

We demonstrate conversion of an excited triplet state to an excited singlet state through resonance energy transfer.
Abstract
The ability to convert between molecular spin states is of utmost importance in materials chemistry. Förster-type energy transfer is based on dipole-dipole interactions and can therefore theoretically be used to convert between molecular spin states. Here, a molecular dyad that is capable of transferring energy from an excited triplet state to an excited singlet state is presented. The rate of conversion between these states was shown to be 36 times faster than the rate of emission from the isolated triplet state. This dyad provides the first solid proof that Förster-type triplet-to-singlet energy transfer is possible, revealing a method to increase the rate of light extraction from excited triplet states.
INTRODUCTION
The insights gained from the last century of research within the fields of energy and electron transfer using organic molecules have been successfully conveyed into organic electronic devices (1, 2). The ability to control the yield and rate of processes occurring in electronically excited states has resulted in efficient and stable devices, which are now commercially available. However, an important parameter that is still not completely understood is the conversion between states with different spin quantum numbers. For instance, the spin angular momentum quantum number is of practical importance in organic light-emitting diodes (OLEDs) because emission from singlet states is efficient, whereas emission from triplet states is quantum-mechanically forbidden. This situation seriously affects OLED performance because statistically, 75% of all formed excited states are triplets (3–5).
Although emission of light from triplet states (phosphorescence) is quantum-mechanically forbidden, it does occur at a very low rate. The rate of phosphorescence has been enhanced by introducing rare-earth metals, which increase the spin-orbit coupling, making the transition quantum-mechanically less forbidden. Organometallic complexes containing metals such as iridium successfully increase the rate of emission from the triplet states (6). However, the excited-state lifetime is still long, which causes efficiency roll-off in OLEDs (7). Recently, the concept of thermally populating the first excited singlet state (from which emission can occur efficiently) from the lower-lying triplet state was proposed (8, 9), offering another approach to address the low rate of emission from triplet states. This approach, referred to as thermally activated delayed fluorescence (TADF), enables triplet-to-singlet conversion without the need for rare-earth metals (8–11). However, although the heavy-atom effect and TADF markedly enhance the rate of emission from excited triplet states, neither of these methods can compete with the rate performance of emission from singlet states (fluorescence). Thus, development of conceptually new methods to convert excited triplet states to excited singlet states is of pivotal importance from both a fundamental viewpoint and for production of high-efficiency organic electronic devices.
Förster-type resonance energy transfer is an essential tool to manipulate electronically excited states of organic molecules (12). Because the resonance energy mechanism is based on the electrostatic interaction between two oscillating dipoles, the total spin angular momentum quantum number of the involved states should be of no consequence. Experiments in the 1960s (13), in which freely diffusing donors (in their triplet excited state) and acceptors (in their singlet excited state) were used, were successfully explained by the Förster model. However, this picture of a spin angular momentum–independent energy transfer mechanism was recently challenged using materials with linked donor and acceptor moieties (14). Furthermore, the notion of an angular momentum conservation requirement is also present in textbooks on molecular photochemistry (15). The heritage of this idea is that the possibility of Förster-type energy transfer to occur has started to be discussed in terms of angular momentum conservation (16–22). Note that a requirement for energy transfer to occur is a finite oscillator strength from the triplet excited state of the donor, which can be achieved by increasing the spin-orbit coupling in the donor (heavy metals increase spin-orbit coupling, breaking down selecting rules for singlet-to-triplet transitions).
Here, we show that it is possible to transfer excited-state energy through the Förster mechanism without conserving the spin angular momentum. A donor-bridge-acceptor dyad (DBA) was synthesized in which the donor and acceptor moieties are rigidly fixed within the molecular framework. Intramolecular energy transfer from the donor to the acceptor units in DBA was proved using time-resolved emission spectroscopy (TRES), and the rate constant of the energy transfer matches well with that calculated using the Förster model. The rate constant of triplet-to-singlet energy transfer was 36 times faster than that of phosphorescence. Triplet-to-singlet energy transfer can thus be used to substantially increase the rate of emission from excited triplet states, which is of practical importance in organic electronics.
