

Abstract
Engineered solid–liquid interfaces will play an important role in the development of future energy storage and conversion (ESC) devices. In the present study, defective graphene oxide (GO) and reduced graphene oxide (rGO) structures were used as engineered interfaces to tune the selectivity and activity of Pt disk electrodes. GO was deposited on Pt electrodes via the Langmuir–Blodgett technique, which provided compact and uniform GO films, and these films were subsequently converted to rGO by thermal reduction. Electrochemical measurements revealed that both GO and rGO interfaces on Pt electrodes exhibit selectivity toward the oxygen reduction reaction (ORR), but they do not have an impact on the activity of the hydrogen oxidation reaction in acidic environments. Scanning transmission electron microscopy at atomic resolution, along with Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), and scanning electron microscopy (SEM), revealed possible diffusion sites for H2 and O2 gas molecules and functional groups relevant to the selectivity and activity of these surfaces. Based on these insights, rGO interfaces are further demonstrated to exhibit enhanced activity for the ORR in nonaqueous environments and demonstrate the power of our ex situ engineering approach for the development of next-generation ESC devices.
Keywords: graphene oxide, reduced graphene oxide, interface, electrocatalysis, oxygen reduction reaction, hydrogen oxidation reaction, selectivity
Introduction
The solid–liquid interface (SLI) is a crucial part of every energy storage and conversion (ESC) device, and yet despite decades of research is still poorly understood at the atomic level. Most of the chemical and electrochemical processes that take place in ESC devices depend on the nature of the SLI. Usually, the formation of the interface is spontaneous and is dictated by the chemical composition of the electrode, surface electronic structure, surface crystal planes, surface functional groups, and chemical composition of the electrolyte. The structure of the SLI, in turn, determines many of its important properties, in particular its activity, stability, and selectivity. It is therefore highly questionable why we would leave these important properties at the mercy of a spontaneous process. In this respect, surface engineering of metal catalysts can play a significant role in tuning the chemical and physical properties of the interface in an advantageous way. Surface engineering approaches are often divided into in situ or ex situ methods, where the presence of various additives in the electrolyte of batteries, supercapacitors, or fuel cells leads to in situ formation of the SLI and where the pretreatment of electrode materials to yield an engineered interface (e.g., a film or coating) represents an ex situ approach to tuning the SLI composition.
Graphene-based derivatives are a particularly attractive class of materials for ex situ design of engineered interfaces in ESC devices, as they exhibit superior properties such as high Young’s modulus, optical transparency, tunable electronic conductivity, and, most interestingly, the ability to directly modify their chemical functionality and surface morphology. Owing to these properties, graphene derivatives have found use in ESC applications as a conductive additives, supporting materials, gas diffusion layers, current collectors, insertion materials, and even as active cathode materials.1−3 Just focusing on its application at the interface, we have previously shown that fluorinated reduced graphene oxide can be used as an engineered barrier on metallic lithium to prevent the formation of high surface area lithium (dendrites) in Li metal batteries,4 and other groups have demonstrated graphene oxide (GO)5 and reduced graphene oxide (rGO)6 interfaces on Li electrodes as dendrite growth suppressors as well. Further, we have used derivatives of graphene as an interface at the sulfur cathode in the Li–S battery system to prevent polysulfide diffusion.7,8 Despite extensive application in battery applications, only a handful of examples exist of the use of graphene derivatives for ex situ engineering of metal electrode interfaces in fuel cell applications and electrocatalysis in general.9,10 A likely reason for this is that the graphene deposition methods used in battery applications lack the necessary control for sensitive electrocatalysis applications.
The most appropriate deposition methods for electrocatalysis applications are those that enable the formation of films with precisely defined thickness and crystal quality. Layer thickness is of particular importance for conversion reactions of gaseous species, as the permeability of a membrane is inversely proportional to its thickness, which in principle stipulates the flux of gas molecules available for the electrochemical reaction. Among the many processing techniques that enable ex situ interface modification with graphene, only two enable the deposition of high-quality graphene-based films with monolayer-level control: direct synthesis (e.g., chemical vapor deposition, CVD) and the Langmuir–Blodgett technique.11 Several groups have demonstrated CVD growth of graphene on metal electrodes for electrochemical applications,12,13 with one of the inherent properties of such interfaces having been shown to be that they are impermeable to gases other than H2.14 For example, Fu et al.15 demonstrated that Pt(111) surfaces modified with CVD-grown graphene act as selective interfaces for hydrogen underpotential deposition (HUPD) while blocking the adsorption of OH–, SO42–, and Cl–. In contrast to graphene, GO and rGO membranes exhibit inherent defects whose sizes are on the order of the size of gas molecules, rendering them gas-permeable,16 and can exhibit either inherent17 or engineered18 selectivity toward specific gaseous species due to their ability to be readily functionalized. As GO and rGO are only accessible via solution-based chemical synthesis routes, CVD-based approaches are impractical for ex situ interfacial engineering. Instead, uniform deposition of metal substrates can be achieved by using the Langmuir–Blodgett technique.19 Despite having been demonstrated before as a viable path to monolayer deposition of GO and rGO, the Langmuir–Blodgett technique has never been used for electrochemical applications, presenting an opportunity to expand the utility of this technique into a new technology area.
