

Abstract
The inhibition of ornithine aminotransferase (OAT), a pyridoxal 5′-phosphate-dependent enzyme, has been implicated as a treatment for hepatocellular carcinoma (HCC), the most common form of liver cancer, for which there is no effective treatment. From a previous evaluation of our aminotransferase inhibitors, (1S,3S)-3-amino-4-(perfluoropropan-2-ylidene)cyclopentane-1-carboxylic acid hydrochloride (1) was found to be a selective and potent inactivator of human OAT (hOAT), which inhibited the growth of HCC in athymic mice implanted with human-derived HCC, even at a dose of 0.1 mg/kg. Currently, investigational new drug (IND)-enabling studies with 1 are underway. The inactivation mechanism of 1, however, has proved to be elusive. Here we propose three possible mechanisms, based on mechanisms of known aminotransferase inactivators: Michael addition, enamine addition, and fluoride ion elimination followed by conjugate addition. On the basis of crystallography and intact protein mass spectrometry, it was determined that 1 inactivates hOAT through fluoride ion elimination to an activated 1,1′-difluoroolefin, followed by conjugate addition and hydrolysis. This result was confirmed with additional studies, including the detection of the cofactor structure by mass spectrometry and through the identification of turnover metabolites. On the basis of this inactivation mechanism and to provide further evidence for the mechanism, analogues of 1 (19, 20) were designed, synthesized, and demonstrated to have the predicted selective inactivation mechanism. These analogues highlight the importance of the trifluoromethyl group and provide a basis for future inactivator design.
Graphical Abstract
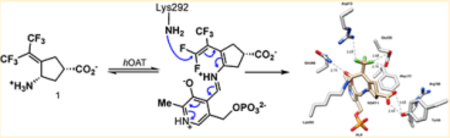
INTRODUCTION
Liver cancer is the second leading cause of cancer death worldwide;1 hepatocellular carcinoma (HCC) accounts for 90% of liver cancer in the United States.2 Current treatments for liver cancer consist of surgery, radiation, and chemotherapy; however, HCC is highly resistant to both radiation3 and chemotherapy4,5 The development of HCC has been shown to correlate with the activation of the Wnt/β-catenin signaling pathway in the liver.6 In addition to its diverse important physiological functions in the liver, β-catenin acts as a proto-oncogene to regulate transcriptional programs governing the initiation and progression of cancer,7 generally as a result of mutations in members of the Wnt/β-catenin pathway. Activation or mutation of the Wnt/β-catenin signaling pathway and the development of HCC in the liver correlate with the upregulation of ornithine aminotransferase (OAT), glutamate transporter GLT-1, and glutamine synthetase.8,9 The conditional knockout of β-catenin in the liver significantly reduces the level of those three proteins and blocks l-glutamine synthesis as a result of the loss of OAT activity.10 OAT converts ornithine and α-ketoglutarate to glutamate-γ-semialdehyde, which cyclizes to Δ−1-pyrroline-5-carboxylate (P5C) and l-glutamate (Figure 1a).11–13 The l-glutamate produced is transported away from the cell (by the GLT-1 transporter), so it does not become toxic to the cell and is converted to nonessential amino acid l-glutamine by l-glutamine synthetase. Glutamine metabolism plays a role in the uptake of essential amino acids and redox biology; l-glutamine also supports anabolic processes that stimulate cell proliferation.13 Tumor cells require enhanced amounts of l-glutamine; therefore, they optimize l-glutamine metabolism to sustain proliferation.14 Generating l-glutamine from ornithine via OAT provides cancer cells with an additional source of l-glutamine that is distinct from canonical de novo l-glutamine synthesis. OAT is overexpressed in HCC; therefore, inhibiting OAT has been suggested to be a novel treatment for liver cancer and was shown to be effective in mice.15,16 We previously demonstrated that compound 1 was a potent inactivator of hOAT, and the administration of 1 to human-derived HCC infected mice resulted in a decrease in tumor size and in the HCC biomarker protein, α-fetoprotein.15
Figure 1.

Catalytic mechanism of human ornithine aminotransferase (hOAT) and earlier-designed aminotransferase inactivators 1 and 2.
To rationally develop new mechanism-based inactivators of OAT, an understanding of the mechanism by which 1 inactivates OAT is essential. Compound 1 was originally designed as an analogue of a potent inactivator of γ-aminobutyric acid aminotransferase (GABA-AT), namely, CPP-115 (2), and was intended to inhibit the conversion of ornithine to P5C.17 Mechanistically, the incubation of ornithine with OAT leads to aldimine A (Scheme 1). Deprotonation of the α-proton and tautomerization yield C via B; intermediate C undergoes cyclization to form P5C (D) and pyridoxamine-5′-phosphate (PMP). Compound 1 is a mechanism-based enzyme inactivator (MBEI) of hOAT. A MBEI is an inactive compound that is converted to an active species by the target enzyme’s normal catalytic machinery. This active intermediate can form a covalent bond with the target enzyme itself or can bind tightly.18 Because of their increased selectivity toward a specific enzyme, MBEIs can exhibit fewer off-target side effects.
Scheme 1.

