

Abstract
Two N-heterocyclic carbene (NHC) ligands provide orthogonal chemoselectivity during the Pd-catalyzed Suzuki-Miyaura (SM) cross coupling of chloroaryl triflates. The use of SIPr [SIPr = 1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazol-2-ylidene] leads to selective cross coupling at chloride, while the use of SIMes [SIMes = 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene] provides selective coupling at triflate. With most chloroaryl triflates and arylboronic acids, ligand-controlled selectivity is high (≥10:1). The scope of this methodology is significantly more general than previously reported methods for selective SM coupling of chloroaryl triflates using phosphine ligands. Density functional theory (DFT) studies suggest that palladium’s ligation state during oxidative addition is different with SIMes compared to SIPr.
Graphical Abstract
INTRODUCTION
Palladium-catalyzed cross couplings are among the most widely used strategies for C—C bond formation during the preparation of diverse classes of products.1 The Suzuki-Miyaura (SM) reaction is particularly attractive due to its mild reaction conditions, its high functional group and moisture tolerance, and the low toxicity and good commercial availability of boronic acids and esters.1 Sequential cross coupling reactions can be used to prepare polyfunctionalized arenes,2 which are ubiquitous in pharmaceuticals, agrochemicals, materials, and natural products. However, controlling the site selectivity of sequential cross coupling reactions can be challenging when multiple (pseudo)halides are present. A particularly attractive strategy for manipulating selectivity is through catalyst or ligand choice.3
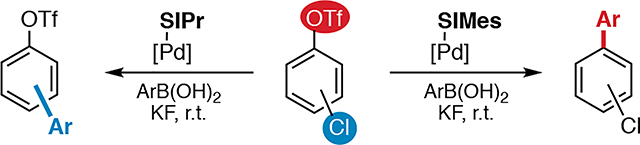
For example, aryl chlorides and triflates are known to react with Pd(0) under similar conditions. The only previously known system for ligand-controlled divergent SM cross coupling of chloroaryl triflates was initially reported by Fu (Scheme 1A and B).4,5,6 The SM coupling of 1 with 2 favors reaction at C—Cl over C—OTf with the bulky ligand PtBu3. Conversely, the less hindered PCy3 effects reaction at triflate. However, this methodology has limited synthetic utility. Selective cross coupling at C—OTf using PCy3 under the reported conditions is specific to the reaction between 1 and 2; the use of other chloroaryl triflates or boronic acids leads to poor yields and/or selectivity (Scheme 1B).7 A detailed scope of the chloride-selective cross coupling with PtBu3 under the original conditions has not been reported.8 As such, alternative methods are needed for selective SM cross coupling of chloroaryl triflates.
Scheme 1.

Chemodivergent Cross Coupling of Chloroaryl Triflates
An elegant procedure for triflate-selective coupling of chloroaryl triflates with organozinc reagents was recently described (Scheme 1C).7 However, a general method to achieve the opposite selectivity—cross coupling of aryl chlorides in the presence of triflates—has not been detailed. Moreover, organozinc reagents have disadvantages compared to organoboron reagents related to functional group tolerance and commercial availability.1
Herein we demonstrate that the use of NHC ligands enables chemodivergent SM coupling of chloroaryl triflates. A Pd/SIPr precatalyst provides selective reaction at chloride, while a Pd/SIMes precatalyst favors reaction at triflate. In contrast to the limited scope reported for the prior phosphine ligand-controlled method,4,7 this NHC ligand-controlled SM coupling is general to a variety of chloroaryl triflates and arylboronic acids.
RESULTS AND DISCUSSION
Initial Catalytic Results with NHC Ligands.
Inspired by recent reports of the high reactivity of Pd(II) precatalysts bearing NHC ligands,9,10 we investigated the selectivity of Pd/NHC complexes 3–7 (Figure 1) in SM reactions of chloroaryl triflates.
Figure 1.

Pd/NHC catalysts used in this work.
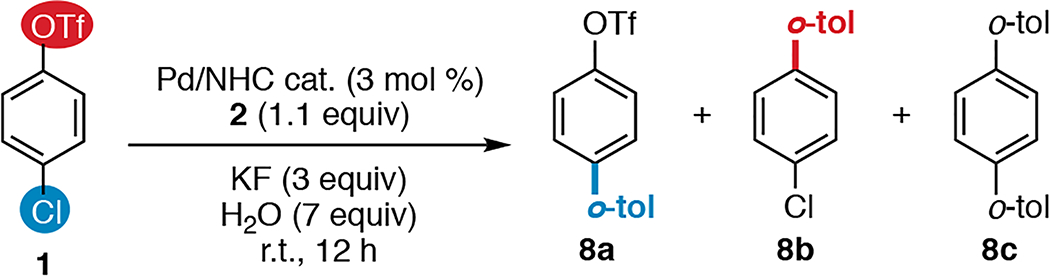
Gratifyingly, the use of Hazari’s10 air-stable Pd/SIPr precatalyst 3 or PEPPSI™-SIPr11 complex 7 provides high selectivity for coupling at C—Cl in the room-temperature SM reaction of 1 with 2 in THF (Table 1, entries 1–2). Precatalyst 4 bearing the related unsaturated ligand IPr gives comparable selectivity to 3, albeit with reduced yield of the desired product (entry 3). In stark contrast, however, however, catalysts containing the moderately smaller ligands SIMes and IMes provide the opposite selectivity under otherwise identical conditions (entries 4–5).12,13,14 For example, the Pd/SIMes-catalyzed reaction of 1 with 2 yields 8b in high yield, resulting from selective C—OTf activation (entry 4). Only small amounts of products from C—Cl activation (8a and 8c) are detected with this catalyst.
Table 1.
Evaluation of NHC ligands for selective Pd-catalyzed SM coupling.a
 | ||||||
|---|---|---|---|---|---|---|
| entry | cat. | NHC | solvent | 8a (%) | 8b (%) | 8c (%) |
| 1 | 3 | SIPr | THF | 85 | 5 | 13 |
| 2 | 7 | SIPr | THF | 55 | 7 | 9 |
| 3 | 4 | IPr | THF | 68 | 4 | 16 |
| 4 | 5 | SIMes | THF | 3 | 90 | 3 |
| 5 | 6 | IMes | THF | 2 | 79 | 13 |
| 6 | 3 | SIPr | PhMe | 92 | 5 | 2 |
| 7 | 5 | SIMes | PhMe | 8 | 13 | 2 |
| 8 | 3 | SIPr | DMF | 1 | 6 | n.d. |
| 9 | 5 | SIMes | DMF | n.d. | 57 | n.d. |
| 10 | 3 | SIPr | 1,4-dioxane | 93 | 5 | 8 |
| 11 | 5 | SIMes | 1,4-dioxane | 7 | 84 | 2 |
| 12b | 3 | SIPr | THF | trace | trace | n.d. |
| 13c | 3 | SIPr | THF | trace | trace | n.d. |
| 14b | 5 | SIMes | THF | n.d. | trace | n.d. |
| 15c | 5 | SIMes | THF | trace | 7 | n.d. |
| 16d | -- | SIPr | THF | n.d. | n.d. | n.d. |
| 17d | -- | SIMes | THF | n.d. | n.d. | n.d. |
| 18e | 9 | -- | THF | 1 | 2 | 0 |
GC yields calibrated against undecane as internal standard. Estimated error in total mass balance ±6%. Entries 1–11 are the average of two runs. n.d. = not detected.
Without KF.
Without H2O.
No Pd catalyst; free SIPr or SIMes was added (3 mol %).
9 = [(η3-1-tBu-indenyl)Pd(Cl)]2.
The use of toluene instead of THF minimizes unwanted diarylation (8c) with SIPr (entry 6), but poor conversion is observed with SIMes in toluene (entry 7). Dimethylformamide (DMF) is an effective solvent for the reaction with SIMes (entry 9) but not with SIPr (entry 8), while 1,4-dioxane is an effective solvent with both ligands (entries 10–11). Control reactions demonstrate that catalyst, KF, and water are each necessary for both the triflate- and the chloride-selective cross couplings using SIMes and SIPr, respectively (entries 8–13). Furthermore, only trace cross-coupling products are detected under ligand-free conditions using [(η3-1-tBu-indenyl)Pd(Cl)]2 (9, entry 14).
Scope of Arylboronic Acids.
The divergent selectivities displayed by SIMes and SIPr are general to the cross coupling of 1 with diverse arylboronic acids (Scheme 2).15,16 In most cases, ≤5% of the minor product and ≤5% of a diarylated product are observed. Pd/SIMes 5 catalyzes coupling at triflate using electron-rich (8b, 11b–14b), -neutral (10b), and -deficient (15b–20b) arylboronic acids. An unprotected phenol (13b), benzylic alcohol (14b), aldehyde (20b), and a chloro substituent (17b) are tolerated on the boronic acid coupling partner. This scope is notable because the previously reported conditions for triflate-selective SM cross coupling using Pd/PCy3 are ineffective with representative electron-rich and -deficient arylboronic acids (see Scheme 1B).7
Scheme 2.

