

Abstract
Many disease resistance genes that have been transferred from wild relatives to cultivated wheat have played a significant role in wheat production worldwide. Ae. umbellulata is one of the species within the genus Aegilops that have been successfully used as sources of resistance genes to leaf rust, stem rust and powdery mildew. The objectives of the current work was to validate the map position of a major QTL that confers resistance to the stem rust pathogen races Ug99 (TTKSK) and TTTTF with an independent bi-parental mapping population and to refine the QTL region with a bulk segregant analysis approach. Two F2 bi-parental mapping populations were developed from stem rust resistant Ae. umbellulata accessions (PI 298905 and PI 5422375) and stem rust susceptible accessions (PI 542369 and PI 554395). Firstly, one of the two populations was used to map the chromosome location of the resistance gene. Later on, the 2nd population was used to validate the chromosome location in combination with a bulk segregant analysis approach. For the bulk segregant analysis, RNA was extracted from a bulk of leaf tissues of 12 homozygous resistant F3 families, and a separate bulk of 11 susceptible homozygous F3 families derived from the PI 5422375 and PI 554395 cross. The RNA samples of the two bulks and the two parents were sequenced for SNPs identification. Stem rust resistance QTL was validated on chromosome 2U of Ae. umbellulata in the same region in both populations. With bulk segregant analysis, the QTL position was delimited within 3.2 Mbp. Although there were a large number of genes in the orthologous region of the detected QTL on chromosome 2D of Ae. tauschii, we detected only two Ae. umbellulata NLR genes which can be considered as a potential candidate genes.
Introduction
Wheat (Triticum aestivum L.) is used as a main source of protein and starch for human consumption. However, its production is threatened by many diseases including stem rust caused by Puccinia graminis f. sp. tritici (Pgt), which is known to cause severe yield losses in susceptible cultivars. Ug99, the stem rust race group discovered in Uganda in 1998, was found to be virulent on widely deployed resistance genes, and the majority of the world’s wheat germplasm is susceptible to this race group [1]. The emergence and continued evolution of Ug99 has pressed the research community to evaluate available genetic resources for resistance and rapidly develop Ug99-resistant cultivars. Since the cultivated wheat gene pool has a narrow genetic base for resistance to Ug99 and up to 90% of world’s wheat cultivars are considered Ug99 susceptible [2], identifying resistance genes from wild relatives and introgressing them into cultivated wheat is a tractable strategy for improving disease resistance. At least 30 wheat stem rust resistance genes are currently available to be in breeding for stem rust resistance in wheat [2]. Since 2011, a total of five Ug99 resistance genes have been introgressed from Ae. tauschii Coss., Ae. searsii Feldman & Kislev ex K. Hammer, and Ae. geniculata into wheat (Sr51, Sr53, SrTA1662, SrTA10171, SrTA10187) [3, 4, 5, 6].
Many wild relatives of cultivated wheats have been used as sources of disease resistance genes over the last century, and those deployed genes have also played a significant role in wheat production worldwide [7]. Among wild relatives of wheat, Aegilops umbellulata has been successfully used as a source of resistance genes to leaf rust, stem rust and powdery mildew [8–10]. Most recently, a major QTL conferred resistance to the Pgt races TTKSK (Ug99) and TTTTF was mapped on chromosome 2U of Ae. umbellulata using genotyping-by-sequencing (GBS) SNP markers [11].
RNA-sequencing is an efficient and cost-effective method of identifying SNPs in transcribed genomic regions especially for non-model species with a little or no available genomic resources [12–14]. The recent development of methods of SNPs detection in the transcriptome allowed the combination of bulk segregant analysis (BSA) [15] and sequencing to fine map genic regions for traits of interest. This process, known as bulked segregant analysis RNA-seq (BSR-Seq), has been used for identifying candidate genes and gene cloning purpose for cereals such as maize [16, 17] and hexaploid wheat [18, 19]. BSR-Seq method has also the ability to identify differentially expressed genes (DFGs). However, BSR-Seq may not be effective for complex traits controlled by many minor genes or traits influenced by environmental factors [20]. Most recently, a major QTL conferred resistance to the Pgt races TTKSK (Ug99) and TTTTF was mapped on chromosome 2U of Ae. umbellulata using genotyping-by-sequencing (GBS) SNP markers [11]. As part of the validation and refined mapping of this previously detected stem rust resistance QTL in Ae. umbellulata, two approaches were followed in the current work; independent mapping in another bi-parental population and BSR-Seq. Therefore, the objectives of the current work are to: 1) Confirm the position of previously detected major QTL from Ae. umbellulata 2) map the QTL identified on chromosome 2U to a shorted genetic region 3) identify candidate resistance genes, and 4) to generate the transcriptome assembly for Ae. umbellulata.
Materials and methods
Genetic materials
Two F2 Ae. umbellulata populations were developed from crossing of resistant and susceptible accessions. The 1st population comprised 140 F2 individuals that were obtained from crossing stem rust resistant accession PI 298905 and susceptible accession PI 542369. PI 298905 was resistant to both Pgt races TTTTF and TTKSK with seedling infection type ‘2-’ whereas PI 542369 was susceptible to both races with seedling infection type ‘3+’. Similarly, the 2nd population consisted of a total of 154 F2 individuals derived from a cross made between accessions PI 542375 (resistant to races TTTTF and TTKSK) and PI 554395 (susceptible to races TTTTF and TTKSK). F3 families of both populations were also screened with race TTKSK from the Ug99 stem rust race group in a Biosafety level 3 (BSL-3) facility.