Catalytic Mechanism of Ornithine Conversion by Ornithine Aminotransferase
On the basis of known inactivation mechanisms of other aminotransferase inactivators, three of the possible mechanisms for the inactivation of hOAT by 1 are shown in Scheme 2.17 All of the mechanisms start with the condensation of 1 with the active-site PLP to form 3. The first mechanism (Michael addition mechanism) results from the direct nucleophilic attack of lysine on an activated Michael acceptor (Scheme 1, pathway a). This mechanism is initiated by the deprotonation of 3 and tautomerization via 4 to intermediate 5, which could be attacked by Lys292, yielding Michael adduct 6. This mechanism is related to the inactivation of GABA-AT by vigabatrin.19
Scheme 2.

Possible Pathways for the Inactivation of hOAT by 1
A second possible mechanism (enamine addition mechanism) results in an enamine addition product (Scheme 2, pathway b). An alternative tautomerization of 3 could result in conjugated enamine 7, which could transiminate back with Lys292, yielding the original Lys292-PLP aldimine (9) and reactive enamine 8. Enamine addition of 8 to 9 would then yield enamine adduct 10. This mechanism is reminiscent of the inactivation of GABA-AT by (1R,3S,4S)-3-amino-4-fluorocyclopentane-1-carboxylic acid with GABA-AT20 and ornithine aminotransferase.21
A third inactivation mechanism (elimination–addition mechanism) involves a vinylogous elimination of fluoride ion to give activated conjugated olefin 11 (Scheme 2, pathway c). Intermediate 11 is attacked by Lys292 to produce intermediate 12, which aromatizes and hydrolyzes to 13 (alternative mechanisms could be drawn in which the remaining two fluoride ions are eliminated prior to aromatization and hydrolysis). Other mechanisms could be drawn that involve the elimination of multiple fluoride ions from both trifluoromethyl groups. Numerous α-halomethyl- and vinyl-substituted amino acids have been shown to inactivate various PLP-dependent enzymes, such as γ-cystathionase (α-trifluoromethyl-alanine),22 aminocyclopropane-1-carboxylic acid synthase (α-vinyl glycine),23 alanine racemase,24a tryptophan synthase24b (α-halovinyl glycine), and lysine decarboxylase (α−1′-fluorovinyllysine).24c Although eflornithine (α-difluoromethyl-ornithine), a difluoromethyl-containing inactivator of ornithine decarboxylase, has been shown to undergo fluoride ion elimination and mostly active-site cysteine addition, it was not via a vinylogous fluoride ion elimination or with an aminotransferase.25 The inactivation of GABA-AT by 226 and the corresponding cyclopentene analogue27 were shown to proceed via a conjugated difluoromethylene group, but only water attacked, not an active-site residue (14, Scheme 3).
Scheme 3.

Mechanism of Inactivation of GABA-AT by 2
Previous research in the Silverman laboratory has used radiolabeled substrates and inhibitors,19 crystallography,26,27 and more recently, metabolomics26 to determine the mechanism of action of GABA-AT inhibitors. With the improved sensitivity of mass spectrometers, minute quantities of metabolites can be identified and quantified in a targeted or untargeted fashion, while covalent modifications of the intact protein can also be detected (through intact protein mass spectrometry) or characterized (by top-down proteomics, linking an intact mass and sequence-level data). Bottom-up proteomics (the digestion of the protein followed by detection of the fragments by LC–MS/MS and subsequent database searches and bioinformatics analyses) has been used in the detection of eflornithine bound to ornithine decarboxylase (ODC);25 however, the use of top-down proteomics or intact protein mass spectrometry to determine the mechanism of an MBEI has, to the best of our knowledge, not been reported.
To determine if any of the aforementioned mechanisms are responsible for the inactivation of hOAT by 1, we obtained a crystal structure of the inactivated enzyme and intact protein mass spectrometry data and further confirmed the mechanism by fluoride ion release data, targeted and untargeted mass spectrometry, the partition ratio, and the design of a new class of inactivators. To fully characterize the mechanism and inactivated enzyme structure, the combination of crystallography and mass spectrometry was essential.
RESULTS
Crystal Structure of hOAT Bound to 1.
The structure of hOAT inactivated by 1 was solved by molecular replacement using a monomer from a previously reported structure of hOAT (PDB entry 1OAT) after the deletion of all water molecules and ligand atoms. In space group P3221, one asymmetric unit was found to contain three monomers. Two monomers form a biological assembly as a homodimer; the other monomer forms a homodimer with a monomer present in another asymmetric unit. The final model was refined to a resolution of 1.75 Å with Rfree/Rwork values of 20.18/17.31%. Final refinement statistics are listed in Supporting Information (SI) Table S1. A simulated annealing omit map (Fo−Fc at 2.5σ), generated by deleting only 1 while keeping PLP and Lys292, was superimposed on the ternary adduct. The observed density around 1 supports the density interpretation and the position and orientation of the ternary adduct within the active site (SI Figure S1). The solved hOAT-1 structure (Figure 2) has been deposited in the Protein Data Bank (PDB code: 6OIA).
Figure 2.

