

Summary
During neural tube closure and spinal cord development, many cells die in both the central and peripheral nervous systems (CNS and PNS, respectively). However, myeloid-derived professional phagocytes have not yet colonized the trunk region during early neurogenesis. How apoptotic cells are removed from this region during these stages remains largely unknown. Using live imaging in zebrafish, we demonstrate that neural crest cells (NCC) respond rapidly to dying cells and phagocytose cellular debris around the neural tube. Additionally, NCCs have the ability to enter the CNS through motor exit point transition zones and clear debris in the spinal cord. Surprisingly, NCC phagocytosis mechanistically resembles macrophage phagocytosis and their recruitment towards cellular debris is mediated by interleukin-1β. Taken together, our results reveal a role for NCCs in phagocytosis of debris in the developing nervous system before the presence of professional phagocytes.
Keywords: neural crest cells, phagocytosis, macrophages, neurogenesis, zebrafish, neural development
eTOC blurb
Neural crest cells clear cellular debris left behind from cell death that occurs during early embryonic development, thus revealing an unexpected source of phagocytic activity prior to the arrival of professional phagocytes in the developing zebrafish nervous system.
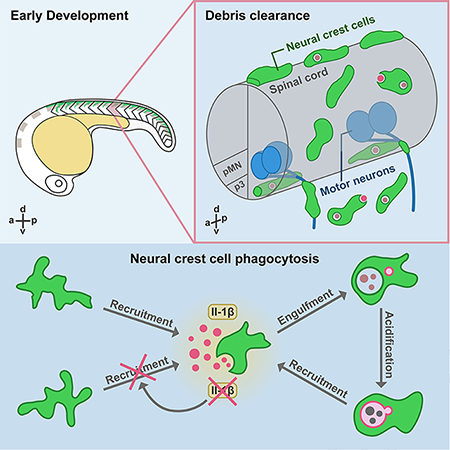
Graphical Abstract
Introduction
Cell death is prevalent during development in multicellular organisms and is important for the removal of unnecessary cells and tissues as well as for the correction of developmental errors (Arya and White, 2015). During vertebrate embryogenesis, dying cells are present during neural tube closure and spinal cord development (Schlüter, 1973; Weil, Jacobson and Raff, 1997; Buss, Sun and Oppenheim, 2006; Massa et al., 2009). Therefore, a rapid and efficient phagocytic response is crucial for the clearance of this debris. In mouse, chicken and zebrafish, a group of primitive macrophages derived from the yolk sac migrate into the developing brain, differentiate into microglia, and contribute to ongoing neurogenesis by clearing apoptotic debris (Cuadros et al., 1993; Herbomel, Thisse and Thisse, 1999; Bertrand et al., 2013). Yolk-sac-derived macrophages, however, do not infiltrate the trunk of developing embryos until the formation of the circulatory system at embryonic day (E)10.5 in mice (McGrath et al., 2003; Bertrand et al., 2013; Stremmel et al., 2018) and 35 hours post fertilization (hpf) in zebrafish (Herbomel, Thisse and Thisse, 1999, 2001). However, neural tube closure and motor axon pathfinding start at E9 and 16 hpf in mice and fish, respectively (Beattie, 2000; Wang et al., 2014), prior to the colonization of macrophages in the trunk region. Although cell death during neural tube closure has been extensively studied, very little is known about the clearance of debris in the trunk during the earliest stages of neural development.
NCCs are a conserved cell lineage in vertebrate embryos that differentiate into a variety of cell types, including pigment cells and neurons and glia of the PNS (Roberto Mayor and Theveneau, 2013). After neural tube closure, a subset of trunk NCCs delaminate and migrate ventral-medially around the perimeter of the spinal cord in segmentally-organized streams (R. Mayor and Theveneau, 2013; Vega-Lopez, Cerrizuela and Aybar, 2017). When motor neurons in the CNS send their axons out of the spinal cord, NCCs reach motor exit points (MEP) and migrate ventrally along motor axons (R. Mayor and Theveneau, 2013; Vega-Lopez, Cerrizuela and Aybar, 2017). Previous studies in mice demonstrate a correlation between the presence of apoptotic debris and migratory NCCs (Massa et al., 2009; Yoshida et al., 2013). Therefore, given the spatiotemporal correlations of NCC migration, the presence of dead cells, and the absence of macrophages at early developmental stages, we hypothesized that migratory NCCs may have the ability to clear cellular debris during early developmental stages.
Here, using live imaging in zebrafish, we demonstrate that NCCs are phagocytic during early development. We show that NCCs migrate away from their segmentally-restricted paths towards dead cells and phagocytose debris. They rapidly internalize this debris and form phosphatidylinositol 3-monophosphate (PI(3)P)-enriched engulfment vesicles that mature into lysosome-associated-membrane-protein-1-positive (Lamp1+) acidic phagolysosomes. Intriguingly, NCCs even migrate into the ventral spinal cord through MEP transition zones (TZ) and phagocytose debris in the CNS. Interestingly, both cell corpses and phagocytic NCCs express interleukin-1β (Il-1β), and blocking Il-1β signaling with an Il-1β receptor antagonist or a Caspase-1 inhibitor significantly impaired NCC recruitment to cellular debris after injury, demonstrating that activated Il-1β is required for NCC recruitment. Together, our findings reveal a role for migratory NCCs in phagocytosing debris in the CNS and PNS during early development.
Results
NCCs, but not macrophages, colonize the trunk during early development
Although primitive macrophages are generated in the first wave of yolk sac hematopoiesis as early as 15 hours post fertilization (hpf) in zebrafish (Lieschke et al., 2001), previous studies demonstrate that these early macrophages do not populate the trunk until 35 hpf or later (Herbomel, Thisse and Thisse, 1999, 2001). To understand the temporal dynamics of macrophage colonization of the trunk, we crossed Tg(mpeg1:GFP) transgenic fish, where mpeg1 regulatory sequences drive GFP expression in macrophages and microglia, with Tg(olig2:DsRed) transgenic fish, where olig2 regulatory sequences label spinal cord neurons and glia, and counted the number of mpeg1+ macrophages in the trunk of these embryos at 24, 36, and 48 hpf (Figure 1A). In these studies, we observed an increase macrophage number in both the yolk and trunk between 24 and 36 hpf, and then again from 36 to 48 hpf (Figure 1A & B). To better characterize macrophage distribution, we plotted their individual locations into 2D histograms and found that at 24 hpf, macrophages resided in the yolk extension (Figure 1C, n = 9 fish). By 36 hpf, most macrophages remained in and around the yolk extension, while a few of started to appear near the notochord and the ventral spinal cord, which is referred to as the dorsal trunk below (Figure 1C, n = 9 fish). Ultimately, by 48 hpf, macrophages fully colonized the trunk with an equal distribution in both the yolk extension and dorsal trunk (Figure 1C, n = 9 fish).
Figure 1. NCCs, but not macrophages, colonize the trunk region during early development.

(A) Trunk lateral views of Tg(olig2:DsRed);Tg(mpeg1:GFP) embryos at 24, 36 and 48 hpf. Arrowheads denote mpeg1+ macrophages. (B) Macrophage quantification in a 0.27 mm2 region in the trunk of embryos at 24, 36 and 48 hpf (mean ± SD). (C) 2D distribution of macrophages in the trunk of embryos at 24, 36 and 48 hpf (n = 9 fish in each condition). Diagram on the left shows the region quantified. All images used for quantification are aligned based on the position of the ventral edge of the spinal cord (dashed line). (D) Lateral view of a Tg(sox10:TagRFP);Tg(bactin2:Gal4);Tg(UAS:secA5-YFP) embryo at 20 hpf. Blue shading denotes dorsal trunk. Filled arrowheads denote dead cells in the dorsal trunk. Open arrowheads denote debris in the ventral trunk/yolk extension. (E) Quantification of the percentage of somites with secA5+ cells in 20 and 36 hpf embryos (n = 6/9 fish for 20/36 hpf). Dashed lines mark the ventral edge of the spinal cord. Scale bars, 50 μm.
Because we did not observe any macrophages in the dorsal trunk in 24 hpf embryos, we sought to determine whether dead cells were present at this stage. To visualize apoptotic cells in live animals, we created a transgenic line, Tg(bactin2:Gal4), which has Gal4 factor the control of β-actin regulatory sequences and crossed it with Tg(UAS:secA5-YFP) zebrafish, to drive expression of secreted human Annexin V protein fused to YFP (secA5-YFP), which binds to the membrane of apoptotic cells (van Ham et al., 2010). In Tg(bactin2:Gal4);Tg(UAS:secA5-YFP) embryos at 20 hpf, we observed secA5+ cells in the yolk extension and intermediate cell mass (Figure 1D) (Sarvothaman et al., 2015; Stachura and Traver, 2016). Additionally, we also found secA5+ cells in the dorsal trunk (Figure 1D) at both 20 and 36 hpf (Figure 1D). Quantification of the percentage of somites with secA5+ cells in the dorsal trunk at 20 and 36 hpf (n = 65/56 somites in 6/9 embryos for 20/36 hpf) showed that the presence of dead cells was robust and consistent among embryos at these stages (Figure 1E). Using a newly created line, Tg(sox10:TagRFP), where TagRFP is expressed in NCCs under sox10 regulatory sequences, we found that sox10+ NCCs were spatially correlated with apoptotic cells near the spinal cord (Figure 1D), which is consistent with a previous study demonstrating the presence of apoptotic cells around the neural tube during NCC migration in mice (Massa et al., 2009). These data demonstrate that migratory NCCs colonize the dorsal trunk in zebrafish when macrophages are absent but apoptotic debris is present.
The association of migratory NCCs with dead cells at early developmental stages (Figure 1D) prompted us to investigate whether NCCs expressed genes implicated in phagocytosis. To do this, we performed RNA-sequencing of foxd3+/sox10+ NCCs collected from the trunk of Gt(foxd3:mCherry);Tg(sox10:mEGFP) embryos at 36 and 72 hpf, which represent developmental stages before and after the full colonization of macrophages into the trunk (Figure 1C) (Simoes-Costa and Bronner, 2015). Using Gene Ontology (GO) enrichment analysis and KEGG pathway analysis (Ashburner et al., 2000; Kanehisa and Goto, 2000; Yu et al., 2012; The Gene Ontology Consortium, 2019), we found that 36 hpf NCCs were highly enriched in cellular components related to debris clearance such as the endoplasmic reticulum, lysosomes, endosomes and V-ATPase (Figure S1A & B). Lysosome and endosome pathways were also up-regulated (Figure S1C & D). Moreover, many genes required for phagocytosis were highly expressed in 36 hpf NCCs (Figure S1E & F; see also Table S2) (Ashburner et al., 2000; Villani et al., 2019). Taken together, we hypothesize that NCCs have the capacity to clear debris at developmental stages before the appearance of professional phagocytes.
NCCs migrate away from their segmental stream of origin to engulf cellular debris
To monitor the migration and behavior of NCCs and investigate whether they contribute to debris clearance during development, we performed in vivo, time-lapse imaging. To do this, we used Tg(sox10:nls-Eos) embryos which express nuclear-localized Eos to track the movement of individual NCCs and a Tg(nkx2.2a:nls-mCherry) transgene to label lateral floorplate cells as a reference for the location of the ventral spinal cord (Figure 2A & B) (Kucenas, Snell and Appel, 2008). In time-lapse movies of Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryos from 19 to 40 hpf, we observed that the majority of newly-delaminated trunk NCCs migrated ventrally around the neural tube, reaching MEP TZs in the middle of each hemi-segment, and then migrated along motor axons (Figure 2A & B), as previously described (Banerjee et al., 2011; Vega-Lopez, Cerrizuela and Aybar, 2017). We also observed approximately 1 to 2 NCCs per hemi-segment undergo apoptosis during their migration (Figure 2B, Movie S1). When cell death occurred, individual NCCs migrated away from their innate streams and moved towards these dying cells, and NCCs that associated with dying cells were observed both dorsal and ventral to MEP TZs (Figure 2B & Movie S1; see also Figure S2A & Movie S2).
Figure 2. A subset of NCCs engulf cellular debris in the developing PNS.