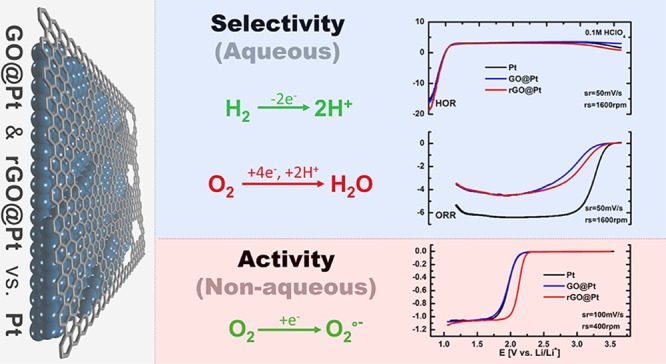
Herein, we make use of Pt disk electrodes modified ex situ by compact Langmuir–Blodgett GO and rGO films (GO@Pt and rGO@Pt, respectively) to demonstrate control over the selectivity of the interface for the hydrogen oxidation reaction (HOR) vis-à-vis the oxygen reduction reaction (ORR) in aqueous electrolytes. Both GO@Pt and rGO@Pt surfaces exhibit strong inhibition for the ORR and minimal differences in HOR activity relative to bare Pt. This selectivity is likely driven by the intrinsic defect structure of the GO and rGO layers, which provides sufficient access to the underlying Pt surface to enable H2 diffusion while excluding O2. Furthermore, the importance of surface functionalities in mediating outer-sphere electrochemical reactions was shown via the use of the ORR in nonaqueous media. Two orders of magnitude increase in ORR activity was observed on rGO@Pt relative to GO@Pt and bare Pt, which is likely driven by differences in the double layer structure deriving from changes in substrate–solvent interactions. These results point to the decisive role engineered solid–liquid interfaces can have in governing reaction kinetics and demonstrate that our modified Langmuir–Blodgett approach for the deposition of GO and rGO films offers a viable pathway for ex situ engineering of electrochemical interfaces to enable next-generation ECS technologies.
Results and Discussion
As discussed above, uniform monolayers are required to test the selectivity of ex situ engineered interfaces, and to this end, we developed a reproducible method for covering the whole surface area of a Pt disk electrode with GO. GO monolayers were deposited on Pt polycrystalline disk electrodes by transferring them from a Langmuir trough using the modified Langmuir–Blodgett (LB) approach shown in Figure 1a and Figure S1. Rather than dipping the samples at the traditional 90° relative to the air–water interface, we have employed a 30° dipping angle to minimize strain on the monolayer during deposition (see also Figure S2).19 To obtain a large area, near 100% coverage GO monolayers, we dipped at high surface pressures, as depicted by the blue dot in Figure 1b. Figure 1b shows a typical surface pressure (Π) versus area isotherm for GO on water in a Langmuir trough. As the barriers on the Langmuir trough are compressed, the GO monolayer experiences three distinct compression regimes, as described previously.19 The high surface pressure plateau marked with the blue dot in Figure 1b is characteristic of GO sheets that overlap strongly and wrinkle at the edges, leading to a highly compressed monolayer. As shown in Figure 1d, near 100% coverage was achieved with very few pinholes in the GO film. Dipping at surface pressures just below the high surface pressure plateau (red dot in Figure 1b) leads to gaps between the sheets (Figure 1c). Dark areas or irregular lines in Figure 1d indicate wrinkled GO, mostly evident where two sheets overlap at the edges of individual flakes. To confirm this assignment of wrinkled structures on the edges, we also performed atomic force microscopy (AFM) on locations where two GO flakes overlap (Figure S3a), and wrinkles with height in the order of 10 nm are clearly observed. More importantly, SEM analysis (Figure 1d) shows no visible holes in the GO monolayer interface on the Pt disk electrode. More SEM and optical images showing large area coverage and uniformity of the GO monolayers are presented in Figures S4 and S5, and energy-dispersive X-ray spectroscopy (EDS, Figure S6) shows that only Pt, C, and O species are detectable on these samples, confirming the high purity of the process without any deposition of residual impurities. For all further experiments, GO was deposited in the same surface pressure regime (blue dot, Figure 1b) to obtain fully covered disk electrodes.
Figure 1.

(a) Schematics of Langmuir–Blodgett trough depicting compression barriers and the holder for dipping the Pt disk electrodes. The holder is angled at 30° relative to the air–water interface, and the graphene oxide (GO) films were transferred in an upstroke direction. (b) A typical Π–Area isotherm for the GO monolayer on the Langmuir trough measured with Wilhelmy plate positioned parallel to barriers. (c) Representative SEM depicting GO sheets on a Pt disk dipped at the surface pressure corresponding to the red dot in (b). (d) Representative SEM depicting GO sheets on a Pt disk dipped at the surface pressure corresponding to the blue dot in (b). The dark areas (flake boundary) consist of regions where GO sheets overlap, and arrows point to possible diffusion sites for gaseous reactants to access the underlying Pt.
Pt electrodes prepared with GO were also annealed at 600 °C in a 4% H2/Ar atmosphere to generate thermally reduced GO (rGO), providing a facile means of modifying the conductivity and chemical functionality of the ex situ engineered interface. This temperature was chosen as lower annealing temperatures were insufficient to quantitatively reduce all oxygen functional groups of GO (XPS, Figure S7), and higher annealing temperatures have been previously shown to drive diffusion of carbon into the Pt crystal lattice during heating followed by carbon surface precipitation during cooling.20 Furthermore, studies have shown that most of the O functional groups (epoxides) are thermally reduced at temperatures between 150 and 450 °C, although hydroxyl and carbonyl groups can be stable up to 1000 °C.21 Thermogravimetric analysis (TGA) of bulk GO (Figure S8) indicates that the greatest weight loss occurs between 50 and 600 °C, indicating that 600 °C is indeed the optimal annealing temperature. SEM imaging after thermal reduction (Figure S9) indicates no microscopic changes of the film during thermal reduction, which was further confirmed by scanning tunneling microscopy (STM) (Figure S10) and AFM (Figure S11) imaging. All images indicate large area coverage and uniformity of the rGO monolayers on the Pt disk electrode after thermal treatment.
