

Abstract
Purpose:
KIT mutations (KIT+) are common in core binding factor (CBF) AML and have been associated with varying prognostic significance. We sought to define the functional and clinical significance of distinct KIT mutations in CBF pediatric AML.
Experimental Design:
Following transfection of exon 17 (E17) and exon 8 (E8) mutations into HEK293 and Ba/F3 cells, KIT phosphorylation, cytokine independent growth, and response to tyrosine kinase inhibitors (TKI) were evaluated. Clinical outcomes of patients treated on COG AAML0531 (), a phase III study of gemtuzumab ozogamicin (GO), were analyzed according to mutation status (KIT+ vs wild type KIT (KIT−)) and mutation location (E8 vs. E17).
Results:
KIT mutations were detected in 63/205(31%) patients; 22 (35%) involved only E8, 32(51%) only E17, 6(10%) both exons, and 3(5%) alternative exons. Functional studies demonstrated that E17, but not E8, mutations result in aberrant KIT phosphorylation and growth. TKI exposure significantly impacted growth of E17, but not E8, transfected cells. KIT+ CBF AML patients had comparable overall survival (OS) to that of KIT− (78%, vs. 81%, p=0.905) but higher relapse rates (RR 43% vs. 21%, p=0.005). E17 KIT+ outcomes were inferior to KIT- patients [disease free survival (DFS) 51% vs. 73%, p=0.027; RR 21% vs. 46%, p=0.007)] although GO abrogated this negative prognostic impact. E8 mutations lacked significant prognostic impact and GO failed to significantly improve outcome.
Conclusion:
E17 mutations impact prognosis in CBF AML, as well as response to GO and TKIs, thus clinical trials utilizing both agents should be considered for KIT+ patients.
Summary of Translational Relevance:
Patients with core binding factor (CBF) AML generally have a good prognosis, while co-occurrence of KIT mutations in this group has been associated with varying prognostic significance. This study evaluated the functional properties of KIT mutations occurring in the immunoglobulin (IgG) and tyrosine kinase domains (TKDs) and their prognostic impact. We found that mutations in the IgG domain (exon 8) have no functional or prognostic impact. In contrast, mutations in the TKD (exon 17) resulted in aberrant KIT phosphorylation and were associated with inferior clinical outcomes. Importantly, exon 17 mutations also conferred sensitivity to KIT-directed tyrosine kinase inhibitors (TKI) in vitro. Moreover, when exon 17-positive patients received gemtuzumab ozogamicin in combination with chemotherapy, their negative prognostic impact was abrogated. Patients with exon 17 mutated CBF AML represent a higher risk subgroup of CBF AML and may benefit from TKIs or CD33-targeted therapy.
INTRODUCTION
The cytogenetic abnormalities t(8;21)(q22;q22) and inv(16)(p13.1q22)/t(16;16)(p13.1;q22) [hereafter referred to as inv(16)], commonly referred to as core binding factor (CBF) acute myeloid leukemia (AML), are observed in 20–30% of pediatric AML cases and confer favorable outcome with overall survival (OS) in the range of 85%.(1,2) However, disease outcomes with initial therapy are not favorable in all cases,(1,3,4) thus efforts continue to further risk stratify such patients to improve clinical response. The KIT proto-oncogene encodes a transmembrane glycoprotein type III receptor tyrosine kinase (RTK).(5–7) Mutations in KIT serve as potent oncogenic events in a number of malignancies, namely gastrointestinal stromal tumors (GIST), melanomas, and mastocytosis.(8–10) Oncogenic mutations often effect domains critical to RTK activity, such as the juxtamembrane domain (JMD) in exon 11 as seen in GIST, or the tyrosine kinase domain (TKD), and result in ligand independent activation.(7,11,12) Among malignancies with KIT mutations (KIT+), inhibition of aberrantly activated KIT has been achieved with a subset of tyrosine kinase inhibitors (TKI) with KIT targeting potential; these medications have proven to be quite effective and, in some cases, are considered the front-line therapeutic approach for KIT-mutated disease.(13,14)
KIT mutations are enriched in CBF AML and published adult series report a prevalence of 12–46% of t(8;21) and 9–53% of inv(16) AML.(15–19) Within pediatric CBF AML, KIT+ disease is seen in approximately 20–40% of patients.(2,17,19–22) We previously screened CBF AML samples from pediatric patients enrolled on serial trials and identified KIT mutations in approximately 20% of patients. Within that heterogeneously treated population, KIT+ disease lacked prognostic significance overall and within cytogenetic subtypes.(19) Data regarding the prognostic significance of KIT+ CBF AML is conflicting, particularly in children. While some pediatric and dual adult/pediatric studies have failed to show a prognostic impact,(2,20,21,23,24) other pediatric series and meta-analyses dispute such findings, suggesting prognostic impact may differ in the context of certain KIT mutations or cytogenetic subgroups.(15,17,22,25–27)
Given this discrepancy and the uncertainty regarding the functional significance of KIT mutations, we sought to investigate the functional impact of distinct mutation subsets as well as TKI response in an in vitro model. Moreover, we defined the prognostic impact, both overall and by mutation subtype, of pediatric KIT+ CBF AML patients treated on the pediatric phase III de novo AML study COG AAML0531, where the primary aim entailed randomization to the CD33-targeted antibody drug conjugate gemtuzumab ozogamicin (GO). Thus, our analysis also provides insight into the therapeutic efficacy of CD33 targeting agents in KIT+ CBF AML. As GO has previously been found to be particularly effective in CBF AML,(1,28–30) we hypothesized that it may also provide therapeutic benefit in KIT+ disease. This large-scale analysis of pediatric CBF AML patients treated with a uniform chemotherapy backbone provides valuable insight into the distinct functional properties of KIT mutations and their prognostic significance, both overall, and in the context of GO treatment.
MATERIALS AND METHODS
Pediatric patients identified as having CBF AML and enrolled on COG AAML0531 were eligible for this study. All samples from patients enrolled on AAML0531 were eligible for our correlative study if written consent for biology studies was obtained. The trial was conducted in accordance with the Declaration of Helsinki. The institutional review boards of all participating institutions approved the clinical protocol and this study was approved by the COG Myeloid Disease Biology Committee. Details of the treatment protocol have been described previously.(1) Patients with CBF AML enrolled on AAML0531 were randomly assigned to one of 2 treatment arms, Arm A (No GO) which was conventional chemotherapy only and Arm B (GO) which included a dose of GO (3 mg/m2) on day 6 of induction I and day 7 of intensification II. Given the historically favorable outcome of CBF AML,(1) CBF patients enrolled on the study received 5 cycles of chemotherapy and the front-line use of hematopoietic stem cell transplant was prohibited.
Cytogenetic data was available for 949/1022 (93%) patients enrolled on AAML0531. A diagnosis of CBF AML was made if either t(8;21)(q22;22) or inv(16)(p13;q22)/t(16;16)(p13;q22) was detected in diagnostic bone marrow (BM) or peripheral blood (PB) specimens by either conventional cytogenetics and/or fluorescent in situ hybridization techniques. Karyotype was confirmed by central review in 100% of cases.
KIT mutational analysis:
Samples from diagnostic and remission BM or PB underwent DNA extraction using the AllPrep Extraction Kit (Qiagen). The integrity of the samples was confirmed by visualization on a 0.8% agarose gel. All samples then underwent targeted exome capture sequencing, which included the entire coding region of KIT, as previously described.(31) Samples from relapse specimens underwent analysis of mutations of exon 8 and 17 using amplification by genomic PCR as previously described.(19)
CD117 expression analysis:
Flow cytometry was used to determine the CD117 (KIT) mean fluoresence intensity (MFI) of myeloid progenitor cells, as defined by CD45 low and side scatter. BM cells were incubated with CD117-PE (clone 104D2; BD Biosciences), C45-peridinin chlorophyll protein (clone 2D1; BD Biosciences), and CD34-allophycocyanin (clone HPCA-2; BD Biosciences). Cells were then incubated followed by red cell lysis and then washed, following which flow cytometric data collected as previously described.(32,33) The CD117 linear MFI was determined without knowledge of any other clinical characteristics. Ba/F3 cell lines also underwent CD117 expression evaluation utilizing the antibody and procedures described above.
Generation of mutants:
KIT D419G, D419del, V559D, L576P, D816V, and N822K mutations were engineered into samples pLXSNneoKIT using the QuikChange mutagenesis kit (Agilent Technologies) and were confirmed by sequence analysis.