Phenotyping mapping populations and QTL mapping
Both F2 individuals and F3 families of both populations were assessed for reaction to both races TTTTF (isolate 01MN84A-1-2) and TTKSK (04KEN156/04). Experimental procedures for inoculation, incubation, and disease assessment were conducted according to previously described methods [21]. The seedlings were inoculated approximately 10 days after planting when the primary leaves were fully expanded and the second leaves had started elongating. Stem rust seedling infection types were scored on the 0–4 scale [22] 12 to 14 days after inoculation, and plants with infection types 0–2 were considered resistant whereas plants with infection types 3–4 were considered susceptible.
QTL mapping was carried out for the two populations described above. QTL mapping was facilitated by GBS SNP markers identified from sequence data from the two populations following the same procedures mentioned in [11]. In our previous work QTL identification was done only for the 1st population using GBS SNPs. In the current work, however, the QTL mapping was carried out with GBS SNPs common between the two populations using composite interval mapping (CIM) in the RQTL package. Both F2 individuals and F3 families that were derived from F2s of both mapping populations were used for QTL analysis. To compare the similarity of the QTL mapped with the two populations, we inferred the QTL region from the GBS marker-based consensus map previously developed from these two populations [23].
Bulk segregant samples, RNA-Seq library preparation and RNA sequencing
The 2nd population (derived from PI 5422375 and PI 554395 cross) was used for bulk segregant analysis. F3 families were grown to differentiate heterozygous from homozygous F2 plants. A total of twenty plants from each F3 family were assessed at the seedling stage with Pgt isolates 01MN84A-1-2 (TTTTF) according to previously described methods [21]. Families were classified as homozygous resistant, heterozygotes, and homozygous susceptible based on response to the Pgt isolates tested. For bulk segregant analysis (BSA), 12 and 11 homozygous F3 families were selected to make resistant bulk and susceptible bulks based on their response to race TTTTF, respectively. Leaf tissues were collected from about 4 weeks old seedlings of F3 families for RNA extraction.
Total RNA was extracted from leaf tissues collected from homozygous resistant and susceptible families and the two parents using the RNeasy Plant Mini Kit (QIAGEN) according to the manufacture’s instruction. At least 500ng RNA of four samples (two bulks and the two parents PI 5422375 and PI 554395) was submitted to the University of Minnesota Genomics Center (Saint Paul, MN) for quality assessment, standard cDNA library preparation and RNA sequencing on Illumina HiSeq 2500 to generate 125 bp paired-end (PE) sequence reads for all four samples. Libraries were created using dual indexed TruSeq-stranded RNA library preparation kits. All libraries were pooled and sequenced across 3 lanes. The pooled libraries were Caliper XT size-selected to have inserts of approximately 200 bp. The reads were concatenated across lanes, and a single fastq file per sample for each read was generated. The average quality scores were above Q30 for all libraries.
De novo transcript assembly, SNPs identification and bulk frequency ratio (BFR) calculation
Sequence read quality was evaluated using the FastQC program (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The Trimmomatic program [24] was used to clean the reads and remove the Illumina paired-end adaptor sequences requiring an average quality score of 15, and a minimum length of reads of 30 base-pairs. Only paired-end reads that passed the cleaning steps were used for the subsequent analysis. We used Trinity assembly program [25] for de novo assembling the reads from the two parents. SNPs identification was done using two approaches: de novo assembly of the parents as a pseudo-reference, and a reference-based approach using the Ae. tauschii reference sequence [26].
In the de novo assembly approach, SNPs were identified from both the two parental assemblies (PI 5422375 and PI 554395 accessions) using the sort, merge, and pileup programs in combination with VarScan software [27]. We used the susceptible parent assembly as a pseudo reference to call SNPs from short reads of the pools. Fisher exact tests with a 0.05 significance level were applied to identify SNPs that showed frequency differences between the two pools. However, only SNPs that were common between the parental SNPs and the two pools were considered as true SNPs. Only reads with base phred quality scores of 20 or more, and requiring a minimum number of 6 reads were considered for SNPs for both pools. Then the parental SNPs were identified from the bulks based on SNP positions by merging the parental SNPs with SNPs identified from the pools. Additionally, bulk frequency ratio was calculated based on the allele frequency of the resistant allele in the pools to identify SNPs-linked to stem rust resistance. Bulk frequency ratio (BFR) is defined as the allele frequency ratio of the resistant allele (resistant parent allele) in the resistant bulk compared to the resistant allele frequency in the susceptible bulk. BFR ratio of four was used as the minimum threshold to identify potential candidate SNPs.
Reference-based identification of SNPs using Ae. tauschii as a reference sequence
SNP calling was also completed using Ae. tauschii v4.0 as a reference. SNP calling was done for the bulks and the parents separately. For the two bulks, the short reads were aligned against the Ae. tauschii reference sequence using BWA and the SNPs were called using samtools [28] and Varscan softwares. SNP quality filtering parameters were set at; minimum read depth at a position to make a call 10, minimum supporting reads at a position to call variants 6, minimum average base quality for variant-supporting reads 20, and average variant frequency of 0.2. The default was used for the remaining parameters. Similarly, to call SNPs from the two parents, the RNA-Seq short reads of both parents were aligned against the Ae. tauschii v4.0 reference sequence using BWA and SNPs were called with samtools software with default set. VCFtools was used to filter out false SNPs with the following filtering criteria: minor allele frequency of >0.20, phered-quality score of 20, minimum read depth of 6 and average mapping quality (MQ) of 30.
