

Abstract
Recurrent clonal genetic alterations are the hallmark of Acute Lymphoblastic Leukemia (ALL) and govern the risk stratification, response to treatment and clinical outcome. In this retrospective study conducted on ALL patient samples, the purpose was to estimate the copy number alterations (CNAs) in ALL by digitalMLPA (dMLPA), validation of the dMLPA data by conventional MLPA and RT-PCR, and correlation of CNAs with Minimal Residual Disease (MRD) status. The ALL patient samples (n = 151; B-ALL, n = 124 cases and T-ALL, n = 27 cases) were assessed for CNAs by dMLPA for detection of sub-microscopic CNAs and ploidy status. This assay allowed detection of ploidy changes and CNAs by multiplexing of karyotyping probes and probes covering 54 key gene targets implicated in ALL. Using the dMLPA assay, CNAs were detected in ~89% (n = 131) of the cases with 66% of the cases harboring ≥3 CNAs. Deletions in CDKN2A/B, IKZF1, and PAX5 genes were detectable in a quarter of these cases. Heterozygous and homozygous gene deletions, and duplications were observed in genes involved in cell cycle control, tumor suppression, lineage differentiation, lymphoid signaling, and transcriptional regulators with implications in treatment response and survival outcome. Distinct CNAs profiles were evident in B-ALL and T-ALL cases. Additionally, the dMLPA assay could reliably identify ploidy status and copy number-based gene fusions (SIL-TAL1, NUP214-ABL, EBF1-PDGFRB). Cases of B-ALL with no detectable recurrent genetic abnormalities could potentially be risk stratified based on the CNA profile. In addition to the commonly used gene deletions for risk assessment (IKZF1, EBF1, CDKN2A/B), we identified a broader spectrum of gene alterations (gains of- RUNX1, LEF1, NR3C2, PAR1, PHF6; deletions of- NF1, SUZ12, MTAP) that significantly correlated with the status of MRD clearance. The CNAs detected by dMLPA were validated by conventional MLPA and showed high concordance (r = 0.99). Our results demonstrated dMLPA to be a robust and reliable alternative for rapid detection of key CNAs in newly diagnosed ALL patients. Integration of ploidy status and CNAs detected by dMLPA with cytogenetic and clinical risk factors holds great potential in further refinement of patient risk stratification and response to treatment in ALL.
Keywords: copy number alterations, CNAs, B-cell acute lymphoblastic leukemia, T-cell acute lymphoblastic leukemia, digital multiplex ligation-dependent probe amplification, dMLPA, MLPA, MRD
Introduction
Acute lymphoblastic leukemia (ALL), the most common childhood cancer manifests as a clinically and genetically heterogenous malignancy. Both B-cell and T-cell ALL are primarily initiated by recurrent chromosomal translocations and driven by accumulation of distinct constellations of gross and sub-microscopic somatic genetic alterations including copy number alterations (CNAs), aneuploidy, structural variants, and DNA sequence mutations (1–4). Several of these lesions are important determinants of the risk of treatment failure and disease relapse but a universal agreement for inclusion of these genetic alterations in clinical risk stratification is still lacking. With current treatment protocols, long-term survival approaches 90%; however, a fraction of patients with favorable genetics relapse and refractory disease still poses a significant clinical challenge due to resistance of the recurrent disease to chemotherapy. Therefore, a more robust comprehensive genetic profiling can potentially facilitate better risk stratification and appropriate therapy.
The specific genetic subtypes in B-ALL are associated with distinct prognostic outcomes e.g., t(9;22) BCR-ABL1 translocation, MLL gene rearrangements, hypodiploidy, and intrachromosomal amplification of chromosome 21 (iAMP21) predict poor outcome and genetic hallmarks such as ETV6-RUNX1 and hyperdiploid ALL predict favorable outcome (1, 5). An integrated cytogenetic and molecular genetics approach has been proposed and validated in small cohorts for improved risk stratification in B-ALL and classification of patients into clinically relevant subtypes with potentially important implications for therapy (6–9). In addition to gross chromosomal rearrangements, partial, or complete deletions of the IKZF1, CDKN2A/B, EBF1, and RB1 genes have emerged as independent predictors of poor outcome in B-ALL (10–12).
In the past two decades, genomic-profiling approaches have provided important insights into the genetic basis of ALL. ALL harbors several recurring regions of genetic alterations, most commonly focal submicroscopic or cryptic lesions that are not evident on conventional cytogenetic analysis (13). Many of the genes altered in ALL encode proteins with roles in key cellular pathways, including lymphoid development and differentiation (e.g., PAX5, IKZF1, EBF1, LEF1, and VPREB), cell-cycle regulation and tumor suppression [CDKN2A, CDKN2B (INK4/ARF), TP53, PTEN, and RB1], lymphoid signaling (BTLA, CD200, and TOX), transcription factors and transcriptional coregulators (ERG, TBL1XR1, and CREBBP), regulation of apoptosis (BTG1), and drug receptor (NR3C1) (10). Importantly, studies have also identified genetic lesions that define novel subtypes of ALL with distinct gene expression profiles, such as subtypes characterized by rearrangement of CRLF2 (14, 15), intrachromosomal amplification of chromosome 21 (16), focal deletions of ERG (17), and NOTCH1 (18). Several of these alterations described are clinically relevant in terms of risk stratification of ALL patients.
Recently, a next-generation sequencing-based Multiplex Ligation-dependent Probe Amplification (MLPA) variant, digitalMLPA (dMLPA) was developed to detect sub-microscopic CNAs, which might be missed by existing conventional, and high-resolution technologies (19). The available molecular diagnostic tools for detection of these key genetic abnormalities to distinguish various subtypes of ALL include conventional karyotyping, FISH, aCGH, SNP arrays, and conventional MLPA which are tedious and time-consuming. dMLPA allows multiplexing of several targets for accurate copy number detection of multiple genomic targets implicated in ALL. The aim of our study was to evaluate the utility of dMLPA in screening of ALL patients for key CNAs. Here, we present (1) the spectrum of chromosomal abnormalities, and CNAs including gene alterations among B-ALL and T-ALL patients, (2) validation of dMLPA data by conventional MLPA and RT-PCR, (3) correlation of CNAs with Minimal Residual Disease (MRD) status and integration of CNAs with risk stratification in ALL. As far as we know, our study demonstrated the utility of dMLPA in the detection of CNAs in ALL for the first time in Indian patients.
Materials and Methods
Patients
In this retrospective study, 151 consecutive samples of ALL patients registered at Dr. BRA IRCH, All India Institute of Medical Sciences, New Delhi, India from June 2017 to November 2018 were included. The diagnosis of ALL was made as per the updated criteria of WHO classification of myeloid neoplasms and acute leukemia (20). Waiver for informed consent was obtained from the Institute Ethics Committee (IEC-573/03.11.2017).