Cell Culture and Transduction:
HEK293T cells (ATCC) were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), L-glutamine, and penicillin/streptomycin. Ba/F3 cells (DSMZ) were maintained in RPMI 1640 supplemented with 10% FBS, 10 ng/mL IL-3, L-glutamine, and penicillin/streptomycin. Ba/F3 cells were passaged 3–6 times following thaw and prior to transduction. For virus generation, HEK293T cells were seeded at 6×105 cells per well and incubated overnight, with media replacement prior to transfection. The transfection complex (135µL Gibco® Opti-MEM +10µL FuGENE 6 Transfection Reagent) and 3.4µg of KIT retroviral expression vector was incubated at room temperature for 15 minutes. The complex was added to the cells and viral supernatant collected and filtered after 48 hours. For transduction, Ba/F3 cells (DSMZ) were split to 4×105 cells/mL and viral supernatant was added to 1×106 cells at 24 hours, centrifuged at 2500 rpm for 90 minutes at 300C and incubated overnight. Cells were then washed with phosphate buffered saline (PBS) to remove viral particles and re-suspended in fresh media and at 24 hours post-virus removal were selected with 500 μg/mL Geneticin. Mycoplasma testing was performed on the Ba/F3 cells using the MycoAlert Mycoplasma Detection Kit (Lonza).
Immunoblot Analysis:
Cells were washed in PBS and lysed in Triton-x Lysis Buffer supplemented with protease and phosphatase inhibitors. Lysate concentrations were measured using the Pierce BCA™ Protein Assay. Protein was subjected to electrophoresis on a 4–12% Bis-Tris Gel with MES buffer according to manufacturer guidelines using the Invitrogen Bolt. Following electrophoresis, proteins were transferred to a nitrocellulose membrane on the Invitrogen iBlot and underwent blocking using iBind solution. Antibody probing was performed using iBind with anti-KIT (Cell Signaling), anti-Phospho-KIT (Cell Signaling), anti-β-actin AC-15 (Sigma-Aldrich,), HRP Goat anti-Mouse IgG (H+L) secondary (ThermoFisher), and HRP Goat anti-Rabbit IgG (H+L) secondary (ThermoFisher). Blots were developed in SuperSignal West Pico Chemiluminescent Substratea and imaged with Bio-Rad ChemiDoc™ MP.
Cell Proliferation/IL-3 withdrawal:
Cells were expanded after being washed three times and then plated at a density of 5×105 cells/ml in non-IL-3 containing medium. Viable cell counts were obtained using trypan blue staining on a Countess II (ThermoFisher).
Drug Sensitivity:
Following transfection, Ba/F3 cells were exposed to dasatinib (Selleckchem) and crenolanib (Arog Pharmaceuticals). Cells were plated in triplicate and cultured in the presence of drug for 72 hours, after which cell viability was assessed using CellTiter-Glo reagent on a BioTek Synergy H4 Hyrbid plate reader using Gen5 2.1 software. Experiment were repeated in triplicate with the median results shown. Numerical IC50 values were generated using non-linear best-fit regression analysis with Prism6 software (GraphPad).
Statistical methods:
Clinical outcome data from AAML0531 was current as of December 31, 2014. The median days follow up for all eligible patients alive at last contact for the clinical trial included in our analysis was 1946 days (range 0–2908 days). For the purpose of this study, patients were defined as being in complete remission (CR) if they had <5% blasts and absence of extramedullary disease after completion of induction I course. The Kaplan-Meier method was used to estimate survival outcomes.(34) OS was defined as the time from study entry or from end of course I, for patients in CR, to death. Event-free survival (EFS) was defined as the time from study entry until death, induction failure, or relapse of any type. Disease-free survival (DFS) was defined from the end of course I for patients in CR until relapse or death from any cause. Relapse rate (RR) was defined as the time from end of induction I for patients in CR to relapse, where deaths in absence of relapse were considered competing events.(35) The significance of predictor variables was tested using log-rank statistic for OS, EFS, and DFS and the Gray statistic for RR.(36,37) Patients lost to follow-up were censored at time of last point of contact. The significance of observed difference in proportions was analyzed by the chi-square test between patient groups and the Fisher’s exact test was used if data were sparse. The Kruskal-Wallis test was used to determine the significance between differences in medians of the groups.
RESULTS
Study population
All CBF AML patients enrolled on COG AAML0531 were eligible for this study. Of the 247 eligible CBF patients, 205 (83%) [109 t(8;21) and 96 inv(16)] had diagnostic BM or PB specimens available for KIT analysis by targeted exome capture and complete clinical information available.
Prevalence and type of KIT mutations at diagnosis
KIT mutations were detected in 63 (31%) samples. Mutations were confirmed to be somatic in all evaluable samples (n=54, 86%) where both a diagnostic and remission sample were available, thus no germline KIT mutations were detected in our cohort. Among all KIT mutations detected, 22 (35%) involved exon 8 (E8) only, 32(51%) involved exon 17 (E17) only, 6 (10%) involved both exons, 1(1%) entailed a dual E17/exon 13 mutation, and an additional 3 (5%) patients had mutations detected outside the 2 hot-spot regions (Figure 1, Supplemental Figure S1A). Mutations occurring in E8, the extracellular IgG domain, included indels (n=24), which were largely inframe, or single nucleotide variants (SNVs) (n=8) (Figure 1). E17 mutations, occurring in the activation loop of the TKD, were mostly SNVs; the most common loci involved were D816 and N822, and included a variety of amino acid substitutions (Figure 1). Mutations located outside E8 and 17 were located in exon 11 (E11; n=2), exon 13 (E13; n=1), both of which make up the JMD, and in exon 16 (E16; n=1), the TKD, a region in which mutations are rarely reported outside of GIST and melanomas. The E11 and E16 mutations were isolated KIT abnormalities. In total, 15 (24%) of KIT+ patients were found to harbor multiple mutations (n = 13 with 2 mutations and n=2 with 3 mutations). Breakdown among E8 mutations by CBF translocation demonstrated that 13 (46%) occurred in t(8;21) and 15 (54%) inv(16) samples (Supplemental Figure S1B). E17 mutations demonstrated a predominance of t(8;21) with 24 (63%) occurring in t(8;21) and 14 (37%) occurring in inv(16) patients (Supplemental Figure S1C). The median variant allele frequency (VAF) of the KIT mutation detected was 0.32 (range 0.04–0.83). Among our KIT+ cohort there were 10 patients with documented relapse with available specimen for KIT sequencing. Five patients had no detectable E8 or E17 KIT mutation at the time of relapse, with diagnostic VAF available in 4 specimens (range 0.15–0.45). An additional 5 patients were found to have retained KIT mutations at the time of relapse (E8, n=4; E17, n=1), with diagnostic VAF available in 4 specimens (range 0.13–0.7).
Figure 1.

KIT mutations detected in CBF AML patients by targeted exome capture. Mutations most commonly occurred in exon 8, which is part of the extracellular immunoglobulin domain, and exon 17, part of the tyrosine kinase domain. Mutations detected include single nucleotide variants (SNVs) and small indels, most of which were in-frame. Dotted lines indicate exon boundaries.