To evaluate the structural quality of GO and rGO materials deposited on Pt electrodes, Raman spectroscopy was used to assess the relative amounts of disordered (D band at ∼1345 cm–1) and graphitic (G band at 1610 cm–1) materials present in the crystal lattice. We must emphasize that Raman spectra were recorded using the same GO on the Pt disk (GO@Pt) electrode, which was thermally treated after initial measurements to generate an rGO on the Pt disk (rGO@Pt) surface. Raman spectra of GO@Pt surfaces (Figure 2a) are similar to those of self-standing GO that have been previously published.22 However, a relatively large blue shift of the G band compared to other similar GO-based materials can be clearly seen. A slight blue shift (∼13 cm–1) is common for GO as compared to graphene due to the presence of oxidized sp3-hybridized domains;23 however, the blue shift observed here (∼30 cm–1) is too large to be explained by this phenomenon alone. One possible explanation is that there is an additional interaction at the GO–Pt interface that further modifies the vibrational response of graphitic domains in GO. After reduction of GO to rGO, the G band of rGO broadens significantly (Figure 2b), which is due to the graphite to amorphous carbon transition observed previously.23 Interestingly, despite this broadening, the G band for the rGO@Pt surface remains at the same position as observed for GO@Pt (1610 cm–1), which supports our hypothesis that there is an additional interaction between the Pt substrate and graphitic domains in the deposited rGO material. Despite this difference in peak broadness, the D to G band intensity ratios (ID/IG) for GO@Pt and rGO@Pt surfaces were quite similar (0.70 and 0.71, respectively), which is expected for distorted graphene materials such as GO and rGO. Additionally, both surfaces exhibit significant spectral intensity at 2673 cm–1 (the so-called 2D band, also known as the G′ band), which Kaniyoor and Ramaprabhu previously attributed to the presence of wrinkles (extended surface defects) in wrinkled few-layer graphene synthesized from graphite.22 This is consistent with our assignment of the irregular black lines in the SEM image in Figure 1d as wrinkles formed the edges of GO flakes on the Pt disk electrode due to the high LB compression regime employed in this work (Figure 1b, blue dot).
Figure 2.

Raman spectroscopy analysis of the D, G, and 2D bands for (a) GO@Pt and (b) rGO@Pt. The rGO@Pt surface in (b) was generated via thermal reduction of the same GO@Pt surface analyzed in (a).
Although Raman spectroscopy suggests relatively similar bulk structures of GO@Pt and rGO@Pt surfaces, local defect chemistries that are not distinguishable via Raman analysis can drive significant differences in activity and stability. In particular, identifying the presence and atomic structure of oxygen-rich domains in GO is important because these structural regions become even more defective during thermal reduction as C is stripped off the GO lattice as evolved CO2 gas. Atomic resolution scanning transmission electron microscopy (STEM) imaging of self-standing GO films reveals a highly defective structure, consisting of point defects (black dots on STEM image in Figure 3a,b) and areas of brighter intensity that likely correspond to oxygen functionalities.24 Some of the regions are also intact honeycomb patterns, characteristic of ideal 2D graphene lattices (upper part of Figure 3b), consistent with the graphitic signal measured with Raman spectroscopy, as well as defective 2D graphitic regions connected in pentagons, heptagons, and sometimes even octagons as also described previously.25 To confirm that the bright parts/dots of Figure 3b are indeed oxygen functionalities, electron energy loss spectroscopy (EELS) was performed in these brighter areas (red square in Figure 3a) and compared to carbon-rich areas (gray square in Figure 3a). The high loss EELS spectrum in Figure 3c reveals the presence of oxygen K-edge signal, which is missing in the high loss EELS spectrum in Figure 3d. This is clear evidence that some regions are rich with oxygen functionalities, while other regions represent mostly 2D carbon structure.
Figure 3.
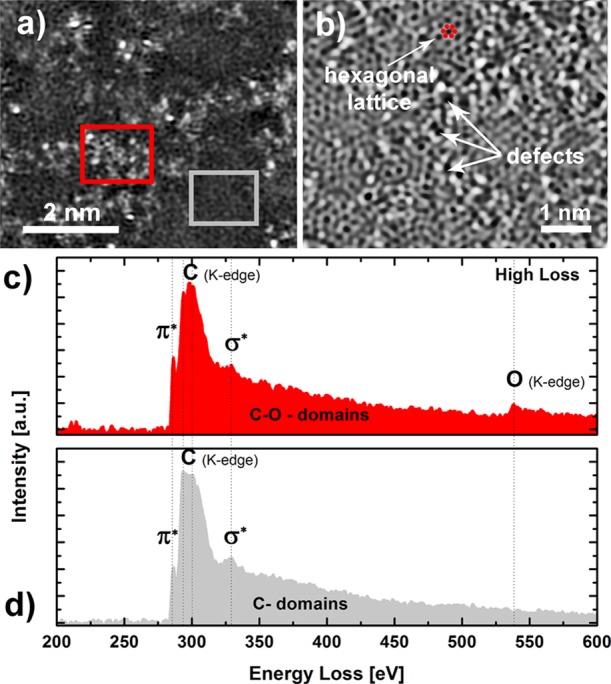
(a) HAADF-STEM image of the graphene oxide (GO). (b) Inverted BF-STEM image of GO with arrows pointing to defects—possible diffusion sites—and a domain with hexagonal lattice. (c) Electron energy loss (EEL) spectrum of carbon and oxygen K-edge of GO from the oxygen-rich red rectangular area marked on HAADF-STEM image (a). (d) EEL spectrum of carbon K-edge of GO from gray rectangular area marked on HAADF-STEM image (a).
Having confirmed the presence of oxygen-rich domains in as-prepared GO, it is critical to understand the chemical nature of these functional groups and how they evolve during conversion to rGO, as the surface chemistry of GO@Pt versus rGO@Pt surfaces will determine their ultimate electrochemical activity and stability. To gain insights into the surface chemistry of GO and rGO surfaces, X-ray photoelectron spectroscopy (XPS) analysis was performed. XPS survey spectra (Figures S12 and S13) indicate the same elemental composition as was observed with EDS (Figure S6), confirming the high purity of the GO and rGO layers. Deconvolution of the C 1s core level spectra for GO@Pt surfaces (Figure 4a) reveals at least five different carbon environments: C=C [sp2] (284.6 eV), C–C [sp3] (285.2 eV), C–O (286.3 eV), C=O (287.4 eV), and O—C=O (289.4 eV). Note that C–O functionality can be assigned to C–OH (hydroxyl) or C–O–C (epoxy) functional groups. A similar distribution of carbon functionalities was observed previously for GO films supported on silicon nitride.26 In contrast, thermal reduction of GO@Pt to rGO@Pt yields surfaces with only three different carbon functionalities (Figure 4b): C=C [sp2] (284.6 eV), C–O (286.3 eV), and C=O (287.3 eV). Further, the concentration of C–O and C=O functionalities dropped substantially from 15.1 to 3.4 at % and from 19.8 to 0.2 at %, respectively (for detailed quantification, see Table S1). This indicates that reduction of oxygen functionalities was almost quantitative. Reduction also results in a significant decrease (but not total removal) of the O 1s peak intensity, which is further consistent with extensive but incomplete GO reduction (Figure S13).