Risk Stratification and Treatment Regimen
The patients were risk stratified as standard (age between 1 and 10 years, TLC < 50 × 109/L, absence of high risk cytogenetics and CNS involvement and good prednisolone response on day 8 post induction chemotherapy i.e., blasts in peripheral blood <1 × 109/L), intermediate (age>10 years/ TLC > 50 × 109/L/ organ enlargement and absence of any high risk cytogenetic) and high risk groups (high risk cytogenetics, CNS involvement, poor early prednisolone response, and MRD positivity post induction).
The treatment protocol used for the pediatric patients (below 18 years) was according to the Indian Childhood Collaborative Leukemia (ICICLE; Clinical Trials Registry-India Reference Number, unpublished CTRI/2015/12/006434) Group protocol. Briefly, the induction regimen for standard risk group consisted of three chemotherapeutic agents (prednisolone, vincristine, and L-asparaginase). Daunomycin was added as the fourth drug for intermediate to high risk patients. The follow up of these patients depended on MRD status and the treatment included consolidation, maintenance, delayed intensification, and maintenance chemotherapy for two years. The adult patients were treated according to the MCP-841 protocol (21). Patients who failed to achieve complete remission or relapsed were given re-induction therapy according to the UK-ALL XII protocol (22). Patients positive for BCR-ABL additionally received Imatinib mesylate (>1 year 340 mg/m2/day; not exceeding 600 mg/day).
The MRD status was assessed at the end of induction chemotherapy using 10-color multi-parameter flow cytometry (Gallios, Beckman Coulter, Brea, CA). Stain-lyse-wash method was used with fluorochrome-tagged antibodies namely, CD81, CD45, CD34, CD58, CD19, HLA-DR, CD10, CD20, CD123, CD38 (Beckman Coulter, Miami, FL, USA). At least, one million cells were acquired in each case and list mode files were analyzed by Kaluza software version 3.0 (Beckman Coulter, Brea, CA, USA).
RT-PCR for Detection of Balanced Translocations
RNA was isolated using Tri Reagent (SIGMA-ALDRICH) as per the manufacturer's instructions. First strand cDNA was generated using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, Massachusetts, United States) as per the manufacturer's instructions. The product of the first strand cDNA synthesis was used directly in PCR for the detection of fusion transcripts including BCR-ABL1, ETV6-RUNX1, KMT2A-AFF1, E2A-PBX1, and STIL-TAL1 (23).
DigitalMLPA for Detection of Copy Number Alterations and Ploidy Status
DNA was isolated from either bone marrow or peripheral blood collected at the time of diagnosis using Genomic DNA Purification Kit (GeneJET, ThermoFisher Scientific, Waltham, Massachusetts, United States). For performing digitalMLPA, the modified well-established MLPA protocol was combined with Illumina next generation sequencing Miseq platform for amplicon quantification as described earlier (19). Probes for detection of both B-ALL and T-ALL associated Copy Number Alterations (CNAs) were included in this digitalMLPA D007 ALL probemix, which can multiplex >600 probes in a single reaction and simultaneously perform copy number analysis of 54 key target genes. In addition, 198 probes in the same probemix were included for karyotyping and data normalization purposes and a set of 128 control probes were included for quality control purposes.
Briefly, 100 ng of each DNA sample (in a total volume of 4 μl) sample was mixed with 2 μl of a unique barcode solution (MRC Holland), followed by DNA denaturation at 98°C for 10 min. After denaturation, a mixture of 1.25 μl dMLPA probe mix (MRC Holland) and 1.25 μl dMLPA buffer (MRC Holland) was added to each sample, and reactions were incubated overnight at 60°C for hybridization. Probes were ligated by incubating the reactions with 32 μl of a ligase master mix containing the ligase-65 enzyme (MRC Holland) and buffers (MRC Holland) at 48°C for 30 min, followed by heat inactivation of the ligase-65 enzyme at 98°C for 5 min and an additional incubation at 65°C for 20 min. PCR amplification of the ligated probes was performed on a calibrated Veriti 96-well thermocycler (Applied Biosystems). The PCR-amplified products were pooled and the library was loaded onto an Illumina MiSeq sequencer (Illumina, San Diego, CA) for quantification using the MiSeq Reagent Kit version 3 (150 cycles; Illumina). Sixteen healthy control DNA samples were included in the first validation run and for subsequent runs eight reference controls were included. Relative peak ratios between 0.8 and 1.2 were considered normal, while values below or above indicated losses or gains of genetic material, respectively. Data analysis was performed as described previously (19). Finally, heatmaps were generated comparing each dMLPA probe in each test sample against that in a reference sample to determine copy number changes for the indicated gene target.
Validation by Conventional MLPA
Conventional MLPA experiments were performed with DNA isolated from bone marrow or peripheral blood samples. Several SALSA MLPA probemixes were used for CNAs identification in these genes (MRC Holland, Amsterdam, The Netherlands). These probemixes included probes for IKZF1, CDKN2A/B, PAX5, EBF1, ETV6, BTG1, RB1, and PAR1 region (P335 & P202 kit), iAMP21-ERG (P327 probemixes) and a probe mix for T-ALL (P383) containing several different targets. The results of MLPA analysis were interpreted by assigning deletion status to the indicated genes for each sample. For each probe the status was assigned as “deleted,” “normal,” or “amplified” depending on the ratio obtained relative to the established normal range (mean ± 2SD or mean ± 3SD). Relation dosage quotient values below 0.8 were considered deletion and above 1.2 as amplification. If more than 50% of the probe ratios in a particular region indicated a copy number alteration of the genetic material, the result for that gene was designated abnormal. Deletion of either CDKN2A or CDKN2B was considered as CDKN2A/B locus to be deleted. For deletion in the PAR1 region of chromosome X or Y, we have considered it synonymous with P2RY8-CRLF2 as described earlier (6).
Statistical Analysis
The significance of any change in DNA copy number for MLPA, and data from dMLPA, were compared using Pearson's correlation test as appropriate. All analyses were performed using SIGMAPLOT version 13.0 (SYSTAT Software, Inc. USA). Chi-square test and Fisher's exact test were used to correlate various parameters with the cytogenetic profile and CNAs. p < 0.05 were considered statistically significant. The FDR values and heat map were derived using R statistical software.
Results
In ALL samples, an average of 900 single reads of 100 nucleotides was generated with minimal variability relative to reference samples that showed a standard deviation below 0.1 (maximum 0.06) across all target probes. Of the 151 ALL samples, three were excluded from the analysis as high variability in read ratios was observed (Figure 1). This could be attributed to the presence of impurities in the DNA samples. The characteristics of ALL patients including gender, age, total leukocyte counts, laboratory parameters (n = 148), and clinical features (n = 92) are mentioned in Table 1.
Figure 1.
