


Abstract
A common mechanism for inducibly controlling protein function relies on reconstitution of split protein fragments using chemical or light-induced dimerization domains. A protein is split into fragments that are inactive on their own, but can be reconstituted after dimerization. As many split proteins retain affinity for their complementary half, maintaining low activity in the absence of an inducer remains a challenge. Here, we systematically explore methods to achieve tight regulation of inducible proteins that are effective despite variation in protein expression level. We characterize a previously developed split Cre recombinase (PA-Cre2.0) that is reconstituted upon light-induced CRY2-CIB1 dimerization, in cultured cells and in vivo in rodent brain. In culture, PA-Cre2.0 shows low background and high induced activity over a wide range of expression levels, while in vivo the system also shows low background and sensitive response to brief light inputs. The consistent activity stems from fragment compartmentalization that shifts localization toward the cytosol. Extending this work, we exploit nuclear compartmentalization to generate light-and-chemical regulated versions of Cre recombinase. This work demonstrates in vivo functionality of PA-Cre2.0, describes new approaches to achieve tight inducible control of Cre DNA recombinase, and provides general guidelines for further engineering and application of split protein fragments.
INTRODUCTION
Since the first understanding that split fragments of a protein could complement each other, allowing restoration of biological activity (1–3), methods utilizing complementation or reconstitution of split protein fragments have been widely used for sensor and protein control applications. Although some split protein fragments have sufficient affinity to assemble on their own, protein fragments that do not self-assemble can be fused to proteins that interact, allowing conditional reconstitution of activity due to the increased proximity (4,5).
When utilized in live cells, split proteins have most commonly been used as reporters to detect interacting proteins. Some of the earliest split protein sensors were transcription factors with modular DNA binding and activation domains that could be split and functionally reconstituted through a protein–protein interaction (6). Another early-developed split protein sensor was ubiquitin, which when reconstituted can allow binding of a ubiquitin protease and coincident cleavage of a reporter (4). Subsequently, split forms of many other reporter proteins have been developed, including split DHFR, GFP and other fluorescent proteins, Cre recombinase, beta-lactamase and luciferase (5,7–9).
While split proteins have been most widely adopted for use as reporters of protein interaction, similar designs can also be used to control protein activity using light or a chemical switch to induce proximity. In this scenario, each half of a split enzyme or other protein is attached to one partner of a protein pair that conditionally dimerizes. Chemically induced dimerization systems, in which domains are brought together by a chemical ligand (10), have been applied to a range of protein targets. More recently, light induced dimerization systems have also been developed that accomplish similar functions, but with the benefits of spatial regulation and reversibility (11,12). Such split protein approaches have been used to conditionally control activity of proteins such as Cre recombinase (13–15), transcriptional activators (16–19), and proteases (20–22). In a recent study, a computational scoring approach to facilitate design of ideal protein split sites was developed and used to conditionally control activity of several split signaling proteins (23).
Two main strategies have been used to regulate biological function with split proteins. A protein with independent separable domains can be split between these domains, as in the case of a transcription factor DNA binding domain and activation domain (6). In this case, the modular domains do not interact and are functionally independent, and the dimerization simply serves to tether the activation domain in proximity to the DNA binding element. In many other cases, however, a protein does not have independent domains. Splitting such proteins yields fragments that may retain substantial affinity even under non-inducing conditions. Affinity of these fragments is required to reconstitute protein function, but can contribute to background activity. A key parameter for tight inducible control of these proteins is the concentration of the protein fragments in the cell: self-assembly under non-induced conditions is more likely to occur as the protein concentration of the fragments increases. To circumvent this issue, several different strategies have been taken. First, fragment interfaces can be disrupted to reduce interaction affinity (4,22). Second, protein expression level can be precisely controlled. Third, an additional layer of inducible control can be added to the regulated system (24–26).
While a number of different strategies have been found to be effective, few detailed studies of parameters affecting activity of inducible split proteins have been performed. Here, we systematically examine the effects of modulating expression and localization of split protein fragments used to generate a light activatable Cre DNA recombinase (PA-Cre2.0) (14). We find that high light activity and low dark background can be achieved by expressing one half of the split Cre system at extremely low levels. With increased expression, background activity of PA-Cre2.0 increases, but remains remarkably low compared to other split Cre recombinase systems. In vivo, PA-Cre2.0 also shows remarkably low dark activity and robust response to short light inputs. We establish that the low dark background of PA-Cre2.0 results from alterations in nuclear-cytosolic shuttling in one of the split Cre fragments, resulting in a reduced fraction of protein in the nucleus as overall protein expression increases. This work suggests a design strategy for achieving tight light regulation of inducible dimerization systems through tuning nuclear import/export signals to reduce sensitivity to expression level differences. Extending this approach, we demonstrate dual control of Cre recombinase activity by controlling nuclear localization or abundance using a small molecule and assembly of split fragments using light, providing more complex control of DNA recombination for a diverse range of applications.
MATERIALS AND METHODS
Cloning and mutagenesis
Full sequences of constructs used in this manuscript and primers used for cloning are provided in Supplementary Table S1. All CreN constructs contain residues 19–104 of Cre DNA recombinase, while CreC constructs contain residues 106–343. The constructs mCh-IRES-CRY2(L348F)-CreN (p688) and mCh-IRES-CIB1-CreC (p645) were described previously (14). The constructs mCh-IRES-CRY2FL(wt)-CreN (p221), CRY2FL(+NLS)-mCherry (p950) and the loxP-STOP-loxP-EGFP reporter are described in Kennedy et al., (27). The DIO-EGFP reporter (p1053) was generated by digesting pcDNA3.1+ with EcoRI and EcoRV enzymes, then inserting a DIO-EGFP cassette (containing loxP2272-loxP-invertedEGFP-loxP2272-loxP) from pAAV-CAG-DIO-EGFP using EcoRI and AfeI sites.
To generate KM10, CIB1-CreC-IRES-CRY2(L348F)-CreN-IRES-mCh, an intermediate construct (p941, CIB1-CreC-IRES-CRY2(L348F)-CreN-stop-mCh) was first generated. To generate this, CIB1-CreC(N1) was digested with EcoRV and XmaI, and CIB1-CreC-IRES-CRY2(L348F)-CreN cut at EcoRV-XmaI sites from plasmid GFP-P2A-CIB1-CreC-IRES-CRY2(L348F)-CreN was inserted (both plasmids are described in Taslimi et al 2016). Next, the IRES element from p688 was polymerase chain reaction (PCR)-amplified (primers 1889f/1890r), and ligated into p941 cut with XmaI. To generate p985 (CRY2(L348F)-CreN-IRES-mCh), KM10 was digested with XhoI and religated to excise the CIB1-CreC-IRES insert. To generate KM60 (CRY2(L348F)-CreN-IRES-CIB1-CreC-IRES-mCh), first the CIB1-CreC-IRES insert was cut out from KM10 using AfeI and SalI and blunt-ended with T4 DNA polymerase. This insert was ligated into a version of p985 with the KpnI site mutated, cut with ClaI (blunt fill in). To generate p1116, CIB1-CreC-IRES-mCh, KM10 was digested with KpnI then religated, excising the IRES-CRY2(L348F)-CreN insert. To generate KM10 ΔpA, KM10 was digested with SbfI and AflII (blunt fill) and ligated to an insert containing IRES-mCherry amplified with 1677f/687r then digested with SbfI and EcoRV.
