


Abstract
Faithful inheritance of DNA methylation across cell division requires DNMT1 and its accessory factor UHRF1. However, how this axis is regulated to ensure DNA methylation homeostasis remains poorly understood. Here we show that SET8, a cell-cycle-regulated protein methyltransferase, controls protein stability of both UHRF1 and DNMT1 through methylation-mediated, ubiquitin-dependent degradation and consequently prevents excessive DNA methylation. SET8 methylates UHRF1 at lysine 385 and this modification leads to ubiquitination and degradation of UHRF1. In contrast, LSD1 stabilizes both UHRF1 and DNMT1 by demethylation. Importantly, SET8 and LSD1 oppositely regulate global DNA methylation and do so most likely through regulating the level of UHRF1 than DNMT1. Finally, we show that UHRF1 downregulation in G2/M by SET8 has a role in suppressing DNMT1-mediated methylation on post-replicated DNA. Altogether, our study reveals a novel role of SET8 in promoting DNA methylation homeostasis and identifies UHRF1 as the hub for tuning DNA methylation through dynamic protein methylation.
INTRODUCTION
In mammals, DNA methylation predominantly occurs at cytosine-C5 in the context of CpG dinucleotides and plays a critical role in transcriptional regulation, heterochromatin formation, X chromosome inactivation, imprinting and genome stability (1–4). Although three active DNA methyltransferases, DNMT3A, DNMT3B and DNMT1, act coordinately to establish and maintain patterns of DNA methylation in a given cell, DNMT1 is mainly responsible for converting hemimethylated CpG substrates generated during DNA replication to symmetrically methylated molecules, a process known as DNA maintenance methylation (5–7). An essential player in DNA maintenance methylation by DNMT1 is UHRF1, a multi-structural and functional nuclear protein (8,9). UHRF1 is loaded onto replication forks in S phase by its ability to bind hemi-methylated CpGs (products of newly replicated DNA) via a unique SRA domain (10–12) and the histone H3 tail with di- or tri-methylated K9 (H3K9me2/3) and/or methylated DNA ligase 1 via a joined action of the tandem tudor domain and a PHD domain (13–18). Replication-fork associated UHRF1 in turn ubiquitinates histone H3 (19,20). DNMT1 is recruited to replication forks by a combinatorial interaction with UHRF1, PCNA and ubiquitinated H3 (20,21). However, how this maintenance machinery axis is regulated is less well understood.
Many previous studies have focused on regulation of DNMT1 expression and activity on DNA methylation. For example, DNMT1 stability and activity have been shown to be regulated by post-translational modification including phosphorylation, acetylation and methylation and by RNA (22–26). However, less is known on control of DNA methylation by regulation of UHRF1. In this regard, as a factor that recruits and activates DNMT1, UHRF1 is perhaps a more sensitive target for regulation of DNA methylation (27). In supporting this idea, recent studies have identified UHRF1 as a critical target for reprogramming global DNA methylation in naïve embryonic stem (ES) cells and during oogenesis (28,29). In somatic cells, high UHRF1 expression in S phase is known to correlate with cell proliferation, and is often deregulated in cancer cells (30). However, it is unclear how UHRF1 is regulated in somatic cells to ensure faithful DNA methylation inheritance in cell division.
SET8 (also known as PR-SET7, SETD8 and KMT5A) is known to catalyze the monomethylation of histone H4 Lys20 (31,32) and non-histone proteins including p53 (33). SET8 protein expression is tightly regulated through cell cycle. It is degraded by proteasome at G1/S transition (34–37). Consequently, SET8 is highly expressed in G2/M and G1. In mammalian cells, depletion of SET8 results in premature chromatin compaction, G2/M arrest and defect in DNA damage repair (34,35,37). However, it is not known if SET8 regulates DNA methylation.
In this study, we show that SET8 regulates UHRF1 in a cell-cycle-dependent manner through a methylation-dependent protein degradation. In addition, SET8 also regulates DNMT1. We present evidence that SET8 is a key regulator of DNA methylation, ensuring DNA methylation homeostasis by controlling the axis of DNA maintenance methylation, especially UHRF1.
MATERIALS AND METHODS
Cell culture
HCT116 cells were cultured in McCoy's 5A medium (GIBCO) containing 10% fetal bovine serum (FBS) (Intergen) and 1% Pen-Strep. HeLa, HEK293T, NIH3T3 and MCF7 cells were grown in DMEM (GIBCO) supplemented with 10% fetal bovine serum (FBS) (Intergen) and 1% Pen-Strep (GIBCO). Human HFL1 fibroblast cells were maintained in F12K medium (GIBCO) containing 10% fetal bovine serum (FBS) (Intergen) and 1% Pen-Strep (GIBCO).
Plasmids and antibodies
Plasmids pcDNA3.1-FLAG-SET8, pcDNA3.1-FLAG-UHRF1, pcDNA3.1-DNMT1, pcDNA3.1-HA-SET8, pcDNA3.1-His-Ub, pcDNA3.1-Myc-LSD1, pcDNA3.1-Myc-USP7 and pPYCAGIP-FLAG-UHRF1 were constructed in our laboratory as previously described (16,38,39). All mutants were generated by PCR-based point mutagenesis strategy and verified by DNA sequencing. The following antibodies were used in this study: Pan-Kme antibody (Abbkine Abm40195), UHRF1 (proteintech 21402-1-AP), monoclonal DNMT1 (homemade), monoclonal LSD1 (homemade), SET8 (Cell Signaling Technology C18B7), DNMT3A (Santa Cruz sc-20703), p53 (Santa Cruz sc-126), Ub (Santa Cruz sc-8017), GAPDH (Abmart M20006L), β-ACTIN (Sigma A5441), H3 (Abcam ab1791), H3S10P (Epitomics 1173-1), FLAG (Sigma 7425/1804), Myc (Abmart 20002 mouse), and BrdU (Sigma B8434). The following secondary antibody were used: Alexa Fluor 680 goat anti-rabbit IgG (Jackson ImmunoResearch 111-625-144), Alexa Fluor 790 goat anti-mouse IgG (Jackson ImmunoResearch 115-655-146), Alexa Fluor 594 goat anti-rabbit IgG (Jackson ImmunoResearch, 111-585-003) and Alexa Fluor 488 goat anti-mouse IgG (Jackson ImmunoResearch, 115-545-003).
Transfection
Transient transfection of protein- or shRNA-encoding plasmids were performed using lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. For siRNA transfection, cells were transfected twice at 24-h intervals with the indicated siRNA using Lipofectamine 2000 (Invitrogen) following manufacturer's instructions.
LSD1 and DNMT3A/3B double knockout by CRISPR-Cas9
The LSD1-KO and DNMT3A/3B-DKO HeLa cell lines were obtained by CRISPR-Cas9 essentially as described (40) with the guide RNAs listed below. GAATAGCAGAGACTCCGGA for LSD1; GCTACCACGCCTGAGCCCGT for DNMT3A; AGACTCGATCCTCGTCAACG for DNMT3B.
Immunostaining and western blot
For immunostaining, HeLa or other cells were washed with PBS buffer twice, fixed with 4% paraformaldehyde for 30 min at 37°C, and permeabilizated with 1% Triton in PBS for 15 min at 4°C. Cells were washed with PBST and incubated in 5% BSA in PBST for 1 h at 37°C before addition of an antibody at appropriate dilution (1:1000 for FLAG-antibody, 1:1000 for UHRF1 (Proteintech, 21402-1-AP), 1:200 for DNMT1 and 1:5000 for 5mC antibody) and further incubated for 2 h at 37°C or 4°C overnight. The cells were then washed with PBST and incubated with fluorconjugated secondary antibodies and 4′,6′-diamidino-2-phenylindole (DAPI) for 1 h at 37°C. Following PBST washing, the images were captured with Olympus microscope. Western blot analysis for various proteins was carried out essentially as described (41).
EdU staining assay
The EdU staining assay for identification of S phase cells was performed according to the Cell-Light TM EdU Fluorescent Detection Kit (RiBoBio, Guangzhou, China; C10310) with a slight modification. In brief, cells grown on 48-well plates were labeled with 20 μM EdU (5-ethynyl-2’-deoxyuridine) for approximately 2 h at 37°C, washed with 1× PBS twice and fixed with 4% paraformaldehyde for 20 min at 4°C before neutralization with glycine (2 mg/ml). Then the cells were permeabilized, blocked and incubated with antibodies as described above. Finally, the Edu were stained by Apollo reaction buffer at 37°C for 30 min. Slides were washed by methanol once and PBS twice before fluorescent imaging.