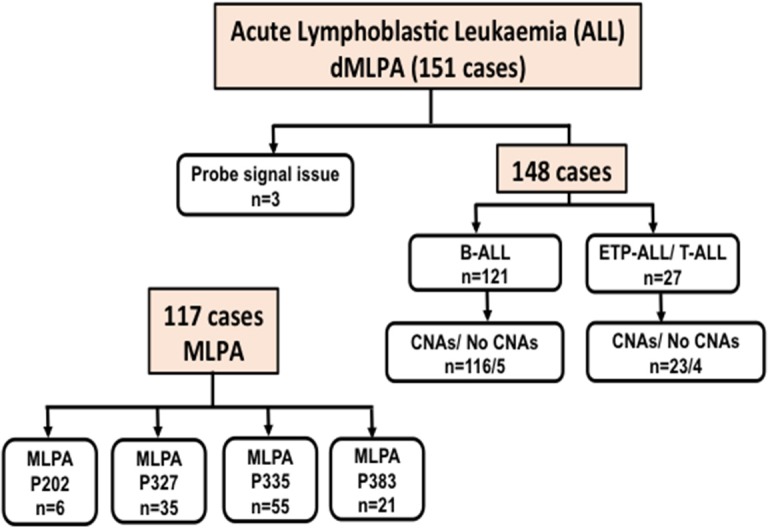
Flow-chart showing the number of Acute Lymphoid Leukemia (ALL) cases including B-cell and T-cell ALL analyzed for Copy Number Alterations (CNAs) by digitalMLPA and conventional MLPA. P202, P327, P335, and P383 indicate the conventional MLPA probemixes used and the number of cases analyzed.
Table 1.
Baseline characteristics of ALL patients evaluated by dMLPA.
| Characteristics | Number (%) |
|---|---|
| Total | 148 |
| GENDER | |
| Male | 109 (73.6) |
| Female | 39 (26.4) |
| AGE RANGE (YEARS) | |
| ≤1 | 9 (6) |
| 2–10 | 83 (56) |
| 10–18 | 41 (28) |
| >18 | 15 (10) |
| TLC, ×109/L | |
| Median | 144 (17.2) |
| Range | 0.06–533.6 |
| ≤50 | 99 (68.75) |
| >50 | 45 (31.25) |
| PERCENT BLAST | |
| Median | 85 |
| IMMUNOPHENOTYPE | |
| Pro-B ALL | 3 |
| Common-ALL | 103 |
| Pre-B ALL | 1 |
| T-ALL | 23 |
| ETP-ALL | 4 |
| KARYOTYPE (B-ALL) | 51/121 |
| MRD STATUS (B-ALL; n = 92) | |
| MRD positive | 25 |
| MRD negative | 67 |
Distribution Frequency of CNAs in ALL
The frequency distribution of CNAs detected in B-ALL (n = 121) were predominantly heterozygous or homozygous deletions in IKZF1 (22%) and CDKN2A/B (22%) followed by PAX5 (19%) (Figure 2A, Supplementary Table S1). Deletions were also detected in MLLT3 (14%), MTAP (14%), ETV6 (14%), and deletions in other gene targets were observed in <10% of the B-ALL cases. Gene duplications were observed in RUNX1 (~32% cases), ERG (~30% cases), PHF6 (20%), and PTEN (19%) genes and the following genes showed only duplication and no deletion was noted in any patient sample- TOX, ABL1, NUP214, NOTCH, PTEN, LMO1, LMO2, CD44, SPRED1, NF1, PTPN2, and PHF6 genes (Figure 2A). In the 27 T-ALL cases (18% of total ALL cases), deletions were noted in 59% (n = 16) cases primarily in the CDKN2A/B gene locus followed by MTAP (~33%), 15% in MLLT3/ PTEN/ JAK2, and <10% in PAR1, IKZF1, EZH2, EBF1, and PHF6 (Figure 2A; Supplementary Table S1).
Figure 2.

Frequency distribution of CNAs in ALL cases analyzed by dMLPA. (A) Gene deletions detected in samples of B-ALL (filled red bar) and T-ALL (Empty red bar) are shown in the left panel and gene duplications (filled and empty green bars, respectively) in the right panel. (B) Bar diagram showing percentage of cases with 0, 1, 2, and ≥3 CNAs, GA represents recurrent genetic chromosomal abnormalities.
Of the 148 ALL samples, no CNAs were detected in 9 samples (~6%) [B-ALL (n = 5), T-ALL (n = 2), and ETP-ALL (n = 2)]. Noticeably, 66% of the cases showed ≥3 CNAs, ~10% had 2 CNAs and ~13% had only 1 CNA (Figure 2B). Interestingly, in ~5% (n = 8 cases) of the samples, which harbored recurrent chromosomal abnormalities including BCR-ABL1 (n = 1), KMT2A-AFF1 (n = 2), and E2A-PBX (n = 5), no CNAs were detected. Copy number alterations identified were on an average of 5.6 somatic CNAs per sample (5.8 in B-cell precursor ALL and 3.8 in T-cell ALL) for the 54 gene targets covered.
Spectrum of CNAs and Prognostic Significance in B-ALL
For risk evaluation, B-ALL cases (~82% of total ALL cases) were categorized by cytogenetic risk groups as patients with good-risk (GR) genetic abnormalities (n = 33, ~27%), poor-risk (PR) abnormalities (n = 25; ~21%), and the remaining 52% (n = 63) of the cases as intermediate cytogenetic risk (IR) group (Figure 3). In the category of Good Risk (GR) genetic abnormalities (High hyperdiploidy and ETV6-RUNX1), 16% (n = 19) of the B-ALL cases were assigned Hyperdiploidy status as determined by the karyotyping probes in the dMLPA assay. Only chromosomes for which all karyotyping probes showed increased read ratios of greater than the cut-off of 1.3 and above were considered as heterozygous gain or >1.75 as homozygous gain whereas partial chromosomal gain were excluded. Concordance of hyperdiploidy status could be demonstrated in three cases where simultaneous conventional karyotyping data was available (Supplementary Table S2). Non-random gain of chromosomes X, 4, 6, 10, 14, 17, 18, and 21 was commonly observed with exceptions of gain of chromosomes 1, 5, 7, and 8 in a few cases (Supplementary Figure S1). In cases with hyperdiploidy, isolated CNAs were noted as deletions in CDKN2A/B, MTAP, IKZF1, CASP8AP2, ERG, RAG2, ETV6, CTCF, TBL1XR1, and VPREB1 genes, which could potentially introduce heterogeneity for risk stratification in this good risk category (Supplementary Table S2).
Figure 3.