Clinical and Biological Characteristics for CBF AML patients with KIT mutations
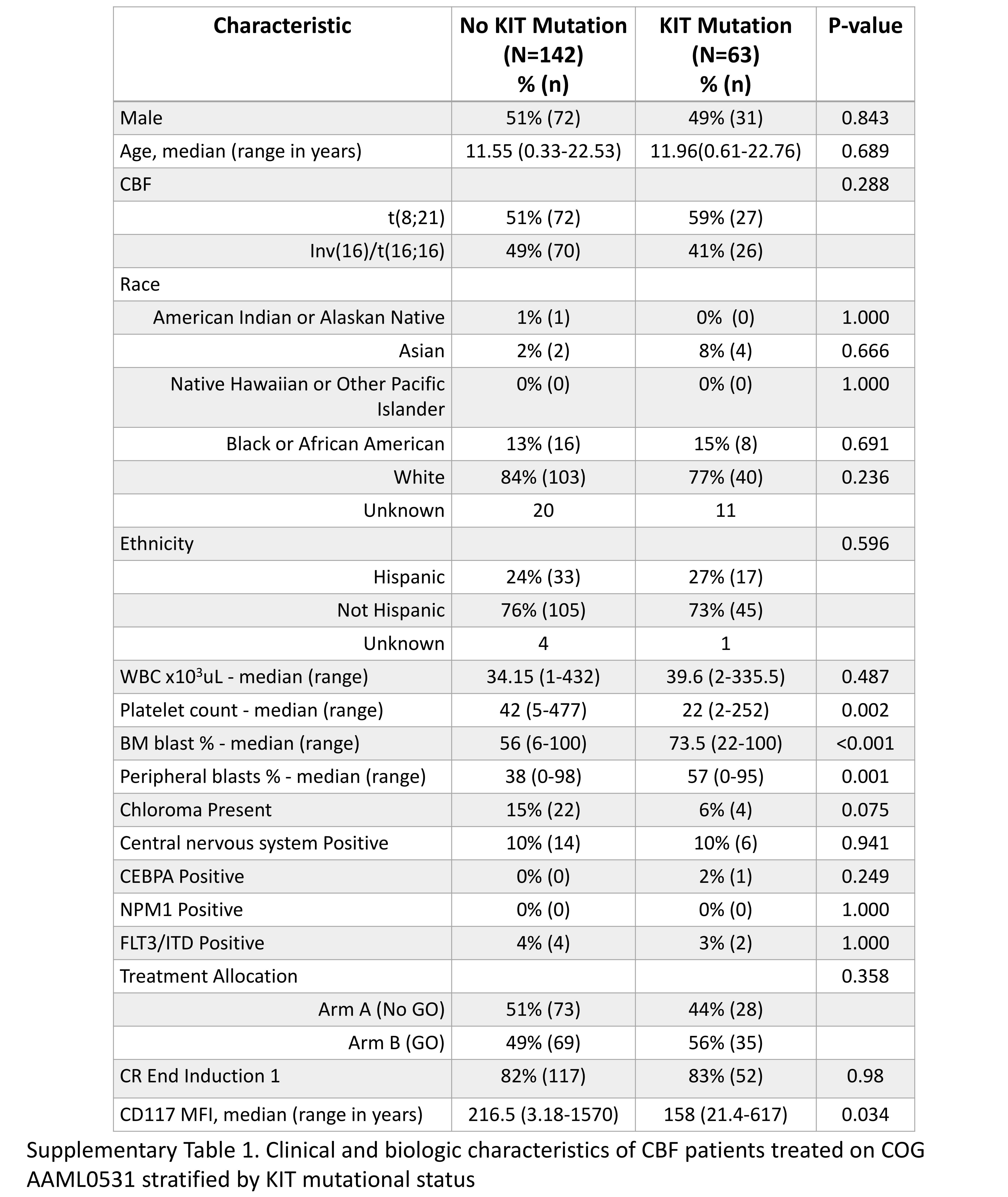
Clinical characteristics and CR rates of the 63 KIT+ CBF AML patients were compared to the 142 CBF patients with wild-type KIT (KIT-). There were no significant differences in sex, race, ethnicity and median age for the 2 groups (Supplementary Table S1). KIT+ patients had significantly higher BM blast percentage and peripheral blasts. There were no significant differences in treatment assignment for KIT+ vs. KIT− patients. Rates of induction CR were also comparable (KIT+ 83% vs. KIT− 82%, p=0.980, Supplementary Table S1). There was a broad range of CD117 surface expression among the CBF patients overall (range 3.18–1570), with a median CD117 MFI of 199, which was significantly higher compared to that of the non-CBF patients treated on AAML0531 (n=795) who had a median MFI of 74.55 (range 2.59–894; p<0.001). Among the CBF cohort, median CD117 expression MFI in KIT− patients was 216.5 (range 3.18–1560) which was significantly compared to 158 (range 21.4–617) in KIT+ patients (p=0.034, Supplementary Table S1). Patients with E8 mutations had a median expression of 178 (range 58.2–342), which was not significantly different than KIT− patients (p=0.568). In contrast, E17+ patients had a median CD117 expression of 132.5 (range 21.4–617), which was significantly lower than those with KIT− CBF AML (p=0.008).
Functional Evaluation of KIT Mutations
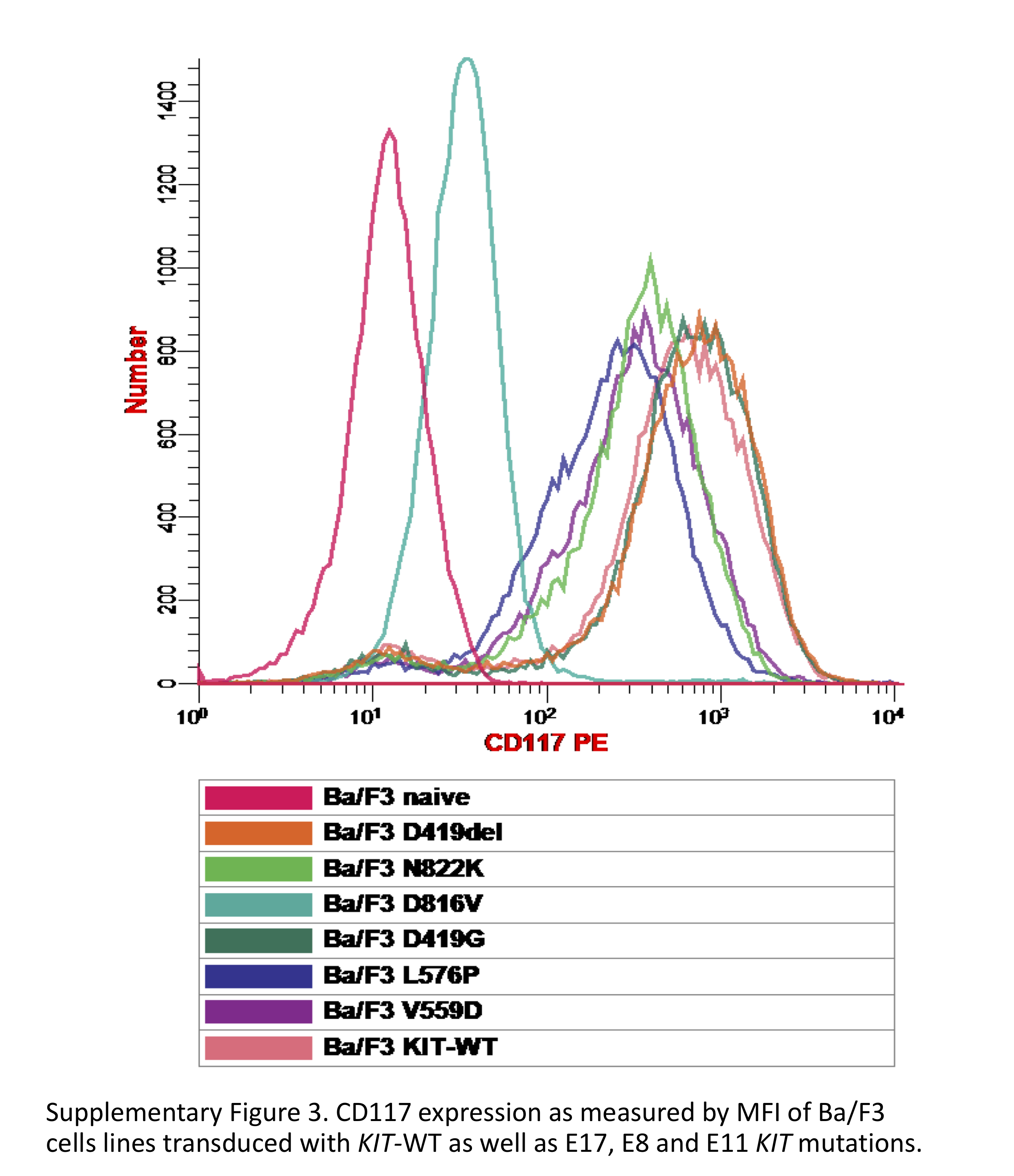
HEK293 cells transduced with E8 mutations (D419G, D419del) had no evidence of KIT phosphorylation and did not demonstrate IL-3 independent proliferation following transduction into Ba/F3 cells (Figure 2A, B). Conversely, transduction of the E17 mutations (D816V, N822K) into HEK293 cells resulted in autonomous receptor phosphorylation and IL-3 independent proliferation in Ba/F3 cells (Figure 2A, B). The D816V mutation resulted in more potent KIT phosphorylation as well increased detection of the immature (125 kDa) vs mature (145kDa) form of KIT, and corresponding activating potential in the Ba/F3 cells, compared to N822K (Figure 2A, B). Transduction of the E11 mutations (V559D, L576P) also resulted in aberrant KIT phosphorylation (Supplementary Figure S2A). Following transduction into the Ba/F3 cells, the L576P demonstrated late much slower growth compared to the E17 mutations and no IL-3 independent growth was observed in the V559D (Supplementary Figure S2B). Evaluation of CD117 expression of cell lines demonstrated a range of MFI (Supplemental Figure S3). Ba/F3 cells transduced with the E8 mutations demonstrated the highest CD117 expression with an MFI of 536.13 and 544.49 for D419del and D419G respectively. E17 mutant Ba/F3 transduced cells demonstrated lower CD117 expression comparatively, with the MFI of D816V and N822K found to be 36.64 and 290.62 respectively, which corresponded with what was seen in patient samples.
Figure 2.