Scope of arylboronic acids in the NHC ligand-controlled chemodivergent SM coupling.a
aIsolated yields. Unless noted, ≤5% of minor monoarylated products and ≤5% of diarylated products were detected by crude GC. b~10% of diarylated product by crude GC. c~6% of minor monoarylated product by crude GC. dSolvent = DMF. eSolvent = 1,4-dioxane. fSolvent = THF.
Conversely, Pd/SIPr complex 3 catalyzes selective cross coupling of 1 at chloride using both electron-rich and -deficient arylboronic acids. The Pd/SIPr-catalyzed cross coupling of 1 with 4-vinylphenyl boronic acid to form 22a is chemoselective for both the electrophile (preferential reaction at C—Cl) and the nucleophile (SM coupling is favored over Heck coupling).17
Scope of Chloroaryl Triflates.15,16
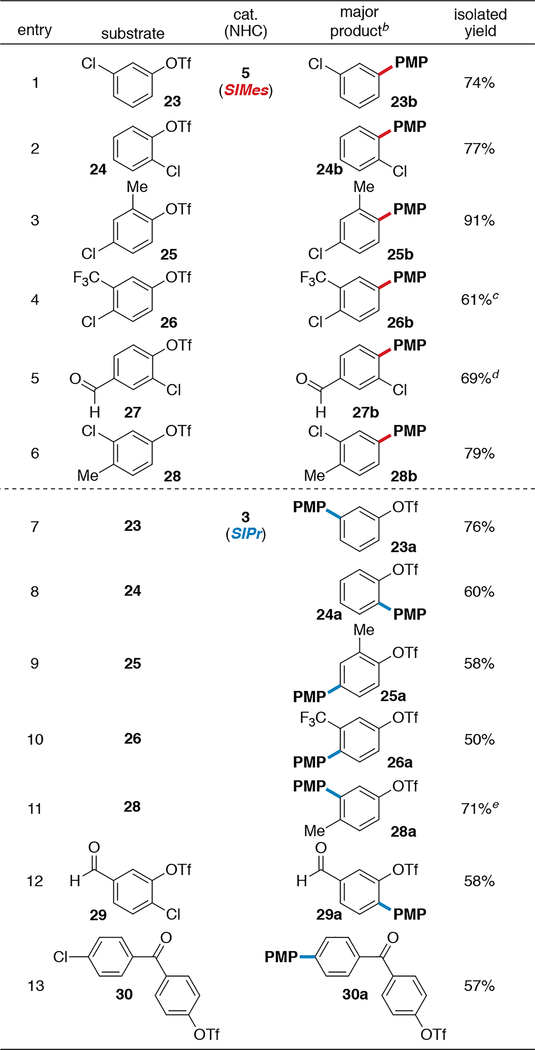
In addition to para-chlorophenyl triflate 1, the meta and ortho analogs undergo triflate- (Table 2, entries 1–2) or chloride-selective (entries 7–8) cross coupling with catalysts 5 and 3, respectively. Even in the presence of steric bias (entries 3, 10, 11) or additional electron-withdrawing or -donating substituents (entries 3–6, 9–13), selectivity remains ligand-controlled. However, conversions and selectivities are poor for some electronically biased substrates that were evaluated (see Supporting Information).15 The expected ligand-based selectivity is retained when chloride and triflate are connected to different aryl rings (entry 13).
Table 2.
Scope of chloroaryl triflates in the NHC ligand-controlled chemodivergent SM coupling.a
|
aGeneral procedure: chloroaryl triflate (1 equiv), p-methoxyphenylboronic acid (1.25–1.5 equiv), 3 or 5 (3 mol %), KF (3 equiv), H2O (3.5–7.0 equiv), THF, DMF, toluene, 1,4-dioxane, or 1,4-dioxane/toluene (1:1), 4–12 h, 25–60 °C. PMP = p-methoxyphenyl. Isolated yields. bUnless noted, ≤5% of minor monoarylated products and ≤5% of diarylated products were detected by crude GC. c~28% of diarylated product by crude GC. d~6% of diarylated product by crude GC. e~19% of minor monoarylated product by crude GC.
DFT Studies.
Having established that SIPr and SIMes ligand-controlled divergent selectivity is general to a variety of chloroaryl triflates and arylboronic acids, we next turned to DFT calculations to understand the origin of selectivity in this system. Calculations were performed at the CPCM(THF) M06/BS2//M06L/BS1 level of theory (see Computational Methods for details). Neutral transition structures were located using monoligated [Pd(SIPr)] for oxidative addition at both C—Cl and C—OTf of 1 (Figure 2A, top). Consistent with experiment, [Pd(SIPr)] is predicted to favor reaction at C—Cl by 4.6 kcal mol−1 (compare TS31a and TS31b). However, the same computational method predicts that [Pd(SIMes)] should also favor reaction at C—Cl, by 9.5 kcal mol−1 (compare TS32a and TS32b). The latter result strongly contradicts the observed experimental preference for cross coupling at C—OTf using SIMes. A variety of other DFT methods were evaluated (see SI), and nearly all provide the same prediction that reaction at C—OTf is disfavored with [Pd(SIMes)].
Figure 2.

Calculated transition structures for oxidative addition involving SIPr and SIMes.
This disagreement between computational and experimental results strongly suggests that monoligated [Pd(SIMes)] is not responsible for oxidative addition of C—OTf. Instead, one possibility is that oxidative addition at triflate takes place through a bisligated [Pd(SIMes)2] transition structure. This possibility is analogous to the current mechanistic understanding of Fu’s phosphine ligand-controlled SM coupling of chloroaryl triflates.4 DFT calculations by Schoenebeck and Houk,18 as well as studies by Sigman,6 showed that selectivity in Fu’s system relates to palladium’s ligation state by phosphine during oxidative addition. PdL2, favored with PCy3, reacts at the C—OTf site of 1 while PdL, favored with PtBu3, reacts at C—Cl (Scheme 1A and 1B). This relationship between ligation state and selectivity was explained by the distortion-interaction model.18,19 PdL2 is more electron-rich than PdL and thus has a stronger interaction with the more electrophilic C—O site compared to C—Cl. It may be possible that SIMes behaves analogously to PCy3, and that [Pd(SIMes)2] is responsible for the selectivity observed with this ligand. However, our attempted calculations to locate sterically congested transition structures involving [Pd(SIMes)2] have all failed to converge.
An alternative explanation for the triflate selectivity observed with SIMes is that oxidative addition involves an anionic species [Pd(SIMes)X]− (X = small anionic ligand).20 Indeed, DFT calculations predict that an anionic transition structure involving [Pd(SIMes)(OH)]− (TS33a) favors reaction at triflate by 1.5 kcal mol−1 (Figure 2B). Formation of a palladium hydroxide species is plausible under the catalytic conditions, which include both water and base (KF).21 The preference for [Pd(SIMes)OH]− to react at OTf is consistent with previously reported calculations using phosphine ligands. Anionic [Pd(PtBu3)X]− (X = F− or PhB(OH)O−) has been shown computationally to favor oxidative addition at C—OTf over C—Cl.18,22 Interestingly however, previous studies suggest that involvement of putative [Pd(PtBu3)X]− is only favored in certain polar solvents like DMF and MeCN.23 In THF or other nonpolar solvents, the active catalyst is [Pd(PtBu3)] and preferential SM cross coupling at C—Cl is observed.22 As such, if [Pd(SIMes)X]− is the active catalyst in THF, it suggests that SIMes is better than PtBu3 at stabilizing anionic Pd. Further studies will be needed to distinguish between mechanisms involving [Pd(SIMes)2], [Pd(SIMes)X]−, or other active catalysts.24
Limitations to Selective Reaction at Chloride: Heteroaryl Substrates.