Cell cycle synchronization
For synchronization of cells at the G1/S border, active growing HCT116/HFL1/HeLa/HEK293T cells were treated with 1 ug/ml aphidicolin for 24 h. For synchronization of cells at S phase, the aphidicolin-treated cells were released in the fresh culture medium for 4 h. For synchronization of cells at G2/M phase, the cells were treated with 50 ng/ml nocodazole for 18 h.
Co-immunoprecipitation
For co-immunoprecipitation of exogenous proteins, the indicated plasmids were transfected into HEK293T cells. Cells were collected 48 h after transfection and lysed in IP Lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1× protease inhibitor cocktail and 1 mM DTT). The lysates were cleared by centrifugation at 12 000 rpm for 20 min at 4°C. The supernatant was diluted with binding buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 8% glycerol, 1 mM EDTA, 1× protease inhibitor cocktail and 1 mM DTT) until the concentration of Triton X-100 decreased to 0.2%. The extracts were then directly incubated with anti-FLAG M2-affinity beads (Bimake, Houston, TX, USA) or specific antibody as indicated for 4 h at 4°C. For co-immunoprecipitation of endogenous proteins, antibodies were added at a concentration of 1 μg/mg of lysates and incubated overnight at 4°C, followed by antibody-protein complex capture with Protein A/G Sepharose beads (Santa Cruz). After extensive washing with washing buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 1 mM EDTA, 1× protease inhibitor cocktail and 1 mM DTT), complexes were boiled in 1× SDS loading buffer and analyzed by western blot.
In vivo ubiquitination assay
For detection of ubiquitination on UHRF1 or DNMT1, HEK293T cells were transfected with His-Ub, FLAG-UHRF1 or mutants, or FLAG-DNMT1 and HA-SET8 WT/mutant as indicated. MG132 (10 μg/ml) was added 8 h before sample collection. At 48 h posttransfection, cells were washed with PBS twice and lysed in denaturing IP Lysis buffer (150 mM NaCl, 0.1% NP40 or Triton X-100, 25 mM Tris–HCl, pH 8.0, 5 mM EDTA, 10% glycerol and 1% SDS) and treated at 100°C for 20 min to inactivate deubiquitinating enzymes. The lysates were cleared by centrifugation at 12 000 rpm for 20 min. The supernatant was diluted with denaturing binding buffer without SDS to make final SDS concentration 0.1%, and then directly incubated with anti-FLAG M2-affinity beads (Bimake, Houston, TX) for 8 h. After extensive washing with IP washing buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 1 mM EDTA), the immunoprecipited proteins were boiled in 1× SDS loading buffer and analyzed by SDS-PAGE and western blot with antibodies as indicated.
Identification of methylation and ubiquitination sites
For identification of SET8-catalyzed methylation sites on UHRF1, FLAG-UHRF1 and HA-SET8 were co-expressed in HEK293T cells and cells were treated with MG132 for 8 h before harvested for IP of FLAG-UHRF1 48 h posttransfection. The bead-bound FLAG-UHRF1 proteins were digested with trypsin and resulting peptides were analyzed by mass spectrometry essentially as described (42). For identification of Ub sites, HeLa cells were treated with MG132 8 h before harvested for IP of endogenous UHRF1 proteins. The identification of Ub sites was essentially as described (43).
In vivo methylation/demethylation assay
For in vivo methylation assay, FLAG-UHRF1 or its mutants or FLAG-DNMT1 were co-transfected with HA-SET8 or its mutant or Myc-LSD1 as indicated. MG132 (10 μg/ml) was added 8 h before cells were harvested at 48 h posttransfection. Cells were washed with PBS twice and lysed in denaturing IP Lysis buffer (150 mM NaCl, 0.1% NP40 or Triton X-100, 25 mM Tris–HCl, pH 8.0, 5 mM EDTA, 10% glycerol and 1% SDS). Methylation was detected by IP-western analysis as above using a Pan-me antibody.
In vitro methylation assay
In vitro methylation of UHRF1 was performed with either proteins expressed and purified from HEK293T cells or Escherichia coli. For former, FLAG-SET8 and its mutant proteins and FLAG-UHRF1 and its mutant proteins were individually expressed and purified from HEK293T cells using M2 anti-FLAG beads essentially as described. In vitro methylation assay was carried out in a 20 μl reaction system with either 0.4 μg FLAG-UHRF1 or mutant proteins and 0.4 μg FLAG-SET8 or mutants in the buffer (50 mM Tris–HCl at pH 8.0, 10 mM DTT and 10 mM MgCl2) containing 0.1 mM S-adenosyl-methionine (SAM; Sigma) for 1 h at 30°C. Afterwards the methylation reaction was stopped by boiling with SDS loading buffer and the reaction mixture was loaded onto a 8% SDS PAGE gel, with methylation on UHRF1 detected by western blot using a Pan-me antibody.
For methylation assay with recombinant proteins, GST-SET8 and its mutant proteins and GST-UHRF1 were individually expressed and purified from E. coli. Recombinant polynucleosomes H3.1 (No. 31466) was purchased from Active Motif and added in reaction to promote SET8 activity (31). In vitro methylation assay was carried out in a 20 μl reaction system with either 1 μg GST-UHRF1 or mutant proteins and 1 μg GST-SET8 or mutants in the buffer (50 mM Tris–HCl at pH 8.0, 10% glycerol, 20 mM KCl, 5 mM MgCl2, 1 mM DTT and 1 mM PMSF) containing 0.1 mM S-adenosyl-methionine (SAM; Sigma) and 0.5 μg nucleosomes for 2 h at 30°C. Afterwards the methylation reaction was stopped by boiling with SDS loading buffer and the reaction mixture was loaded onto a 8% SDS PAGE gel, with methylation on UHRF1 detected by western blot using a Pan-me antibody.
FACS analysis
For FACS analysis, cells were fixed in 70% ethanol and cellular DNA was stained with propidium iodide (Sigma 81845) before analyzed by a FACS Calibur flowcytometer (Becton Dickinson).
HPLC analysis of 5mC
Preparation of genomic DNA, DNA hydrolysis and measurement of dCMP and 5me-dCMP by HPLC were performed as described (18).
LC–MS analysis of 5mC
Genomic DNA was extracted from cultured cells as described (18). The extracted DNA was digested to nucleosides with 1.0 U DNase I, 2.0 U calf intestinal phosphatase, and 0.005 U snake venom phosphodiesterase I at 37°C overnight and then subjected to LC–MS analysis as described (44).
Protein stability analysis
Measurement of UHRF1 and DNMT1 protein stability with block of new protein synthesis by cycloheximide was performed essentially as described (45).
Statistical analysis
All experiments were performed at least three independent times unless otherwise indicated. Statistical analysis and graphs were generated using excel software. All statistical analyses were performed using an unpaired/paired two sided t-test. The level of significance was set at P < 0.05, P < 0.01, P < 0.001 and P < 0.0001 for three independent experiments.
RESULTS
SET8 is required for downregulation of UHRF1 proteins in G2/M
Although UHRF1 is generally deregulated in cancer cells, it was shown previously that UHRF1 proteins were specifically detected in the S phase of normal mouse thymocytes (46). Given its essential role in DNMT1-mediated maintenance methylation, we are interested in cell-cycle-regulated expression of UHRF1 and its role in DNA methylation. We confirmed that UHRF1 protein is readily detected in S phase but not in other phases of cell cycle in NIH3T3 cells (Supplementary Figure S1A and B). Similarly, click chemistry detection of 5-ethyl-2'-deoxyuridine (EdU), a thymidine analog that was used to label S phase cells, together with immunostaining of UHRF1 revealed an elevated level of UHRF1 in the S phase of human HFL1 fibroblast cells (Figure 1A). We also enriched HFL1 cells in G1/S phase by aphidicolin treatment, in S phase by release of aphidicolin treated cells in fresh medium for 4 h, and in G2/M phase by nocodazole treatment. Subsequent western blot analysis showed a decreased level of UHRF1 in G2/M in compared to that detected in G1/S or S phase (Figure 1B). The G2/M arrest by nocodazole was evident by a substantial increase of serine 10-phosphorylated histone H3 (H3S10P). Similar results were observed in HeLa, 293T and HCT116 cells, suggesting downregulation of UHRF1 in G2/M is a general event.
Figure 1.