To generate the myc-tagged constructs, for p926, CRY2(L348F)-CreN was PCR-amplified to add a N-terminal myc tag (primers 1683f/1685r, then 1684f/1685r), then cloned into p688, replacing CRY2(L348F)-CreN at SacI and XmaI sites. To generate p1108, the L348F mutation in CRY2 was mutagenized to wild-type (L348L) using one-step Phusion mutagenesis protocols from New England Biolabs. Briefly, oligos containing the 348L substitution (1145r/2109f) were used to amplify the entire plasmid using Phusion High-Fidelity DNA Polymerase (NEB), then the PCR products were treated by T4 Polynucleotide Kinase (NEB) in the presence of ATP at 37°C for 30 min before self-ligation using T4 Quick Ligase (NEB).
To generate p1133, CRY2(L348F) was PCR amplified (primers 654f/639r) using p985 as template and digested with NheI and XmaI, then ligated into pmCherryN1 at NheI and XmaI sites. To generate p1089 (mCh-CRY2(L348F)-CreN), a NheI-XmaI fragment containing CRY2(L348F)-CreN was ligated at NheI/XmaI sites into a modified version of pmCherryC1 (Clontech) in which the linker between BsrGI and SacI had been replaced by the sequence agggaggtggaggtgctagc, containing a NheI site in place of SacI. To generate CW53, CRY2FL was isolated from p1108 at EcoRI and NotI sites, then ligated into p1089 at the same sites. To generate p1097, mCh-CRY2(L348F)-CreC, the NotI-XmaI fragment from CIB1-CreC (containing CreC) was inserted into p1089 digested with NotI and XmaI. To generate p1103 (mCh-CRY2(L348F)-NLS-CreN), a nuclear localization signals (NLS) was first added to the C-terminus of CRY2(L348F) by PCR (primers 1677f/2130r) using p688 as template. This fragment was ligated into p688 cut with SacI and NotI. To generate p1105, the CRY2(L348F)-NLS-CreN insert from p1103 was cut out (EcoRI and XmaI sites) and ligated into p1097 digested with EcoRI and XmaI. To generate CW184, the C-terminus of p1089 was modified by PCR to mutate the putative NES sequence using primers 1290f/2985f then 1290f/2986r. The insert was digested with NotI and XmaI and ligated into p1089 at the same sites.
To generate p1087 (mCh-CIB1-CreN), a NotI-XmaI fragment containing CreN was isolated from p688 and ligated into KM69 at NotI/XmaI sites. To generate KM69 (mCh-CIB1-CreC(106)-IRES-Cry2(L348F)-CreN), the mCh-CIB1-CreC(106)-IRES-CRY2(L348F)-CreN insert was isolated from p1071 (SpeI/XmaI sites). The insert was ligated into pmCherryC1 cut with NheI and XmaI. To generate p1117 (mCh-CIB1-CreC), KM69 was digested with KpnI and SmaI to remove the IRES-CRY2(L348F)-CreN fragment, then religated. To generate KM91 (mCh-IRES-Magnet-PA-Cre), a SnaBI-SacI fragment from p688 (containing the mCherry-IRES insert) was ligated into Magnet PA-Cre (pcDNA3.1 backbone) (15). To generate p1115 (CIB1-CreN), a NotI-XmaI fragment containing CreN was ligated into CIB1-CreC(N1) at NotI/XmaI sites. To generate p1088/KM75 (mCh-IRES-CRY2(L348F)-CreC), a NotI-XmaI fragment containing CreC was ligated into p688 at NotI/XmaI sites.
To generate CW40, CRY2(L348F)-CreN was PCR-amplified from p1089 (primers 2197f/2198r), then digested with KpnI and XhoI for cloning into pcDNA-NES-PhoCI-mCherry (Campbell lab, Addgene #87690) at KpnI and XhoI sites. CW59 was generated by PCR amplifying (primers 2264f/1081r) the PhoCl-CRY2(L348F) insert from CW40. The insert was cut with XhoI and EcoRI and ligated into p688 previously cut with XhoI and EcoRI. CW57 was made using primers 2278f/2279r to amplify a human estrogen receptor ligand binding domain insert, which was digested with SacI and ligated into CW59 cut with SacI. To generate CW79, the ecDHFR insert from pBMN DHFR(DD)-YFP (Wandless lab, Addgene #29325) was PCR amplified (primers 2561f/2562r) to add ClaI and SalI ends. The insert was ligated into p688 cut with ClaI and SalI.
To generate p989, CRY2(L348F)-CreN-P2A-CIB1-CreC-IRES-mCh, p688 was amplified by PCR (1689f/1924r, then 1689f/1925r) to add a P2A sequence at the C-terminus of CRY2(L348F)-CreN. This insert was digested with EcoRI and KpnI, then ligated into p985 digested with EcoRI and KpnI to generate intermediate clone p986. In a second step, KM10 was PCR amplified (primers 1926f/1890r) and the insert digested with KpnI and XmaI, then ligated into p986 digested with KpnI and XmaI. To generate AAV constructs, myc-CRY2(L348F)-CreN or CIB1-CreC were cloned into AAV-CaMKII-GFP at KpnI and BsrGI sites (where GFP was removed).
Cell culture experiments
HEK293T cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C with 5% CO2. To test split constructs, 1 μg (or indicated amounts) of the loxP-STOP-loxP-EGFP Cre recombinase reporter (or DIO-EGFP reporter, where indicated) and each split fragment were transfected into HEK293T cells on 12-well plates using standard calcium phosphate transfection methods. Nonspecific ‘stuffer’ DNA was included in transfections as needed to ensure all transfection wells received equal amounts of DNA. Cells were wrapped in aluminum foil after transfection and kept in dark for 24 h before blue light treatment (461 nm delivered from custom-built LED arrays, 7.4–18 mW/cm2). For extended light treatment, 2 s pulses were delivered every 3 min, or as indicated in figures. Dark samples were kept in the dark throughout the experiment. Cells were imaged to quantify reporter fluorescence 24 h after initial onset of light treatment (48 h after transfection), unless specified otherwise.
For immunoblotting, cells were harvested 24 h after transfection, washed in 1× phosphate-buffered saline (PBS), and lysed in 2× Laemmli sample buffer with boiling. Proteins were separated by electrophoresis on an sodium dodecyl sulphate-polyacrylamide gelelectrophoresis gel and transferred to nitrocellulose membranes, followed by probing with primary (anti-Cre recombinase, Novusbio NB100-56135; anti-tubulin, Li-COR, 926-42213; anti-myc tag, Sigma M4439) and secondary (goat anti-rabbit IR-Dye 800CW, LiCOR, 926-32211; goat anti-mouse IR-Dye 680RD, Li-COR, 926-68070) antibodies. An Odyssey FC Imager (Li-COR) was used to visualize labeled immunoblots.
Cre recombinase quantification and statistical comparison
Quantification of Cre DNA recombinase activity was carried out in live cells using a Leica DM IL LED Fluo microscope with L5 ET and TX2 ET filters. Images were taken with an iPhone7 (Apple) equipped with an iDu microscope adaptor (iDu Optics) and visualized using ImageJ. The percent of transfected cells (typically expressing mCherry) showing EGFP reporter activity was quantified from images using CellProfiler (https://cellprofiler.org) (28) or manual count, and is indicated in graphs as ‘% Cre recombination’. Significance for experiments comparing two populations was determined using a two-tailed unpaired student's t-test.
