-independent TCR was also proposed in yeast (8–10).
Rad2/XPG is a DNA-repair 3′-endonuclease involved in NER. In human, mutations in XPG gene constitute one of the complementation groups found in patients with XP or combined XP/CS syndromes. Together with Rad1–10/XPF-ERCC1 5′-endonuclease, Rad2/XPG ensures the dual incision of the damaged DNA in both NER subpathways. Crystal structure of catalytic core of Rad2 in complex with DNA substrate has been reported (11). In addition to its nuclease activity, a non-catalytic function for human XPG in coordinated recognition of stalled transcription together with CSB, human homolog of yeast Rad26, has been proposed in TCR initiation (12). Previously, links of human XPG with transcription of nuclear receptor (NR)-dependent genes have been suggested, since XPG formed a stable complex with TFIIH allowing transactivation (13) and was present on active promoters and distal regions of NR-dependent genes (14,15). XPG roles have been also proposed in other DNA repair mechanisms, such as the processing of RNA-DNA hybrids into DNA double-strand breaks (16) or a non-catalytic function in homologous recombination (17).
Recently, we identified a novel link between Rad2/XPG and the Mediator complex (18). Mediator is a multisubunit coactivator complex conserved from yeast to human cells (19–22). In human, Mediator subunits have been involved in many diseases including several types of cancer (23,24). This complex plays a crucial role in Pol II transcription regulation. It is recruited to regulatory regions via direct interactions with specific transcription factors. Mediator is also in direct contact with Pol II serving as a functional bridge between specific regulators and Pol II basal transcription machinery. Mediator acts in cooperation with general transcription factors (GTFs) in preinitiation complex (PIC) assembly on core promoters (25). It has been shown recently that depletion or inactivation of Kin28 kinase subunit of TFIIH, one of the GTF, stabilizes a transient association of Mediator with the PIC assembled on core promoters (26,27). A modular organization of Mediator in head, middle, tail and Cdk8 kinase modules contributes to Mediator function. Crystallographic models have been reported for several yeast Mediator submodules (28), the head module (29–31) and more recently for core Mediator composed of head and middle modules (32). Our recent discovery of a Mediator link to NER suggested the Mediator functions beyond the transcription process per se (18). Yeast two-hybrid and coimmunoprecipitation experiments show that the essential Med17 Mediator subunit interacts with Rad2/XPG DNA repair protein. Binding of human MED17 to XPG was also observed in vitro (33). Genome-wide location analyses revealed that Rad2 was associated with upstream activating sequences (UAS) in the absence of exogenous genotoxic stress and that Rad2 occupancy of UAS was highly correlated with that of Mediator. In addition, Rad2 was enriched on transcribed regions of class II genes, correlating with Pol II occupancy. Interestingly, Rad2 occupancy of Pol II-transcribed genes was strongly decreased in rpb1–1 Pol II mutant when transcription was rapidly stopped, demonstrating that Rad2 binding to the chromatin was transcription-dependent. This decrease in Rad2 occupancy was observed on UAS and transcribed regions. However, no growth phenotypes, except UV sensitivity, or transcriptional effects were observed in the rad2Δ context suggesting that Rad2 does not play a major role in the transcriptional process in yeast. On the contrary, specific med17 Mediator mutants were UV-sensitive in a GGR-deficient genetic background. This UV sensitivity of med17 mutants was correlated with reduced Rad2 occupancy of Pol II-transcribed genes, and a concomitant decrease of the interaction between Mediator and Rad2 protein.
RNA polymerase II is the main component of the transcription machinery and the first complex that encounters the DNA damage in TCR (4–7). Rpb9 Pol II subunit is non-essential in yeast. Rpb9 modulates transcription start site (TSS) selection (34) and is required, together with TFIIS, to stimulate the intrinsic RNA cleavage activity of Pol II (35). In addition, Rpb9 subunit was proposed to be involved in an alternative, Rad26-independent TCR process, since in yeast its deletion increased the UV sensitivity of rad26 strain in a GGR-deficient context (36,37). It should be noted that Pol II interacts with Rad2/XPG in yeast and in human cells. Indeed, XPG protein was reported to coimmunoprecipitate with Pol II in crude extracts from undamaged HeLa cells (12). In yeast, we showed that Rad2 also coimmunoprecipitated with Pol II (18). Mediator is well known to directly interact with Pol II and these contacts are essential for transcription regulation (38–44). It remains to be determined how Mediator link to Rad2/XPG is related to Pol II and how Rad2 recruited by Mediator to UAS arrives on transcribed regions in regards to physical interactions and functional interplay between Mediator, Rad2/XPG and Pol II, three crucial components of nuclear machineries. It is also unknown if Mediator implication in TCR is Rad26- and/or Rpb9-dependent. Our understanding of the functional dynamics between TCR-related proteins/complexes is important to address one of the key biological questions on a complex cooperation between transcription and DNA repair in vivo on a genomic scale.
In this work, we used kin28 TFIIH, med17 Mediator and rpb9 Pol II mutants to precisely decipher the functional interplay between Mediator, Rad2 and Pol II. We show that in a kin28 mutant, in which a transient association of Mediator with Pol II and the PIC is stabilized on core promoters, Rad2 genome-wide occupancy shifts towards the core promoters following that of Mediator but is decreased on transcribed regions. We then performed extensive mutational analysis of Rad2-interacting domain of Med17 and showed that specific med17 mutations are involved in UV sensitivity, reduce Rad2 recruitment to transcribed regions and lead to uncoupling of Rad2 with Mediator and Pol II. Finally, we deleted Rpb9 Pol II subunit involved in TCR, which leads to allele-specific colethality with UV-sensitive Mediator mutants. Rpb9 deletion also leads to Rad2 stabilization on regulatory regions. Taken together, our data provide new insights into the functional interplay between Mediator, Rad2 and Pol II. We propose that Rad2 loading to regulatory regions bound by Mediator and its association with transcribed regions enriched in elongating Pol II could be connected and dependent on its dynamic interactions with Mediator and Pol II. Importantly, our work contributes to our understanding of a complex relation between transcription and DNA repair machineries, as well as Mediator implication in this link.
MATERIALS AND METHODS
Strains and plasmids
All Saccharomyces cerevisiae strains, plasmids and oligonucleotides used in this study can be found in Supplementary Tables S1–3.
To generate yeast strains allowing to test MED17 mutations, strains with med17 deletion complemented by a URA-selectable plasmid carrying the wild-type (WT) MED17 (med17Δ/MED17 URA) were transformed with a TRP-selectable plasmid carrying the WT or mutated MED17. Transformed clones obtained on medium without tryptophan were grown for 2 days on a non-selective medium (YPD) at 25°C and then replica-plated on 5-FOA containing medium. After 3 days, plates were replicated again on 5-FOA containing medium, and finally on medium without tryptophan after 3 more days. The plasmids were checked by polymerase chain reaction (PCR) and sequencing.
Random mutagenesis of Med17 and mutation screening
Briefly, we amplified Med17 (382–681) domain in the presence of 7 mM MgCl2 and 0.1 (A) or 0.5 mM (B) MnCl2 for 30 PCR cycles. PCR product were purified and mixed with a linearized yeast expression vector pVV204 including the coding sequence of Med17 without its C-terminal domain and TRP auxotrophic marker. The mix was co-transformed in a rad7Δ med17Δ/MED17 URA yeast strain and plated to have 8000–10 000 colonies. MED17 URA vector was then chased by replicating plates on 5-FOA containing medium. Selected colonies were finally replicated on three YPD plates, one kept at 30°C, one at 37°C and one treated with 30 J/m2 UVC before being incubated at 30°C in the dark. We looked for clones with either UV-sensitivity or temperature sensitivity phenotypes. Selected clones were streaked on medium without tryptophan (Casamino Acids medium supplemented with adenine and uracil, CAU), and their phenotypes were confirmed by spotting assay. The genomic DNA was then prepared and transformed in DH10b competent cells, using Ampicillin as selectable marker. Mutated vector was purified and retransformed in rad7Δ med17Δ/MED17 URA strain, before counterselecting MED17 URA vector on 5-FOA, in order to verify that the observed phenotypes were due to Med17 mutations. When it was the case, the plasmids were sequenced to determine Med17 mutations.
Site-directed mutagenesis of Med17
To generate specific mutations, we designed mutagenesis primers with 15–20 nt overhangs surrounding the targeted base mutation in the forward and reverse orientation. Two PCR products were amplified, one with primers Med17-F-attB1 and reverse mutagenesis primer, the other with primer Med17-R-attB2 and forward mutagenesis primer, using Phusion Flash Mastermix (ThermoFisher), according to manufacturer's instructions (15 cycles) and 100 ng pVV204-MED17 plasmid as a template. PCR products were separated on an agarose gel, the desired gel bands were excised, pooled and purified using Qiaquick Gel Extraction Kit (Qiagen), according to manufacturer's instructions. A total of 20 μl of the combined PCR products were mixed with an equal amount of Phusion Flash Mastermix, and seven PCR cycles were run without primers. A total of 25 pmol Med17-F-attB1 and Med17-R-attB2 (2.5 μl of a 10 μM stock), mixed with 5 μl of Phusion Flash Mastermix, were then added to the PCR mix and 15 PCR cycles were run. A total of 5 μl of the reaction were deposited on gel to confirm that the desired PCR product was obtained. The other 45 μl were purified using Qiaquick PCR Purification Kit (Qiagen).
For truncations of MED17 or RAD2, attB-containing primers were designed to amplify the desired portion of MED17 or RAD2. Products were amplified using Phusion Flash Mastermix (ThermoFisher) for 30 cycles, and 100 ng YPH500 genomic DNA as a template.
PCR products containing attB sites were inserted in pDONR201 plasmid using Gateway BP Clonase II Enzyme mix (ThermoFisher). The obtained donor plasmid was then used to transfer mutated Med17 cassette in pVV212 (for GAL4-DBD fusion constructs), pVV213 (for GAL4-AD fusion constructs) or pVV204 (for yeast expression plasmids with TRP auxotrophic marker) Gateway Destination plasmids, using Gateway LR Clonase II Enzyme mix (ThermoFisher), according to manufacturer's instructions.
Yeast two-hybrid assay
For yeast two-hybrid assay, haploid strains (Y187 and Y190) were transformed with constructs expressing the desired gene in fusion with Gal4 DNA-binding domain (with TRP auxotrophic marker) or Gal4 activating domain (with LEU auxotrophic marker), respectively. Clones growing on -trp or -leu medium, respectively, were selected and the presence of the plasmid was verified by PCR.