Crystal structure of hOAT inactivated by 1 (PDB code: 6OIA).
Within the active site of hOAT, a ternary adduct was observed with covalent bonds formed between the syn-allylic carbon of what was initially the hexafluoropropan-2-ylidine group and Lys292 and between 1 and PLP. The crystal structure suggests that 1 facilitates the inactivation of hOAT through pathway c (Scheme 2). The PLP is bound to the inhibitor through a Schiff base, while the inactivator is covalently bound through an amide linkage. The syn-trifluoromethyl group (syn to the amino group) underwent exclusive fluoride ion elimination, conjugate addition, and hydrolysis to form the amide linkage. A major difference between the observed adduct in Figure 2 and 13 (Scheme 2) is the presence of an sp3-hybridized carbon atom adjacent to the trifluoromethyl group (Figure 2) rather than the sp2-hybridized carbon atom adjacent to the trifluoromethyl group shown in 13. The PLP-imine π system is out of conjugation by 46.5° with the pyridine π system, which results in the breakage of the normally strong iminium proton–phenoxide hydrogen bond.
Intact Protein Mass Spectrometry of 1 and hOAT.
The intact protein mass spectrometry of recombinant hOAT revealed that untreated enzyme is unmodified and exists in one form (m/z 769.97, [M + 60H]+60) (Figure 3A, bottom). Mass spectra were deconvoluted to determine an observed average mass of 46 139 Da, in close agreement with the theoretical mass of 46 138.5 Da (Figure 3B). Further analysis of intact hOAT protein inactivated by 1 revealed that the product found in the crystal structure was indeed covalently bound to the enzyme. When hOAT was inactivated by 1, the unmodified enzyme and two additional species were observed by mass spectrometry (m/z 777.80, [M+60H]+60; m/z 773.88, [M +60H]+60) (Figure 3A, top). One species corresponds to a mass shift of +464.4–469.6 Da, consistent with the +465 Da adduct observed in the crystal structure (Figure 3B). The second adduct observed was formed from the loss of PLP by hydrolysis of the +465 Da species (theoretical mass 46 373.7 Da; calculated mass 46 373 Da) (Figure 3B). It is unclear as to whether hydrolysis occurs during inactivation or results from the weak formic acid used in the liquid chromatography prior to mass spectrometry. No mass shifts consistent with the Michael addition (Scheme 2, pathway a) or enamine addition (Scheme 2, pathway b) pathways were observed. To the best of our knowledge, this is the first successful intact protein mass spectrometry data with an inactivated aminotransferase.
Figure 3.

(A) Expanded view of the intact mass spectrum of untreated hOAT (bottom) and 1-inactivated hOAT (top) with the region containing the highlighted [M + 60H]60+ adduct annotated to show theoretical native hOAT (in blue), 1·hOAT-PLP (in orange), and 1·hOAT (in cyan). The mass-to-charge ratios of the observed ion species is provided. (B) Deconvoluted mass spectra for unmodified hOAT and 1-inactivated hOAT with theoretical masses (red), calculated masses, and associated standard deviations (black).
Structure 13 in Scheme 2 indicates that the cofactor should be in the PMP form. The cofactor form cannot be discerned from the crystal structure; however, the sp3 carbon adjacent to the unmodified trifluoromethyl group suggests that PLP is the cofactor form in the adduct. To determine the identity of the cofactor, previously desalted hOAT, thereby removing any excess or unbound inhibitor, metabolites, and PLP, was heated to 37 °C with trifluoroacetic acid to hydrolyze imine bonds. After size exclusion filtration, the filtrate was analyzed by untargeted metabolomics (+ESI HRMS) to detect PLP and/or PMP (Figure 4). Only PLP was detected by mass spectrometry (m/z 248.0315); neither PMP nor adducts or fragments thereof were observed. Standards of PLP and PMP were monitored by HRMS and MS/MS to confirm the metabolite detection and retention time (SI Figure S3). This high-resolution mass spectrometry-based method represents a new technique for analyzing the PLP/PMP composition, which previously was accomplished through the tedious synthesis and incorporation of tritium-labeled PLP into enzymes.17,26
Figure 4.

Inactivated samples were treated with trifluoroacetic acid and filtered through a 30 kDa MWCO filter. The flowthrough was then processed for LC–MS. The TFA-treated flowthrough (B) and a non-TFA-treated control (A) were run in an untargeted fashion. Extracted ion chromatograms for PLP (248.02–248.04 m/z) and PMP (249.04–249.06 m/z) are shown. See Supporting Information Figure S3 for the mass spectra of PLP and PMP standards.
Design and Synthesis of Monotrifluoromethyl Analogues.
If the adduct in Figure 2 were indeed the complex that formed, then only the syn-trifluoromethyl group (syn to the amino group) would be necessary for inactivation. Both isomeric monotrifluoromethyl compounds were previously assessed with hOAT,16 and both compounds inactivated hOAT similarly (KI of 0.21 and 0.17 mM and kinact of 0.20 and 0.15 min−1 for 15a and 15b, respectively; Figure 5A). Given that hOAT has a large active site,28 it is possible that after initial deprotonation bond rotation occurs, resulting in identical intermediates (Figure 5B). Monotrifluoromethyl-containing inactivators that restrict rotation, however, may be able to verify our proposed mechanism. We have recently shown that the introduction of an enone adjacent to the α-amino group vastly improves kinact·27 If only one trifluoromethyl group is necessary for inactivation, we hypothesized that replacing the other trifluoromethyl group with an ester group could help to verify the mechanism (a syn-CF3 should be active while an anti-CF3 should not), but the electron-withdrawing nature of the enone should further lower the pKa of the α-amino proton. Furthermore, the analysis of the active site of hOAT bound with 1 indicates that there is an arginine and a glutamate in the region occupied by the second inactive CF3 group. This arginine (Arg413) is known to be flexible and is hypothesized to break its salt bridge with Glu235 upon binding of α-ketoglutarate.29,30 The introduction of an ester would possibly provide additional electrostatic interactions with the enzyme, thus decreasing KI (Figure 5C). We therefore synthesized syn- and anti-monotrifluoromethyl esters 19 and 20, respectively (Figure 5C).
Figure 5.