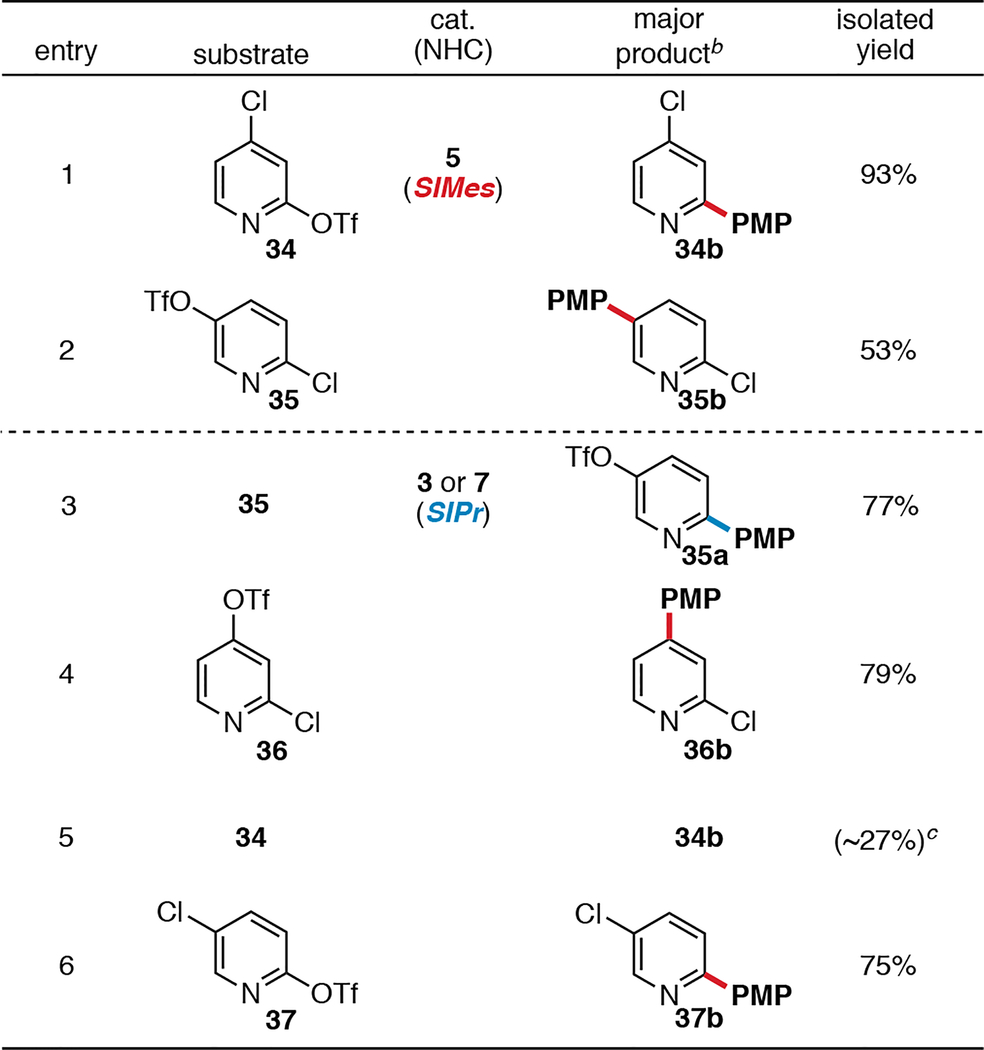
To test the limits of the NHC ligand-controlled selectivity, chloropyridyl triflates were examined as substrates for the SM reaction (Table 3). All else being equal, the reactivity of pyridine C—X bonds toward Pd(0) generally follows the order C2 > C4 > C3/C5.25 We were interested in whether the strong electronic biases of this substrate class would erode the NHC ligand-controlled selectivity. Furthermore, the mechanism of oxidative addition of chloropyridines at Pd(0) may be different from that of chloroarenes, which could affect selectivity.26 The results of the reactions of 34 and 35 with Pd/SIMes indicate that selectivity for triflate using SIMes remains ligand-controlled (entries 1–2). Selective cross-coupling at triflate of substrate 35 occurs even though pyridine electronics should favor reaction at the C2—Cl bond. Interestingly, however, the selectivity with SIPr is less predictable. Selective Pd/SIPr-catalyzed cross coupling at chloride only occurs with 35, the substrate that is most heavily electronically biased for reaction at C—Cl. With this substrate, cross coupling at C2—Cl takes place preferentially over reaction at C5—OTf to give 35a (entry 3). However, with substrates 36, 34, and 37, Pd/SIPr-catalyzed cross coupling takes place selectively at C—OTf (entries 4–6). The results with substrate 36 are particularly intriguing: reaction at chloride should be expected based on both the electronic bias of the pyridine ring and SIPr’s usual preference for reaction at chloride. However, coupling takes place selectively at C4—OTf. Further study is needed to understand these incongruous results and their possible relationship to differences in the mechanism of oxidative addition of chloroarenes compared to chloropyridines.26
Table 3.
Scope and limitations of the NHC ligand-controlled chemodivergent SM coupling of chloropyridyl triflates.a
|
aGeneral procedure: chloropyridyl triflate (1 equiv), p-methoxyphenylboronic acid (1.25–1.5 equiv), 3, 5, or 7 (3 mol %), KF (3 equiv), H2O (3.5–14.0 equiv), THF (entries 1, 2, 5), toluene (entries 3, 4), or THF/toluene (2:1, entry 6), 12 h, r.t. PMP = p-methoxyphenyl. b≤5% of the minor monoarylated products and ≤5% of diarylated products were detected by crude GC. cProduct not isolated; crude GC yield.
CONCLUSIONS
This work provides an improved method for chemodivergent Suzuki-Miyaura cross coupling of chloroaryl triflates. Although phosphine ligands (PtBu3 and PCy3) have been previously shown to control selectivity in the cross coupling of 1 with 2, the Pd/phosphine-catalyzed method is narrow in scope.4,7 In contrast, the use of Pd/NHC catalysts enables selective coupling with diverse chloroaryl triflates and arylboronic acids. Pd/SIPr favors cross coupling at chloride, while Pd/SIMes favors reaction at triflate.
DFT studies suggest that, with SIPr, oxidative addition takes place preferentially at chloride through a neutral monoligated transition state. However, monoligated [Pd(SIMes)] is not responsible for oxidative addition of C—OTf. Instead, triflate selectivity with SIMes may be attributed to oxidative addition at bisligated [Pd(SIMes)2] or [Pd(SIMes)X]−. Further study is needed to distinguish between these and other mechanistic possibilities for oxidative addition with Pd/SIMes.
The Pd/SIMes system remains selective for reaction of triflate even with electronically biased chloropyridyl triflate substrates. However, the Pd/SIPr-catalyzed cross coupling of chloropyridyl triflates does not clearly fit a pattern dictated by pyridine electronics or SIPr’s typical preference for chloride. The incongruous results with chloropyridyl triflates and SIPr may relate to differences in the mechanism for oxidative addition of chloropyridines versus chloroarenes.26 This possibility is the subject of ongoing study in our laboratory.
EXPERIMENTAL AND COMPUTATIONAL DETAILS
Computational Methods.
Calculations were performed with Gaussian 09.27 An ultrafine integration grid and the keyword 5d were used for all calculations. Unless otherwise specified in Section II-D of the Supporting Information, geometry optimizations of stationary points were carried out in the gas phase with the M06L28 functional with BS1 (BS1 = the SDD29 pseudopotential for Pd, the 6–31+G(d) basis set for O and Cl, and the the 6–31G(d) basis set for all other atoms). Frequency analyses were carried out at the same level to evaluate the zero-point vibrational energy and thermal corrections at 298 K. Gibbs free energy values are reported after applying Cramer and Truhlar’s anharmonic correction to frequencies that are less than 100 cm−1.30 The nature of the stationary points was determined in each case according to the appropriate number of negative eigenvalues of the Hessian matrix. Forward and reverse intrinsic reaction coordinate (IRC) calculations were carried out on the optimized transition structures to ensure that the TSs indeed connect the appropriate reactants and products.31 Multiple conformations were considered for all structures, and the lowest energy conformations are reported. Unless otherwise specified in Section 1D of the Supporting Information, single point energy calculations were performed on the gas-phase optimized geometries using the M06 functional with BS2 (BS2 = the SDD pseudopotential for Pd and the 6–311++G(2d,p) basis set for all other atoms). Bulk solvent effects in tetrahydrofuran were considered implicitly in the single point energy calculations through the CPCM continuum solvation model.32 Images of optimized structures were generated with CYLview.33
General Materials and Methods.
PEPPSI™-SIPr (7) and Pd(OAc)2 were obtained from Sigma-Aldrich and used as received. (η3-1-tBu-indenyl)2(μ-Cl)2Pd2, (η3-1-tBuindenyl)Pd(IMes)(Cl), and (η3-1-tBu-indenyl)Pd(IPr)(Cl) were synthesized from a literature procedure.10 NMR spectra were recorded at 298 K on a Bruker DRX 500 MHz (500.233 MHz for 1H, 125.795 MHz for 13C, 470.639 MHz for 19F) or a Bruker DPX 300 MHz (300.172 MHz for 1H) spectrometer. 1H and 13C NMR chemical shifts are reported in parts per million (ppm) relative to TMS, with the residual solvent peak used as an internal reference [1H NMR: CHCl3 (7.26 ppm) or C6D5H (7.16 ppm); 13C NMR: CDCl3 (77.16 ppm) or C6D6 (128.06 ppm)]. 19F chemical shifts are reported in ppm relative to hexafluorobenzene (−164.9 ppm). Multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublets (dd), doublet of doublets of doublets (ddd), triplet (t), triplet of doublets (td), triplet of triplets (tt), quartet (q), and multiplet (m). GC data were collected using a Shimadzu GC-2010 Plus with a flame ionization detector equipped with a SH-Rxi-5ms capillary column (15 m x 0.25 mm ID x 0.25 μm df). HRMS data were collected on an Agilent 6538 UHD-QTOF or a Bruker MicroTOF. LRMS (GC-MS) data were collected on an Agilent 7890A/5975C GC/MSD system equipped with an Agilent J&W VF-5ms column (30 m x 0.25 mm x 0.25 μm). Melting points were measured using a Thomas-Hoover “Uni-Melt” capillary melting point apparatus. Flash column chromatography was performed on SiliCycle silica gel 60 (40–63 μm particle size) and thin layer chromatography was performed on SiliCycle TLC plates pre-coated with extra hard silica gel 60 F254.