Image analysis
For localization studies, HEK293T cells were seeded onto coverslips in 12-well plates, transfected with specified plasmid DNA, then harvested after 24 h. Coverslips were fixed in 4% paraformaldehyde, washed in 1 × PBS, then mounted on a glass slide using Fluoromount-G (Southern Biotech). Cells were imaged on an Andor Dragonfly 301 spinning disc imaging system with Olympus IX73 base and four-line ILE laser merge module and controller. Images were acquired using a 60× UPlanSApo 1.35 NA oil objective, and collected on a 1024 × 1024 pixel Andor iXon EM-CCD camera. Data acquisition and analysis were performed with Fusion (Andor) and ImageJ software.
After initial image collection, ImageJ 1.45s was used for all image analysis. For nuclear:cytoplasmic ratio quantification, confocal image Z-stacks were compiled into a maximal projection. Total fluorescence in either the nucleus or cytosol was calculated by manually selecting either the nucleus or cytosolic region of interest and quantifying average fluorescence and area within these compartments. A separate region of the image was used to calculate background, which was subtracted from the average fluorescence. Average fluorescence was multiplied by area and calculated for nuclear vs cytosolic regions, then these were divided to obtain a ratio of nuclear:cytoplasmic protein. To quantify nuclear concentration versus total cell fluorescence for Figure 2G, the mean intensity of fluorescence in the nucleus was background subtracted and reported on the X-axis. The total cell fluorescence on the Y-axis was defined by multiplying each cell area (manually selected) by the background subtracted average fluorescence intensity. Values depicted for each are arbitrary units. Significance for experiments comparing two populations was determined using a two-tailed unpaired student's t-test.
Figure 2.

CRY2 L348F mutant shows reduced nuclear localization. (A) Reduced dark activity of CRY2(L348F)-CreN is not due to reduced overall expression. Shown is an immunoblot of cells (anti-myc antibody) expressing myc-tagged versions of wild-type or L348F CRY2-CreN. Loading control, Ponceau S total protein reference band. (B) Localization of mCherry-fused versions of CRY2 (wild-type) or CRY2 L348F. The L348F mutation reduces the percent of expressed protein localized to the nucleus. Graph at right shows quantification of the ratio of nuclear:cytosolic protein (average and error, s.e.m, n = 7 cells; *, P < .05, student's t-test). Scale bars, 10 μm. (C) mCherry-fused versions of L348F CRY2-CreN show even further reduced nuclear localization, compared to wt. Mutation of a putative ‘NES’ in CreN (CreN ΔNES) or addition of an extra NLS (far right, CRY2-L348F-NLS) restores nuclear localization. Graph at right shows quantification of nuclear:cytosolic protein (average and error, s.e.m., n = 7 cells (10 cells for ΔNES); *, P < .05, student's t-test). Scale bars, 10 μm. (D) Quantification of Cre recombinase activity. Addition of an extra NLS (to generate CRY2(L348F)-NLS-CreN) results in enhanced dark activity, with minimal effects on light activity. Graph represents average and range from two independent experiments. (E) Localization of switched CreN and CreC versions. While CRY2(L348F) combined with CreN shows reduced nuclear localization, switched CRY2(L348F)-CreC shows strong nuclear localization. The CIB1 construct shows strong nuclear localization with either CreC or CreN. Scale bars, 10 μm. (F) Quantification of Cre recombinase activity in cells expressing CRY2(L348F)-CreN/CIB1-CreC (‘original’) or CRY2(L348F)-CreC/CIB1-CreN (‘switched’) versions of PA-Cre. Both mCherry-fused and non-fused versions of CIB1-CreN were assayed and results combined. Data represents average and error (s.d., n = 4–6 independent experiments; P < .05, student's t-test). (G) Quantification of nuclear fluorescence relative to total cell fluorescence. HEK293T cells were transfected with increasing amounts of wt or L348F mCh-CRY2-CreN. The concentration of mCherry fluorescent signal in the nucleus, relative to total protein abundance, was quantified. Each dot represents an individual cell, arbitrary units. Red triangles, wt; blue circles, L348F. At all concentrations, wt expressing cells show higher nuclear signal compared with L348F. (H) Model of CRY2(L348F)-CreN nuclear trafficking. The CreN fragment contains a sequence recognized as a NES, while the L348F mutation appears to impair CRY2 nuclear localization. Together this results in a protein with reduced nuclear occupancy.
Production of AAVs for in vivo injection
To generate AAVs for injection, we used the AAV-DJ strain. HEK293T cells were co-transfected with each AAV plasmid along with helper plasmids (pDJ and pHelper) using calcium phosphate transfection. Seventy-two hours post-transfection cells were harvested, lysed and purified over an iodixanol gradient column (2 h at 400 000 × g in a Beckman Type80Ti rotor). The virus was collected, dialyzed to remove excess iodixanol and aliquoted and stored at −80°C until use.
In vivo studies in mouse
Adult Ai14 mice containing an integrated CAG-loxP-STOP-loxP-tdTomato cassette at the Gt(ROSA)26Sor locus were stereotactically injected with a 1:1:1 volume/volume mixture of AAV constructs (AAV-CaMKII-myc-CRY2(L348F)-CreN, AAV-CaMKII-CIB1-CreC, and AAV-hSyn-EYFP) into the dentate gyrus. AAV-CaMKII-myc-CRY2(L348F)-CreN (titer 9.52 × 1011 GC/ml) and AAV-CaMKII-CIB1-CreC (titer 1.53 × 1012 GC/ml) were packaged at the University of Colorado. AAV-hSyn-EYFP virus was obtained from the University of North Carolina viral core (https://www.med.unc.edu/ genetherapy/vectorcore/) with a titer of 3.3 × 1012 GC/ml. For injection, mice were anesthetized with 2% isoflurane, skin prepped and opened and a small hole was created in the skull at designated coordinates using a high-speed drill and burr. A total of 300 nl viral solution was injected into the dentate gyrus. After injection, a fiber optic was implanted above the injection site and secured using meta-bond dental cement. Animals were left to recover for 3 weeks post-surgery. Animals were connected to a patch cable and a 473 nm laser light source to provide light to brain tissue. The output at the end of the patch cable was measured at 15 mW prior to connection to implanted fiber. Fiber optic implants were made in house with the minimum acceptable efficiency of 80% (29). Animals were treated with light (1 s pulse of light through the implanted fiber optic or no light for control treated animals. One week after light treatment, mice were transcardially perfused with 4% paraformaldehyde. The brains were collected, sectioned and imaged using a Leica DM6 fluorescent microscope.