Sequence assembly characterization and functional annotation
Both de novo assemblies of Ae. umbellulata accessions were aligned against local genomic and cDNA databases of hexaploid wheat, Ae. tauschii and barley. Transcripts were also annotated using BLAST homology searches against the UniProt databases within the Trinotate functional annotation site (https://trinotate.github.io/). Briefly, the likely coding regions were predicted using Transcoder software and then the predicted proteins were searched in the protein database by BLAST. In addition, protein domains were searched using “hmmer” software [29]. The predicted proteins and protein domain search results were integrated into coding region selection. Finally, all search results were uploaded to “sqlite” database to extract annotation reports with Trinotate suite.
We also assessed if there were genes that contained a nucleotide-binding site and leucine-rich repeat (NLR) domain in the QTL region. For this purpose, all contigs from the resistant parent assembly that were mapped on chromosome 2D of Ae. tauschii through blast approach were used to identify contigs potentially containing NLR domains in this region. Motif Alignment and Search Tool (MAST) in MEME suite [30] and NLR-parser, a java program that identifies NLRs from MAST motifs output [31], were used to identify contigs that comprised NLR motifs using a set 20 previously characterized amino acid sequences that contain NB-LRR domains [32], and six-frame translated amino acid sequences derived from contig sequences that were mapped on chromosome 2D of Ae. tauschii.
Results
QTL mapping using two bi-parental mapping populations
GBS markers common across the two mapping populations were used to identify stem rust resistance QTL. Detailed procedures of GBS marker development have been reported previously for these bi-parental populations [23]. QTL mapping was done separately for each population. A major stem rust resistance QTL was detected on chromosome 2U of Ae. umbellulata at the same region with both mapping populations (Fig 1, Table 1). Although the segregation of response to P. graminis f. sp. tritici races TTTTF and TTKSK significantly deviated from a 1:2:1 ratio in the 1st population (data not shown), the segregation pattern within segregating F2:3 families was in agreement with a 3:1 ratio for a single gene model (TTTTF, Χ2 = 0.46 p = 0.50; TTKSK, Χ2 = 0.42 p = 0.52). Majority of the SNPs linked to stem rust resistance in both populations also explained over 50% of the phenotypic variation suggesting that the resistance to each race is conferred by a single gene.
Fig 1. Chromosome and logarithm of odds (LOD) profile of stem resistance QTL detected for races TTTTF and TTKSK in two the Ae. umbellulata F2:3 populations.

A) 1st population. B) 2nd population.
Table 1. QTL detected for resistance to two Pgt races on chromosome 2U with two populations of Ae umbellulata with composite interval (CIM) and multiple QTL mapping methods (MQM).
| Linked marker | Consensus position (CM) | Method | population | LOD | Race | Phenotypic variance (%) |
|---|---|---|---|---|---|---|
| TP34301 | 87.89 | CIM | 1st population | 30.8 | TTTTF | 72.69 |
| TP44300 | 89.24 | CIM | 2nd population | 21.78 | TTTTF | 52.94 |
| TP44300* | 89.24 | CIM | 2nd population | 19.16 | TTKSK | 53.42 |
| TP5701 | 89.47 | MQM | 1st population | 17.78 | TTTTF | 72.33 |
| TP5701 | 89.47 | MQM | 1st population | 5.9 | TTKSK | - |
| TP27197 | 87.56 | MQM | 2nd population | 24.78 | TTKSK | - |
| TP13266 | 95.0⁺ | MQM | 1st population | 18.03 | TTTTF | 78.7 |
| TP13266 | 95.0⁺ | MQM | 1st population | 5.65 | TTKSK | 52.43 |
*QTL flanking markers
⁺ = map position based on 1st population
Comparing QTL interval size between the two populations, the interval size in the 1st population (derived from accession PI 298905 and PI 542369 cross) was larger (5.86 cM based on 1.5-lod interval and 0.95 Bayes confidence interval analyses) than that of the 2nd population (accession PI 5422375 and PI 554395 cross) which was estimated to be 1.68 cM. GBS marker TP34301 (consensus position: 87.89 cM) that was linked with the QTL in the 1st population was only 1.35 cM away from another GBS marker TP44300 (consensus position: 89.24 cM) linked with the QTL (based on race TTTTF) in the 2nd population, and both markers are shown in bold face in Fig 2. The markers flanking the entire region of the QTL (83.7–89.5 cM) are colored red. Based on the two populations, the QTL is bounded by GBS markers TP17463 (83.7 cM) and TP24305 (89.5 cM) within a 5.8 cM interval. Thus, the target for fine mapping with BSR-Seq approach is this 5.8 cM QTL region.
Fig 2. Stem resistance QTL region mapped from two Ae. umbellulata bi-parental populations.