General Procedure for Synthesis of Chloroaryl Triflates.
Chloroaryl triflates were prepared according to a modified literature procedure.34 To an oven-dried 250 mL round-bottom flask equipped with a stir bar was added the chlorophenol (1 equiv). The flask was then sealed with a septum and subjected to 3 evacuation-refill cycles using N2. Degassed pyridine (2 equiv) and CH2Cl2 were added by syringe. The solution was cooled to 0 °C and trifluoromethanesulfonate anhydride (1.2 equiv) was added dropwise over 15 minutes. The solution was allowed to warm to room temperature and stir for 16 hours under N2. The reaction was quenched with 5% aqueous HCl (2/3 of the volume of CH2Cl2 used) and diluted with Et2O (2/3 of the volume of CH2Cl2 used). The layers were separated, and the aqueous layer was extracted with Et2O (3 x 2/3 of the volume of CH2Cl2 used). The combined organic layers were washed with 10% aqueous NaHCO3 (1 x the volume of CH2Cl2 used) and brine (1 x 2/3 of the volume of CH2Cl2 used), dried over MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography on silica gel or by taking up the residue in a nonpolar solvent and filtering through a silica plug.
4-Chlorophenyl trifluoromethanesulfonate (1): Chloroaryl triflate 1 was prepared according to the general procedure using 4-chlorophenol (2.57 g, 20 mmol, 1.0 equiv), pyridine (3.2 mL, 40 mmol, 2.0 equiv), CH2Cl2 (30 mL), and Tf2O (4 mL, 24 mmol, 1.2 equiv). The crude residue was purified by flash column chromatography on silica gel (Rf = 0.45 in 100% hexanes) to yield 1 as a pale yellow oil (3.40 g, 65% yield). 1H NMR (500 MHz, CDCl3, δ): 7.43 (d, J = 9.1 Hz, 2H), 7.23 (d, J = 9.1 Hz, 2H); 13C{1H} NMR (126 MHz, CDCl3, δ): 148.0, 134.5, 130.5, 122.9, 118.9 (q, 1JCF = 320.7 Hz); 19F NMR (470 MHz, CDCl3, δ): −72.7. Spectral data are consistent with those previously reported.35
3-Chlorophenyl trifluoromethanesulfonate (23): Chloroaryl triflate 23 was prepared according to the general procedure using 3-chlorophenol (1.93 g, 15 mmol, 1 equiv), pyridine (2.4 mL, 30 mmol, 2.0 equiv), CH2Cl2 (20 mL), and Tf2O (3 mL, 18 mmol, 1.2 equiv). The crude residue was purified by flash column chromatography on silica gel (Rf = 0.45 in 100% hexanes) to yield 23 as a colorless oil (3.15 g, 81% yield). 1H NMR (500 MHz, CDCl3, δ): 7.38–7.42 (multiple peaks, 2H), 7.30–7.32 (m, 1H), 7.18–7.23 (m, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 149.7, 135.9, 131.1, 129.0, 122.2, 119.9, 119.0 (q, 1JCF = 320.2 Hz); 19F NMR (470 MHz, CDCl3, δ): −72.7. Spectral data are consistent with those previously reported.35
2-Chlorophenyl trifluoromethanesulfonate (24): Chloroaryl triflate 24 was prepared according to the general procedure using 2-chlorophenol (1.29 g, 10 mmol, 1 equiv), pyridine (1.6 mL, 20 mmol, 2.0 equiv), CH2Cl2 (15 mL), and Tf2O (2 mL, 12 mmol, 1.2 equiv). The crude residue was purified by flash column chromatography on silica gel (Rf = 0.32 in 100% hexanes) to yield 24 as a colorless oil (1.75 g, 67% yield). 1H NMR (500 MHz, CDCl3, δ): 7.56–7.51 (m, 1H), 7.38–7.31 (multiple peaks, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 145.9, 131.5, 129.4, 128.5, 127.4, 123.2, 118.7 (q, 1JC-F = 320.7 Hz); 19F NMR (470 MHz, CDCl3, δ): −73.5. Spectral data are consistent with those previously reported.35
4-Chloro-2-methylphenyl trifluoromethanesulfonate (25): Chloroaryl triflate 25 was prepared according to the general procedure using 4-chloro-2-methylphenol (0.5 g, 3.5 mmol, 1 equiv), pyridine (0.56 mL, 7 mmol, 2.0 equiv), CH2Cl2 (5 mL), and Tf2O (0.71 mL, 4.2 mmol, 1.2 equiv). The crude residue was purified by flash column chromatography on silica gel (Rf = 0.43 in 100% hexanes) to yield 25 as a colorless oil (1.21 g, 41% yield). 1H NMR (300 MHz, CDCl3, δ): 7.31 (d, J = 2.1 Hz, 1H), 7.24 (m, 1H, overlaps with solvent), 7.18 (d, J = 7.2 Hz, 1H), 2.37 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 147.0, 134.0, 133.0, 132.2, 127.9, 122.7, 118.8 (q, 1JCF = 319.8 Hz), 16.5; 19F NMR (470 MHz, CDCl3, δ): −73.7. HRMS (ESI/Q-TOF) m/z: [M - H]− Calcd for C8H5ClF3O3S− 272.9606; Found 272.9612.
4-Chloro-3-(trifluoromethyl)phenyl trifluoromethanesulfonate (26): Chloroaryl triflate 26 was prepared according to the general procedure using 4-chloro-3-(trifluoromethyl)phenol (487.4 mg, 2.5 mmol, 1 equiv), pyridine (0.4 mL, 5 mmol, 2 equiv), CH2Cl2 (5 mL), and Tf2O (0.5 mL, 3 mmol, 1.2 equiv). The crude residue was purified by dissolving in pentane and filtering through a silica plug to remove solid impurities before drying in vacuo to yield 26 as a colorless oil (676.4 mg, 83% yield). 1H NMR (500 MHz, CDCl3, δ): 7.62 (d, J = 8.8 Hz, 1H), 7.61 (d, J = 3.1 Hz, 1H), 7.43 (dd, J = 8.8, 3.1 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 147.5, 133.6, 132.8, 130.7 (q, 2JCF = 32.9 Hz), 126.0, 121.6 (q, 1JCF = 274.5 Hz), 121.3 (q, 3JCF = 5.6 Hz), 118.9 (q, 1JCF = 322.2 Hz); 19F NMR (470 MHz, CDCl3, δ): −73.5, −63.4. Anal. Calcd for C8H3ClF6O3S: C, 29.24; H, 0.92. Found: C, 29.21; H, 0.92.
2-Chloro-4-formylphenyl trifluoromethanesulfonate (27): Chloroaryl triflate 27 was prepared according to the general procedure using 3-chloro-4-hydroxybenzaldehyde (187.9 mg, 1.2 mmol, 1 equiv), pyridine (0.3 mL, 3.6 mmol, 2 equiv), CH2Cl2 (2.9 mL), and Tf2O (0.29 mL, 3.6 mmol, 1.2 equiv). The crude residue was purified by flash column chromatography on silica gel (Rf = 0.36 in 95% hexanes, 5% acetone) to yield 27 as a colorless oil (131.6 mg, 38% yield). 1H NMR (500 MHz, CDCl3, δ): 10.01 (s, 1H), 8.06 (d, J = 2.0 Hz, 2H), 7.88 (d, J = 8.5, 2.0 Hz, 2H), 7.56 (d, J = 8.5 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3, δ): 188.9, 149.3, 136.5, 132.1, 129.4, 128.8, 123.9, 118.6 (q, 1JCF = 320 Hz); 19F NMR (470 MHz, CDCl3, δ): −73.2. Spectral data are consistent with those previously reported.36
3-Chloro-4-methylphenyl trifluoromethanesulfonate (28): Chloroaryl triflate 28 was prepared according to the general procedure using 3-chloro-4-methylphenol (171.1 mg, 1.2 mmol, 1 equiv), pyridine (0.29 mL, 1.8 mmol, 2 equiv), CH2Cl2 (3 mL), and Tf2O (0.3 mL, 1.8 mmol, 1.2 equiv). The crude residue was purified by dissolving in hexanes and filtering through a silica plug to remove solid impurities before drying in vacuo to yield 28 as a colorless oil (198 mg, 60% yield). 1H NMR (500 MHz, CDCl3, δ): 7.30 (s, 1H), 7.24 (d, J = 8.6 Hz, 1H, overlaps with solvent), 7.18 (d, J = 8.6 Hz, 1H), 2.37 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 146.9, 134.0, 133.0, 132.1, 127.8, 122.7, 118.8 (q, 1JCF = 320.2 Hz), 16.4. 19F NMR (470 MHz, CDCl3, δ): −73.7. Anal. Calcd for C8H6ClF3O3S: C, 34.99; H, 2.20; Found: C, 35.19; H, 2.22.