Haploid strains growing on -trp and -leu agar plates were scraped and resuspended in sterile water. Y187 and Y190 derived strains were mated together by spotting 2.5 μl of each suspension on a YPD plate and incubating at 30°C overnight. Patches were replica-plated on -trp -leu plates and incubated for 3 days at 30°C. Sufficient amounts of diploid yeasts were scraped and resuspended in synthetic defined (SD) medium supplemented with 40 mg/l Alanine (SD+2A). Optical Density at 600nm of the suspension was measured, and the suspension was diluted to obtain an OD600 of 0.1. 10 μl of this dilution were then spotted on agar plates containing either SD+2A medium supplemented with 10, 25 or 50 mM 3-amino-1,2,4-triazole (3AT), or -leu -trp medium, and incubated for 3 days at 30°C.
When indicated, X-gal staining was done as follows: for one 120 mm square petri dish, 10 ml 1% Agarose in water was mixed with 10 ml phosphate buffer (made from 6.15 ml 1M K2HPO4 and 3.85 ml 1M KH2PO4 aqueous solutions), both prewarmed at 50°C. 1.2 ml N,N-dimethylformamide (DMF), 0.2 ml 10% sodium dodecyl sulphate (SDS) solution (in ddH2O) and 0.2 ml of 4% X-Gal solution (in DMF, kept at −20°C) were successively added to the mix. The mix was then poured onto yeast spots. As soon as solidified, the plate was incubated at 30°C for 24 h. Pictures were taken using an office scanner, plate open in direction of the scanner. Luminosity and contrast were adjusted using ImageJ software.
Spotting assay
Yeast were grown on YPD plates for 2–3 days at 25°C. Sufficient amounts of cells were scraped and resuspended in water. Optical Density at 600 nm of the suspension was measured, and the suspension was diluted to obtain an OD600 of 0.5. Serial dilutions were then led to obtain suspensions with OD600 of 0.05, 0.005 and 0.0005. A total of 5 μl of each dilution was spotted on YPD agar plates, sometimes supplemented with 4-NQO or hydroxyurea as indicated. Once dried, spots were irradiated with the indicated UV dose using a UV Stratalinker 1800 (Stratagene). Plates were then incubated at 25°C, 30°C or 37°C for 3 days, as indicated. Pictures were taken using an office scanner, plate open in direction of the scanner. Luminosity and contrast were adjusted using ImageJ software.
Coimmunoprecipitation
A total of 100 ml exponentially growing cells were collected by centrifugation, washed and cell lysis was performed by bead-beating for 30 min at 4°C in WB+ buffer (10% Glycerol, 50 mM Hepes-KOH pH 7.5, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.05% NP-40, 1 mM dithiothreitol (DTT), 1 mM PMSF, cOmplete protease inhibitor cocktails (Roche)), as described previously (18). Protein concentration was measured using Bradford method, taking bovine serum albumin (BSA) as reference.
Protein extracts were then used for co-IP as follows: 50 μl Dynabeads pan-mouse IgG were washed three times in phosphate-buffered saline (PBS) containing 0.1% BSA, and incubated 1 h with antibodies (70 ng/μl anti-HA (12CA5), 30 ng/μl anti-Myc (9E10), 100 ng/μl anti-Rpb1-CTD (8WG16) or 80 ng/μl anti-Flag (M2)) at 4°C. Beads were washed again three times in PBS/0.1% BSA, then two times in WB+ buffer. A total of 1.5 mg of proteins were added to beads and WB+ buffer was added to adjust volume to have the same volume in all samples (at least 50 μl). Beads and proteins were incubated together for 3 h at 4°C with constant agitation (1300 rpm). Beads were then washed four times in WB+ buffer. A total of 40 μl SDS Sample buffer (15% Glycerol, 3% SDS, 75 mM Tris–HCl, pH 6.8, 15 mM EDTA) was finally added to beads, and samples (with beads) were kept at −80°C until further analysis.
Just prior to SDS-polyacrylamide gel electrophoresis analysis, samples were thawed, supplemented with 5 μl 1M DTT and incubated at 95°C for 2 min. Separation was done on 8% bis-acrylamide gels in Tris-Glycine-SDS buffer, and proteins were transferred on Amersham Protran 0.2 NC membranes (GE Healthcare) for western blotting. Membranes were preblocked overnight in Tris-buffered-saline supplemented with 0.5% Tween 20 (TBS-T) and 5% Milk, then incubated 1 h with the indicated antibody in TBS-T with 2% Milk. After three washes in TBS-T, membranes were incubated 45 min in TBS-T with 2% Milk containing secondary antibodies (HRP-anti Mouse-IgG (H+L) (Promega)). After three more washes in TBS-T, detection was carried out using Amersham ECL or ECL-Prime reagents (GE Healthcare). Imaging was done using a Fusion FX7 imaging system. Luminosity and contrast were adjusted using ImageJ software.
The relative intensity of immune staining was quantified using ImageJ software. The intensity of immune staining for coimmunoprecipitated protein signals relative to the WT was normalized against immunoprecipitation signals. The mean values and standard deviation (indicated by error bars) of three or four independent experiments were calculated.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was done as previously described (18) except for a sonication step performed on a S220 focused-ultrasonicator (Covaris) in 1 ml milliTube (Covaris), for two cycles of 3 min spaced by a 30 s rest time. Each cycle consisted of 150W pulses for 10% of the time (duty factor 10). Sonicated lysates were then transferred to a new 2 ml safe-lock tube and the centrifugation was performed at 15 000 g for 20 min. Supernatant was collected and combined with 300 μl FA/SDS/PMSF (50mM HEPES-KOH, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% sodium deoxycholate, 1% Triton X100, 0.1% SDS, 1 mM PMSF). Sonicated chromatin was divided in 250 μl aliquots, snap-frozen in liquid nitrogen and kept at −80°C until further use.
Immunoprecipitation, DNA precipitation and library preparation were done on an IP-Star compact automated system (Diagenode) using built-in programs and following manufacturer's instructions, with the following exceptions (Denby Wilkes et al., in preparation), except for kin28-ts and the corresponding WT samples prepared as previously described (18).
IP was done using the ChIP_IPure_200_D program, at 22°C, with 1 h ‘Ab coating’ (Slow speed), 3 h ‘IP reaction’ (Medium speed) and 5 min ‘washes’ (Fast speed). ‘Beads wash buffer’ was PBS + 0.1% BSA, FA/SDS adjusted to 500 mM NaCl was used for ‘IP Wash 1’ and ‘IP Wash 2’, ‘IP Wash 3’ was done with IP buffer (Tris 10 mM pH8, LiCl 0.25M, EDTA 1 mM, NP40 0.5%, Na-Deoxycholate 0.5%), ‘IP Wash 4’ was done with TE (Tris 10 mM pH 8, EDTA 1 mM). ‘Elution buffer’ was the elution buffer (A+B) of Auto iPure kit V2 (Diagenode). ‘Ab coating mix’ was anti-HA (12CA5, 35 ng/μl), anti-Myc (9E10, 15 ng/μl) or anti-Rpb1-CTD (8WG16, 50 ng/μl) completed to 100 μl with ‘Beads wash buffer’. ‘Sample’ was 200 μl sheared chromatin, supplemented with 4 μl of 5% BSA and 4 μl 50X Protease Inhibitor Cocktail (prepared by dissolving one cOmplete tablet in 1 ml ddH2O). A total of 20 μl DiaMag protein A-coated magnetic beads (Diagenode) were used per sample.
After program completion, strips containing the eluates were warmed to redissolve precipitated SDS, eluates were recovered using a magnetic stand, 5 μl of 5M NaCl was added to eluates and they were incubated for 4 h at 65°C to reverse crosslink. A total of 1 μl RNase A (1 mg/ml, ThermoFisher) was added to eluates and they were then incubated for 30 min at 37°C. DNA purification was done on IP-Star system using Auto iPure kit V2 (Diagenode), with iPure program selected (50 μl elution), following manufacturer's instructions.
Three biological replicates were used for ChIP experiments analysed by qPCR and then combined for sequencing. Library preparation for ChIP-seq was performed on IP-Star system using MicroPlex Library Preparation Kit v2 (Diagenode), following manufacturer's instructions.
ChIP-seq data analysis
ChIP-seq data were analysed using the following procedure. Reads were first trimmed with cutadapt (v1.12, http://dx.doi.org/10.14806/ej.17.1.200) then mapped on S. cerevisiae genome (University of California at Santa Cruz [UCSC] version sacCer3) using bowtie2 (v2.2.1, (45)). Files were converted using Samtools (v1.16, (46)) and deepTools (v2.4.2–4-99ec5d, (47)). Read counts were first normalized in RPM (Reads Per Million of mapped reads) then by qPCR data, on a set of selected regions, using the ratio between WT and mutant strains as previously described (48). The number of mapped reads for each ChIP-seq experiment and normalization coefficients are indicated in Supplementary Table S5. Input DNA and DNA from ChIP with an untagged strain were used as negative controls.
The transcribed regions were determined using the TSS and TES (Transcription End Sites) of mRNA genes taken from (49,50) (n = 5337). In each experiment, transcribed regions were grouped in deciles based on their average Pol II occupancy signal in the WT. Med15 Mediator subunit and Rad3 TFIIH subunit peaks were called from the data in (18,25,48) using MACS2 (v2.1.10) and filtered (fold-change >2.5 and P-value < 1e-10) (n = 561 for Med15 and n = 2411 for Rad3). Peaks that were further than 1 kb away from a TSS were discarded. Promoter regions (n = 4068) were defined as corresponding intergenic regions in tandem or in divergent orientation in yeast genome excluding intergenic regions encompassing Pol III-transcribed genes. In order to avoid potential biases that could arise from low enrichment of Rad2 ChIP, we focused our analysis on the most Pol II-enriched decile. All correlations were calculated with Spearman method (51). All figures were prepared using R packages (https://www.R-project.org/). Data visualization package for R ggplot2 was used to prepare Supplementary Figures S9 and 10 (52).
RESULTS
Rad2 is blocked on promoter regions in a kin28 mutant
To analyse Rad2-Mediator-Pol II functional interplay, we sought to determine if Rad2 occupancy depends on the transient contact between Mediator and Pol II on the core promoters. We therefore used a temperature-sensitive mutant of Kin28 TFIIH subunit, kin28-ts16 (53). In this mutant, shifting from 25°C to 37°C decreased Pol II CTD phosphorylation that regulates the promoter escape of the enzyme. Previously, Mediator stabilization on core promoters where the PIC is assembled was evidenced after depletion or inhibition of Kin28 (26,27,54,55).