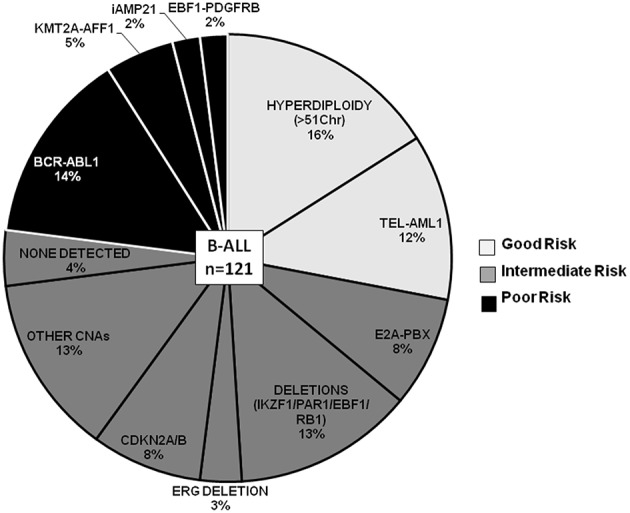
B-ALL cases categorized by recurrent cytogenetic abnormalities and CNAs. Pie chart depicting the frequency distribution of B-ALL cases with chromosomal abnormalities and CNAs. The various categories are distributed by risk groups and are color-coded as good risk (light gray), intermediate risk (dark gray), and poor risk (black).
The other GR category comprised 12% (n = 14 cases) of the B-ALL cases with ETV6-RUNX1 genetic abnormality (Figure 3), wherein predominant CNAs were observed as gene deletions in the ETV6 gene indicating rearrangements and fusions (Figure 4). In addition, deletions were also observed in PAX5 (3 cases), IGHM (6 cases), VPREB1 (6 cases), EBF1, and MYB (2 cases) genes. No case with ETV6-RUNX1 harbored deletions in IKZF1, PAR1, or BTG1. An isolated case within this category showed a deletion in the RB1 gene. Two cases with a deletion in the EBF1 gene showed co-occurrence of deletions in NR3C2, PAX5, RAG2, and VPREB1 or deletions in multiple other genes (CD200, BTLA, CASP8AP2, and MYB). Also, gene duplications were detected in RUNX1, ERG, PHF6, and PTEN genes in a few cases (Figure 4).
Figure 4.

B-ALL cases categorized by cytogenetic abnormalities showing heterogeneous distribution of CNAs. Heat map showing recurrent chromosomal abnormalities in Good risk (GR) category included ETV6-RUNX1 (hyperdiploidy cases not shown), Intermediate risk (IR) represented by E2A-PBX (samples with IKZF1, EBF1, RB1, ERG, and CDKN2A/B deletions not shown) and Poor risk (PR) included KMT2A-AFF1, BCR-ABL1 (cases with p210 and p190 isoforms), EBF1-PDGFRB (indicated by P), and iAMP21 (indicated by i). Also included in this heat map are CNAs in B-ALL cases with no detectable cytogenetic abnormalities (indicated by No GA). (The color key shows a gradient from red to blue wherein red indicates homozygous deletion, yellow and orange indicate heterozygous deletion and blue indicates duplication. The gene names are labeled on the right hand side and the patient sample IDs with or without genetic abnormality are indicated in the bottom row).
In the poor risk (PR) category, the dominant CNAs detected in B-ALL with BCR-ABL genetic abnormalities (14%; n = 17 cases) (Figure 3) were deletions in IKZF1 (~82%; n = 14 cases) and CDKN2A/B locus (~53%; n = 9 cases) (Figure 4). Majority of the cases showed partial or complete deletion of the exonic region of IKZF1 gene but in one case heterozygous deletion in the genomic region immediately upstream of IKZF1 gene was noted. Mutually exclusive deletions in BTG1, IGHM, PAX5, and VPREB1 genes were also present in almost 25% of the cases. The BCR-ABL isoform (p210) was detected in the five samples and the remaining 12 samples showed the p190 isoform. Interestingly, the CNA profile observed in the cases with p190 isoform were more heterogenous as compared to the cases with p210; the latter harbored only IKZF1 or CDKN2A/B gene deletions (Figure 4). One case also harbored a rare NUP214-ABL1 intrachromosomal rearrangement, a rare entity seen in B-ALL.
In the other poor risk (PR) category of B-ALL with KMT2A-AFF1 (5%; n = 6 cases) genetic abnormalities (Figure 3), gene deletions were detected in IKZF1, CDKN2A/B, TP53, IKZF3, and VPREB1 genes. No gene deletions were detected in three cases. Interestingly, four cases harbored gene duplications in DMD and PHF6 genes (Figure 4). One case of B-ALL with iAMP21 amplification showed gene deletions in RB1, TP53, CTCF, CD200, and BTLA (Figure 4; Supplementary Figure S2). Intrachromosomal EBF1-PDGFRB rearrangement was detected by dMLPA in two cases, which also carried deletions in SUZ12, BTG1, BTLA, and TP53 genes associated with poor risk (Figure 4; Supplementary Figure S2).
Contrary to the GR category with ETV6-RUNX1 genetic abnormality, cases in the intermediate risk category with E2A-PBX (8%; n = 10 cases) (Figure 3) chromosomal abnormality rarely showed any gene deletions. Three out of ten cases showed deletions in RB1 and PAX5 genes with co-occurrence of TBL1XR1 or BTG1 or CDKN2A/B, CASP8AP2, and JAK2 gene deletions (Figure 4).
The remaining ~42% cases with no detectable cytogenetic abnormalities were classified as intermediate-risk group and further evaluated based on CNAs (loss or gain) detected by dMLPA. Of these, 16% (n = 20 cases) showed deletions in IKZF1/PAR1/EBF1/RB1/ERG, and 8% (n = 10) of these cases carried deletions in CDKN2A/B locus, which are associated with poor risk (Figure 3). Within this subgroup, one case also harbored a NUP214-ABL1 intrachromosomal rearrangement, a rare entity seen in B-ALL. The remaining 13% (n = 16 cases) displayed substantial heterogeneity in the CNAs detected (Figure 4). Strikingly, ~50% of these cases showed duplications in RUNX1 and ERG and two cases in DMD, MYB, and CASP8AP2. Four cases harbored a deletion in the VPREB1 gene, and two cases showed gene deletions in EZH2 and EPHA1. Interestingly, intragenic amplification of exon 2–5 of the PAX5 gene was detected in one patient sample that was also MRD positive.
The CNAs (both gene deletions and duplications) were correlated with post-induction MRD status, which was available for 92 cases of which 27% (n = 25) cases were MRD positive (≥0.01%) and the remaining 73% (n = 67) were MRD negative (Table 1). In addition to the previously described CNAs of prognostic relevance (IKZF1, CDKN2A, BTG1, EBF1, PAX5, ETV6, RB1), we observed a correlation between deletions in NF1 and SUZ12 and duplications in LEF1, NR3C2, RUNX1, and PAR1 region and MRD positivity (Table 2). A trend of correlation was also seen for PHF6 and CASP8AP2 gene duplication and MRD positivity.