(A) Schematic diagram showing NCC migration in a 22 hpf embryo. (B) Images from a 21 h time-lapse movie of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo starting at 19 hpf. Asterisks denote the location of MEP TZs. Arrowheads denote dead NCCs. NCCs that migrated towards debris are outlined in yellow and cyan. (C) Images from a 35 hpf Tg(sox10:Eos) embryo treated with LysoTracker Deep Red. Boxed region is magnified on the right. Arrows denote an engulfment vesicle. Region outlined with dashed box is magnified in D. (D) Quantification of fluorescent intensity across the engulfment vesicle shown in C. (E) Measurement of diameters of NCC engulfment vesicles (mean ± SD, n = 95 vesicles). (F) Images from a 21 h time-lapse movie starting at 19 hpf of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo treated with LysoTracker Red DND-99. A NCC denoted (arrows) migrated towards and engulfs LysoTracker+ debris (arrowheads). Note that the white color of the engulfment vesicle indicates co-localization of that NCC and the corpse. Dashed line marks the ventral edge of the spinal cord. (G) Quantification of the number of engulfing NCCs per hemi-segment (mean ± SD, n = 17 fish, 3–6 somites per fish). Scale bars, 20 μm.
We next examined whether NCCs that migrated towards dying cells engulfed them. In order to better visualize NCC behavior, we used a Tg(sox10:Eos) line to label NCC cytoplasm with Eos, a photoconvertible protein that when exposed to ultraviolet (UV) light, shifts its emission wavelength from a neutral state emitting green fluorescence (516 nm), to an anionic state emitting red fluorescence (581 nm) (Prendergast et al., 2012). Recent studies demonstrate that the Eos protein settles in a pH-dependent equilibrium between these two states: the neutral form (green) favors lower pH (6–8), and the anionic state (red) favors physiological pH (8–10) (Berardozzi et al., 2016; Turkowyd et al., 2017). Considering the acidification that occurs during apoptosis and inside engulfment vesicles (Gottlieb, 1996; Levin, Grinstein and Canton, 2016), we hypothesized that healthy and apoptotic NCCs could be differentially labeled by the two forms of Eos protein. To verify this, we exposed Tg(sox10:Eos) embryos to UV light at 20 hpf, photoconverting existing Eos protein in NCCs to the anionic state (red), and performed time-lapse imaging. As expected, NCCs labeled with high levels of photoconverted (red) Eos switched to yellow upon apoptosis (Figure S2B & C). Yellow NCC corpses were then quickly engulfed by neighboring red NCCs which formed large engulfment vesicles with green debris inside (Figure 2C; see also Figure S2B & C). Moreover, these engulfment vesicles were positive for LysoTracker Deep Red (Figure 2C & D), a dye that labels acidic organelles (Fogel, Thein and Mariani, 2012), and measured approximately 2 to 8 μm in diameter (Figure 2E, n = 95 vesicles), similar to the size of phagosomes in professional phagocytes (Champion, Walker and Mitragotri, 2008). From these data, we conclude that migratory NCCs engulf apoptotic neighbors and form acidic engulfment vesicles.
We next asked whether these NCCs could engulf non-NCC debris. To do this, we treated Tg(sox10:Eos);Tg(nkx2.2a:nls-mCherry) embryos with 20 μM LysoTracker Red DND-99, which stains both dying cells and acidic organelles (Fogel, Thein and Mariani, 2012), and time-lapse imaged from 19 to 40 hpf. In these movies, we observed NCCs migrate away from motor axons towards LysoTracker+ debris located between two spinal motor nerves in neighboring somites (Figure 2F; Movie S3). In one instance, we observed a NCC first send dynamic protrusions towards LysoTracker+ debris and then quickly engulf it (Figure 2F; Movie S3). When NCCs tried to engulf large debris, like dead muscle fibers, they exhibited a behavior similar to frustrated phagocytosis, where one or many NCCs circled the dead cell for hours without successful engulfment (Figure S2D) (Cannon and Swanson, 1992). In spite of being highly active in response to cellular debris, NCCs do not engulf frequently in healthy embryos. Quantification of NCCs with engulfment vesicles showed that 1 to 2 cells per hemi-segment between 24 to 36 hpf had engulfment vesicles (Figure 2G, n = 17 fish), which is about 5% to 10% of all NCCs in a hemi-segment. Taken together, these data demonstrate that NCCs can migrate away from motor axons and engulf debris.
Engulfing NCCs have distinct migratory patterns but are not lineage restricted
Previous studies show that trunk NCCs migrate in segmentally-organized streams along motor axons in each somite (Figure 2A for schematic diagram) (Banerjee et al., 2011; Vega-Lopez, Cerrizuela and Aybar, 2017). Our data, however, demonstrate that individual NCCs can migrate away from motor axons towards dead cells (Figure 2B & F; see also Figure S2A). To better understand these unique migratory patterns, we tracked the migration of individual NCCs with or without engulfment vesicles for 10 hours after they reached MEP TZs and observed that NCCs with vesicles did not migrate along axons (5/5) as most trunk NCCs did (5/5) (Figure 3A). Occasionally, we also observed them cross somite boundaries and interact with axons and debris in neighboring somites (2/5) (Figure 3A; See also Figure S2A). Plotting the migration data from these movies into a directional histogram showed that 90% of the NCCs without vesicles were located 60–90° below the edge of the ventral spinal cord, along axonal bundles (Figure 3B, n = 10). In contrast, the majority of NCCs that contained engulfment vesicles were distributed in a 30°−150° angle below the ventral spinal cord with only 20% of them following axonal bundles (Figure 3B, n = 10). Similarly, when we traced these NCCs 5 hours prior to when they reached MEP TZs, we found that 92% NCCs without engulfment vesicles came from a 30–60° angle dorsal to the spinal cord (Figure 3B, n = 13), as previously described (Honjo and Eisen, 2005). In contrast, those with engulfment vesicles reached MEP TZs from various directions, ranging from 30–150° (Figure 3B, n = 10). Interestingly, we never observed NCCs migrate dorsally after reaching the MEP TZ.
Figure 3. Migratory patterns and lineage tracing of engulfing NCCs.

(A) Trajectories of NCCs with (red, n = 5 cells) or without (blue, n = 5 cells) engulfment vesicles during a 10 h period after they reached MEP TZs (yellow dots). (B) Circular histogram showing directions of NCCs 5 h before (top, n = 13 cells for each group) or 10 hours after (bottom, n = 10 cells for each group) they reached MEP TZs (yellow dot). (C&D) Lineage tracing of engulfing NCCs that were photoconverted at 20 hpf (arrowheads). Arrows in C denote engulfment vesicles. Color-coded arrowheads in D denote daughter cells derived from individual photoconverted NCCs. (E) Quantification of the lineage of engulfing NCCs photoconverted at 20 hpf (n = 56 cells). Grey background shows location of the neural tube and motor axons in A & B. MEP, motor exit point. Scale bars, 20 μm.
We next asked whether these NCCs were lineage restricted. To answer this question, we photoconverted engulfing NCCs in Tg(sox10:Eos) embryos at 20 hpf and tracked them using in vivo imaging. At 50 hpf, we found these NCCs had differentiated into pigment cells (Figure 3C & D) and motor nerve-associated glia (Figure 3D; see also Figure S2E). Among all the NCCs we were able to track (49/56 cells), 79.5% migrated laterally to the skin and had a pigment cell morphology, 16.3% migrated medially and associated with motor nerves where Schwann cells and dorsal root ganglion (DRG) neurons and glia reside, and 4.1% died during our imaging (Figure 3E). These data indicate that engulfing NCCs are not lineage restricted.
NCCs form PI(3)P+ and Lamp1+ phagosomes after engulfment
Because NCCs engulf cellular debris in a manner similar to macrophages, we sought to determine whether NCC engulfment was mechanistically similar to phagocytosis. To investigate this, we determined whether NCC engulfment vesicles acidified progressively, which is required for the degradation of internalized debris (Kinchen and Ravichandran, 2008). To visualize the acidification process of NCC vesicles, we treated Tg(sox10:Eos) embryos with neutral red, a pH-sensitive dye that stains acidic lysosomes (Herbomel, Thisse and Thisse, 2001). In these embryos, we observed NCCs form phagocytic cups that then developed into circular vacuoles (Figure 4A; Movie S4). Approximately 40 minutes after the formation of engulfment vesicles, neutral red staining gradually appeared inside the newly formed compartments (Figure 4A; Movie S4). A similar acidification process was also observed using LysoTracker (Figure S3A). Therefore, we conclude that NCC engulfment vesicles are progressively acidified in a manner similar to phagosome maturation in professional phagocytes.
Figure 4. The formation of PI(3)P+ and Lamp1+ phagosomes after NCC engulfment.

(A) Images from a time-lapse movie starting at 19 hpf in a Tg(sox10:nls-Eos) embryo treated with neutral red at 16 hpf. Arrows denote a NCC engulfment vesicle that was gradually stained by neural red. (B) Images from a time-lapse movie of a 24 hpf Tg(sox10:Gal4);Tg(UAS:GFP-FYVE) embryo showing the fusion of scattered PI(3)P signals (arrowheads) with an engulfment vesicle (arrows). Schematic diagrams are shown below. (C) Images of an engulfment vesicle from a time-lapse movie of a 28 hpf Tg(sox10:Gal4);Tg(UAS:GFP-FYVE) embryo treated with neutral red. (D) Quantification of PI(3)P+ vesicle formation and normalized neutral red brightness over time (mean ± SD, n = 6 vesicles). Dashed line indicates time points with a small sample size. (E) Images from a time-lapse movie of a Tg(sox10:TagRFP);Tg(sox10:lamp1-GFP) embryo starting at 20 hpf. Arrowheads denote gradual enrichment of Lamp1-GFP in a NCC engulfment vesicle. (F) Orthogonal views of a Lamp1-GFP+ vesicle in a NCC. Scale bars, 10 μm in A, B, E and F; 2 μm in C.
Next, we asked whether these vesicles acidify similarly to that of phagosomes in professional phagocytes. To investigate this, we first determined if NCC engulfment vesicles were PI(3)P+, which is the characteristic phosphoinositide transferred to the membrane of newly formed phagosomes via fusion with early endosomes, and is crucial during early phagosome progression (Fratti et al., 2001; Vieira et al., 2001). To visualize PI(3)P activity in NCCs, we crossed Tg(sox10:Gal4) adults to a PI(3)P reporter line, Tg(UAS:GFP-FYVE) (Rasmussen et al., 2015). Time-lapse imaging of these embryos from 20 to 40 hpf revealed labeling of dynamic PI(3)P+ signals resembling early endosomes in NCCs (Figure 4B). When NCCs formed phagocytic cups and engulfed debris, PI(3)P signals fused with engulfment vesicles within 2 minutes after the sealing of NCC vesicles, resulting in the formation of bright PI(3)P+ spherical structures reminiscent of early phagosomes (Figure 4B) (Levin, Grinstein and Canton, 2016). After 4 to 12 minutes, PI(3)P signals quickly defused from engulfment vesicles and turned back into small spherical structures (Figure 4B). In addition to trunk NCCs, we also observed the formation of PI(3)P+ engulfment vesicles in cranial NCCs (Figure S3C). This rapid and transient accumulation of PI(3)P to NCC engulfment vesicles closely resembles PI(3)P dynamics during phagosome maturation in cultured macrophages (Kamen et al., 2008).
During phagosome maturation in macrophages, PI(3)P depletion is accompanied by the fusion of early phagosomes with late endosomes, which have a more acidic luminal pH (Levin, Grinstein and Canton, 2016). To determine whether NCC phagosomes have similar temporal regulation of PI(3)P activity and acidification, we treated Tg(sox10:Gal4);Tg(UAS:GFP-FYVE) embryos with neutral red to visualize acidic organelles and imaged from 20 to 40 hpf. In these movies, we observed large PI(3)P+ vesicles become neutral-red+ upon the loss of the PI(3)P signal (Figure 4C and Movie S5). Quantification of PI(3)P+ vacuole formation and neutral red fluorescent intensity over time showed that neutral red accumulation began during the depletion of PI(3)P on the NCC engulfment vesicle membrane (Figure 4D, n = 6 vesicles), which is similar to the transition from early to late phagosomes in macrophages (Levin, Grinstein and Canton, 2016). Moreover, acidification of NCC engulfment vesicles occurred within 10 minutes after the initial recruitment of PI(3)P (Figure 4D, n = 6 vesicles). This rapid acidification resembles phagosome pH regulation in alternatively activated M2 macrophages (Galli, Borregaard and Wynn, 2011; Canton et al., 2014).