To determine genome-wide Mediator, Pol II and Rad2 distribution in kin28 mutant, we performed ChIP followed by high throughput sequencing (ChIP-seq). Med17 Mediator subunit, Rad2 and Rpb1 Pol II subunit occupancy were analysed after a transfer of the WT and kin28-ts mutant cells to 37°C for 75 min. ChIP followed by qPCR were also performed on selected UASs, core promoters and transcribed regions (Supplementary Figure S1). Bioinformatic analyses of Mediator (Med17 subunit) profile in kin28 mutant showed the appearance of peaks corresponding to core promoter regions that aligned with TFIIH (Rad3 subunit) peaks (black arrows on Figure 1A), whereas these peaks were low or absent in the WT strain (Figure 1A and C), in agreement with previous observation of Mediator stabilization in Kin28-depleted or inhibited contexts (26,27,54,55). Metagene (or average density profile) analysis was performed for Rad3 TFIIH peaks, corresponding to core promoter regions, and Med15 Mediator peaks, corresponding to UASs. Average density profiles show an increase in Med17 enrichment on UAS (Figure 1B, upper panel) and a clear enrichment of Med17 in the vicinity of Rad3 TFIIH peaks, corresponding to core promoter regions (Figure 1C, upper panel). This suggests the stabilization of a Mediator-containing intermediate on the core promoters, due to the decrease in Pol II phosphorylation (26,27,54,55). Mediator occupancy ratios between the kin28 mutant and the WT clearly indicate this Mediator stabilization on the core promoters (Figure 1D and Supplementary Figure S2, ratio panels). Interestingly, our results show clear changes in Rad2 distribution in kin28 mutant compared to the WT (Figure 1 and Supplementary Figure S2). Specific examples on Figure 1A illustrate an increase in Rad2 occupancy peaks on regulatory regions in kin28 mutant and a shift of Rad2 distribution in this mutant towards the core promoters defined by Rad3 TFIIH subunit peaks (black arrows on Figure 1A). Metagene analysis demonstrated that Rad2 association was increased on UAS in the vicinity of Mediator enrichment peaks defined in the WT context (Figure 1B). Rad2 occupancy in kin28 mutant was also increased on core promoters. Our analysis showed that Rad2 distribution was shifted towards the core promoters in the kin28 context, following the Mediator stabilization on these regions, although to a lesser extent than Mediator (Figure 1C and D, note also an asymmetric Rad2 distribution in kin28-ts on UAS in Figure 1B). Compared to Mediator, which was stabilized mostly on core promoters, the highest Rad2 occupancy ratio between the kin28 mutant and the WT was located between the UAS and the core promoter (Figure 1D and Supplementary Figure S2, ratio panels). Importantly, the maximal Rad2 occupancy ratio between kin28 mutant and the WT was similar to that of Mediator (∼2-fold, Figure 1D). Finally, the presence of Rad2 was reduced on transcribed regions on a genomic scale, together with a global decrease in Pol II association (Figure 2A; Supplementary Figures S1 and 3). Indeed, Pol II occupancy was strongly decreased in kin28-ts16 mutant compared to the WT strain (Figure 1A; Supplementary Figures S1 and 3). We noted that a global decrease in Pol II occupancy was less pronounced at the beginning of transcribed regions, when the Pol II occupancy ratios between kin28-ts mutant and the WT were analysed (Supplementary Figures S2, ratio panels and Supplementary Figure S3, right panels). This fact is consistent with Pol II promoter escape defect in kin28 mutants (26,27,54,55). Figure 2A (middle panel) clearly shows major changes of Rad2 genome-wide occupancy in kin28 mutant compared to the WT with an increase on UASs and core promoters and a decrease within transcribed regions.
Figure 1.
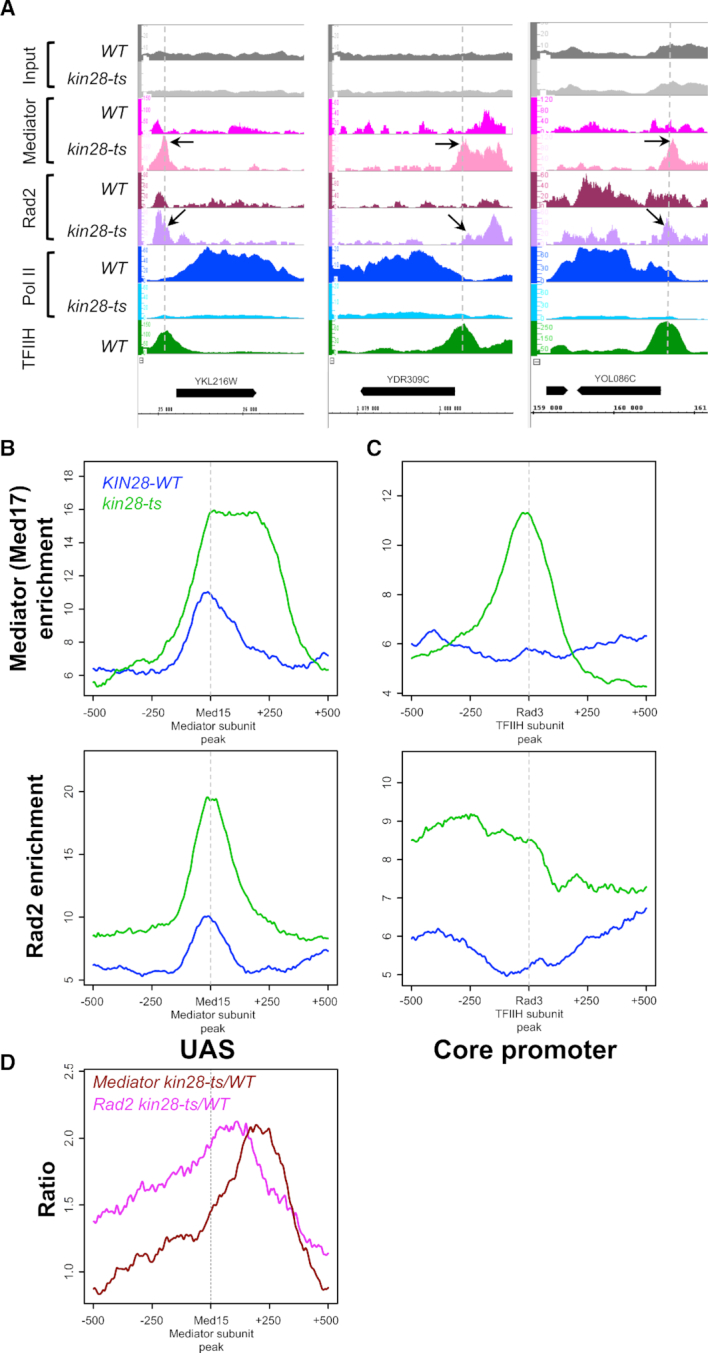
Effects of kin28-ts mutation on genome-wide Mediator and Rad2 occupancy. (A) Examples of ChIP-seq tag density profile of Mediator (Med17), Rad2, Pol II (Rpb1) and TFIIH (Rad3) in WT and kin28-ts context following a 75-min shift from 25°C to 37°C. ChIP-seq was done on the chromatin from exponentially growing yeasts expressing tagged versions of Med17 (Myc) and Rad2 (HA) in WT or kin28-ts context. Mapped reads were extended to 150 bp, and the number of reads for each position of the genome was counted to determine tag density. Densities were scaled per millions reads, and normalization step was performed as described in ‘Materials and Methods’ section. Alignment of Med17 Mediator peaks present in kin28-ts mutant, and low or absent in WT strain, with Rad3 TFIIH peaks in core promoter regions is indicated by the vertical dashed lines. The black arrows point to the increase of Mediator on core promoter regions and increase of Rad2 on UAS and core promoter regions. (B andC) Average tag density in Med17 Mediator ChIP (upper panel) and Rad2 ChIP (lower panel), around Med15 Mediator peaks (B, UAS, 1000 bp window) and Rad3 TFIIH peaks (C, Core promoter, 1000 bp window). Average tag density in WT strains is indicated in blue, whereas average tag density in kin28-ts strains is indicated in green. (D) Mediator (in brown) and Rad2 (in pink) occupancy ratio between kin28-ts mutant and the WT around Med15 Mediator peaks (UAS, 1000 bp window). The maximum of the Rad2 occupancy ratios is located between UAS and the maximum for Mediator occupancy ratios.
Figure 2.

Effects of kin28-ts mutation on genome-wide Mediator, Rad2 and Pol II occupancy on transcribed regions and Mediator/Rad2/Pol II interactions. (A) Average tag density in Med17 Mediator ChIP (upper panel), Rad2 ChIP (middle panel) and Rpb1 Pol II ChIP (lower panel) on transcribed regions (scaled windows for 500 bp before TSS, between TSS and TES, and 500 bp after TES, 10% Pol II most-enriched regions in WT). Average tag density in WT strains is indicated in blue, whereas average tag density in kin28-ts strains is indicated in green. The black arrows point to the increase in Rad2 signal on regulatory and core promoter regions in kin28 mutant compared to the WT. (B) Effect of kin28-ts mutation on Mediator/Rad2/Pol II interactions. Western blot analysis of Mediator interaction with Rad2 and Pol II. Crude extracts were prepared from yeasts expressing tagged versions of Med17 (Myc) and Rad2 (HA) in the WT strain and kin28-ts mutant following a 75-min shift at 37°C, after reaching exponential phase at 25°C. Samples were immunoprecipitated with α-Rpb1 (Pol II) antibody (IP). Immunoprecipitates and Inputs were analysed by western blotting with α-Myc, α-HA and α-Rpb1 antibodies. The effect of kin28-ts mutation on Ser5 phosphorylation of Pol II CTD was verified by western blotting with an antibody against Ser5P-CTD (H14, Abcam). The intensity of immune staining for coimmunoprecipitated Med17 or Rad2 signals relative to the WT was normalized against immunoprecipitation signals and is displayed in the bottom panels. The mean values and standard deviation (indicated by error bars) of three independent experiments are shown. The asterisk represents a significant difference between the WT and the mutant at P-value < 0.05 in a Student's t-test.