Figure 4.

C 1s XPS core level spectra with spectral deconvolution for (a) GO@Pt and (b) rGO@Pt surfaces. Corresponding contact angle measurements for (c) GO@Pt and (d) rGO@Pt surfaces measured via the sessile drop technique.
The reduction of oxygen functionalities for the rGO@Pt interface relative to GO@Pt manifests in a corresponding difference in the wettability of both surfaces, as demonstrated by contact angle measurements (Figure 4c,d). Sessile drop measurements reveal 69 and 100° contact angles for GO@Pt and rGO@Pt surfaces, respectively, indicating that the rGO monolayers are much more hydrophobic than the GO monolayers. The rGO monolayers are more hydrophobic after heat treatment because the functional groups on the GO are reduced and the sp3 carbon is converted to sp2 carbon, as evidenced by the XPS measurements. Both GO and rGO are more hydrophobic than the unmodified Pt surface after heat treatment in 4% H2/Ar, which exhibits a contact angle that spreads with time and varies from 34 to 48°, stabilizing at 48° after a few minutes (Figure S14). Note that in contrast, the drops on GO and rGO monolayers were very stable. Wettability can have an impact on transport and effective diffusivity of reagents to the Pt catalyst27 and further suggests that these surfaces may exhibit differences in their electrochemical behavior.
The above insights into the atomic-scale defect structure and chemical functionality of both GO@Pt and rGO@Pt surfaces provide a foundation from which to rationalize the electrochemical response of these engineered interfaces and ultimately understand the origins of their activity and selectivity. We begin by analyzing the intrinsic cyclic voltammetry (CV) responses of the interfaces in an Ar-saturated 0.1 M HClO4 electrolyte, shown in Figure 5c. While the adsorption of hydrogen (HUPD) between 0.1 and 0.4 V is hindered relative to bare Pt (30 and 50% for GO@Pt and rGO@Pt, respectively), the adsorption of hydroxyl species (OHad) between 0.7 and 0.9 V is significantly more blocked (60 and 90% for GO@Pt and rGO@Pt, respectively). This indicates a significant selectivity toward smaller species, that is, HUPD as compared to OHad. As these species are believed to be intermediates in the hydrogen oxidation reaction (HOR, reaction 1) and oxygen reduction reaction (ORR, reaction 2), respectively, we consequently anticipate a pronounced selectivity for these two reactions as well.
| 1 |
| 2 |
Figure 5.

Selectivity of the HOR and ORR on bare Pt, GO@Pt, and rGO@Pt disk electrodes. Polarization curves for (a) HOR and (b) ORR. (c) Cyclic voltammograms for bare Pt (black line), GO@Pt (blue line), and rGO@Pt (red line) in 0.1 M HClO4. (d) Schematic of rGO@Pt surface exhibiting the selectivity toward the HOR and ORR. (e) Cyclic voltammetry and (f) polarization curves of O2 electrochemistry in 0.15 M TBAPF6/DME saturated with 10% O2 in Ar on bare Pt (black line), GO@Pt (blue line), and rGO@Pt surfaces (red line). Sweep and rotation rates are noted as appropriate for each curve.
Indeed, polarization curves for the HOR in 0.1 M HClO4 (Figure 5a) clearly demonstrate that the measured reaction rates are basically the same for GO@Pt, rGO@Pt, and bare Pt electrodes. A similar response was also observed previously with a calix[4]arene-modified Pt surface,28 which was attributed to the small number of Pt catalytic sites necessary to push the HOR into the diffusion-controlled regime. Along the same lines, we can conclude that both GO and rGO layers allow enough access of H2 to the Pt surface that the reaction is not measurably hindered under our experimental conditions. The HOR most likely takes place on those parts of the surface where H2 can penetrate the GO/rGO overlayer, with the point defects or wrinkles at the grain boundaries demonstrated with SEM, STEM, and Raman spectroscopy serving as the most likely access points for H2 to the Pt surface. These findings indicate that our engineered catalyst with GO and rGO interfaces is still highly active toward the HOR.
In contrast to the HOR, the ORR is a far more complex reaction that requires a larger ensemble of Pt sites for it to take place. Close inspection of ORR polarization curves in Figure 5b reveals significant deactivation of GO@Pt and rGO@Pt catalysts. The inhibition of the ORR is so pronounced that the theoretical diffusion-limited currents expected for the disk geometric area were not observed. Thus, it is reasonable to say that GO and rGO interfaces selectively inhibit the ORR on the Pt electrode while allowing the HOR to proceed unhindered. The reason for this selectivity is likely that the defects in the atomic structure of rGO and GO and/or wrinkles in GO and rGO films on the Pt electrode are of the correct size to render them permeable for H2/HUPD but not for O2/OHad species. A similar explanation was also assumed for self-assembled calix[4]arene monolayers on the Pt electrodes28 where access of reactants was possible either through the macromolecular ring or between individual molecules. To underscore the importance of defects in selectivity of the GO- and rGO-modified interfaces, we also tested the HOR and ORR activities on CVD-grown graphene on Pt. Due to the extremely low density of defects, this interface exhibited almost no activity for either of the reactions (Figures S15 and S16), indicating that no selectivity can be achieved with nondefective 2D graphene structures. Defects in the 2D structure of GO and rGO as well as wrinkles between individual GO and rGO flakes are therefore required for selectivity of the interface-engineered electrode, as shown in the scheme in Figure 5d.