2-Chloro-5-formylphenyl trifluoromethanesulfonate (29): Chloroaryl triflate 29 was prepared according to the general procedure using 4-chloro-3-hydroxybenzaldehyde (388.3 mg, 2.5 mmol, 1 equiv), pyridine (0.4 mL, 5 mmol, 2 equiv), CH2Cl2 (5 mL), and Tf2O (0.5 mL, 3 mmol, 1.2 equiv). The crude residue was purified by dissolving in hexanes and Et2O and filtering through a silica plug to remove solid impurities before drying in vacuo to yield 29 as a yellow oil (243.4 mg, 34% yield). 1H NMR (500 MHz, CDCl3, δ): 10.09 (s, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.85 (s, 1H), 7.73 (d, J = 8.4 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 188.9, 146.5, 136.6, 134.0, 132.4, 130.1, 123.3, 118.7 (q, 1JCF = 322.7 Hz); 19F NMR (470 MHz, CDCl3, δ): −73.2. HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C8H4ClF3O4S+ 287.9471; Found 287.9475. The product gradually decomposes in ambient conditions.
4-(4-Chlorobenzoyl)phenyl trifluoromethanesulfonate (30): Chloroaryl triflate 30 was prepared according to the general procedure using 4-chloro-4’-hydroxybenzophenone (500 mg, 2.15 mmol, 1 equiv), pyridine (0.34 mL, 4.3 mmol, 2 equiv), CH2Cl2 (3 mL), and Tf2O (0.43 mL, 2.58 mmol, 1.2 equiv). The crude residue was purified by flash column chromatography on silica gel (Rf = 0.48 in 90% hexanes, 10% acetone) to yield 30 as a colorless oil (203.9 mg, 26% yield). 1H NMR (500 MHz, CDCl3, δ): 7.88 (d, J = 8.8 Hz, 2H), 7.75 (d, J = 8.5 Hz, 2H), 7.49 (d, J = 8.6 Hz, 2H), 7.41 (d, J = 8.6 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3, δ): 193.6, 152.2, 139.8, 137.4, 135.1, 132.1, 131.5, 129.1, 121.7, 118.9 (q, 1JCF = 321.4 Hz); 19F NMR (470 MHz, CDCl3, δ): −72.7. Spectral data are consistent with those previously reported.37 The product gradually decomposes in ambient conditions.
4-Chloropyridin-2-yl trifluoromethanesulfonate (34): Chloropyridyl triflate 34 was prepared according to the general procedure using 4-chloro-2-hydroxypyridine (321.3 mg, 2.5mmol, 1 equiv), pyridine (0.40 mL, 5 mmol, 2 equiv), CH2Cl2 (5 mL), and Tf2O (0.50 mL, 3 mmol, 1.2 equiv). The crude residue was purified by dissolving in hexanes and filtering through a silica plug to remove solid impurities before drying in vacuo to yield 34 as a yellow oil (418.6 mg, 64% yield). 1H NMR (500 MHz, CDCl3, δ): 8.32 (d, J = 5.4 Hz, 1H), 7.40 (dd, J = 5.4, 1.6 Hz, 1H), 7.21 (d, J = 1.6 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 156.3, 149.2, 148.1, 124.9, 118.7 (q, 1JCF = 320.7 Hz), 115.9; 19F NMR (470 MHz, CDCl3, δ): −73.0. HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C6H3ClF3NO3S+ 260.9474; Found 260.9484.
6-Chloropyridin-3-yl trifluoromethanesulfonate (35): Chloroaryl triflate 35 was prepared according to the general procedure using 2-chloro-5-hydroxypyridine (155.4 mg, 1.2 mmol, 1 equiv), pyridine (0.29 mL, 3.6 mmol, 2 equiv), CH2Cl2 (3 mL), and Tf2O (0.3 mL, 1.8 mmol, 1.2 equiv). The crude residue was purified by dissolving in hexanes and filtering through a silica plug to remove solid impurities before drying in vacuo to yield 35 as a white solid (235.4 mg, 75% yield). 1H NMR (500 MHz, CDCl3, δ): 8.39 (d, J = 2.9 Hz, 1H), 7.61 (d, J = 8.8, 2.9 Hz, 1H), 7.45 (d, J = 8.8 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 151.1, 145.8, 142.8, 132.0, 125.8, 118.8 (q, 1JCF = 321.9 Hz); 19F NMR (470 MHz, CDCl3, δ): −72.3. Spectral data are consistent with those previously reported.7
2-Chloropyridin-4-yl trifluoromethanesulfonate (36): Chloroaryl triflate 36 was prepared according to the general procedure using 2-chloro-4-hydroxypyridine (453.4 mg, 3.5 mmol, 1 equiv), pyridine (0.56 mL, 7 mmol, 2 equiv), CH2Cl2 (3 mL), and Tf2O (0.7 mL, 4.2 mmol, 1.2 equiv). The crude residue was purified by dissolving in hexanes and filtering through a silica plug to remove solid impurities before drying in vacuo to yield 36 as a colorless oil (677.5 mg, 74% yield). 1H NMR (500 MHz, CDCl3, δ): 8.52 (d, J = 5.6 Hz, 1H), 7.31 (d, J = 1.9 Hz, 1H), 7.21 (dd, J = 5.6, 1.9 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 157.1, 153.6, 152.0, 118.6 (q, 1JCF = 322.4 Hz), 117.2, 115.3. 19F NMR (470 MHz, CDCl3, δ): −72.6. Anal. Calcd for C6H3ClF3NO3S: C, 27.55; H, 1.16; N, 5.35. Found: C, 27.27; H, 1.13; N, 5.26.
5-Chloropyridin-2-yl trifluoromethanesulfonate (37): Chloroaryl triflate 37 was prepared according to the general procedure using 5-chloro-2-hydroxypyridine (453.4 mg, 3.5 mmol, 1 equiv), pyridine (0.56 mL, 7 mmol, 2 equiv), CH2Cl2 (3 mL), and Tf2O (0.7 mL, 4.2 mmol, 1.2 equiv). The crude residue was purified by dissolving in hexanes and filtering through a silica plug to remove solid impurities before drying in vacuo to yield 37 as a colorless oil (460.7 mg, 68% yield). 1H NMR (500 MHz, CDCl3, δ): 8.35 (d, J = 2.5 Hz, 1H), 7.86 (dd, J = 8.7, 2.5 Hz, 1H), 7.16 (d, J = 8.7 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 154.0, 147.6, 140.7, 132.6, 118.7 (q, 1JCF = 320.6 Hz), 116.4. 19F NMR (470 MHz, CDCl3, δ): −73.0. Spectral data are consistent with those previously reported.38
Synthesis of (η3-1-tBu-indenyl)Pd(SIPr)Cl (3): In a nitrogen-atmosphere glovebox, (η3-1-tBu-indenyl)2(μ-Cl)2Pd2 (100 mg, 0.16 mmol, 1 equiv), SIPr (125 mg, 0.32 mmol, 2 equiv), and a 1:1 mixture of THF/Et2O (5 mL) were combined in a 20 mL vial equipped with a magnetic stir bar. The vial was sealed with a PTFE-lined cap and the reaction was allowed to stir for 2 h at room temperature. The vial was removed from the glovebox, opened to air, and the contents filtered through celite to afford a deep red filtrate. The filtrate was concentrated under vacuum, triturated with hexanes, and the residue was purified by recrystallization from DCM/hexanes to yield 3 as orange-red needles (225 mg, 80% yield). 1H NMR (500 MHz, C6D6, δ): 7.23 (dd, J = 15.3, 7.6 Hz, 2H), 7.15 (d, 3H, overlaps with solvent), 7.07 (dd, J = 7.7, 1.4 Hz, 2H), 6.80 (td, J = 7.6, 1.0 Hz, 1H), 6.39 (t, J = 7.4 Hz, 1H), 6.06 (d, J = 3.0 Hz, 1H), 5.83 (d, J = 7.4 Hz, 1H), 5.20 (d, J = 3.0 Hz, 1H), 3.72–3.38 (multiple peaks, 8H), 1.57 (d, J = 6.6 Hz, 6H), 1.42 (s, 9H), 1.18 (d, J = 6.6 Hz, 6H), 1.13 (d, J = 6.8 Hz, 6H), 1.06 (d, J = 6.8 Hz, 6H); 13C{1H} NMR (125 MHz, C6D6, δ): 204.6, 147.8, 147.5, 139.3, 139.0, 137.2, 129.2, 124.7, 124.5, 124.2, 119.4, 117.0, 116.0, 107.4, 64.0, 53.7, 35.0, 34.9, 34.2, 32.0, 30.0, 29.4, 28.9, 28.6, 26.7, 26.5, 25.7, 24.4, 23.5, 23.1, 20.9, 18.0, 14.3, 11.7; HRMS (ESI/Q-TOF) m/z: [M − Cl]+ Calcd for [C40H53N2Pd]+ 667.3238; Found 667.3253.