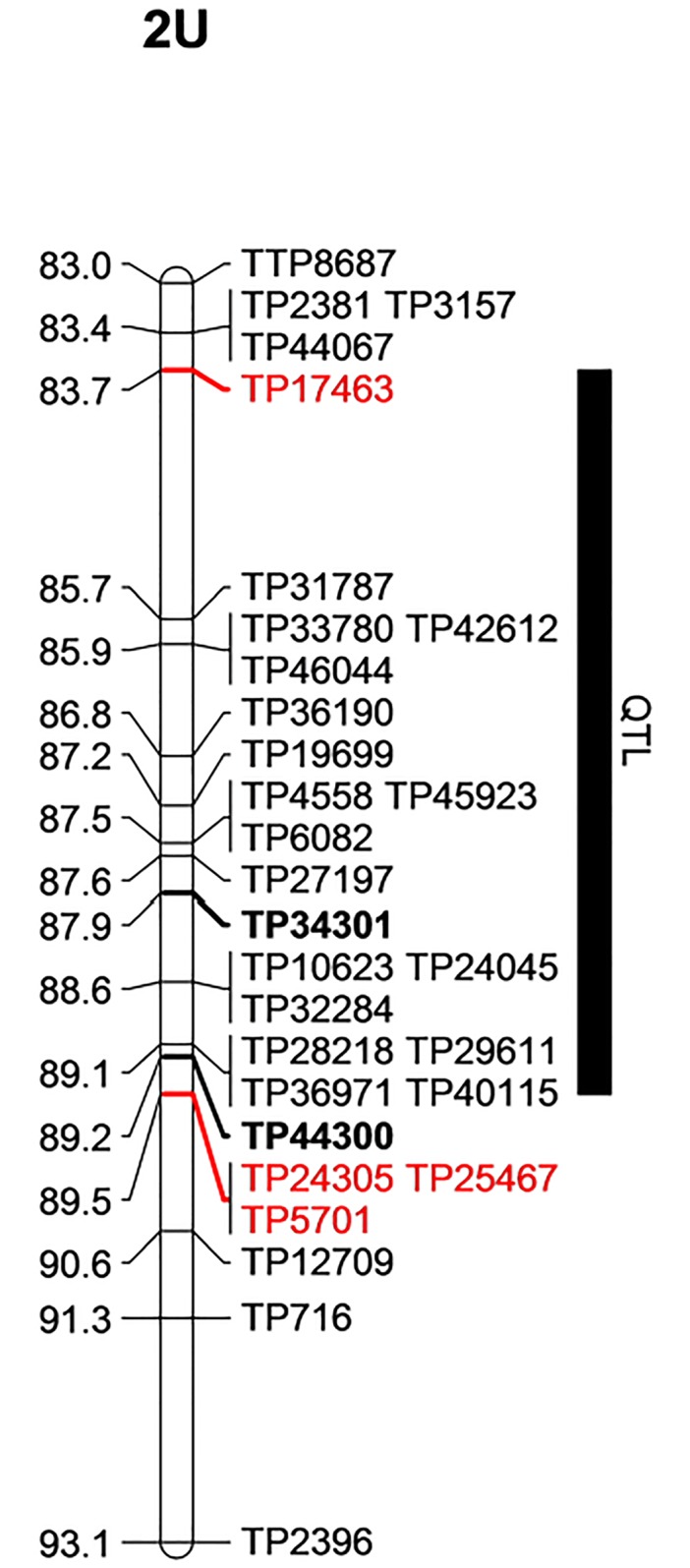
The SNP context sequences of all GBS markers were aligned against the pseudomolecule of the Ae. tauschii reference, and all markers linked with resistance against Pgt races TTTTF and TTKSK in both populations were within 7.89 Mbp (566677717–574572533 bp) on chromosome 2D of Ae. tauschii (Fig 3).
Fig 3. Physical map position of the stem resistance QTL based on Ae. tauschii reference sequence.

Transcriptome assemblies of Ae umbellulata
The number of contigs were 201,588 and 239,727 for the de novo assembly of resistant and susceptible parents, respectively (Table 2). After clustering with cd-hit program [33], the contig number was reduced to 164,456 and 194,020 for the resistant and susceptible accessions with N50 values 1,548 and 1,829, respectively.
Table 2. Basic statistics of the primary assemblies generated for PI 5422375 and PI 554395 genotypes of Ae. umbellulata.
| Genotype | Contigs | Contig mean size (bp) | N50 | GC% |
|---|---|---|---|---|
| PI 5422375 | 201,588 | 889.5 | 1,548 | 48.17 |
| PI 554395 | 239,727 | 1061.50 | 1,829 | 47.58 |
Aegilops tauschii as a reference for SNP identification
Using Ae. tauschii as a reference genome sequence, we identified a total of 400,327 SNPs, and the highest number of SNPs was recorded on chromosome 2D (correspond to 2U of Ae. umbellulata) whereas the lowest number of SNPs was found on chromosome 4D (Fig 4). Out of these, three SNPs within the QTL region (566.7–574.6 Mbp) showed large allele frequency differences between the two bulks. These three SNPs were located within 0.3 Mbp and two of them were within 14 bp. Most importantly, one of these three SNPs (position 567,944,494 bp) had the largest bulk frequency ratio (BFR) of 97.08 of all SNPs in the dataset, and another SNP (position 567,944,392 bp) was also ranked 3rd with BFR value of 89.29 (S1 Table). These two SNPs are very close (0.39 Mbp) to the GBS markers TP13266 and TP28218 that were linked to stem rust resistance QTL in the 1st and the 2nd mapping population, respectively.