Table 2.
Correlation of Minimal Residual Disease status with Deletions and Duplications in individual genes/loci.
| CNA in genes | MRD − | MRD+ | Fisher's exact (p) | FDR (p. adj.) | CNA in Genes | MRD − | MRD+ | Fisher's exact (p) | FDR (p. adj.) |
|---|---|---|---|---|---|---|---|---|---|
| IKZF1 deletion | 13 | 6 | 0.77 | 1 | LEF1 gain | 2 | 6 | 0.005* | 0.08 |
| IKZF wt | 54 | 19 | LEF1 wt | 64 | 19 | ||||
| CDKN2A/B deletion | 18 | 2 | 0.08 | 0.7 | NR3C2 gain | 3 | 6 | 0.010* | 0.08 |
| CDKN2A/B wt | 48 | 22 | NR3C2 wt | 62 | 18 | ||||
| BTG1 Deletion | 4 | 1 | 1 | 1 | RUNX1 gain | 14 | 12 | 0.015* | 0.08 |
| BTG1 wt | 61 | 24 | RUNX1 wt | 50 | 11 | ||||
| EBF1 deletion | 3 | 0 | 0.55 | 0.95 | PAR1 gain | 10 | 9 | 0.042* | 0.17 |
| EBF1 wt | 58 | 25 | PAR1 wt | 56 | 16 | ||||
| PAX5 deletion | 16 | 3 | 0.38 | 0.86 | PHF6 gain | 10 | 8 | 0.081 | 0.25 |
| PAX5 wt | 49 | 20 | PHF6 wt | 57 | 17 | ||||
| ETV6 deletion | 13 | 4 | 0.77 | 1 | CASP8AP2 gain | 6 | 6 | 0.092 | 0.25 |
| ETV6 wt | 53 | 21 | CASP8AP2 wt | 57 | 19 | ||||
| Rb1 deletion | 3 | 0 | 0.56 | 0.95 | PTPN2 gain | 2 | 3 | 0.122 | 0.28 |
| Rb1 wt | 64 | 25 | PTPN2 wt | 65 | 22 | ||||
| MLLT3 deletion | 12 | 2 | 0.33 | 0.86 | MYB gain | 7 | 6 | 0.177 | 0.35 |
| MLLT3 wt | 54 | 23 | MYB wt | 58 | 19 | ||||
| MTAP deletion | 16 | 2 | 0.14 | 0.79 | PTEN gain | 9 | 6 | 0.224 | 0.4 |
| MTAP wt | 50 | 23 | PTEN wt | 58 | 19 | ||||
| NF1+SUZ12 del. | 0 | 2 | 0.06 | 0.7 | TOX gain | 5 | 4 | 0.261 | 0.42 |
| NF1+SUZ12 wt | 61 | 19 | TOX wt | 59 | 21 | ||||
| EPHA1 deletion | 2 | 2 | 0.3 | 0.86 | ADD3 gain | 3 | 3 | 0.34 | 0.47 |
| EPHA1 wt | 64 | 23 | ADD3 wt | 63 | 22 | ||||
| EZH2 deletion | 2 | 2 | 0.3 | 0.86 | IGHM gain | 10 | 6 | 0.35 | 0.47 |
| EZH2 wt | 64 | 23 | IGHM wt | 49 | 16 | ||||
| VPREB deletion | 13 | 7 | 0.41 | 0.86 | DMD gain | 6 | 4 | 0.446 | 0.55 |
| VPREB wt | 53 | 18 | DMD wt | 61 | 20 | ||||
| CD200/BTLA deletion | 4 | 1 | 1 | 1 | PDGFRB gain | 3 | 0 | 0.5 | 0.57 |
| CD200/BTLA wt | 62 | 23 | PDGFRB wt | 64 | 25 | ||||
| TBL1XR1 deletion | 5 | 1 | 1 | 1 | NR3C1 gain | 4 | 0 | 0.57 | 0.61 |
| TBL1XR1 wt | 62 | 23 | NR3C1 wt | 62 | 24 | ||||
| IGHM deletion | 8 | 3 | 1 | 1 | LMO1 gain | 4 | 2 | 0.668 | 0.67 |
| IGHM wt | 49 | 16 | LMO1 wt | 61 | 23 |
and bold p < 0.05 were considered statistically significant.
p values showing a trend of correlation are shown in bold.
Frequency Distribution and Prognostic Value of CNAs in T-ALL
Intrachromosomal fusions were detected in ~19% cases, with five cases showing a SIL-TAL1 fusion and two cases showed NUP214-ABL genetic rearrangement (Figure 5A; Supplementary Figure S3). All cases with SIL-TAL1 fusion and one case with NUP214-ABL also carried CDKN2A/B gene deletions (Figure 5B). Deletions in CDKN2A/B, IKZF1, and EZH2 were mutually exclusive. Two cases showed NF1/ SUZ12 deletions associated with poor risk. Four cases including two cases of ETP-ALL did not show any CNAs (not shown in the heat map Figure 5B).
Figure 5.

T-ALL cases categorized by cytogenetic abnormalities showing distribution of CNAs. (A) T-ALL cases sub-categorized by chromosomal abnormalities and CNAs in a pie-chart. (B) Heat map showing the CNAs distribution in T-ALL cases. The color codes indicate red as homozygous deletion, orange indicate heterozygous deletion and green indicates duplication.
Validation of dMLPA by Conventional MLPA and RT-PCR
The CNAs detected by dMLPA were confirmed by conventional MLPA assays (P202, P335, P327, and P383) as per the availability of samples (n = 117). The reference range data showed a normal distribution in each case, and a narrow variation in the mean and SD in the reference samples (Mean ± 2SD or 99% CI, Mean ± 3SD or 95% CI; p = 0.05), dosage quotient ranging from 0.8 to 1.2 (representative read ratios, Supplementary Figure S4). The degree of concordance between the conventional MLPA and dMLPA assays for the detection of listed genes indicating the number of samples tested is shown in Supplementary Table S3. The Pearson's correlation coefficient was calculated to determine the efficiency of dMLPA and conventional MLPA and a positive correlation (r = 0.99) between the two assay methods was observed. Heterozygous deletion in EZH2 gene was detected in three cases by conventional MLPA as opposed to one case by dMLPA.
Chromosomal abnormalities including BCR-ABL1, KMT2A-AFF1, E2A-PBX, and ETV6-RUNX1 were detected by RT-PCR and a few positive cases were also confirmed by FISH (FISH data was available for the following number of cases BCR-ABL1 = 12/17; KMT2A-AFF1 = 5/6; E2A-PBX = 7/10; and ETV6-RUNX1 = 9/14). Intrachromosomal gene fusion, STIL-TAL1 detected in five T-ALL cases was also validated by RT-PCR and conventional MLPA (Supplementary Figure S4). Also, positive cases for NUP214-ABL1 were confirmed by conventional MLPA (P383 probemix).