Synthesis of (η3-1-tBu-indenyl)Pd(SIMes)Cl (5): In a nitrogen-atmosphere glovebox, (η3-1-tBu-indenyl)2(μ-Cl)2Pd2 (100 mg, 0.16 mmol, 1 equiv), SIMes (98 mg, 0.32 mmol, 2 equiv), and a 1:1 mixture of THF/Et2O (5 mL) were combined in a 20 mL vial equipped with a magnetic stir bar. The vial was sealed with a PTFE-lined cap and the reaction was allowed to stir for 2 h at room temperature. The vial was removed from the glovebox, opened to air, and the contents filtered through celite to afford a deep red filtrate. The filtrate was concentrated under vacuum, triturated with hexanes, and the residue was purified by recrystallization from DCM/hexanes to yield 5 as red crystals (198 mg, 87% yield). 1H NMR (500 MHz, C6D6, δ): 7.09 (d, J = 7.8 Hz, 1H), 6.80 (s, 2H), 6.76 (td, J = 8.0, 1.1 Hz, 1H), 6.68 (s, 2H), 6.57 (td, J = 7.8, 0.8 Hz, 1H), 6.48 (d, J = 7.4 Hz, 1H), 5.88 (d, J = 2.8 Hz, 1H), 5.05 (d, J = 2.6 Hz, 1H), 3.13 (m, 4H), 2.39 (s, 6H), 2.27 (s, 6H), 2.15 (s, 6H), 1.42 (s, 9H); 13C{1H} NMR (125 MHz, C6D6, δ): 203.8, 139.2, 139.0, 137.8, 136.6, 129.6, 129.5, 125.2, 122.9, 119.6, 117.7, 115.5, 108.3, 64.2, 50.7, 34.2, 29.7, 21.0, 19.1, 18.7; HRMS (ESI/Q-TOF) m/z: [M − Cl]+ Calcd for [C34H41N2Pd]+ 583.2299; Found 583.2287.
General Procedure for Chemoselective SM Coupling.
Pd catalyst 3, 5, or 7 (3 mol %), KF (3 equiv), and boronic acid (1–1.5 equiv) were combined in a 4 mL vial. The vial was transferred into a nitrogen-atmosphere glovebox, and solvent and chloroaryl triflate substrate (1 equiv) were added. The vial was sealed with a cap equipped with a PTFE-lined septum and removed from the glovebox. Within 60 seconds, degassed water was added through the septum cap, and the puncture hole in the septum was sealed with grease. The reactions were stirred at room temperature for 12 h unless otherwise indicated. The reaction mixture was diluted with Et2O and filtered through celite. The product was purified according to procedures A, B, or C as follows. Purification Procedure A: The crude filtrate was concentrated onto silica under vacuum and dry-loaded onto a silica column for purification by flash column chromatography. Purification Procedure B: The crude filtrate was concentrated under vacuum and purified by flash column chromatography or preparatory thin layer chromatography. Following isolation, the product was stored under nitrogen or in a desiccator to avoid hydrolysis. Purification Procedure C: The crude filtrate was concentrated under vacuum. The residue was dissolved in a minimal amount of acetonitrile or DMSO/acetone and purified by reverse-phase preparatory HPLC using H2O/MeCN (note: no acidic additives were added to the mobile phase in order to avoid hydrolysis of the product). The aqueous fractions were extracted with pentane, dried over MgSO4, filtered, concentrated to a small volume, and passed over a plug of silica pretreated with 1% triethylamine/99% hexanes. The silica plug was rinsed with pentane and the collected solution was concentrated under vacuum. Following isolation, the product was stored under nitrogen or in a desiccator to avoid hydrolysis.
General Procedure for Larger Scale Chemoselective SM Coupling.
Pd catalyst 3 or 5 (1.5 mol %), KF (3 equiv), and boronic acid (1.1 equiv) were combined in a 20 mL vial. The vial was transferred into a nitrogen-atmosphere glovebox, and solvent and chloroaryl triflate substrate (1 equiv) were added. The vial was sealed with a rubber septum and removed from the glovebox. Within 60 seconds, degassed water was added through the septum, and the puncture hole in the septum was sealed with grease. The reactions were stirred vigorously at room temperature for 12 h. The reaction mixture was diluted with Et2O and filtered through celite. The crude filtrate was dissolved in MeCN, then filtered to remove solid precipitates. The crude material was then purified by flash column chromatography.
4’-Chloro-2-methyl-1,1’-biphenyl (8b): Product 8b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 2-tolylboronic acid (68.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.62 in 100% hexanes) provided 8b as a colorless oil (76.2 mg, 94% yield). 1H NMR (500 MHz, CDCl3, δ): 7.38 (d, J = 8.4 Hz, 2H), 7.28–7.21 (multiple peaks, 5H, overlaps with solvent), 7.19 (d, J = 6.9 Hz, 1H), 2.25 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 140.8, 140.5, 135.4, 133.0, 130.7, 130.6, 129.8, 128.4, 127.7, 126.0, 20.5. Spectral data are consistent with those previously reported.4 Larger scale reaction: Product 8b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 437.5 μL, 2.5 mmol, 1 equiv), 2-tolylboronic acid (373.9 mg, 2.75 mmol, 1.1 equiv) KF (435.6 mg, 7.5 mmol, 3 equiv), 5 (18.6 mg, 0.03 mmol, 1.5 mol %), degassed H2O (312.5 μL, 17.25 mmol, 7 equiv), and THF (5 mL). Purification by flash column chromatography (Rf = 0.62 in 100% hexanes) provided 8b as a colorless oil (366.9 mg, 72% yield). Spectral data are identical to those reported above.
4-Chloro-1,1’-biphenyl (10b): Product 10b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), phenylboronic acid (61.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (1.6 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.69 in 100% hexanes) provided 10b as a white solid (66.2 mg, 88% yield). 1H NMR (500 MHz, CDCl3, δ): 7.58–7.51 (multiple peaks, 4H), 7.48–7.40 (multiple peaks, 4H), 7.37 (tt, J = 7.5, 1.3 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 140.1, 139.8, 133.5, 129.05, 129.02, 128.5, 127.7, 127.1. Spectral data are consistent with those previously reported.4
4’-Chloro-3-methyl-1,1’-biphenyl (11b): Product 11b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 3-tolylboronic acid (68.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.69 in 100% hexanes) provided 11b as a white solid (75.7 mg, 93% yield). 1H NMR (500 MHz, CDCl3, δ): 7.52 (d, J = 8.6 Hz, 2H). 7.40 (d, J = 8.6 Hz, 2H), 7.38–7.31 (multiple peaks, 3H), 7.18 (d, J = 6.9 Hz, 1H), 2.43 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 140.1, 139.9, 138.7, 133.4, 129.0, 128.9, 128.54, 128.48, 127.9, 124.2, 21.7. Spectral data are consistent with those previously reported.39
4-Chloro-4’-methoxy-1,1’-biphenyl (12b): Product 12b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (91.2 mg, 0.6 mmol, 1.5 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.41 in 3% EtOAc, 97% hexanes) provided 12b as a white solid (78.7 mg, 90% yield). 1H NMR (500 MHz, CDCl3, δ): 7.49 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.5 Hz, 2H), 7.38 (d, J = 8.5 Hz, 2H), 6.98 (d, J = 8.8 Hz, 2H), 3.85 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 159.5, 139.4, 132.8, 132.7, 129.0, 128.2, 128.1, 114.5, 55.5. Spectral data are consistent with those previously reported.22
4’-Chloro-[1,1’-biphenyl]-4-ol (13b): Product 13b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 4-hydroxyphenylboronic acid (82.8 mg, 0.6 mmol, 1.5 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.48 in 20% EtOAc, 10% benzene, 70% hexanes) provided 13b as an off-white solid (46.4 mg, 57% yield). 1H NMR (500 MHz, CDCl3, δ): 7.46 (d, J = 9.3 Hz, 2H), 7.44 (d, J = 9.3 Hz, 2H), 7.37 (d, J = 8.6 Hz, 2H), 6.90 (d, J = 8.6 Hz, 2H), 4.71 (s, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 155.4, 139.3, 133.0, 132.9, 129.0, 128.4, 128.1, 115.9. Spectral data are consistent with those previously reported.40
(4’-Chloro-[1,1’-biphenyl]-4-yl)methanol (14b): Product 14b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 4-(hydroxymethyl)phenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.3 in 20% EtOAc, 10% benzene, 70% hexanes) provided 14b as a white solid (52.9 mg, 60% yield). 1H NMR (500 MHz, CDCl3, δ): 7.56 (d, J = 8.3, 2H). 7.52 (d, J = 8.6, 2H), 7.45 (d, J = 8.3, 2H), 7.41 (d, J = 8.6, 2H), 4.75 (d, J = 4.0 Hz, 2H), 1.66 (s, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 140.3, 139.4, 139.3, 133.5, 129.0, 128.3, 127.5, 127.2, 65.1. Spectral data are consistent with those previously reported.41