SET8 negatively controls the level of UHRF1 protein and is responsible for UHRF1 downregulation in G2/M. (A) UHRF1 is highly expressed in the S phase of cell cycle in human HFL1 fibroblast cells. HFL1 cells were cultured with addition of 5-ethyl-2'-deoxyuridine (EdU) for 2 h, followed by detection of EdU by click chemistry and UHRF1 by immunostaining. In a representative experiment, 65 out of 68 counted UHRF1 highly expressed cells were cells in S phase. (B) Western blot analysis of the levels of SET8, UHRF1 and DNMT1 proteins in different stages of cell cycle in four different cell lines. (C and D) Western blot and RT-qPCR analyses showing that knockdown of SET8 increased the level of UHRF1 proteins and had no effect on the level of UHRF1 mRNA in HFL1 cells (C) and HeLa cells (D). (E) Immunostaining showing that knockdown of SET8 by siRNA increased the number of non-S phase cells with highly expression of UHRF1. In a representative experiment, in the control 95 out of 100 UHRF1-highly expressed cells were in S phase, whereas only 75 out of 100 were in S phase with SET8 knockdown. (F) Western blot analysis showing that knockdown of SET8 by two different SET8-specific shRNAs impaired UHRF1 downregulation in G2/M phase of cell cycle in HFL1 and HeLa cells. (G) IP-western analysis showing an interaction between ectopically expressed UHRF1 and SET8.
To define the mechanism(s) responsible for UHRF1 downregulation in the G2/M phase, we noticed from literature that the level of SET8 proteins was low in S phase and high in G2/M (34–37), displaying an inverse correlation with that of UHRF1. Indeed, in all the cell lines examined, we observed this inverse relationship between SET8 and UHRF1 in G2/M phase (Figure 1B). We therefore investigated if SET8 could regulate UHRF1 expression. As shown in Figure 1C, knockdown of SET8 in HFL1 cells by two distinct siRNAs resulted in an elevated level of UHRF1 proteins. As reported, we observed that SET8 knockdown led to an increased G2/M population (data not shown, but see Figure 7E). Quantitative RT-PCR analysis showed that knockdown of SET8 did not affect the level of UHRF1 mRNA. Similarly, knockdown of SET8 in HeLa cells by siRNAs also resulted in an elevated level of UHRF1 proteins but not its mRNA (Figure 1D), suggesting that SET8 may downregulate UHRF1 through a post-transcriptional mechanism. We confirmed by using shRNAs and immunostaining assay that knockdown of SET8 resulted in elevated UHRF1 protein in HeLa cells (Supplementary Figure S1C).
Figure 7.
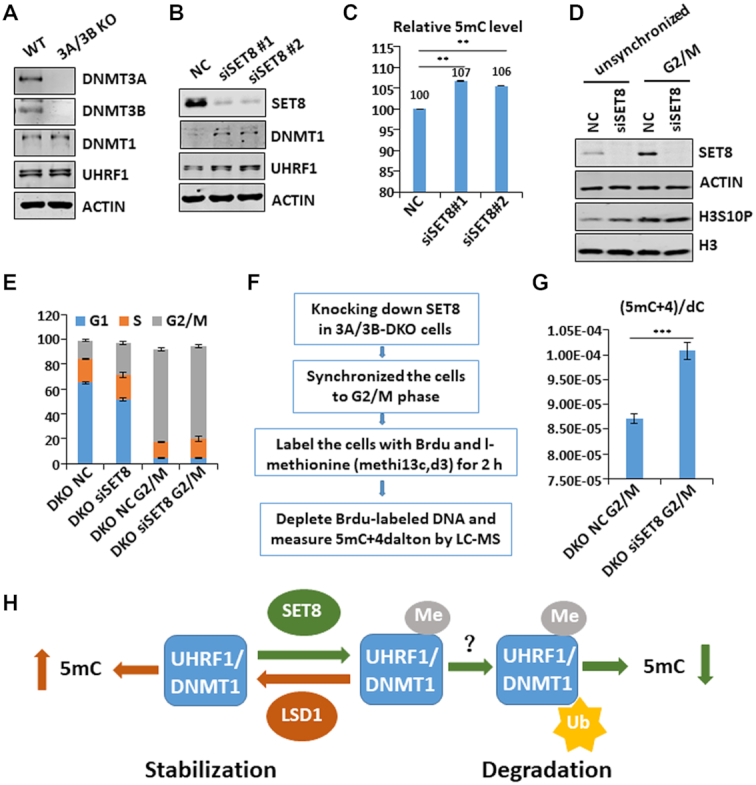
SET8 regulates global DNA methylation independent of DNMT3A/3B and suppresses DNA methylation in the G2/M phase of cell cycle. (A) Western blot analysis of DNMT3A/3B DKO HeLa cells showing that loss of DNMT3A and DNMT3B did not affect the levels of UHRF1 and DNMT1. (B) Western blot analysis showing that knockdown of SET8 in DNMT3A/3B DKO cells led to elevated levels of UHRF1 and DNMT1 proteins. (C) Quantitative HPLC analysis showing that knockdown of SET8 in DNMT3A/3B DKO cells resulted in elevated global levels of DNA methylation. **P< 0.01. (D) Western blot analysis showing successful knockdown of SET8 and enrichment of G2/M cells by nocodazole treatment. (E) Cell cycle analysis of the cells in (D) by FACS. (F) Strategy for analyzing DNA methylation in G2/M phase cells. (G) Quantitative analysis of the levels of DNA methylation in G2/M phase derived from I-methionine. (H) Working model showing that SET8 and LSD1 play an opposite role in regulating global DNA methylation and do so by controlling UHRF1 and DNMT1 protein stability through a methylation-mediated protein degradation pathway.
As SET8 is high in G2/M phase in which UHRF1 is low, we next examined if SET8 plays a role in downregulating UHRF1 in G2/M. While in control HFL1 cells nearly all UHRF1-positive cells are also the EdU-positive S phase cells (95 out of 100 cells), about 25% of UHRF1-positive cells are non-S phase cells upon SET8 knockdown (Figure 1E). By cell synchronization followed by western blot analysis, we observed that knockdown of SET8 in HFL1 cells more significantly elevated the level of UHRF1 in G2/M phase than that in S phase (Figure 1F, left panel). Similarly, knockdown of SET8 in HeLa cells also more significantly elevated the level of UHRF1 in G2/M phase (Figure 1F, right panel). These results suggest that SET8 is required for downregulation of UHRF1 in G2/M phase.
To test if SET8 interacts with UHRF1, we carried out co-immunoprecipitation (co-IP) assay with ectopically expressed proteins in HEK293T cells. The representative results in Figure 1G showed co-IP of HA-SET8 with FLAG-UHRF1. Furthermore, co-IP assay with endogenous proteins in HEK293T cell extracts confirmed an interaction between UHRF1 and SET8 (Supplementary Figure S2A).
SET8 downregulates UHRF1 through a methylation-mediated, ubiquitin-dependent degradation
We next tested if SET8 downregulates UHRF1 through a proteasome-dependent degradation pathway. To this end, we showed that ectopic overexpression of a FLAG-tagged mouse SET8 in HeLa cells markedly diminished the level of endogenous UHRF1 proteins (Figure 2A). Addition of MG132, a proteasome inhibitor, 8 h before immunostaining blocked SET8-induced UHRF1 downregulation (Figure 2A). Importantly, SET8 induced UHRF1 degradation in a methylase activity-dependent manner, as ectopic expression of SET8(1–320), a truncated and methylase-deficient SET8 mutant, did not downregulate UHRF1 (Figure 2B). We confirmed that ectopic overexpression of SET8 led to a substantial reduction of endogenous UHRF1 proteins in both HeLa and 293T cells (Figure 2C) and had no effect on the level of UHRF1 mRNA (Figure 2D). Furthermore, ectopic overexpressed SET8 downregulated UHRF1 proteins but not mRNA in a methylase activity-dependent manner, as no reduction of UHRF1 proteins was observed with truncated SET8(1–320) and a Tyr322-to-Ala SET8 mutant (SET8-Y332A) (Figure 2E and F). Like wild-type SET8, the SET8(1–320) and Y332A mutants interacted with UHRF1 in co-IP assay (Supplementary Figure S2B), suggesting that their failure to induce UHRF1 degradation is not due to impaired interaction.
Figure 2.