RESULTS AND DISCUSSION
Molecular design
Molecules in their excited states can interact with each other in a variety of different ways. Förster-type energy transfer is unique because the interacting molecules can be located fairly far apart (up to 50 to 100 Å). Other types of interactions such as Dexter-type energy transfer or electron transfer need more or less direct orbital overlap to occur (23). To examine energy transfer through a dipole-dipole mechanism, we synthesized a DBA molecule (Fig. 1A; see section S1 for synthesis and characterization details). Design criteria used for DBA were the following: (i) The donor emission should overlap with the acceptor absorption to allow energy transfer through a dipole-dipole mechanism to occur. (ii) The bridging unit should contain no π-electrons or lone electron pairs to minimize electron tunneling between the donor and acceptor. (iii) The bridging unit should be rigid to avoid direct contact between the donor and acceptor. Furthermore, considering possible applications, energy transfer should occur with minimal spectral energy loss. The acceptor should therefore be a highly fluorescent molecule with a reasonably small Stokes shift to minimize spectral energy loss in the triplet-to-singlet conversion. Because the most important spectral feature of the dyad components is a considerable overlap between the donor emission and acceptor absorption, both functional components were selected with great care. An IrIII complex (D) was chosen as the donor (24) and perylene (A), which has a fluorescence quantum yield close to unity and small Stokes shift (25), was selected as the acceptor. The bridging unit was of pivotal importance to spatially separate the donor and acceptor moieties. We chose bicyclo[2.2.2]octane as the bridging unit because its rigid structure permits free rotation only along the long axis of the molecule, meaning it can behave as a stable linker for the functional components of the dyad.
Fig. 1. Structure and energy levels of the dyad.
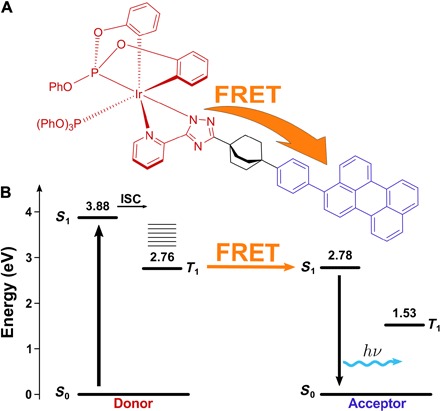
(A) The DBA molecule used in this study. (B) Jabłoński diagram showing triplet-to-singlet energy transfer via dipole-dipole interactions, i.e., the Förster-type resonance energy transfer (FRET) mechanism.
Assessing the preserved integrity of the components in the dyad
When evaluating the efficiency of energy transfer, the emission of the dyad was compared to the emission of the individual components. For this comparison to be accurate, it is important that the integrity of the donor and acceptor chromophores in the dyad is preserved. If the properties of A and D are not conserved in DBA, then the system should not be considered as one molecule containing two chromophores but rather as one chromophore, and the Jabłoński diagram in Fig. 1B would not be valid. Figure 2 shows the absorption spectra of A, D, and DBA. Donor D exhibits a relatively weak transition around 320 nm, and A displays the characteristic vibronic pattern of perylene-containing molecules. Figure 2 also depicts the arithmetic sum of the D and A absorption spectra. A close-to-perfect overlap between the sum of D and A absorption spectra and the absorption spectrum of DBA was observed, indicating that the integrity of the components in the dyad was preserved. Thus, the ground- and excited-state energy levels of D and A are retained in the dyad, making it an excellent model system to study possible triplet-to-singlet energy transfer.
Fig. 2. Ultraviolet-visible absorption of the studied molecules.
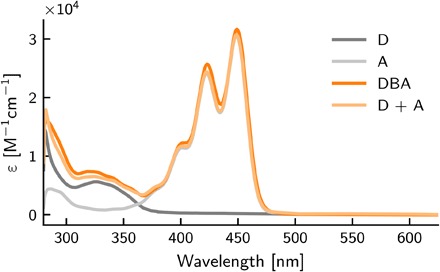
Absorption spectra of donor (D), acceptor (A), and DBA dissolved in toluene. Also shown is the mathematical sum of the donor and acceptor spectra (D + A).