Discussion
In this study, the potential of detection of Copy Number Alterations by dMLPA assay and its integration with cytogenetic profiling for an improved genetic risk stratification of ALL patients was evaluated. After categorization of patients into cytogenetic risk groups as determined by recurrent chromosomal abnormalities, CNA profiles obtained by dMLPA could further aid in refining the classification of risk groups and prediction of clinical outcome. The previous study by Benard-Slagter et al. (19) demonstrated dMLPA as a robust and reliable tool for genetic characterization of ALL patients. We found this to be the case as we were able to detect CNAs in ~89% of our cases with dMLPA assay as opposed to conventional MLPA where almost 50% of the cases harboring rare (<10% cases) yet relevant CNAs are missed as limited targets are covered and multiple assays have to be performed. Moreover, dMLPA could detect ≥3 CNAs in 66% of our cases whereas that reported by conventional MLPA were shown in only 10% of the cases (6). Our study provides a validation of dMLPA assay and takes the previous study by Benard-Slagter et al. (19) to the next level for its potential translation into routine diagnostics and genetic risk stratification of ALL.
CNAs detected by dMLPA were validated by available MLPA assays that showed high concordance between the two methods [as also shown by (19)]. The multiplexing of ~600 probes in a single reaction of dMLPA rarely resulted in false positive or false negative results as confirmed by conventional MLPA assays. Indeed, a wide range of genomic targets could be analyzed for CNAs with less hands-on time and easier data analysis. With a limit of detection of CNAs in samples carrying 25–30% of neoplastic cells (19), in two B-ALL cases the blast percent was borderline (20–30%), which could have resulted in failure to detect CNAs.
Distinct CNA profiles were evident in B-ALL and T-ALL with both mutually exclusive and co-occurring genes. The dominance pattern of key genes affected in B-ALL cases was similar to earlier studies from India (IKZF1, CDKN2A/B, PAX5, ETV6) (8, 9, 12) and different from that reported by other groups where deletions in CDKN2A/B and ETV6 were dominant (6). The new gene targets such as BTLA, CD200, VPREB1 which are associated with inferior event-free and overall survival; DMD with relapse, TBL1XR1 and TP53 with chemo-resistance; EZH2, PAR1 region, and P2RY8-CRLF2 with poor prognosis; CASP8AP2, NF1, and SUZ12 associated with poor response to induction therapy are included in this dMLPA panel and useful for further risk stratification in cytogenetically good risk B-ALL cases. In contrast, cases with T-ALL but not B-ALL harbored deletions in PTEN, PHF6, and TOX genes. Inactivation of PTEN frequently occurs in T-ALL and may be associated with chemotherapy resistance and poor prognosis. The CNA profiles obtained using the dMLPA assay were potentially relevant in facilitating risk stratification of cases without recurrent or detectable genetic abnormalities and classical genetic alterations in our cases.
In the cases with hyperdiploidy, isolated CNAs were noted as deletions in CDKN2A/B, IKZF1, CASP8AP2, ERG, RAG2, ETV6, CTCF, TBL1XR1, and VPREB1 genes, which could potentially introduce heterogeneity in this good risk category. As described by Benard-Slagter et al. (19) a rare intragenic deletion of ERG exons 5–9 was detected in two of our cases and a single deletion in exon 5 was also noted. Few cases harbored partial chromosomal deletions in Chromosomes 7 and 9 but we did not detect any case of hypodiploidy. Absence of deletions in CDKN2A/B, IKZF1, PAR1, and BTG1 in cases with ETV6-RUNX1 genetic abnormality indicated certain genetic predisposition of the combination of gene targets, which might be affected with the primary genetic abnormality. Two cases with a deletion in the EBF1 gene showed co-occurrence of deletions in NR3C2, PAX5, RAG2, and VPREB1 or deletions in multiple other genes (CD200, BTLA, CASP8AP2, MYB) influencing the risk stratification of these good risk category. Unlike reported in the previous study by Benard-Slagter et al. (19), which showed the presence of homozygous deletion of CD200 and heterozygous deletion of BTLA, we identified a case of ALL relapse with heterozygous deletion of CD200 and a homozygous deletion of BTLA. One case of B-ALL with iAMP21 amplification showed homozygous deletion of CD200 and BTLA and gene deletions in RB1, TP53, CTCF as has been reported earlier (24). An isolated case of intragenic amplification of PAX5 gene with MRD positive status may define a novel, relapse-prone subtype of B-cell precursor ALL with a poor outcome but this need to be validated in larger cohort.
B-ALL with t(9;22) BCR-ABL1 chromosomal translocation is epidemiologically more prevalent in adults (~25%) and accounts for 2–4% of cases in children. These patients belong to the high risk category depending on the age, WBC count and response to therapy. In the majority of childhood cases, a p190 BCR-ABL1 fusion transcript is observed and p210 is rare. The distinct CNA profile observed in patients with p190 and p210 isoforms could be further evaluated using dMLPA. The cases with p210 isoform primarily harbored deletions in IKZF1 gene whereas samples with p190 isoform had IKZF1 plus profile (CDKN2A/B, PAX5, and other genes) associated with MRD-dependent very-poor prognostic profile in BCP ALL.
Several other common gene deletions, such as CDKN2A/2B, IKZF1, PAX5, ETV6, RB1, and BTG1 have also been described in ALL patients (6, 25). Deletions in the IKZF1 gene have been associated with relapse and poor clinical outcome in several studies. In ~80% of pediatric and 60–90% of adult BCR-ABL1 positive ALL cases, partial or complete gene deletions of IKZF1 are detected. Eighty percent of the B-ALL with BCR-ABL1 chromosomal abnormality showed IKZF1 deletions in our cohort. Partial gene deletions of IKZF1 frequently affect exons 4–7 and was seen in majority of our cases (n = 10), but a rare smaller deletion, affecting a region upstream of IKZF1 gene was detected which was not reported by Benard-Slagter et al. (19). Also, deletions of IKZF1 exon 4–8 associated with poor outcome although are rare but were seen in two of our samples in which this gene was affected. Earlier studies have reported the prognostic relevance of deletions in IKZF1 and EBF1 genes which are associated with MRD positivity (6, 9, 12). In our retrospective study, since IKZF1 deletions were primarily detected in BCR-ABL1 positive B-ALL cases, these cases were risk stratified into the poor risk category and were given intensive chemotherapy. This could have resulted in skewing of the correlation between IKZF1 and other gene deletions and MRD clearance in our cases. Individual poor risk chromosomal abnormalities were too rare to reliably test for correlation.