RESULTS
PA-Cre recombinase activity correlates with CRY2-CreN expression level
We previously generated a photoactivatable Cre DNA recombinase (PA-Cre2.0) in which N- and C-terminal fragments of Cre recombinase were fused to interacting partner proteins, Arabidopsis thaliana photoreceptor cryptochrome 2 (CRY2) and CIB1, a protein that interacts with light-stimulated CRY2 (14). In dark, CRY2 and CIB1 have low affinity and the fused N- and C-terminal fragments of Cre do not functionally assemble. In light, the fragments are brought into proximity through light-induced dimerization, promoting assembly and functional reconstitution of recombinase activity. To increase the sensitivity of the system to allow high activity in response to brief light input, we also included a L348F mutation on CRY2 that extends the photoexcited state, thus allowing extended interaction of CRY2 with CIB1 after light exposure (14). While PA-Cre2.0 showed low background and robust light response, our initial studies did not examine how activity would be altered by differences in expression of split protein fragments or determine the optimal expression level of the system. One drawback of many split protein systems is that the affinity of the N- and C-terminal fragments, required for induced assembly and reconstitution of activity, can contribute to background activity in an expression-dependent manner (11,22). Although our initial choice of split Cre recombinase fragments (CreN, residues 19–104; CreC, residues 106–343) showed minimal self-assembly when expressed at moderate levels in the cell (14), we had not tested at very high expression.
As a first step to test and improve the system, we sought to equalize expression of the CreN and CreC fragments by generating a bicistronic vector using a P2A cleavable peptide tag. We avoided tagging the CIB1-CreC fragment (i.e. CIB1-CreC-P2A-CRY2-CreN) as the cistron upstream of the P2A peptide retains a 18-residue tag at the C-terminus, and CIB1-CreC was previously found to be intolerant of an epitope tag at this site (14). Unfortunately, placement of a P2A tag downstream of CRY2(L348F)-CreN (p989) substantially reduced recombinase activity (Supplementary Figure S1). We next tested bicistronic vectors in which the CIB1-CreC and CRY2(L348F)-CreN fragments were separated by an IRES (internal ribosome entry site) element. A version containing CIB1-CreC upstream, KM10 (CIB1-CreC-IRES-CRY2(L348F)-CreN), showed good functionality in a single construct (Figure 1A and B). Using this construct, we were able to achieve ∼80% recombination rate in cells treated with just three pulses of light (4s duration each) spaced 45 min apart (Supplementary Figure S2). However, we also observed that the CRY2-CreN fragment in this construct was expressed at a much lower level than the CIB1-CreC fragment (Figure 1C), potentially limiting the system. Because the cistron downstream of an IRES element often expresses at lower levels (30), and since CRY2 is often expressed more poorly than CIB1, we hypothesized that switching the order of the proteins within the IRES construct (e.g. CRY2(L348F)-CreN-IRES-CIB1-CreC, KM60) may equalize fragment expression and improve function. This configuration resulted in higher levels of expressed CRY2-CreN compared with CIB1-CreC (Figure 1C), but no functional improvement, showing elevated dark background with no improvement in light activity (Figure 1B).
Figure 1.

Alteration of expression level of CRY2-CreN and CIB1-CreC fragments causes changes in dark background, with minimal effects on light-induced activity. (A) Schematic showing combined constructs used in (B) and (C). (B) Quantification of Cre recombinase activity in HEK293T cells. Cells expressing CRY2(L348F)-CreN in front of an IRES element (KM60) show higher dark background compared with KM10. Cells were transfected with indicated plasmids and a loxP-STOP-loxP-EGFP reporter, then kept in dark and harvested at 48 h. Light-treated samples were exposed to a 4 s pulse of light at 24 h. Graph shows average and error (s.d., n = 4–6 independent experiments). (C) Immunoblot comparing expression of Cre fragments in KM10 versus KM60 (anti-Cre antibody, top panel; anti-tubulin, bottom panel). (D) Immunoblots of dual plasmid system. HEK293 cells were transfected with indicated constructs and immunoblotted with an anti-Cre antibody (top). Bottom panel shows anti-tubulin loading control. Control lane consisted of non-transfected cells. (E) Quantification of Cre recombinase activity in HEK293T cells transfected with indicated constructs and light-treated as in (B). Protein expression level as determined in (D) is indicated below the graph, with ‘+’ (low expression) or ‘+++’ (high expression). Graph shows average and error (s.d., n = 3–5 independent experiments). (ns, not significant; *, P < .05, student's t-test). (F) Comparison of expression level and activity of HEK293T cells expressing a combined PA-Cre2.0 construct (KM10), or an identical version (ΔpA) with a mutant polyadenylation signal. Expression of the Cre fragments is greatly reduced in the ΔpA version. Dark background activity is significantly reduced (*, P < .05), but no significant difference in light-stimulated activity is observed. Graph shows average and error (s.d., n = 3–6 independent experiments). (G) Quantification of Cre recombinase activity with different amounts of wt or L348F CRY2-CreN. All samples contained 1 μg EGFP Cre reporter and 1 μg p645 (CIB1-CreC) and were quantified 48 h post-transfection. Light samples received a 4 h light exposure at 24 h (2 s pulse every 1 min, 461 nm). Samples labeled ‘high’ were transfected with 1 μg of L348F or wt CRY2-CreN; ‘low’ samples contained 50 ng. Graph shows average and range of two independent experiments.
Given the significant differences in dark activity resulting from altering the ratio of CRY2(L348F)-CreN and CIB1-CreC, we undertook a systematic characterization of the effect of modulating expression level of each split protein half. Using separate plasmids to express each fragment, we tested the CreN or CreC components (CRY2(L348F)-CreN or CIB1-CreC), positioning each fragment either upstream or downstream of an IRES element to manipulate expression level. To quantify activity, we included a mCherry fluorescent reporter in the opposing position (i.e. mCherry-IRES-CIB1-CreC) (Figure 1D). Thus, we tested high-expressing CreC (CIB1-CreC-IRES-mCh, p1116, +++), low-expressing CreC (mCh-IRES-CIB1-CreC, p645, +), high-expressing CreN (CRY2(L348F)-CreN-IRES-mCh, p985, +++) and low-expressing CreN (mCh-IRES-CRY2(L348F)-CreN, p688, +). Altering the expression level of CIB1-CreC had no significant effect on either dark or light activity (Figure 1E). In contrast, increasing CRY2(L348F)-CreN expression resulted in a significant increase in dark background activity, with minimal effect on light activity. Combined, these results suggested a correlation between increased CRY2(L348F)-CreN expression and dark background.
Since our studies indicated we could maintain high light activity but reduce dark activity by reducing CRY2(L348F)-CreN expression, we explored whether we could decrease expression of this fragment further. A construct (ΔpA) identified during cloning that lacked much of the polyA termination signal results in nearly undetectable levels of CRY2-CreN protein as assayed by immunoblot (Figure 1F), due to the loss of much of the 3′ UTR that acts to stabilize the mRNA. Despite the substantially lower levels of expressed protein, ΔpA showed similar light-induced activity as KM10 and minimal dark background activity (1.3% dark recombination, compared with 5.1% for KM10). These results suggested that high levels of expression of PA-Cre2.0 were not required for maintaining high activity.
The minimal change to light and dark activity over such a large range of expression levels was surprising, given that the split protein fragments are expected to retain moderate affinity for each other in order to reassemble. As expression increases, the fragments would be expected to interact increasingly, resulting in higher dark background activity. Interestingly, when we had originally characterized the PA-Cre2.0 system, we observed that dark background was significantly reduced when using L348F CRY2-CreN compared with wt CRY2-CreN, however we had not compared activity of these constructs at different expression levels. When we reduced the expression of wt CRY2-CreN in cells, both L348F and wt versions showed similar activity (Figure 1G). Thus, while PA-Cre with wt CRY2 was highly dependent on expression level, the L348F variant appeared to confer activity that was remarkably buffered from expression level differences.