Triplet-to-singlet energy transfer
To assess whether the dyad can transfer energy, we determined emission quantum yields (Φ), as well as excited-state lifetimes (τ) (Fig. 3, A to C). Upon exciting D, the initially formed excited singlet state is rapidly converted to a triplet state (Fig. 1B) in high yield (24). The observed lifetime of 41.5 μs and the emission quantum yield of 62% are therefore solely related to the process of phosphorescence. The rate of phosphorescence calculated from τ and Φ was 1.49 × 104 s−1. A exhibited a much shorter lifetime (3.1 ns), as expected for the quantum-mechanically allowed process of fluorescence, and an emission quantum yield of 85%. The excited-state lifetime of both D and A could be explained with a single exponential fit. However, this was not the case for the dyad, which required two exponentials [τDBA_1 (lifetime of the short-lived component) = 3.7 ns; τDBA_2 (lifetime of the long-lived component) = 290 ns] to explain the observed emission decay. τDBA_1 is similar to the lifetime of A and can therefore be explained by direct excitation of the acceptor moiety in DBA (see section S2). τDBA_2 is two orders of magnitude larger than τDBA_1 but still much shorter than the lifetime of D. The shortened lifetime in DBA when exciting the donor moiety does not prove that an energy transfer event has occurred. Instead, the decrease of the lifetime could be caused by other decay pathways of the excited triplet state of the donor moiety opening up in the dyad. To prove that the emission at long time scales occurs because of triplet-to-singlet energy transfer, TRES was performed. In a TRES experiment, emission decay is measured for each emission wavelength, covering the whole emission profile (Fig. 3D). This offers the possibility to compare the spectral envelope of the decay at short and long times after excitation. Figure 3E shows normalized emission spectra of D and A, as well as snapshots of dyad emission 0 to 7 and 15 to 1500 ns after excitation. The two snapshots represent the emission profiles of the short- and long-lived components of the emission decay of DBA, and both resemble the emission profile of A. Thus, at both short and long time scales, emission occurs from the excited singlet state of acceptor moiety in DBA, proving that triplet-to-singlet energy transfer has occurred. The long-lived component in the emission decay represents the rate of energy transfer, which, together with the emission quantum yield of DBA, was used to calculate the rate constant of energy transfer to 5.33 × 105 s−1 (see section S2 for details). The rate of energy transfer in DBA is thus 36 times faster than the rate of phosphorescence. Furthermore, the rate of energy transfer depends on the intrinsic rate of emission of D and the energy overlap between D and A. Triplet-to-singlet energy transfer thus constitutes a general method to enhance the rate of emission of any phosphorescent material.
Fig. 3. Emission properties of the studied molecules.
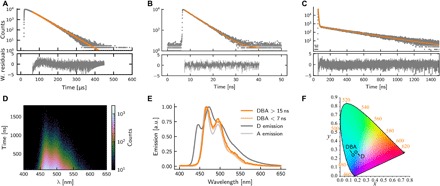
Time-resolved emission of the (A) donor, (B) acceptor, and (C) DBA dissolved in toluene, excited at 320, 405, and 320 nm, respectively. (D) Time-resolved emission spectrum of DBA. (E) Spectra at defined time intervals after DBA excitation together with donor and acceptor emission spectra. (F) Emission from D and DBA plotted in a CIE diagram. a.u., arbitrary units.
We have so far shown that an energy transfer event from the excited triplet state of the donor moiety to the excited singlet state of the acceptor moiety can occur in DBA. To prove that the most plausible mechanism for this energy transfer is a Förster-type dipole-dipole mechanism, we compared the experimentally obtained rate constant (kT = 5.33 × 105 s−1) with a theoretically derived value. The main parameters for Förster-type energy transfer are the overlap between donor emission (Fig. 3E) and acceptor absorption (Fig. 2) and the distance and relative angle of the responsible transition dipole moments. The bridging unit is stiff but able to rotate in solution. Assuming free rotation of the carbon─carbon single bond in the bridging unit, the average rate constant of energy transfer in DBA was calculated to be 3.35 × 105 s−1 (see section S3 for details). This value agrees well with that measured experimentally, confirming the occurrence of fast triplet-to-singlet energy transfer.
The energy transfer process in DBA is a one-way process driven by the enthalpy decrease of the system. It is instructive to quantify how much energy is lost in the unidirectional energy transfer step, i.e., how much energy is expended in the triplet-to-singlet conversion. Figure 3F presents a CIE diagram of the emission colors of D and DBA (calculated from the data in Fig. 3E). The hue of the emission from DBA is in fact bluer than that of D despite the energy required for the triplet-to-singlet transfer to occur. The reason for the bluer emission of DBA than that of D is the narrower emission bandwidth of A compared to that of D. Förster-type resonance energy transfer can thus be used to increase the rate of emission without an apparent loss of photon energy. This is because strong fluorophores generally have a small Stokes shift and narrower emission profile compared to those of phosphorescent metal-organic compounds.