(A) Previously developed monotrifluoromethyl inhibitors and their associated activities.15 (B) Similarities in the activities of both syn- and anti-monotrifluoromethyl inhibitors can be explained by a bond rotation after the formation of the 1,1′-difluoroolefin. (C) Elaboration of the anti-CF3 of 1 to an ester might provide additional hydrogen bonding interactions with the enzyme.
The synthesis of 19 and 20 (Scheme 4) was initiated with the addition of methyl 3,3,3-trifluoropropionate to ketone 21. Although methyl 3,3,3-trifluoropropionate had been used in the Knoevenagel condensation with aldehydes using various Lewis acids, its use with ketones was less prevalent.31,32 The exposure of ketone 21 and methyl 3,3,3-trifluoropropionate to Bu2BOTf cleanly resulted in addition; however, elimination of the alcohol was not possible. When TiCl4 (2 equiv) was used, followed by the addition of triethylamine, a mixture of isomers 22 and 23 was obtained in a modest yield. Careful chromatography allowed the separation of 22 and 23. Deprotection of the p-methoxybenzyl group from 22 and 23 with ceric ammonium nitrate followed by hydrolysis yielded pure 19 (syn) and 20 (anti), respectively.
Scheme 4.

Synthesis of Trifluoromethyl Esters 19 and 20
Assay with hOAT.
The activity of the inhibitors was tested in a previously developed coupled enzyme hOAT assay.16,33 syn isomer 19 proved to be similarly potent to the inactivator of hOAT as 1. kinact was similar to that of 1 (0.10 min−1 for 19 versus 0.08 min−1 for 1). KI, however, was more than double for 19 when compared to that for 1 (130 μM versus 54 μM, respectively). This could indicate that the methyl ester is sterically too large for the active site and thus the binding is slightly reduced. The anti-CF3 ester (20) was shown to be inactive against hOAT (Ki > 2 mM). This result indicates that a syn-CF3 or a rotatable bond is required for inactivation via pathway c (Scheme 2). Both compounds were inactive against GABA-AT.
Intact Protein Mass Spectrometry of 19.
Intact protein mass spectrometry of hOAT inactivated by 19 showed an observed mass of 46 362.83 ± 0.78 Da (SI Figure S3). This mass corresponds to a mass shift of +224.3 Da, which is consistent with the loss of a trifluoromethyl group and the formation of an enzyme–inhibitor amide bond, as depicted in Figure 5C.
Computer Modeling.
Molecular docking simulations were employed to help visualize the active intermediates.34 Molecular docking showed that when intermediate 11 (Scheme 2) was docked into hOAT, the 1,1′-difluoro-olefin was located in an area close to that of Lys292 (Figure 6A). The syn-CF3 19-PLP adduct also docks effectively into the active site in a similar manner and is consistent with the pathway c mechanism in Scheme 2 (Figure 6B). The anti-CF3 adduct (20-PLP), however, does not dock into the active site, consistent with the mechanism of action.
Figure 6.

Molecular docking of intermediate 11 (A) and 19-PLP (B) into hOAT.
Partition Ratio of 1.
The partition ratio, which is the ratio of the compound acting as a substrate (and transaminating) relative to the compound inactivating the enzyme, is calculated by titrating the enzyme with varying equivalents of inactivator with a known amount of hOAT (Figure 7).26,27 The linear relationship was extrapolated to yield the exact equivalents required to inactivate the enzyme completely. Non-pseudo-firstorder kinetics were observed (x = 20–40 equiv), most likely arising from product (metabolite) inhibition and consistent with the turnover of other known aminotransferase enzymes.26,27 From this, we determined the partition ratio of 1 to be 12 ± 1 (i.e., it requires 13 equiv of 1 to completely inactivate hOAT, with 12 being converted to product and 1 leading to the inactivation of the enzyme).
Figure 7.

Titration of hOAT with 1 to obtain a partition ratio of 12 ± 1. The number of equiv of 1 required for inactivation is derived from the x intercept (y = 0%) of the linear regression of the initial linear (first-order) relationship (x = 0–10 equiv); the non-pseudo-first-order kinetics and deviation from linearity (x = 20–30 equiv) are possibly a result of product inhibition. The partition ratio is the number of equiv of 1 for inactivation minus 1.
Fluoride Ion Release and Metabolomics of 1.
The number of fluoride ions released per enzyme turnover can be detected using a fluoride ion selective electrode.26 In the absence of α-ketoglutarate, the enzyme can turn over only once because the PLP cofactor is converted to PMP, which cannot return to PLP. When, for example, CPP-115 (2) was tested under these conditions, it was found that two fluoride ions were released per enzyme active site.26 Under these conditions, when hOAT is inactivated with an excess of 1, 79 equiv of fluoride ions was released per enzyme active site (SI Table S3). This indicates that the turnover of the inhibitor must regenerate PLP about 78 times prior to inactivation. A mechanism consistent with this observation is shown in Scheme 5.
Scheme 5.