Interestingly, there is also a slight difference in ORR activity between the two surfaces, with lower kinetic currents observed for the ORR on GO@Pt as compared with rGO@Pt. This indicates that the rGO interface exhibits a higher activity for the ORR than GO, despite the fact that both electrodes are relatively inactive as compared to the pure Pt disk electrode (black line, Figure 5b). At 0.9 V, rGO@Pt and GO@Pt are 5 and 10 times less active than bare Pt, respectively. In addition to possible differences in defect populations, this difference in activity between the two surfaces may also be driven by differences in the population of surface functional groups on GO@Pt and rGO@Pt as measured by XPS. The higher fraction of oxygen functionalities on the GO surface relative to rGO may result in stronger electrostatic interactions with the solvent at the GO@Pt surface, leading to an increased immobilization of the solvent at the interface and thus to a decrease of available sites for the reaction, leading to an overall lower reaction rate.
To further explore whether differences in surface functionality and their interaction with the solvent can drive differences in activity as well, GO@Pt and rGO@Pt interfaces were also tested for their ORR activity in a nonaqueous environment. In aprotic solvents, the ORR is a single electron reaction that leads to superoxide anion (O2–) formation per reaction 3:29
| 3 |
CV and RDE experiments were performed in 0.15 M tetrabutylammonium hexafluorophosphate (TBAPF6) in 1,2-dimethoxyethane (DME), as it has been previously shown that the ORR in TBAPF6 in DME is an outer-sphere process.29 This means that the reactant O2 does not come into direct contact with the electrode surface prior to electron transfer, but rather, electron transfer occurs via the tunneling of the electron through the solvation shell of the reactant and possible additional molecular species that are adsorbed or otherwise immobilized at the electrode surface. As a result, we assume that the effects of surface defects should be minimized as compared with the ORR in aqueous media, and surface functional groups play a more important role due to the presence or absence of steric effects from solvent interactions with oxygen functionalities on the surface that can modify the double layer structure and the resulting electron transfer kinetics as shown previously.29
In Figure 5e, the 250 mV difference between the anodic and cathodic peaks clearly shows that the ORR is irreversible for both bare Pt and GO@Pt surfaces. In contrast, a much smaller difference between the oxidation and reduction peaks (60 mV) is observed for rGO@Pt surfaces, indicating reversible ORR activity. In RDE experiments, this yields a 200 mV lower overpotential for the ORR on rGO@Pt as compared to the other surfaces, which corresponds more than two orders of magnitude higher activity compared to bare Pt and GO@Pt surfaces. Because of the high concentration of the supporting electrolyte and the charge neutrality of the reactant O2, we assume that double layer effects on the electrode kinetics, such as diminishing of the effective electrode potential or changes in the reactant concentration at the outer Helmholtz plane (OHP) compared to outside the diffuse double layer, can be largely excluded. Instead, a more likely reason for the higher activity on rGO@Pt surfaces is a different distribution and/or orientation of the solvent molecules in the double layer formed at each surface, effectively leading to changes in the distance of the OHP from the electrode surface or to a different density of states at the OHP. Both will effectively change the probability of electron transfer through tunneling at a specific potential, hence affecting the activity. Based on contact angle experiments, rGO@Pt surfaces are significantly more hydrophobic than GO@Pt or bare Pt, which supports the proposed differences in the interaction between the solvent and the three surfaces. XPS analysis further suggests that the contrasting electrode–solvent interactions come from the nature and abundance of the surface functional groups on the different electrodes, with the rGO interface exhibiting a primarily C=C [sp2] functionalities as compared to the large portion of oxygenated functional groups on GO surfaces (Figure 4a,b). These differences in surface functionalization are likely significant enough to change the orientation of the solvent molecules at the interface and modify the electron transfer, yielding the significant differences in activity observed. Importantly, the enhanced activity of rGO@Pt-engineered interfaces points to an important opportunity for ECS technologies involving nonaqueous ORR, for example, Li–air batteries, as engineering of surface functionality may make it possible to significantly enhance reaction rates and/or reversibility in these systems.
Conclusions
We have demonstrated that the activity and selectivity of electrochemical interfaces can be successfully controlled via ex situ engineering with defective GO and rGO layers. Successful deposition of uniform, compact, and high coverage GO monolayers onto Pt disk electrodes was achieved using a modified Langmuir–Blodgett technique, and these GO@Pt surfaces were able to be further transformed to rGO@Pt via thermal annealing. Both interfaces were subject to detailed spectroscopic, microscopic, surface, and electrochemical studies, which revealed significant differences in both the surface defect structure and chemical functionality. Electrochemical studies revealed that ex situ formed GO and rGO@Pt surfaces introduce selective inhibition of the ORR in aqueous electrolytes while exhibiting no impact on HOR activity. This unique behavior likely derives from the intrinsic GO and rGO defect structures, as well as to wrinkles that are formed between individual GO and rGO flakes on top of the Pt electrode. Differences in ORR activity between GO and rGO@Pt surfaces further suggested the role of surface functional groups in mediating electrochemical reactions. Indeed, for the outer-sphere ORR in nonaqueous media, rGO@Pt surfaces exhibited two orders of magnitude higher activity toward the ORR than GO@Pt and bare Pt, which likely derives from differences in the double layer structure due to substrate–solvent interactions that result from differences in surface chemistry. The concept of ex situ interface engineering for selectivity developed in this work points to a new methodology for addressing a wide variety of issues in both aqueous and nonaqueous systems, for example, cathode degradation during the shutdown/startup conditions in aqueous proton exchange membrane fuel cells and metal–air conversion chemistries for nonaqueous battery applications. We believe that the development and synthesis of ex situ engineered interfaces, coupled with systematic studies of their structure, activity, and stability, will lead to rapid progress in the development of next-generation energy storage and conversion technologies.
Experimental Methods
Chemicals and General Purification and Synthesis Procedures
Graphene oxide (GO) was synthesized according to the modified Hummers method as previously described.30 H2SO4 (ACS reagent, 95.0–98.0%, Sigma-Aldrich), H3PO4 (BioReagent, 85%, Sigma-Aldrich), KMnO4 (Sigma-Aldrich), graphite flakes (Sigma-Aldrich), and H2O2 (30–35%, Fisher Science Education) were used without further purification. Milli-Q water was used in an isolation/purification method.