To determine if a part of the observed changes in Rad2, Mediator and Pol II chromatin binding could be explained by modified interactions between these protein/complexes, we performed coimmunoprecipitation experiments with crude extracts from kin28-ts mutant compared to the WT (Figure 2B). We confirmed a large decrease in Ser5-phosphorylated Pol II in kin28 mutant by western blot analysis of crude extracts prepared from the cells after a transfer to a non-permissive temperature. Pol II was immunoprecipitated via its Rpb1 subunit and coimmunoprecipitated Rad2 and Mediator (Med17 subunit) were analysed in both contexts. We observed that less Rad2 was coimmunoprecipitated in kin28 mutant. Interestingly, more Mediator was coimmunoprecipitated with Pol II in kin28 mutant. Our results show that interactions between Mediator, Rad2 and Pol II are modified when Ser5 phosphorylation of Pol II is impaired. Namely, we observed a significant increase for Mediator-Pol II contact and a significant decrease for Pol II-Rad2 interaction (Figure 2B, bottom panels for quantification). We verified that Mediator or Rad2 CoIP signals were insensitive to DNAse treatment (Supplementary Figure S4).
We next investigated a potential link of Rad2 to TFIIH which is one of the PIC components, since Rad2 interacts with TFIIH in NER (56,57). We performed coimmunoprecipitation experiments with crude extracts from kin28-ts mutant and the WT (Supplementary Figure S5A). TFIIH was immunoprecipitated via its Rad3 subunit and coimmunoprecipitated Rad2 was analysed in both contexts. We observed that Rad2 was coimmunoprecipitated with TFIIH in the absence of genotoxic stress and that the similar amount of Rad2 was coimmunoprecipitated both in the WT and kin28 mutant. In line with a strong transcriptional defect in kin28 mutant, our ChIP experiments show that Rad3 TFIIH subunit occupancy on core promoters was strongly decreased in this mutant compared to the WT (Supplementary Figure S5B).
Taken together, our results show that Rad2 chromatin binding in kin28 mutant is influenced by both Mediator stabilized on core promoter regions and Pol II whose occupancy is decreased within the gene bodies. This suggests that Rad2 recruitment to upstream regulatory regions and its loading to transcribed regions could be connected and that Rad2 distribution on the chromatin depends on its interactions with Mediator and Pol II.
Mutational analysis of Med17 C-terminal domain interacting with Rad2
To investigate precisely the role of Mediator in Rad2 recruitment, we analysed the details of Mediator-Rad2 interactions and their implications in Mediator/Rad2/Pol II functional relationships. To identify med17 mutants specifically affecting the interaction with Rad2 in vivo, we first used a yeast two-hybrid approach (Y2H) to determine interaction domains in Med17 and Rad2 proteins. The Med17-interacting fragment of RAD2 gene encoded amino acids 549–857 of its protein product (18). We analysed two series of Med17 truncation mutants for their interactions with Rad2 fragment and Med22 Mediator subunit (Figure 3A and B). We conclude that Med17 domain (amino acids 381–681) is necessary and sufficient for Med17 interaction with Rad2 fragment. Further truncations likely affect Med17 secondary structure, also impairing the interactions with Med22 and Med11 Mediator subunits.
Figure 3.

Mutation analysis of Med17 region interacting with Rad2 fragment. (A andB) Serial truncations of Med17 were assayed for interaction with Med22 and a Rad2 fragment (549–857) identified in a yeast two-hybrid screening using Med17 as bait. (A) Gal4 DNA binding (Gal4-DBD) domain, alone (empty) or in fusion with full length Med17, 5 C-terminal truncations or 5 N-terminal truncations of Med17, was co-expressed with Gal4 Activation Domain (Gal4 AD), alone or in fusion with Med22 or residues 549 to 857 of Rad2. Yeast cells were spotted on SD+2A medium supplemented with 25 mM 3-AT, grown for 3 days (left panel) and then stained with X-Gal for 24h (middle panel). Residues 346–687 appear necessary and sufficient to have an interaction of Med17 with both Med22 and Rad2 fragment. (B) This minimal interaction domain was subjected to further N-terminal or C-terminal truncations, and N-terminal and C-terminal fragments of this domain were also tested for the interaction with Med22 and Rad2 fragment, using the same protocol as in panel A. (C) Pile histogram for occurrence of mutations affecting each residue in Med17 region interacting with Rad2, and the associated UV sensitivity phenotype. A graph for a complete Med17 protein is shown on Supplementary Figure S6D. The number of mutation occurrences is indicated for each amino acid position. Only mutants predicted to express full length Med17 were considered. The colours correspond to the associated phenotypes as follows: UV-sensitive in green, mild UV-sensitive in clear green and UV-insensitive in pink. More frequently mutated residues F437, M442, I541 were more frequently associated with UV sensitivity (in green), whereas K517 and nearby residues were more frequently found with temperature-sensitive only mutants (UV-insensitive in pink). (D) Med17 residues represented in Supplementary Figure S6E are highlighted in the structure of Mediator head module from Robinson and colleagues (PDB ID: 4GWP). Residues, mutations of which impaired or reduced Y2H Med17 interaction with Rad2 fragment specifically are in green (M442, K446, I541, N639). Residues, mutations of which impaired Y2H interaction of Med17 with Med22, Med11 and Rad2 fragment (K433, I445, I562) are in red. Residues, mutations of which impaired Y2H Med17 interaction with Med11 and Rad2 fragment, but not Med22, are in yellow (S409, V457, T509, I647) with the exception of M442. Other residues were not depicted. (E) Spotting assay to determine UV and temperature sensitivity of yeast strains expressing either WT or mutant versions of Med17 in rad7Δ context. Yeast cells were spotted on YPD agar plates, irradiated or not with 30 J/m2 UV-C (254 nm) and incubated at 30°C or 37°C for 3 days.
We performed a similar analysis to identify which Rad2 fragment is the most important for the interaction with Med17. We found that Rad2 catalytic I domain (amino acids 756–857) is necessary and sufficient for the interaction with Med17 (Supplementary Figure S6A and B).
We then performed a random mutational screening of Med17 C-terminal domain based on in vivo phenotypes as described in Materials and Methods (Supplementary Figure S6C). We identified and sequenced 88 new med17 mutants, which were UV-sensitive (UVs) and/or temperature-sensitive (ts) in a rad7Δ GGR-deficient context. This set of mutants completed a collection of 27 previously characterized med17 mutants (Supplementary Table S4, (48)).
Among over 100 med17 mutants, UVs phenotype was systematically associated with ts phenotype (Supplementary Table S4). This suggests that the UVs phenotype in med17 mutants is closely related to ts phenotypes. Conversely, not all Med17 ts mutants were UVs, suggesting that the UV sensitivity of Mediator mutants is not the systematic indirect consequence of transcription defects per se. Moreover, UV-sensitive phenotypes were observed only in GGR-deficient context, but not in a context when both NER subpathways are functional. In most cases, several mutations co-occurred in med17 mutants and we identified positions that were more frequently mutated and generally associated with UV sensitivity (Figure 3C and Supplementary Figure S6D, residues indicated in green).
Direct mutagenesis allowed us to identify point med17 mutations that specifically affected Med17-Rad2 interaction in Y2H system (Supplementary Figure S6E). Interestingly, the corresponding residues are closely located on the S. cerevisiae Mediator head structure (31) (Figure 3D). It should be noted that these med17 point mutations do not modify Rad2 or Pol II coimmunoprecipitation with Mediator (Supplementary Figure S6F) and do not lead to UVs or ts phenotype (Supplementary Figure S6G and data not shown), suggesting that the Y2H system is extremely sensitive to detect Med17-Rad2 interaction changes and that stronger defects with additional mutations are required for changes in CoIP and for UV sensitivity phenotypes of med17 mutants.
We identified several cases where one additional mutation distinguished a UV-sensitive mutant from a non UV-sensitive mutant (Figure 3E). In particular, we found that med17-Q444P mutant is temperature-sensitive but does not have a pronounced UV-sensitive phenotype in rad7Δ context, whereas med17-Q444P/M442L is both ts and strongly UVs. The latter mutant is also more sensitive to 4-NQO, further showing its sensitivity to genotoxic stress (Supplementary Figure S6H). Another example is the double mutant med17-M442V/V670E, which is both UVs and ts, whereas med17-M442V and med17-V670E are neither UVs nor ts. Interestingly, M442 is one of the most frequently mutated positions in our screening, and in all but one case the corresponding mutants are UVs (Figure 3C and Supplementary Figure S6D). Mutations in M442 also decreased Med17-Rad2 interaction in Y2H system. Two mutants carrying med17-Q444P single and med17-Q444P/M442L double mutations were therefore selected for further study. It should be noted that med17-M442L single mutant did not have any ts or UVs phenotypes (Supplementary Figure S6G).
med17 UV-sensitive mutations lead to uncoupling between Rad2 occupancy and those of Mediator and Pol II genome-wide
We first examined the effect of Med17 mutations on Mediator interaction with Rad2 and with Pol II by CoIP experiments. Med5-Myc subunit was used to immunoprecipitate Mediator and the coimmunoprecipitation of Rad2 and Rpb1 Pol II subunit were analysed by western blotting. We found that significantly less Rad2 was coimmunoprecipitated with Mediator in the UVsmed17-Q444P/M442L mutant compared to the WT and med17-Q444P (Figure 4A, lower left panel for quantification). Surprisingly, we found that an increased amount of Rpb1 was coimmunoprecipitated with Mediator in med17-Q444P and med17-Q444P/M442L mutants compared to the WT strain (Figure 4A, lower right panel for quantification). This suggests that Mediator-Pol II interaction is modified in these mutants independently of their UV sensitivity. Together with the absence of ts or UVs phenotypes in med17-M442L single mutant, no effect on Rad2 or Pol II CoIP was observed (Supplementary Figure S7A).
Figure 4.

Effects of med17 mutations on Mediator interactions with Rad2 and Pol II and on genome-wide Mediator, Rad2 and Pol II occupancy. (A) Western blot analysis of Mediator interaction with Rad2 and Pol II in standard growth conditions. Crude extracts were prepared from yeasts expressing tagged (+) or untagged (−) versions of Med5 (Myc) in MED17-wt, med17-Q444P or med17-Q444P/M442L context in exponential phase and samples were immunoprecipitated with α-Myc antibody (IP Myc). Immunoprecipitates and Inputs were analysed by western blotting with α-Myc, α-HA and α-Rpb1 antibodies. The intensity of immune staining for coimmunoprecipitated Rad2 or Pol II signals relative to the WT was normalized against immunoprecipitation signals and is displayed in the bottom panels. The mean values and standard deviation (indicated by error bars) of four independent experiments are shown. The asterisk represents a significant difference between the WT and the mutant at P-value < 0.05 in a Student's t-test. The Rad2 CoIP signal in med17-M442L/Q444P mutant was also significantly different from med17-Q444P. (B) The tag densities in the Pol II ChIP-seq experiments were calculated for the Pol II-transcribed mRNA genes. Tag densities were normalized relative to qPCR data on a set of selected genes. Each point on the plot corresponds to one transcribed region. A linear regression (dotted line) for ChIP-seq density in the med17-Q444P or med17-Q444P/M442L mutant versus ChIP-seq density in WT or in med17-Q444P versus med17-Q444P/M442L and an R2 linear regression coefficient are indicated. The dashed line corresponds to y = x. (C) Pair-wise SCC of ChIP-seq data between med17 mutants and WT were calculated for Rad2 and Mediator on intergenic regions (left column, intergenic regions for Pol II-transcribed genes in tandem or in divergent orientation, excluding intergenic regions encompassing Pol III-transcribed genes) or for Rad2 and Pol II on transcribed regions (right column). The colours correspond to the scale for SCC indicated in Supplementary Figures S9 and 10. (D) Average tag density in Med5 Mediator ChIP (upper left panel), Rad2 ChIP (lower panels) and Rpb1 Pol II ChIP (upper right panel), around Med15 peaks corresponding to UAS (left panels, 1000 bp window), and on transcribed regions of 10% Pol II most-enriched genes (right panels, scaled windows for 500 bp before TSS, between TSS and TES, and 500 bp after TES). Average tag density in WT, med17-Q444P and med17-Q444P/M442L strains is indicated in blue, green and orange, respectively.