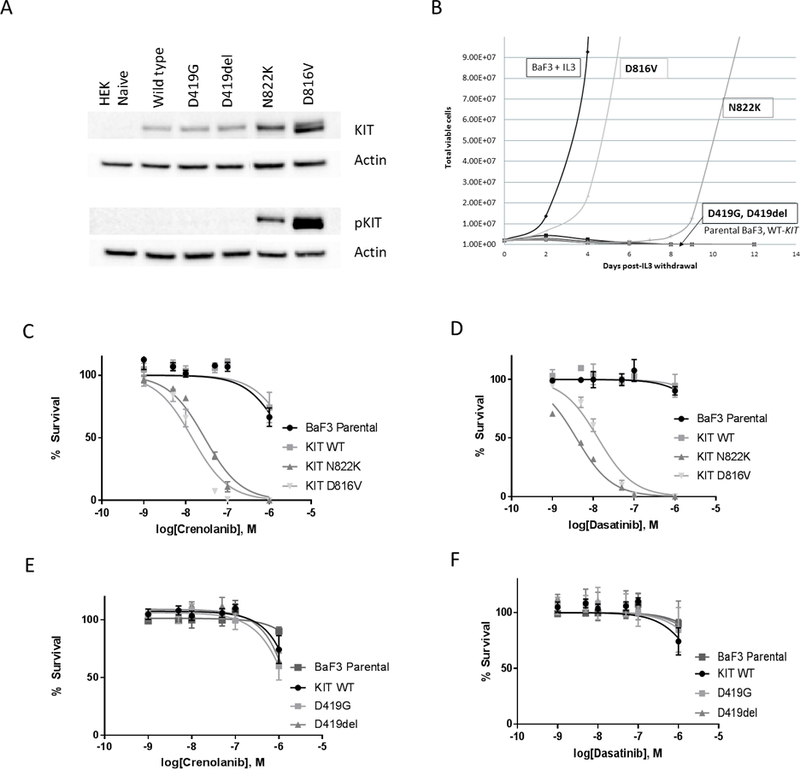
In vitro evaluation of functional effect of KIT mutations in exon 8 and exon 17. (A) KIT phosphorylation following transfection of the mutations in exon 8 (D419G, D419del) and exon 17 (D816V, N822K) into HEK293 cell line. (B). IL3 withdrawal experiments in the Ba/F3 cell line confirmed the transforming potential of the E17 mutations D816V and N822K, both of which demonstrated growth following withdrawal of IL3. (C) Cytotoxicity following crenolanib exposure in Ba/F3 transduced cells with D816V and N822K mutations; (D) Cytotoxicity following dasatinib exposure in Ba/F3 transduced cells with D816V and N822K; (E) Cytoxicity following crenolanib exposure in Ba/F3 transduced cells D419del and D419G mutations, (F) Cytotoxicity following dasatinib exposure in Ba/F3 transduced cells with D419del and D419G mutations.
We subsequently evaluated the cytotoxic effect of dasatinib and crenolanib on transduced cells. Neither parental Ba/F3 nor those transduced with WT-KIT demonstrated any response to the TKIs with IC50s >10,000 nM (Figure 2C–F). However, Ba/F3 cells transduced with either of the E17 variants were uniquely susceptible to both TKIs. D816V-transduced cells had an IC50 of 14.3 and 13 nM when exposed to crenolanib and dasatinib respectively (Figure 2C, D). The N822K-transduced cells had an IC50 of 3.8 and 23 nM following dasatinib and crenolanib exposure, respectively (Figure 2C, D). Ba/F3 cells transduced with the D419G and D419del mutations and cultured in the presence of IL-3 demonstrated no particular sensitivity to the TKIs (Figure 2E, F). Transduction of the V559D and L576P mutations into HEK293 cells exhibited sensitivity to inhibition of phosphorylation with dasatinib, but not crenolanib (Supplementary Figure S2C–F).
Prognostic significance of KIT mutations in CBF AML
Five-year OS for KIT+ vs. KIT− CBF AML was comparable (KIT+ 78±12% vs. KIT− 81±7% (p=0.905, Table 1). However, KIT+ patients who achieved CR had inferior DFS compared to KIT− patients (55±14% vs. 73±8%, respectively, p=0.040, Figure 3A) and higher RR (43±14% vs. 21±8% for KIT− patients, p=0.005, Figure 3B). Analysis of outcome by cytogenetic subtype did not demonstrate any differences, with exception that KIT+ patients with t(8;21) had higher RR (36±19% vs. 11±9% for KIT−, p=0.007; Table 1). When treated with conventional chemotherapy only, KIT+ patients had inferior outcomes compared to KIT− (EFS 44±20% vs. 70±11%, p=0.021; DFS 43±20% vs. 69±12%, p=0.034; RR 57±21% vs. 28±12% vs. p=0.013; Table 1). However, this effect was not observed in the GO arm (Table 1) suggesting that GO abrogated the negative prognostic impact of KIT+ disease.
Table 1.
Clinical and biologic characteristics of CBF patients treated on COG AAML0531 stratified by KIT mutational status
| Characteristic | No KIT Mutation (N = 142) | KIT Mutation (N = 63) | P-value |
|---|---|---|---|
| Male,N (%) | 72 (51%) | 31 (49%) | 0.843 |
| Age,median (range in years) | 11.55 (0.33–22.53) | 11.96 (0.61–22.76) | 0.689 |
| CBF | 0.288 | ||
| t (8;21) | 72 (51%) | 27 (59%) | |
| Inv(16)/t(16;16) | 70 (49%) | 26 (41%) | |
| Race | |||
| American Indian or Alaskan Native | 1 (1%) | 0 (0%) | 1.000 |
| Asian | 2 (2%) | 4 (8%) | 0.666 |
| Native Hawaiian or Other Pacific Islander | 0 (0%) | 0 (0%) | 1.000 |
| Black or African American | 16 (13%) | 8 (15%) | 0.691 |
| White | 103 (84%) | 40 (77%) | 0.236 |
| Unknown | 20 | 11 | |
| Ethnicity | 0.596 | ||
| Hispanic | 33 (24%) | 17 (27%) | |
| Not Hispanic | 105 (76%) | 45 (73%) | |
| Unknown | 4 | 1 | |
| WBC×103uL – median (range) | 34.15 (1–432) | 39.6 (2–335.5) | 0.487 |
| Platelet count – median (range) | 42 (5–477) | 22 (2–252) | 0.002 |
| BM blast % - median (range) | 56 (6–100) | 73.5 (22–100) | <0.001 |
| Chloroma present | 38 (0–98) | 57 (0–95) | 0.001 |
| Central nervous system Positive | 22(15%) | 4 (6%) | 0.075 |
| CEBPA Positive | 0 (0%) | 1 (2%) | 0.249 |
| NPM1 positive | 0 (0%) | 0 (0%) | 1.000 |
| FLT3/ITD Positive | 4 (4%) | 2 (3%) | 1.000 |
| Treatment Allocation | 0.358 | ||
| Arm A (No GO) | 73 (51%) | 28 (44%) | |
| Arm B (GO) | 69 (49%) | 35 (56%) | |
| CR End Induction 1 | 117 (82%) | 52 (83%) | 0.98 |
Figure 3.
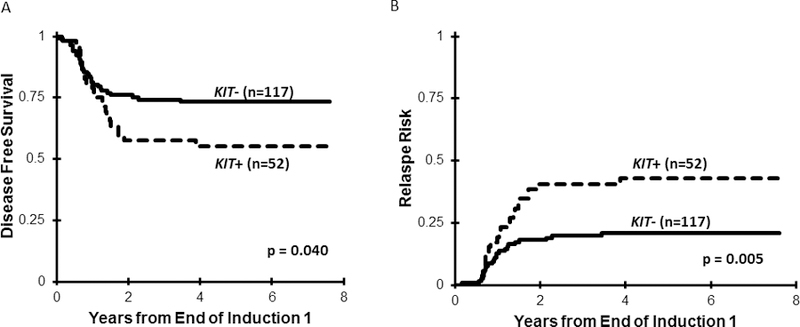
(A) Disease-free survival among the entire cohort of CBF AML patients according to KIT mutational status; (B) Relapse risk for the cohort of CBF AML patients according to KIT mutational status.