L348F mutation of PA-Cre2.0 alters nuclear localization to reduce dark background
To further investigate the low dark background of PA-Cre2.0, we focused on the L348F mutation. When generating PA-Cre2.0, we had added this mutation, which extends the photocycle, to allow reconstitution of the split fragments for a longer period of time in response to a short light pulse (14). Given that the CRY2 photocycle phenotype would not be expected to manifest in dark, the exceptionally low background activity in dark (Figure 1G) was unexpected, suggesting L348F was also affecting CRY2 in another way. We first hypothesized that the mutation could destabilize the CRY2-CreN fragment, but observed no difference in steady-state protein levels between wt and L348F versions (Figure 2A).
We next examined whether the L348F substitution affects CRY2 subcellular localization, as both CRY2-CreN and CIB-CreC fragments must localize to the nucleus to catalyze DNA recombination. When designing the PA-Cre, to enable nuclear localization of each fragment we used versions of CRY2 and CIB1 that retain endogenous NLS found near the C-terminus of each protein (27). Indeed, while mCherry-tagged wt CRY2 was strongly nuclear, L348F CRY2 showed a reduced proportion of protein in the nucleus (Figure 2B). These results suggest that the L348F substitution could alter exposure of the CRY2 NLS, or interfere with its interaction with import machinery.
We next examined the localization of the full length fragment (CRY2-CreN fusion), using mCherry-tagged versions. With both L348F and wt CRY2, the presence of the CreN fragment resulted in further reduced nuclear localization (Figure 2C), suggesting adding this domain either reduced nuclear import or increased nuclear export. Using NetNES 1.1 prediction software (31), we identified a predicted nuclear export consensus sequence (NES) ‘LQARGLAV’ at residues 78–85 of Cre recombinase, which resides in the CreN fragment. We hypothesized that this motif and the L348F mutation act additively to restrict levels of CRY2(L348F)-CreN in the nucleus, resulting in the low background activity observed. Indeed, mutating the critical ‘LxV’ of the predicted NES to ‘LQARGAAA,’ to yield CRY2(L348F)-CreN(ΔNES) resulted in significantly increased nuclear localization (Figure 2C). When we added a strong SV40 NLS to CRY2(L348F)-CreN (Figure 2C), we also observed an increase in nuclear localization, along with a corresponding increase in dark background activity (Figure 2D). Likewise, switching the CreN and CreC domains of the constructs, to generate mCherry-CRY2(L348F)-CreC (p1097) and mCherry-CIB1-CreN (p1087), resulted in constructs with predominantly nuclear localization (Figure 2E), but also higher dark background compared with the original system (Figure 2F).
To more precisely explore the differences between wt or L348F versions of CRY2-CreN, we imaged the mCherry-tagged fragments and quantified the concentration of protein in the nucleus as total protein expression increased (Figure 2G). With both wt and L348F CRY2, the concentration of protein in the nucleus increased as total cell fluorescence increased. At all expression levels, the concentration of L348F CRY2-CreN in the nucleus was lower than wt CRY2-CreN. These results support that the reduced dark background of L348F is due to reduced nuclear localization and buffering of protein in the cytosol as protein expression levels increase overall. In Figure 2H we propose a model of PA-Cre2.0 nuclear trafficking. The effect of combining a L348F CRY2 protein with reduced nuclear occupancy with a CreN fragment that promotes nuclear export leads to a CreN version that is predominantly cytosolic, but that undergoes nuclear shuttling. Thus, at any given time, some fraction of the protein would be available in the nucleus to induce recombination with light. Because the majority of protein is buffered in the cytosol, we speculate that such a design is less sensitive to expression level differences, resulting in universally low dark background activity.
Comparison of PA-Cre2.0 with Magnet-actuated PA-Cre
We hypothesized that the unique design of PA-Cre2.0 may provide an advantage over other split systems that are strongly nuclear localized, such as the Magnet PA-Cre system (15), which has a strong SV40 NLS attached to each Cre half. Mag PA-Cre uses a similar split fragment approach, but uses nMag/pMag (‘Magnet’) photodimerizers and a different Cre split site (15). To directly compare these two systems, we placed Mag PA-Cre downstream of a mCherry-IRES element (i.e. mCh-IRES-MagPA-Cre, KM91) and used a EGFP DIO (Double-floxed Inverse Open reading frame) Cre reporter (32), allowing quantification of Cre activity by recording the number of mCherry-expressing cells with EGFP expression. We tested the responses of both systems in side-by-side experiments in dark, or after exposure to 4 s or 4 h blue light (Figure 3).
Figure 3.
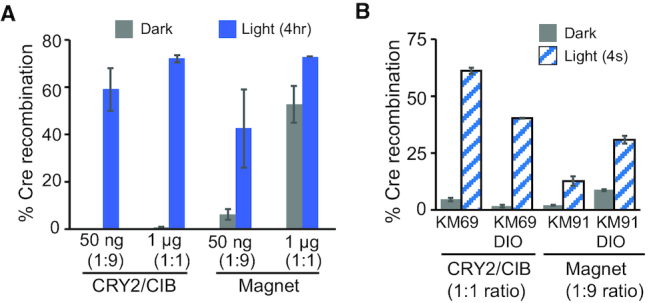
Functional comparison of CRY2/CIB1 PA-Cre2.0 with Mag PA-Cre. (A) Comparison of PA-Cre2.0 and Mag PA-Cre activity at different expression levels. HEK293T cells were transfected with either 1 μg of each indicated Cre system (KM69 or KM91) and 1 μg DIO-EGFP reporter (1:1 ratio), or with 50 ng each Cre system and 450 ng reporter (1:9 ratio). Samples were treated with dark or 4 h light at 24 h, before quantifying at 48 h. Graph indicates average and range of two independent experiments. (B) Comparison of PA-Cre2.0 and Mag PA-Cre responses to a 4 s light pulse. HEK293T cells were transfected with 1 μg of PA-Cre2.0 (KM69) and 1 μg DIO-EGFP or loxP-STOP-loxP-EGFP reporter (1:1), or 50 ng of Mag PA-Cre (KM91) and 450 ng of each reporter (1:9). Samples were treated with dark or light as in (A) and quantified at 48 h. Graph indicates average and range of two independent experiments.
Because different dimerizer-induced split protein systems have different optimal expression levels, it is essential to explore a range of expression levels when comparing different systems. Previous studies with Mag PA-Cre indicated that the optimal dynamic range of the system required substantially lower expression, as these prior experiments used much lower amounts of transfected Mag PA-Cre DNA (typically, a 1:9 ratio of Mag PA-Cre to Cre reporter was used) (15). We tested equivalent conditions as previously used, using a 1:9 ratio of Mag PA-Cre relative to the Cre reporter and transfected similar amounts of DNA (50 ng MagPA-Cre, 450 ng DIO reporter) (Figure 3A). In turn, we tested expression of CRY2/CIB1 PA-Cre2.0 under similar expression conditions (50 ng PA-Cre2.0 and 450 ng reporter). We compared these results at low expression levels with results at higher expression using 20-fold more DNA (1 μg PA-Cre and 1 μg reporter). At the higher expression level, the Mag PA-Cre showed high background, but this was dramatically reduced at the lower (optimized) expression level. The CRY2/CIB-actuated PA-Cre2.0 system performed equally well at both high (1:1 ratio Cre:reporter, 1μg PA-Cre2.0) and low (1:9 ratio, 50 ng PA-Cre2.0) levels. At the low (1:9) expression level, Mag PA-Cre showed higher dark background and reduced light-induced stimulation compared with PA-Cre2.0, resulting in lower overall dynamic range (ratio of activity in light/dark was 6.8 for Mag PA-Cre, compared with 154.5 for CRY/CIB PA-Cre2.0). When compared using expression levels optimized for each system (1:1 ratio for PA-Cre2.0; 1:9 ratio for Mag PA-Cre), PA-Cre2.0 also showed enhanced dynamic range and higher levels of recombination in response to a short 4 s light pulse (Figure 3B). We also note that these results showing high activity of CRY2/CIB1 PA-Cre2.0 with the DIO reporter indicate this system is able to catalyze recombination events at non-canonical loxP sites, which has not been previously demonstrated.