Fig 4. Distribution of SNPs identified from Ae. umbellulata assembly on Ae. tauschii chromosomes.

A total of 1,237 SNPs with BFR greater or equal to four with no heterozygotes in the two bulks were obtained. Out of these, 81% (1003) SNPs were on chromosome 2D of Ae. tauschii and the remaining 234 SNPs were distributed on the remaining six chromosomes (Fig 5). Moreover, out of 129 SNPs with BFR >20, a total of 95% (123) SNPs were on chromosome 2D. Only 4.6% (6) SNPs were false positive. Among the SNPs on chromosome 2D, a majority with high BFR values mapped in the QTL region detected with the bi-parental QTL mapping approach (Fig 6).
Fig 5. Distribution of SNPs with bulk frequency ratio higher than 4 on Ae. tauschii chromosomes.

Fig 6. Distribution of SNPs with bulk frequency ratio higher than 4 on chromosome 2D Ae. tauschii chromosome.

Therefore, we predicted, based on GBS QTL mapping and bulk segregant analysis, the stem rust resistance QTL identified with Ae. umbellulata bi-parental mapping populations is in the orthologous region of 566.68–569.96 Mbp on the physical map of chromosome 2D of Ae. tauschii. The size of this refined region is approximately 3.2 Mbp. All genes in this region were extracted from the Ae. tauschii transcribed region, and there were a total of 111 predicted genes in the region (S2 Table).
For the purpose of assessing the presence of genes that contain a nucleotide-binding site and leucine-rich repeat (NLR) domain in the QTL region, contigs of the resistant parent assembly that mapped on chromosome 2D of Ae. tauschii were used. Among 34,906 resistant contigs that had a significant blast hit on chromosome 2D, a total of 147 contigs contain an NLR domain (partial domains included). Out of these, only four contigs are in the predicted gene region, of which DN39311_c0_g2_i1, DN39311_c0_g2_i4 and DN39311_c0_g2_i5 are isoforms of one gene and mapped on the same physical position (567.98 Mbp) on chromosome 2D. The remaining NLR contig, DN16173_c0_g1_i1, was mapped 0.94 Mbp away from the former (at 568.93 Mbp on chromosome 2D of Ae. tauschii physical map). Interestingly, from BSR-Seq analysis, two SNPs with the highest BFR (97.08 and 89.29) for the two bulks (resistant and susceptible) were identified at 567.94 Mbp on chromosome 2D suggesting that these three isoforms represent potential candidate genes for the QTL that we mapped with the bi-parental populations (Fig 7; S3 Table). The similarity searches in the NCBI database for one of the three contigs (DN39311_c0_g2_i1, DN39311_c0_g2_i2, DN39311_c0_g2_i4 and N39311_c0_g2_i5) retrieved CDS that encoded NBS-LRR disease resistance protein in barley (rga S-127 gene) and hexaploid wheat (NBS-LRR resistance protein CIN14 mRNA) as top hits. Similarly, the identity search for DN16173_c0_g1_i1 retrieved Brachypodium distachyon disease resistance protein RGA2-like (LOC100839373) but none of the hits was NBS-LRR protein.
Fig 7. Distribution of SNPs with NLR domain on chromosome 2D of Ae. tauschii chromosome.

De novo SNPs identification
Using the susceptible parent assembly as a reference, a total of 103,970 unique SNPs were identified with default settings of samtools and VarScan softwares. Out of these, 99,170 SNPs had significant best hits in the Ae. tauschii reference sequence and 98.8% (97,987) of the SNPs had known physical positions. The remaining 1% mapped to unmapped scaffolds. Chromosome 2U had the highest number of SNPs whereas chromosome 4U had the least number of SNPs, and the number of markers were between the two extremes for the remaining six chromosomes (Fig 8). The blast similarity search of the susceptible parent assembly in the Ae. tauschii local data base retrieved hits for a total of 203,271 contigs. Out of these, a total of 201,065 had known physical map positions and the remaining contigs mapped to unmapped scaffolds. In terms of distribution of contigs across chromosomes, although the lowest number of contigs was for chromosome 4D, generally the seven chromosomes can be grouped broadly in two. Chromosomes 2D, 3D, 5D and 7D each compose greater numbers of contigs (29,000–33,000) whereas chromosomes 1D, 4D and 6D possess lower numbers of contigs (23,000–26,000) (Fig 9). Similar patterns of contig distribution across barley chromosomes was also observed through integrating SNP-containing contigs into the barley reference assembly (Table 3).
Fig 8. Distribution of SNPs identified from susceptible parent’s assembly with de novo approach on Ae tauschii chromosomes.

Fig 9. Distribution of contigs in the susceptible parent’s assembly on the Ae. tauschii chromosomes.

Table 3. Distribution of SNP-containing contigs from susceptible Ae. umbellulata accession PI 554395 assembly across barley chromosomes.
| Chr | #Ae umbellulata contigs | Maximum gap size (cM) |
|---|---|---|
| 1H | 249 | 8.57 |
| 2H | 449 | 8.34 |
| 3H | 258 | 11.69 |
| 4H | 231 | 10.99 |
| 5H | 311 | 15.00 |
| 6H | 274 | 7.15 |
| 7H | 346 | 5.54 |
| Total | 2118 |
SNPs were also called for the two pools using the de novo assembly approach. A total of 25,931 unique SNPs were obtained with the default setting. In order to identify the parental SNPs in the two pools, we merged the parental SNP dataset with that of the bulks’ SNP data sets using the parental SNP positions to link the two SNP data sets. A total of 12,311 parental SNPs were identified from the bulks and merged with output from blasting the susceptible parent assembly with the Ae. tauschii reference sequence. Of these, a total of 11, 764 had significant hits in the reference sequence of Ae. tauschii.