To analyse how med17 mutations affected Mediator, Pol II and Rad2 distribution on the yeast genome, we performed ChIP-seq experiments for Mediator (Med5 subunit), Pol II (Rpb1 subunit) and Rad2 in the med17 mutant and the WT strains. Occupancy of these components was also determined in med17 mutants compared to the WT using ChIP followed by qPCR on a set of selected UASs, promoters and transcribed regions (Supplementary Figure S8). Our ChIP experiments show that med17-M442L single mutation does not significantly affect Mediator, Pol II or Rad2 chromatin occupancy (Supplementary Figure S7B, C and D). Our results indicate a strong transcriptional defect in both med17-Q444P and med17-Q444P/M442L mutants. Genome-wide analysis shows a global 3-fold decrease in Pol II occupancy in both med17 mutants compared to the WT (the slope of regression line is equal to 0.33 and 0.32 for med17-Q444P and med17-Q444P/M442L, with a high R2 equal to 0.97 and 0.92, respectively) (Figure 4B, upper and middle panels). This global effect on Pol II occupancy was very similar between the two mutants, with a high R2 coefficient equal to 0.97 and the slope of the regression line equal to 0.97 (Figure 4B, lower panel).
In the WT context, Rad2 and Mediator colocalize on UAS and their genome-wide occupancies are highly correlated (18). To evaluate how med17 mutants affect Rad2 occupancy on intergenic regions, we calculated Spearman correlation coefficients (SCC) between Mediator and Rad2 occupancy on these regions in mutant and the WT strains (Supplementary Figure S9). SCC provides a statistical measure of the strength of a relationship between two sets of data. We observed a decrease of this Rad2-Mediator correlation on intergenic regions in med17 mutants compared to the WT, especially in med17-Q444P/M442L. SCC was equal to 0.74 for the WT that is considered as strong and decreased to 0.69 and 0.58 for med17-Q444P and med17-Q444P/M442L, respectively (Figure 4C, left column). For Rad2 genomic occupancy, we observed lower correlation between med17-Q444P/M442L and the WT or med17-Q444P (Supplementary Figure S9). A similar correlation analysis was then performed to compare Rad2 and Pol II occupancy on transcribed regions in med17 mutants and the WT (Figure 4C, right column, and Supplementary Figure S10). We observed a decrease in Rad2–Pol II correlation in med17 mutants. The correlation between Rad2 and Pol II decreases in both mutants, with a much stronger decrease in the double mutant, which is strongly UVs (SCC equal to 0.81, 0.64 and 0.48 for the WT, med17-Q444P and med17-Q444P/M442L, respectively).
Average density analysis was then done to compare Mediator, Rad2 and Pol II distributions in med17 mutants and the WT (Figure 4D). Our analysis shows some increase of Mediator binding in the single and further in the double mutant and also an increase in Rad2 association to UAS in the med17 mutants (Figure 4D, left panels). The position of Mediator and Rad2 peaks remains largely unchanged and no shift to core promoters was observed (Supplementary Figure S11). In accordance with our linear regression analysis, a large decrease in Pol II occupancy was observed on transcribed regions for both med17 mutants (Figure 4D, upper right panel, and Supplementary Figure S12). We noted some differences in Pol II profiles between med17 mutants with the enzyme accumulation at the beginning of transcribed regions in the double mutant, as illustrated by Pol II occupancy ratios between med17-Q444P/M442L mutant and the WT on transcribed regions for all Pol II enrichment quantiles (Supplementary Figure S12A). Importantly, Rad2 occupancy on transcribed regions of 10% Pol II most-enriched genes was decreased in med17 mutants, with a stronger decrease observed in the UVs double mutant (Figure 4D, lower right panel and Supplementary Figure S12B).
Using a single Med17 mutant (Q444P) with ts phenotype and a double mutant with additional M442L mutation leading to strong UV sensitivity phenotype, we revealed an uncoupling of Rad2 and Mediator on UASs and especially of Rad2 and Pol II on transcribed regions and a decrease in Rad2 presence within the gene bodies on the genomic scale, particularly pronounced in the double mutant. Thus, our results clearly show that specific Med17 Mediator mutations that modify Mediator interactions with Rad2 and Pol II affect Rad2 presence on transcribed regions genome-wide. Importantly, Pol II occupancy is globally decreased in a similar way in both mutants, demonstrating that Pol II alone could not explain Rad2 loading to transcribed regions and suggesting a connection between Rad2 binding to UAS and its loading to transcribed regions.
Rpb9 deletion is colethal in combination with med17 UV-sensitive mutations
To further investigate the UV sensitivity phenotypes of Mediator mutants, we performed genetic interaction analyses of Mediator and NER components. The phenotype of med17 UVs mutants is only visible in rad7Δ GGR-deficient context, suggesting a role for Mediator in TCR. We therefore tested whether med17 mutants would be epistatic with the deletion of rad26, the homolog of CSB and the most prominent TCR factor. It was the case in GGR-proficient background (18). Surprisingly, we found that med17 UVs mutants had synthetic UV sensitivity phenotype with rad26 deletion in rad7Δ GGR-deficient context (Figure 5A and Supplementary Figure S13A). Indeed, a combination of med17 UVs mutations with rad26 deletion in rad7Δ context leads to an increase in UV sensitivity compared to the med17 UVsrad7Δ or rad26Δ rad7Δ. For example, med17-Q444P/M442L rad26Δ rad7Δ mutant is more UV-sensitive than med17-Q444P/M442L rad7Δ or MED17 rad26Δ rad7Δ. This suggests that the UV sensitivity of med17 mutants could be at least in part independent of Rad26-related TCR.
Figure 5.
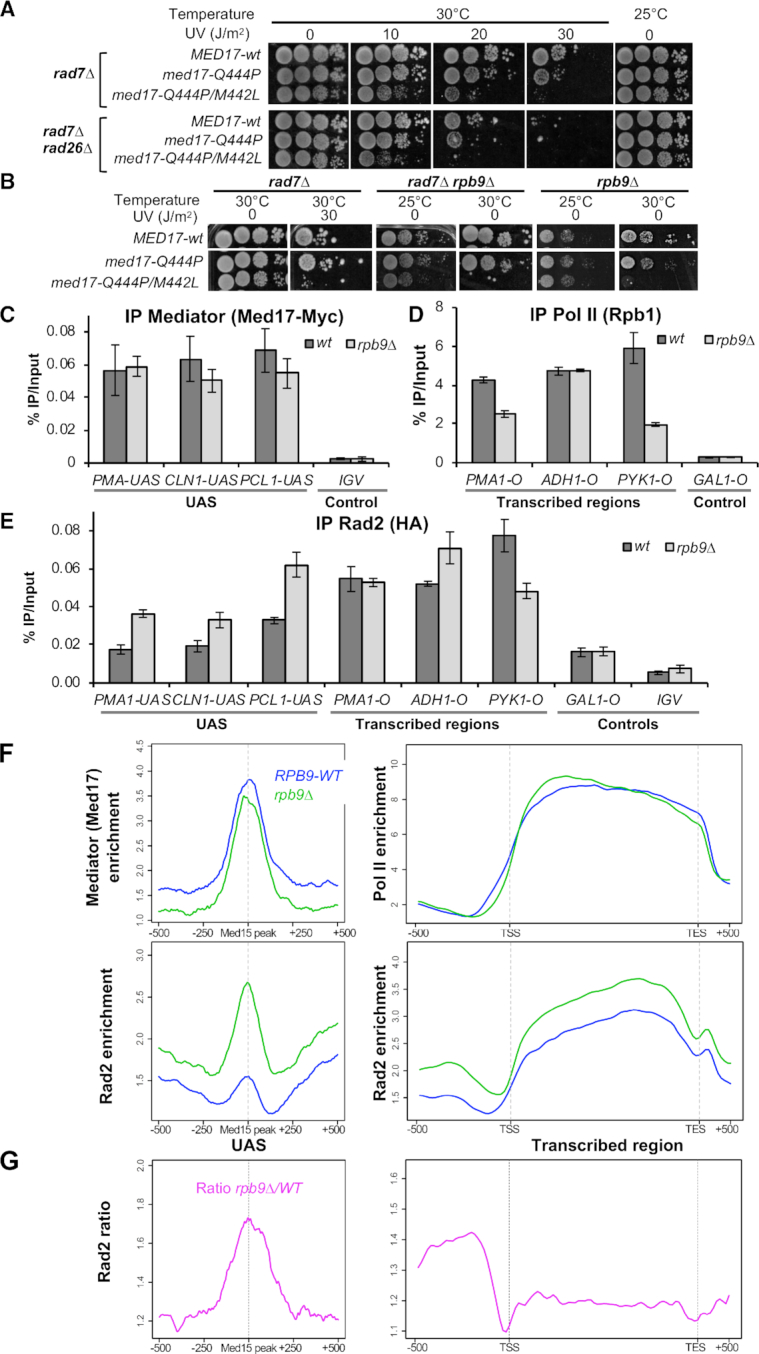
Effects of rpb9 deletion on the viability of Med17 mutants and on Mediator, Rad2 and Pol II chromatin occupancy. (A) Spotting assay to determine UV and temperature sensitivity of yeast strains expressing WT or mutant versions of Med17 in rad7Δ or rad7Δ rad26Δ contexts. Yeast cells were spotted on YPD agar plates, irradiated or not with 10, 20 or 30 J/m2 UV-C (254 nm) and incubated at 30°C or 25°C for 3 days. (B) Spotting assay to determine UV and temperature sensitivity of yeast strains expressing WT or mutant versions of Med17 in rad7Δ, rad7Δ rpb9Δ or rpb9Δ contexts. Yeast cells were spotted on YPD agar plates, irradiated or not with 30 J/m2 UV-C (254 nm) and incubated at 30°C or 25°C for 3 days. Med17 mutants unable to grow after UV treatment in rad7Δ context are also unable to grow at 30°C, in the absence of UV treatment, in rad7Δ rpb9Δ and rpb9Δ contexts. (C, D andE) Quantitative ChIP analysis of Mediator, Pol II and Rad2 occupancies under standard growth conditions. Sonicated chromatin from exponentially growing yeasts expressing tagged versions of Med17 (Myc) and Rad2 (HA) in WT or rpb9Δ context was precipitated using α-Myc (C), α-Rpb1 (D) or α-HA (E) antibodies. Quantitative PCR was performed on the precipitated DNA, using primer pairs designed to amplify either regions in open reading frames (O) or in upstream activating sequences (UAS). Relative quantity of an amplicon was determined by comparing the obtained Ct to a standard curve made on the same qPCR plate. Quantities were reported to qPCR performed on Input DNA and are expressed as a percentage. GAL1-O and IGV (non-transcribed intergenic region on chromosome V) amplicons were used as controls. The indicated value is the mean of three biological replicates, and error bars represent the standard deviation. (F and G) Effects of rpb9 deletion on genome-wide Mediator, Rad2 and Pol II occupancy. (F) Average tag density in Med17 Mediator ChIP (upper left panel), Rad2 ChIP (lower panels) and Rpb1 Pol II ChIP (upper right panel), around Med15 peaks corresponding to UAS (left panels, 1000 bp window), and on transcribed regions of 10% Pol II most-enriched genes (right panels, scaled windows for 500 bp before TSS, between TSS and TES, and 500 bp after TES). Average tag density in WT and rpb9Δ strains is indicated in blue and green, respectively. (G) Rad2 occupancy ratio between rpb9 mutant and the WT is shown in pink around Med15 peaks corresponding to UAS (left panel) and on transcribed regions of 10% Pol II most-enriched genes (right panel).