Proposed Mechanism for the Turnover of 1 by hOAT
As depicted in Scheme 5, when hOAT was inactivated with 20 equiv of 1 in the presence of α-ketoglutarate, metabolite 32 (m/z 257.024, [M – H]−1) was detected by mass spectrometry (SI Figure S4). Ketone 34, which corresponds to the transamination of 1 (m/z 275.015, [M – H]−1), was not detected, even with a highly sensitive and targeted selective ion monitoring (SIM) scan for the theoretical ion. Furthermore, pathway b would release PMP, which also was not detected.
DISCUSSION
Bis(trifluoromethyl) compound 1 is an inactivator of hOAT that has been shown to be a potential treatment for HCC.16 To develop more potent mechanism-based inactivators of hOAT, it is important to determine the mechanism of 1. Although 1 was developed from CPP-115 (2), a potent inactivator of both GABA-AT and hOAT, 1 is highly selective for hOAT over GABA-AT.16 In GABA-AT, Phe351 crowds the active site, whereas this position in hOAT has a glycine residue, resulting in a larger active site, which is responsible for the selective binding of 1 to hOAT. This is further verified through molecular modeling, while intermediate 11 (Scheme 2) (also intermediate 37, Scheme 6) docks effectively into the active site of hOAT (Figure 6A), forming the prerequisite electrostatic interactions with Tyr55 and failing to dock into the active site of GABA-AT.
Scheme 6.

Revised Inactivation Mechanism of 1 with hOAT
On the basis of what is known about aminotransferase inhibitors,17 we postulated three reasonable mechanisms: Michael addition (pathway a, Scheme 2), enamine addition (pathway b), and elimination–addition (pathway c). From the crystal structure (Figure 2) and intact protein mass spectrometry (Figure 3), we were able to conclude that the elimination–addition mechanism was most likely. Intriguingly, Arg413 and Glu235, which in the native enzyme form a salt bridge with one another, have dissociated in the presence of 1, favoring instead hydrogen bonds with the trifluoromethyl moiety (Figure 2). Of note is the loss of an interaction between Arg180 and the inhibitor carboxylate, a common interaction observed in other hOAT inhibitor crystal structures. Instead, hydrogen bonds were observed between the inactivator carboxylate and Tyr55 and between the amide carbonyl and Gln266. Arg180 has been shown to bind to the carboxylate of other hOAT inhibitors such as 5-fluoromethylornithine35,36 and 3-amino-4-fluorocyclopentane-1-carboxylic20 and has previously been reported to be responsible for the correct positioning of the terminal amino group of l-ornithine within the active site to facilitate transamination.36 The loss of this interaction with 43 (Scheme 6) suggests that the inhibition of hOAT can be achieved without any interaction with Arg180. Rather, the interaction with Tyr55, one of several residues responsible for substrate recognition, and/or Arg413 and Glu235 might be of more importance for the selective inhibition of hOAT. The hydrogen bond to Tyr55 is not unprecedented because it is seen in the crystal structure of gabaculine with hOAT.37 There was also a difference observed in the orientation of Phe177, when compared with other inhibited structures of hOAT. For inhibitors where an interaction is observed between the inhibitor and Arg180, Phe177 is positioned so as not to obstruct the inhibitor–Arg180 interaction.
However, because the interaction with Arg180 is absent here, Phe177 was observed to be in an alternate orientation, possibly stabilizing the ternary adduct by increasing the surrounding hydrophobicity. The overall orientation of binding of this carboxylic acid ligand is unconventional, possibly because of its bulkiness. Another potentially important interaction for adduct formation is hydrogen bonding between the unmodified trifluoromethyl group and Glu235 and Arg413.38 Finally, the PLP-imine π system is out of conjugation by 46.5° with the pyridine π system, which results in the breakage of the normally strong iminium proton–phenoxide hydrogen bond. This broken π system is a relatively common feature in PLP-dependent enzymes, as evidenced by the X-ray crystal structure of CPP-115 and OV329 with GABA-AT26,27 and gabaculine and l-canaline with hOAT.37 A table of seven PLP-dependent enzymes with PLP-imine pyridine dihedral angles of up to 50° is in the Supporting Information (Table S2). The breaking of this hydrogen bond could be a result of bond rotations forced upon the complex by the formation of the newly formed lysine covalent bond.
It is apparent from the crystal structure (Figure 2) that the carbon atom bound to the α-trifluoromethyl group is sp3 hybridized instead of sp2-hybridized as shown in 13 (Scheme 2). This alters the mechanism from that predicted in Scheme 2 because an sp3-hybridized carbon atom adjacent to the trifluoromethyl group would require that upon inactivation the cofactor would be in the PLP form. High-resolution mass spectrometry was used to identify the cofactor form as PLP (Figure 4), consistent with the crystal structure. Intact protein mass spectrometry ruled out the other mechanisms because only two species were observed. One species was observed to be expected adduct 43, while the other was the adduct with the loss of PLP from 43. We could not determine whether this hydrolysis occurred as a result of the formic acid used in liquid chromatography or during inactivation.
On the basis of the intact protein mass spectrometric data and the crystal structure, a revised inactivation mechanism by 1 is shown in Scheme 6. The condensation of 1 with PLP yields aldimine 35. The base-catalyzed elimination of the fluoride ion via 36 gives 37. The base in this reaction is most likely Lys292, as has been suggested in GABA-AT that a conserved lysine accomplishes this deprotonation.17 Toney and co-workers have shown that the formation of quinonoid intermediates, such as 36, is likely accomplished with aminotransferase enzymes.39,40 Furthermore, additional direct evidence has revealed the UV absorbance of the quinonoid species with both OAT (λmax = 490 nm)41 and adenosylmethionine-8-amino-7-oxononanoate aminotransferase (λmax = 540 nm).42 It is therefore likely that 36, along with subsequent quinonoid species, form transiently.
The formation of intermediate 37 was verified by metabolomics, which identified 32 as the major metabolite (SI Figure S4). The partition ratio and the number of equivalents of 1 converted to product per inactivation event are higher than those observed for both CPP-11526 and OV32927 with GABA-AT. Given the continuous release of fluoride ion detected in the absence of α-ketoglutarate, it is likely that turnover proceeds through a fluoride ion elimination mechanism, which would regenerate PLP, rather than through transamination to yield 38 and PMP, neither of which was observed. Intermediate 37 could undergo attack by Lys292 at the PLP imine bond (pathway a), followed by hydrolysis to give 32 (92% of the time based on the partition ratio of 12), or Lys292 could undergo conjugate addition at the difluoromethylenyl group of 37 (pathway b) to give 40 (via 39) 8% of the time. The hydrolysis of 40 gives 43 (via possible intermediates 41 and 42), one of the observed species by intact protein mass spectrometry, and the hydrolysis of 43 gives 44, the other species observed by intact protein mass spectrometry. To obtain the sp3-hybridized center at the carbon adjacent to the α-trifluoromethyl group, enolate 42 must be protonated stereoselectively. Given that the iminium proton has broken its phenoxide hydrogen bond and is 2.7 Å from the enol alkene (Figure 8) and because of the geometric constraints of the newly formed covalent bond to Lys292 and the lack of rotatable bonds, we propose an intramolecular stereoselective proton transfer (42) to produce 43, as depicted in Scheme 6.
Figure 8.

