

Summary
Drug-like inhibitors are often designed by mimicking cofactor or substrate interactions with enzymes. However, as active sites are comprised of conserved residues, it is difficult to identify the critical interactions needed to design selective inhibitors. We are developing an approach, named RADD (Resistance Analysis During Design), which involves engineering point mutations in the target to generate active alleles and testing compounds against them. Mutations that alter compound potency identify residues that make key interactions with the inhibitor and predict target-binding poses. Here, we apply this approach to analyze how diaminotriazole-based inhibitors bind spastin, a microtubule-severing AAA (ATPase associated with diverse cellular activities) protein. The distinct binding poses predicted for two similar inhibitors were confirmed by a series of X-ray structures. Importantly, our approach not only reveals how selective inhibition of the target can be achieved but also identifies resistance-conferring mutations at the early stages of the design process.
Graphical Abstract
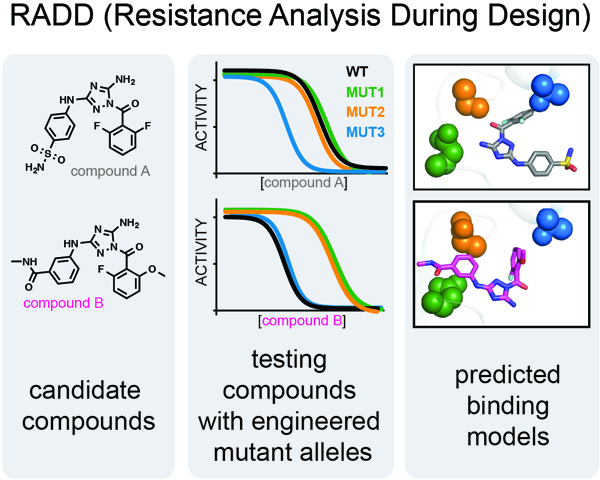
Pisa et al. describes how engineered point mutations in protein active sites can be used to predict the binding modes of chemical inhibitors. These data can guide inhibitor optimization and can identify cognate resistance-conferring mutations at the start of the inhibitor design process.
Introduction
Enzyme active sites are common binding sites for chemical inhibitors, as compounds can mimic substrates or co-factors to compete for occupancy (Copeland, 2013). Active sites are typically comprised of conserved structural motifs and amino acid sequences and their overall steric and stereoelectronic features can be similar across enzymes within a protein family (Wendler et al., 2012, Endicott et al., 2012). For some protein families, such as kinases, a wealth of high-resolution structural data for how different chemical scaffolds interact with residues in conserved active sites has enabled the design of selective chemical inhibitors (Ferguson and Gray, 2018). However, the relatively low resolution (3–4 Å) of structures for many proteins, including members of AAA (ATPases associated with diverse cellular activities) family (Erzberger and Berger, 2006), can limit their use for rational inhibitor design (Davis et al., 2008) and additional approaches are needed to identify the key interactions determining inhibitor potency and specificity (Erlanson et al., 2019).
Proteins in the AAA (ATPases associated with diverse cellular activities) family carry out critical tasks in various cellular processes including DNA unwinding and replication, protein unfolding or membrane remodelling (Bleichert et al., 2017; McCullough et al., 2018; van den Boom and Meyer, 2018). For a few AAA proteins, chemical inhibitors have been identified by screening compound libraries (Anderson et al., 2015; Chou et al., 2011; Firestone et al., 2012; Kawashima et al., 2016; Magnaghi et al., 2013). In most cases, the inhibitor binding sites have been mapped to the AAA domain, the core enzymatic module of AAA proteins (Wendler et al., 2012), either in the active site (Anderson et al., 2015; Cupido et al., 2019; Magnaghi et al., 2013) in an allosteric site (Magnaghi et al., 2013, Banerjee et al., 2016; Pohler et al., 2018). Structural models for a few inhibitor-bound AAA proteins are also now available (Banerjee et al., 2016; Boyaci et al., 2016; Pisa et al., 2019; Tang et al., 2019). However, for many AAA proteins the key inhibitor-target interactions needed for the design of selective chemical inhibitors are not known.
We have recently focused on spastin, a microtubule-severing AAA protein whose functions have been linked to several cellular processes including nuclear envelope reformation and cytokinesis (Connell et al., 2009; Vietri et al., 2015). In addition, blocking spastin function has been shown to reduce amyloid-β toxicity in a model for Alzheimer’s disease (Zempel et al., 2013). Therefore, chemical inhibitors would be valuable tools to probe spastin functions in normal physiology and disease. We recently designed spastazoline, a potent and selective inhibitor of spastin (Cupido et al., 2019). To design this pyrazolylpyrrolopyrimidine-based inhibitor, we analyzed compound activity against biochemically active mutant alleles of spastin. We reasoned that mutant alleles that alter the potency of compounds would reveal key compound-target interactions and guide the selection of robust inhibitor-protein binding models. From a collection of heterocyclic scaffolds that could mimic key hydrogen-bonding interactions made by adenine in the AAA active site, we identified a pyrazolyl-based scaffold. Testing this compound against wild-type and mutant spastin alleles revealed key interactions that we used to rank order different solutions from computational docking. We used the selected inhibitor-spastin binding model to design modifications of the core scaffold and generated spastazoline, the potent and selective inhibitor of spastin (Cupido et al., 2019). Structural models we generated by X-ray crystallography confirmed the predicted binding models (Pisa et al., 2019). However, it remains unclear if our approach, which we now name RADD (for Resistance Analysis During Design), can be used to identify binding site interactions of inhibitors based on different chemical scaffolds and if the target-binding modes we predict are accurate.
Here, we focus on applying RADD to diaminotriazole-based compounds, which are chemically unrelated to spastazoline. Testing compound activity against wild-type and mutant spastin alleles identified key interactions that contribute to inhibitor binding. Our approach also indicated that a more potent derivative binds spastin in a distinct pose, essentially oriented ~180° relative to the starting compound. High-resolution X-ray structures of these two different diaminotriazole-based compounds confirmed the inhibitor-spastin interactions predicted by RADD. Together, these data suggest how biochemical analyses of resistance against chemical inhibitors can be leveraged to determine the key inhibitor-protein interactions in conserved active sites and can establish potential resistance-conferring mutations in the target at the start of inhibitor design process.
Results
Using RADD to examine the binding of a diaminotriazole-based inhibitor to spastin.
In our efforts to identify different chemical scaffolds that inhibit AAA proteins we found a diaminotriazole-based compound -an inhibitor of cyclin-dependent and Janus JNJ-7706621 (Lin et al., 2005; Malerich et al., 2010)- that blocks the ATPase activity of spastin and related AAA proteins katanin and fidgetin-like 1 (Cupido et al., 2019). To characterize the binding of the JNJ-7706621 inhibitor (hereafter compound 1, Figure 1A) to spastin, we first tested the inhibition of the steady-state ATPase activity of wild-type spastin at a single ATP concentration (Drosophila melanogaster spastin, aa 209–758, hereafter spastin-WT, IC50: 5.8 ± 0.6 µM, 0.5 mM MgATP, average ± s.d., n=3, Figure 1B, C - black trace). Further analyses of spastin-WT inhibition, across a range of ATP concentrations, revealed that compound 1 increased the apparent K1/2 for ATP without altering the kcat (Figure 1D and Figure S1A), indicating an ATP competitive mechanism of inhibition that is consistent with compound 1 binding in spastin’s nucleotide-binding site.
Figure 1. Using RADD to analyze binding of compound 1 to spastin.