Finally, to determine if NCC phagosomes fuse with lysosomes and ultimately mature into phagolysosomes similar to those described in macrophages (Levin, Grinstein and Canton, 2016), we investigated whether these vesicles were enriched in Lamp1, an integral membrane protein crucial for phagolysosome maturation (Binker et al., 2007; Huynh et al., 2007; Levin, Grinstein and Canton, 2016). To do this, we created a transgenic line, Tg(sox10:lamp1-GFP), which expresses an established Lamp1-GFP fusion protein in NCCs (Rasmussen et al., 2015). Time-lapse imaging of these embryos from 20 to 40 hpf showed that Lamp1-GFP was gradually enriched in NCC engulfment vesicles (Figure 4E), indicating a fusion process with lysosomes similar to phagolysosome biogenesis in macrophages (Binker et al., 2007; Huynh et al., 2007). Interestingly, we found that Lamp1-GFP formed a variety of ultrastructures within NCC phagocytic vesicles cells (Figure 4F; see also Figure S3B), suggesting complex membrane topologies, consistent with published results showing multiple layers of Lamp1+ membrane inside lysosomes of cultured cells (System and Klumperman, 2015). Taken together, we conclude that NCCs can migrate towards dying cells and phagocytose debris, which results in the formation of PI(3)P+ and Lamp1+ phagosomes. We therefore call these cells phagocytic NCCs.
Cell ablation induces NCC phagocytosis
Our data demonstrate that NCCs phagocytose debris like professional phagocytes. Therefore, we next asked whether cell death triggers the NCC phagocytic response. To investigate this, we ablated single migratory NCCs in the dorsal trunk using a pulsed nitrogen dye laser in Tg(sox10:Gal4);Tg(UAS:GFP-FYVE) embryos at 24 hpf (Figure 5A; Movie S6). Time-lapse imaging showed that multiple NCCs around the ablation site formed PI(3)P+ phagosomes starting approximately 10 minutes post ablation (mpa) (Figure 5A; Movie S6). The NCC vesicle number peaked at 30 to 40 mpa and reduced to baseline levels by 50 to 60 mpa (Figure 5B; Movie S6). Notably, not every NCC responded to laser-induced damage and sometimes a single NCC formed multiple PI(3)P+ vesicles (Figure 5A). Quantification of NCC phagocytosis within a 100 μm2 region around the ablation site showed that in 100 mpa, 52.9% of the NCCs formed PI(3)P+ phagosomes and 4.6% of them engulfed at least 4 times (Figure 5C, n = 5 fish).
Figure 5. Laser ablation-induced NCC phagocytosis.

(A) Images from a time-lapse movie of a Tg(sox10:Gal4);Tg(UAS:GFP-FYVE) embryo after ablation of a NCC (outlined in yellow) at 24 hpf. Arrowheads denote NCC engulfment vesicles. Cyan and red arrowheads denote newly-formed and pre-existing vesicles, respectively, in each frame, correlating with the quantification in B. A NCC that engulfed twice is outlined in cyan. (B) Quantification of phagosome formation in panel A. Top panel shows phagosome count per frame fitted with a Gaussian curve (R square = 0.7075). Bottom panel shows the duration of each PI(3)P+ vesicle. (C) Quantification of the number of times NCCs phagocytose in a 100 μm2 region around the ablation site in 100 mpa (mean ± SD, n = 5 fish). (D & E) Quantification of the number of NCC phagosomes per frame before and after cell ablation (D), or distal and proximal to ablation sites (E) (n = 50 frames). mpa, minutes post ablation. Scale bar, 10 μm.
To confirm that the induction of phagosome formation in NCCs was directly correlated to ablation, we quantified the number of PI(3)P+ vesicles in each time frame for 100 minutes, before and after the ablation within the same 100 μm2 region around the ablation site (Figure 5D). We found that more phagosomes formed post-ablation as compared to pre-ablation (Figure 5D). Similarly, the majority of NCC phagosomes formed in a 100 μm2 region proximal to the ablation site (Figure 5E). Therefore, we conclude that NCCs phagocytose debris after cell ablation.
NCCs and macrophages clear debris during distinct developmental stages
NCCs phagocytose dead cells before macrophage colonization of the trunk. Therefore, we wanted to investigate the temporal dynamics of NCC and macrophage-mediated debris clearance during development. To do this, we time-lapse imaged Tg(sox10:Eos) and Tg(mpeg1:GFP) embryos and quantified phagocytic events between 22 and 44 hpf. Our results showed that before 36 hpf, the majority of debris in the trunk was cleared by NCCs, whereas macrophages phagocytosed more actively at later stages (Figure S4A & B). This time difference correlated with the presence of active trunk NCC migration between 16 to 36 hpf and increased abundance of macrophages in the dorsal trunk after 36 hpf (Figure S4C, n = 7 fish).
In these movies, we also noticed a difference in the migratory speed between NCCs and macrophages. Quantification of the average velocity of phagocytic NCCs and macrophages showed that phagocytic NCCs migrated 0.3 μm/min (Figure S4D, n = 13 cells), while macrophages migrated much more rapidly, with an average velocity of 1.3 μm/min (Figure S4D, n = 10 cells), identical to the macrophage migratory speed reported in previous studies (Grabher et al., 2006). This difference in cell motility indicates that the phagocytic NCCs identified in our study are not macrophages.
NCCs migrate into the spinal cord and clear CNS debris
Our findings demonstrate that NCCs phagocytose cellular debris around the spinal cord. However, during these developmental stages, there is also a significant amount of debris inside the spinal cord (van Ham et al., 2010; Shklover, Levy-Adam and Kurant, 2015) and how this debris is cleared, is not well understood. A previous finding from the lab showed that NCCs can migrate into the ventral spinal cord after reaching motor axons (Smith et al., 2016). This led us to hypothesize that cell death in the spinal cord recruits NCCs into the CNS to phagocytose debris. To investigate this, we imaged MEP TZs in Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryos from 22 to 40 hpf and captured NCCs migrating into the spinal cord (Figure 6A; see also Movie S7). Once inside the CNS, these cells migrated both around and underneath floorplate cells (Figure 6A; see also Movie S7).
Figure 6. NCCs migrate into the spinal cord and phagocytose CNS debris.

(A) Images from a time-lapse movie of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo starting at 20 hpf. Arrows denote a NCC nuclei crossing into the CNS, which is magnified below. (B) Quantification of the ratio of CNS-located NCCs per hemi-segment after ablation of 2 floorplate cells (mean ± SD, n = 8/10 fish for control/ablated). (C) Images of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry);Tg(mnx1:mCerulean) embryo at 20 hpf after ablation of 2 nkx2.2a+ floorplate cells. Left: z projection; middle: 90-degree rotated image; right: schematic view of the rotated image, illustrating the locations of floorplate cells (fp), motor neurons (mn) and NCCs in the CNS (cN) and PNS (pN). (D) Quantification of the ratio of CNS-located NCCs in embryos with radial glial ablation (n = 5 fish) or DMSO-treated controls (n = 6 fish). The data includes NCCs in 3 hemi-segments per fish. (E) Quantification of the number of NCCs entering the CNS per hemi-segment in DMSO and MTZ-treated embryos within a 20 h time window (mean ± SD). (F & G) Quantification of the number of NCCs per hemi-segment (F) and the ratio of proliferating NCCs (G) in DMSO and MTZ-treated embryos between 20 to 40 hpf (mean ± SD). (H) Distribution of the length of time NCCs spent in the CNS (n = 15 cells). (I) Images from a time-lapse movie of a Tg(sox10:Eos);Tg(gfap:NTR-mCherry) embryo. Arrows denote a NCC engulfment vesicle filled with radial glia debris. Dashed lines mark the ventral edge of the spinal cord. Scale bars, 10 μm.
We then asked whether inducing cell death in the spinal cord would increase NCC migration into the CNS. To do this, we generated Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry);Tg(mnx1:mCerulean) embryos and laser ablated two nkx2.2a+ floorplate cells at 20 hpf. We then imaged these embryos for 20 hours post ablation (hpa) and found more NCCs in the spinal cord when compared to control embryos (Figure 6B & C; see also Movie S7). Interestingly, these CNS-located NCCs always migrated underneath motor neuron cell bodies (Figure 6A & C; Movie S7). These findings demonstrate that cell death in the CNS induces NCC migration into the spinal cord.
Considering that laser ablation might cause damage and lead to increased NCC entry, we examined the effect of drug-induced CNS cell death. We made use of Tg(gfap:NTR-mCherry) embryos, which when treated with a pro-drug, Metronidazole (MTZ), results in the specific loss of radial glial cells (Johnson et al., 2016; Smith et al., 2016). In Tg(sox10:nls-Eos);Tg(gfap:NTR-mCherry) embryos treated with 15 mM MTZ in 1% DMSO starting from 10 hpf, we observed more NCCs in the spinal cord between 20 to 40 hpf when compared to DMSO-treated controls (Figure 6D & E, n = 6/5 fish for mock/drug). This increase in the number of CNS-located NCCs was attributed to cell migration because the total number of NCCs and their proliferation rate remained unchanged (Figure 6F & G, n = 6/5 fish for mock/drug). However, when we delayed MTZ treatment to 30 hpf and imaged from 40 to 60 hpf, we rarely observed NCCs entering the CNS (Figure 6E, n = 6/7 fish for mock/drug), suggesting that NCCs can only migrate into the spinal cord before 40 hpf, corresponding with the time frame when they are highly phagocytic (Figure S4A & B). Based on these results, we conclude that NCCs can be recruited into the spinal cord by inducing cell death in the CNS.
To understand the behavior of these CNS-located NCCs, we traced 15 individual NCCs after they entered the CNS and found that 60% of them spent approximately 2 hours in the ventral spinal cord before exiting back into the periphery, while 13.3% NCCs stayed inside for more than 10 hours (Figure 6H). When we traced all the CNS-located NCCs until the end of our movies, we found that 75.0% of them ultimately returned to the periphery (n = 52 cells).
Lastly, to examine whether CNS-located NCCs also phagocytosed cellular debris, we treated Tg(sox10:Eos);Tg(gfap:NTR-mCherry) embryos with MTZ starting at 10 hpf and imaged from 20 to 40 hpf. In these movies, we observed single NCCs migrate into the spinal cord through the MEP TZ, form engulfment vesicles filled with mCherry+ debris, and bring them back into the periphery (Figure 6I). Taken together, we conclude that NCCs can migrate into the spinal cord in response to CNS cell death, phagocytose debris.
Il-1β recruits NCCs to debris after damage
Interleukin-1β (Il-1β) is a proinflammatory cytokine secreted by macrophages and CNS neurons and glia after injury and infection and is crucial for the recruitment of professional phagocytes (Srinivasan, 2004; Lopez-Castejon and Brough, 2011; van der Vaart et al., 2014; Tsarouchas et al., 2018). Therefore, we asked whether Il-1β signaling was involved in NCC recruitment.
Il-1β is cleaved from pro-Il-1β by activated Caspase-1 (or zebrafish homolog Caspase A) before it can be secreted as a pro-inflammatory cytokine (Masumoto et al., 2003; Lopez-Castejon and Brough, 2011; Vojtech et al., 2012). To investigate the role of Il-1β signaling in NCC recruitment, we treated Tg(sox10:nls-Eos);Tg(olig2:DsRed) embryos at 12 hpf with 75 μM Ac-YVAD-cmk, a Caspase-1 inhibitor (Masumoto et al., 2003; Tsarouchas et al., 2018), to block the secretion of Il-1β, then laser ablated two olig2+ motor neurons at 20 hpf, and performed time-lapse imaging (Figure 7A & C). In DMSO-treated controls, we observed NCC recruitment to a 50 μm x 50 μm region surrounding the MEP TZs where motor neurons were ablated within 4 to 8 hpa (Figure 7C & D). In contrast, fewer NCCs were recruited after ablation in YVAD-treated embryos (Figure 7D). It is important to note that YVAD treatment alone did not influence NCC migration (Figure 7D). Because YVAD inhibits the cleavage of pro-Il-1β (Mathiak et al., 2000), we conclude that bioactive Il-1β is required for NCC recruitment during phagocytosis.