CONCLUSION
To conclude, the first example of intramolecular triplet-to-singlet energy transfer was presented here. The energy transfer was 36 times faster than the rate of donor phosphorescence. The results were fully explained using the theory of Förster-type resonance energy transfer. Furthermore, the transfer could be achieved without an apparent loss of photon energy. These results demonstrate that triplet-to-singlet transfer is possible and can be used to increase the rate of emission from phosphorescent compounds. Our results are thus of direct practical importance in the field of organic electronics, where the slow emission of light from triplet states is problematic.
MATERIALS AND METHODS
General
All starting materials were purchased from Sigma-Aldrich Chemical Co. and used without further purification unless otherwise noticed. All moisture- and oxygen-sensitive reactions were carried out using Schlenk techniques in oven-dried glassware. Solvents used for moisture- and oxygen-sensitive reactions were dried using an MBraun MB SPS-800 solvent purification system and, if necessary, degassed by freeze-pump-thaw cycles and stored over 4-Å molecular sieves under argon atmosphere. Flash chromatography was performed by a Teledyne CombiFlash EZ prep using liquid chromatography–mass spectrometry (LC-MS)–grade solvents and normal-phase silica with a mesh size of 230 to 400, a particle size of 40 to 63 μm, and a pore size of 60 Å.
1H NMR (nuclear magnetic resonance) spectra were recorded on a Varian spectrometer at 400 MHz and on a Bruker spectrometer at 800 MHz, 13C NMR spectra were recorded at 101 and 201 MHz, and 31P NMR spectra were recorded at 162 MHz using CDCl3 and dimethyl sulfoxide (DMSO)–d6 as solvents. J-coupling values are given in hertz, and chemical shifts are given in parts per million using tetramethysilane, with 0.00 ppm as an internal standard. MS was performed on an Agilent 7820A equipped with HP-5MS column coupled to a 5977E MSD unit or on an Agilent 1290 Infinity System in tandem with an Agilent Quadrupole 6120 LC/MS detector. High-resolution MS was obtained from an Agilent 1290 infinity LC system equipped with an autoSampler in tandem with an Agilent 6520 Accurate Mass Q-TOF LC/MS.
Photophysical characterization
Absorbance spectra were measured using a Perkin Elmer LAMBDA 650 spectrophotometer. Emission and excitation measurements were performed using an FLS1000 (Edinburgh Instruments) spectrofluorometer. Quantum yields were measured using an integrated sphere (Edinburgh Instruments) and relative method using quinine sulfate (Sigma-Aldrich) in a 0.1-N solution of H2SO4 (quantum yield of quinine sulfate, 0.546), as an average of three samples. Fluorescence lifetime measurements were determined with a time-correlated single-photon counting system (FLS1000) using picosecond-pulsed diode lasers (405 nm) and pulsed LEDs (320 nm) as excitation sources. Phosphorescence lifetime was determined with a time-resolved single-photon counting (multichannel scaling) system with a microsecond flashlamp as excitation source (FLS1000). For lifetime measurements, the samples were prepared using a solvent degassed by freeze-pump-thaw cycles. The samples for quantum yield measurements were prepared in a glove box. Samples were dissolved in EMSURE-grade toluene (Sigma-Aldrich); samples prepared in the glove box were dissolved in anhydrous toluene (Sigma-Aldrich) purged with argon for 30 min. Transient absorption measurements were performed on an LP980 (Edinburgh Instruments) spectrometer with a Spectra-Physics Nd:YAG laser coupled to a Spectra-Physics primoscan optical parametric oscillator as excitation source (320 nm). All sample concentrations, for both emission and lifetime measurements, were set to a maximum of 0.1 absorbance unit (13.5 × 10−6 M for the DBA) to avoid inner filter effects.