In vivo validation of PA-Cre2.0
The consistent activity afforded by PA-Cre2.0 in cell culture, with minimal background even at higher expression levels, suggested the system would be well-suited for use in vivo. We thus tested the system with viral delivery in vivo in mouse brain, expressing each Cre half in an AAV vector and using a CaMKII promoter to impart neuronal expression (pAAV-CaMKII-myc-CRY2(L348F)-CreN and pAAV-CaMKII-CIB1-CreC). To visualize induced recombinase activity in mouse, we used Ai14 Cre reporter mice, which contain an integrated loxP-flanked STOP cassette preventing transcription of a CAG promoter-driven tdTomato reporter (33). These mice express tdTomato fluorescent protein upon Cre mediated-recombination.
Ai14 mice were stereotactically injected in the dentate gyrus with virus comprising each of the two Cre halves, as well as a hSyn-EYFP virus to mark the site of injection, as shown in Figure 4A. Mice were implanted with an optical fiber targeted directly above the injection site (Figure 4B). Three weeks after injection and implantation, mice were either exposed to a 1 s pulse of blue (473 nm) light via a patch cable connected to the implanted fiber, or no light for control animals. Minimal expression of tdTomato was observed in brains from control mice not exposed to the blue light pulse (Figure 4C, top row). Brains from animals treated with a 1 s light pulse showed tdTomato expression in CaMKII positive neurons directly below the implanted fiber (Figure 4C, bottom row). These results indicate low background recombination can be achieved in vivo using virally delivered components. In addition, Cre recombination can be effectively triggered in vivo as well as in vitro (cell culture) by a very brief light input (a single 1 s light pulse).
Figure 4.

In vivo validation of PA-Cre2.0. (A) AAVs (hSyn-EYFP, CaMKII-mycCRY2(L348F)-CreN and CaMKII-CIB1-CreC) were mixed at a 1:1:1 ratio and injected into the dentate gyrus of Ai14 reporter mice. (B) Mice were implanted with an optical fiber (dashed outline in Figure 2C) over hippocampus. (C) Representative images from control animals (no light pulse, top row) or animals exposed to a 1 s pulse of blue light (473 nm) (‘Light’, bottom row). EYFP (green) marks virally infected neurons, with DAPI nuclear staining highlighted in blue. Minimal tdTomato expression (red) was observed in control animals (n = 3) that did not receive light, while light-treated animals (n = 3) showed Cre-dependent tdTomato expression in CaMKII positive cells in brain. Scale bar, 400 μm.
Manipulation of split Cre fragment nuclear localization for tunable activity control
Our results indicated that expressing both Cre halves at high levels in the nucleus is a major contributor to dark background activity, presumably due to the affinity of the two split Cre fragments. We hypothesized that we could achieve tighter control of activity by combining two inducible systems, wherein abundance or nuclear accumulation of the CRY2-CreN fragment is manipulated by a first inducible system, followed by light-induced fragment reconstitution. In theory, by restricting the amount of CRY2-CreN fragment available for recombination until an inducible signal, we could achieve very tightly controlled recombination activity with very low background. Such an approach has previously been used to improve dynamic range of split and allosterically-controlled CRISPR switches (24,34).
To first test this concept, we fused the CRY2(L348F)-CreN fragment to a destabilized Escherichia coli DHFR that is unstable unless bound to the small molecule trimethoprim (TMP) (35). Previous studies demonstrated effective use of this domain to destabilize intact Cre recombinase in mouse brain, with induced Cre function after delivery of TMP, a relatively inexpensive compound that passes the blood-brain barrier (36). We generated ecDHFR-CRY2(L348F)-CreN and tested activity of the recombinase with or without prior addition of TMP (Figure 5A and B). In the absence of TMP, we observed minimal activity in dark or light (0.2% of cells showing recombination in dark, 2.3% in light). With 10 μM TMP, we observed low background in dark (0.6%) but 29% of cells underwent recombination after a 30 min light treatment (delivered 20 min to 2 h after TMP addition).
Figure 5.
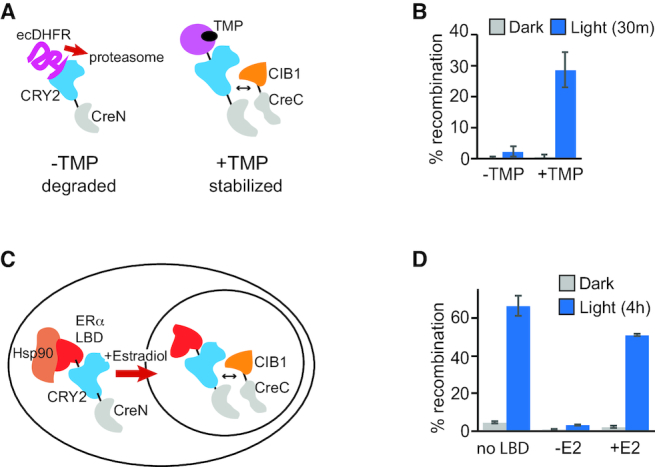
Light and chemical regulation of Cre recombinase activity. (A) Schematic of TMP-regulated PA-Cre, in which destabilized Escherichia coli DHFR is fused to the CRY2(L348F)-CreN fragment. (B) HEK293T cells were transfected with the constructs indicated in (A) (CW79, 1 μg; p645, CIB1-CreC, 1 μg), along with a loxP-STOP-loxP-EGFP reporter (1 μg). Twenty-four hours after transfection, TMP (10 μM) was added to indicated samples. After 30–120 min, samples were maintained in dark or exposed to light (461 nm pulse, 2 s every 2 min for 30 min) then quantified at 48 h. Graph represents average and error (s.d., n = 3 independent experiments). (C) Schematic of estradiol-and-light-regulated Cre recombinase, containing an ERα-LBD fused to CRY2(L348F)-CreN. (D) HEK293T cells were transfected with the constructs indicated in (C) (CW57, 1 μg; p645, CIB1-CreC, 1 μg), and a loxP-STOP-loxP-EGFP reporter (1 μg). Twenty-four hours after transfection, 17-β-estradiol (400 nM) was added to indicated samples, then samples were kept in dark or exposed to 4 h light (461 nm pulse, 2 s every 3 min), then quantified at 48 h. Control samples labeled ‘No LBD’ contained p688, p645 and the reporter. Graph represents average and range of two independent experiments.