Figure 7. NCC recruitment towards damage is mediated by Il-1β signaling.

(A & B) Illustrations of experimental design in D and E, respectively. (C) Images from a time-lapse movie of a Tg(sox10:nls-Eos);Tg(olig2:DsRed) embryo before and after ablation of two motor neurons (asterisks). Dashed box indicates the region where the numbers of NCCs are counted in D and E. (D) Quantification of the number of NCCs in DMSO or YVAD-treated embryos after ablation (Numbers in the legends denote the number of embryos quantified. Same for E). (E) Quantification of the number of NCCs in control embryos or embryos treated with IL-1Ra after motor neuron ablation. (F) Images from 20 hpf Tg(sox10:TagRFP) embryos labeled with an Il-1β antibody; NCC protrusions (arrows), NCC debris (filled arrowheads), and NCC engulfment vesicles (open arrowheads). (G) In 20 hpf Tg(sox10:TagRFP) embryos labeled with an Il-1β antibody, NCC vesicles are filled with Il-1β+ debris (arrowheads). (H) Images from Tg(sox10:TagRFP);il1b:GFP-F embryos at 20 hpf. Open arrowheads denote phagocytic NCCs that are il-1β+. Filled arrowheads denote il-1β+ debris inside a NCC vesicle. Scale bars, 20 μm in C, 10 μm in F, G & H.
To further validate the function of Il-1β in NCC recruitment, we treated Tg(sox10:nls-Eos);Tg(olig2:DsRed) embryos with an IL-1 receptor antagonist (IL-1Ra) to block the detection of Il-1β by NCCs (Vojtech et al., 2012), and performed the laser ablation experiment described above (Figure 7B). Strikingly, we found that NCC recruitment was abolished with IL-1Ra treatment, while their normal migration remained intact in embryos without ablation (Figure 7E). Based on these results, we conclude that Il-1β signaling is critical for the recruitment of NCCs towards debris.
Next, to determine the source of Il-1β used by NCCs, we labeled 20 hpf Tg(sox10:TagRFP) embryos with an antibody specific to Il-1β and found Il-1β+ puncta on the tip of NCC extensions (Figure 7F), suggesting that NCCs may either express Il-1β or associate with Il-1β+ debris. To clarify this, we ablated 2 to 3 NCCs in each hemi-segment of Tg(sox10:TagRFP) embryos and then labeled for Il-1β. Interestingly, we observed co-localization between Il-1β labeling and a variety of NCC structures, including NCC corpses, engulfment protrusions (Figure 7F), and debris engulfed by NCCs (Figure 7G). These results demonstrate that dying cells and their debris contain high levels of Il-1β.
Professional phagocytes secrete Il-1β in response to injury or infection to recruit more phagocytes (Madej et al., 2017). Therefore, we next examined whether NCCs also express Il-1β. We genetically labeled the membrane of cells expressing Il-1β by transiently injecting an established il1b:GFP-F construct into Tg(sox10:TagRFP) embryos (Nguyen-Chi et al., 2014). Under physiological conditions at 20 hpf, we observed that a subset of NCCs with engulfment vesicles was GFP-F+ (Figure 7H). We also detected robust GFP-F signal inside NCC engulfment vesicles, supporting our observations that NCCs phagocytose Il-1β+ cellular debris (Figure 7H). Based on these results, we conclude that Il-1β is expressed by both dying cells and NCCs.
Discussion
During development, dead cells must be removed via phagocytosis (Arya and White, 2015). Because myeloid-derived professional phagocytes are not always present or sufficient for clearing developmental debris, previous studies demonstrate that many non-professional phagocytes contribute to debris clearance during embryogenesis, including glia in the embryonic Drosophila CNS, zebrafish skin epithelial cells, satellite glial precursors and neural progenitor cells in mice, as well as retinal cells in the developing human retina (Kurant et al., 2008; Wu et al., 2009; Lu et al., 2011; Francisco-Morcillo et al., 2014; Rasmussen et al., 2015). In this study, we demonstrate that NCCs also phagocytose debris in the developing PNS and CNS using a mechanism similar to macrophage phagocytosis.
NCCs as a distinct type of phagocyte
Other non-professional phagocytes are mostly stationary and have limited phagocytic ability (Shklover, Levy-Adam and Kurant, 2015). However, our results show that NCCs are highly motile and respond to cell death and injury more that 100 μm away from their innate paths, which also enables them to phagocytose many types of debris, including other NCCs, CNS neurons and glia, and muscle cells.
In our movies, we noted another difference between NCCs and other non-professional phagocytes: the nearest NCC was not always the first cell to respond to cell death. In contrast, studies demonstrate that other non-professional phagocytes show less heterogeneity and mostly phagocytose adjacent debris (Shklover, Levy-Adam and Kurant, 2015). This finding raises an interesting possibility: only a subgroup of NCCs respond to cell death and phagocytose debris, which is also supported by the distinct migratory pattern of these NCCs. However, results from our cell ablation assay provide evidence that the phagocytic population can be drastically increased by inducing damage, suggesting that a large proportion of NCCs are capable of phagocytosis. Additionally, our lineage tracing results demonstrate that phagocytic NCCs become a variety of NCC derivatives. Taken together, we propose an alternative explanation to the heterogeneity in NCC phagocytic response: early NCCs have variations in their sensitivities to phagocytic signals and under physiological conditions, only the most sensitive cells respond to debris during development, while under injury or disease conditions, other NCCs with low sensitives can be activated aid in clearance.
Developmental role of NCC phagocytosis
Although cell death during neural tube closure has been studied extensively (Schlüter, 1973; Mirkes, 2002; Massa et al., 2009; Yamaguchi et al., 2011), the mechanism of debris clearance remains unknown. Our study demonstrates that migratory NCCs clear cellular corpses in the trunk of developing embryos. In addition to trunk NCCs, we also observed cranial NCCs phagocytosing debris (Figure S3C). Interestingly, live imaging in mice showed that apoptotic cells are cleared both during and after closure of the cranial neural tube, prior to the maturation of professional phagocytes (Yamaguchi et al., 2011). Given that massive migration of cranial NCCs in mice occurs during neurulation (Theveneau and Mayor, 2012), we hypothesize that NCCs also remove dead cells during neural tube closure in mice.
Our study raises another intriguing question: whether inhibition of NCC phagocytosis causes developmental defects? Unfortunately, many targetable components in the phagocytosis pathway are also required for cell migration, which limits us from blocking NCC phagocytosis without impairing their motility and development. Given that the level of NCC phagocytosis can be significantly increased after damage, we hypothesize that it is important for correcting developmental errors. Recently, a study in zebrafish bubblebrain (blb) mutants demonstrated that Slc37a2, a solute carrier transporter, is required for phagosomal shrinkage in professional phagocytes and maintenance of their phagocytic ability (Villani et al., 2019). Since slc37a2 is highly expressed in NCCs (Figure S1F) and we observed phagosomal shrinkage in NCC phagocytosis (data not shown), manipulation of Slc37a2 could be a potential future approach to block NCC phagocytosis and examine its developmental role. Moreover, although migratory NCCs efficiently phagocytose dead cells, whether other non-professional phagocytes exist and contribute to debris clearance during early developmental stages remain to be investigated.
Our results also demonstrate that NCCs are capable of entering the spinal cord and phagocytosing CNS debris. In certain disease conditions, Schwann cells are observed inside the spinal cord (BLAKEMORE, 1976; Duncan, Hammang and Gilmore, 1988; Duncan and Hoffman, 1997). Given that Schwann cells de-differentiate and phagocytose axonal debris after nerve injury (Jessen and Mirsky, 2016), we hypothesize that, in certain neurodegenerative diseases or after injury, Schwann cells may behave like NCCs, reactivate phagocytic pathways, and migrate into the spinal cord to clear debris.
The role of Il-1β in NCC phagocytosis
The function of Il-1β as a pro-inflammatory cytokine has been widely studied, with most studies focusing on its secretion and regulation within the innate immune system (Lopez-Castejon and Brough, 2011). Our data demonstrate that Il-1β signaling is critical for NCC recruitment during development. However, we note that the level of Il-1β expression in NCCs after cell ablation is significantly lower than that which has been shown in professional phagocytes after bacterial infection (Bernut et al., 2014; Nguyen-Chi et al., 2014). Given that our cell ablation is precise compared to whole-body bacterial infection, the low level of Il-1β release we observe supports the hypothesis that Il-1β secretion is dependent upon the strength of the inflammatory stimulus (Lopez-Castejon and Brough, 2011). In addition, we observe NCCs phagocytosing Il-1β+ debris, indicating that Il-1β can be released by both phagocytic NCCs and dead cells. Similarly, a recent study showed that zebrafish epidermal cells also express Il-1β after fin fold amputation (Hasegawa et al., 2017). These findings suggest that regulated Il-1β secretion might be a universal mechanism to initiate immune responses.
STAR★Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Materials Availability Statements
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sarah Kucenas (sk4ub@virginia.edu). Plasmids and zebrafish lines generated in this study will be distributed upon request to other investigators under a Material Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Zebrafish Husbandry
All animal studies were approved by The University of Virginia Institutional Animal Care and Use Committee (Protocol No. 3782). Adult zebrafish were maintained at 28°C at a density of 8–10 fish/L. Zebrafish embryos were produced by pairwise mating and raised at 28.5°C in 10 cm petri dishes filled with egg water (6 g Instant Ocean/20 L RO water), and staged by hours post fertilization (hpf) (Kimmel et al., 1995). Embryos used for live imaging after 24 hpf were treated with 0.004% phenylthiourea (PTU) in egg water to reduce pigmentation. Embryos and larvae were anesthetized using Tricaine. Euthanasia used an overdose of Tricaine. Because zebrafish sex cannot be determined until 25 days post-fertilization (TAKAHASHI, 1977), the sex of animals used for experiments was unknown.
Zebrafish Transgenic Lines
The following published zebrafish strains were used in this study: AB*, Tg(sox10(4.9):Eos)w9, Tg(sox10(4.9):nls-Eos)w18 (McGraw et al., 2012), Tg(sox10(7.2):mEGFP)sl3 (Kirby et al., 2006), Tg(olig2:DsRed)vu19 (Shin et al., 2003), Gt(foxd3:mCherry)ct110R (Hochgreb-Hägele and Bronner, 2013), Tg(gfap:NTR-mCherry)sc129 (Johnson et al., 2016; Smith et al., 2016), Tg(4xUAS:EGFP-2xFYVE)la214 (Rasmussen et al., 2015), Tg(pBH-UAS:secA5-YFP) (van Ham et al., 2010), Tg(sox10:Gal4-VP16,cmlc2:EGFP)sq9 (Lee et al., 2013) and Tg(mpeg1:EGFP)gl22 (Ellett et al., 2011). The following lines are created in this study: Tg(nkx2.2a(3.5):nls-mCherry)uva2, Tg(mnx1:mCerulean3)uva3, Tg(sox10(4.9):TagRFP)uva5, Tg(sox10(4.9):lamp1-GFP, cmlc2:EGFP)uva8 and Tg(bactin2:Gal4-VP16,cmlc2:EGFP)uva33. See methods for details about the generation of transgenic lines. Table S1 denotes abbreviations used for each strain and summarizes what each transgene labels. All the strains above are used as stable, germline transgenic lines in this study. For the maintenance of transgenic lines, animals were outcrossed at least every other generation to ensure genetic diversity. Mosaic transgenesis of il1b:GFPFTASE was generated by injection of reporter construct il1b:GFP-FTASE into zebrafish embryos at one-cell stage (Nguyen-Chi et al., 2014).