(A) Chemical structure of compound 1 (JNJ-7706621). (B) Schematic shows the AAA domain (gray box) of the recombinant spastin construct used for ATPase assays. The positions of three variability hotspot residues are highlighted (colored arrows). The first and last residues of the construct are also indicated (aa 209–758). (C) Compound 1 concentration-dependent inhibition of the steady-state ATPase activity of spastin wild-type (WT) and three constructs with mutations at variability hot-spot residues (Q488V, N527C and T692A). Graphs show average values ± s.d. (n = 3) fit to a sigmoidal dose-response equation. IC50 values: spastin-WT: 5.7 ± 0.6 µM; -Q488V: 8.9 ± 0.5 µM; -N527C: 6.0 ± 1.0 µM and -T692A: 0.9 ± 0.1 µM; average ± s.d., n = 3; all ATPase assays were performed at 0.5 mM MgATP. (D) ATP concentration-dependence of the steady-state ATPase activity of spastin at different concentrations of compound 1, analyzed using an NADH-coupled assay. Average values (n = 3, error bars denote s.d.) were fit to the Michaelis-Menten equation for cooperative enzymes (see Methods). Enzyme activity parameters determined for each condition are provided in Figure S1A. (E) Concentration-dependent effect of compound 1 on the heat-induced denaturation of spastin-AAA constructs analyzed by differential scanning fluorimetry. Observed ∆Tm: -WT ~4.5 °C; -N527C ~4.3 °C; -Q488V ~4.5 °C; -T692A ~6.2 °C, 100 µM compound 1, average values are shown, error bars denote range, n = 2. (F) Schematic for predicted interactions between compound 1 and the variability hotspot residues based on analyses of the inhibition of spastin alleles (filled circle, predicted interaction; dashed circles, predicted weak or no interaction). (G) Computational docking model (ribbon and stick representation) of compound 1 in the active site of spastin that is consistent with the observed activity of the compound against mutant spastin alleles (for additional data see Figure S2 and Methods). Predicted hydrogen-bonding network between the diaminotriazole core of compound 1 and spastin’s N-loop motif is shown (black dashed lines). The spastin variability hotspot residues (Gln-488, Asn-527, and Thr-692) are also shown. The distance between the compound and the side chain of the sensor-II variability hotspot residue is indicated (gray dotted line).
The first step of RADD is to analyze compound activity against alleles that have mutations in the putative compound binding site of the target enzyme but retain biochemical activity. We have found that active mutant alleles of spastin can be engineered in three structural motifs in spastin’s active site (the N-loop, the P-loop and the sensor-II motif) by substituting the relatively less conserved residues, which we named variability hot-spots, with residues found at equivalent positions in related AAA proteins (Cupido et al., 2019). Here, we focus on three mutant alleles (Q488V, N527C and T692A) in spastin’s variability hotspots that we have previously characterized (Cupido et al., 2019). We found that a sensor-II mutation (T692A) increased the potency of compound 1 by ~7-fold (Figure 1C). In contrast, mutations in the N-loop (Q488V) and the P-loop (N527C) hot-spot residues led to less than 2-fold changes in inhibitor potency (Figure 1C). To further characterize binding of compound 1 to spastin mutant alleles, we used differential scanning fluorimetry. For these assays we focused on a truncated construct including only spastin’s AAA domain (aa 445–758, Drosophila melanogaster spastin, hereafter, spastin-AAA, Figure S1B, C) that we have characterized (Pisa et al., 2019). A dose-dependent increase in the melting temperature of spastin-AAA constructs in presence of compound 1 was observed (Figure 1E and Figure S1D–G). Gratifyingly, the spastin-AAA-T692A construct was stabilized by compound 1 more substantially than the wild-type, N527C and Q488V constructs (∆Tm: T692A ~6.2 °C; WT ~4.5 °C; N527C ~4.3 °C, Q488V ~ 4.5 °C, 100 µM compound 1, DMSO was used as a vehicle control, Figure 1E and Figure S1D–G) indicating that compound 1 binds spastin-AAA-T692A with higher affinity compared to the other alleles. These data suggest that compound 1 interacts with the side chain of the sensor-II hot-spot residue in spastin’s active site and does not substantially interact with those of the N-loop and P-loop hot-spot residues (Figure 1F).
Next, we performed computational docking of compound 1 on a conformational ensemble of apo spastin (pdbID: 3B9P) obtained from molecular dynamics simulations (see Methods for additional details). These analyses suggested several possible orientations of compound 1 in the active site (Figure S2) with computed docking scores within ~2 kcal/mol. In four of the five top-scoring models, compound 1 establishes hydrogen-bonding interactions with the protein backbone of the spastin N-loop (Figure S2). In one of the poses, the diaminotriazole core establishes contacts with the N-loop of spastin in a manner that is analogous to ATP (pose 5, Figure 1G and Figure S2I, J). We noted that in this pose compound 1 is also proximal to the sensor-II residue (Thr-692; side chain to compound distance: <4 Å), while the side chains of variability hot-spot residues in the N-loop (Gln-488) and P-loop (Asn-527) are positioned further away from the compound (side chain to compound distance: >4 Å, Figure 1G). This pose, in which the difluorobenzoyl substituent of the compound 1 is proximal to the Thr-692 sensor-II side chain and the phenylsulfonamide substituent points out from the AAA domain towards the bulk solvent (Figure 1G), is consistent with the changes in activity of compound 1 observed against mutant spastin alleles. Therefore, we rejected the other poses with similar or even more favorable docking scores as they were not consistent with the mutant activity analyses. This model of compound 1 bound in the active site of spastin is different from our RADD models for pyrazolylaminoquinazoline-based spastin inhibitors, which we have confirmed by X-ray crystallography (Cupido et al., 2019; Pisa et al., 2019). Importantly, mutant alleles conferring resistance to the pyrazolylaminoquinazoline-based inhibitors are different from those that alter potency of compound 1, consistent with these compounds binding differently to spastin.
Crystal structure of spastin-N527C bound to compound 1.
To test the binding pose for compound 1 predicted by our RADD approach, we sought to determine the crystal structure of the spastin-compound 1 complex. Our trials with the spastin-AAA wild-type (WT) construct in the presence of compound 1 did not yield any diffracting crystals. As increased thermal stability of proteins has been shown to improve likelihood of crystallization (Dupeux et al., 2011), we examined the stability of spastin-AAA constructs against heat-induced denaturation using differential scanning fluorimetry and determined their melting temperature (Tm). Spastin-AAA-WT has a Tm of ~38.5 °C, the sensor-II mutation (T692A) does not substantially alter stability (Tm ~39 °C) and the N-loop mutation (Q488V) causes a modest reduction of this construct’s melting temperature (Tm ~34.5 °C)(Figure S3A). Remarkably, we found that the P-loop mutation (N527C) stabilizes spastin in the differential scanning fluorimetry assay (Tm ~42 °C)(Figure S3A). As compound 1 inhibits the ATPase activity of the N527C allele of spastin with nearly identical potency as the wild-type protein (Figure 1C), we set up crystallization trials of spastin-AAA-N527C in presence of compound 1.
We obtained diffracting crystals and determined the structure of the spastin-compound 1 complex at ~2.30 Å resolution using molecular replacement (Table S1). We observed a density, in the pocket between the large and small AAA subdomain (Figure 2A, B and Figure S3B, C) and could assign this to the compound. In this model, the diaminotriazole core of compound 1 is positioned to form a network of three hydrogen bonding interactions with the backbone atoms of residues Asp-484 and Ala-486 in spastin’s N-loop (Figure 2C and Figure S3C). The phenylsulfonamide substituent is proximal to a hydrophobic residue in the large AAA subdomain (Leu-531) at the entrance of the ATP-binding site and the difluorobenzoyl moiety contacts the side chain of Thr-692, the variability hotspot residue in the sensor-II motif (Figure 2D). In particular, one fluorine atom in the difluorobenzoyl moiety is oriented towards the Thr-692 hydroxyl group (fluorine to hydroxyl oxygen distance: ~3.2 Å, Figure 2D). In contrast, the N-loop (Gln-488) and P-loop (Cys-527) variability hot-spot residues are located >4 Å away from the compound (Figure 2D), which is consistent with mutations at these sites not having a substantial effect on the inhibition of spastin’s ATPase activity by compound 1. As the Cys-527 residue is not proximal to compound 1, we hypothesize that the binding mode of compound 1 is not altered by the N527C mutation and is likely similar in the WT protein. Together, these structural data match our RADD model for compound 1 bound in the active site of spastin.
Figure 2. Crystal structure of the spastin variability hot-spot mutant (N527C) bound to compound 1 matches the RADD model.

(A) Structural model of the spastin-AAA-N527C-compound 1 complex (surface and stick representation, generated using UCSF Chimera). Small and large AAA subdomains are indicated (black and gray). (B) Compound 1 and the corresponding simulated annealing composite omit electron density map contoured at 3σ is shown (mFo – DFc, green mesh). (C) Compound 1 in spastin’s active site (surface and stick representation). Predicted hydrogen-bonding interactions are also shown (black dashed lines). (D) View of compound 1 in spastin’s active site highlighting the N-loop and the variability hotspot residues in the P-loop and the sensor-II motifs. Distance between the side chains of variability hot-spot residues (Gln-488, Cys-527 and Thr-692) and compound 1 are indicated (gray dotted lines).