We then focused on Rpb9 Pol II subunit that was previously suggested to be required for Rad26-independent TCR (36,58). Given the strong link of Mediator with Pol II, we tested whether med17 mutants would be epistatic with rpb9 deletion. We found that the introduction of med17 double mutation Q444P/M442L in rad7Δ rpb9Δ context completely impaired growth at 30°C and moderately slowed growth at 25°C (Figure 5B). In contrast, med17-Q444P single mutant grew as a WT MED17 in this context (Figure 5B). Similar observations were made with other med17 UVs and non-UVs mutants (Supplementary Figure S13B). All tested med17 UVs mutants were colethal with rpb9 deletion at 30°C, whereas none of the non-UVs mutants were. Similar phenotypes were also observed in RAD7 rpb9Δ context (right panels, Figure 5B and Supplementary Figure S13B). This suggests a specific defect of med17 UVs mutants that is enhanced by rpb9 deletion, in line with a functional link between Mediator and TCR-related Rpb9 Pol II subunit.
Next, we analysed Mediator, Pol II and Rad2 occupancy on UAS and transcribed regions in rpb9Δ context using ChIP followed by qPCR on selected regions. We showed that rpb9 deletion did not affect the Med17 recruitment to UASs (Figure 5C). Surprisingly, we did not observe a strong decrease of Pol II recruitment to transcribed regions, but rather gene-specific changes (Figure 5D). Similarly, we did not find a strong decrease of Rad2 recruitment to transcribed regions, but rather gene-specific effects (Figure 5E). However, we found a stronger Rad2 ChIP signal on UASs. These results indicate that changes in Pol II composition could affect Rad2 chromatin binding both on UAS and transcribed regions.
To obtain a genome-wide view of rpb9 mutation effect on Rad2, Mediator and Pol II occupancy, we performed ChIP-seq experiments in rpb9Δ and the WT strains. On the genomic scale, a clear stabilization of Rad2 on UASs was observed (Figure 5F, lower left panel). We did not detect any major changes in Mediator occupancy in rpb9 mutant (Figure 5F, upper left panel). No changes were observed for the position of Mediator and Rad2 peaks (Supplementary Figure S13C and D). Surprisingly, a metagene analysis of Pol II distribution on transcribed regions demonstrated a slight change in Pol II profile with the enzyme stabilization at the beginning of the transcribed regions and a downstream shift on the promoters in rpb9 mutant compared to the WT (Figure 5F, upper right panel and Supplementary Figure S14A). This potentially points out on more complex and gene-specific effects of rpb9 deletion on different steps of transcriptional cycle. Rad2 occupancy on transcribed regions was slightly increased in rpb9 mutant compared to the WT, especially on the Pol II most-enriched regions (Figure 5F, lower right panel and Supplementary Figure S14B). No increase in Rad2 protein level was observed in rpb9 mutant compared to the WT (data not shown). Our analysis of Rad2 occupancy ratio between rpb9 mutant and the WT clear shows that the highest increase occurs at UASs (Figure 5G; Supplementary Figures S13C-D and 14B). The slight increase (1.2-fold) observed in other regions might be due to the normalization coefficient which was difficult to define in this particular case with gene-specific effects. Our ChIP-seq results indicate some gene-specific effects on Pol II occupancy in rpb9Δ context (Supplementary Figure S15A). Examples of Pol II ChIP-seq distribution on individual genes illustrate gene-specific effects of rpb9 deletion on Pol II occupancy (Supplementary Figure S15B, left panel). Examples of Rad2 ChIP-seq distribution demonstrate that gene-specific changes in Rad2 occupancy on transcribed regions do not follow exactly the changes in Pol II occupancy, and illustrate the Rad2 stabilization on UAS (Supplementary Figure S15B, right panel). Further studies using different approaches are needed to precisely define complex mechanistic consequences of the absence of Rpb9 subunit on gene-specific transcription and TCR.
In line with the implication of Medaitor-Rad2-Pol II interplay in Rad2 loading, our results thus show that deletion of Rpb9 Pol II subunit changes Rad2 chromatin binding and especially increases Rad2 occupancy on Mediator-bound UASs where no Pol II enrichment was detected.
DISCUSSION
This work directly addresses a functional interplay between Mediator, Rad2 and Pol II, three nuclear components related to TCR, and provides significant advance in a genomic view of Rad2 loading to the chromatin. (i) We provide genetic and genomic analyses helping to understand how Mediator, essential transcriptional coregulator, and RNA polymerase II contribute to loading of Rad2 DNA repair protein to the chromatin. These data also help to understand a complex relationship between transcription and DNA repair machineries, as well as Mediator implication in this link. (ii) An extensive mutational analysis allowed us to identify specific residues of Med17 Mediator subunit involved in UV sensitivity. Our results strongly support a conclusion that the UV sensitivity of Mediator mutants is not the systematic indirect consequence of transcriptional defects per se. (iii) Finally, this work provides new information on the genetic interactions between Mediator and TCR-related components in yeast and suggests that the UV sensitivity of Mediator mutants is at least in part independent of Rad26-related TCR pathway and functionally linked to Rpb9 Pol II subunit.
Implication of Mediator and Pol II in Rad2 recruitment to regulatory and transcribed regions
The results presented in this work provide insights into functional dynamics on a genomic scale between Mediator, Rad2 and Pol II that is important for Rad2 loading to regulatory regions bound by Mediator and on transcribed regions enriched in elongating Pol II in the yeast S. cerevisiae. Using a mutant affected in Pol II phosphorylation and therefore Mediator chromatin binding, as well as Mediator or Pol II mutants, we showed how the presence of Rad2 on the yeast genome is influenced by both Mediator and Pol II.
We demonstrated that inhibition of Pol II Ser5 phosphorylation in kin28-ts mutant, which leads to stabilization of Mediator on core promoters, results in major changes in Rad2 genome-wide occupancy and Rad2 enrichment on core promoters. Thus, Rad2 genome-wide distribution shifts from UASs towards the core promoters in this mutant, following Mediator stabilization on core promoters and decreases on transcribed regions, together with Pol II decrease. Importantly, the amplitude of Rad2 stabilization in the mutant was similar to that of Mediator (Figure 1D). Rad2 distribution does not follow exactly that of Mediator stabilized mostly on core promoters: the highest Rad2 occupancy ratio between the kin28 mutant and the WT was located between the UAS and the core promoter (Figure 1D and Supplementary Figure S2). This observation could be explained by the fact that both Mediator and Pol II would influence Rad2 occupancy near the core promoters and that the stability of transient interactions with the chromatin might differ between Mediator and Rad2. Moreover, other PIC components assembled on core promoters could influence the Rad2 occupancy on these regions. For example, we showed that Rad2 interacts with TFIIH in the absence of genotoxic stress. In kin28 mutant, this interaction remained unchanged and TFIIH occupancy on core promoters was strongly decreased. The Rad2 ChIP signal that we detect on core promoters in kin28-ts mutant is thus a result of its complex and probably dynamic interplay with Mediator, Pol II and TFIIH within the PIC. Observed changes in Rad2 distribution in kin28 mutant cannot be explained only by the transcriptional defects in this mutant since it was previously shown that an arrest of transcription in rpb1–1 mutant led to a decrease of Rad2 on both UAS and transcribed regions (18). In addition to our ChIP-seq results, co-immunoprecipitation experiments revealed important modifications of interactions between Rad2, Pol II and Mediator when Pol II Ser5 phosphorylation was impaired. Mediator-Pol II interaction was increased in kin28 mutant, in line with the fact that this phosphorylation promotes Mediator-Pol II dissociation (59,60). An opposite effect was observed for Rad2-Pol II interaction, emphasizing the importance of this phosphorylation step in the interplay between Rad2, Pol II and Mediator. It would be interesting to define how dynamic physical interactions between Mediator, Rad2 and Pol II and their interactions with the chromatin are.
The two Mediator mutants we chose to study in details (med17-Q444P and med17-Q444P/M442L) present different UVs phenotypes and modifications of Rad2 occupancy. Importantly, Pol II occupancy is globally decreased in a very similar way in both mutants (Figure 4B, lower panel), demonstrating that Pol II alone could not explain the difference in Rad2 loading to transcribed region. For Rad2 genome-wide association, the strongly UVs mutant med17-Q444P/M442L shows lower Rad2 occupancy on transcribed regions and lower correlation between Mediator and Rad2 on promoter regions and especially between Rad2 and Pol II on transcribed regions. In this med17 UV-sensitive mutant, Rad2 interaction with Mediator is reduced and Mediator interaction with Pol II is increased, in line with implication of Mediator/Rad2/Pol II network. Our CoIP and ChIP experiments show that a decrease in Mediator-Rad2 interaction is accompanied by a decrease in Rad2 presence on transcribed regions and an uncoupling of Rad2 with Mediator on UASs and with Pol II on transcribed regions. These results are consistent with a link between Mediator, Rad2 and Pol II and an abnormal Rad2 occupancy on transcribed regions when Mediator–Rad2 interaction is affected.