Supplementary Material
Acknowledgments
Funding: K.B. acknowledges the European Research Council (ERC-2017-StG-757733), the Swedish Research Council (2016-03354), and the Swedish Foundation for Strategic Research (ICA14-0018) for financial support. Author contributions: A.C. performed the synthesis and chemical characterization. M.H. and C.Y. performed the photophysical characterization. A.C., M.N.I., L.E., and U.M. performed the single-crystal diffraction and structure determination. K.B. devised the conceptual idea and supervised the synthesis and photophysical characterization. All authors contributed to writing the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaaw5978/DC1
Section S1. Synthesis and characterization
Section S2. Details of the experimental determination of the rate of energy transfer
Section S3. Details of the simulation of the rate of energy transfer
Section S4. X-ray diffraction of 19 and DBA
Scheme S1. Synthesis of the DBA.
Table S1. Excited-state lifetimes (fractional contributions in parentheses) and associated emission quantum yields for D, A, and DBA in toluene solutions.
Fig. S1. Emission quantum yield of A, measured using an integrated sphere.
Fig. S2. Transient absorption spectroscopy and decay of D in toluene when excited at 320 nm.
Fig. S3. Transient absorption spectroscopy of DBA in toluene solution when excited at 320 nm.
Fig. S4. Transient absorption decays of DBA when excited at 320 nm.
Fig. S5. The simulated rate of energy transfer as a function of dihedral angle (orange line).
Fig. S6. Lifetime of DBA with different excitation intensity.
Fig. S7. Structure of the ligand (19), as solved by x-ray diffraction.
Fig. S8. Structure of DBA (20), as solved by x-ray diffraction.
Fig. S9. 1H NMR (400 MHz, CDCl3), 6.
Fig. S10. 13C NMR (101 MHz, CDCl3), 6.
Fig. S11. 1H NMR (800 MHz, CDCl3), 15.
Fig. S12. 13C NMR (201 MHz, CDCl3), 15.
Fig. S13. 31P NMR (162 MHz, CDCl3), 15.
Fig. S14. 1H NMR (800 MHz, CDCl3), 17.
Fig. S15. 13C NMR (201 MHz, CDCl3), 17.
Fig. S16. 1H NMR (800 MHz, DMSO-d6), 18.
Fig. S17. 13C NMR (201 MHz, DMSO-d6), 18.
Fig. S18. 1H NMR (800 MHz, DMSO-d6), 19.
Fig. S19. 13C NMR (201 MHz, DMSO-d6), 19.
Fig. S20. 1H NMR (800 MHz, CDCl3), 20.
Fig. S21. 13C NMR (201 MHz, CDCl3), 20.
Fig. S22. 31P NMR (162 MHz, CDCl3), 20.
Fig. S23. HRMS (ESI+), 6.
Fig. S24. HRMS (ESI+), 15.
Fig. S25. HRMS (ESI+), 17.
Fig. S26. HRMS (ESI+), 18.
Fig. S27. HRMS (ESI+), 19.
Fig. S28. HRMS (ESI+), 20.
REFERENCES AND NOTES
- 1.F. So, Organic Electronics: Materials, Processing, Devices and Applications (CRC Press, 2009). [Google Scholar]
- 2.S. Ogawa, Organic Electronics Materials and Devices (Springer, 2015). [Google Scholar]
- 3.P. Klán, J. Wirz, Photochemistry of Organic Compounds: From Concepts to Practice (Wiley, 2009). [Google Scholar]
- 4.Roest M. R., Oliver A. M., Paddon-Row M. N., Verhoeven J. W., Distance dependence of singlet and triplet charge recombination pathways in a series of rigid bichromophoric systems. J. Phys. Chem. A 101, 4867–4871 (1997). [Google Scholar]
- 5.Baldo M. A., O’Brien D. F., You Y., Shoustikov A., Sibley S., Thompson M. E., Forrest S. R., Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 395, 151–154 (1998). [Google Scholar]
- 6.D. J. Gaspar, E. Polikarpov, OLED Fundamentals: Materials, Devices, and Processing of Organic Light-Emitting Diodes (CRC Press, 2015). [Google Scholar]