SET8 downregulates UHRF1 through methylation-mediated ubiquitination and proteasome degradation. (A) Immunostaining showing that addition of MG132 blocked downregulation of UHRF1 induced by ectopically expressed SET8 in HeLa cells. (B) Immunostaining showing that unlike the wild-type SET8, the SET8(1–320) mutant failed to downregulate UHRF1 when ectopically expressed in HeLa cells. (C and D) Western blot and RT-qPCR analyses showing that overexpression of SET8 diminished the level of UHRF1 proteins (C) and had no effect on the level of UHRF1 mRNA (D) in HeLa cells and HEK293T cells. (E) and (F) Western blot and RT-qPCR analyses showing that overexpression of wild-type but not the enzymatic inactive Y332A and truncated SET8 mutants diminished the level of UHRF1 proteins and had no effect on the level of UHRF1 mRNA in HEK293T cells. (G) In vitro methylation assay showing that recombinant SET8 methylated recombinant UHRF1 in a methylase activity-dependent manner. The methylation was detected by western blot using a Pan-Kme antibody. (H) Western blot analysis showing that SET8 enhanced UHRF1 methylation in a methylase activity-dependent manner. FLAG-UHRF1 was co-expressed with wild-type or SET8 mutants in HEK293T cells as indicated. FLAG-UHRF1 protein was immunoprecipitated and methylation was detected by western blotting using a Pan-Kme antibody. (I) SET8 promoted UHRF1 ubiquitination. FLAG-UHRF1 was co-expressed with or without HA-SET8 and with His-Ub. The cells were treated with MG132 for 8 h before harvest. The FLAG-UHRF1 was enriched by immunoprecipitation with anti-FLAG antibody followed by western blot analysis using anti-His antibody. (J) Western blot analysis showing that SET8 enhanced UHRF1 ubiquitination in a methylase activity-dependent manner. The experiments were performed essentially as in I).
The finding that SET8 downregulates UHRF1 proteins in a methylase activity-dependent manner raises the question if SET8 methylates UHRF1. To test if SET8 directly methylates UHRF1, we expressed and purified recombinant GST-fused UHRF1 and SET8 and its mutants from bacteria and performed in vitro methylation assay. As SET8 is known to only catalyze monomethylation on lysine, a pan-Kme antibody was used to detect potential methylation of UHRF1 by SET8. As shown in Figure 2G, we found that recombinant SET8, but not SET8(1–320) and Y332A mutants, was able to increase UHRF1 methylation. A basal level of monomethylation was detected in GST-UHRF1, presumably due to methylation during expression in E. coli. To test if SET8 promotes UHRF1 methylation in mammalian cells, we co-expressed FLAG-UHRF1 with either SET8 or its enzymatic-deficient mutants in HEK293T cells. The FLAG-UHRF1 proteins were then immunoprecipitated and the status of methylation was analyzed by western blotting. The results in Figure 2H show that co-expression of wild-type but not mutant SET8 increased the level of UHRF1 methylation. Altogether, these results indicate that SET8 is able to methylate UHRF1 both in vitro and in vivo.
As SET8 induces UHRF1 degradation in a proteasome-dependent manner, we analyzed if SET8 promotes UHRF1 ubiquitination. Indeed, ectopic co-expression of SET8 markedly promoted UHRF1 ubiquitination (Figure 2I). Consistent with a methylase-dependent degradation of UHRF1, we found that SET8 promoted UHRF1 ubiquitination in a methylase activity-dependent manner (Figure 2J).
Methylation at K385 is required for ubiquitination at K500 and UHRF1 degradation
To identify SET8-catalyzed methylation site(s) in UHRF1, we co-expressed FLAG-UHRF1 and SET8 in HEK293T cells and analyzed subsequently purified FLAG-UHRF1 by mass spectrometry (Figure 3A). This analysis allowed us to detect a momomethylation on residue K385 when co-expressed with SET8 (Figure 3B). To validate if SET8 methylates UHRF1 on K385, we generated an UHRF1 K385-to-arginine (K385R) mutant. Subsequent in vitro methylation (using FLAG-tagged proteins expressed and purified from HEK293T cells) and in vivo assays demonstrated that SET8 enhanced methylation on wild-type UHRF1 but not K385R mutant (Figure 3C and D), indicating that SET8 methylates UHRF1 primarily on K385. By dot blot analysis of control UHRF1 and K385me1 peptides, we validated that the pan-Kme antibody recognized K385me1 but not the unmodified control peptide (Supplementary Figure S2C).
Figure 3.

Identification and characterization of UHRF1 methylation and ubiquitination sites. (A) Strategy for identification of UHRF1 methylation site(s). Note that a peptide with monomethylated K385 was identified from FLAG-UHRF1 co-expressed with SET8. (B) Mass spectrometry profile of the K385me1-containing peptide. (C) In vitro methylation assay showing that SET8 methylated UHRF1 in a K385 site-dependent manner. FLAG-SET8, FLAG-UHRF1 or FLAG-UHRF1-K385R mutant was individually expressed in HEK293T cells, purified by anti-FLAG M2 beads and subjected to in vitro methylation assay. (D) SET8 enhanced UHRF1 methylation in vivo in a K385-dependent manner. Wild-type or UHRF1 K385R mutant was co-expressed with or without SET8 in HEK283T cells and the methylation on UHRF1 was detected by IP-western blot analysis. (E) Strategy and summary of UHRF1 ubiquitination sites identified by mass spectrometry. (F) Western blot analysis showing that K385A and K500A mutants were resistant to SET8-induced UHRF1 degradation. HeLa cells stably expressing FLAG-UHRF1 or various mutants were established first and then transfected with a SET8-expressing plasmid. (G) Immunostaining showing that ectopically expressed HA-SET8 downregulated wild-type FLAG-UHRF1 but not K385A and K500A mutants in the corresponding stable cell lines. (H) Ubiquitination assay showing that K385A and K500A mutants were resistant to SET8-induced ubiquitination of UHRF1. (I) Western blot analysis showing that the K385R mutant was not downregulated in G2/M phase.
As SET8 promotes UHRF1 ubiquitination and degradation, we next wished to identify potential ubiquitination site(s) on UHRF1. We treated HeLa cells with MG132 to block degradation of ubiquitinated proteins by proteasome. We then purified UHRF1 by IP and identified the ubiqutination sites by mass spectrometry using a previously established protocol (43). This effort allowed us to identify K50, K500 and K692 as sites of ubiquitination (unpublished observation) as summarized in Figure 3E.
To test the roles of aforementioned methylation and ubiquitination sites in SET8-induced degradation, we generated corresponding K to alanine (A) mutants of UHRF1 constructs and HeLa cell lines stably expressing FLAG-tagged wild-type or UHRF1 mutant proteins. Subsequent experiments showed that ectopic overexpression of SET8 resulted in marked reduction of wild-type UHRF1, K50A and K692A mutant proteins (Figure 3F). However, K385A and K500A mutant proteins were not downregulated by SET8 (Figure 3F), suggesting that K385 methylation and K500 ubiquitination are both required for SET8-induced UHRF1 degradation. In support of this idea, immunostaining experiments in Figure 3G showed that ectopic overexpression of SET8 downregulated FLAG-tagged wild-type but not K385A and K500A mutants. Furthermore, ubiquitination assay showed that co-expression of SET8 induced UHRF1 ubiquitination but not that of K385A and K500A mutants (Figure 3H), suggesting that K385 methylation by SET8 is required for subsequent K500 ubiquitination and eventually UHRF1 degradation.
To exclude the potential structural perturbation of K-to-A mutations being responsible for resistance of the K385A and K500A mutants to SET8-induced degradation, we also generated UHRF1 K385R and K500R mutants and stable HeLa cells expressing these proteins (Supplementary Figure S3A). We confirmed that ectopically expressed SET8 downregulated FLAG-UHRF1 but not K385R and K500R mutants (Supplementary Figure S3B). Furthermore, cell cycle analysis revealed that, while FLAG-tagged wild-type UHRF1 was downregulated in G2/M phase, K385R was not (Figure 3I). Finally, SET8 interacted with K385R and K500A mutants, indicating that these sites are not required for SET8 and UHRF1 interaction (Supplementary Figure S3C).
SET8 also regulates DNMT1 in a methylase activity-dependent manner
SET7/9 has been shown previously to regulate DNMT1 protein stability through direct methylation of DNMT1 (22,25). Having demonstrated that SET8 regulates UHRF1 stability, we wondered if SET8 also regulates DNMT1. As shown in Figure 4A, we found that knockdown of SET8 in HEK293T cells resulted in elevation of both UHRF1 and DNMT1 proteins but not their mRNAs. On the other hand, ectopic expression of SET8 downregulated both UHRF1 and DNMT1 proteins but not their mRNAs (Figure 4B). Like UHRF1, SET8-induced DNMT1 downregulation could be blocked by addition of MG132 (Figure 4C) and was dependent on SET8 methylase activity (Figure 4D and E). The interaction between SET8 and DNMT1 was detected by co-IP (Figure 4F). Furthermore, SET8 promoted DNMT1 methylation in a methylase activity-dependent manner (Figure 4G). Importantly, SET8 also promoted DNMT1 ubiquitination in a methylase activity-dependent manner (Figure 4H). Altogether, these data suggest that SET8 directly methylates DNMT1, which in turn leads to DNMT1 ubiquitination and subsequent degradation by proteasome.