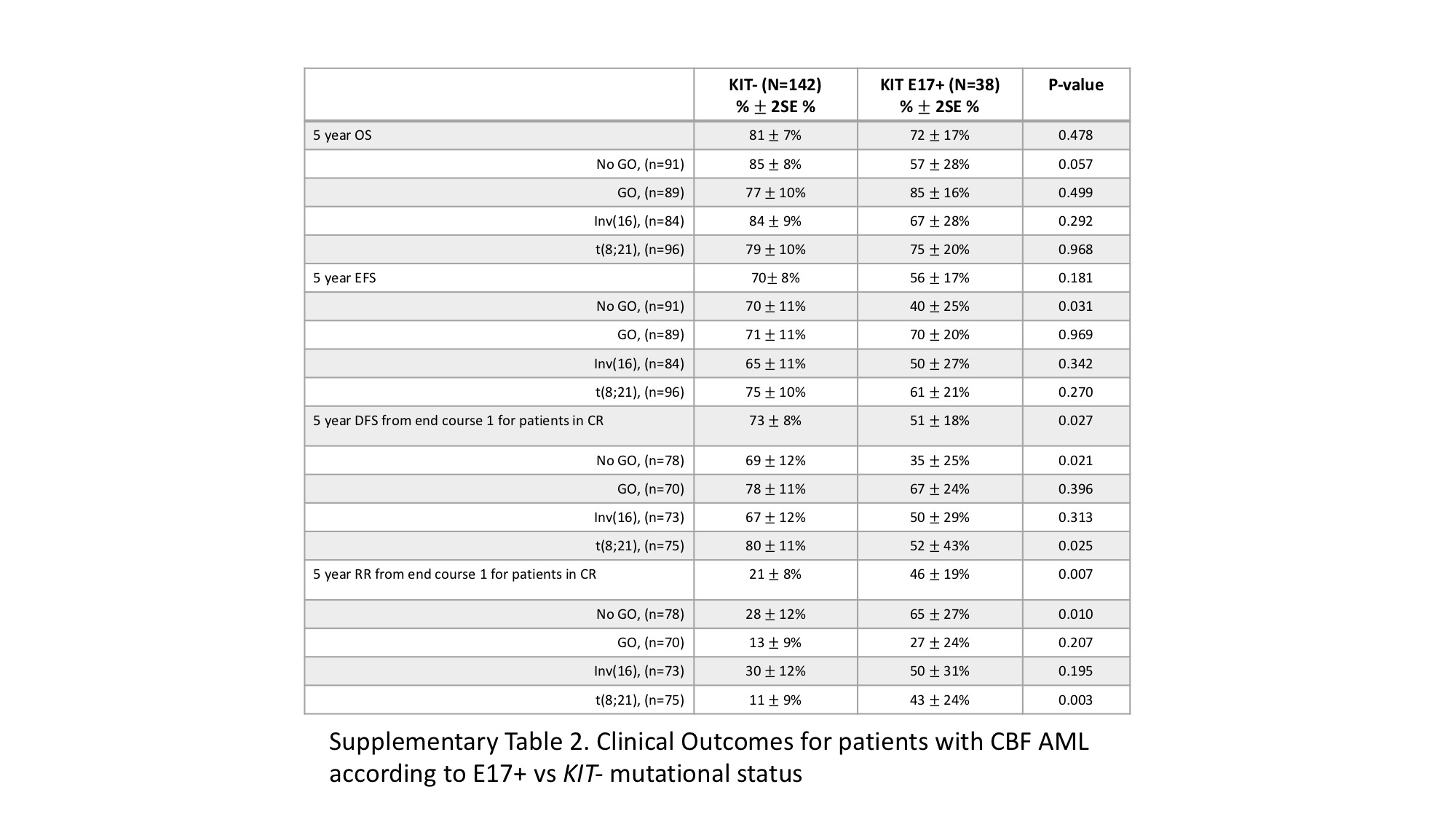
Given the distinct functional properties of E8 vs. E17 mutations, we analyzed the outcomes among CBF patients according to the location of the KIT mutation. For E8 mutations (E8+), we compared E8+ only patients to KIT−. However, given activating potential of E17 mutations, we also included patients with dual E17/E8 mutations as E17+. Patients (n=3) with KIT mutations that only occurred outside E8 or E17 were excluded from the outcome analysis given the small size of this cohort. While OS was comparable for E17+ vs. KIT− (72±17% vs. 81±7%; p=0.478; Supplementary Table S2), E17+ patients experienced inferior DFS of 51±18% vs. 73±8% for KIT− (p=0.027; Figure 4A), with a corresponding RR of 46±19% vs. 21±8%, respectively (p=0.007; Figure 4B). This effect was most pronounced in the E17+ t(8;21) subset, with this cohort experiencing DFS of 52±23% vs. 80±11% for KIT− (p=0.025) with a corresponding RR of 43±24% vs. 11±9%, respectively (p=0.003; Supplementary Table S2). Analysis by treatment arm demonstrated that GO recipients experienced similar outcomes, regardless of E17+ status, with similar OS among the cohorts (Supplementary Table S2). Corresponding DFS and RR were also comparable among GO recipients (DFS E17+ 67±24% vs. KIT− 78±11%, p=0.396, RR E17+ 27±24% vs. KIT− 13±9%, p=0.207; Figure 4C and 4D). In contrast, among patients treated on the No GO arm, E17+ patients experienced a DFS of 35±25% vs. 69±12% in KIT− patients (p=0.021), with a corresponding RR of 65±27% vs. 28±12% respectively (p=0.010; Supplementary Table S2). We subsequently analyzed the differential impact of GO within the E17+ cohort. Those receiving GO experienced a DFS of 67±24% vs. 35±25% for No GO recipients (p=0.161; Figure 4C, Supplementary Table S3), with a corresponding RR of 27±24% vs. 65±27, respectively (p=0.062; Figure 4D, Supplementary Table S3), demonstrating a trend towards improved disease outcomes with GO for E17+ patients. Among patients receiving standard chemotherapy, E17+ patients experienced an OS of 57±28% vs. 85±8% in KIT− patients (p=0.057; Supplementary Table S3). This suggests that following relapse, although some E17+ patients could be successfully salvaged, those who did not receive GO-augmented up-front therapy had a trend towards inferior survival.
Figure 4.

Association of E17 mutations with outcomes in CBF AML. (A) DFS in CBF patients for KIT E17+ compared to KIT− patients; (B) RR in CBF patients for KIT E17+ compared to KIT− patients; (C) DFS for CBF patients treated on the GO-containing arm according to KIT E17 status, with the E17+ patients stratified by treatment arm of standard chemotherapy (No GO) compared to GO containing regimen (GO); (D) RR for the CBF patients treated on the GO-containing arm according to KIT E17 status, with the E17+ patients stratified by treatment arm of standard chemotherapy (No GO) compared to GO containing regimen (GO).
E8+ patients experienced an OS of 91±12% compared to 81±7% (p=0.302) for KIT− patients (Supplementary Table S4). DFS in E8+ patients was 60±22% vs. 73±8% in KIT− patients (p=0.305; Figure 5A) with a corresponding RR of 40±23% vs. 21±8%, respectively (p=0.072; Figure 5B). Despite the less significant clinical effect of E8 mutations observed compared to E17, we also sought to analyze whether GO had differential impact in this cohort. OS among GO recipients was 100±0% in E8+ vs. 77±10% for KIT− patients (p=0.092). For GO recipients, this translated into a DFS of 64±29% for E8+ vs. 78±11% (p=0.456) in KIT− patients (Figure 5C) with a corresponding RR of 36±31% vs. 13±9%, respectively (p=0.082; Figure 5D). For CBF AML patients treated with standard chemotherapy (No GO arm), similar outcomes were seen for E8+ vs. KIT− patients across OS, DFS and RR (Supplementary Table S5). Analysis of the use of GO did not result in any signal of improvement in outcome for E8+ patients. In contrast, E17+ CBF AML treated with GO experienced similar DFS and RR to KIT− patients (Supplementary Table S2). Taken together, while GO did not result in the similar significant clinical impact in E8+ patients as compared to E17+, there was a trend towards reduced relapse and improved outcomes among all KIT+ GO recipients.
Figure 5.

Association of E8 mutations with outcomes in CBF AML. (A) DFS in CBF patients for KIT E8+ compared to KIT− patients; (B) RR in CBF patients for KIT E8+ compared to KIT− patients; (C) DFS for CBF patients treated on the GO-containing arm according to KIT E8 status, with the E8+ patients stratified by treatment arm of standard chemotherapy (No GO) compared to GO containing regimen (GO); (D) RR for the CBF patients treated on the GO-containing arm according to KIT E8 status, with the E8+ patients stratified by treatment arm of standard chemotherapy (No GO) compared to GO containing regimen (GO).
DISCUSSION
We retrospectively examined the prevalence and prognostic significance of KIT mutations in a cohort of 205 children with CBF AML enrolled on AAML0531 and demonstrated a mutation rate of 31% and, overall, an association of KIT+ status with decreased EFS and DFS and increased RR. The negative prognostic impact observed was primarily driven by E17 mutations, which were associated with higher rates of disease recurrence and inferior DFS. In the context of GO therapy, E17+ outcomes were comparable to that of KIT− CBF AML patients. GO therapy resulted in a trend towards reduced relapse and improved OS, EFS, and DFS in E17+ patients compared to those who received standard chemotherapy only. Importantly, this trend towards clinical improvement with GO therapy was not as pronounced in E8+ disease. The similar outcome achieved in the No GO arm for E8+ vs. KIT− patients underscores the less significant clinical impact of this mutation subtype compared to E17. Further, we showed that E17 mutations resulted in autonomous activation of KIT signaling and were sensitive to the cytotoxic effects of TKIs.
This study is significant as it contrasts with our previous large pediatric CBF AML analysis in which KIT mutations lacked prognostic significance. However, in that earlier analysis, the study cohort was heterogeneously treated on serial POG/CCG/COG trials with regimens of variable intensity. Our present study reflects a more uniformly treated CBF AML patient population. It is also significant in that it is the first study to suggest that GO provides therapeutic benefit, specifically for E17+ patients, but potentially all KIT + CBF AML.