Crystal structure of the 1-hOAT inactivation adduct shows the possible stereoselective protonation of the enolate by the PLP iminium proton. Distances are shown in angstroms.
We postulated a similar turnover mechanism to occur with 3-amino-4-fluorobutanoic acid and GABA-AT,43 which had a partition ratio of 7.6 and released a total of 6.7 fluoride ions. Using14 C-labeled inhibitor, we previously showed that both turnover and inhibition yielded products in which fluoride ions had been eliminated; a bifurcated mechanism also was suggested to occur after fluoride ion elimination. However, this radioactive approach relies on a comparison of standards of each metabolite with the14 C metabolites isolated from inactivation or turnover. The use of mass spectrometric techniques, as employed here, to determine specific metabolites and the form of the coenzyme streamlines the identification and removes the need to synthesize and work with radioactive materials.
If the above mechanism is indeed valid, then only one trifluoromethyl group is required. Compound 19 was shown to inactivate hOAT, while 20 was inactive. This further supports the involvement of the trifluoromethyl group in inactivation and that it must be oriented syn to the amino group. Intact protein mass spectrometry of hOAT inactivated by 19 showed a loss of a trifluoromethyl group and covalent attachment to hOAT through an amide bond. These results are further supported by the molecular docking of 19-PLP, as shown in Figure 6B, whereas 20-PLP does not dock.
CONCLUSIONS
Our results provide a novel mechanism for the inactivation of hOAT. On the basis of intact protein mass spectrometry and crystallographic data, 1 is shown to inactivate hOAT via the elimination of a fluoride ion and Lys292 attack on activated intermediate 37 (Scheme 6, pathway b). A mechanism by which a trifluoromethyl group undergoes vinylogous fluoride ion elimination followed by nucleophilic attack is novel for aminotransferase enzymes, and 1 and 19 constitute a new class of aminotransferase mechanism-based inactivators that appear to utilize that mechanism. In this study, we used targeted mass spectrometry to aid in PLP/PMP identification, which shortcuts previous approaches involving radioactive labeling. We currently are carrying out IND-enabling studies to bring 1 into clinical trials for the treatment of hepatocellular carcinoma.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health (grant R01 DA030604 to R.B.S. and grant P30 DA018310 to N.L.K.) for financial support. This work made use of the IMSERC at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology (IIN). X-ray diffraction data collection used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. The use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (grant 085P1000817). We thank Dr. Joseph Brunzelle at LS-CAT for help on data collection.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b03254.
Methods, syntheses, all spectra, and crystallographic data (PDF)
REFERENCES
- (1).World Cancer Factsheet; World Health Organization, 2014, accessed on 7/20/2016, https://www.cancerresearchuk.org/sites/default/files/cs_report_world.pdf.
- (2).Liver cancer; American Cancer Society, 2016, accessed on 7/20/2016, http://www.cancer.org/acs/groups/cid/documents/webcontent/003114-pdf.pdf.
- (3).Han S-X; Zhu Q; M J-L; Lv Y; Zhao J; Huang C; Jia X; Ou W; Guo H-T Apoptin sensitizes radiation-induced cell death via classic mitochondrial, caspase and p53-dependent signaling in hepg2 cells. Mol. Med. Rep 2010, 3, 59–63. [DOI] [PubMed] [Google Scholar]
- (4).Perez MJ; Castano B; Jimenez S; Serrano MA; Gonzalez-Buitrago JM; Marin JJG Role of vitamin c transporters and biliverdin reductase in the dual pro-oxidant and anti-oxidant effect of biliary compounds on the placental-fetal unit in cholestasis during pregnancy. Toxicol. Appl. Pharmacol 2008, 232, 327. [DOI] [PubMed] [Google Scholar]
- (5).Hong L; Han Y; Zhang H; Zhao Q; Wu K; Fan D Drug resistance-related mirnas in hepatocellular cancer. Exp. Rev. Gastro-enterol. Hepatol 2014, 8, 283–288. [DOI] [PubMed] [Google Scholar]
- (6).Thompson MD; Monga SPS Wnt/β-catenin signaling in liver health and disease. Hepatology 2007, 45, 1298–1305. [DOI] [PubMed] [Google Scholar]