Finally, deletion of Rpb9 Pol II subunit involved in TCR increases genome-wide Rad2 occupancy mostly on regulatory regions (UAS) bound by Mediator, while devoid of Pol II enrichment. In this mutant, a global change in Pol II profile with gene-specific effects is accompanied to some extent by changes in Rad2 occupancy on transcribed regions. Our results demonstrate that deletion of Rpb9 Pol II subunit affects Rad2 chromatin binding both on regulatory and transcribed regions. In particular, our data suggest that mutations of Pol II enriched on transcribed regions could influence the presence of Rad2 on UASs, consistent with a link between Rad2 loading to regulatory regions bound by Mediator and that to Pol II-transcribed regions.
Taken together, our results suggest that Rad2 loading to the chromatin is affected by changes in Mediator/Rad2/Pol II interfaces and that Rad2 interplay with Mediator and Pol II contributes to its loading to the chromatin. This study is focused on Mediator, Rad2 and Pol II, but we do not exclude that additional factors might contribute to Rad2 chromatin binding. As PIC is assembled at core promoters, additional PIC components like TFIIH might influence Rad2 presence on the chromatin. We propose that Rad2 is first recruited to the UAS by Mediator. Different mechanisms might be proposed for Rad2 recruitment to transcribed regions: two independent events for its loading to UASs and transcribed regions and/or Rad2 loading to transcribed regions connected to its recruitment to UASs. At this stage, we could not exclude the possibility that several mechanisms might co-exist. The exact molecular events remain to be elucidated, but one of the potential mechanisms would be an implication of dynamic interactions between Rad2, Mediator and Pol II on core promoters to connect Rad2 recruitment on UASs and transcribed regions.
Interaction interface between Med17 and Rad2
Using Y2H approach, we identified interaction domains between Med17 Mediator subunit and Rad2 protein. Interestingly, the Med17 381–681 domain interacting with Rad2 is similar to the Med17 fragment with a globular structure that was crystallized in complex with Med11-Med22 C-terminal helices (30). We also specified a 100 amino-acid domain of Rad2 interacting with Med17. This domain is within a crystal structure of catalytic core of Rad2 that, however, lacks a linker between the two catalytic domains of the protein, making difficult the prediction of possible conformation of the Med17-interacting part (11).
We observed that all Med17 residues affecting Med17–Rad2 Y2H interaction are quite closely located according to S. cerevisiae Mediator head structure (31) (Figure 3D). This suggests a potential Med17–Rad2 interaction interface, at least in the yeast two-hybrid assay. Recent structural studies also suggest that this region of Med17 may be involved in the interaction with Med14 (32,44,61–63). Therefore, it is possible that the identified mutants may affect Mediator conformation. The location of Med17 residues affecting Med17–Rad2 Y2H interaction on these structures clearly shows that there is enough space for interactions between this Med17 region and Rad2.
Based on our identification of Med17 domain interacting with Rad2, we performed an extensive mutagenesis analysis demonstrating that the UV sensitivity and temperature-sensitivity of Med17 Mediator mutants are connected. It is highly unlikely that the UV sensitivity of Mediator mutants could be explained by transcriptional defects per se. Indeed, the same med17 mutants are UV-sensitive in GGR-deficient background, but UV-insensitive when both TCR and GGR pathways are functional. Thus, the same transcriptional defect leads to UV sensitivity only when GGR pathway is deficient. Moreover, Mediator med17 mutations affecting transcription do not systematically lead to UV sensitivity. A similar and global effect on transcription was observed for UV-sensitive and UV-insensitive med17 mutations and lower Pol II occupancy could not explain their UV sensitivity. We identified specific residues of Med17 (F437, M442, I541) mutations of which were frequently associated with UV sensitivity. We focused our study on M442L mutation, which in association with Q444P strongly increases the UV sensitivity of the yeast cells. Importantly, mutated residues are localized within the conserved domains of Med17 based on multiple sequence alignments and secondary structure features (64). These residues correspond to similar amino acids in human Med17 protein, suggesting that molecular mechanisms might extend to human cells.
Synthetic genetic interactions between Mediator and components of transcription-coupled repair
Our results are particularly interesting as they provide important genetic information on the yeast TCR pathway. This work suggests that the UV sensitivity of Mediator mutants is at least in part independent of Rad26-related pathway and functionally linked to Rpb9 Pol II subunit. We show a synthetic increase in UV sensitivity between med17 UVs mutations and rad26 deletion in GGR-deficient background suggesting that Mediator involvement is not exactly in the same pathway as Rad26. In the context of deletion of Rpb9 involved in transcription and TCR as a Pol II subunit, we observe a synthetic colethality with med17 UVs mutations suggesting a functional link of Mediator with Rpb9 Pol II subunit. All these synthetic genetic interactions of Mediator with Rad26 and Rpb9 TCR components, that are specific to UVsmed17 alleles, might suggest a particular Mediator-dependent mechanism, but further studies are needed to elucidate molecular events involved. It should be noted that in yeast other proteins have been proposed to be also involved in TCR suggesting that more than two potential TCR pathways might exist (8–10). Many questions remain to be answered to understand in vivo mechanisms governing TCR process on a genomic level. Future studies with UV-treated cells will help to elucidate the Mediator involvement in these mechanisms in relation to TCR proteins.
Recent study showed that rpb9Δ, and other Pol II mutations, impaired replication fork progression, suggesting that Pol II itself can help to resolve transcription-replication conflicts (65). Preliminary results suggest that med17 UVs mutants are sensitive to hydroxyurea (Supplementary Figure S6H). It would therefore be interesting to determine whether Rpb9 and Mediator may be involved in the removal of stalled Pol II from damages and/or conflicts with the replication machinery.
In conclusion, the work presented here provides new information on functional dynamics between essential components of transcription and nucleotide excision repair related to TCR, including Mediator coregulator complex, Rad2 DNA repair protein and RNA polymerase II. This work thus contributes to our understanding of the complex interplay between transcription and DNA repair machineries on the genomic scale.
DATA AVAILABILITY
The ChIP-seq data have been deposited to the Array Express under the accession number E-MTAB-7081.
Supplementary Material
ACKNOWLEDGEMENTS
We thank C. Thermes, E. Van Dijk and Y. Jaszczyszyn for performing the high-throughput sequencing of ChIP samples, the SPI (CEA/Saclay) for monoclonal antibodies, M. Werner, A. Goldar, S. Marcand, A. Verger and R. Guerois for fruitful discussions, M. Werner for critical reading of the manuscript.
Notes
Present address: Adrien Georges, PARCC, Hôpital Européen Georges Pompidou, 56 Rue Leblanc, 75015, Paris, France.
Present address: Elizaveta Novikova, IBENS, ENS, PSL Research University, 75005, Paris, France.
Present address: Diyavarshini Gopaul, IGH, CNRS, Université de Montpellier, 34396, Montpellier, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale de la Recherche [ANR-14-CE10–0012-01]; Fondation ARC [n°SL220130607079, PGA1 RF20170205342]; Fondation ARC [n° PDF20131200577 to A.G.]; La Ligue Nationale Contre le Cancer (to D.G.). Funding for open access charge: Agence Nationale de la Recherche [ANR-14-CE10–0012-01].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lagerwerf S., Vrouwe M.G., Overmeer R.M., Fousteri M.I., Mullenders L.H.. DNA damage response and transcription. DNA Repair (Amst). 2011; 10:743–750. [DOI] [PubMed] [Google Scholar]
- 2. Marteijn J.A., Lans H., Vermeulen W., Hoeijmakers J.H.. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014; 15:465–481. [DOI] [PubMed] [Google Scholar]
- 3. Hanawalt P.C., Spivak G.. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008; 9:958–970. [DOI] [PubMed] [Google Scholar]
- 4. Mullenders L. DNA damage mediated transcription arrest: step back to go forward. DNA Repair (Amst). 2015; 36:28–35. [DOI] [PubMed] [Google Scholar]
- 5. Svejstrup J.Q. Mechanisms of transcription-coupled DNA repair. Nat. Rev. Mol. Cell Biol. 2002; 3:21–29. [DOI] [PubMed] [Google Scholar]
- 6. Svejstrup J.Q. Contending with transcriptional arrest during RNAPII transcript elongation. Trends Biochem. Sci. 2007; 32:165–171. [DOI] [PubMed] [Google Scholar]
- 7. Vermeulen W., Fousteri M.. Mammalian transcription-coupled excision repair. Cold Spring Harb. Perspect. Biol. 2013; 5:a012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li W., Selvam K., Ko T., Li S.. Transcription bypass of DNA lesions enhances cell survival but attenuates transcription coupled DNA repair. Nucleic Acids Res. 2014; 42:13242–13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tatum D., Li W., Placer M., Li S.. Diverse roles of RNA polymerase II-associated factor 1 complex in different subpathways of nucleotide excision repair. J. Biol. Chem. 2011; 286:30304–30313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verhage R.A., van Gool A.J., de Groot N., Hoeijmakers J.H., van de Putte P., Brouwer J.. Double mutants of Saccharomyces cerevisiae with alterations in global genome and transcription-coupled repair. Mol. Cell Biol. 1996; 16:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mietus M., Nowak E., Jaciuk M., Kustosz P., Studnicka J., Nowotny M.. Crystal structure of the catalytic core of Rad2: insights into the mechanism of substrate binding. Nucleic Acids Res. 2014; 42:10762–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sarker A.H., Tsutakawa S.E., Kostek S., Ng C., Shin D.S., Peris M., Campeau E., Tainer J.A., Nogales E., Cooper P.K.. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne Syndrome. Mol. Cell. 2005; 20:187–198. [DOI] [PubMed] [Google Scholar]
- 13. Ito S., Kuraoka I., Chymkowitch P., Compe E., Takedachi A., Ishigami C., Coin F., Egly J.M., Tanaka K.. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol. Cell. 2007; 26:231–243. [DOI] [PubMed] [Google Scholar]
- 14. Le May N., Fradin D., Iltis I., Bougneres P., Egly J.M.. XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol. Cell. 2012; 47:622–632. [DOI] [PubMed] [Google Scholar]