Aqueous EC Experiments
HClO4 (0.1 M) was prepared from 7 g of 70% perchloric acid (OmniTrace Ultra, EDM) (7 g) and 500 mL of Milli-Q water and then purified electrochemically.
Nonaqueous EC Experiments
All experiments were performed in dried glassware under an Ar atmosphere unless stated otherwise. 1,2-Dimethoxyethane (DME) (CHROMASOLV, for HPLC grade, Sigma-Aldrich) was dried over activated basic Al2O3, filtered, and distilled over liquid Na/K alloy. Caution: All steps involving Na/K alloy should be carried out with extreme caution under strict exclusion of air or moisture, under inert gas and appropriate personal protection (hood, blast shields, face shield, protective, and fire-resistant clothing) should be used and worn at all times. Tetrabutylammonium hexafluorophosphate (TBAPF6) (for electrochemical analysis, Sigma-Aldrich) was dried in vacuum (∼10–2 mbar) at 100 °C for 24 h. TBAPF6 (0.15 M) in DME was prepared in a glovebox and used without further purification.
Langmuir–Blodgett Deposition and GO Reduction
Before introducing GO to the Langmuir trough (KSV Nima medium size, 354 × 75 × 4 mm), the trough was thoroughly cleaned with DI water, acetone, isopropanol, and again with Milli-Q water. Any remaining contaminants were aspirated from the surface until barrier compression resulted in a surface pressure (Π) of <0.3 mN/m, as measured by a Pt Wilhelmy plate. The spreading solution consisted of a 0.04 mg/mL GO suspension in a 1:5 mixture of Milli-Q water–methanol. Approximately 30–40 mL of the spreading solution was deposited dropwise using a syringe pump at a rate of 6 mL/h. The methanol was then allowed to evaporate for at least 1 h before depositing GO onto substrates. Clean, annealed Pt electrodes with surfaces protected by Milli-Q water drops were secured onto a custom-built stainless steel holder (Figure 1a and Figure S1). The holder was designed such that the crystals were dipped at 30° relative to the air–water interface to improve dipping results, as is discussed previously and in the Supporting Information.19 After lowering the sample into the trough, the barriers were compressed at 50 mm/min to surface pressures corresponding to the region in the isotherm with high sheet-edge overlap, as depicted by the blue dot in Figure 1b. The surface pressure was held constant during upstroke dips, which were performed at 0.3 mm/min after allowing for a 2 min stabilization period. The rGO interface on Pt electrodes was prepared by thermal reduction from the GO interface on Pt. GO at the Pt electrode was exposed to a constant flow of 4% H2/Ar at 600 °C for 10 min and let to slowly cool down.
Scanning Electron Microscope
SEM images were taken on an FEI Magellan at 1 kV.
Contact Angle Measurements
Measurements were taken on a dynamic contact angle analyzer (First Ten Angstroms).
Raman Spectroscopy
The Raman spectra were acquired using a Renishaw inVia confocal Raman microscope, and 532 nm wavelength laser was used for excitation.
X-ray Photoelectron Spectroscopy (XPS) Measurements
XPS measurements were performed using a SPECS PHOIBOS 150 hemispherical energy analyzer with a monochromated Al Kα X-ray source. All measurements were performed at room temperature under ≤6 × 10–10 mbar vacuum. Survey spectra were measured using a pass energy of 40 eV at a resolution of 0.2 eV/step and a total integration time of 0.1 s/point. Core level spectra were measured using a pass energy of 20 eV at a resolution of 0.05 eV/step and a total integration time of 0.5 s/ point. As carbon is the species of interest in this study, adventitious carbon at 284.8 eV cannot be reliably utilized as a charge reference. Instead, samples were charge-referenced against the Pt 4f7/2 peak at 71.2 eV. Deconvolution was performed using CasaXPS software with a Shirley-type background and 70–30 Gaussian–Lorentzian peak shapes for all peaks but sp2 carbon, which was fit using an asymmetric peak shape. All carbon species exhibit a slight shift to higher binding energy than expected, which is likely due to several factors. First, we have previously observed a ∼0.4 eV shift to higher binding energy for graphene supported on Pt(111) surfaces relative to graphite,31 and a similar shift to higher binding energy is likely present for the GO and rGO@Pt materials in this study as well. Although the GO and rGO layers are quite thin, we also cannot rule out a small degree of differential charging, especially given the lower conductivity of GO relative to Pt, which would induce a small shift to positive binding energy as well.
Scanning Transmission Electron Microscopy (STEM)
Probe Cs-corrected scanning transmission electron microscopy (STEM) was performed with a Jeol ARM 200 CF with cold FEG and equipped with a Jeol Centurio EDXS system with a 100 mm2 SDD detector and Gatan GIF Quantum ER dual EELS system. Graphene oxide samples were dispersed in Milli-Q water and transferred to lacey carbon-coated Cu TEM grids. To minimize the sample damage, 80 kV accelerating voltages were used in this study. High-angle annular dark field (HAADF) and bright field (BF) images were collected simultaneously.
Electrochemistry
Aqueous
Pt electrodes of 6 mm in diameter were used in the experiments. Pt electrodes were annealed before each experiment using RF induction heating at 1100 °C under a 4% H2/Ar stream. For the ORR in aqueous electrolytes, pure O2 was used as a reagent. The aqueous experiments were performed in a glass cell in a three-electrode setup. A Ag/AgCl reference electrode was employed, and the potential was recalculated with respect to the reversible hydrogen electrode (RHE) scale.
Nonaqueous
For the ORR in nonaqueous electrolytes, 10% O2/Ar mixture was used as a reagent. Water content in electrolytes was measured using Karl Fischer titration (Mettler-Toledo) placed inside an Ar-filled glovebox with a H2O level below 0.5 ppm. Water concentration of supporting electrolytes was measured before each experiment and was always <0.2 ppm (method detection limit). All experiments were carried out inside of an Ar-filled glovebox and in a home-designed glass cell with ∼35 mL of electrolyte. A three-electrode system was employed with Pt wire as a counter electrode and Ag/Ag+ nonaqueous quasi-reference electrode (QRE) reference electrode separated by a Luggin capillary. The counter electrode compartment was separated from the main compartment by a frit. The Ag/Ag+ QRE reference was sealed in a separate compartment divided from the rest of the cell by a Vycor tip. The working electrode was tightly held in a Kel-F collet and sealed by a PTFE u-cap to ensure that only the defined surface was in contact with the electrolyte. The reference potential was calibrated by measurement of the ferrocene redox couple. For the experiments in organic solvents, all potentials were recalculated versus the standard Li/Li+ couple. The Pine Instruments rotators used in the RDE measurements were set to 400 rpm unless stated otherwise. Autolab potentiostats were used in the electrochemical measurements, and IR drop correction was used in all the experiments.