- (7).Lucero OM; Dawson DW; Moon RT; Chien A A re-evaluation of the ″oncogenic″ nature of wnt/β-catenin signaling in melanoma and other cancers. Curr. Oncol. Rep 2010, 12, 314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zucman-Rossi J; Benhamouche S; Godard C; Boyault S; Grimber G; Balabaud C; Cunha AS; Biolac-Sage P; Perret C Differential effects of inactivated axin1 and activated β-catenin mutations in human hepatocellular carcinomas. Oncogene 2007, 26, 774–780. [DOI] [PubMed] [Google Scholar]
- (9).Cadoret A; Ovejero C; Terris B; Souil E; Levy L; Lamers WH; Kitajewski J; Kahn A; Perret C New targets of β-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [DOI] [PubMed] [Google Scholar]
- (10).Sekine SL; Y-A B; Bedolli M; Feng S; Hebrok M Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 2006, 43, 817–825. [DOI] [PubMed] [Google Scholar]
- (11).de Lope CR; Tremosini S; Forner A; Reig M; Bruix J Management of HCC. J. Hepatol 2012, 56, S75–S87. [DOI] [PubMed] [Google Scholar]
- (12).Brosnan ME; Brosnan JT Hepatic glutamate metabolism: A tale of 2 hepatocytes. Am. J. Clin. Nutr 2009, 90, 857S–861S. [DOI] [PubMed] [Google Scholar]
- (13).Wise DR; Thompson CB Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci 2010, 35, 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Medina MA Glutamine and cancer. J. Nutr 2001, 131, 2539s–2542s. [DOI] [PubMed] [Google Scholar]
- (15).Zigmond E; Ben Ya’acov A; Lee H; Lichtenstein Y; Shalev Z; Smith Y; Zolotarov L; Ziv E; Kalman R; Le HV; Lu H; Silverman RB; Ilan Y Suppression of hepatocellular carcinoma by inhibition of overexpressed ornithine aminotransferase. ACS Med. Chem. Lett 2015, 6, 840–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Miyasaka Y; Enomoto N; Nagayama K; Izumi N; Marumo F; Watanabe M; Sato C Analysis of differentially expressed genes in human hepatocellular carcinoma using suppression subtractive hybridization. Br. J. Cancer 2001, 85, 228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Silverman RB Design and mechanism of GABA-amino-transferase inactivators. Treatments for epilepsies and addictions. Chem. Rev 2018, 118, 4037–4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Silverman RB Mechanism-based enzyme inactivators. Methods Enzymol 1995, 249, 240–283. [DOI] [PubMed] [Google Scholar]
- (19).Nanavati SM; Silverman RB Mechanisms of inactivation of gamma-aminobutyric-acid aminotransferase by the antiepilepsy drug gamma-vinyl GABA (vigabatrin). J. Am. Chem. Soc 1991, 113, 9341–9349. [Google Scholar]
- (20).Storici P; Qiu J; Schirmer T; Silverman RB Mechanistic crystallography. Mechanism of inactivation of γ-aminobutyric acid aminotransferase by (1R,3S,4S)-3-amino-4-fluorocyclopentane-1-carboxylic acid as elucidated by crystallography. Biochemistry 2004, 43, 14057–14063. [DOI] [PubMed] [Google Scholar]
- (21).Mascarenhas R; Le HV; Clevenger KD; Lehrer HJ; Ringe D; Kelleher NL; Silverman RB; Liu D Selective targeting by a mechanism-based inactivator against pyridoxal 5′-phosphate-dependent enzymes: Mechanisms of inactivation and alternative turnover. Biochemistry 2017, 56, 4951–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Silverman RB; Abeles RH Mechanism of inactivation of γ-cystathionase by β,β,β-trifluoroalanine. Biochemistry 1977, 16, 5515–5520. [DOI] [PubMed] [Google Scholar]
- (23).Capitani G; Tschopp M; Eliot AC; Kirsch JF; Grutter MG Structure of acc synthase inactivated by the mechanism-based inhibitor l-vinylglycine. FEBS Lett 2005, 579, 2458–2462. [DOI] [PubMed] [Google Scholar]
- (24).(a) Thornberry NA; Bull HG; Taub D; Wilson KE; Giménez-Gallego G; Rosegay A; Soderman DD; Patchett AA Mechanism-based inactivation of alanine racemase by 3-halovinylglycines. J. Biol. Chem 1991, 266, 21657–21665. [PubMed] [Google Scholar]; (b) Xu Y; Abeles RH Inhibition of tryptophan synthase by (1-fluorovinyl)glycine. Biochemistry 1993, 32, 806–811. [DOI] [PubMed] [Google Scholar]; (c) McCune CD; Beio ML; Sturdivant JM; de la Salud-Bea R; Darnell BM; Berkowitz DB Synthesis and deployment of an elusive fluorovinyl cation equivalent: access to quaternary α-(1’-fluoro)vinyl amino acids as potential PLP enzyme inactivators. J. Am. Chem. Soc 2017, 139, 14077–14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Poulin R; Lu L; Ackermann B; Bey P; Pegg AE Mechanism of the irreversible inactivation of mouse ornithine decarboxylase by alpha-difluoromethylornithine. Characterization of sequences at the inhibitor and coenzyme binding sites. J. Biol. Chem 1992, 267, 150–158. [PubMed] [Google Scholar]