Figure 4.

SET8 negatively regulates DNMT1 stability through methylation-mediated protein degradation. (A) Western blot and RT-qPCR analyses showing that knockdown of SET8 resulted in elevated levels of both UHRF1 and DNMT1 proteins but not mRNA in HEK293T cells. (B) Western blot and RT-qPCR analyses showing that ectopic overexpression of SET8 reduced the levels of both UHRF1 and DNMT1 proteins but not mRNA in HeLa cells. (C) Immunostaining assay showing that addition of MG132 blocked SET8-induced downregulation of DNMT1 in HeLa cells. (D) Western blot analysis showing that ectopic overexpression of SET8 reduced the levels of both UHRF1 and DNMT1 proteins in a methylase activity-dependent manner. (E) Immunostaining assay showing that ectopic overexpression of SET8 reduced the level of DNMT1 proteins in a methylase activity-dependent manner. (F) IP-western blot analysis showing that SET8 interacted with both UHRF1 and DNMT1. (G) IP-western blot analysis showing that SET8 promoted DNMT1 methylation in a methylase activity-dependent manner. (H) IP-western blot analysis showing that SET8 promoted DNMT1 ubiquitination in a methylase activity-dependent manner.
LSD1 stabilizes both UHRF1 and DNMT1
As lysine methylation is often dynamically controlled by methylases and demethylases (47), we tested if LSD1, which often demethylates mono- and di-methylated proteins including DNMT1 (48,49), demethylates and stabilizes UHRF1. We found that knockdown of LSD1 in either HeLa or HCT116 cells by siRNAs markedly reduced the level of UHRF1 proteins (Figure 5A) but not its mRNA (Figure 5B). Consistent with a previous report (49), we found that knockdown of LSD1 also markedly reduced the level of DNMT1 proteins (Figure 5A) and had no effect on DNMT1 mRNA level (Figure 5B). By immunostaining we confirmed that transfection of HeLa cells with two distinct LSD1-specific shRNAs resulted in marked reduction of DNMT1 proteins (Figure 5C) and UHRF1 proteins (Figure 5D). To examine this further, we generated LSD1-knockout HeLa cell lines by CRISPR-Cas9 technology (Supplementary Figure S4A). A successful disruption of both alleles of LSD1 gene was validated in two independent clones by sequencing of genomic DNA (Supplementary Figure S4B) and by western blot analysis (Figure 5E). Interestingly, we found that, unlike the situation in acute LSD1 knockdown by siRNAs, LSD1 knockout only slightly reduced the global level of DNMT1 and UHRF1 proteins in HeLa cells (Figure 5E). However, protein stability analysis revealed a substantially reduced half-life of both DNMT1 and UHRF1 in LSD1 knockout cells (Figure 5F). This paradox could be explained by a ∼2-fold increase of UHRF1 and DNMT1 mRNAs in LSD1-KO cells, likely as a compensatory mechanism for unstable UHRF1 and DNMT1 proteins (Supplementary Figure S4C). Together these data indicate that, in contrast to SET8, the demethylase LSD1 acts to stabilize both UHRF1 and DNMT1.
Figure 5.

LSD1 protects both UHRF1 and DNMT1 from degradation. (A) Western blot analysis showing that knockdown of LSD1 markedly downregulated the levels of both UHRF1 and DNMT1 proteins in HeLa and HCT116 cells. (B) RT-qPCR analysis showing that knockdown of LSD1 did not affect the levels of both UHRF1 and DNMT1 mRNAs in HeLa and HCT116 cells. (C and D) Immunostaining assay showing that knockdown of LSD1 by shRNA transfection significantly reduced the level of DNMT1 (C) and UHRF1 proteins (D) in HeLa cells. (E) Western blot analysis of DNMT1, UHRF1 and DNMT3A in control and two LSD1-knockout HeLa cell lines. (F) Protein stability analysis showing a reduced half-life for UHRF1 and DNMT1 in LSD1-knockout cells. Note that p53 is very unstable compared to UHRF1 and DNMT1. (G) IP-western blot analysis showing that LSD1 was able to demethylate UHRF1 in a demethylase activity-dependent manner. (H) IP-western analysis showing that endogenous LSD1 and UHRF1 in HEK293T nuclear extracts interacted. (I) IP-western analysis showing that ectopically expressed LSD1 interacted with both DNMT1 and UHRF1 in a similar extent.
As LSD1 was reported to stabilize DNMT1 by demethylation (49), we examined if LSD1 could also demethylate UHRF1. We co-expressed FLAG-UHRF1 with Myc-tagged wild-type LSD1 or mutants (N535A and K661A) deficient for demethylase activity, and examined their effect on UHRF1 methylation by western blot analysis using Pan-Kme antibody. As shown in Figure 5G, co-expression of wild-type LSD1 but not the mutants reduced the level of UHRF1 methylation. We thus conclude that LSD1 most likely stabilizes UHRF1 through its ability to demethylate UHRF1. Consistent with this idea, a protein-protein interaction between UHRF1 and LSD1 could be detected for both endogenous UHRF1 and LSD1 (Figure 5H) and ectopically expressed proteins (Figure 5I) by co-IP. In addition, the interaction between LSD1 and UHRF1 is comparable to that detected between LSD1 and DNMT1 (Figure 5I).
SET8 and LSD1 oppositely control the global level of DNA methylation
Having established that SET8 and LSD1 oppositely regulate protein stability of UHRF1 and DNMT1, we next determined if they also oppositely regulate the global level of DNA methylation. In this regard, targeted deletion of LSD1 in mice resulted in embryonic lethality and impaired global DNA methylation (49). To test if SET8 regulates DNA methylation, we first ectopically expressed wild-type and SET8 mutants in HeLa cells and examined their effect on global DNA methylation by immunostaining using anti-5mC antibody. The representative results in Figure 6A showed a significant reduction of DNA methylation in cells expressing wild-type SET8. However, no reduction in DNA methylation was observed in cells expressing mutant SET8. Thus, SET8 inhibits DNA methylation and this activity correlates with its ability to induce UHRF1 and DNMT1 degradation. To test if endogenous SET8 regulates DNA methylation, we knocked down SET8 in HEK293T cells by siRNAs and measured the level of 5mC in genomic DNA by quantitative HPLC analysis three days after transfection. Representative results showed that knockdown of SET8 not only led to an increase of UHRF1 and DNMT1 proteins (Figure 6B, left panel), but also an ∼9–12% increase of the 5mC level (Figure 6B, right panel). Similarly, knockdown of SET8 in HeLa cells led to an increase of both UHRF1 and DNMT1 proteins and increase of global level of 5mC (Figure 6C). An increased level of 5mC in SET8 knockdown cells was also confirmed by measurement of 5mC using LC–MS (Supplementary Figure S5A and B). We also observed by immunostaining that knockdown of SET8 in NIH3T3 cells by transfected SET8-specific shRNAs led to increased DNA methylation (Supplementary Figure S5c). Thus, endogenous SET8 has a role in suppression of global level of DNA methylation.
Figure 6.

Both SET8 and LSD1 play a role in control of global DNA methylation. (A) Immunostaining assay using anti-5mC antibody showing that ectopic overexpression of SET8 impaired the global level of DNA methylation in a methylase activity-dependent manner. (B) Quantitative HPLC analysis showing that knockdown of SET8 in HEK293T cells resulted in elevated global levels of DNA methylation (right panel). Western blot analysis in the left panel validated that knockdown of SET8 resulted in elevated levels of UHRF1 and DNMT1 proteins. *P< 0.05. (C) Quantitative HPLC analysis showing that knockdown of SET8 in HeLa cells resulted in elevated global levels of DNA methylation (right panel). Western blot analysis in the left panel showing that knockdown of SET8 resulted in elevated levels of UHRF1 and DNMT1 proteins. **P< 0.01; ***P< 0.001. (D) Quantitative HPLC analysis showing that ectopic overexpression of LSD1 but not its demethylase inactive mutant in HEK293T cells resulted in elevated global levels of DNA methylation (right panel). Western blot analysis in the left panel showing that ectopic overexpression of LSD1 but not its mutant elevated the levels of UHRF1 and DNMT1 proteins. *P< 0.05. (E) Quantitative HPLC analysis showing that knockdown of LSD1 in HeLa and HCT116 cells resulted in substantial reduction of global DNA methylation (right panel). Western blot analysis in the left panel validated that knockdown of LSD1 resulted in substantial reduction of both UHRF1 and DNMT1 proteins. *P< 0.05, **P< 0.01; ***P< 0.001. (F) Quantitative HPLC analysis showing reduced global levels of DNA methylation in LSD1 knockout HeLa cell lines. *P< 0.05.