As a second approach, we tested a method to trap CRY2(L348F)-CreN in the cytosol and allow release with light, by tethering to a NES-PhoCl construct. PhoCl is an engineered protein derived from a mMaple fluorescent reporter that undergoes self-cleavage with 400 nm light application (37). We reasoned that fusion to NES-PhoCl could trap the CRY2-CreN half in the cytosol in dark, but allow release with UV light. However, we were unable to observe any improvement in dynamic range of the system using this approach (Supplementary Figure S3). Because 400 nm light can stimulate CRY2-CRY2 self-association, we speculate that the PhoCl light treatment could affect CRY2 nuclear entry, but did not investigate this further.
In a similar approach to trap one of the fragments in the cytosol, we next tested fusion of the CRY2(L348F)-CreN fragment to a human estrogen receptor alpha ligand-binding domain (ERα-LBD). Steroid hormone LBDs such as ERα-LBD are effectively trapped in the cytosol in the absence of ligand due to interaction with Hsp90. Addition of hormone releases the protein from Hsp90 and allows nuclear import (38). The ERα-fused construct, ER-CRY2(L348F)-CreN, showed minimal activity in light or dark in the absence of 17-ß-estradiol, with 8-fold reduced dark background compared with CRY2(L348F)-CreN (Figure 5C and D). In samples exposed to estradiol but maintained in dark, background was also reduced (2.9% recombination with ER fusion, compared with 4.2% without ER fusion). In turn, the addition of estradiol plus light led to high activity, comparable to activity obtained without the ER fusion. These results indicate that use of the ER-LBD-fused CRY2-CreN fragment can further reduce dark background, while enabling high levels of induced recombination with light plus hormone. Such a system (replacing ER LBD with a tamoxifen-responsive variant) (39) may be useful for in vivo studies, as it reduces potential dark background during long term expression and removes the requirement for samples to be maintained in dark at all times prior to adding the ligand.
DISCUSSION
In this work, we systematically explored methods for achieving tight control of activity of a split protein, photoactivatable Cre DNA recombinase, that would be effective over a wide range of protein expression levels. Photoactivatable Cre recombinase (PA-Cre2.0) consists of split Cre protein fragments that are brought in proximity by a light-dependent interaction between the plant photoreceptor CRY2 (containing a L348F mutation), and its binding partner CIB1. We observed that activity of PA-Cre2.0 was relatively insensitive to differences, in particular, in expression of the CRY2(L348F)-CreN fragment. We found that insensitivity was due to reduced nuclear localization of this fragment caused by a combination of two factors: weakening of the endogenous CRY2 nuclear localization by the L348F mutation, and a cryptic NES identified in the CreN fragment. With this combination, the protein is expected to shuttle into and out of the nucleus, but is predominantly retained in the cytosol, resulting in steady but lower nuclear levels than observed with the higher background wild-type protein.
Our study provides new results showing that only very low levels of protein and light are needed to achieve high levels of Cre recombinase activity. We demonstrated that high light induction (up to ∼80% of cells showing recombination) could be achieved using just three 4 s light pulses, spaced 45 min apart. The system is also highly sensitive, even when the fragments are expressed at very low levels. A RNA-destabilized version of PA-Cre2.0 (ΔpA) that produced protein levels that were nearly undetectable by immunoblot nevertheless maintained high induced activity and low background in functional activity tests. Our results also unequivocally demonstrate that Cre recombinase split at residue 105 is fully competent to catalyze recombination events at non-canonical loxP sites such as used in DIO/FLEx recombinase reporters. A previous study (15) had proposed that photoactivatable Cre recombinase split at residue 105 was not competent to induce recombination at non-canonical loxP sites, however we believe the discrepancy was due to utilization of the prior-characterized system at non-optimal expression levels. As we demonstrate here, Cre recombinase split at residue 105 functioned equally well with a DIO reporter using alternate loxP sites (loxP2272), as with a loxP-specific recombinase reporter.
We followed up our in vitro characterization of PA-Cre2.0 with functional testing in vivo in rodent brain. In these experiments, due to packaging size limits for AAV vectors, we packaged each fragment into a separate AAV, then combined the two for injection in the brain. Our in vivo results, which used a completely different approach to deliver protein (viral transduction) and tested the system in a different cell type and organism, showed very similar performance as compared with the cell culture studies, with minimal background in dark, high induced activity, and sensitive response to a single 1 s light pulse. As the CRY2/CIB1 interaction can be stimulated by two-photon excitation (14,27), we expect PA-Cre2.0 can also be stimulated with multiphoton activation, although did not test this specifically.
As the combination of light and nuclear localization are important for activity, we extended our findings to develop two additional photoactivatable Cre approaches that add chemical regulation increasing the selectivity of the system. A number of recent studies have incorporated dual control mechanisms for achieving tighter inducible regulation, compared with single-chemical or light-regulated systems (24–26,34). For example, an approach combining two inducible systems was previously demonstrated to reduce background of a nuclear import photoswitch (26). Here, we generated a light-and-estrogen regulated recombinase in which estrogen regulates nuclear entry of the CRY2-CreN fragment, while light is required for split fragment reconstitution. Likewise, a version of CRY2-CreN fused to a destabilization domain showed low background and high activity in the presence of light and a stabilizing drug. We expect these new versions of PA-Cre2.0 to offer further versatility for use in vivo. While this work focuses on ways to induce Cre recombinase activity, with additional engineering these approaches can also be used to generate versatile sensors based on Cre recombinase activity. For example, by fusing a NLS-deleted version of the CRY2-CreN fragment to a protein that conditionally localizes to the nucleus, cells that show nuclear import of this protein within a specific time defined by light can be permanently marked and tracked.
An important implication of our study is that for optimal inducible regulation of a split nuclear enzyme, a strong NLS is not always desired, and may in fact be detrimental. Indeed, when we added a SV40 NLS to the CRY2-CreN fragment, we obtained high background. Likewise, when we tested an alternate Magnet-actuated PA-Cre system (15) that contains a strong NLS on each fragment, we observed high background activity as expression increased. We hypothesize that manipulating the balance of nuclear import and export could be useful not only with Cre recombinase, but also for achieving tight regulation of other split protein systems that are specifically localized to the nucleus or cytosol. In this manner, proteins can be engineered that inhabit both compartments, but are tuned to be predominantly localized in one or the other compartment as needed, resulting in consistent function despite expression level variation in different cell types and organisms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Tom Wandless for providing construct pBMN DHFR(DD)-YFP (Addgene, #29325), and Robert Campbell for providing plasmid pcDNA-NES-PhoCl-mCherry (Addgene, #87690).
FUNDING
National Institutes of Health (NIH) [R01GM100225]. Funding for open access charge: NIH [R01GM100225].
Conflict of interest statement. None declared.