- (26).Lee H; Doud EH; Wu R; Sanishvili R; Juncosa JI; Liu D; Kelleher NL; Silverman RB Mechanism of inactivation of gamma-aminobutyric acid aminotransferase by (1S,3S)-3-amino-4-difluoromethylene-1-cyclopentanoic acid (CPP-115). J. Am. Chem. Soc 2015, 137, 2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Juncosa JI; Takaya K; Le HV; Moschitto MJ; Weerawarna PM; Mascarenhas R; Liu D; Dewey SL; Silverman RB Design and mechanism of (S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid, a highly potent gaba aminotransferase inactivator for the treatment of addiction. J. Am. Chem. Soc 2018, 140, 2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lee H; Juncosa JI; Silverman RB Ornithine aminotransferase versus gaba aminotransferase: Implications for the design of new anticancer drugs. Med. Res. Rev 2015, 35, 286–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liu W; Peterson PE; Carter RJ; Zhou X; Langston JA; Fisher AJ; Toney MD Crystal structures of unbound and aminooxyacetate-bound escherichia coli gamma-aminobutyrate aminotransferase. Biochemistry 2004, 43, 10896–10905. [DOI] [PubMed] [Google Scholar]
- (30).Markova M; Peneff C; Hewlins MJ; Schirmer T; John RA Determinants of substrate specificity in omega-aminotransferases. J. Biol. Chem 2005, 280, 36409–36416. [DOI] [PubMed] [Google Scholar]
- (31).Liu Y; Lai H; Rong B; Zhou T; Hong J; Yuan C; Zhao S; Zhao X; Jiang B; Fang Q Titanium-mediated direct carbon-carbon double bond formation to α-trifluoromethyl acids: A new contribution to the knoevenagel reaction and a high-yielding and stereoselective synthesis of α-trifluoromethylacrylic acids. Adv. Synth. Catal 2011, 353, 3161–3165. [Google Scholar]
- (32).Ramachandran PV; Parthasarathy G; Gagare PD Bis-exo-2-norbornylboron triflate for stereospecific enolization of 3,3,3-trifluoropropionates. Org. Lett 2010, 12, 4474–4477. [DOI] [PubMed] [Google Scholar]
- (33).Juncosa JI; Lee H; Silverman RB Two continuous coupled assays for ornithine-delta-aminotransferase. Anal. Biochem 2013, 440, 145–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jones G; Willett P; Glen RC; Leach AR; Taylor R Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- (35).Bolkenius FN; Knodgen B; Seiler N Dl-canaline and 5-fluoromethylornithine. Comparison of two inactivators of ornithine aminotransferase. Biochem. J 1990, 268, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Storici P; Capitani G; Muller R; Schirmer T; Jansonius JN Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine. J. Mol. Biol 1999, 285, 297–309. [DOI] [PubMed] [Google Scholar]
- (37).Shah SA; Shen BW; Brünger AT Human ornithine aminotransferase complexed with L-canaline and gabaculine: Structural basis for substrate recognition. Structure 1997, 5, 1067–1075. [DOI] [PubMed] [Google Scholar]
- (38).Howard JAK; Hoy VJ; O’Hagan D; Smith GT How good is fluorine as a hydrogen bond acceptor? Tetrahedron 1996, 52, 12613–12622. [Google Scholar]
- (39).Griswold WR; Toney MD Role of the pyridine nitrogen in pyridoxal 5′-phosphate catalysis: Activity of three classes of PLP enzymes reconstituted with deazapyridoxal 5′-phosphate. J. Am. Chem. Soc 2011, 133, 14823–14830. [DOI] [PubMed] [Google Scholar]
- (40).Griswold WR; Fisher AJ; Toney MD Crystal structures of aspartate aminotransferase reconstituted with 1-deazapyridoxal 5′-phosphate: Internal aldimine and stable l-aspartate external aldimine. Biochemistry 2011, 50, 5918–5924. [DOI] [PubMed] [Google Scholar]
- (41).John RA; Morgan PH; Fowler LJ Comparison of aspartate-aminotransferase with other aminotransferases by absorption-spectrum analysis. Biochem. Soc. Trans 1984, 12, 430–432. [DOI] [PubMed] [Google Scholar]
- (42).Eiden CG; Maize KM; Finzel BC; Lipscomb JD; Aldrich CC Rational optimization of mechanism-based inhibitors through determination of the microscopic rate constants of inactivation. J. Am. Chem. Soc 2017, 139, 7132–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Silverman RB; Chamberlain Roscher CL Mechanism-based inactivation of γ-aminobutyric acid aminotransferase by 3-amino-4-fluorobutanoic acid. Bioorg. Med. Chem 1996, 4, 1521–1535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.