Using RADD to examine binding of compound 1 derivatives to spastin.
To further test our structural models, we synthesized and tested analogs of compound 1. We modified the difluorobenzoyl and phenylsulfonamide substituents while keeping the diaminotriazole core, as it forms a key hydrogen-bonding network with the backbone of spastin’s active site residues (Figure 3A and Figure S4A). Changes of the difluorobenzoyl moiety result in modest (~2-fold) changes in potency (compound 2: IC50 = 2.77 ± 0.39 µM, average ± s.d., n=3, Figure 3A, B). In contrast, modifications of the phenylsulfonamide can lead to >10-fold higher potency. In particular, we identified compound 3, in which a meta-N-methylamide group replaces the para-sulfonamide, which inhibits spastin with submicromolar potency (IC50 = 0.45 ± 0.03 µM, average ± s.d., n=3, Figure 3A, B). We combined substitutions from compounds 2 and 3 and synthesized compound 4. This compound inhibits spastin-WT ~40-fold more potently than compound 1 (IC50 = 0.23 ± 0.02 µM, average ± s.d., n=3, Figure 3A, B).
Figure 3. Using RADD to characterize binding of a potent diaminotriazole-based inhibitor to spastin.

(A) Chemical structures of compounds 1–4. (B) Compound concentration-dependent inhibition of the steady-state ATPase activity of spastin wild-type (WT). Graph shows average values ± s.d. (n = 3) fit to a sigmoidal dose-response equation. For comparison, the curve for compound 1 (data from Figure 1C) is also shown (gray dashed line). Compound 2: IC50 = 2.77 ± 0.39 µM; Compound 3: IC50 = 0.45 ± 0.03 µM; Compound 4: IC50 = 0.23 ± 0.02 µM (average ± s.d., n=3, 0.5 mM MgATP). (C) Compound 4 concentration-dependent inhibition of the steady-state ATPase activity of three spastin constructs with mutations at variability hot-spot residues (Q448V, N527C and T692A). Graph shows average values ± s.d. (n = 3) fit to a sigmoidal dose-response equation. For comparison, the corresponding curve for spastin-WT is shown (dashed line, data from Figure 3B). IC50 values: spastin-Q488V: 2.60 ± 0.15 µM; -N527C: 1.11 ± 0.21 µM and -T692A: 0.22 ± 0.04 µM; average ± s.d., n = 3, 0.5 mM MgATP. (d) Concentration-dependent effect of compound 4 on the heat-induced denaturation of spastin-AAA constructs analyzed by differential scanning fluorimetry. Observed ∆Tm: -WT ~6.7 °C; -N527C ~4.5 °C; - Q488V ~5.0 °C; -T692A ~7.2 °C, 100 µM compound 4, average values are shown, error bars denote range, n = 2. (E) Schematic for the expected compound 4 interactions with variability hotspot residues in the active site of spastin (filled circles, predicted interaction; dashed circle, predicted weak or no interaction). Variability hot-spot residues in three nucleotide-binding motifs are shown (N-loop, green; P-loop, orange; sensor-II, blue). (F) Sequence alignment of nucleotide-binding motifs (N-loop, P-loop and sensor-II) in four AAA proteins. Variability hot-spot residues are highlighted (green, orange and blue rectangles). (G) Percentage steady-state ATPase activity of four AAA proteins in the presence of compound 4 (2 µM, 0.5 mM MgATP, average values are shown, error bars denote range, n=2).
To examine how compound 4 binds spastin, we used our RADD approach and tested the inhibitor against spastin constructs with mutations at variability hot-spot residues in the N-loop, P-loop and sensor-II motifs (Figure 3C). Surprisingly, we observe a different pattern of compound 4 activity against these mutant alleles in comparison to compound 1. We found that the mutation in the N-loop (Q488V) reduces compound 4 potency ~11-fold in comparison to the wild-type protein (Figure 3C). Similarly, the P-loop mutation (N527C) reduces the potency ~4-fold (Figure 3C). We note that the same N loop and P loop mutations did not substantially alter the potency of compound 1 (Figure 1C). In contrast, the mutation of sensor-II variability residue (T692A), which sensitizes spastin to inhibition by compound 1, did not change the potency of compound 4 (Figure 3C). We also examined the direct binding of compound 4 to spastin-AAA mutant alleles using differential scanning fluorimetry. We found that compound 4 stabilizes WT and T692A constructs more substantially than the N-loop (Q488V) and P-loop (N527C) constructs (Figure 3D and Figure S4B–E). Taken together, these analyses indicate that compound 4 binds in the active site of spastin differently than compound 1 (Figure 3E).
To examine specificity of compound 4, we tested the inhibitor against three AAA proteins related to spastin (katanin, fidgetin-like 1 and VPS4B, Figure 3F and Figure S5). We found that compound 4 at 2 µM concentration potently inhibits spastin but does not substantially inhibit the other three AAA proteins (Figure 3G). These analyses suggest that interactions of compound 4 with the unique combination of N-loop and P-loop variability residues (Gln-488 and Asn-527) in spastin contribute to the compound’s potency as well as specificity against other AAA proteins. However, available structure-activity relationships for diaminotriazole-based compounds suggest that compound 4 likely inhibits several kinases (Lin et al., 2005; Malerich et al., 2010). Further optimization of diaminotriazole-based spastin inhibitors should be possible using our RADD models.
Crystal structure of spastin-WT bound to compound 4.
To examine if the spastin-compound 4 interactions predicted by the RADD approach were indeed correct, we set up crystallization trials. For these experiments we chose the spastin-AAA-WT construct instead of spastin-AAA-N527C as compound 4 inhibits the wild type protein with higher potency. Gratifyingly, we obtained crystals of spastin-AAA-WT in presence of the inhibitor and determined the structure of the spastin-AAA-WT-compound 4 complex at ~1.94 Å resolution (Table S1). We observed a well-defined density in the active site that was assigned to the compound (Figure 4A, B). In this structural model, the diaminotriazole core of compound 4 is positioned to form hydrogen-bonding interactions with backbone amides of the residues Ala-486 and Asp-484 in spastin’s N-loop (Figure 4C, D).
Figure 4. Crystal structure of the spastin-WT-compound 4 complex.

(A) Structural model of the spastin-AAA-WT-compound 4 complex (surface and stick representation). (B) Compound 4 (stick representation) and the corresponding simulated annealing composite omit electron density map contoured at 3σ is shown (mFo – DFc, green mesh). (C and D) Compound 4 in spastin’s active site. Predicted hydrogen-bonding interactions are shown (C and D, black dashed lines). Side chains of the variability hotspot residues in the N loop (Gln-488, green), P loop (Asn-527, orange), and sensor-II (Thr-692, blue) motifs as well as the backbone atoms of the hinge residue (Ser-649) are shown (D). (E) Overlay of spastin-AAA-WT-compound 4 and spastin-N527C-compound 1 complexes. Variability hot-spot residues (Gln-488, Asn-527 and Thr-692) are indicated.
Remarkably, as predicted by the RADD approach, there are several differences between the binding modes of compound 1 and 4 in spastin’s active site. First, the diaminotriazole scaffold of compound 4 is positioned in the active site in an orientation ~180° relative to the diaminotriazole core of compound 1. The exocyclin 5-amino group of the core scaffold in compound 4 points out of the pocket into the bulk solvent, whereas in the spastin-compound 1 complex it faces the inside of the ATP binding site (Figure 4E). Second, compound 4’s methoxy-fluorobenzoyl substituent is positioned between the P-loop and the sensor-II helix in a nearly perpendicular orientation to the difluorobenzoyl moiety of compound 1 (Figure 4E). Third, the phenyl-N-methylamide substituent of compound 4 is positioned in the ATP pocket and forms a π-stacking interaction with the side chain amide of the N-loop variability residue (Gln-488). In addition, the N-methylamide group of compound 4 is oriented between spastin’s N-loop and P-loop motifs such that it can form hydrogen bonding interactions with the backbone amide nitrogen of the N-loop residue (Gln-488) and the backbone carbonyl oxygen of the Ser-649 residue in the hinge motif (Figure 4D). These interactions are missing in spastin-compound 1 structure and may contribute to more potent binding of compound 4 to spastin. Taken together, the compound-target interactions predicted by our RADD approach match the high-resolution X-ray data for the spastin-AAA-WT-compound 4 complex.