To examine whether LSD1 also regulates global DNA methylation under the same context, we first ectopically overexpressed wild-type LSD1 and enzymatic inactive K661A mutant in HEK293T cells. We found that expression of wild-type LSD1 increased the level of UHRF1 and DNMT1 proteins, whereas expression of K661A mutant had no effect (Figure 6D, left panel), suggesting that LSD1 stabilizes both UHRF1 and DNMT1 through its demethylase activity. Measurement of the level of 5mC showed that overexpression of wild-type LSD1 but not K661A mutant enhanced the global level of DNA methylation (Figure 6D, right panel). Consistent with a role of LSD1 in enhancing global DNA methylation, we found that knockdown of LSD1 reduced the level of 5mC in both HeLa and HCT116 cells (Figure 6E). Reduced DNA methylation was also detected in two independent LSD1 knockout HeLa cell lines (Figure 6F), although to a less extent, possibly due to a less severe reduction of UHRF1 and DNMT1 proteins in LSD1-KO cells (Figure 5E).
SET8 and LSD1 control global DNA methylation most likely via regulation of UHRF1 than DNMT1
As both UHRF1 and DNMT1 are regulated by SET8 and LSD1, it raises the question if UHRF1 or DNMT1 is the primary target for regulation of global DNA methylation. A previous study indicates that DNMT1 is often excessively expressed and UHRF1 could be a more sensitive target for DNA demethylation in cancer cells (27). To determine how the levels of UHRF1 and DNMT1 proteins influences DNA methylation, we progressively knocked down the expression of UHRF1 or DNMT1 by using an increasing concentration of UHRF1- or DNMT1-specific siRNA and determined the levels of 5mC by HPLC analysis. In agreement with the previous report, we observed that, while increased knockdown of DNMT1 and UHRF1 in HeLa cells was associated with progressively reduced DNA methylation, DNA methylation is more sensitive to reduction of UHRF1 than DNMT1 (Supplementary Figure S6A). This is not unique to HeLa cells, as the same results were observed in MCF7 cells (Supplementary Figure S6B) and HCT116 cells (data not shown).
SET8 controls DNA methylation independent of DNMT3A/3B
Although knockdown of SET8 did not affect the level of DNMT3A and DNMT3B proteins, we could not completely rule out the possibility that SET8 may regulate global DNA methylation via controlling DNMT3A/3B activity. To test this possibility, we generated a DNMT3A/3B double knockout HeLa cell line by CRISPR-Cas9 (Figure 7A). Like the parental cells, knockdown of SET8 in DNMT3A/3B-DKO cells led to increased levels of UHRF1 and DNMT1 proteins (Figure 7B), whereas ectopic expression of SET8 downregulated UHRF1 and DNMT1 in a methylase activity-dependent manner (Supplementary Figure S7A and B). Importantly, measurement of 5mC by quantitative HPLC analysis demonstrated that knockdown of SET8 in DNMT3A/3B DKO cells led to increased DNA methylation comparable to that observed in parental cells (Figure 7C). Thus, SET8 regulates global DNA methylation independent of DNMT3A/3B.
Downregulation of UHRF1 in G2/M phase by SET8 suppresses post-replication DNA methylation
As UHRF1 is downregulated in G2/M phase by SET8, we hypothesized this downregulation would potentially suppress post-replication DNA methylation by DNMT1. As shown recently, despite being mainly a maintenance methylation enzyme, DNMT1 possesses de novo methylase activity that should be tightly controlled to ensure accurate DNA methylation inheritance. To interrogate the potential physiological function of downregulated UHRF1 in G2/M phase, we determined if disruption of this regulation affects DNA methylation in G2/M phase by DNMT1. We first established conditions to knock down SET8 in DNMT3A/3B-DKO cells and enriched G2/M population by nocodazole treatment (Figure 7D). The resulting G2/M arrest was confirmed by FACS analysis (Figure 7E). To specifically measure DNA methylation in G2/M phase, we devised a protocol as illustrated in Figure 7F, in which DNMT3A/3B-DKO cells were treated without or with siSET8 and arrested in the G2/M phase. The cells were then cultured in medium with 5-bromo-2′-deoxyuridine (BrdU) and I-methionine, which could be converted to the methyl donor SAM with an increase +4 Da in mass, for 2 h. Genomic DNA was then prepared and sheared to 0.5–1 kb fragments by sonication. The addition of BrdU allowed us to deplete DNA derived from a small fraction of S phase cells in the G2/M-arrested cells. G2/M-specific DNA methylation was then measured by quantification of 5mC+4 Da nucleotide by LC–MS. As shown in Figure 7G, we found that knockdown of SET8 led to a consistent 15% increase of DNA methylation in the G2/M phase.
DISCUSSION
Within the DNA maintenance methylation axis consisting of DNMT1 and UHRF1, it was shown previously that DNMT1 protein stability is regulated by dynamic methylation and demethylation by SET7/9 and LSD1, respectively (22,49). This regulation is believed to be critical for DNA methylation, as LSD1-knockout mice are embryonic lethal and correlate with reduced DNMT1 protein and impaired global DNA methylation (49). However, the SET7/9-null mice are viable and not known for defect in DNA methylation (50,51). Here, we show that both UHRF1 and DNMT1 are regulated by SET8 through a methylation-mediated protein degradation pathway. Importantly, SET8 has a role in controlling the global level of DNA methylation and is responsible for down-regulation of UHRF1 in the G2/M phase of cell cycle. Furthermore, we show that LSD1 also regulates UHRF1 protein stability. As DNA methylation is more sensitive to reduction of UHRF1 proteins than that of DNMT1, our data suggest that SET8 and LSD1 are more likely to oppositely control the global level of DNA methylation through dynamic methylation of UHRF1.
Although the initial idea that SET8 might regulate UHRF1 was based on their opposite expression patterns in the cell cycle (34,35,37,46) (Figure 1), SET8 also emerged as a hit in our screening for lysine methylases that were able to downregulate UHRF1 by immunostaining assay (data not shown). We validated by both gain and loss of functional assays that SET8 downregulates UHRF1 through proteasome-mediated degradation in a methylation-dependent manner (Figures 1 and 2). We identified K385 as the site for SET8-catalyzed UHRF1 methylation by mass spectrometry. Although our significant efforts have so far failed to generate a UHRF1 K385me1-specific antibody, a Pan-Kme antibody is able to recognize this modification. Because SET8 failed to promote UHRF1 methylation both in vivo and in vitro when K385 was mutated, K385 is the primary, if not the sole, site for SET8-catalyzed UHRF1 methylation. Since both K385A and K385R UHRF1 mutants were resistant to SET8-induced degradation, K385 methylation is clearly required for SET8-induced UHRF1 degradation. Furthermore, as the K385R mutant was not downregulated in G2/M phase, it implies that SET8 is primarily responsible for downregulation of UHRF1 in the G2/M phase of cell cycle.
Consistent with a proteasome-dependent degradation of UHRF1, we show that SET8 promotes UHRF1 ubiquitination in a methylase activity-dependent manner (Figure 2J). However, how K385 methylation promotes UHRF1 ubiquitination is currently unknown. It does not appear to promote UHRF1 intrinsic ubiquitin E3 ligase activity, as SET8 is able to induce degradation of E3-deficient UHRF1 (data not shown). In addition, it does not appear to depend on L3MBTL3 (data not shown), a methyl-binding protein that has been shown to mediate degradation of methylated DNMT1 (52). Previous study has revealed a role of USP7 in promoting UHRF1 stability and the M phase specific-kinase CDK1-cyclin B in promoting UHRF1 degradation by phosphorylating S652 of UHRF1 (53). However, SET8 does not appear to promote UHRF1 degradation by inhibiting the interaction between UHRF1 and USP7 (Supplementary Figure S8A). Furthermore, SET8-induced UHRF1 degradation and its interaction with UHRF1 are independent of UHRF1 S652 phosphorylation (Supplementary Figure S8B and C). Thus, future work is needed to elucidate how K385 methylation induces UHRF1 ubiquitination.
As SET8 also methylates DNMT1 and induces DNMT1 degradation through proteasome in a methylase activity-dependent manner, we believe that methylation by SET8 is likely to promote DNMT1 ubiquitination and degradation. SET7/9 has been shown to induce DNMT1 degradation by methylating human DNMT1 at K142 (25) and K1094 (49). Previous studies have identified methyl degrons in multiple proteins including EZH2 (54) and SOX2 (55). In both cases, site-specific methylation in these proteins generates a methyl degron, which recruits a unique E3 ligase or complex to promote ubiquitination and proteasome-mediated degradation (56). It is of interest to determine whether a similar mechanism is involved in SET8-induced degradation of UHRF1 and DNMT1.