- 15. Le May N., Mota-Fernandes D., Velez-Cruz R., Iltis I., Biard D., Egly J.M.. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell. 2010; 38:54–66. [DOI] [PubMed] [Google Scholar]
- 16. Sollier J., Stork C.T., Garcia-Rubio M.L., Paulsen R.D., Aguilera A., Cimprich K.A.. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell. 2014; 56:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trego K.S., Groesser T., Davalos A.R., Parplys A.C., Zhao W., Nelson M.R., Hlaing A., Shih B., Rydberg B., Pluth J.M. et al.. Non-catalytic Roles for XPG with BRCA1 and BRCA2 in homologous recombination and genome stability. Mol. Cell. 2016; 61:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eyboulet F., Cibot C., Eychenne T., Neil H., Alibert O., Werner M., Soutourina J.. Mediator links transcription and DNA repair by facilitating Rad2/XPG recruitment. Genes Dev. 2013; 27:2549–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kornberg R.D. Mediator and the mechanism of transcriptional activation. Trends Biochem. Sci. 2005; 30:235–239. [DOI] [PubMed] [Google Scholar]
- 20. Malik S., Roeder R.G.. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat. Rev. Genet. 2010; 11:761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poss Z.C., Ebmeier C.C., Taatjes D.J.. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 2013; 48:575–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soutourina J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018; 19:262–274. [DOI] [PubMed] [Google Scholar]
- 23. Schiano C., Casamassimi A., Rienzo M., de Nigris F., Sommese L., Napoli C.. Involvement of Mediator complex in malignancy. Biochim. Biophys. Acta. 2014; 1845:66–83. [DOI] [PubMed] [Google Scholar]
- 24. Spaeth J.M., Kim N.H., Boyer T.G.. Mediator and human disease. Semin. Cell Dev. Biol. 2011; 22:776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eychenne T., Werner M., Soutourina J.. Toward understanding of the mechanisms of Mediator function in vivo: Focus on the preinitiation complex assembly. Transcription. 2017; 8:328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jeronimo C., Langelier M.F., Bataille A.R., Pascal J.M., Pugh B.F., Robert F.. Tail and kinase modules differently regulate core mediator recruitment and function in vivo. Mol. Cell. 2016; 64:455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Petrenko N., Jin Y., Wong K.H., Struhl K.. Mediator undergoes a compositional change during transcriptional activation. Mol. Cell. 2016; 64:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lariviere L., Seizl M., Cramer P.. A structural perspective on Mediator function. Curr. Opin. Cell Biol. 2012; 24:305–313. [DOI] [PubMed] [Google Scholar]
- 29. Imasaki T., Calero G., Cai G., Tsai K.L., Yamada K., Cardelli F., Erdjument-Bromage H., Tempst P., Berger I., Kornberg G.L. et al.. Architecture of the Mediator head module. Nature. 2011; 475:240–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lariviere L., Plaschka C., Seizl M., Wenzeck L., Kurth F., Cramer P.. Structure of the Mediator head module. Nature. 2012; 492:448–451. [DOI] [PubMed] [Google Scholar]
- 31. Robinson P.J., Bushnell D.A., Trnka M.J., Burlingame A.L., Kornberg R.D.. Structure of the mediator head module bound to the carboxy-terminal domain of RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:17931–17935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nozawa K., Schneider T.R., Cramer P.. Core Mediator structure at 3.4 A extends model of transcription initiation complex. Nature. 2017; 545:248–251. [DOI] [PubMed] [Google Scholar]
- 33. Kikuchi Y., Umemura H., Nishitani S., Iida S., Fukasawa R., Hayashi H., Hirose Y., Tanaka A., Sugasawa K., Ohkuma Y.. Human mediator MED17 subunit plays essential roles in gene regulation by associating with the transcription and DNA repair machineries. Genes Cells. 2015; 20:191–202. [DOI] [PubMed] [Google Scholar]
- 34. Ziegler L.M., Khaperskyy D.A., Ammerman M.L., Ponticelli A.S.. Yeast RNA polymerase II lacking the Rpb9 subunit is impaired for interaction with transcription factor IIF. J. Biol. Chem. 2003; 278:48950–48956. [DOI] [PubMed] [Google Scholar]
- 35. Ruan W., Lehmann E., Thomm M., Kostrewa D., Cramer P.. Evolution of two modes of intrinsic RNA polymerase transcript cleavage. J. Biol. Chem. 2011; 286:18701–18707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li S., Smerdon M.J.. Rpb4 and Rpb9 mediate subpathways of transcription-coupled DNA repair in Saccharomyces cerevisiae. EMBO J. 2002; 21:5921–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gaillard H., Tous C., Botet J., Gonzalez-Aguilera C., Quintero M.J., Viladevall L., Garcia-Rubio M.L., Rodriguez-Gil A., Marin A., Arino J. et al.. Genome-wide analysis of factors affecting transcription elongation and DNA repair: a new role for PAF and Ccr4-not in transcription-coupled repair. PLoS Genet. 2009; 5:e1000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koleske A.J., Young R.A.. An RNA polymerase II holoenzyme responsive to activators. Nature. 1994; 368:466–469. [DOI] [PubMed] [Google Scholar]
- 39. Nonet M.L., Young R.A.. Intragenic and extragenic suppressors of mutations in the heptapeptide repeat domain of Saccharomyces cerevisiae RNA polymerase II. Genetics. 1989; 123:715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Plaschka C., Lariviere L., Wenzeck L., Seizl M., Hemann M., Tegunov D., Petrotchenko E.V., Borchers C.H., Baumeister W., Herzog F. et al.. Architecture of the RNA polymerase II-Mediator core initiation complex. Nature. 2015; 518:376–380. [DOI] [PubMed] [Google Scholar]
- 41. Robinson P.J., Trnka M.J., Bushnell D.A., Davis R.E., Mattei P.J., Burlingame A.L., Kornberg R.D.. Structure of a complete Mediator-RNA polymerase II Pre-Initiation complex. Cell. 2016; 166:1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Soutourina J., Wydau S., Ambroise Y., Boschiero C., Werner M.. Direct interaction of RNA polymerase II and mediator required for transcription in vivo. Science. 2011; 331:1451–1454. [DOI] [PubMed] [Google Scholar]
- 43. Thompson C.M., Koleske A.J., Chao D.M., Young R.A.. A multisubunit complex associated with the RNA polymerase II CTD and TATA-binding protein in yeast. Cell. 1993; 73:1361–1375. [DOI] [PubMed] [Google Scholar]
- 44. Tsai K.L., Yu X., Gopalan S., Chao T.C., Zhang Y., Florens L., Washburn M.P., Murakami K., Conaway R.C., Conaway J.W. et al.. Mediator structure and rearrangements required for holoenzyme formation. Nature. 2017; 544:196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S. . The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Eyboulet F., Wydau-Dematteis S., Eychenne T., Alibert O., Neil H., Boschiero C., Nevers M.C., Volland H., Cornu D., Redeker V. et al.. Mediator independently orchestrates multiple steps of preinitiation complex assembly in vivo. Nucleic Acids Res. 2015; 43:9214–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Malabat C., Feuerbach F., Ma L., Saveanu C., Jacquier A.. Quality control of transcription start site selection by nonsense-mediated-mRNA decay. Elife. 2015; 4:e06722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pelechano V., Wei W., Steinmetz L.M.. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature. 2013; 497:127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hollander M., Wolfe D.A.. Nonparametric Statistical Methods. 1973; NY: John Wiley & Sons. [Google Scholar]
- 52. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2009; NY: Springer-Verlag. [Google Scholar]
- 53. Cismowski M.J., Laff G.M., Solomon M.J., Reed S.I.. KIN28 encodes a C-terminal domain kinase that controls mRNA transcription in Saccharomyces cerevisiae but lacks cyclin-dependent kinase-activating kinase (CAK) activity. Mol. Cell Biol. 1995; 15:2983–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jeronimo C., Robert F.. Kin28 regulates the transient association of Mediator with core promoters. Nat. Struct. Mol. Biol. 2014; 21:449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wong K.H., Jin Y., Struhl K.. TFIIH phosphorylation of the Pol II CTD stimulates mediator dissociation from the preinitiation complex and promoter escape. Mol. Cell. 2014; 54:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bardwell A.J., Bardwell L., Iyer N., Svejstrup J.Q., Feaver W.J., Kornberg R.D., Friedberg E.C.. Yeast nucleotide excision repair proteins Rad2 and Rad4 interact with RNA polymerase II basal transcription factor b (TFIIH). Mol. Cell. Biol. 1994; 14:3569–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Habraken Y., Sung P., Prakash S., Prakash L.. Transcription factor TFIIH and DNA endonuclease Rad2 constitute yeast nucleotide excision repair factor 3: implications for nucleotide excision repair and Cockayne syndrome. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:10718–10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen X., Ruggiero C., Li S.. Yeast Rpb9 plays an important role in ubiquitylation and degradation of Rpb1 in response to UV-induced DNA damage. Mol. Cell Biol. 2007; 27:4617–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schneider M., Hellerschmied D., Schubert T., Amlacher S., Vinayachandran V., Reja R., Pugh B.F., Clausen T., Kohler A.. The nuclear Pore-Associated TREX-2 complex employs mediator to regulate gene expression. Cell. 2015; 162:1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sogaard T.M., Svejstrup J.Q.. Hyperphosphorylation of the C-terminal repeat domain of RNA polymerase II facilitates dissociation of its complex with mediator. J. Biol. Chem. 2007; 282:14113–14120. [DOI] [PubMed] [Google Scholar]
- 61. Robinson P.J., Trnka M.J., Pellarin R., Greenberg C.H., Bushnell D.A., Davis R., Burlingame A.L., Sali A., Kornberg R.D.. Molecular architecture of the yeast Mediator complex. Elife. 2015; 4:e08719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tsai K.L., Tomomori-Sato C., Sato S., Conaway R.C., Conaway J.W., Asturias F.J.. Subunit architecture and functional modular rearrangements of the transcriptional mediator complex. Cell. 2014; 157:1430–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schilbach S., Hantsche M., Tegunov D., Dienemann C., Wigge C., Urlaub H., Cramer P.. Structures of transcription pre-initiation complex with TFIIH and Mediator. Nature. 2017; 551:204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bourbon H.M. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 2008; 36:3993–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Felipe-Abrio I., Lafuente-Barquero J., Garcia-Rubio M.L., Aguilera A.. RNA polymerase II contributes to preventing transcription-mediated replication fork stalls. EMBO J. 2015; 34:236–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ChIP-seq data have been deposited to the Array Express under the accession number E-MTAB-7081.