To date, the prognostic impact of KIT mutations in CBF AML remains controversial. Some studies suggest that KIT mutations are associated with inferior remission duration and overall survival.(4,18,24,38–41) However, in other analyses KIT mutations lack prognostic significance.(3,19,21,42–44) Ayatollahi et al, in a meta-analysis of 22 published series, demonstrated that KIT mutations were associated with inferior prognosis, with this effect most significant in t(8;21) as compared to inv(16) disease.(26) A second recent meta-analysis by Chen et al found that outcomes were inferior in KIT + CBF AML overall, and specifically in KIT + t(8;21).(15) Four additional pediatric studies found that KIT mutations adversely affected OS, DFS and RR in pediatric t(8;21) AML.(17,22,27,45) Conversely, a more recent series of t(8;21) pediatric CBF AML patients enrolled on multiple cooperative group clinical trials failed to demonstrate prognostic significance of KIT + disease.(2) Importantly, KIT+ patients enrolled on that study received high doses of cytarabine and anthracyclines during induction and it was hypothesized that this might explain the failure to detect a difference in outcome.(2) Unlike the study by Klein et al and our previous study cohort, the uniform conventional chemotherapy backbone used in our present study cohort may have facilitated our ability to detect differences in outcome for KIT+ disease. Similar to the 4 other pediatric studies in which prognostic significance was noted,(17,22,27,45) our study population was treated with a standardized MRC based conventional chemotherapy backbone. Additionally, our large cohort size allowed us to detect differences in outcome according to the location of the KIT mutation
Previous studies demonstrate that GO improves outcome in CBF AML(1,28–30) and our series also supports this finding, demonstrating that among CBF patients the prognostic impact of KIT mutations was abrogated with addition of GO. Among GO recipients, KIT+ and KIT− patients experienced equivalent outcomes; this effect was most evident in E17+ patients. Importantly, the clinical benefit of GO in this patient population suggests that the inferior outcome observed with KIT+ CBF AML patients can be mitigated with appropriate intensification of therapy and without the use of HSCT. The mechanism of determinants of response to GO is an area of ongoing investigation and has included investigation into mediating aspects including expression of CD33 and cytogenetic or mutational profile.(29,32,46) Although CBF AML is associated with lower CD33 expression compared to other AML subgroups, this group of patients still experiences improved outcomes with GO.(32,46,47) In a prior analysis of CD33 expression for this trial, we demonstrated that 79% of patients with CBF AML had CD33 MFI below the median CD33 expression as defined for the entire cohort of de novo AML patients.(32) Strategies that involve intensification of therapy in CBF AML, such as high dose anthracyclines and higher dose cytarabine, have historically resulted in improved outcomes.(48–50) As CBF patients are known to be chemo-responsive, our results may also support that intensification of therapy for the E17+ subgroup of CBF patients with GO can result in further improvement in outcome despite lower expression of the CD33 cell surface target. As multiple aspects of the disease, in addition to CD33 expression, may mediate GO response (e.g. mutational profile, SNP genotype) (29,32,51,52), future analyses that comprehensively study factors that predict GO response may aid in our understanding as to the differential GO effect seen in our study cohort.
We also provide biologic rationale for the discrepancies in clinical outcomes observed, demonstrating the functional significance of activating E17 abnormalities. The recent study by Yui et al supports our findings demonstrating in adult CBF AML, that only E17 mutations conferred inferior prognosis, with the D816 mutation demonstrating the strongest impact on prognosis.(25) Kim et al also reported on the adverse prognostic impact of D816 mutations, which were most pronounced in t(8;21) AML.(39)
We recognize that the prognostic effect of KIT mutations in our study may have been influenced by therapy, as patients included in our analysis included GO and No GO recipients. Although due to the relatively large cohort size for a pediatric study, we were able to analyze patients according to treatment arm in order to delineate the impacts of GO and KIT mutations. However, a more definitive analysis on the impact of GO in CBF KIT+ patients will require additional patients as our study was not powered to detect that difference. While our large cohort size allowed us to detect differences in outcome according to the location of the KIT mutation, limitations in the number of patients with each distinct mutation did not allow us to analyze the prognostic impact of the distinct E17 mutations. Additionally, the number of patients in our study cohort with relapse specimen available limited further analysis regarding the clonality of KIT mutations in CBF AML.
As next generation sequencing identifies a greater number of mutations in a variety of oncogenic genes, it is important to understand the functional and clinical consequences of noted abnormalities and relation to clonality. Although our relapse cohort is too small to draw conclusions regarding the clonality of KIT mutations and presence in the dominant clone, they do suggest occurrence of dynamic patterns of clonal evolution with regard to KIT mutations that occur between diagnosis and relapse in CBF patients, and are in line with the findings presented by Faber et al.(53) Our data regarding the functional implications of KIT mutations suggest that E17 mutations result in aberrant phosphorylation of KIT and subsequent oncogenic growth, with the D816V demonstrating even more potent transformation capacity compared to the N822K alteration. This is line with previous studies that have demonstrated that D816 mutations result in structural alterations of the activation loop, resulting in ligand-independent auto-activation.(54) Our findings also support the prior study by Omori et al which demonstrated a more potent proliferative effect of D816V compared to N822K mutations.(55) Similar to Omori et al, following transduction we also observed higher levels of KIT detected in the exon 17 mutations compared to WT-KIT, as well as in the exon 8 and 11 mutations we tested.(55) Further, we report that transfection of the D816V resulted in higher levels of the immature form of KIT when compared to the other KIT mutations and WT-KIT. This aligns with data derived from analysis of GIST, which has shown that activating exon 11 KIT mutations result in higher levels of the immature form of KIT, which is constitutively phosphorylated and retained within the intracellular compartment.(56)
We show that E17 mutations, in addition to serving as activating events, may serve as a substrate for TKIs with activity against mutant KIT. The apoptotic effect of crenolanib and dasatinib observed in E17+ transduced cells is in line with other in vitro studies, and supports that E17 mutations can serve as therapeutic targets in AML.(57,58) In contrast, neither of the E8 mutations we investigated had a functional impact and both were insensitive to the cytotoxic effects of TKIs. We also demonstrate that mutations in exon 11, which compromise the JMD and is critical to KIT activation, result in aberrant phosphorylation. These mutations resulted in a weaker phosphorylation signal as compared to the E17 mutations, and were not found to be transforming events, which is likely a function of less potent phosphorylation. However, E11 mutations are amenable to TKI therapy, as mutations at these sites have been the targets of effective TKI therapy in many solid tumors.(8,9) Taken together, these in vitro findings suggest that intensification of therapy with TKIs should be studied further in patients with E17 as well as rare E11, but not E8, mutations. We acknowledge that response to TKIs in KIT mutant AML may be mediated by multiple mechanisms. While levels of cell surface KIT expression have been shown to be a mechanism that can mediate KIT-inhibitor mediated cytotoxicity, higher levels of KIT expression do not necessarily result in enhanced toxicity (59–61). We demonstrated that cell surface KIT expression was lowest in the E17+ patients, and significantly lower than KIT− patients. We observed similar findings in the Ba/F3 transduced cell lines with E17+ mutations demonstrating lower CD117 expression compared to E8 mutations and KIT-WT. These findings suggest that KIT mutations are not always associated with higher cell surface KIT expression, and that aberrantly activated KIT signaling is likely to be the most important target of anti-KIT TKIs. Notably, aberrant intracellular retention of constitutively phosphorylated immature KIT, as was seen in the D816V mutation, is a common mechanism for KIT activating mutations in GIST and has been shown to be amenable to imatinib treatment, as the drug blocked mutant KIT phosphorylation and restored protein maturation.(56) Ongoing clinical trials are currently investigating the role of TKIs in CBF AML, regardless of KIT mutational status, and will provide important information regarding the efficacy of TKIs in this population and how KIT mutational status impact TKI response. Addition of TKIs may be an important therapeutic strategy as the limits of intensification with conventional cytotoxic agents has been reached in pediatric AML therapy and TKIs have been shown to be safe when combined with intensive chemotherapy.(62–64)
Our study is significant in that it reflects, within the context of homogenously treated pediatric CBF AML population, that KIT mutations are associated with inferior clinical outcome and that this effect is largely due to the impact of E17+ disease, a finding supported by our in vitro functional studies. Importantly, not all KIT mutations are equally amenable to TKI targeting and this study illustrates the importance of defining the functional significance of a particular RTK mutation and the impact of targeted therapeutics on such abnormalities. In the context of KIT+ CBF AML, future clinical trials that include GO, as well as TKI therapy for activating KIT mutations, namely E17+ disease, are prudent, and can further the goal of improving outcome for this higher risk group of pediatric CBF AML patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This research was supported by the following funding: St. Baldrick’s Career Development Award (JP), Hyundai Hope On Wheels (JP, KT), St. Baldrick’s Scholar Award (KT), COG Chairs grant U10 CA180886–01 and U10CA98543, NCTN Statistics and Data Center Grant D, NCI R01 CA 114563 (SM), Andrew McDonough B+ Foundation, and St. Baldrick’s Foundation.