In agreement with the general concept that lysine methylation is dynamic, we find that LSD1 stabilizes UHRF1 by demethylation of UHRF1 (Figure 5). Furthermore, SET8 and LSD1 oppositely regulate global DNA methylation (Figure 6). This finding raises the question if LSD1 controls global DNA methylation through its regulation of DNMT1 or UHRF1 or both. In our gain and loss of functional assays, we found that LSD1 regulates the levels of DNMT1 and UHRF1 proteins to a similar extent. In fact, we found that SET8 also regulates both DNMT1 and UHRF1 to a similar extent. Given that knockdown of SET8 did not affect the levels of DNMT3A and DNMT3B and that knockdown of SET8 elevated global DNA methylation in DNMT3A/3B-DKO cells (Figure 7), we conclude that SET8 controls the global level of DNA methylation through regulating UHRF1 and DNMT1, the axis of maintenance methylation. In this regard, LSD1 and SET8 are well suited for regulating the global level of DNA methylation, with their opposite function in control of UHRF1 and DNMT1 protein stability. However, for the following reasons, we believe that both SET8 and LSD1 are most likely to control global DNA methylation through regulation of UHRF1 than DNMT1. First, we found that the global level of DNA methylation is more sensitive to the change of UHRF1 proteins than DNMT1 in two different cell lines (Supplementary Figure S6). This finding is consistent with an elegant study by Baylin and his colleagues, showing that moderate reduction of UHRF1 but not DNMT1 can lead to reduction of global DNA methylation (27). Second, under the physiological condition UHRF1 is likely more sensitive to changes of cellular levels of SET8 proteins than DNMT1, as the level of UHRF1 proteins but not DNMT1 inversely correlates with that of SET8 in cell cycle (Figure 1).
In principle, accurate inheritance of DNA methylation requires not only a stringent fidelity of DNA maintenance methylation but also suppression of do novo methylation (5). Being a maintenance enzyme with strong preference for hemi-methylated CpG sites, DNMT1 also possesses de novo methylase activity in vitro and in vivo (57–59). A recent study demonstrated that Stella is required to safeguard the oocyte methylome by preventing de novo methylation mediated by DNMT1 (29), providing a clear example for the functional importance of suppressing DNMT1 de novo methylase activity in promoting accurate inheritance of DNA methylation. Although it is well documented that DNA maintenance methylation by UHRF1/DNMT1 axis is coupled to DNA replication, recent studies revealed a fraction of DNMT1-mediated methylation is delayed post-replication for hours (60). This observation raises the question if DNMT1 also catalyzes DNA methylation in G2/M, presumably due to its de novo activity. As UHRF1 is downregulated by SET8 in G2/M, we are particularly interested in knowing whether this downregulation contributes to global control of DNA methylation by SET8. We designed experiments to specifically measure DNA methylation in G2/M and we found that knockdown of SET8 increases DNA methylation in G2/M by ∼15% (Figure 7). Thus, we speculate that downregulation of UHRF1 by SET8 in G2/M phase is likely to prevent excessive DNA methylation by suppressing de novo methylation by DNMT1.
In sum, we propose a working model in Figure 7H that SET8 and LSD1 oppositely regulate global DNA methylation by regulating the axis of maintenance methylation, especially UHRF1, through control of their protein stability. This regulation has a role in preventing excessive DNA methylation, thus ensuring accurate inheritance of DNA methylome across cell division. As disruption of DNA methylation is a vital approach for clinical treatment of cancer, our study suggests compounds targeting UHRF1 could be promising for cancer therapy.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Cheng-Ming Chiang (University of Texas Southwestern Medical Center) for critical reading of the manuscript. We thank members of Wong's laboratory for valuable discussion.
Author contributions: J.L. and J.W. conceived and supervised the study. H.Z., Q.G., S.T., J.Y., M.H.and J.L. performed essentially all biochemical, cellular and molecular experiments. C.L. and H.W. performed 5mC measurement by LC–MS. Y.Z. and L.L. carried out identification of UHRF1-methylation sites by mass spectrometry, J.Q. performed identification of Ub-modified sites in UHRF1. H.Z., J. Li and J.W. wrote the manuscript with assistant from Q.G.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Science and Technology of China (MOST) [2017YFA054201 to J.W.]; National Natural Science Foundation of China [31730048, 81530078 and 31571325 to J.W.]. Funding for open access charge: MOST.
Conflict of interest statement. None declared.
REFERENCES
- 1. Law J.A., Jacobsen S.E.. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. 2010; 11:204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen T., Li E.. Establishment and maintenance of DNA methylation patterns in mammals. Curr. Top. Microbiol. Immunol. 2006; 301:179–201. [DOI] [PubMed] [Google Scholar]
- 3. Suzuki M.M., Bird A.. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. 2008; 9:465–476. [DOI] [PubMed] [Google Scholar]
- 4. Xu G.L., Wong J.M.. Oxidative DNA demethylation mediated by Tet enzymes. Natl. Sci. Rev. 2015; 2:318–328. [Google Scholar]
- 5. Jones P.A., Liang G.. Rethinking how DNA methylation patterns are maintained. Nat. Rev. 2009; 10:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen T., Li E.. Structure and function of eukaryotic DNA methyltransferases. Curr. Top. Dev. Biol. 2004; 60:55–89. [DOI] [PubMed] [Google Scholar]
- 7. Gujar H., Weisenberger D.J., Liang G.. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes (Basel). 2019; 10:10020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bostick M., Kim J.K., Esteve P.O., Clark A., Pradhan S., Jacobsen S.E.. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007; 317:1760–1764. [DOI] [PubMed] [Google Scholar]
- 9. Sharif J., Muto M., Takebayashi S., Suetake I., Iwamatsu A., Endo T.A., Shinga J., Mizutani-Koseki Y., Toyoda T., Okamura K. et al.. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007; 450:908–912. [DOI] [PubMed] [Google Scholar]
- 10. Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M.. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008; 455:818–821. [DOI] [PubMed] [Google Scholar]
- 11. Avvakumov G.V., Walker J.R., Xue S., Li Y., Duan S., Bronner C., Arrowsmith C.H., Dhe-Paganon S.. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008; 455:822–825. [DOI] [PubMed] [Google Scholar]
- 12. Hashimoto H., Horton J.R., Zhang X., Bostick M., Jacobsen S.E., Cheng X.. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008; 455:826–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karagianni P., Amazit L., Qin J., Wong J.. ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol. Cell Biol. 2008; 28:705–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nady N., Lemak A., Walker J.R., Avvakumov G.V., Kareta M.S., Achour M., Xue S., Duan S., Allali-Hassani A., Zuo X. et al.. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J. Biol. Chem. 2011; 286:24300–24311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arita K., Isogai S., Oda T., Unoki M., Sugita K., Sekiyama N., Kuwata K., Hamamoto R., Tochio H., Sato M. et al.. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:12950–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu X., Gao Q., Li P., Zhao Q., Zhang J., Li J., Koseki H., Wong J.. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 2013; 4:1563. [DOI] [PubMed] [Google Scholar]
- 17. Rothbart S.B., Dickson B.M., Ong M.S., Krajewski K., Houliston S., Kireev D.B., Arrowsmith C.H., Strahl B.D.. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013; 27:1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao Q., Zhang J., Chen R., Wang L., Li B., Cheng H., Duan X., Zhu H., Wei W., Li J. et al.. Dissecting the precise role of H3K9 methylation in crosstalk with DNA maintenance methylation in mammals. Nat. Commun. 2016; 7:12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nishiyama A., Yamaguchi L., Sharif J., Johmura Y., Kawamura T., Nakanishi K., Shimamura S., Arita K., Kodama T., Ishikawa F. et al.. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 2013; 502:249–253. [DOI] [PubMed] [Google Scholar]