METHOD DETAILS
Generation of transgenic lines
All constructs were generated using the Tol2kit Gateway-based cloning system (Kwan et al., 2007). Vectors used for making the expression constructs were p5E-bactin2, pME-nls-mCherry, pME-Gal4-VP16 and p3E-polyA (Kwan et al., 2007), pME-mCerulean3, pME-TagRFP (Don et al., 2017), p5E-nkx2.2a(−3.5) (Pauls et al., 2007), p5E-sox10(−4.9) (Carney et al., 2006), p5E-mnx1 (Jao, Appel and Wente, 2012) and pME-lamp1-GFP (Rasmussen et al., 2015), as well as pDestTol2pA2 and pDesTol2CG2 destination vectors (Kwan et al., 2007). Corresponding p5E, pME vectors and p3E-polyA were inserted into destination vectors through LR reactions (Kwan et al., 2007). Final constructs were amplified and sequenced to confirm the insertions. To generate stable transgenic lines, plasmid DNAs were microinjected at a concentration of 24 ng/μL in combination with 36 ng/μL Tol2 transposase mRNA at the one cell stage and screened for founders (Kawakami, 2004).
In vivo Imaging
For imaging, embryos were manually dechorionated, anesthetized with 3-aminobenzoic acid ester (Tricaine), immersed in 0.8% low-melting point agarose and mounted laterally in 35 mm glass bottom petri dishes (Fisher, Greiner Bio-One). After mounting, petri dishes were filled with egg water containing Tricaine. PTU was used to reduce pigmentation when larvae were older than 24 hpf when mounted. A 25X multi-immersion objective (NA = 0.8), 40X water objective (NA = 1.1) or a 63X water objective (NA = 1.2) mounted on a motorized Zeiss AxioObserver Z1 microscope equipped with a Quorum WaveFX-XI (Quorum Technologies Inc.) or Andor CSU-W (Andor Oxford Instruments plc.) spinning disc confocal system was used to capture all images except for orthogonal views of Lamp1-GFP+ vesicles. Time intervals for time-lapse imaging were set at 2 min, 5 min or 10 min depending on the experiment. Image processing was performed with MetaMorph and Fiji (ImageJ) to enhance brightness and contrast. Movies in the supporting information were annotated using Fiji plugin Manual Tracking. Images of Lamp1-GFP+ vesicles in NCCs (Figure 4F; Figure S3B) were taken using a Zeiss (Jena, Germany) LSM880 scanning laser confocal microscopes equipped with a 63x water objective and Airyscan detectors.
Eos photoconversion and lineage tracing
For whole-embryos photoconversion, Tg(sox10:Eos) embryos were mounted for imaging as described above and then exposed to UV light using a DAPI filter for 20 s with a Zeiss Axiozoom microscope at 20 hpf. Single-cell photoconversion and lineage tracing were performed using a nitrogen-pulsed MicroPoint laser (Andor) attached to the spinning disk confocal systems mentioned above, a dye (wavelength 404 nm) and a 40x water immersion objective. A region of interest (ROI) was created inside cells of interest to ensure precise photo-conversion. MicroPoint laser power was set between 3 to 8 depending on the location of cells. Successful photoconversion was confirmed immediately by imaging with both red and green filter sets. For lineage tracing, individual NCCs in Tg(sox10:Eos) embryos with engulfment vesicles larger than 4 μm were photoconverted at 20 hpf and time-lapse imaged immediately afterwards.
Cell ablation
For radial glial ablation, Tg(sox10:nls-Eos);Tg(gfap:NTR-mCherry) embryos were immersed in 15 mM Metronidazole (MTZ) solution in egg water with 1% DMSO starting at 10 h prior to imaging (at 10 hpf or 30 hpf) (Johnson et al., 2016; Smith et al., 2016). Fresh MTZ solution with Tricaine was applied when embryos were mounted for imaging (at 20 or 40 hpf). Control embryos were immersed in 1% DMSO in egg water. Single-cell laser ablation was performed using a nitrogen-pulsed MicroPoint laser (Andor) attached to the spinning disk confocal system mentioned above, a coumarin dye (wavelength 435 nm) and a 40x water immersion objective. To ablate individual cells, a ROI was first created inside the cell of interest to ensure precise ablation. MicroPoint laser was then fired within the ROI using a laser power between 20 to 40 depending on the location of the cell to ablate. Successful laser ablation was confirmed by the disappearance of the fluorescence of the cell (Lewis and Kucenas, 2014). Immediately after the ablation, conditions of cells and tissue around the ablation site were carefully examined. Embryos with nonspecific damage caused by excessive laser power were excluded from experiments and/or quantification.
Vital dye staining
LysoTracker Red DND-99 (Thermo Fisher Scientific) and LysoTracker Deep Red (Thermo Fisher Scientific) were diluted to final concentrations of 10 μM with egg water. Embryos were manually dechorionated at 18 hpf and immersed in LysoTracker solution for 1 h in the dark at 25°C. Embryos were then rins ed for 3 times with fresh egg water and immediately mounted for imaging. For neutral red staining, embryos were manually dechorionated at 16 hpf and incubated in 2.5 μg/ml neutral red (Sigma-Aldrich) dissolved in egg water at 25°C in the dark for 4 h. After incubation, embryos were rinsed for 3 times with fresh egg water and immediately mounted for imaging.
Immunohistochemistry
Dechorionated embryos were fixed with 4% PFA at 20 hpf, permeabilized in 1mg/ml collagenase for 8 min, followed by a 5 min wash with PBSTx (1% TritonX-100, 1x PBS) (Smith et al., 2014). Embryos were then pre-blocked in 5% goat serum/PBSTx for 1 hour, incubated in primary antibody with 5% goat serum/PBSTx for 1 hour at 24 °C and overnight at 4 °C. Embryos were washed extensively with 1x PBSTx at 24 °C and incubated in secondary antibody for 1 hour at 24 °C and overnight at 4 °C. After antibody incubation, embryos were washed extensively with 1x PBSTx and stored in 50% glycerol/PBS at 4 °C until imaging (Smith et al., 2014). Antibodies used were: rabbit anti-Il-1β (1:200, Proteintech) (Tsarouchas et al., 2018) and Alexa 488 goat anti-rabbit (1:600, ThermoFisher).
Chemical treatments
Ac-YVAD-cmk (YVAD) (Sigma) was dissolved in DMSO to a stock concentration of 10 mM. For treatment, the stock solution was diluted with egg water to a working concentration of 75 μM with 1% DMSO (Chan and Yager, 2002; Tsarouchas et al., 2018). At 10 hpf, embryos were manually dechorionated, immersed in the YVAD solution and incubated at 25°C for 12 hours. Embryo s were then mounted for imaging and cell ablation in fresh YVAD solution (with Tricaine) from 20 to 40 hpf. Control siblings were treated with 1% DMSO in egg water. For IL-1Ra experiments, embryos were manually dechorionated at 16 hpf and treated with 10 μM IL-1Ra (Sigma) in egg water (or egg water only as a control) at 25°C (Mesureur et al., 2017). Embryos were then mounted for imaging and cell ablation from 20 to 36 hpf.
Cell dissociation for flow cytometry
For NCC dissociation, 36 hpf and 72 hpf Gt(foxd3:mCherry);Tg(sox10:mEGFP) embryos were chilled in egg water on ice and the anterior halves of the embryos were removed using a scalpel. Trunk pieces were placed in calcium-free Ringer’s solution with 2.5mM EDTA and rocked for 15 minutes at 4 °C. The trunk pieces were washed three times with chilled Dulbecco’s PBS (D-PBS) and then transferred to microcentrifuge tubes along with 100ul D-PBS. A pellet pestle was used to break up the trunk pieces and then 30ul Liberase TM was added. The samples were incubated for 15 minutes at 28.5 °C, 1ml 1x Trypsin with 5% EDTA sol ution was added, and the samples were incubated for 15 more minutes at 28.5 °C. The samples were pipetted into 5ml D-PBS with 1% BSA and then passed through a 40 μm cell strainer and a syringe plunger was used to gently mash the samples into a petri dish. The contents of the petri dish were passed through a new cell strainer and the samples were then transferred to a microcentrifuge tube and were washed 2x with D-PBS+BSA. Three samples of cells (1750 ± 750) at each stage were used for RNA-sequencing. Total RNA was extracted using the RNeasy Micro Kit (QIANGEN) followed by cDNA preparation using Smart-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara). For library preparation, we used the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB).
RNA-seq analysis
Abundance of transcripts from RNA-seq datasets were first imported into “DESeq2” pipeline using the R package “tximport” (Love, Huber and Anders, 2014; Soneson, Love and Robinson, 2016). R package “AnnotationDbi” was used to acquire ENTREZ IDs and Gene Symbols (Pagès H, Carlson M, Falcon S, 2019). Normalized FPKM counts were generated using the “fpkm()” function in the “DESeq2” package and were used for making Figure S1E & F (Love, Huber and Anders, 2014). Differentially expressed genes were analyzed using the “DESeq2” package (abs(log2FoldChange) > 1 & padj < 0.1) (Love, Huber and Anders, 2014). Lists of differentially expressed genes were then used to perform functional analysis using the “clusterProfiler” package (Yu et al., 2012). Genes with padj == NA were removed to exclude genes with low counts. Gene set enrichment analysis (GSEA) for Gene Ontology and KEGG pathway were then performed using functions “gseGO()” and “gseKEGG()”, respectively (Figure S1C & D) (Kanehisa and Goto, 2000; Yu et al., 2012; Hancock, Zvelebil and Stevens, 2014). Functions “enrichGO()” and “enrichKEGG()” were used to perform over-representation tests (Figure S1A & B) (Yu et al., 2012). Raw sequencing data is available on GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135237. Transcript abundance files are available on Mendeley Data: http://dx.doi.org/10.17632/htdnfjb22c.3.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of NCC numbers and behavior
For quantification of engulfing NCCs (Figure 2G), z-stack images from time-lapse movies of Tg(sox10:Eos) embryos from 24 to 36 hpf were used to detect NCCs with engulfment vesicles. Engulfing NCCs in 3–6 hemi-segments were quantified in each fish. For quantification of CNS located NCCs, z-stack images from 20 h time-lapse movies taken with a 25X multi-immersion objective were used to detect NCC entrance into the spinal cord. Distortion of the nucleus indicated passage through MEP TZs (Smith et al., 2016). The number of NCCs with CNS experience divided by total NCC number in the field of view (185 μm x 185 μm) was used to calculate the percentage of NCCs that have entered the CNS. For the number of NCCs entering the CNS per hemi-segment, the number of entering events were counted and divided by the number of hemi-segments in the field of view. Quantification of NCC recruitment after cell ablation was done with composite z image stacks compiled using MetaMorph software. Individual z images were sequentially observed and cells were counted within the entire z stack.
Quantification of the formation of PI(3)P+ vesicles and neutral red intensity
Individual z images were sequentially observed to determine time point 0 as when GFP-FYVE fluorescence on NCC vesicles became the brightest. For quantification of the roundness of GFP-FYVE vesicles and neutral red intensity over time, square ROIs were created around individual NCC vesicles in Fiji. Circularity (4π*area/perimeter^2) and Aspect Ratio of GFP-FYVE signals in the ROIs were first calculated using the measurement function in Fiji. Roundness values, defined as Circularity/Aspect Ratio, were used to distinguish between large, ring-like, vesicle signals and small, granule-like, endosome signals. Neutral red intensity was calculated within the same ROI using Mean Gray Value measurement and then normalized to [0,1] using min-max scaling.
Statistical analysis
GraphPad Prism was used for all statistical analyses. Two-way ANOVA followed by Tukey’s multiple comparison test was used for quantification of NCC recruitment. Unpaired student’s t-test or, for multiple comparisons, one-way ANOVA followed by Tukey’s multiple comparison test were used for all other quantifications. A confidence interval of 95% was used to determine the level of significance. Other statistical details, such as sample size, p-value and dispersion, are labeled on the figures or can be found in the legends.