Identification of SNPs associated with stem rust resistance using de novo assembly
The susceptible parent assembly was aligned against the Ae. tauchii genome reference sequence, and a total of 36, 973 unique contigs had hits on chromosome 2D, and then these unique contigs were again merged with the 25, 931 SNPs identified using the susceptible parent as a pseudo reference. Out of these, a total of 6,346 SNPs were from chromosome 2D, of these, 2,222 SNPs had BFR values of four and above (S4 Table). The number of SNPs with BFR values of 20 and above was 394. There were a total of three SNPs that are scored both in parents and bulks with BFR values of 4 and above in the identified QTL region (562.3–574.6 Mbp on chr 2D). The SNPs within the contig DN19592_c0_g1_i showed the highest BFR values and mapped within the QTL region 0.01 Mbp (at position 562305371 on chr 2D) away from TP12079 that was linked with the QTL. There were a total of nine SNPs in this contig with high BFR values. The 2nd SNP DN19324_c0_g1_i2 (position on chromosome 2D: 566490221 pb) with BFR value of 119 was mapped at position 566.49 which is also in the QTL region. The 3rd contig DN2794_c0_g1_i1 (574564921 bp) with a SNP associated with stem rust resistance had a BFR value of 6.77.
Clustering the Ae umbellulata assemblies into genes
To predict the number of genes in Ae. umbellulata, the two assemblies were aligned against the high confidence gene sets of hexaploid wheat, Ae. tauschii and barley. Contigs from Ae umbellulta that shared a high confidence CDS with hexaploid wheat were considered as isoforms of a gene. Out of the total 204,037 contigs from the resistant parent assembly, 59% (120,819) transcripts had hits in the hexaploid wheat gene model IWGSC RefSeq Annotation v1.0. These hits were grouped into the gene model identification numbers they matched. The resistant parent hits matched a total 43,276 genes in the 137,914 high confidence genes of hexaploid wheat. Out of these, a total of 42,807 genes had known chromosome information in the hexaploid wheat gene annotation file. Similarly, out of 239,727 transcripts in the susceptible parent assembly, 52% (125,293) transcripts were assigned into 44,027 unique high confidence genes in hexaploid wheat. The database comprised a total of 137,056 high confidence genes of hexaploid wheat. The similarity search in the 39, 622 high confidence gene set of Ae. tauschii retrieved a total of 22,685 and 23,111 unique genes for resistant and susceptible accession assemblies. Similarly, integrating the resistant parent into the 26,159 high confidence gene model of barley, Ae. umbellulata transcripts matched a total of 18,669 unique genes.
Annotation of transcriptome
For the two assemblies, 40% and 39% of all transcripts matched records in Uniprot for the resistant parent and susceptible parents, respectively (S5 Table). Gene Ontology (GO) terms were assigned to transcripts matching GO-annotated records in the Uniprot database to characterize the biological functions from sequence similarity. The distribution of the GO terms across the three domains were highly comparable for the two assemblies (S1 Fig).
Discussion
Wild relatives of wheat have been used as sources of resistance genes in wheat improvement programs for many decades. Ae. umbellulata is one of those wild species that has been a useful source of resistance genes. Leaf rust resistance Lr9 was the 1st gene that was transferred to wheat 60 years ago from Ae. umbellulata, the secondary gene pool of wheat [34]. Ae. umbellulata has also been identified as a source of resistance to stem rust [35, 25, 11], powdery mildew, Hessian fly and green bug [8]. In the current work a new stem rust resistance gene from Ae. umbellulata was mapped on chromosome 2U, and its map position was validated with independent mapping bi-population, and finally the region was fine-mapped via integration of bi-parental QTL mapping method with the bulk segregant RNA-Seq analysis (BSR-Seq) approach.
BSR-Seq has been recently applied for fine-mapping gene region in cereal crops such as wheat [18, 19] and maize [16, 17]. It is a powerful method to enrich gene region with SNPs derived only from expressed portion of a genome thereby facilitates identification the actual gene itself. Using SNP markers that showed large allele frequency difference between resistance and susceptible bulks, the QTL that was linked with stem rust resistance in two Ae umbellulata bi-parental populations was validated and refined with the BSR-Seq approach in this report.