Acknowledgments
This work was supported as part of the Joint Center for Energy Storage Research (JCESR), an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences. XPS, Raman, and nonaqueous electrochemical measurements were performed at the Electrochemical Discovery Laboratory, a JCESR facility at Argonne National Laboratory. The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (“Argonne”). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under contract no. DE-AC02-06CH11357. Work related to electrochemistry in aqueous electrolytes was supported by the U.S. Department of Energy, Basic Energy Science, Materials Science and Engineering Division. Financial support from the Slovenian Research Agency (ARRS) through the Research Core Funding Programme P1-0175, Project BI-US/18-19-041 is also fully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.9b13391.
Additional figures, SEM, AFM, STM, and optical images, evolved gas analysis, contact angle measurements, XPS, EDS, CVs, and RDE experiments (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hou J.; Shao Y.; Ellis M. W.; Moore R. B.; Yi B. Graphene-Based Electrochemical Energy Conversion and Storage: Fuel Cells, Supercapacitors and Lithium Ion Batteries. Phys. Chem. Chem. Phys. 2011, 13, 15384–15402. 10.1039/c1cp21915d. [DOI] [PubMed] [Google Scholar]
- Ye M.; Zhang Z.; Zhao Y.; Qu L. Graphene Platforms for Smart Energy Generation and Storage. Joule 2018, 2, 245–268. 10.1016/j.joule.2017.11.011. [DOI] [Google Scholar]
- Bai L.; Zhang Y.; Tong W.; Sun L.; Huang H.; An Q.; Tian N.; Chu P. K. Graphene for Energy Storage and Conversion: Synthesis and Interdisciplinary Applications. Electrochem. Energy Rev. 2019, 1–36. 10.1007/s41918-019-00042-6. [DOI] [Google Scholar]
- Bobnar J.; Lozinšek M.; Kapun G.; Njel C.; Dedryvère R.; Genorio B.; Dominko R. Fluorinated Reduced Graphene Oxide as a Protective Layer on the Metallic Lithium for Application in the High Energy Batteries. Sci. Rep. 2018, 8, 5819. 10.1038/s41598-018-23991-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. J.; Xia X. H.; Wang X. L.; Gu C. D.; Tu J. P. Graphene Oxide Modified Metallic Lithium Electrode and Its Electrochemical Performances in Lithium-Sulfur Full Batteries and Symmetric Lithium-Metal Coin Cells. RSC Adv. 2016, 6, 66161–66168. 10.1039/c6ra13039a. [DOI] [Google Scholar]
- Zhang Y. J.; Xia X. H.; Wang D. H.; Wang X. L.; Gu C. D.; Tu J. P. Integrated Reduced Graphene Oxide Multilayer/Li Composite Anode for Rechargeable Lithium Metal Batteries. RSC Adv. 2016, 6, 11657–11664. 10.1039/c5ra25553h. [DOI] [Google Scholar]
- Vizintin A.; Patel M. U. M.; Genorio B.; Dominko R. Effective Separation of Lithium Anode and Sulfur Cathode in Lithium-Sulfur Batteries. ChemElectroChem 2014, 1, 1040–1045. 10.1002/celc.201402039. [DOI] [Google Scholar]
- Vizintin A.; Lozinšek M.; Chellappan R. K.; Foix D.; Krajnc A.; Mali G.; Drazic G.; Genorio B.; Dedryvère R.; Dominko R. Fluorinated Reduced Graphene Oxide as an Interlayer in Li–S Batteries. Chem. Mater. 2015, 27, 7070–7081. 10.1021/acs.chemmater.5b02906. [DOI] [Google Scholar]
- Ambrosi A.; Chua C. K.; Bonanni A.; Pumera M. Electrochemistry of Graphene and Related Materials. Chem. Rev. 2014, 114, 7150–7188. 10.1021/cr500023c. [DOI] [PubMed] [Google Scholar]
- Yan D.; Li Y.; Huo J.; Chen R.; Dai L.; Wang S. Defect Chemistry of Nonprecious-Metal Electrocatalysts for Oxygen Reactions. Adv. Mater. 2017, 29, 1606459. 10.1002/adma.201606459. [DOI] [PubMed] [Google Scholar]
- El-Kady M. F.; Shao Y.; Kaner R. B. Graphene for Batteries, Supercapacitors and Beyond. Nat. Rev. Mater. 2016, 1, 16033. 10.1038/natrevmats.2016.33. [DOI] [Google Scholar]
- Brownson D. A. C.; Banks C. E. CVD Graphene Electrochemistry: The Role of Graphitic Islands. Phys. Chem. Chem. Phys. 2011, 13, 15825–15828. 10.1039/c1cp21978b. [DOI] [PubMed] [Google Scholar]
- Ambrosi A.; Bonanni A.; Sofer Z.; Pumera M. Large-Scale Quantification of CVD Graphene Surface Coverage. Nanoscale 2013, 5, 2379–2387. 10.1039/c3nr33824j. [DOI] [PubMed] [Google Scholar]
- Bunch J. S.; Verbridge S. S.; Alden J. S.; van der Zande A. M.; Parpia J. M.; Craighead H. G.; Mceuen P. L. Impermeable Atomic Membranes From Graphene Sheets. Nano Lett. 2008, 8, 2458–2462. 10.1021/nl801457b. [DOI] [PubMed] [Google Scholar]