DATA AND CODE AVAILABILITY
Data and Code Availability Statements
The neural crest RNA-Seq data generated in this study has been deposited onto GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135237. Transcript abundance files of the RNA-Seq results are available at Mendeley Data: http://dx.doi.org/10.17632/htdnfjb22c.3.
Supplementary Material
Movie S1. Related to Figure 2B. A 20 h time-lapse movie of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo starting at 20 hpf shows two NCCs (dots) associated with cellular debris.
Movie S7. Related to Figure 6. A 17 h time-lapse movie starting at 20 hpf of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo, after ablation of 2 nkx2.2a+ lateral floorplate cells (asterisks denote where the ablation was performed). NCCs crossing the CNS-PNS boundary were circled in yellow.
Figure S1. Phagocytosis related genes are up-regulated in 36 hpf NCCs. Related to Figure 1. (A) Gene Ontology over-representation test of up-regulated genes in 36 hpf NCCs shows enrichment in endoplasmic reticulum (shaded in blue) and lysosome (shaded in red) related cellular components. (B) KEGG pathway over-representation test of genes differentially expressed in 36 hpf v.s. 72 hpf NCCs shows that lysosome (shaded in red) and endocytosis (shaded in blue) pathways are upregulated. (C & D) Results of GSEA show that (C) endosome genes and (D) the lysosome pathway are up-regulated in 36 hpf NCCs. (E) Density plot of 36 (red) and 72 hpf (blue) NCC RNA-Seq dataset in FPKM. The vertical line shows the FPKM cutoff used in F. (F) X-Y dot plot of NCC RNA-Seq data set. Genes critical to phagocytosis are highlighted in red. GSEA, gene set enrichment analysis.
Figure S2. NCC migration and engulfment of PNS debris. Related to Figures 2 & 3. (A) Images from a time-lapse movie of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo. Starts denote estimated locations of MEP TZs. Filled arrowheads denote NCCs that are going to die. A NCC (outlined in yellow) migrated towards NCC debris (open arrowheads) on the neighboring nerve and was then photoconverted from green to magenta. (B & C) Color change of NCC Eos protein after cell death and engulfment. Images from two time-lapse movies of photoconverted NCCs in Tg(sox10:Eos) embryos. Arrows denote red NCCs before death. Open arrowheads denote yellow/green NCC debris after cell death. Filled arrowheads denote green NCC debris inside neighboring NCCs. (D) Two examples of NCCs performing behaviors similar to frustrated phagocytosis in two time-lapse movies of Tg(sox10:Eos) embryos. Arrows denote NCCs circling large muscle corpses (shaded in green in the first example). (E) Lineage tracing of engulfing NCCs in Tg(sox10:Eos) embryos that were photoconverted at 20 hpf (arrowheads). Scale bars, 20 μm.
Figure S3. Characteristics of NCC phagosomes. Related to Figure 4. (A) Images from a time-lapse movie of a Tg(sox10:Eos) embryo after LysoTracker Red DND-99 treatment. Arrows denote a NCC engulfment vesicle gradually stained by LysoTracker. (B) Orthogonal views of NCC phagosomes in Tg(sox10:TagRFP);Tg(sox10:lamp1-GFP) embryos at 27 hpf. (C) Left: bright field image of the head of a Tg(sox10:Gal4);Tg(UAS:GFP-FYVE) embryo at 20 hpf with a schematic diagram. Boxed region denotes magnified views on the right. Right: images from a time-lapse movie starting at 20 hpf. Arrows denote a cranial NCC formed a PI(3)P+ engulfment vesicle. Scale bars, 10 μm in A & C, 4 μm in B.
Figure S4. Characterization of NCCs and macrophages phagocytosis. Related to Figure 5. (A) Quantification of phagocytic events performed by NCCs and macrophages between 22 and 44 hpf (mean ± SD). (B) Histogram of data in A fitted with Gaussian distribution (R2 = 0.7573/0.6164 for NCCs/Macrophages). (C) Quantification of the number of macrophages in a 0.073 mm2 region in the dorsal trunk of Tg(mpeg1:GFP) embryos over time (mean ± SD, n = 7 fish). (D) Quantification of the average velocity of phagocytic NCCs (n = 13 cells) and macrophages (mean ± SD, n = 10 cells). For NCCs, only their migration before reaching debris were calculated. 65–400 min of time-lapse movies of each cell were used for quantification.
Movie S2. Related to Figure 2B & Figure 3A. A 18 h time-lapse movie starting at 20 hpf of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo. A NCC (blue dot) migrated towards dead cells (red dot) on the neighboring nerve. To better visualize the interaction between this NCC and debris, the NCC was photoconverted from green to magenta. The movement of green debris is clearly associated with the migration of the photoconverted NCC.
Movie S3. Related to Figure 2F. A 14 h time-lapse movie starting at 19 hpf of a Tg(sox10:Eos);Tg(nkx2.2a:nls-mCherry) embryo treated with LysoTracker Red DND-99. A NCC (triangle) migrated towards LysoTracker-positive debris (circled) and engulfed it.
Movie S4. Related to Figure 4A. A 2 h time-lapse movie of a Tg(sox10:Eos) embryo treated with neutral red shows NCC (green) engulfment vesicles were stained by neutral red (magenta) gradually.
Movie S5. Relate to Figure 4C. A 1.5 h time-lapse movie of a 28 hpf Tg(sox10:Gal4);Tg(UAS:FYVE-GFP) embryo treated with neutral red shows temporal dynamics of PI(3)P signal and neutral red staining in NCC phagosomes (arrowheads).
Movie S6. Related to Figure 5. A 10 h time-lapse movie of a Tg(sox10:Gal4);Tg(UAS:FYVE-GFP) starting at 19 hpf. Arrowhead denotes a PI(3)P+ NCC engulfment vesicle before ablation of a NCC (circled in red). hba, hours before ablation; hpa, hours post ablation.
Highlights.
Neural crest cells phagocytose debris during early development in the PNS
They migrate into the spinal cord through motor exit points to clear CNS debris
Their recruitment towards debris is mediated by interleukin-1β signaling
Maturation of neural crest phagosomes is similar to that in professional phagocytes
Acknowledgements
We would like to thank members of the Kucenas and Ravichandran laboratories for valuable discussions, Dr. John Lukens for helpful advice, Lori Tocke for zebrafish care, and the following investigators for their generous gifts of fish lines and reagents: Dr. Alvaro Sagasti, Tg(4xUAS:EGFP-2xFYVE)la214 and pME-lamp1-GFP construct; Dr. Georges Lutfalla, il1b:GFP-FTASE construct. This work was funded by the National Institutes of Health (NIH): NS072212 (SK) and The University of Virginia Brain Institute (SK).
Footnotes
Declaration of Interests:
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Arya R and White K (2015) ‘Cell death in development: Signaling pathways and core mechanisms’, Seminars in Cell & Developmental Biology. Academic Press, 39, pp. 12–19. doi: 10.1016/J.SEMCDB.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M et al. (2000) ‘Gene Ontology: tool for the unification of biology’, Nature Genetics, 25(1), pp. 25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S. et al. (2011) ‘A novel role for MuSK and non-canonical Wnt signaling during segmental neural crest cell migration’, Development, 138(15), pp. 3287–3296. doi: 10.1242/jcs.96677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie CE (2000) ‘Control of motor axon guidance in the zebrafish embryo.’, Brain research bulletin, 53(5), pp. 489–500. doi: 10.1016/S0361-9230(00)00382-8. [DOI] [PubMed] [Google Scholar]
- Berardozzi R et al. (2016) ‘Arginine 66 Controls Dark-State Formation in Green-to-Red Photoconvertible Fluorescent Proteins’, Journal of the American Chemical Society, 138(2), pp. 558–565. doi: 10.1021/jacs.5b09923. [DOI] [PubMed] [Google Scholar]
- Bernut A et al. (2014) ‘Mycobacterium abscessus cording prevents phagocytosis and promotes abscess formation.’, Proceedings of the National Academy of Sciences of the United States of America. National Academy of Sciences, 111(10), pp. E943–52. doi: 10.1073/pnas.1321390111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JY et al. (2013) ‘Three pathways to mature macrophages in the early mouse yolk sac Three pathways to mature macrophages in the early mouse yolk sac’, Blood, 106(9), pp. 3004–3011. doi: 10.1182/blood-2005-02-0461. [DOI] [PubMed] [Google Scholar]
- Binker MG et al. (2007) ‘Arrested maturation of Neisseria -containing phagosomes in the absence of the lysosome-associated membrane proteins, LAMP-1 and LAMP-2’, Cellular Microbiology, 9(9), pp. 2153–2166. doi: 10.1111/j.1462-5822.2007.00946.x. [DOI] [PubMed] [Google Scholar]
- BLAKEMORE WF (1976) ‘Invasion of Schwann Cells Into the Spinal Cord of the Rat Following Local Injections of Lysolecithin’, Neuropathology and Applied Neurobiology, 2(1), pp. 21–39. doi: 10.1111/j.1365-2990.1976.tb00559.x. [DOI] [Google Scholar]
- Buss RR, Sun W and Oppenheim RW (2006) ‘ADAPTIVE ROLES OF PROGRAMMED CELL DEATH DURING NERVOUS SYSTEM DEVELOPMENT’, Annual Review of Neuroscience. Annual Reviews, 29(1), pp. 1–35. doi: 10.1146/annurev.neuro.29.051605.112800. [DOI] [PubMed] [Google Scholar]
- Cannon GJ and Swanson JA (1992) ‘The macrophage capacity for phagocytosis.’, Journal of cell science, 101 ( Pt 4, pp. 907–13. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1527185. [DOI] [PubMed] [Google Scholar]
- Canton J et al. (2014) ‘Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages.’, Molecular biology of the cell. American Society for Cell Biology, 25(21), pp. 3330–41. doi: 10.1091/mbc.E14-05-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney TJ et al. (2006) ‘A direct role for Sox10 in specification of neural crest-derived sensory neurons.’, Development (Cambridge, England), 133(23), pp. 4619–30. doi: 10.1242/dev.02668. [DOI] [PubMed] [Google Scholar]
- Champion JA, Walker A and Mitragotri S (2008) ‘Role of Particle Size in Phagocytosis of Polymeric Microspheres’, Pharmaceutical Research. Springer US, 25(8), pp. 1815–1821. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DW and Yager TD (2002) ‘Preparation and imaging of nuclear spreads from cells of the zebrafish embryo’, Chromosoma, 107(1), pp. 39–60. doi: 10.1007/s004120050280. [DOI] [PubMed] [Google Scholar]
- Cuadros MA et al. (1993) ‘First appearance, distribution, and origin of macrophages in the early development of the avian central nervous system.’, The Journal of comparative neurology, 330(1), pp. 113–29. doi: 10.1002/cne.903300110. [DOI] [PubMed] [Google Scholar]
- Don EK et al. (2017) ‘A Tol2 Gateway-Compatible Toolbox for the Study of the Nervous System and Neurodegenerative Disease’, Zebrafish, 14(1), pp. 69–72. doi: 10.1089/zeb.2016.1321. [DOI] [PubMed] [Google Scholar]
- Duncan ID., Hammang JP. and Gilmore SA. (1988) ‘Schwann cell myelination of the myelin deficient rat spinal cord following X-irradiation’, Glia. John Wiley & Sons, Ltd, 1(3), pp. 233–239. doi: 10.1002/glia.440010309. [DOI] [PubMed] [Google Scholar]
- Duncan ID and Hoffman RL (1997) ‘Schwann cell invasion of the central nervous system of the myelin mutants.’, Journal of anatomy. Wiley-Blackwell, 190 ( Pt 1)(Pt 1), pp. 35–49. doi: 10.1046/J.1469-7580.1997.19010035.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellett F et al. (2011) ‘mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish.’, Blood. American Society of Hematology, 117(4), pp. e49–56. doi: 10.1182/blood-2010-10-314120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel JL, Thein TZT and Mariani FV (2012) ‘Use of LysoTracker to Detect Programmed Cell Death in Embryos and Differentiating Embryonic Stem Cells’, Journal of Visualized Experiments, (68). doi: 10.3791/4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco-Morcillo J et al. (2014) ‘Ontogenetic Cell Death and Phagocytosis in the Visual System of Vertebrates’, Developmental Dynamics. John Wiley & Sons, Ltd, 243(10), pp. 1203–1225. doi: 10.1002/dvdy.24174. [DOI] [PubMed] [Google Scholar]