4-Chloro-4’-fluoro-1,1’-biphenyl (15b): Product 15b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 4-fluorophenylboronic acid (78.4 mg, 0.56 mmol, 1.4 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (1.6 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.59 in 100% hexanes) provided 15b as a white solid (49.6 mg, 60% yield). 1H NMR (500 MHz, CDCl3, δ): 7.51 (dd, J = 8.5, 5.8 Hz, 2H), 7.47 (d, 2H, J = 8.5 Hz), 7.40 (d, J = 8.5 Hz, 2H), 7.13 (t, J = 8.5 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3, δ): 162.8 (d, 1JCF = 247.0 Hz), 138.9, 136.3 (d, 4JCF = 3.2 Hz), 133.6, 129.1, 128.7 (d, 3JCF = 8.2 Hz), 128.4, 115.92 (d, 2JCF = 21.76 Hz); 19F NMR (470 MHz, CDCl3, δ): −115.17. Spectral data are consistent with those previously reported.42
Methyl 4’-chloro-[1,1’-biphenyl]-4-carboxylate (16b): Product 16b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), (4-(methoxycarbonyl)phenyl)boronic acid (108.0 mg, 0.6 mmol, 1.5 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.41 in 5% EtOAc, 95% hexanes) provided 16b as a white solid (87.7 mg, 89% yield). 1H NMR (500 MHz, CDCl3, δ): 8.11 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 3.95 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 144.6, 138.6, 134.6, 130.4, 129.4, 129.3, 128.7, 127.1, 52.4. Spectral data are consistent with those previously reported.43
4,4’-Dichloro-1,1’-biphenyl (17b): Product 17b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 4-chlorophenylboronic acid (68.8 mg, 0.44 mmol, 1.1 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and DMF (0.8 mL), with a reaction time of 7 h at 60 °C, followed by purification procedure A. Purification by flash column chromatography (Rf = 0.62 in 100% hexanes) provided 17b as a white solid (81.7 mg, 92% yield). 1H NMR (500 MHz, CDCl3, δ): 7.48 (d, J = 8.4 Hz, 4H), 7.40 (d, J = 8.4 Hz, 4H); 13C{1H} NMR (125 MHz, CDCl3, δ): 138.6, 133.9, 129.2, 128.4. Spectral data are consistent with those previously reported.44
4-Chloro-4’-(trifluoromethyl)-1,1’-biphenyl (18b): Product 18b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), (4-(trifluoromethyl)phenyl)boronic acid (87.5 mg, 0.44 mmol, 1.1 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.67 in 100% hexanes) provided 18b as a white solid (82.2 mg, 80% yield). 1H NMR (500 MHz, CDCl3, δ): 7.70 (d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.3 Hz, 2H), 7.53 (d, J = 8.5 Hz, 2H), 7.45 (d, J = 8.5 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3, δ): 143.5, 138.2, 134.5, 129.7 (q, 2JCF = 32.4 Hz), 129.2, 128.5, 127.3, 125.9 (q, 3JCF = 3.7 Hz), 124.2 (q, 1JCF = 272.5 Hz); 19F NMR (470 MHz, CDCl3, δ): −62.5. Spectral data are consistent with those previously reported.45
4’-Chloro-[1,1’-biphenyl]-4-carbonitrile (19b): Product 19b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), (4-cyanophenyl)boronic acid (70.7 mg, 0.48 mmol, 1.2 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and DMF (0.8 mL), with a reaction temperature of 60 °C, followed by purification procedure A. Purification by flash column chromatography (Rf = 0.33 in 5% EtOAc, 95% hexanes) provided 19b as a white solid (52.9 mg, 62% yield). 1H NMR (500 MHz, CDCl3, δ): 7.73 (d, J = 8.6 Hz, 2H), 7.65 (d, J = 8.6 Hz, 2H), 7.52 (d, J = 8.6 Hz, 2H), 7.46 (d, J = 8.6 Hz, 2H); 13C{1H} NMR (125 MHz CDCl3, δ): 144.6, 137.8, 135.2, 132.9, 129.5, 128.7, 127.8, 118.9, 111.5. Spectral data are consistent with those previously reported.46
4’-Chloro-[1,1’-biphenyl]-4-carbaldehyde (20b): Product 20b was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), (4-formylphenyl)boronic acid (75.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.26 in 5% EtOAc, 95% hexanes) provided 20b as a white solid (68.2 mg, 79% yield). 1H NMR (500 MHz, CDCl3, δ): 10.06 (s, 1H), 7.96 (d, J = 8.3 Hz, 2H), 7.72 (d, J = 8.3 Hz, 2H), 7.57 (d, J = 8.6 Hz, 2H) 7.46 (d, J = 8.6 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3, δ): 191.9, 146.1, 138.3, 135.6, 134.9, 130.5, 129.4, 128.8, 127.7. Spectral data are consistent with those previously reported.47
3-Chloro-4’-methoxy-1,1’-biphenyl (23b): Product 23b was prepared according to the general procedure using 3-chlorophenyl trifluoromethanesulfonate (23, 70.5 μL, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (91.2 mg, 0.6 mmol, 1.5 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (1.6 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.52 in 5% EtOAc, 95% hexanes) provided 23b as a white solid (64.8 mg, 74% yield). 1H NMR (500 MHz, CDCl3, δ): 7.53 (dd, J = 1.9, 1.9 Hz 1H), 7.50 (d, J = 8.8 Hz, 2H), 7.42 (ddd, J = 7.9, 1.9, 1.9 Hz, 1H), 7.34 (dd, J = 7.9, 7.9 Hz, 1H), 7.27 (ddd, J = 7.9, 1.9, 1.9 Hz, 1H, overlaps with solvent), 6.98 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 159.6, 142.7, 134.6, 132.3, 129.9, 128.2, 126.8, 126.6, 124.8, 114.3, 55.4. Spectral data are consistent with those previously reported.48
2-Chloro-4’-methoxy-1,1’-biphenyl (24b): Product 24b was prepared according to the general procedure using 2-chlorophenyltrifluoromethanesulfonate (24, 69.5 μL, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (91.2 mg, 0.6 mmol, 1.5 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and DMF (0.8 mL) with a reaction time of 4 h at 60 °C followed by purification procedure A. Purification by flash column chromatography (Rf = 0.51 in 5% EtOAc, 95% hexanes) provided 24b as a pale yellow oil (67.8 mg, 77% yield). 1H NMR (300 MHz, CDCl3, δ): 7.48–7.18 (multiple peaks, 6H, overlaps with solvent), 6.96 (d, J = 8.3 Hz, 2H), 3.84 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 159.3, 140.3, 132.8, 132.0, 131.6, 130.8, 130.1, 128.4, 127.0, 113.67, 55.5. Spectral data are consistent with those previously reported.7
4-Chloro-4’-methoxy-2-methyl-1,1’-biphenyl (25b): Product 25b was prepared according to the general procedure using 4-chloro-2-methylphenyl trifluoromethanesulfonate (25, 74.5 μL, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and DMF (0.8 mL) with a reaction time of 4 h at 60 °C followed by purification procedure A. Purification by flash column chromatography (Rf = 0.64 in 10% CH2Cl2, 20% toluene, 70% hexanes) provided 25b as a pale yellow oil (84.9 mg, 91% yield). 1H NMR (300 MHz, CDCl3, δ): 7.25–7.15 (multiple peaks, 4H), 7.11 (d, J = 8.1 Hz, 1H), 6.93 (dd, J = 9.0, 4.9 Hz, 2H), 3.84 (s, 3H), 2.23 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 158.9, 140.2, 137.6, 133.3, 132.7, 131.3, 130.4, 130.2, 126.0, 113.8, 55.5, 20.6. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C14H14ClO 233.0728; Found 233.0728.
4-Chloro-4’-methoxy-3-(trifluoromethyl)-1,1’-biphenyl (26b): Product 26b was prepared according to the general procedure using 4-chloro-3-(trifluoromethyl)phenyl trifluoromethanesulfonate (26, 131.4 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.8 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure A. The crude residue was purified by flash column chromatography (Rf = 0.48 in 5% EtOAc, 95% hexanes) to yield 26b as a white solid (70.0 mg, 61% yield); m.p. 47–48 °C [uncorrected, measured against benzoic acid (109–112 °C)]. 1H NMR (500 MHz, CDCl3, δ): 7.85 (d, J = 2.0 Hz, 1H), 7.63 (dd, J = 8.4, 2.0 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.50 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 160.1, 139.9, 131.9, 131.2, 130.9, 130.5, 128.8 (q, 2JCF = 31.9 Hz), 128.3, 125.8 (q, 3JCF = 5.5 Hz), 123.1 (q, 1JCF = 272.6 Hz), 114.7, 55.6; 19F NMR (470 MHz, CDCl3, δ): −62.6. HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C 14H10ClF3O 286.0372; Found 286.0373.