Footnotes
Scientific section designation: Clinical Trials: Targeted Therapy
Conflict of interest disclosure: The authors have no conflicts of interest to disclose.
References:
- 1.Gamis AS, Alonzo TA, Meshinchi S, Sung L, Gerbing RB, Raimondi SC, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol 2014;32(27):3021–32 doi 10.1200/JCO.2014.55.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klein K, Kaspers G, Harrison CJ, Beverloo HB, Reedijk A, Bongers M, et al. Clinical Impact of Additional Cytogenetic Aberrations, cKIT and RAS Mutations, and Treatment Elements in Pediatric t(8;21)-AML: Results From an International Retrospective Study by the International Berlin-Frankfurt-Munster Study Group. J Clin Oncol 2015;33(36):4247–58 doi 10.1200/JCO.2015.61.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen C, Hills RK, Lamb K, Evans C, Tinsley S, Sellar R, et al. The importance of relative mutant level for evaluating impact on outcome of KIT, FLT3 and CBL mutations in core-binding factor acute myeloid leukemia. Leukemia 2013;27(9):1891–901 doi 10.1038/leu.2013.186. [DOI] [PubMed] [Google Scholar]
- 4.Paschka P, Du J, Schlenk RF, Gaidzik VI, Bullinger L, Corbacioglu A, et al. Secondary genetic lesions in acute myeloid leukemia with inv(16) or t(16;16): a study of the German-Austrian AML Study Group (AMLSG). Blood 2013;121(1):170–7 doi 10.1182/blood-2012-05-431486. [DOI] [PubMed] [Google Scholar]
- 5.Paschka P, Marcucci G, Ruppert AS, Mrozek K, Chen H, Kittles RA, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol 2006;24(24):3904–11 doi 10.1200/JCO.2006.06.9500. [DOI] [PubMed] [Google Scholar]
- 6.d’Auriol L, Mattei MG, Andre C, Galibert F. Localization of the human c-kit protooncogene on the q11-q12 region of chromosome 4. Hum Genet 1988;78(4):374–6. [DOI] [PubMed] [Google Scholar]
- 7.Renneville A, Roumier C, Biggio V, Nibourel O, Boissel N, Fenaux P, et al. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia 2008;22(5):915–31 doi 10.1038/leu.2008.19. [DOI] [PubMed] [Google Scholar]
- 8.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279(5350):577–80. [DOI] [PubMed] [Google Scholar]
- 9.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24(26):4340–6 doi 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 10.Chatterjee A, Ghosh J, Kapur R. Mastocytosis: a mutated KIT receptor induced myeloproliferative disorder. Oncotarget 2015;6(21):18250–64 doi 10.18632/oncotarget.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SY, Kang JJ, Lee HH, Kang JJ, Kim B, Kim CG, et al. Mechanism of activation of human c-KIT kinase by internal tandem duplications of the juxtamembrane domain and point mutations at aspartic acid 816. Biochem Biophys Res Commun 2011;410(2):224–8 doi 10.1016/j.bbrc.2011.05.111. [DOI] [PubMed] [Google Scholar]
- 12.Chan PM, Ilangumaran S, La Rose J, Chakrabartty A, Rottapel R. Autoinhibition of the kit receptor tyrosine kinase by the cytosolic juxtamembrane region. Mol Cell Biol 2003;23(9):3067–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21(23):4342–9 doi 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 14.Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29(21):2904–9 doi 10.1200/JCO.2010.33.9275. [DOI] [PubMed] [Google Scholar]
- 15.Chen W, Xie H, Wang H, Chen L, Sun Y, Chen Z, et al. Prognostic Significance of KIT Mutations in Core-Binding Factor Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. PLoS One 2016;11(1):e0146614 doi 10.1371/journal.pone.0146614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia 2006;20(6):965–70 doi 10.1038/sj.leu.2404188. [DOI] [PubMed] [Google Scholar]
- 17.Shimada A, Taki T, Tabuchi K, Tawa A, Horibe K, Tsuchida M, et al. KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): a study of the Japanese Childhood AML Cooperative Study Group. Blood 2006;107(5):1806–9 doi 10.1182/blood-2005-08-3408. [DOI] [PubMed] [Google Scholar]
- 18.Cairoli R, Beghini A, Grillo G, Nadali G, Elice F, Ripamonti CB, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 2006;107(9):3463–8 doi 10.1182/blood-2005-09-3640. [DOI] [PubMed] [Google Scholar]
- 19.Pollard JA, Alonzo TA, Gerbing RB, Ho PA, Zeng R, Ravindranath Y, et al. Prevalence and prognostic significance of KIT mutations in pediatric patients with core binding factor AML enrolled on serial pediatric cooperative trials for de novo AML. Blood 2010;115(12):2372–9 doi 10.1182/blood-2009-09-241075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goemans BF, Zwaan CM, Miller M, Zimmermann M, Harlow A, Meshinchi S, et al. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia 2005;19(9):1536–42 doi 10.1038/sj.leu.2403870. [DOI] [PubMed] [Google Scholar]
- 21.Shih LY, Liang DC, Huang CF, Chang YT, Lai CL, Lin TH, et al. Cooperating mutations of receptor tyrosine kinases and Ras genes in childhood core-binding factor acute myeloid leukemia and a comparative analysis on paired diagnosis and relapse samples. Leukemia 2008;22(2):303–7 doi 10.1038/sj.leu.2404995. [DOI] [PubMed] [Google Scholar]
- 22.Manara E, Bisio V, Masetti R, Beqiri V, Rondelli R, Menna G, et al. Core-binding factor acute myeloid leukemia in pediatric patients enrolled in the AIEOP AML 2002/01 trial: screening and prognostic impact of c-KIT mutations. Leukemia 2014;28(5):1132–4 doi 10.1038/leu.2013.339. [DOI] [PubMed] [Google Scholar]
- 23.Qin YZ, Zhu HH, Jiang Q, Jiang H, Zhang LP, Xu LP, et al. Prevalence and prognostic significance of c-KIT mutations in core binding factor acute myeloid leukemia: a comprehensive large-scale study from a single Chinese center. Leuk Res 2014;38(12):1435–40 doi 10.1016/j.leukres.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 24.Wang D, Qiao C, Xiao M, Geng Z, Shang Z, He J, et al. Integrative analysis of prognostic factors in Chinese core binding factor leukemia. Biochem Biophys Res Commun 2012;428(3):411–5 doi 10.1016/j.bbrc.2012.10.069. [DOI] [PubMed] [Google Scholar]
- 25.Yui S, Kurosawa S, Yamaguchi H, Kanamori H, Ueki T, Uoshima N, et al. D816 mutation of the KIT gene in core binding factor acute myeloid leukemia is associated with poorer prognosis than other KIT gene mutations. Ann Hematol 2017;96(10):1641–52 doi 10.1007/s00277-017-3074-y. [DOI] [PubMed] [Google Scholar]
- 26.Ayatollahi H, Shajiei A, Sadeghian MH, Sheikhi M, Yazdandoust E, Ghazanfarpour M, et al. Prognostic Importance of C-KIT Mutations in Core Binding Factor Acute Myeloid Leukemia: A Systematic Review. Hematol Oncol Stem Cell Ther 2017;10(1):1–7 doi 10.1016/j.hemonc.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Dou H, Wang X, Huang Y, Lu L, Bin J, et al. KIT mutations correlate with adverse survival in children with core-binding factor acute myeloid leukemia. Leuk Lymphoma 2018;59(4):829–36 doi 10.1080/10428194.2017.1361025. [DOI] [PubMed] [Google Scholar]
- 28.Burnett AK, Hills RK, Milligan D, Kjeldsen L, Kell J, Russell NH, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol 2011;29(4):369–77 doi 10.1200/JCO.2010.31.4310. [DOI] [PubMed] [Google Scholar]
- 29.Castaigne S, Pautas C, Terre C, Raffoux E, Bordessoule D, Bastie JN, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet 2012;379(9825):1508–16 doi 10.1016/S0140-6736(12)60485-1. [DOI] [PubMed] [Google Scholar]
- 30.Loke J, Khan JN, Wilson JS, Craddock C, Wheatley K. Mylotarg has potent anti-leukaemic effect: a systematic review and meta-analysis of anti-CD33 antibody treatment in acute myeloid leukaemia. Ann Hematol 2015;94(3):361–73 doi 10.1007/s00277-014-2218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolouri H, Farrar JE, Triche T Jr., Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 2017. doi 10.1038/nm.4439. [DOI] [PMC free article] [PubMed]