- Fratti RA et al. (2001) ‘Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest.’, The Journal of cell biology. Rockefeller University Press, 154(3), pp. 631–44. doi: 10.1083/jcb.200106049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli SJ, Borregaard N and Wynn TA (2011) ‘Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils’, Nature Immunology. Nature Publishing Group, 12(11), pp. 1035–1044. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb RA (1996) ‘Cell acidification in apoptosis’, Apoptosis. Kluwer Academic Publishers, 1(1), pp. 40–48. doi: 10.1007/BF00142077. [DOI] [Google Scholar]
- Grabher C. et al. (2006) ‘Birth and life of tissue macrophages and their migration in embryogenesis and inflammation in medaka’, Journal of Leukocyte Biology, 81(1), pp. 263–271. doi: 10.1189/jlb.0806526. [DOI] [PubMed] [Google Scholar]
- van Ham TJ et al. (2010) ‘Live imaging of apoptotic cells in zebrafish.’, The FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 24(11), pp. 4336–4342. doi: 10.1096/fj.10-161018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock JM, Zvelebil MJ and Stevens R (2014) ‘Gene Ontology Consortium’, Dictionary of Bioinformatics and Computational Biology, 25(may), pp. 25–29. doi: 10.1002/9780471650126.dob0273.pub2. [DOI] [Google Scholar]
- Hasegawa T et al. (2017) ‘Transient inflammatory response mediated by interleukin-1β is required for proper regeneration in zebrafish fin fold’, eLife, 6, pp. 1–22. doi: 10.7554/eLife.22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbomel P, Thisse B and Thisse C (1999) ‘Ontogeny and behaviour of early macrophages in the zebrafish embryo.’, Development (Cambridge, England), 126(17), pp. 3735–45. [DOI] [PubMed] [Google Scholar]
- Herbomel P, Thisse B and Thisse C (2001) ‘Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process.’, Developmental biology, 238(2), pp. 274–88. doi: 10.1006/dbio.2001.0393. [DOI] [PubMed] [Google Scholar]
- Hochgreb-Hägele T and Bronner ME (2013) ‘A novel FoxD3 gene trap line reveals neural crest precursor movement and a role for FoxD3 in their specification.’, Developmental biology. NIH Public Access, 374(1), pp. 1–11. doi: 10.1016/j.ydbio.2012.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honj Y. and Eise JS. (2005) ‘Slow muscle regulates the pattern of trunk neural crest migration in zebrafish’, Development, 132(20), pp. 4461–4470. doi: 10.1242/dev.02026. [DOI] [PubMed] [Google Scholar]
- Huynh KK et al. (2007) ‘LAMP proteins are required for fusion of lysosomes with phagosomes’, The EMBO Journal, 26(2), pp. 313–324. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao L-E, Appel B and Wente SR (2012) ‘A zebrafish model of lethal congenital contracture syndrome 1 reveals Gle1 function in spinal neural precursor survival and motor axon arborization’, Development, 139(7), pp. 1316–1326. doi: 10.1242/dev.074344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR and Mirsky R (2016) ‘The repair Schwann cell and its function in regenerating nerves’, The Journal of Physiology. John Wiley & Sons, Ltd (10.1111), 594(13), pp. 3521–3531. doi: 10.1113/JP270874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K et al. (2016) ‘Gfap-positive radial glial cells are an essential progenitor population for later-born neurons and glia in the zebrafish spinal cord’, Glia, 64(7), pp. 1170–1189. doi: 10.1002/glia.22990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamen LA et al. (2008) ‘SHIP-1 increases early oxidative burst and regulates phagosome maturation in macrophages.’, Journal of immunology (Baltimore, Md. : 1950). American Association of Immunologists, 180(11), pp. 7497–505. doi: 10.4049/JIMMUNOL.180.11.7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M and Goto S (2000) ‘KEGG: kyoto encyclopedia of genes and genomes.’, Nucleic acids research, 28(1), pp. 27–30. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10592173 (Accessed: 22 March 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakam K. (2004) ‘Transgenesis and Gene Trap Methods in Zebrafish by Using the Tol2 Transposable Element’, Methods in Cell Biology. Academic Press, 77, pp. 201–222. doi: 10.1016/S0091-679X(04)77011-9. [DOI] [PubMed] [Google Scholar]
- Kimmel CB et al. (1995) ‘Stages of embryonic development of the zebrafish’, Developmental dynamics: an official publication of the American Association of Anatomists, 203(3), pp. 253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kinchen JM and Ravichandran KS (2008) ‘Phagosome maturation: going through the acid test.’, Nature reviews. Molecular cell biology. NIH Public Access, 9(10), pp. 781–95. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby BB et al. (2006) ‘In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development’, Nature neuroscience, 9(12), pp. 1506–1511. doi: 10.1038/nn1803. [DOI] [PubMed] [Google Scholar]
- Kucenas S, Snell H and Appel B (2008) ‘nkx2.2a promotes specification and differentiation of a myelinating subset of oligodendrocyte lineage cells in zebrafish.’, Neuron glia biology. NIH Public Access, 4(2), pp. 71–81. doi: 10.1017/S1740925X09990123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurant E et al. (2008) ‘Six-Microns-Under Acts Upstream of Draper in the Glial Phagocytosis of Apoptotic Neurons’, Cell. Cell Press, 133(3), pp. 498–509. doi: 10.1016/J.CELL.2008.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KM et al. (2007) ‘The Tol2kit: A multisite gateway-based construction kit forTol2 transposon transgenesis constructs’, Developmental Dynamics, 236(11), pp. 3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Lee RTH. et al. (2013) ‘An exclusively mesodermal origin of fin mesenchyme demonstrates that zebrafish trunk neural crest does not generate ectomesenchyme’, Development, 140(14), pp. 2923–2932. doi: 10.1242/dev.093534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin R, Grinstein S and Canton J (2016) ‘The life cycle of phagosomes: formation, maturation, and resolution’, Immunological Reviews, 273(1), pp. 156–179. doi: 10.1111/imr.12439. [DOI] [PubMed] [Google Scholar]
- Lewis GM and Kucenas S (2014) ‘Perineurial Glia Are Essential for Motor Axon Regrowth following Nerve Injury’, Journal of Neuroscience, 34(38), pp. 12762–12777. doi: 10.1523/JNEUROSCI.1906-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieschke GJ et al. (2001) ‘Morphologic and functional characterization of granulocytes and macrophages in embryonic and adult zebrafish’, Blood, 98(10), pp. 3087–3096. doi: 10.1182/blood.V98.10.3087. [DOI] [PubMed] [Google Scholar]
- Lopez-Castejon G and Brough D (2011) ‘Understanding the mechanism of IL-1β secretion’, Cytokine and Growth Factor Reviews. Elsevier Ltd, 22(4), pp. 189–195. doi: 10.1016/j.cytogfr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W and Anders S (2014) ‘Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2’, Genome Biology. BioMed Central, 15(12), p. 550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z et al. (2011) ‘Phagocytic activity of neuronal progenitors regulates adult neurogenesis’, Nature Cell Biology. Nature Publishing Group, 13(9), pp. 1076–1083. doi: 10.1038/ncb2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej MP et al. (2017) ‘Different regulation of interleukin-1 production and activity in monocytes and macrophages: Innate memory as an endogenous mechanism of IL-1 inhibition’, Frontiers in Pharmacology, 8(JUN), pp. 1–12. doi: 10.3389/fphar.2017.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa V. et al. (2009) ‘Apoptosis is not required for mammalian neural tube closure.’, Proceedings of the National Academy of Sciences of the United States of America, 106(20), pp. 8233–8. doi: 10.1073/pnas.0900333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto J et al. (2003) ‘Caspy, a zebrafish caspase, activated by ASC oligomerization is required for pharyngeal arch development’, Journal of Biological Chemistry, 278(6), pp. 4268–4276. doi: 10.1074/jbc.M203944200. [DOI] [PubMed] [Google Scholar]
- Mathiak G et al. (2000) ‘Caspase-1-inhibitor ac-YVAD-cmk reduces LPS-lethality in rats without affecting haematology or cytokine responses.’, British journal of pharmacology. Wiley-Blackwell, 131(3), pp. 383–6. doi: 10.1038/sj.bjp.0703629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor Roberto and Theveneau E (2013) ‘The neural crest’, Development, 140(11), pp. 2247–2251. doi: 10.1242/dev.091751. [DOI] [PubMed] [Google Scholar]
- Mayor R and Theveneau E (2013) ‘The neural crest’, Development, 140(11), pp. 2247–2251. doi: 10.1242/dev.091751. [DOI] [PubMed] [Google Scholar]
- McGrath KE et al. (2003) ‘Circulation is established in a stepwise pattern in the mammalian embryo.’, Blood. American Society of Hematology, 101(5), pp. 1669–76. doi: 10.1182/blood-2002-08-2531. [DOI] [PubMed] [Google Scholar]
- McGraw H et al. (2012) ‘Postembryonic neuronal addition in Zebrafish dorsal root ganglia is regulated by Notch signaling’, Neural Development, 7(1), p. 23. doi: 10.1186/1749-8104-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesureur J et al. (2017) ‘Macrophages, but not neutrophils, are critical for proliferation of Burkholderia cenocepacia and ensuing host-damaging inflammation’, PLOS Pathogens. Edited by Tobin DM. Public Library of Science, 13(6), p. e1006437. doi: 10.1371/journal.ppat.1006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkes PE. (2002) ‘2001 Warkany lecture: To die or not to die, the role of apoptosis in normal and abnormal mammalian development’, Teratology, 65(5), pp. 228–239. doi: 10.1002/tera.10049. [DOI] [PubMed] [Google Scholar]
- Nguyen-Chi M et al. (2014) ‘Transient infection of the zebrafish notochord with E. coli induces chronic inflammation’, Disease Models & Mechanisms, 7(7), pp. 871–882. doi: 10.1242/dmm.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès H, Carlson M, Falcon S, L. N (2019) AnnotationDbi: Manipulation of SQLitebased annotations in Bioconductor, R package version 1.46.0. doi: 10.18129/B9.bioc.AnnotationDbi. [DOI] [Google Scholar]
- Pauls S et al. (2007) ‘Function and regulation of zebrafish nkx2.2a during development of pancreatic islet and ducts’, Developmental Biology. Academic Press, 304(2), pp. 875–890. doi: 10.1016/J.YDBIO.2007.01.024. [DOI] [PubMed] [Google Scholar]
- Prendergast A et al. (2012) ‘The metalloproteinase inhibitor Reck is essential for zebrafish DRG development.’, Development (Cambridge, England). Company of Biologists, 139(6), pp. 1141–52. doi: 10.1242/dev.072439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen JP et al. (2015) ‘Vertebrate epidermal cells are broad-specificity phagocytes that clear sensory axon debris.’, The Journal of neuroscience : the official journal of the Society for Neuroscience. Society for Neuroscience, 35(2), pp. 559–70. doi: 10.1523/JNEUROSCI.3613-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarvothaman S et al. (2015) ‘Apoptosis: Role in myeloid cell development’, Blood Research, 50(2), pp. 73–79. doi: 10.5045/br.2015.50.2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter G (1973) ‘Ultrastructural observations on cell necrosis during formation of the neural tube in mouse embryos.’, Zeitschrift fur Anatomie und Entwicklungsgeschichte, 141(3), pp. 251–64. Available at: http://www.ncbi.nlm.nih.gov/pubmed/4767584 (Accessed: 31 July 2018). [DOI] [PubMed] [Google Scholar]
- Shin J et al. (2003) ‘Neural cell fate analysis in zebrafish using olig2 BAC transgenics’, Methods in Cell Science. Kluwer Academic Publishers, 25(1/2), pp. 7–14. doi: 10.1023/B:MICS.0000006847.09037.3a. [DOI] [PubMed] [Google Scholar]