We used comparative mapping to infer the map positions of QTL-linked GBS markers and RNA-Seq derived SNPs using recently released Ae tauschii reference sequence v.1.0, and de novo assemblies of Ae. umbellulata. From the integration of the SNP context sequences from RNA-Seq and GBS data into a pseudomolecule of Ae. tauschii, the QTL-associated GBS markers were mapped in the same region with the RNA-Seq SNPs that showed the highest BFR ratio, suggesting the suitability of BSR-Seq for validating and identifying candidate genes detected originally as QTL in classical mapping. Once the gene region was confidently known, by combining information from the physical positions of all GBS markers that were associated with the resistance gene in two bi-parental populations and RNA-Seq SNPs with high BFR) the size of gene interval was approximately 3.2 Mbp based on physical map of chromosome 2D of Ae. tauschii. Given stem rust resistance genes are known to encode nucleotide-binding site and leucine rich repeat (NLR) proteins [36–38], and the QTL identified from Ae. umbellulata bi-parental populations fits into features of a qualitative gene, exploring the presence of NLR genes in the predicted region is useful to identify short lists of potential candidate genes. In this way, five contigs with NLR domains were discovered from the resistant parent assembly that had hits within this 3.2 Mbp gene region, of which four NLR contigs are variants of a single gene. The similarity search of the four variants of this gene indicated that the gene encodes disease resistance NLR proteins in barley and hexaploid wheat suggesting the NLR contigs represent a potential candidate gene for the major stem resistance QTL we mapped on chromosome 2U of Ae umbelluata. Moreover, this result agrees with our previous hypothesis that the gene identified from Ae umbellulata is a novel gene as none of the candidate NLR contigs had similarity to the known major disease resistance genes in barley and hexaploid wheat.
Two approaches were followed for the identification of SNPs that showed differences between the resistant and susceptible bulks to fine map the QTL region: de novo method and orthologous reference-based approach. Although SNPs were effectively identified with the de novo approach where de novo assemblies were used as a pseudo-references to call SNPs from RNA-Seq data, orthologous reference sequence-based approach enabled us to precisely identify SNPs that are closer to the region of the gene mapped with GBS markers with two bi-parental mapping populations. This indicated that with the absence of a species reference sequence the use of closely related species reference sequence is an advantageous alternative for SNPs identification. SNPs with high BFR from both methods mapped at the same region, but the resolution was better in the cross-species reference-based approach as we identified more SNPs with higher BFR that mapped closer to the GBS marker position.
In summary, our study demonstrated that BSR-Seq is cheap and efficient approach not only precisely validating the map position of previously identified QTL but also identification of potential candidate genes is possible as the approach provides additional SNPs in the mapping interval. BSR-Seq could also be utilized for other wild relatives of crop species that are difficult to propagate and self-fertilize.
Supporting information
(TIF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability
All relevant data are within the paper and its Supporting Information files. Data is also deposited in the NCBI database (submission number: SUB5918868; accession number: PRJNA560000).
Funding Statement
Funded with the financial support from United States Department of Agriculture-Agricultural Research Service.
References
- 1.Singh RP, Hodson DP, Huerta-Espino J, Jin Y, Njau P, Wanyera R, et al. Will stem rust destroy the world’s wheat crop? Adv. Agron. 2008; 98:271–309. [Google Scholar]
- 2.Singh RP, Hodson DP, Jin Y, Lagudah ES, Ayliffe MA, Bhavani S, et al. Emergence and spread of new races of wheat stem rust fungus: continued threat to food security and prospects of generic control. Phytopathol. 2015; 105:872–884. [DOI] [PubMed] [Google Scholar]
- 3.Liu W.X., Jin Y., Rouse M., Friebe B., Gill B., and Pumphrey M.O. Development and characterization of wheat-Ae. searsii Robertsonian translocations and a recombinant chromosome conferring resistance to stem rust. Theor Appl Genet. 2011; 122:1537–1545. 10.1007/s00122-011-1553-4 [DOI] [PubMed] [Google Scholar]
- 4.Liu W., Rouse M., Friebe B., Jin Y., Gill B., and Pumphrey M.O. Discovery and molecular mapping of a new gene conferring resistance to stem rust, Sr53, derived from Aegilops geniculata and characterization of spontaneous translocation stocks with reduced alien chromatin. Chromosome Res. 2011; 19:669–82. 10.1007/s10577-011-9226-3 [DOI] [PubMed] [Google Scholar]
- 5.Olson E. L., Rouse M. N., Pumphrey M. O., Bowden R. L., Gill B. S., and Poland J. A. Simultaneous transfer, introgression, and genomic localization of genes for resistance to stem rust race TTKSK (Ug99) from Aegilops tauschii to wheat. Theor Appl Genet, 2013; 126:1179–1188. 10.1007/s00122-013-2045-5 [DOI] [PubMed] [Google Scholar]
- 6.Olson E. L., Rouse M. N., Pumphrey M. O., Bowden R. L., Gill B. S., and Poland J. A. Introgression of stem rust resistance genes SrTA10187 and SrTA10171 from Aegilops tauschii to wheat. Theor Appl Genet. 2013; 126: 2477–2484. 10.1007/s00122-013-2148-z [DOI] [PubMed] [Google Scholar]
- 7.Wulf B, Moscou M. Strategies for transferring resistance into wheat: from wide crosses to GM cassettes. Frontiers in Plant Science 2014; 5:1–11. 10.3389/fpls.2014.00692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill BS, Sharma HC, Raup WJ, Browder LE, Hatchet JW, Harvey JH, et al. Evaluation of Aegilops species for resistance to wheat powdery mildew, wheat leaf rust and Hessian fly and green bug. Plant Dis.1985; 69:314–316. [Google Scholar]