- 32.Pollard JA, Loken M, Gerbing RB, Raimondi SC, Hirsch BA, Aplenc R, et al. CD33 Expression and Its Association With Gemtuzumab Ozogamicin Response: Results From the Randomized Phase III Children’s Oncology Group Trial AAML0531. J Clin Oncol 2016;34(7):747–55 doi 10.1200/JCO.2015.62.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walter RB, Alonzo TA, Gerbing RB, Ho PA, Smith FO, Raimondi SC, et al. High expression of the very late antigen-4 integrin independently predicts reduced risk of relapse and improved outcome in pediatric acute myeloid leukemia: a report from the children’s oncology group. J Clin Oncol 2010;28(17):2831–8 doi 10.1200/JCO.2009.27.5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaplan ELMP. Nonparametric estimation from incomplete observations. Journal of the American Statistical Association 1958;53(282):457–81. [Google Scholar]
- 35.RL KJaP. The Statistical Analysis of Failure Time Data Hoboken, JJ: John Wiley & Sons, Inc; 2002. [Google Scholar]
- 36.Peto RPaJ. Asymptomatically Efficient Rank Invariant Test Procedures Journal of the Royal Statistical Society: Series A 1972;135(2):185–207. [Google Scholar]
- 37.RJ G A Calls of K-sample tests for comparing the cumulative incidence of a competing risk. The Annals of Statistics 1988;16:1141–54. [Google Scholar]
- 38.Krauth MT, Eder C, Alpermann T, Bacher U, Nadarajah N, Kern W, et al. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: frequency and impact on clinical outcome. Leukemia 2014;28(7):1449–58 doi 10.1038/leu.2014.4. [DOI] [PubMed] [Google Scholar]
- 39.Kim HJ, Ahn HK, Jung CW, Moon JH, Park CH, Lee KO, et al. KIT D816 mutation associates with adverse outcomes in core binding factor acute myeloid leukemia, especially in the subgroup with RUNX1/RUNX1T1 rearrangement. Ann Hematol 2013;92(2):163–71 doi 10.1007/s00277-012-1580-5. [DOI] [PubMed] [Google Scholar]
- 40.Park SH, Lee HJ, Kim IS, Kang JE, Lee EY, Kim HJ, et al. Incidences and Prognostic Impact of c-KIT, WT1, CEBPA, and CBL Mutations, and Mutations Associated With Epigenetic Modification in Core Binding Factor Acute Myeloid Leukemia: A Multicenter Study in a Korean Population. Ann Lab Med 2015;35(3):288–97 doi 10.3343/alm.2015.35.3.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jung HA, Maeng CH, Park S, Kim SJ, Kim K, Jang JH, et al. Prognostic factor analysis in core-binding factor-positive acute myeloid leukemia. Anticancer Res 2014;34(2):1037–45. [PubMed] [Google Scholar]
- 42.Riera L, Marmont F, Toppino D, Frairia C, Sismondi F, Audisio E, et al. Core binding factor acute myeloid leukaemia and c-KIT mutations. Oncol Rep 2013;29(5):1867–72 doi 10.3892/or.2013.2328. [DOI] [PubMed] [Google Scholar]
- 43.Cairoli R, Beghini A, Turrini M, Bertani G, Nadali G, Rodeghiero F, et al. Old and new prognostic factors in acute myeloid leukemia with deranged core-binding factor beta. Am J Hematol 2013;88(7):594–600 doi 10.1002/ajh.23461. [DOI] [PubMed] [Google Scholar]
- 44.Jourdan E, Boissel N, Chevret S, Delabesse E, Renneville A, Cornillet P, et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood 2013;121(12):2213–23 doi 10.1182/blood-2012-10-462879. [DOI] [PubMed] [Google Scholar]
- 45.Tokumasu M, Murata C, Shimada A, Ohki K, Hayashi Y, Saito AM, et al. Adverse prognostic impact of KIT mutations in childhood CBF-AML: the results of the Japanese Pediatric Leukemia/Lymphoma Study Group AML-05 trial. Leukemia 2015;29(12):2438–41 doi 10.1038/leu.2015.121. [DOI] [PubMed] [Google Scholar]
- 46.Khan N, Hills RK, Virgo P, Couzens S, Clark N, Gilkes A, et al. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leukemia 2017;31(5):1059–68 doi 10.1038/leu.2016.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pollard JA, Alonzo TA, Loken M, Gerbing RB, Ho PA, Bernstein ID, et al. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood 2012;119(16):3705–11 doi 10.1182/blood-2011-12-398370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med 2009;361(13):1249–59 doi 10.1056/NEJMoa0904544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prabahran A, Tacey M, Fleming S, Wei A, Tate C, Marlton P, et al. Prognostic markers in core-binding factor AML and improved survival with multiple consolidation cycles of intermediate-/high-dose cytarabine. Eur J Haematol 2018. doi 10.1111/ejh.13089. [DOI] [PubMed]
- 50.Byrd JC, Ruppert AS, Mrozek K, Carroll AJ, Edwards CG, Arthur DC, et al. Repetitive cycles of high-dose cytarabine benefit patients with acute myeloid leukemia and inv(16)(p13q22) or t(16;16)(p13;q22): results from CALGB 8461. J Clin Oncol 2004;22(6):1087–94 doi 10.1200/JCO.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 51.Lamba JK, Chauhan L, Shin M, Loken MR, Pollard JA, Wang YC, et al. CD33 Splicing Polymorphism Determines Gemtuzumab Ozogamicin Response in De Novo Acute Myeloid Leukemia: Report From Randomized Phase III Children’s Oncology Group Trial AAML0531. J Clin Oncol 2017;35(23):2674–82 doi 10.1200/JCO.2016.71.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tarlock K, Alonzo TA, Gerbing RB, Raimondi SC, Hirsch BA, Sung L, et al. Gemtuzumab Ozogamicin Reduces Relapse Risk in FLT3/ITD Acute Myeloid Leukemia: A Report from the Children’s Oncology Group. Clin Cancer Res 2016;22(8):1951–7 doi 10.1158/1078-0432.CCR-15-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet 2016;48(12):1551–6 doi 10.1038/ng.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem 2004;279(30):31655–63 doi 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- 55.Omori I, Yamaguchi H, Miyake K, Miyake N, Kitano T, Inokuchi K. D816V mutation in the KIT gene activation loop has greater cell-proliferative and anti-apoptotic ability than N822K mutation in core-binding factor acute myeloid leukemia. Exp Hematol 2017;52:56–64 10.1016/j.exphem.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 56.Tabone-Eglinger S, Subra F, El Sayadi H, Alberti L, Tabone E, Michot JP, et al. KIT mutations induce intracellular retention and activation of an immature form of the KIT protein in gastrointestinal stromal tumors. Clin Cancer Res 2008;14(8):2285–94 doi 10.1158/1078-0432.CCR-07-4102. [DOI] [PubMed] [Google Scholar]
- 57.Schittenhelm MM, Shiraga S, Schroeder A, Corbin AS, Griffith D, Lee FY, et al. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res 2006;66(1):473–81 doi 10.1158/0008-5472.CAN-05-2050. [DOI] [PubMed] [Google Scholar]
- 58.Kampa-Schittenhelm KM, Frey J, Haeusser LA, Illing B, Pavlovsky AA, Blumenstock G, et al. Crenolanib is a type I tyrosine kinase inhibitor that inhibits mutant KIT D816 isoforms prevalent in systemic mastocytosis and core binding factor leukemia. Oncotarget 2017;8(47):82897–909 doi 10.18632/oncotarget.19970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heo SK, Noh EK, Kim JY, Jeong YK, Jo JC, Choi Y, et al. Targeting c-KIT (CD117) by dasatinib and radotinib promotes acute myeloid leukemia cell death. Sci Rep 2017;7(1):15278 doi 10.1038/s41598-017-15492-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heo SK, Noh EK, Kim JY, Jo JC, Choi Y, Koh S, et al. Radotinib induces high cytotoxicity in c-KIT positive acute myeloid leukemia cells. Eur J Pharmacol 2017;804:52–6 doi 10.1016/j.ejphar.2017.03.040. [DOI] [PubMed] [Google Scholar]
- 61.Herrmann MD, Lennerz JK, Bullinger L, Bartholomae S, Holzmann K, Westhoff MA, et al. Transitory dasatinib-resistant states in KIT(mut) t(8;21) acute myeloid leukemia cells correlate with altered KIT expression. Exp Hematol 2014;42(2):90–100 doi 10.1016/j.exphem.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 62.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017;377(5):454–64 doi 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rollig C, Serve H, Huttmann A, Noppeney R, Muller-Tidow C, Krug U, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol 2015;16(16):1691–9 doi 10.1016/S1470-2045(15)00362-9. [DOI] [PubMed] [Google Scholar]
- 64.Paschka P, Schlenk RF, Weber D, Benner A, Bullinger L, Heuser M, et al. Adding dasatinib to intensive treatment in core-binding factor acute myeloid leukemia-results of the AMLSG 11–08 trial. Leukemia 2018. doi 10.1038/s41375-018-0129-6. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.