Structural models of the spastin sensor-II mutant allele (T692A) bound to compounds 1 and 4.
Our findings show that a sensor-II mutation (T692A) in spastin increases the potency of compound 1 in the ATPase assays but does not substantially alter the potency of compound 4. To examine this difference in binding we determined three additional structures, one of the spastin-AAA-T692A construct with compound 1 (~2.15 Å resolution, Table S1) and two with compound 4 (crystal form A: ~2.10 Å and crystal form B: ~1.93 Å, Table S1).
Refinement of the spastin-AAA-T692A-compound 1 structural model suggested that compound 1 can sample two conformations (Figure 5A and S6A, B). In both conformations, the diaminotriazole core is positioned to form a hydrogen-bonding network with backbone amides of the N-loop residues (Asp-484 and Ala-486). The difluorobenzoyl moieties in each of the two conformers are also nearly co-aligned and are proximal to the sensor-II motif (Figure 5A). Interestingly, we observe a different orientation of the phenylsufonamide moiety in the two conformers. One conformer of compound 1 in the spastin-AAA-T692A structure has the phenylsulfonamide proximal to Leu-531 residue of the large AAA subdomain (conformer 1, Figure 5A and S6B). The binding mode of this conformer is similar to the orientation of compound 1 we observed in the spastin-AAA-N527C-compound 1 co-crystal structure (Figure 5B). In the other conformer, the phenylsulfonamide moiety is aligned proximal to helix 4 of the small AAA subdomain and is positioned to form a hydrogen-bonding interaction between the sulfonamide NH and the backbone carbonyl oxygen of the Arg-662 residue (conformer 2; Figures 5A, S6A, and S6B). These interactions are not observed in the structure of spastin-N527C-compound 1 complex.
Figure 5. Structural models of the sensor-II variability hot-spot mutant allele of spastin (T692A) bound to compounds 1 and 4.

(A) Structural model of the spastin-AAA-T692A-compound 1 complex. View of two compound 1 conformers from the entrance of the ATP pocket is shown (ribbon and stick representation). Simulated annealing composite omit map around the compound contoured at 3σ is shown (mFo – DFc, green mesh). Side chain of the mutated variability hot-spot residue (Ala-692) and the backbone atoms of a residue in the small AAA domain (Arg-662) are indicated. (B) Overlay of spastin-AAA-N527C-compound 1 (carbon atoms in orange) and spastin-AAA-T692A-compound 1 (carbon atoms in cyan) structural models. The side chains of the variability hotspot residue in the sensor-II motif (Thr-692 or Ala-692) are shown. (C) Overlay of AAA domains in spastin-AAA-WT-compound 4 (magenta) and two crystal forms of the spastin-AAA-T692A-compound 4 complex (crystal form A - blue; crystal form B - gray). The displacement of helices induced due to crystal packing is indicated by black arrows. (D) Overlay of spastin-AAA-T692A-compound 4 (crystal form A - blue; crystal form B - gray) and spastin-AAA-WT-compound 4 (magenta) complexes is shown. The N-loop, hinge, P-loop and sensor-II motifs are indicated. Overlays in panels (C) and (D) were generated in Pymol using the Align function with spastin-AAA-WT-compound 4 complex set as the target model. (E-G) Views of sensor-II variability hot-spot residue in the spastin-AAA-T692A-compound 4 (E - form A; F - form B) and spastin-AAA-WT-compound 4 (G) complexes. Distances between the sensor-II residue and the compounds are indicated.
Next, to examine the structural basis for how compound 4 inhibits the wild-type spastin and sensor-II mutant allele (T692A) with nearly identical potency we set up crystallization trials of the spastin-AAA-T692A and compound 4. Interestingly, the trials yielded two crystal forms of the spastin-T692A-compound 4 complex (forms A and B, Table S1). The structural model of the spastin-AAA-T692A construct in the crystal form A has nearly identical protein backbone conformation to that observed in the spastin-AAA-WT-compound 4 complex (Cα root-mean-square-deviation (RMSD) ~0.2 Å, WT vs form A, Figure 5C). In crystal form B, we observe small changes in the periphery of the protein in comparison to the wild-type complex (Cα RMSD ~1.9 Å, WT vs form B, Figure 5C). Interestingly, the inhibitor adopts nearly superimposable conformations in these three structural models (WT and T692A form A and B, Figure 5D). In both crystal forms of the spastin-AAA-T692A-compound 4 complex, the side chain of the sensor-II residue (Ala-692) is positioned >4 Å away from the methoxy group in compound 4’s fluorobenzoyl substituent (Figure 5E, F). In comparison, the structure of spastin-AAA-WT-compound 4 complex reveals that the compound’s methoxy group is proximal (<4 Å) to the sensor-II residue (Thr-692, Figure 5G). Taken together, our X-ray structures of inhibitor-spastin complexes show that mutations in variability hotspot residues do not disrupt the active site conformation and reveal how diaminotriazole-based inhibitors interact with spastin.
Discussion
In this study, we used inhibitor analyses against spastin alleles with engineered mutations in variability hotspot residues to identify key compound-target interactions for diaminotriazole-based spastin inhibitors. Our findings reveal distinct sets of resistance-conferring residues that predict different binding modes for two chemically-related compounds. Our X-ray crystallography models of the inhibitor-bound spastin complexes match the predicted binding modes. Together, these findings suggest a general approach, which we name RADD (Resistance Analysis During Design), for designing potent and selective inhibitors of AAA proteins.
Our RADD inhibitor design approach (Figure 6) involves the following steps. First, mutations of residues in the inhibitor binding site are engineered so that the steric and stereoelectronic properties are altered without disrupting enzyme activity. In particular, we generated active mutant alleles by replacing relatively less conserved residues in spastin’s active site with residues present at equivalent positions in other AAA proteins (Cupido et al., 2019). It will be important to compare the efficiency of our approach to other techniques, such as alanine scanning (Wells and McClendon, 2007), for identifying active mutant alleles. Second, the analyses of inhibitor activity against engineered mutant alleles are used to determine the key inhibitor-target interactions. These interactions can guide the selection of computational docking models as rank ordering of these models is often difficult without additional data that characterize inhibitor binding (Chen, 2015). Our findings indicate that the RADD approach leads to robust inhibitor-target models that can be useful for further optimization of chemical probes for AAA proteins.
Figure 6. Developing compound-target binding models using the RADD approach.

Schematic for the RADD approach. Mutations that maintain biochemical activity (e.g. ATPase) but alter the shape and electrostatics of the inhibitor binding site are identified (left). Analyses of inhibitor activity against the different mutant alleles reveal key inhibitor-protein contacts (middle) and guide the selection of robust binding models (right).
The active site of AAA proteins is composed of a set of conserved residues, which interact with the nucleotide, and a set of less conserved residues that lie in the interior (i.e. N-loop and P-loop variability hot-spots) or at the entrance of the nucleotide pocket (i.e. the sensor-II residues). In principle, compounds can achieve selective inhibition over related AAA proteins by interacting with all or subsets of these residues. Compounds that selectively inhibit spastin, such as the diaminotriazole-based compound 4 and the pyrazolylpyrrolopyrimidine-based spastazoline, interact with the N-loop and P-loop variability residues (glutamine and asparagine), which are unique to spastin in the AAA family. To engineer selective inhibitors of AAA proteins that share common N-loop and P-loop residues, compounds binding in a different orientation, such as the diaminotriazole-based compound 1, will be needed. In these cases, selectivity could be achieved by engineering interactions with other residues at the entrance of the active site (i.e. the sensor-II residues). For example, the AAA proteins katanin and VPS4B share identical N-loop and P-loop hot-spots (leucine and threonine, respectively, Figure S5A) but have different residues at the entrance of the nucleotide binding pocket (tyrosine in VPS4B in comparison to katanin’s leucine, Figure S5A) and sensor-II residues (threonine in katanin and serine in VPS4B, Figure S5A). Using the RADD approach could help identify and prioritize chemical scaffolds that interact with specific residues in a desired target to guide rational design of selective inhibitors.