- 9.Friebe B., Jiang J, Raupp WJ, McIntosh RA and Gill BS. Characterization of wheat alien translocations conferring resistance to diseases and pests: current status. Euphytica 1996; 71:59–87. [Google Scholar]
- 10.Schneider A., Molnar I, and Molnar-Lang M. Utilization of Aegilops (goat grass) species to widen the genetic diversity of cultivated wheat. Euphytica 2008; 163:1–19. [Google Scholar]
- 11.Edae AE, Olivera DP, Jin Y, Poland JA, and Rouse NM. Genotype-by-Sequencing facilitates genetic mapping of a stem rust resistance locus in Aegilops umbellulata, a wild relative of cultivated wheat. BMC Genomics 2016; 17:1039 10.1186/s12864-016-3370-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chepelev I., Wei G, Tang Q and Zhao K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucl Acids 2009; 37:e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canovas A, Rincon G, Islas-Trjo A, Wickamasinghe S and Medrano J. SNP Discovery in the bovine milk transcriptome using RNA-Seq technology. Mamm Genome 2010; 21: 592–598. 10.1007/s00335-010-9297-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Neil S, Emrich S. Assessing De Novo transcriptome assembly metrics for consistency and utility. BMC Genomics 2013; 14:465 10.1186/1471-2164-14-465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michelmore RW, Paran I, Kesseli RV. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci 1991; 88:9828–9832. 10.1073/pnas.88.21.9828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu SZ, Yeh CT, Tang HM, Nettleton D, Schnable PS. Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS One 2012; 7:e36406 10.1371/journal.pone.0036406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L., Li D, Liu SZ, Ma XL, Dietrich CR, Hu HC, et al. The maize glossy13 gene, cloned via BSR-Seq and Seq-walking encodes a putative ABC transporter required for the normal accumulation of epicuticular waxes. PLoS One 2013; 8:e82333 10.1371/journal.pone.0082333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trick M., Adamski NM, Mugford SG, Jiang CC, Febrer M, et al. Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol 2012; 12:14 10.1186/1471-2229-12-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramirez-Gonzalez RH., Segovia V, Bird N, Fenwick P, Holdgate S, et al. RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol 2015; 13: 613–624. [DOI] [PubMed] [Google Scholar]
- 20.Zou C, Wang P, Xu Y. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol 2016; 10:1941–1955. 10.1111/pbi.12559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouse NM, Wanyera R, Njau P, Jin Y. Sources of resistance to stem rust race Ug99 in spring wheat germplasm. Plant Dis. 2011; 95:762–766. 10.1094/PDIS-12-10-0940 [DOI] [PubMed] [Google Scholar]
- 22.Stakman EC, Stewart DM, Loegering WQ. Identification of physiologic races of Puccinia graminis var. tritici. U. S. Dep Agric Agric Res Serv. 1962; E-617. [Google Scholar]
- 23.Edae AE., Olivera DP, Jin Y, Rouse N M. Genotyping-by-Sequencing facilitates a high density consensus linkage map for Aegilops umbellulata, a wild relative of cultivated wheat. G3: Genes/ Genomics/ Genetics 2017; 7(5): 1551–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolger AM., Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30(15):2114–20. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grabherr M, Haas M, Yassour M, Levin J, Thompson D, Amit I, et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol 2013; 29(7): 644–652. https://trinotate.github.io/ Accessed on 05/18/2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo MC., Gu YQ, Puiu D, Wang H, Twardziok SO, Deal KR, et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017; 551:498–502. 10.1038/nature24486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H., Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25(16): 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koboldt D, Chen K, Wylie T, Larson D, McLellan M, Mardis E. et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009; 25(17):2283–2285. 10.1093/bioinformatics/btp373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Research 2011; 39:W29–W37, 10.1093/nar/gkr367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Research 2009; 37:W22–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steuernagel B, Florian J, Kamil W, Jonathan J, Brande W. NLR-parser: rapid annotation of plant NLR complements. Bioinformatics 2015; 31(10):1665–166. 10.1093/bioinformatics/btv005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jupe F, Pritchard L, Etherington GJ, Mackenzie K, Cock P, Wright F, et al. Identification and localization of the NB-LRR gene family within the potato genome. BMC Genomics 2012; 13:75 10.1186/1471-2164-13-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006; 22(13):1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- 34.Sears ER. The transfer of leaf rust resistance from Aegilops umbellulata to wheat. Brookhaven Symp. Biol. 1956; 9: 1–21. [Google Scholar]
- 35.Ozgen M, Yildiz M, Ulukan H, Koyuncu N. Association of gliadin protein pattern and rust resistance derived from Aegilops ubmellulata Zhuk. In winter Triticum durum Desf. Breeding Science 2004; 54:287–290. [Google Scholar]
- 36.Saintenac C., Zhang W, Salcedo A, Rouse N M, Trick HN, Akhunov E, et al. Identification of wheat gene Sr35 that confers resistance to Ug99 stem rust race group. Science 2013; 341: 783–786. 10.1126/science.1239022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellis JG, Lagudah ES, Spielmeyer W, Dodds PN. The past, present and future of breeding rust resistant wheat. Frontiers in Plant Science 2014; 5(641). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang W, Chen S, Abate Z, Nirmala N, Rouse NM, Dubcovsky J, et al. Identification and characterization of Sr13, a tetraploid wheat gene that confers resistance to the Ug99 stem rust race group. PNAS 2017; E9483–E9492. 10.1073/pnas.1706277114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. Data is also deposited in the NCBI database (submission number: SUB5918868; accession number: PRJNA560000).