REFERENCES
- 1. Richards F.M. On the enzymic activity of Subtilisin-Modified ribonuclease. Proc. Natl. Acad. Sci. U.S.A. 1958; 44:162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ullmann A., Jacob F., Monod J.. Characterization by in vitro complementation of a peptide corresponding to an operator-proximal segment of the beta-galactosidase structural gene of Escherichia coli. J. Mol. Biol. 1967; 24:339–343. [DOI] [PubMed] [Google Scholar]
- 3. Villarejo M., Zamenhof P.J., Zabin I.. Beta-galactosidase. In vivo -complementation. J. Biol. Chem. 1972; 247:2212–2216. [PubMed] [Google Scholar]
- 4. Johnsson N., Varshavsky A.. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pelletier J.N., Campbell-Valois F.X., Michnick S.W.. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:12141–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fields S., Song O.. A novel genetic system to detect protein-protein interactions. Nature. 1989; 340:245–246. [DOI] [PubMed] [Google Scholar]
- 7. Wehrman T., Kleaveland B., Her J.H., Balint R.F., Blau H.M.. Protein-protein interactions monitored in mammalian cells via complementation of beta -lactamase enzyme fragments. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:3469–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luker K.E., Smith M.C.P., Luker G.D., Gammon S.T., Piwnica-Worms H., Piwnica-Worms D.. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:12288–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilson C.G., Magliery T.J., Regan L.. Detecting protein-protein interactions with GFP-fragment reassembly. Nat. Methods. 2004; 1:255–262. [DOI] [PubMed] [Google Scholar]
- 10. Spencer D.M., Wandless T.J., Schreiber S.L., Crabtree G.R.. Controlling signal transduction with synthetic ligands. Science. 1993; 262:1019–1024. [DOI] [PubMed] [Google Scholar]
- 11. Liu Q., Tucker C.L.. Engineering genetically-encoded tools for optogenetic control of protein activity. Curr. Opin. Chem. Biol. 2017; 40:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rost B.R., Schneider-Warme F., Schmitz D., Hegemann P.. Optogenetic tools for subcellular applications in neuroscience. Neuron. 2017; 96:572–603. [DOI] [PubMed] [Google Scholar]
- 13. Jullien N., Sampieri F., Enjalbert A., Herman J.-P.P.. Regulation of Cre recombinase by ligand-induced complementation of inactive fragments. Nucleic Acids Res. 2003; 31:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taslimi A., Zoltowski B., Miranda J.G., Pathak G.P., Hughes R.M., Tucker C.L.. Optimized second-generation CRY2–CIB dimerizers and photoactivatable Cre recombinase. Nat. Chem. Biol. 2016; 12:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kawano F., Okazaki R., Yazawa M., Sato M.. A photoactivatable Cre–loxP recombination system for optogenetic genome engineering. Nat. Chem. Biol. 2016; 12:1059–1064. [DOI] [PubMed] [Google Scholar]
- 16. Shimizu-Sato S., Huq E., Tepperman J.M., Quail P.H.. A light-switchable gene promoter system. Nat. Biotechnol. 2002; 20:1041–1044. [DOI] [PubMed] [Google Scholar]
- 17. Motta-Mena L.B., Reade A., Mallory M.J., Glantz S., Weiner O.D., Lynch K.W., Gardner K.H.. An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat. Chem. Biol. 2014; 10:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X., Chen X., Yang Y.. Spatiotemporal control of gene expression by a light-switchable transgene system. Nat. Methods. 2012; 9:266–269. [DOI] [PubMed] [Google Scholar]
- 19. Pathak G.P., Spiltoir J.I., Höglund C., Polstein L.R., Heine-Koskinen S., Gersbach C.A., Rossi J., Tucker C.L.. Bidirectional approaches for optogenetic regulation of gene expression in mammalian cells using Arabidopsis cryptochrome 2. Nucleic Acids Res. 2017; 45:e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gray D.C., Mahrus S., Wells J.A.. Activation of specific apoptotic caspases with an engineered small-molecule-activated protease. Cell. 2010; 142:637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wehr M.C., Laage R., Bolz U., Fischer T.M., Grunewald S., Scheek S., Bach A., Nave K.A., Rossner M.J.. Monitoring regulated protein-protein interactions using split TEV. Nat. Methods. 2006; 3:985–993. [DOI] [PubMed] [Google Scholar]
- 22. Liu Q., Sinnen B.L., Boxer E.E., Schneider M.W., Grybko M.J., Buchta W.C., Gibson E.S., Wysoczynski C.L., Ford C.P., Gottschalk A. et al.. A photoactivatable botulinum neurotoxin for inducible control of neurotransmission. Neuron. 2019; 101:863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dagliyan O., Krokhotin A., Ozkan-Dagliyan I., Deiters A., Der C.J., Hahn K.M., Dokholyan N. V.. Computational design of chemogenetic and optogenetic split proteins. Nat. Commun. 2018; 9:4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nguyen D.P., Miyaoka Y., Gilbert L.A., Mayerl S.J., Lee B.H., Weissman J.S., Conklin B.R., Wells J.A.. Ligand-binding domains of nuclear receptors facilitate tight control of split CRISPR activity. Nat. Commun. 2016; 7:12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zetsche B., Volz S.E., Zhang F.. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015; 33:139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yumerefendi H., Wang H., Dickinson D.J., Lerner A.M., Malkus P., Goldstein B., Hahn K., Kuhlman B.. Light-Dependent cytoplasmic recruitment enhances the dynamic range of a nuclear import photoswitch. ChemBioChem. 2018; 19:1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kennedy M.J., Hughes R.M., Peteya L.A., Schwartz J.W., Ehlers M.D., Tucker C.L.. Rapid blue-light–mediated induction of protein interactions in living cells. Nat. Methods. 2010; 7:973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I., Friman O., Guertin D.A., Chang J., Lindquist R.A., Moffat J. et al.. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006; 7:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sparta D.R., Stamatakis A.M., Phillips J.L., Hovelsø N., van Zessen R., Stuber G.D.. Construction of implantable optical fibers for long-term optogenetic manipulation of neural circuits. Nat. Protoc. 2012; 7:12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mizuguchi H., Xu Z., Ishii-Watabe A., Uchida E., Hayakawa T.. IRES-Dependent second gene expression is significantly lower than Cap-dependent first gene expression in a bicistronic vector. Mol. Ther. 2000; 1:376–382. [DOI] [PubMed] [Google Scholar]
- 31. la Cour T., Kiemer L., Mølgaard A., Gupta R., Skriver K., Brunak S.. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 2004; 17:527–536. [DOI] [PubMed] [Google Scholar]
- 32. Sohal V.S., Zhang F., Yizhar O., Deisseroth K.. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009; 459:698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R. et al.. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2009; 13:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oakes B.L., Nadler D.C., Flamholz A., Fellmann C., Staahl B.T., Doudna J.A., Savage D.F.. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat. Biotechnol. 2016; 34:646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iwamoto M., Björklund T., Lundberg C., Kirik D., Wandless T.J.. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol. 2010; 17:981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sando R., Baumgaertel K., Pieraut S., Torabi-Rander N., Wandless T.J., Mayford M., Maximov A.. Inducible control of gene expression with destabilized Cre. Nat. Methods. 2013; 10:1085–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang W., Lohman A.W., Zhuravlova Y., Lu X., Wiens M.D., Hoi H., Yaganoglu S., Mohr M.A., Kitova E.N., Klassen J.S. et al.. Optogenetic control with a photocleavable protein, PhoCl. Nat. Methods. 2017; 14:391–394. [DOI] [PubMed] [Google Scholar]
- 38. Picard D., Salser S.J., Yamamoto K.R.. A movable and regulable inactivation function within the steroid binding domain of the glucocorticoid receptor. Cell. 1988; 54:1073–1080. [DOI] [PubMed] [Google Scholar]
- 39. Feil R., Brocard J., Mascrez B., LeMeur M., Metzger D., Chambon P.. Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:10887–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.