During inhibitor design many analogs are often generated to optimize and improve the initial starting scaffold. X-ray crystallography is commonly employed to confirm the binding modes of a handful of these analogs in the process. Interestingly, analyses of inhibitor-target complexes suggest that even closely related compounds can bind their target differently and often reorient in the binding site. For example, moving a lactate substituent from ortho to meta position in an indole-based PPARγ induces a ~180° turn over of the ligand in the binding site (Bruning et al., 2007). Similarly, closely related diphenylamide-based inhibitors of p38 MAP kinase bind to the active site in different orientations and can even bind to different conformations of the kinase (Angell et al., 2008). Our results for two diaminotriazole-based inhibitors, which bind the active site of spastin in distinct orientations, suggest that inhibitor activity analyses against protein mutant alleles can help differentiate these binding modes. In some cases, the binding modes of inhibitor analogs can be successfully predicted using computational docking methods (Shoichet and Kobilka, 2012), but these methods can be challenging when the inhibitor binding sites undergo large conformational rearrangements, as it is the case with AAA proteins (Puchades et al., 2017). Testing inhibitors against active mutant alleles can provide experimental validation for poses predicted by computational methods, and help determine the compound interactions and orientation in the active site of the target protein. Therefore, the RADD approach could be useful for monitoring compound binding at multiple stages of inhibitor development, especially in the case of protein families for which high-resolution structures of inhibitor-protein complexes are lacking.
Resistance to chemical inhibitors is a common complication in the clinic (e.g. cancer or HIV therapy) and mutations conferring resistance often emerge in drug targets (Gorre et al., 2001; Wensing et al., 2017). In particular, these resistance-conferring mutations commonly map to residues in drug binding sites that are in direct contact with the inhibitor (Anderson et al., 2015; Furman et al., 2014; Gorre et al., 2001; Wacker et al., 2012). For example, mutations identified in cells resistant to CB-5083 inhibitor of VCP/p97, which entered clinical trials, map to the active site in one of the two AAA domains of the protein (Anderson et al., 2015). Notably, a mutation in the sensor-II variability hot-spot residue (T688A, human VCP/p97) shows >60-fold change in compound potency. Strategies to overcome resistance involve the development of compounds that retain potent binding to the mutant alleles. Our data suggest that using RADD could help identifying active-site inhibitors that have different patterns of resistance-conferring mutations. Integrating this information at early stages of compound design could guide development of drug combinations for which resistance is less likely to arise.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tarun M Kapoor (kapoor@rockefeller.edu).
Experimental model and subject details
Escherichia coli BL21 Rosetta™ (DE3) pLysS cells used for protein expression in this study were grown in 4l Erlenmeyer flasks each containing ~1l LB media supplemented with 34 mg/l chloramphenicol and either 100 mg/l of ampicillin or 50 mg/L kanamycin (for details see STAR Methods - Protein expression and purification).
Method details
Quantification and statistical analysis
Statistical parameters including the number of replicates, mean ± standard deviation (s.d.) or range are reported in respective figure legends.
Data and software availability
The crystal structures of spastin-inhibitor complexes solved in this paper have been deposited in the RCSB protein data bank (PDB: 6P10, 6P11, 6P12, 6P13, 6P14).
Vectors for recombinant protein expression
Vector for expression of D. melanogaster wild-type protein (aa 209–758) was obtained from Dr. Antonina Roll-Mecak (pDEST15-spastin-D.melanogaster) (Roll-Mecak and Vale, 2008). Constructs for expression of mutant spastin alleles (Q488V, N527C and T692A; aa 209–758) were described in earlier work (Cupido et al., 2019).
Constructs for expression of D. melanogaster AAA domain (spastin-AAA, aa 445–758) was described earlier work (Pisa et al., 2019). Briefly, the coding sequence for spastin AAA domain (aa 445–758) was PCR amplified (CloneAmp HiFi, Clontech) from pDEST15-spastin-D.melanogaster (WT, Q488V, N527C or T692A) and cloned into pDEST15 vector using AscI and ApaI sites to generate N-terminal GST-fusion constructs. Full coding sequence for all recombinant constructs was verified by Sanger sequencing. Sequence alignments were performed using ClustalW algorithm in MacVector software (MacVector, Inc).
Protein expression and purification
D. melanogaster spastin (spastin, aa 209–758) wild-type and mutant constructs (Q488V, N527C and T692A) were expressed and purified as described earlier (Roll-Mecak and Vale, 2008). Xl-katanin, Hs-FIGL1 and Hs-VPS4B were purified and assayed as described earlier (Cupido et al., 2019).
D. melanogaster spastin AAA domain (spastin-AAA, aa 445–758) wild-type and mutant constructs (Q488V, N527C and T692A) were expressed and purified described earlier (Pisa et al, 2019). Briefly, E.coli Rosetta cells were grown in Miller’s LB medium to O.D.600 ~ 0.8 at 30°C, chilled to 18°C and the protein expression was indu ced overnight (14–16 hours, 0.5 mM IPTG). The cells were resuspended in lysis buffer (50 mM Tris.HCl pH = 8.0, 400 mM NaCl, 10 mM MgCl2, 1 mM PMSF, 5 mM β-mercaptoethanol, 0.001% triton-X and 1 tablet of Roche cOmplete protease inhibitor cocktail per 50 ml of buffer), supplemented with 0.1 µg / ml lysozyme and 5 U / ml benzonase and lysed using Emulsiflex-C5 homogenizer (Avestin, 5–10 cycles at 10–15 kPsi). The lysate was spun at 45,000 rpm for 45 min at 4°C in Ti-45 rotor using Beckman Coulter Optima LE-80K ultracentrifuge. The clarified supernatant was incubated with glutathione agarose resin for 1 hr. The beads were washed with ~300 ml of wash buffer (50 mM Tris.HCl pH = 8.0, 400 mM NaCl, 10 mM MgCl2, 5 mM β-mercaptoethanol) and the protein was eluted by on-bead cleavage using PreScission protease made in-house. Cleaved protein was diluted two-fold with low salt buffer (20 mM Tris.HCl pH = 8.2, 40 mM NaCl, 5 mM MgCl2, 5 mM β-mercaptoethanol), loaded onto HiTrap™ Q HP (GE Healthcare) and eluted with high salt buffer (20 mM Tris.HCl pH = 8.0, 500 mM NaCl, 5 mM MgCl2, 5 mM β-mercaptoethanol). All constructs eluted around 180–200 mM salt. The protein was further purified over a HiLoad 16/60 Superdex 200 column (GE Healthcare) in size exclusion buffer (20 mM HEPES-NaOH pH = 7.4, 100 mM NaCl, 5 mM MgCl2, 2 mM TCEP, 25 mM (NH4)2SO4) and the desired fractions were concentrated using Amicon 30 kDa MWCO device to 10–20 mg ml−1. The PreScission cleavage did not leave any non-native residues at the N terminus.
Analyses of ATPase activity
Steady-state ATPase activity of spastin (aa 209–758) was determined using the NADH-coupled assay. The time course of fluorescence decrease corresponding to the consumption of the NADH was measured using a Synergy NEO Microplate Reader (λex = 340 nm, 440 nm emission filter). The fluorescence values were plotted against time and fit by linear regression to obtain a rate of fluorescence decrease. The ATPase rate was calculated from the background (no ATP) corrected rates of fluorescence decrease using ADP calibration curve. Percent inhibition of the ATPase activity was calculated by normalizing the rate of background-corrected fluorescence decrease in the presence of compounds to DMSO control. All compound stocks were made using DMSO. Final conditions for spastin wild type and mutants (50–75 nM): 25 mM HEPES-KOH pH 7.5, 225 mM KCl, 20 mM (NH4)2SO4, 5 mM MgCl2, 2.5 mM DTT, 0.005% triton-X, BSA 0.1 mg/ml, 175–200 µM NADH, 1 mM phosphoenolpyruvate, 15–30 U/ml lactate dehydrogenase, 15–30 U/ml pyruvate kinase.
Determining enzyme parameters
Enzyme parameters K1/2, kcat and Hill coefficients (h) for the recombinant enzymes were determined by fitting the rates to the Michaelis-Menten equation for cooperative enzymes (1) using Prism v.6.0 (GraphPad Software Inc).
| (1) |
In this equation Vmax denotes the maximum ATPase rate, h is the Hill coefficient, x denotes ATP concentration and K1/2 is the ATP concentration required for half maximal enzyme rate. We adopted the K1/2 annotation to distinguish this constant from the Michaelis constant KM. Catalytic turnover number (kcat) was calculated by dividing the Vmax value by the concentration of enzyme in the assay.