We successfully detected intrachromosomal fusions with copy number imbalance (SIL-TAL1, NUP214-ABL, EBF1-PDGFRB) in a few cases, which could potentially benefit from treatment with tyrosine kinase inhibitors (26–28). The NUP214-ABL1 fusion gene is demonstrated to be amplified as multiple (5–50) episomal copies in 6% of T-cell acute lymphoblastic leukemia (T-ALL) and has been reported as a rare subtype of B-cell precursor (ALL) that could benefit from tyrosine kinase inhibitors (29–31). EBFI-PDGFRB gene fusion is reported in <1% of B-ALL, Philadelphia like ALL subtype, which also benefit from tyrosine kinase therapy (26, 32). Deletion of the PTEN gene was exclusively detected in T-ALL cases, which is associated with resistance to chemotherapy and early treatment failure (33). Deletion in NF1 and SUZ12 in T-ALL are associated with poor response to induction chemotherapy (34). We also observed NF1 and SUZ12 deletions in two MRD positive cases of B-ALL suggesting that although rare, these can have implications in treatment outcome.
Gene duplication is a recurring phenomenon in cancer and a major driving force in the gain of biological functions that leads to over-expression and alteration of gene expression patterns. This emerged as a strong correlation in our MRD positive cases which predominantly showed gain of copies of several genes including LEF1, RUNX1, PAR1, NR3C2, PHF6, and CASP8AP2. Over-expression of LEF1 has been reported to predict unfavorable outcome and the standard-risk B-ALL patients with high LEF1 expression can possibly benefit from early treatment modifications and alternative molecular therapies, such as agents targeting the Wnt signaling pathway (35). Amplification of RUNX1 is associated with a poor outcome in childhood ALL (16). Currently, FISH with probes directed to RUNX1 is the only reliable method of detection of duplication of 21q with amplification of RUNX1 in ALL. We could reliably detect RUNX1 amplification in 27% of our cases and thus, dMLPA provides an alternate platform for its detection. Gene duplication of the PAR1 region has been reported to be associated with KMT2A-AFF1 cases and a similar profile was also observed in two of our cases (36).
Risk stratification algorithms are regularly revisited for better refinement and monitoring of prognostic factors. Our findings demonstrated the significance of both cytogenetics and CNAs for prognostication. Integration of CNAs with cytogenetic risk stratification allowed identification of subgroups with variable outcomes in the MRD positive high risk group and MRD negative intermediate category because CNAs were detected that are associated with poor prognosis. These intermediate risk group patients can therefore be considered for de-intensification or intensification of chemotherapy. Patients with genetic profiles associated with poor prognosis could be considered for targeted or alternative modalities. MRD status combined with molecular genetic profiling maybe valuable in monitoring and clinical management of these patients. Since chromosomal abnormalities are insufficient to initiate leukemia, the detection of cooperating genetic lesions guide a better risk prognostication, aid in disease monitoring and predict the probability of relapse. However, few limitations remain in this technology such as it is restricted to probe sequence specificity, and silent copy number changes such as inversions or translocations and sequence changes (SNPs, indels) remain undetected. In conclusion, dMLPA is a robust, reliable and a valuable alternative technology for rapid identification of key CNAs in ALL patients, which warrants evaluation in larger cohorts.
Data Availability
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Institute Ethics Committee, All India Institute of Medical Sciences, New Delhi, India. Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author Contributions
DT designed and conducted the study, performed the data interpretation, statistical analysis, and wrote the manuscript. GK and RG conceived the idea, performed the data interpretation, statistical analysis, and wrote the manuscript. SS and AB-S are the team from MRC Holland who provided the reagents and assisted with data analysis. IK performed the technical experiments of dMLPA and MLPA. RA compiled the clinical data. LR and VS performed the statistical analysis. PV and SJ performed the RT-PCR experiments. LK, AS, SB, and RS are the clinical faculty who treated the patients during the course of this study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the MRC Holland team for providing dMLPA and MLPA probemixes.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00871/full#supplementary-material
References
- 1.Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. (2010) 11:429–38. 10.1016/S1470-2045(10)70066-8 [DOI] [PubMed] [Google Scholar]
- 2.Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. (2017) 7:e577. 10.1038/bcj.2017.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. (2017) 129:1113–23. 10.1182/blood-2016-10-706465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Starý J, Zuna J, Zaliova M. New biological and genetic classification and therapeutically relevant categories in childhood B-cell precursor acute lymphoblastic leukemia. F1000Research. (2018) 7:F1000 Faculty Rev–156. 10.12688/f1000research.16074.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. (2012) 26:123–35. 10.1016/j.blre.2012.01.001 [DOI] [PubMed] [Google Scholar]
- 6.Moorman AV, Enshaei A, Schwab C, Wade R, Chilton L, Elliott A, et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. (2014) 124:1434–44. 10.1182/blood-2014-03-562918 [DOI] [PubMed] [Google Scholar]
- 7.Irving JAE, Enshaei A, Parker CA, Sutton R, Kuiper RP, Erhorn A, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood. (2016) 128:911–22. 10.1182/blood-2016-03-704973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta SK, Bakhshi S, Kumar L, Kamal VK, Kumar R. Gene copy number alteration profile and its clinical correlation in B-cell acute lymphoblastic leukemia. Leuk Lymph. (2017) 58:333–42. 10.1080/10428194.2016.1193855 [DOI] [PubMed] [Google Scholar]
- 9.Patkar N, Subramanian PG, Tembhare P, Mandalia S, Chaterjee G, Rabade N, et al. An integrated genomic profile that includes copy number alterations is highly predictive of minimal residual disease status in childhood precursor B-lineage acute lymphoblastic leukemia. Indian J Pathol Microbiol. (2017) 60:177–84. 10.4103/IJPM.IJPM_466_16 [DOI] [PubMed] [Google Scholar]
- 10.Loh ML, Mullighan CG. Advances in the genetics of high-risk childhood b-progenitor acute lymphoblastic leukemia and juvenile myelomonocytic leukemia: Implications for therapy. Clin Cancer Res. (2012) 18:2754–67. 10.1158/1078-0432.CCR-11-1936 [DOI] [PubMed] [Google Scholar]