- Shklover J, Levy-Adam F and Kurant E (2015) Apoptotic Cell Clearance in Development 1st edn, Current Topics in Developmental Biology. 1st edn. Elsevier Inc. doi: 10.1016/bs.ctdb.2015.07.024. [DOI] [PubMed] [Google Scholar]
- Simoes-Costa M and Bronner ME (2015) ‘Establishing neural crest identity: a gene regulatory recipe’, Development, 142(2), pp. 242–257. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ et al. (2014) ‘Contact-Mediated Inhibition Between Oligodendrocyte Progenitor Cells and Motor Exit Point Glia Establishes the Spinal Cord Transition Zone’, PLoS Biology. Edited by Barres BA, 12(9), p. e1001961. doi: 10.1371/journal.pbio.1001961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ et al. (2016) ‘Radial glia inhibit peripheral glial infiltration into the spinal cord at motor exit point transition zones’, Glia, 64(7), pp. 1138–1153. doi: 10.1002/glia.22987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI and Robinson MD (2016) ‘Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences’, F1000Research, 4(2), p. 1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan D (2004) ‘Cell Type-Specific Interleukin-1 Signaling in the CNS’, Journal of Neuroscience, 24(29), pp. 6482–6488. doi: 10.1523/jneurosci.5712-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachura DL and Traver D (2016) Cellular dissection of zebrafish hematopoiesis Third Edit, Methods in Cell Biology. Third Edit. Elsevier Ltd. doi: 10.1016/bs.mcb.2016.03.022. [DOI] [PubMed] [Google Scholar]
- Stremmel C. et al. (2018) ‘Yolk sac macrophage progenitors traffic to the embryo during defined stages of development’, Nature Communications. Nature Publishing Group, 9(1), p. 75. doi: 10.1038/s41467-017-02492-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- System E and Klumperman J (2015) ‘The Complex Ultrastructure of the Endolysosomal System’, Cold Spring Harbor Perspectives in Biology, 6, pp. 1–22. doi: 10.1101/cshperspect.a016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKAHASHI H (1977) ‘Juvenile Hermaphroditism in the Zebrafish, Brachydanio rerio’, <image>, 28(2), pp. 57–65. Available at: https://eprints.lib.hokudai.ac.jp/dspace/bitstream/2115/23605/1/28(2)_P57-65.pdf (Accessed: 31 July 2019). [Google Scholar]
- The Gene Ontology Consortium (2019) ‘The Gene Ontology Resource: 20 years and still GOing strong’, Nucleic Acids Research, 47(D1), pp. D330–D338. doi: 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E and Mayor R (2012) ‘Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration’, Developmental Biology. (Neural Crest), 366(1), pp. 34–54. doi: 10.1016/j.ydbio.2011.12.041. [DOI] [PubMed] [Google Scholar]
- Tsarouchas TM et al. (2018) ‘Dynamic control of proinflammatory cytokines Il-1β and Tnf-α by macrophages in zebrafish spinal cord regeneration’, Nature Communications, 9(1). doi: 10.1038/s41467-018-07036-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkowyd B et al. (2017) ‘A General Mechanism of Photoconversion of Green-to-Red Fluorescent Proteins Based on Blue and Infrared Light Reduces Phototoxicity in Live-Cell Single-Molecule Imaging’, Angewandte Chemie International Edition. Wiley-Blackwell, 56(38), pp. 11634–11639. doi: 10.1002/anie.201702870. [DOI] [PubMed] [Google Scholar]
- van der Vaart M. et al. (2014) ‘Cellular Visualization of Macrophage Pyroptosis and Interleukin-1 Release in a Viral Hemorrhagic Infection in Zebrafish Larvae’, Journal of Virology, 88(20), pp. 12026–12040. doi: 10.1128/jvi.02056-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Lopez GA, Cerrizuela S and Aybar MJ (2017) ‘Trunk neural crest cells: formation, migration and beyond.’, The International journal of developmental biology, 61(1–2), pp. 5–15. doi: 10.1387/ijdb.160408gv. [DOI] [PubMed] [Google Scholar]
- Vieira OV et al. (2001) ‘Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation’, Journal of Cell Biology, 155(1), pp. 19–25. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani A et al. (2019) ‘Clearance by Microglia Depends on Packaging of Phagosomes into a Unique Cellular Compartment’, Developmental Cell. Elsevier Inc., pp. 1–12. doi: 10.1016/j.devcel.2019.02.014. [DOI] [PubMed] [Google Scholar]
- Vojtech LN et al. (2012) ‘Roles of inflammatory caspases during processing of zebrafish interleukin-1β in Francisella noatunensis infection.’, Infection and immunity. American Society for Microbiology (ASM), 80(8), pp. 2878–85. doi: 10.1128/IAI.00543-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L et al. (2014) ‘A conserved axon type hierarchy governing peripheral nerve assembly.’, Development, 141(9), pp. 1875–83. doi: 10.1242/dev.106211. [DOI] [PubMed] [Google Scholar]
- Weil M., Jacobson MD. and Raff MC. (1997) ‘Is programmed cell death required for neural tube closure?’, Current biology : CB, 7(4), pp. 281–4. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9094312 (Accessed: 31 July 2018). [DOI] [PubMed] [Google Scholar]
- Wu H-H et al. (2009) ‘Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor.’, Nature neuroscience, 12(12), pp. 1534–41. doi: 10.1038/nn.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y et al. (2011) ‘Live imaging of apoptosis in a novel transgenic mouse highlights its role in neural tube closure’, Journal of Cell Biology, 195(6), pp. 1047–1060. doi: 10.1083/jcb.201104057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H et al. (2013) ‘Local Apoptosis Modulates Early Mammalian Brain Development through the Elimination of Morphogen-Producing Cells’, Developmental Cell. Elsevier Inc, 27(6), pp. 621–634. doi: 10.1016/j.devcel.2013.11.015. [DOI] [PubMed] [Google Scholar]
- Yu G et al. (2012) ‘clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters’, OMICS: A Journal of Integrative Biology. Mary Ann Liebert, Inc. 140 Huguenot Street, 3rd Floor New Rochelle, NY 10801 USA, 16(5), pp. 284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Related to Figure 2B. A 20 h time-lapse movie of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo starting at 20 hpf shows two NCCs (dots) associated with cellular debris.
Movie S7. Related to Figure 6. A 17 h time-lapse movie starting at 20 hpf of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo, after ablation of 2 nkx2.2a+ lateral floorplate cells (asterisks denote where the ablation was performed). NCCs crossing the CNS-PNS boundary were circled in yellow.
Figure S1. Phagocytosis related genes are up-regulated in 36 hpf NCCs. Related to Figure 1. (A) Gene Ontology over-representation test of up-regulated genes in 36 hpf NCCs shows enrichment in endoplasmic reticulum (shaded in blue) and lysosome (shaded in red) related cellular components. (B) KEGG pathway over-representation test of genes differentially expressed in 36 hpf v.s. 72 hpf NCCs shows that lysosome (shaded in red) and endocytosis (shaded in blue) pathways are upregulated. (C & D) Results of GSEA show that (C) endosome genes and (D) the lysosome pathway are up-regulated in 36 hpf NCCs. (E) Density plot of 36 (red) and 72 hpf (blue) NCC RNA-Seq dataset in FPKM. The vertical line shows the FPKM cutoff used in F. (F) X-Y dot plot of NCC RNA-Seq data set. Genes critical to phagocytosis are highlighted in red. GSEA, gene set enrichment analysis.
Figure S2. NCC migration and engulfment of PNS debris. Related to Figures 2 & 3. (A) Images from a time-lapse movie of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo. Starts denote estimated locations of MEP TZs. Filled arrowheads denote NCCs that are going to die. A NCC (outlined in yellow) migrated towards NCC debris (open arrowheads) on the neighboring nerve and was then photoconverted from green to magenta. (B & C) Color change of NCC Eos protein after cell death and engulfment. Images from two time-lapse movies of photoconverted NCCs in Tg(sox10:Eos) embryos. Arrows denote red NCCs before death. Open arrowheads denote yellow/green NCC debris after cell death. Filled arrowheads denote green NCC debris inside neighboring NCCs. (D) Two examples of NCCs performing behaviors similar to frustrated phagocytosis in two time-lapse movies of Tg(sox10:Eos) embryos. Arrows denote NCCs circling large muscle corpses (shaded in green in the first example). (E) Lineage tracing of engulfing NCCs in Tg(sox10:Eos) embryos that were photoconverted at 20 hpf (arrowheads). Scale bars, 20 μm.
Figure S3. Characteristics of NCC phagosomes. Related to Figure 4. (A) Images from a time-lapse movie of a Tg(sox10:Eos) embryo after LysoTracker Red DND-99 treatment. Arrows denote a NCC engulfment vesicle gradually stained by LysoTracker. (B) Orthogonal views of NCC phagosomes in Tg(sox10:TagRFP);Tg(sox10:lamp1-GFP) embryos at 27 hpf. (C) Left: bright field image of the head of a Tg(sox10:Gal4);Tg(UAS:GFP-FYVE) embryo at 20 hpf with a schematic diagram. Boxed region denotes magnified views on the right. Right: images from a time-lapse movie starting at 20 hpf. Arrows denote a cranial NCC formed a PI(3)P+ engulfment vesicle. Scale bars, 10 μm in A & C, 4 μm in B.
Figure S4. Characterization of NCCs and macrophages phagocytosis. Related to Figure 5. (A) Quantification of phagocytic events performed by NCCs and macrophages between 22 and 44 hpf (mean ± SD). (B) Histogram of data in A fitted with Gaussian distribution (R2 = 0.7573/0.6164 for NCCs/Macrophages). (C) Quantification of the number of macrophages in a 0.073 mm2 region in the dorsal trunk of Tg(mpeg1:GFP) embryos over time (mean ± SD, n = 7 fish). (D) Quantification of the average velocity of phagocytic NCCs (n = 13 cells) and macrophages (mean ± SD, n = 10 cells). For NCCs, only their migration before reaching debris were calculated. 65–400 min of time-lapse movies of each cell were used for quantification.
Movie S2. Related to Figure 2B & Figure 3A. A 18 h time-lapse movie starting at 20 hpf of a Tg(sox10:nls-Eos);Tg(nkx2.2a:nls-mCherry) embryo. A NCC (blue dot) migrated towards dead cells (red dot) on the neighboring nerve. To better visualize the interaction between this NCC and debris, the NCC was photoconverted from green to magenta. The movement of green debris is clearly associated with the migration of the photoconverted NCC.
Movie S3. Related to Figure 2F. A 14 h time-lapse movie starting at 19 hpf of a Tg(sox10:Eos);Tg(nkx2.2a:nls-mCherry) embryo treated with LysoTracker Red DND-99. A NCC (triangle) migrated towards LysoTracker-positive debris (circled) and engulfed it.
Movie S4. Related to Figure 4A. A 2 h time-lapse movie of a Tg(sox10:Eos) embryo treated with neutral red shows NCC (green) engulfment vesicles were stained by neutral red (magenta) gradually.
Movie S5. Relate to Figure 4C. A 1.5 h time-lapse movie of a 28 hpf Tg(sox10:Gal4);Tg(UAS:FYVE-GFP) embryo treated with neutral red shows temporal dynamics of PI(3)P signal and neutral red staining in NCC phagosomes (arrowheads).
Movie S6. Related to Figure 5. A 10 h time-lapse movie of a Tg(sox10:Gal4);Tg(UAS:FYVE-GFP) starting at 19 hpf. Arrowhead denotes a PI(3)P+ NCC engulfment vesicle before ablation of a NCC (circled in red). hba, hours before ablation; hpa, hours post ablation.
Data Availability Statement
Data and Code Availability Statements
The neural crest RNA-Seq data generated in this study has been deposited onto GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135237. Transcript abundance files of the RNA-Seq results are available at Mendeley Data: http://dx.doi.org/10.17632/htdnfjb22c.3.
The neural crest RNA-Seq data generated in this study has been deposited onto GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135237. Transcript abundance files of the RNA-Seq results are available at Mendeley Data: http://dx.doi.org/10.17632/htdnfjb22c.3.