Compound IC50 calculation
For each experiment the measured activity was plotted against concentration of compound and the data were fit using a sigmoidal dose-response curve equation (2) in Prism to determine the IC50. The values from at least three independent experiments were averaged and standard deviations were calculated.
| (2) |
Molecular Dynamics (MD)
Molecular dynamics calculations and ensemble docking were performed as described earlier (Cupido et al., 2019). Briefly, the MD system was set up for D. melanogaster spastin (PDB: 3B9P) using the Protein preparation wizard in Maestro® (version 11.6.013, Schrödinger, New York). First, all non-protein atoms in the PDB file were kept, and the protein loops with no coordinates were modeled using Uniprot: Q8I0P1 as reference sequence. Hydrogen atoms were added to the protein structure, and the protonation states of ionizable residues were assigned for pH 7.0, using the protein preparation wizard in Maestro. This structure was neutralized with KCl ions, additional KCl ions were placed to reproduce a salt concentration of 0.15 M and the system was solvated with a orthorhombic box of simple point charge (SPC) water molecules using the desmond package in Maestro (Desmond Molecular Dynamics System, version 4.6, D. E. Shaw Research).
To generate the MD atomic trajectory, the standard system relaxation protocol provided in Maestro® was used, followed by a 250 ns of NPT (Isothermal-Isobaric ensemble) molecular dynamics (300 K, 1.01325 bar), during which atomic positions were recorded every 50 ps. To maintain the pressure and temperature of the system, isotropic position scaling and the Nosè-Hoover chain thermostat methods 42 were used with relaxation times of 2 ps and 1 ps, respectively. A RESPA integrator scheme was employed (step: 2.0 fs for bonded interactions; 2 fs for van der Waals and short-range electrostatic interactions; 6 fs for long-range electrostatic interactions). Short-range electrostatic interactions were cut off at 9.0 Å.
Ensemble Docking procedure
All non-protein atoms were removed from the 5000 molecular dynamics frames and the spastin structures from each frame were aligned to remove translational and rotational movement of the macromolecule. Starting from 2 ns of MD trajectory, 1 every 10 frames were selected (97 structures total) as representative of the entire trajectory. To remove overlap of the atoms’ van der Waals radii, these structures were minimized using 20 iterations of the Polak-Ribière conjugated gradient method or until the root mean square of the gradient of the energies was < 0.05 kJ mol-1 Å-1, using OPLS2003e force field and a cut-off of 20 Å for the electrostatic interactions and 8 Å for the van der Waals interactions.
Molecular docking was performed on the 97 structures (see above) using the XGlide script in Schrodinger. The major tautomer at pH 7.0 of compound 1 (4-((5-amino-1-(2,6-difluorobenzoyl)-1H-1,2,4-triazol-3-yl)amino)benzene sulfonamide) was calculated using the LigPrep panel in Maestro® and energy minimized. A box of 22 Å was placed, centered approximately at the Gln-488 residue of spastin, and compound 1 (4-((5-amino-1-(2,6-difluorobenzoyl)-1H-1,2,4-triazol-3-yl)amino)benzenesulfonamide) was docked into the 97 spastin structures using Glide XP and the OPLS2003e forcefield with Schrodinger’s GlideScore multi-ligand scoring function44. Receptor grids were generated using a scaling factor of 0.9 for the van Der Waals radii for atom with partial charge <0.25 (absolute value). For ligand docking, the options REWARD_INTRA_HBONDS, SAMPLE_RINGS, HBOND_DONOR_AROMH were set true, the FORCEPLANAR options was set false, and a scaling factor of 0.9 was applied to the van der Waals radii for atom with partial charge <0.15 (absolute value). The binding poses shown in Figure 1g and Figure S2 represent five distinct poses with the highest docking score.
Differential Scanning Fluorimetry
These experiments were carried out on a C1000 Touch Thermal cycler CFX-96 instrument (GE Healthcare). Spastin AAA-WT, -Q488V, -N527C or T692A (aa 445–758) recombinant constructs were diluted to a final concentration of 7–10 µM in 25 mM HEPES-KOH (pH 7.5), 225 mM KCl, 5 mM MgCl2, 20 mM (NH4)2SO4, 2.5 mM DTT, 0.005% triton-X and assayed in a 96-well plate (Hard-shell® HSP9665 Bio-Rad). Compounds were added (0.8 µM to 100 µM, 1–2.5% DMSO). SYPRO® Orange (Sigma S5692, excitation 490 nm, emission 590 nm) was used at 1:500 to 1:1000 dilution. The temperature was linearly increased with a step of 0.5 °C for 55 minutes, from 25 °C to 95 °C and fluorescence readings were taken at each interval. Melting temperatures were recorded as the minimum value of the first derivative of the fluorescence vs temperature curves.
Co-crystallization of Drosophila spastin-AAA constructs (aa 445–758) with compounds
Co-crystallization of spastin with compound 1.
Spastin-AAA constructs (N527C or T692A) were concentrated to ~20 mg/ml and mixed in 1:1 ratio with a saturated solution of compound 1 in size exclusion buffer (20 mM HEPES-NaOH pH = 7.4, 100 mM NaCl, 5 mM MgCl2, 2 mM TCEP, 25 mM (NH4)2SO4). The saturated solution of compound 1 was prepared by dissolving the compound in size exclusion buffer and spinning at 14000 rpm for 10 minutes. The protein-compound complex was then mixed with reservoir solution (0.1 M Na-acetate pH 5–7, 2% PEG-4000, 15% 2-methyl-2,4-pentanediol (MPD)) using the hanging drop method and the crystals were allowed to form at 14–18 °C. The crystals were additionally soaked for ~1 hour in reservoir solution supplemented with 20% MPD and 200 µM of compound 1 prior to freezing in liquid nitrogen.
Co-crystallization of spastin with compound 4.
Spastin-AAA constructs (WT or T692A) were concentrated to 10–15 mg/ml and compound 4 was added (final compound concentration ~200 µM) and the mixture was incubated on ice for ~0.5 h. The protein-compound complex was mixed with reservoir solution (0.1 M Na-acetate pH 5–7, 2% PEG-4000, 15% MPD) supplemented with ~200 µM of compound 4 and the crystals were allowed to form at 14–18 °C using the hanging drop method. Obtained crystals were cryo-protected in a solution of the reservoir supplemented with 20% MPD prior to flesh freezing in liquid nitrogen.
Data collection and refinement
Data for all structures were collected at NSLS-II 17-ID-1 and 2 (AMX and FMX) beamlines at Brookhaven National Laboratory at 100 K, and processed using HKL2000 software (v7.16.1., HKL Research, Inc). The resolution cutoff was determined by considering values of Rmerge (<0.5 in the high resolution shell) and I/σ (>1.5 in the high resolution shell). The structures were solved by molecular replacement with PHASER (McCoy et al., 2007) as part of the PHENIX software package (Adams et al., 2010). All structures were solved from independently obtained data for individual crystals by molecular replacement using apo spastin (3B9P) as a template. The model was rebuilt and refined using COOT (Emsley et al., 2010) and phenix.refine software (Afonine et al., 2012). In all crystals the asymmetric unit contained a single molecule of the protein with the hexameric symmetry embedded in the crystal lattice. During refinement ~10% of reflections were assigned as Rfree for each dataset by the phenix.refine software and not used in any further refinement steps. Data collection and refinement statistics are shown in Table S1. Simulated annealing composite omit maps were computed in PHENIX (Adams et al., 2010). Figures were generated in MacPyMOL (Schrodinger LLC, http://www.pymol.org) and UCSF Chimera (Pettersen et al., 2004).
Chemical synthesis information
Solvents and reagents were purchased from Sigma-Aldrich. Reactions were run in round-bottom flasks or glass vials and stirred with Teflon-coated magnetic stir bars. Solvent evaporation was performed under stream of nitrogen or on rotary evaporator (Büchi) under reduced pressure. Reactions were monitored by thin layer chromatography or LC-MS (Waters Acquity H-Class UPLC/MS with QDa mass spectrometer using water + 0.1% formic acid (solvent A) and acetonitrile + 0.1% formic acid (solvent B). Proton nuclear magnetic resonance spectra were recorded on Bruker Avance II spectrometer (600 MHz). Observed proton absorptions are reported as δ units of parts per million (ppm) relative to tetramethylsilane (δ 0.0). Multiplicities are reported: s (singlet), d (doublet), t (triplet), m (multiplet) or br.s. (broad signal). Coupling constants are reported as a J value in Hertz (Hz).