- 11.Dörge P, Meissner B, Zimmermann M, Möricke A, Schrauder A, Bouquin JP, et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica. (2013) 41:7–11. 10.3324/haematol.2011.056135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta SK, Bakhshi S, Kumar L, Seth R, Kumar R. IKZF1 (IKAROS) deletions in B-ALL and its clinical correlation: a prospective study from a tertiary care centre in Northern India. Leuk Res. (2016) 41:7–11. 10.1016/j.leukres.2015.07.010 [DOI] [PubMed] [Google Scholar]
- 13.Usvasalo A, Savola S, Räty R, Vettenranta K, Harila-Saari A, Koistinen P, et al. CDKN2A deletions in acute lymphoblastic leukemia of adolescents and young adults-An array CGH study. Leuk Res. (2008) 32:1228–35. 10.1016/j.leukres.2008.01.014 [DOI] [PubMed] [Google Scholar]
- 14.Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. (2009) 114:2688–98. 10.1182/blood-2009-03-208397 [DOI] [PubMed] [Google Scholar]
- 15.Mullighan CG, Collins-Underwood JR, Phillips LAA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. (2009) 41:1243–6. 10.1038/ng.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson HM, Broadfield ZJ, Cheung KL, Harewood L, Harris RL, Jalali GR, et al. Amplification of AML1 in acute lymphoblastic leukemia is associated with a poor outcome [9]. Leukemia. (2003) 17:2249–50. 10.1038/sj.leu.2403140 [DOI] [PubMed] [Google Scholar]
- 17.Clappier E, Auclerc MF, Rapion J, Bakkus M, Caye A, Khemiri A, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. (2014) 28:70–7. 10.1038/leu.2013.277 [DOI] [PubMed] [Google Scholar]
- 18.Haydu JE, De Keersmaecker K, Duff MK, Paietta E, Racevskis J, Wiernik PH, et al. An activating intragenic deletion in NOTCH1 in human T-ALL. Blood. (2012) 119:5211–4. 10.1182/blood-2011-10-388504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benard-Slagter A, Zondervan I, de Groot K, Ghazavi F, Sarhadi V, Van Vlierberghe P, et al. Digital multiplex ligation-dependent probe amplification for detection of key copy number alterations in T- and B-cell lymphoblastic leukemia. J Mol Diagnost. (2017) 19659–72. 10.1016/j.jmoldx.2017.05.004 [DOI] [PubMed] [Google Scholar]
- 20.Wenzinger C, Williams E, Gru AA. Updates in the pathology of precursor lymphoid neoplasms in the revised fourth edition of the WHO classification of tumors of hematopoietic and lymphoid tissues. Curr Hematol Malign Rep. (2018) 13:275–88. 10.1007/s11899-018-0456-8 [DOI] [PubMed] [Google Scholar]
- 21.Magrath I, Shanta V, Advani S, Adde M, Arya LS, Banavali S, et al. Treatment of acute lymphoblastic leukaemia in countries with limited resources; lessons from use of a single protocol in India over a twenty year peroid. Eur J Cancer. (2005) 41:1570–83. 10.1016/j.ejca.2004.11.004 [DOI] [PubMed] [Google Scholar]
- 22.Rowe JM, Buck G, Burnett AK, Chopra R, Wiernik PH, Richards SM, et al. Induction therapy for adults with acute lymphoblastic leukemia: results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood. (2005) 106:3760–67. 10.1182/blood-2005-04-1623 [DOI] [PubMed] [Google Scholar]
- 23.Van Dongen JJM, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, Biondi A. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. (1999) 13:1901–192. 10.1038/sj.leu.2401592 [DOI] [PubMed] [Google Scholar]
- 24.Rand V, Parker H, Russell LJ, Schwab C, Ensor H, Irving J, et al. Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell precursor acute lymphoblastic leukemia. Blood. (2011) 117:6848–55. 10.1182/blood-2011-01-329961 [DOI] [PubMed] [Google Scholar]
- 25.Schwab CJ, Chilton L, Morrison H, Jones L, Al-Shehhi H, Erhorn A, et al. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica. (2013) 98:1081–8. 10.3324/haematol.2013.085175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lengline E, Beldjord K, Dombret H, Soulier J, Boissel N, Clappier E. Successful tyrosine kinase inhibitor therapy in a refractory B-cell precursor acute lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica. (2013) 98:e146–8. 10.3324/haematol.2013.095372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attarbaschi A, Morak M, Cario G, Cazzaniga G, Ensor HM, te Kronnie T, et al. Treatment outcome of CRLF2 -rearranged childhood acute lymphoblastic leukaemia: a comparative analysis of the AIEOP-BFM and UK NCRI-CCLG study groups. Br J Haematol. (2012) 158:772–7. 10.1111/j.1365-2141.2012.09221.x [DOI] [PubMed] [Google Scholar]
- 28.Schwab C, Andrews R, Chilton L, Elliott A, Keil G, Richardson S, et al. EBF1-PDGFRB fusion in paediatric acute lymphoblastic leukaemia (ALL): genetic profile and clinical implications. Blood. (2014) 127:2214–8. 10.1182/blood-2015-09-670166 [DOI] [PubMed] [Google Scholar]
- 29.Graux C, Cools J, Melotte C, Quentmeier H, Ferrando A, Levine R, et al. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat Genet. (2004) 36:1084–9. 10.1038/ng1425 [DOI] [PubMed] [Google Scholar]
- 30.Eyre T, Schwab CJ, Kinstrie R, McGuire AK, Strefford J, Peniket A, et al. Episomal amplification of NUP214-ABL1 fusion gene in B-cell acute lymphoblastic leukemia. Blood. (2012) 120:4441–3. 10.1182/blood-2012-09-456517 [DOI] [PubMed] [Google Scholar]
- 31.Duployez N, Grzych G, Ducourneau B, Fuentes MA, Grardel N, Boyer T, et al. NUP214-ABL1 fusion defines a rare subtype of B-cell precursor acute lymphoblastic leukemia that could benefit from tyrosine kinase inhibitors. Haematologica. (2016) 101:e133–4. 10.3324/haematol.2015.136499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weston BW, Hayden MA, Roberts KG, Bowyer S, Hsu J, Fedoriw G, et al. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB –positive acute lymphoblastic leukemia. J Clin Oncol. (2013) 31:e413–6. 10.1200/JCO.2012.47.6770 [DOI] [PubMed] [Google Scholar]
- 33.Zuurbier L, Petricoin EF, Vuerhard MJ, Calvert V, Kooi C, Buijs-Gladdines JGCAM, et al. The significance of PTEN and AKT aberrations in pediatric T-cell acute lymphoblastic leukemia. Haematologica. (2012) 97: 1405–13. 10.3324/haematol.2011.059030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balgobind BV, Van Vlierberghe P, Van Den Ouweland AMW, Beverloo HB, Terlouw-Kromosoeto JNR, Van Wering ER, et al. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood. (2008) 111:4322–8. 10.1182/blood-2007-06-095075 [DOI] [PubMed] [Google Scholar]
- 35.Kühnl A, Gökbuget N, Kaiser M, Schlee C, Stroux A, Burmeister T, et al. Overexpression of LEF1 predicts unfavorable outcome in adult patients with B-precursor acute lymphoblastic leukemia. Blood. (2011) 118:6362–7. 10.1182/blood-2011-04-350850 [DOI] [PubMed] [Google Scholar]
- 36.Bhandari P, Ahmad F, Das BR. Molecular profiling of gene copy number abnormalities in key regulatory genes in high-risk B-lineage acute lymphoblastic leukemia: frequency and their association with clinicopathological findings in Indian patients. Med Oncol. (2017) 34:92. 10.1007/s12032-017-0940-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.