- 7.Schmidbauer S., Hohenleutner A., König B., Chemical degradation in organic light-emitting devices: Mechanisms and implications for the design of new materials. Adv. Mater. 25, 2114–2129 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Uoyama H., Goushi K., Shizu K., Nomura H., Adachi C., Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492, 234–238 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Stranius K., Hertzog M., Börjesson K., Selective manipulation of electronically excited states through strong light-matter interactions. Nat. Commun. 9, 2273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakanotani H., Higuchi T., Furukawa T., Masui K., Morimoto K., Numata M., Tanaka H., Sagara Y., Yasuda T., Adachi C., High-efficiency organic light-emitting diodes with fluorescent emitters. Nat. Commun. 5, 4016 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Yang Z., Mao Z., Xie Z., Zhang Y., Liu S., Zhao J., Xu J., Chi Z., Aldred M. P., Recent advances in organic thermally activated delayed fluorescence materials. Chem. Soc. Rev. 46, 915–1016 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Förster T., Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 437, 55–75 (1948). [Google Scholar]
- 13.Bennett R. G., Schwenker R. P., Kellogg R. E., Radiationless intermolecular energy transfer. II. Triplet→singlet transfer. J. Chem. Phys. 41, 3040–3041 (1964). [Google Scholar]
- 14.Guo D., Knight T. E., McCusker J. K., Angular momentum conservation in dipolar energy transfer. Science 334, 1684–1687 (2011). [DOI] [PubMed] [Google Scholar]
- 15.N. J. Turro, V. Ramamurthy, J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction (Univ. Science Books, 2009). [Google Scholar]
- 16.Skaisgirski M., Guo X., Wenger O. S., Electron accumulation on naphthalene diimide photosensitized by [Ru(2,2′-Bipyridine)3]2+. Inorg. Chem. 56, 2432–2439 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Chang Y.-L., Song Y., Wang Z., Helander M. G., Qiu J., Chai L., Liu Z., Scholes G. D., Lu Z., Highly efficient warm white organic light-emitting diodes by triplet exciton conversion. Adv. Funct. Mater. 23, 705–712 (2013). [Google Scholar]
- 18.Chang Y. L., Gong S., Wang X., White R., Yang C., Wang S., Lu Z. H., Highly efficient greenish-blue platinum-based phosphorescent organic light-emitting diodes on a high triplet energy platform. Appl. Phys. Lett. 104, 173303 (2014). [Google Scholar]
- 19.Chang Y.-L., Lu Z.-H., White organic light-emitting diodes for solid-state lighting. J. Disp. Technol. 9, 459–468 (2013). [Google Scholar]
- 20.Cerdán L., Enciso E., Martín V., Bañuelos J., López-Arbeloa I., Costela A., García-Moreno I., FRET-assisted laser emission in colloidal suspensions of dye-doped latex nanoparticles. Nat. Photonics 6, 621–626 (2012). [Google Scholar]
- 21.Cadranel A., Oviedo P. S., Alborés P., Baraldo L. M., Guldi D. M., Hodak J. H., Electronic energy transduction from {Ru(py)4} chromophores to Cr(III) luminophores. Inorg. Chem. 57, 3042–3053 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Bonn A. G., Wenger O. S., Photoinduced charge accumulation by metal ion-coupled electron transfer. Phys. Chem. Chem. Phys. 17, 24001–24010 (2015). [DOI] [PubMed] [Google Scholar]
- 23.B. Valeur, M. N. Berberan-Santos, Molecular Fluorescence: Principles and Applications (John Wiley & Sons, 2012). [Google Scholar]
- 24.Lin C.-H., Chang Y.-Y., Hung J.-Y., Lin C.-Y., Chi Y., Chung M.-W., Lin C.-L., Chou P.-T., Lee G.-H., Chang C.-H., Lin W.-C., Iridium(III) complexes of a dicyclometalated phosphite tripod ligand: Strategy to achieve blue phosphorescence without fluorine substituents and fabrication of OLEDs. Angew. Chem. Int. Ed. 50, 3182–3186 (2011). [DOI] [PubMed] [Google Scholar]
- 25.I. Berlman, Handbook of Fluorescence Spectra of Aromatic Molecules (Elsevier Science, ed. 2, 1971). [Google Scholar]
- 26.Chang H., Kiesman W. F., Petter R. C., Convenient one-pot preparation of dimethyl bicyclo[2.2.2]octane-1,4-dicarbolylate, a key intermediate for a novel adenosine A1 receptor antagonist. Synth. Commun. 37, 1267–1272 (2007). [Google Scholar]