Abbreviations used in this section: THF = tetrahydrofuran; EtOAc = ethyl acetate; HCl = hydrochloric acid; eq = equivalents; rt = room temperature.
JNJ-7706621 (compound 1) was purchased from MedChemExpress (Cat. No.: HY-10329). Procedures for synthesis of compounds 2, 3 and 4 were similar and based on previously published methods (Malerich et al, 2010). As an example, synthesis of compound 4 is summarized.
Synthesis of compound 4, 3-((5-amino-1-(2-fluoro-6-methoxybenzoyl)-1H-1,2,4-triazol-3-yl) amino)-N-methylbenzamide. Step 1. 3-amino-N-methylbenzamide (0.5 g, 3.3 mmol) and diphenyl cyanocarbonimidate (1.1 eq, 0.87 g, 3.7 mmol) were dissolved in THF (3 mL) in a round bottom glass flask and refluxed overnight. Solvent was evaporated under reduced pressure using Rotary evaporator and the residue was treated with diethylether (2×10 ml) and triturated. Crude product was used directly in the next step. MS-ESI [M+1]: expected 295.3, found 295.2. Step 2. Without further purification, 0.9 g of this intermediate (~3.1 mmol) was dissolved in 1M hydrazine solution in THF (1.5 eq, 4.6 mmol, 4.6 ml) while stirring at rt. The reaction was heated at reflux for 4 h while stirring and then cooled down to rt. The excess solvent was evaporated under reduced pressure, and the product of the reaction was washed with diethylether (3×10 ml) and dried. MS-ESI [M+1]: expected 233.2, found 233.3. Step 3. 30 mg (0.13 mmol) of the crude intermediate from step 2 was suspended in ~1 ml of pyridine and 1.2 eq of 2-fluoro-6-methoxybenzoyl chloride were added dropwise while stirring on ice. The reaction was allowed to warm up to room temperature and the solvent was evaporated under flow of nitrogen. The compound 4 was separated from the crude mixture by silica gel chromatography in hexane/EtOAc. To obtain HCl salt, the purified compound 4 was dissolved in ethylacetate and precipitated by addition of 37% HCl and dried.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial strains | ||
| Escherichia coli BL21 Rosetta™ (DE3) pLysS | Merck | Cat#70954 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ampicillin, sodium salt | Sigma-Aldrich | Cat#A9518–25G |
| Kanamycin sulfate | Sigma-Aldrich | Cat#K1377–25G |
| Chloramphenicol | Fisher Scientific | Cat#BP904–100 |
| 1,4-dithiotreitol (DTT) | GoldBio | Cat#DTT25 |
| β-Nicotinamide adenine dinucleotide, reduced disodium salt (NADH) | Sigma-Aldrich | Cat#N7410 |
| Adenosine 5′-triphosphate disodium salt hydrate | Sigma-Aldrich | Cat#A6419 |
| Bovine Serum Albumin, fraction V | Bioworld | Cat#22070008 |
| Dimethylsulfoxide (DMSO) | Sigma-Aldrich | Cat#276855 |
| L-Glutathione reduced | Sigma-Aldrich | Cat#G6529 |
| D-Lactic Dehydrogenase (LDH) | Sigma-Aldrich | Cat#L3888 |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat#P7626 |
| Phospho(enol)pyruvic acid monopotassium salt (PEP) | Sigma-Aldrich | Cat#P7127 |
| Pyruvate Kinase from rabbit muscle (PK) | Sigma-Aldrich | Cat#P9136 |
| Roche cOmplete, EDTA-free Protease Inhibitor | Sigma-Aldrich | Cat#11873580001 |
| Tris(2-carboxyethyl)phosphine HCl (TCEP) | GoldBio | Cat#TCEP10 |
| Triton™ X-100 | Sigma-Aldrich | Cat#X100–100ML |
| Miller’s LB medium | Formedium | Cat#LMM105 |
| Isopropyl-beta-D-thiogalactoside (IPTG) | GoldBio | Cat#I2481 |
| Glutathione Agarose Resin | GoldBio | Cat#G-250–10 |
| Sodium acetate trihydrate | Sigma-Aldrich | Cat#71188–250G |
| 2-methyl-2,4-pentanediol (MPD) | Hampton Research | Cat#HR2–627 |
| Polyethylene glycol 4000 (PEG-4000) | Hampton Research | Cat#HR2–529 |
| JNJ-7706621 | MedChemExpress | Cat#HY-10329 |
| Deposited Data | ||
| Spastin (template for molecular replacement) | (Roll-Mecak et al. 2008) | PDB: 3B9P |
| Spastin-N527C-compound 1 structure | This paper | PDB: 6P10 |
| Spastin-T692A-compound 1 structure | This paper | PDB: 6P11 |
| Spastin-WT-compound 4 structure | This paper | PDB: 6P12 |
| Spastin-T692A-compound 4 structure (form A) | This paper | PDB: 6P13 |
| Spastin-T692A-compound 4 structure (form B) | This paper | PDB: 6P14 |
| Recombinant DNA | ||
| pDEST15-Dm-spastin (D. melanogaster spastin) | (Roll-Mecak et al. 2008) | N/A |
| pDEST15-Dm-spastin-AAA (D. melanogaster spastin) | (Pisa et al. 2019) | N/A |
| pMAL-c5x-p60 (X. laevis katanin p60) | (Loughlin et al. 2011) | N/A |
| pQE9-His-p97(wt) (M. musculus VCP/p97) | (Meyer et al. 2000), Addgene #14666 | https://www.addgene.org/14666/ |
| pDEST15-FIGL1 (H. sapiens FIGL1) | (Cupido et al, 2019) | N/A |
| pET28-His-SUMO-VPS4B (H. sapiens VPS4B) | (Cupido et al, 2019) | N/A |
| Software and Algorithms | ||
| GraphPad Prism version 6.0 | GraphPad Software | https://www.graphpad.com/ |
| Phenix, version 1.12–2829 | (Adams et al, 2010) | https://www.phenix-online.org/download/ |
| HKL-2000, v7.16.1 | HKL Research, Inc | http://www.hkl-xray.com/ |
| Coot 0.8.8 | (Emsley et al, 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Schrodinger Maestro, release 2016–2 | Schrodinger LLC | https://www.schrodinger.com/ |
| MacPYMOL, v1.8.2.3 Enhanced for Mac OS X | Schrodinger LLC | https://pymol.org/2/ |
| UCSF Chimera package version 1.8.1 | UCSF | https://www.cgl.ucsf.edu/chimera/ |
| MacVector (version 17.0.4) | MacVector, Inc | https://macvector.com |
Significance.
Robust models for inhibitor-target binding are critical for the design of potent and selective chemical probes and drugs. We describe an approach, which we name RADD (Resistance Analysis During Design) that employs data from tests of compound activity against constructs with engineered mutations to identify inhibitor-target interactions. Here, we apply RADD to predict the binding mode for a diaminotriazole-based chemical inhibitor of spastin, a microtubule-severing AAA protein required for cell division and organelle dynamics. We also identify a more potent analog and use our approach to show that it binds the spastin’s active site in a different orientation in comparison to the starting compound. The distinct binding modes of these diaminotriazole-based compounds predicted by RADD match the high-resolution models we generated using X-ray crystallography. Together, our data provide a framework for how mutations in active sites of proteins could facilitate inhibitor design.
Highlights:
Testing compounds against engineered mutant alleles of target leads to binding model.
High-resolution X-ray structures confirm predicted models.
Cognate inhibitor resistance-conferring mutations are identified.
This approach can be applied to multiple chemical scaffolds.