- 20. Qin W., Wolf P., Liu N., Link S., Smets M., La Mastra F., Forne I., Pichler G., Horl D., Fellinger K. et al.. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015; 25:911–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xie S., Qian C.. The Growing Complexity of UHRF1-Mediated Maintenance DNA Methylation. Genes (Basel). 2018; 9:9120600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Esteve P.O., Chin H.G., Benner J., Feehery G.R., Samaranayake M., Horwitz G.A., Jacobsen S.E., Pradhan S.. Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:5076–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Du Z., Song J., Wang Y., Zhao Y., Guda K., Yang S., Kao H.Y., Xu Y., Willis J., Markowitz S.D. et al.. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal. 2010; 3:ra80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bronner C. Control of DNMT1 abundance in epigenetic inheritance by acetylation, ubiquitylation, and the histone code. Sci. Signal. 2011; 4:e3. [DOI] [PubMed] [Google Scholar]
- 25. Esteve P.O., Chang Y., Samaranayake M., Upadhyay A.K., Horton J.R., Feehery G.R., Cheng X., Pradhan S.. A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat. Struct. Mol. Biol. 2011; 18:42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Di Ruscio A., Ebralidze A.K., Benoukraf T., Amabile G., Goff L.A., Terragni J., Figueroa M.E., De Figueiredo Pontes L.L., Alberich-Jorda M., Zhang P. et al.. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature. 2013; 503:371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cai Y., Tsai H.C., Yen R.C., Zhang Y.W., Kong X., Wang W., Xia L., Baylin S.B.. Critical threshold levels of DNA methyltransferase 1 are required to maintain DNA methylation across the genome in human cancer cells. Genome Res. 2017; 27:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. von Meyenn F., Iurlaro M., Habibi E., Liu N.Q., Salehzadeh-Yazdi A., Santos F., Petrini E., Milagre I., Yu M., Xie Z. et al.. Impairment of DNA methylation maintenance is the main cause of global demethylation in naive embryonic stem cells. Mol. Cell. 2016; 62:848–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Y.F., Zhang Z.Q., Chen J.Y., Liu W.Q., Lai W.Y., Liu B.D., Li X., Liu L.P., Xu S.H., Dong Q. et al.. Stella safeguards the oocyte methylome by preventing de novo methylation mediated by DNMT1. Nature. 2018; 564:136–140. [DOI] [PubMed] [Google Scholar]
- 30. Ashraf W., Ibrahim A., Alhosin M., Zaayter L., Ouararhni K., Papin C., Ahmad T., Hamiche A., Mely Y., Bronner C. et al.. The epigenetic integrator UHRF1: on the road to become a universal biomarker for cancer. Oncotarget. 2017; 8:51946–51962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fang J., Feng Q., Ketel C.S., Wang H., Cao R., Xia L., Erdjument-Bromage H., Tempst P., Simon J.A., Zhang Y.. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol. 2002; 12:1086–1099. [DOI] [PubMed] [Google Scholar]
- 32. Nishioka K., Rice J.C., Sarma K., Erdjument-Bromage H., Werner J., Wang Y., Chuikov S., Valenzuela P., Tempst P., Steward R. et al.. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell. 2002; 9:1201–1213. [DOI] [PubMed] [Google Scholar]
- 33. Shi X., Kachirskaia I., Yamaguchi H., West L.E., Wen H., Wang E.W., Dutta S., Appella E., Gozani O.. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell. 2007; 27:636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Centore R.C., Havens C.G., Manning A.L., Li J.M., Flynn R.L., Tse A., Jin J., Dyson N.J., Walter J.C., Zou L.. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell. 2010; 40:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abbas T., Shibata E., Park J., Jha S., Karnani N., Dutta A.. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell. 2010; 40:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oda H., Hubner M.R., Beck D.B., Vermeulen M., Hurwitz J., Spector D.L., Reinberg D.. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol. Cell. 2010; 40:364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu S., Wang W., Kong X., Congdon L.M., Yokomori K., Kirschner M.W., Rice J.C.. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 2010; 24:2531–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang J., Gao Q., Li P., Liu X., Jia Y., Wu W., Li J., Dong S., Koseki H., Wong J.. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 2011; 21:1723–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang Z., Jiang J., Stewart M.D., Qi S., Yamane K., Li J., Zhang Y., Wong J.. AOF1 is a histone H3K4 demethylase possessing demethylase activity-independent repression function. Cell Res. 2010; 20:276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu F., Shi G., Cheng S., Chen J., Wu S.Y., Wang Z., Xia N., Zhai Y., Wang Z., Peng Y. et al.. SUMO suppresses and MYC amplifies transcription globally by regulating CDK9 sumoylation. Cell Res. 2018; 28:670–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang K., Williams K.E., Huang L., Yau P., Siino J.S., Bradbury E.M., Jones P.R., Minch M.J., Burlingame A.L.. Histone acetylation and deacetylation: identification of acetylation and methylation sites of HeLa histone H4 by mass spectrometry. Mol. Cell Proteomics. 2002; 1:500–508. [DOI] [PubMed] [Google Scholar]
- 43. Shi Y., Chan D.W., Jung S.Y., Malovannaya A., Wang Y., Qin J.. A data set of human endogenous protein ubiquitination sites. Mol. Cell Proteomics. 2011; 10:M110 002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yin R., Mao S.Q., Zhao B., Chong Z., Yang Y., Zhao C., Zhang D., Huang H., Gao J., Li Z. et al.. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013; 135:10396–10403. [DOI] [PubMed] [Google Scholar]
- 45. Ding G., Chen P., Zhang H., Huang X., Zang Y., Li J., Li J., Wong J.. Regulation of Ubiquitin-like with Plant Homeodomain and RING Finger Domain 1 (UHRF1) Protein Stability by Heat Shock Protein 90 Chaperone Machinery. J. Biol. Chem. 2016; 291:20125–20135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fujimori A., Matsuda Y., Takemoto Y., Hashimoto Y., Kubo E., Araki R., Fukumura R., Mita K., Tatsumi K., Muto M.. Cloning and mapping of Np95 gene which encodes a novel nuclear protein associated with cell proliferation. Mamm. Genome. 1998; 9:1032–1035. [DOI] [PubMed] [Google Scholar]
- 47. Pradhan S., Chin H.G., Esteve P.O., Jacobsen S.E.. SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics. 2009; 4:383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nicholson T.B., Chen T.. LSD1 demethylates histone and non-histone proteins. Epigenetics. 2009; 4:129–132. [DOI] [PubMed] [Google Scholar]
- 49. Wang J., Hevi S., Kurash J.K., Lei H., Gay F., Bajko J., Su H., Sun W., Chang H., Xu G. et al.. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009; 41:125–129. [DOI] [PubMed] [Google Scholar]
- 50. Campaner S., Spreafico F., Burgold T., Doni M., Rosato U., Amati B., Testa G.. The methyltransferase Set7/9 (Setd7) is dispensable for the p53-mediated DNA damage response in vivo. Mol. Cell. 2011; 43:681–688. [DOI] [PubMed] [Google Scholar]
- 51. Lehnertz B., Rogalski J.C., Schulze F.M., Yi L., Lin S., Kast J., Rossi F.M.. p53-dependent transcription and tumor suppression are not affected in Set7/9-deficient mice. Mol. Cell. 2011; 43:673–680. [DOI] [PubMed] [Google Scholar]
- 52. Leng F., Yu J., Zhang C., Alejo S., Hoang N., Sun H., Lu F., Zhang H.. Methylated DNMT1 and E2F1 are targeted for proteolysis by L3MBTL3 and CRL4(DCAF5) ubiquitin ligase. Nat. Commun. 2018; 9:1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ma H., Chen H., Guo X., Wang Z., Sowa M.E., Zheng L., Hu S., Zeng P., Guo R., Diao J. et al.. M phase phosphorylation of the epigenetic regulator UHRF1 regulates its physical association with the deubiquitylase USP7 and stability. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:4828–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee J.M., Lee J.S., Kim H., Kim K., Park H., Kim J.Y., Lee S.H., Kim I.S., Kim J., Lee M. et al.. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell. 2012; 48:572–586. [DOI] [PubMed] [Google Scholar]
- 55. Fang L., Zhang L., Wei W., Jin X., Wang P., Tong Y., Li J., Du J.X., Wong J.. A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol. Cell. 2014; 55:537–551. [DOI] [PubMed] [Google Scholar]
- 56. Yang Y., Bedford M.T.. Titivated for destruction: the methyl degron. Mol. Cell. 2012; 48:487–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pradhan S., Bacolla A., Wells R.D., Roberts R.J.. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 1999; 274:33002–33010. [DOI] [PubMed] [Google Scholar]
- 58. Hermann A., Goyal R., Jeltsch A.. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004; 279:48350–48359. [DOI] [PubMed] [Google Scholar]
- 59. Arand J., Spieler D., Karius T., Branco M.R., Meilinger D., Meissner A., Jenuwein T., Xu G., Leonhardt H., Wolf V. et al.. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012; 8:e1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Charlton J., Downing T.L., Smith Z.D., Gu H., Clement K., Pop R., Akopian V., Klages S., Santos D.P., Tsankov A.M et al.. Global delay in nascent strand DNA methylation. Nat. Struct. Mol. Biol. 2018; 25:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.