- 27.Al Hussainy R., Verbeek J., van der Born D., Braker A. H., Leysen J. E., Knol R. J., Booij J., Herscheid J. D. M., Design, synthesis, radiolabeling, and in vitro and in vivo evaluation of bridgehead iodinated analogues of N-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}-N-(pyridin-2-yl)cyclohexanecarboxamide (WAY-100635) as potential SPECT ligands for the 5-HT1A receptor. J. Med. Chem. 54, 3480–3491 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Candish L., Standley E. A., Gómez-Suárez A., Mukherjee S., Glorius F., Catalytic access to alkyl bromides, chlorides and iodides via visible light-promoted decarboxylative halogenation. Chem. Eur. J. 22, 9971–9974 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Birch A. M., Birtles S., Buckett L. K., Kemmitt P. D., Smith G. J., Smith T. J. D., Turnbull A. V., Wang S. J. Y., Discovery of a potent, selective, and orally efficacious pyrimidinooxazinyl bicyclooctaneacetic acid diacylglycerol acyltransferase-1 inhibitor. J. Med. Chem. 52, 1558–1568 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Barlind J. G., Bauer U. A., Birch A. M., Birtles S., Buckett L. K., Butlin R. J., Davies R. D. M., Eriksson J. W., Hammond C. D., Hovland R., Johannesson P., Johansson M. J., Kemmitt P. D., Lindmark B. T., Gutierrez P. M., Noeske T. A., Nordin A., O’Donnell C. J., Petersson A. U., Redzic A., Turnbull A. V., Vinblad J., Design and optimization of pyrazinecarboxamide-based inhibitors of diacylglycerol acyltransferase 1 (DGAT1) leading to a clinical candidate dimethylpyrazinecarboxamide phenylcyclohexylacetic acid (AZD7687). J. Med. Chem. 55, 10610–10629 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Kodama K., Kobayashi A., Hirose T., Synthesis and spectral properties of ruthenium(II) complexes based on 2,2′-bipyridines modified by a perylene chromophore. Tetrahedron Lett. 54, 5514–5517 (2013). [Google Scholar]
- 32.Cui X., Charaf-Eddin A., Wang J., Guennic B. L., Zhao J., Jacquemin D., Perylene-derived triplet acceptors with optimized excited state energy levels for triplet–triplet annihilation assisted upconversion. J. Org. Chem. 79, 2038–2048 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Torres É., Berberan-Santos M. N., Brites M. J., Synthesis, photophysical and electrochemical properties of perylene dyes. Dyes Pigments 112, 298–304 (2015). [Google Scholar]
- 34.Avlasevich Y., Müllen K., An efficient synthesis of quaterrylenedicarboximide NIR dyes. J. Org. Chem. 72, 10243–10246 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Weil T., Abdalla M. A., Jatzke C., Hengstler J., Müllen K., Water-soluble rylene dyes as high-performance colorants for the staining of cells. Biomacromolecules 6, 68–79 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Bandak D., Babii O., Vasiuta R., Komarov I. V., Mykhailiuk P. K., Design and synthesis of novel 19F-amino acid: A promising 19F NMR label for peptide studies. Org. Lett. 17, 226–229 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Adcock W., Kok G. B., Polar substituent effects on fluorine-19 chemical shifts of aryl and vinyl fluorides: A fluorine-19 nuclear magnetic resonance study of some 1,1-difluoro-2-(4-substituted-bicyclo[2.2.2]oct-1-yl)ethenes. J. Org. Chem. 50, 1079–1087 (1985). [Google Scholar]
- 38.Case F. H., The preparation of hydrazidines and as-triazines related to substituted 2-cyanopyridines. J. Org. Chem. 30, 931–933 (1965). [Google Scholar]
- 39.Tai S., Marchi S. V., Carrick J. D., Efficient preparation of pyridinyl-1,2,4-triazines via telescoped condensation with diversely functionalized 1,2-dicarbonyls. J. Heterocyclic Chem. 53, 1138–1146 (2016). [Google Scholar]