Acknowledgments
T.M.K. is grateful to the NIH/NIGMS for funding (GM98579 and R35 GM130234–01). T.C. was supported by the EMBO Long-Term fellowship for Postdoctoral studies and by the Kestenbaum Award for Research in Neurodegenerative Diseases. R.P. and N.H.J. were supported by the Tri-Institutional PhD Program in Chemical Biology and the Rockefeller University Graduate Program. N.H.J. was also supported by the NIH T32 GM115327 Chemistry-Biology Interface Training Grant. We thank Dr. Seth Darst, Dr. Elizabeth Campbell and Dr. Michael Oldham for their generous help and advice with crystallography data processing and refinement. We thank Dr. Michael Grasso for help with purification of VPS4B protein. We also thank Dr. Antonina Roll-Mecak (NIH) and Dr. Rebecca Heald (UC Berkeley) for providing plasmids. We are grateful to Dr. Carolina Adura and the staff at the Rockefeller University High-Throughput and Spectroscopy Resource Center and Dr. Deena Oren at the Rockefeller University Structural Biology Center for assistance. The use of the Formulator robot and the Rigaku/MSC microMax 007HF in the Rockefeller University Structural Biology Resource Center was made possible by grants 1S10RR027037–01 and 1S10RR022321–01 from the National Center for Research Resources of the NIH. This research used the 17-ID-1 and 17-ID-2 beamlines of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.
Footnotes
Declaration of Interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, et al. (2015). Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 28, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angell RM, Angell TD, Bamborough P, Bamford MJ, Chung CW, Cockerill SG, Flack SS, Jones KL, Laine DI, Longstaff T, et al. (2008). Biphenyl amide p38 kinase inhibitors 4: DFG-in and DFG-out binding modes. Bioorg Med Chem Lett 18, 4433–4437. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. (2016). 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 351, 871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleichert F, Botchan MR, and Berger JM (2017). Mechanisms for initiating cellular DNA replication. Science 355. [DOI] [PubMed] [Google Scholar]
- Boyaci H, Shah T, Hurley A, Kokona B, Li Z, Ventocilla C, Jeffrey PD, Semmelhack MF, Fairman R, Bassler BL, et al. (2016). Structure, Regulation, and Inhibition of the Quorum-Sensing Signal Integrator LuxO. PLoS Biol 14, e1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JB, Chalmers MJ, Prasad S, Busby SA, Kamenecka TM, He Y, Nettles KW, and Griffin PR (2007). Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure 15, 1258–1271. [DOI] [PubMed] [Google Scholar]
- Chen YC (2015). Beware of docking! Trends Pharmacol Sci 36, 78–95. [DOI] [PubMed] [Google Scholar]
- Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, et al. (2011). Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A 108, 4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell JW, Lindon C, Luzio JP, and Reid E (2009). Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic 10, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland RA (2013). Evaluation of enzyme inhibitors in drug discovery : a guide for medicinal chemists and pharmacologists, 2nd edn (Hoboken, N.J.: Wiley; ). [PubMed] [Google Scholar]
- Cupido T, Pisa R, Kelley ME, and Kapoor TM (2019). Designing a chemical inhibitor for the AAA protein spastin using active site mutations. Nat Chem Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AM, St-Gallay SA, and Kleywegt GJ (2008). Limitations and lessons in the use of X-ray structural information in drug design. Drug Discov Today 13, 831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupeux F, Rower M, Seroul G, Blot D, and Marquez JA (2011). A thermal stability assay can help to estimate the crystallization likelihood of biological samples. Acta Crystallogr D Biol Crystallogr 67, 915–919. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott JA, Noble ME, and Johnson LN (2012). The structural basis for control of eukaryotic protein kinases. Annu Rev Biochem 81, 587–613. [DOI] [PubMed] [Google Scholar]
- Erlanson DA, Davis BJ, and Jahnke W (2019). Fragment-Based Drug Discovery: Advancing Fragments in the Absence of Crystal Structures. Cell Chem Biol 26, 9–15. [DOI] [PubMed] [Google Scholar]
- Ferguson FM, and Gray NS (2018). Kinase inhibitors: the road ahead. Nat Rev Drug Discov [DOI] [PubMed] [Google Scholar]
- Firestone AJ, Weinger JS, Maldonado M, Barlan K, Langston LD, O’Donnell M, Gelfand VI, Kapoor TM, and Chen JK (2012). Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 484, 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, Racchumi J, Xu G, Wu H, Ma J, et al. (2014). Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med 370, 2352–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, and Sawyers CL (2001). Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880. [DOI] [PubMed] [Google Scholar]
- Kawashima SA, Chen Z, Aoi Y, Patgiri A, Kobayashi Y, Nurse P, and Kapoor TM (2016). Potent, Reversible, and Specific Chemical Inhibitors of Eukaryotic Ribosome Biogenesis. Cell 167, 512–524 e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Connolly PJ, Huang S, Wetter SK, Lu Y, Murray WV, Emanuel SL, Gruninger RH, Fuentes-Pesquera AR, Rugg CA, et al. (2005). 1-Acyl-1H-[1,2,4]triazole-3,5-diamine analogues as novel and potent anticancer cyclin-dependent kinase inhibitors: synthesis and evaluation of biological activities. J Med Chem 48, 4208–4211. [DOI] [PubMed] [Google Scholar]
- Magnaghi P, D’Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, et al. (2013). Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol 9, 548–556. [DOI] [PubMed] [Google Scholar]
- Malerich JP, Lam JS, Hart B, Fine RM, Klebansky B, Tanga MJ, and D’Andrea A (2010). Diamino-1,2,4-triazole derivatives are selective inhibitors of TYK2 and JAK1 over JAK2 and JAK3. Bioorg Med Chem Lett 20, 7454–7457. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Frost A, and Sundquist WI (2018). Structures, Functions, and Dynamics of ESCRT-III/Vps4 Membrane Remodeling and Fission Complexes. Annu Rev Cell Dev Biol 34, 85–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pisa R, Cupido T, and Kapoor TM (2019). Designing Allele-Specific Inhibitors of Spastin, a Microtubule-Severing AAA Protein. J Am Chem Soc [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohler R, Krahn JH, van den Boom J, Dobrynin G, Kaschani F, Eggenweiler HM, Zenke FT, Kaiser M, and Meyer H (2018). A Non-Competitive Inhibitor of VCP/p97 and VPS4 Reveals Conserved Allosteric Circuits in Type I and II AAA ATPases. Angew Chem Int Ed Engl 57, 1576–1580. [DOI] [PubMed] [Google Scholar]
- Puchades C, Rampello AJ, Shin M, Giuliano CJ, Wiseman RL, Glynn SE, and Lander GC (2017). Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puleo DE, Kucera K, Hammaren HM, Ungureanu D, Newton AS, Silvennoinen O, Jorgensen WL, and Schlessinger J (2017). Identification and Characterization of JAK2 Pseudokinase Domain Small Molecule Binders. ACS Med Chem Lett 8, 618–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll-Mecak A, and Vale RD (2008). Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 451, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NP, Tran C, Lee FY, Chen P, Norris D, and Sawyers CL (2004). Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 305, 399–401. [DOI] [PubMed] [Google Scholar]
- Shoichet BK, and Kobilka BK (2012). Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol Sci 33, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WK, Odzorig T, Jin W, and Xia D (2019). Structural Basis of p97 Inhibition by the Site-Selective Anticancer Compound CB-5083. Mol Pharmacol 95, 286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J, and Meyer H (2018). VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol Cell 69, 182–194. [DOI] [PubMed] [Google Scholar]
- van Zundert GCP, Hudson BM, de Oliveira SHP, Keedy DA, Fonseca R, Heliou A, Suresh P, Borrelli K, Day T, Fraser JS, et al. (2018). qFit-ligand Reveals Widespread Conformational Heterogeneity of Drug-Like Molecules in X-Ray Electron Density Maps. J Med Chem 61, 11183–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietri M, Schink KO, Campsteijn C, Wegner CS, Schultz SW, Christ L, Thoresen SB, Brech A, Raiborg C, and Stenmark H (2015). Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 522, 231–235. [DOI] [PubMed] [Google Scholar]
- Wacker SA, Houghtaling BR, Elemento O, and Kapoor TM (2012). Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat Chem Biol 8, 235–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA, and McClendon CL (2007). Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Wendler P, Ciniawsky S, Kock M, and Kube S (2012). Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta 1823, 2–14. [DOI] [PubMed] [Google Scholar]
- Wensing AM, Calvez V, Gunthard HF, Johnson VA, Paredes R, Pillay D, Shafer RW, and Richman DD (2017). 2017 Update of the Drug Resistance Mutations in HIV-1. Top Antivir Med 24, 132–133. [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, and Mandelkow EM (2013). Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J 32, 2920–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystal structures of spastin-inhibitor complexes solved in this paper have been deposited in the RCSB protein data bank (PDB: 6P10, 6P11, 6P12, 6P13, 6P14).