- Fu Y.; Rudnev A. V.; Wiberg G. K. H.; Arenz M. Single Graphene Layer on Pt(111) Creates Confined Electrochemical Environment via Selective Ion Transport. Angew. Chem., Int. Ed. 2017, 56, 12883–12887. 10.1002/anie.201705952. [DOI] [PubMed] [Google Scholar]
- Dimiev A. M.; Eigler S.. Graphene Oxide: Fundamentals and Applications; Wiley, 2016; https://doi.org/10.1002/9781119069447. [Google Scholar]
- Pierleoni D.; Minelli M.; Ligi S.; Christian M.; Funke S.; Reineking N.; Morandi V.; Doghieri F.; Palermo V. Selective Gas Permeation in Graphene Oxide-Polymer Self-Assembled Multilayers. ACS Appl. Mater. Interfaces 2018, 10, 11242–11250. 10.1021/acsami.8b01103. [DOI] [PubMed] [Google Scholar]
- Wang S.; Tian Z.; Dai S.; Jiang D. E. Effect of Pore Density on Gas Permeation through Nanoporous Graphene Membranes. Nanoscale 2018, 10, 14660–14666. 10.1039/c8nr02625d. [DOI] [PubMed] [Google Scholar]
- Harrison K. L.; Biedermann L. B.; Zavadil K. R. Mechanical Properties of Water-Assembled Graphene Oxide Langmuir Monolayers: Guiding Controlled Transfer. Langmuir 2015, 31, 9825–9832. 10.1021/acs.langmuir.5b01994. [DOI] [PubMed] [Google Scholar]
- Selman G. L.; Ellison P. J.; Darling A. S. Carbon in Platinum and Palladium. Solubility Determinations and Diffusion at High Temperatures. Platinum Met. Rev. 1970, 14, 14–20. [Google Scholar]
- D’Angelo D.; Bongiorno C.; Amato M.; Deretzis I.; La Magna A.; Fazio E.; Scalese S. Oxygen Functionalities Evolution in Thermally Treated Graphene Oxide Featured by EELS and DFT Calculations. J. Phys. Chem. C 2017, 121, 5408–5414. 10.1021/acs.jpcc.7b00239. [DOI] [Google Scholar]
- Kaniyoor A.; Ramaprabhu S. A Raman Spectroscopic Investigation of Graphite Oxide Derived Graphene. AIP Adv. 2012, 2, 032183 10.1063/1.4756995. [DOI] [Google Scholar]
- Kudin K. N.; Ozbas B.; Schniepp H. C.; Prud’homme R. K.; Aksay I. A.; Car R. Raman Spectra of Graphite Oxide and Functionalized Graphene Sheets. Nano Lett. 2008, 8, 36–41. 10.1021/nl071822y. [DOI] [PubMed] [Google Scholar]
- Wilson N. R.; Pandey P. A.; Beanland R.; Young R. J.; Kinloch I. A.; Gong L.; Liu Z.; Suenaga K.; Rourke J. P.; York S. J.; Sloan J. Graphene Oxide: Structural Analysis and Application as a Highly Transparent Support for Electron Microscopy. ACS Nano 2009, 3, 2547–2556. 10.1021/nn900694t. [DOI] [PubMed] [Google Scholar]
- Jia Y.; Zhang L.; Du A.; Gao G.; Chen J.; Yan X.; Brown C. L.; Yao X. Defect Graphene as a Trifunctional Catalyst for Electrochemical Reactions. Adv. Mater. 2016, 28, 9532–9538. 10.1002/adma.201602912. [DOI] [PubMed] [Google Scholar]
- Yang D.; Velamakanni A.; Bozoklu G.; Park S.; Stoller M.; Piner R. D.; Stankovich S.; Jung I.; Field D. A.; Ventrice C. A. Jr.; Ruoff R. S. Chemical Analysis of Graphene Oxide Films after Heat and Chemical Treatments by X-Ray Photoelectron and Micro-Raman Spectroscopy. Carbon 2009, 47, 145–152. 10.1016/j.carbon.2008.09.045. [DOI] [Google Scholar]
- Fathi H.; Raoof A.; Mansouri S. H. Insights into the Role of Wettability in Cathode Catalyst Layer of Proton Exchange Membrane Fuel Cell; Pore Scale Immiscible Flow and Transport Processes. J. Power Sources 2017, 349, 57–67. 10.1016/j.jpowsour.2017.03.012. [DOI] [Google Scholar]
- Genorio B.; Strmcnik D.; Subbaraman R.; Tripkovic D.; Karapetrov G.; Stamenkovic V. R.; Pejovnik S.; Marković N. M. Selective Catalysts for the Hydrogen Oxidation and Oxygen Reduction Reactions by Patterning of Platinum with Calix [4] Arene Molecules. Nat. Mater. 2010, 9, 998–1003. 10.1038/nmat2883. [DOI] [PubMed] [Google Scholar]
- Genorio B.; Staszak-Jirkovský J.; Assary R. S.; Connell J. G.; Strmcnik D.; Diesendruck C. E.; Lopes P. P.; Stamenkovic V. R.; Moore J. S.; Curtiss L. A.; Markovic N. M. Superoxide (Electro)Chemistry on Well-Defined Surfaces in Organic Environments. J. Phys. Chem. C 2016, 120, 15909–15914. 10.1021/acs.jpcc.5b12230. [DOI] [Google Scholar]
- Marcano D. C.; Kosynkin D. V.; Berlin J. M.; Sinitskii A.; Sun Z.; Slesarev A. S.; Alemany L. B.; Lu W.; Tour J. M. Correction to Improved Synthesis of Graphene Oxide. ACS Nano 2018, 12, 2078–2078. 10.1021/acsnano.8b00128. [DOI] [PubMed] [Google Scholar]
- Strmcnik D.; Castelli I. E.; Connell J. G.; Haering D.; Zorko M.; Martins P.; Lopes P. P.; Genorio B.; Østergaard T.; Gasteiger H. A.; Maglia F.; Antonopoulos B. K.; Stamenkovic V. R.; Rossmeisl J.; Markovic N. M. Electrocatalytic Transformation of HF Impurity to H2 and LiF in Lithium-Ion Batteries. Nat. Catal. 2018, 1, 255–262. 10.1038/s41929-018-0047-z. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.