- 40.Fanni S., Keyes T. E., O’Connor C. M., Hughes H., Wang R., Vos J. G., Excited-state properties of ruthenium(II) polypyridyl complexes containing asymmetric triazole ligands. Coord. Chem. Rev. 208, 77–86 (2000). [Google Scholar]
- 41.John K. D., Salazar K. V., Scott B. L., Baker R. T., Sattelberger A. P., Comparison of the reactivity of M(allyl)3 (M = Rh, Ir) with donor ligands. Organometallics 20, 296–304 (2001). [Google Scholar]
- 42.Chiu Y.-C., Lin C.-H., Hung J.-Y., Chi Y., Cheng Y.-M., Wang K.-W., Chung M.-W., Lee G.-H., Chou P.-T., Authentic-blue phosphorescent iridium(III) complexes bearing both hydride and benzyl diphenylphosphine; control of the emission efficiency by ligand coordination geometry. Inorg. Chem. 48, 8164–8172 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Steren C. A., van Willigen H., Biczók L., Gupta N., Linschitz H., C60 as a photocatalyst of electron-transfer processes: Reactions of triplet C60 with chloranil, perylene, and tritolylamine studied by flash photolysis and FT-EPR. J. Phys. Chem. 100, 8920–8926 (1996). [Google Scholar]
- 44.Halasinski T. M., Weisman J. L., Ruiterkamp R., Lee T. J., Salama F., Head-Gordon M., Electronic absorption spectra of neutral perylene (C20H12), terrylene (C30H16), and quaterrylene (C40H20) and their positive and negative ions: Ne matrix-isolation spectroscopy and time-dependent density functional theory calculations. J. Phys. Chem. A 107, 3660–3669 (2003). [Google Scholar]
- 45.J. R. Lakowicz, Principles of Fluorescence Spectroscopy (Springer US, ed. 3, 2006). [Google Scholar]
- 46.Kabsch W., Automatic indexing of rotation diffraction patterns. J. Appl. Crystallogr. 21, 67–72 (1988). [Google Scholar]
- 47.Sheldrick G. M., A short history of SHELX. Acta Crystallogr. Sect. A: Found. Crystallogr. 64, 112–122 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaaw5978/DC1
Section S1. Synthesis and characterization
Section S2. Details of the experimental determination of the rate of energy transfer
Section S3. Details of the simulation of the rate of energy transfer
Section S4. X-ray diffraction of 19 and DBA
Scheme S1. Synthesis of the DBA.
Table S1. Excited-state lifetimes (fractional contributions in parentheses) and associated emission quantum yields for D, A, and DBA in toluene solutions.
Fig. S1. Emission quantum yield of A, measured using an integrated sphere.
Fig. S2. Transient absorption spectroscopy and decay of D in toluene when excited at 320 nm.
Fig. S3. Transient absorption spectroscopy of DBA in toluene solution when excited at 320 nm.
Fig. S4. Transient absorption decays of DBA when excited at 320 nm.
Fig. S5. The simulated rate of energy transfer as a function of dihedral angle (orange line).
Fig. S6. Lifetime of DBA with different excitation intensity.
Fig. S7. Structure of the ligand (19), as solved by x-ray diffraction.
Fig. S8. Structure of DBA (20), as solved by x-ray diffraction.
Fig. S9. 1H NMR (400 MHz, CDCl3), 6.
Fig. S10. 13C NMR (101 MHz, CDCl3), 6.
Fig. S11. 1H NMR (800 MHz, CDCl3), 15.
Fig. S12. 13C NMR (201 MHz, CDCl3), 15.
Fig. S13. 31P NMR (162 MHz, CDCl3), 15.
Fig. S14. 1H NMR (800 MHz, CDCl3), 17.
Fig. S15. 13C NMR (201 MHz, CDCl3), 17.
Fig. S16. 1H NMR (800 MHz, DMSO-d6), 18.
Fig. S17. 13C NMR (201 MHz, DMSO-d6), 18.
Fig. S18. 1H NMR (800 MHz, DMSO-d6), 19.
Fig. S19. 13C NMR (201 MHz, DMSO-d6), 19.
Fig. S20. 1H NMR (800 MHz, CDCl3), 20.
Fig. S21. 13C NMR (201 MHz, CDCl3), 20.
Fig. S22. 31P NMR (162 MHz, CDCl3), 20.
Fig. S23. HRMS (ESI+), 6.
Fig. S24. HRMS (ESI+), 15.
Fig. S25. HRMS (ESI+), 17.
Fig. S26. HRMS (ESI+), 18.
Fig. S27. HRMS (ESI+), 19.
Fig. S28. HRMS (ESI+), 20.