2-Chloro-4’-methoxy-[1,1’-biphenyl]-4-carbaldehyde (27b): Product 27b was prepared according to the general procedure using 2-chloro-4-formylphenyl trifluoromethanesulfonate (27, 115.4 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.8 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure A. The crude residue was purified by flash column chromatography (Rf = 0.3 in 5% EtOAc, 95% hexanes) to yield 27b as a off-white solid (68.5 mg, 69% yield). 1H NMR (500 MHz, CDCl3, δ): 10.00 (s, 1H), 7.97 (d, J = 1.6 Hz, 1H), 7.80 (dd, J = 7.9, 1.6 Hz, 1H), 7.51 (d, J =7.9 Hz, 1H), 7.43 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 190.7, 160.0, 146.2, 136.3, 133.8, 132.2, 131.3, 130.7, 130.6, 128.0, 113.9, 55.5. Spectral data are consistent with those previously reported.49
3-Chloro-4’-methoxy-4-methyl-1,1’-biphenyl (28b): Product 28b was prepared according to the general procedure using 3-chloro-4-methylphenyl trifluoromethanesulfonate (28, 109.9 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL, 0.5 M) followed by purification procedure A. Purification by flash column chromatography (Rf = 0.63 in 15% EtOAc, 85% hexanes) provided 28b as a white solid (73.4 mg, 79% yield). 1H NMR (500 MHz, CDCl3), δ (ppm): 7.54 (d, J = 1.8 Hz, 1H), 7.49 (d, J = 8.7 Hz, 2H), 7.34 (dd, J = 7.8, 1.8 Hz, 1H), 7.25 (d, J = 7.8 Hz, 1H, overlaps with solvent), 6.97 (d, J = 8.7 Hz, 2H), 3.85 (s, 3H), 2.40 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3), δ (ppm): 159.5, 140.2, 134.8, 134.3, 132.5, 131.3, 128.1, 127.3, 125.0, 114.4, 55.5, 19.8. Spectral data are consistent with those previously reported.50
4-Chloro-2-(4-methoxyphenyl)pyridine (34b): With SIMes: Product 34b was prepared according to the general procedure using 4-chloropyridin-2-yl trifluoromethanesulfonate (34, 104.6 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (91.2 mg, 0.6 mmol, 1.5 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.8 mmol, 6.9 equiv), and THF (0.8 mL, 0.5 M) followed by purification procedure A. The crude residue was purified by flash column chromatography (Rf = 0.32 in 8% EtOAc, 92% hexanes) to yield 34b as a white solid (81.7 mg, 93% yield). 1H NMR (500 MHz, CDCl3, δ): 8.54 (d, J = 5.3 Hz, 1H); 7.94 (d, J = 8.8 Hz, 2H), 7.67 (d, J = 1.7 Hz, 1H), 7.18 (dd, J = 5.3, 1.7 Hz, 1H), 7.00 (d, J = 8.8 Hz, 2H), 3.87 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 161.1, 158.1, 150.5, 144.8, 130.9, 128.5, 121.7, 120.1, 114.4, 55.5. Spectral data are consistent with those previously reported.51 With SIPr: Product 34b was prepared according to the general procedure using using 4-chloropyridin-2-yl trifluoromethanesulfonate (34, 20.9 mg, 0.08 mmol, 1 equiv), 4-methoxyphenylboronic acid (15.2 mg, 0.1 mmol, 1.25 equiv), KF (13.9 mg, .24 mmol, 3 equiv), 3 (1.7 mg, 0.0024 mmol, 3 mol %), degassed H2O (10 μL, 0.56 mmol, 7 equiv), and THF (0.32 mL). Undecane was added as an internal GC standard, and the reaction mixture was diluted with Et2O, filtered through celite, and analyzed by GC, indicating 27% crude GC yield of 34b. The retention time of the product prepared by this method matches that of the analogous reaction with SIMes.
2-Chloro-5-(4-methoxyphenyl)pyridine (35b): Product 35b was prepared according to the general procedure using 2-chloropyridin-5-yl trifluoromethanesulfonate (35, 104.6 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 5 (7.5 mg, 0.012 mmol, 3 mol %), degassed H2O (100 μL, 5.6 mmol, 13.9 equiv), and THF (0.8 mL) followed by purification procedure A. The crude residue was purified by flash column chromatography (Rf = 0.35 in 10% EtOAc, 90% hexanes) to yield 35b as a white solid (47.0 mg, 53% yield). 1H NMR (500 MHz, CDCl3, δ): 8.56 (d, J = 2.6 Hz, 1H), 7.79 (dd, J = 8.4, 2.6 Hz, 1H), 7.48 (d, J = 8.7 Hz, 2H), 7.36 (d, J = 8.4 Hz, 1H), 7.01 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 160.2, 149.8, 147.7, 136.8, 135.4, 129.0, 128.3, 124.3, 114.8, 55.6. Spectral data are consistent with those previously reported.52
2-Chloro-4-(4-methoxyphenyl)pyridine (36b): Product 36b was prepared according to a modified procedure using 2-chloropyridin-4-yl trifluoromethanesulfonate (36, 104.6 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 7 (8.2 mg, 0.012 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and toluene (0.8 mL). The reaction was run at 60 °C, followed by purification procedure A. The crude residue was purified by flash column chromatography (Rf = 0.42 in 20% EtOAc, 80% hexanes) to yield 36b as a white solid (105.5 mg, 79% yield); 1H NMR (500 MHz, CDCl3, δ): 8.38 (d, J = 5.2 Hz, 1H), 7.58 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 1.2 Hz, 1H), 7.39 (dd, J = 5.2, 1.2 Hz, 1H), 7.01 (d, J = 8.8 Hz, 2H), 3.87 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 161.2, 152.4, 151.2, 150.0, 129.2, 128.4, 121.5, 120.0, 114.8, 55.6. Spectral data are consistent with those previously reported.53
5-Chloro-2-(4-methoxyphenyl)pyridine (37b): Product 37b was prepared according to the general procedure using 5-chloropyridin-2-yl trifluoromethanesulfonate (104.6 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.8 mmol, 6.9 equiv), and 2:1 THF/toluene (0.8 mL) purification procedure A. The crude residue was purified by flash column chromatography (Rf = 0.42 in 5% EtOAc, 95% hexanes) to yield 37b as a white solid (100.0 mg, 75% yield); m.p. 90–93 °C [uncorrected, measured against benzoic acid (109–112 °C)]. 1H NMR (500 MHz, CDCl3, δ): 8.59 (d, J = 2.3 Hz, 1H), 7.92 (d, J = 8.7 Hz, 2H), 7.68 (dd, J = 8.8, 2.3 Hz, 1H), 7.61 (d, J = 8.8 Hz, 1H) 7.00 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 160.8, 155.4, 148.5, 136.5, 131.0, 129.9, 128.3, 120.5, 114.4, 55.5. Spectral data are consistent with those previously reported.51
2’-Methyl-[1,1’-biphenyl]-4-yl trifluoromethanesulfonate (8a): Product 8a was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 2-tolylboronic acid (68.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.0120 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and toluene (0.8 mL) followed by purification procedure C. Purification by HPLC provided 8a as a colorless oil (85.1 mg, 67% yield). 1H NMR (500 MHz, CDCl3, δ): 7.39 (d, J = 8.6 Hz, 2H), 7.34–7.23 (multiple peaks, overlaps with solvent, 5H), 7.19 (d, J = 7.3 Hz, 1H), 2.25 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 148.7, 142.6, 140.1, 135.4, 131.2, 130.7, 129.8, 128.2, 126.2, 121.2, 118.9 (q, 1JCF = 320.8 Hz), 20.5; 19F NMR (470 MHz, CDCl3, δ): −72.8. Spectral data are consistent with those previously reported.4 Larger scale reaction: Product 8a was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 437.5 μL, 2.5 mmol, 1 equiv), 2-tolylboronic acid (373.9 mg, 2.75 mmol, 1.1 equiv) KF (435.6 mg, 7.5 mmol, 3 equiv), 3 (21.1 mg, 0.03 mmol, 1.5 mol %), degassed H2O (125 μL, 8.75 mmol, 3.5 equiv), and toluene (5 mL). Purification by flash column chromatography (Rf = 0.70 in 1% Et3N, 99% hexanes) provided 8a as a colorless oil (572.1 mg, 72% yield). Spectral data are identical to those reported above.
[1,1’-Biphenyl]-4-yl trifluoromethanesulfonate (10a): Product 10a was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 74.7 μL, 0.4 mmol, 1 equiv), phenylboronic acid (61.0 mg, 0.44 mmol, 1.1 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.0120 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and toluene (1.6 mL) followed by purification procedure B. The crude residue was purified by flash column chromatography (Rf = 0.59 in 100% pentane) to yield 10a as a white solid (104.4 mg, 86% yield). 1H NMR (500 MHz, CDCl3, δ): 7.65 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 7.7 Hz, 2H), 7.47 (m, 2H), 7.40 (tdd, J = 7.7, 1.4, 1.4 Hz, 1H), 7.35 (d, J = 8.8 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3, δ): 149.1, 141.9, 139.5, 129.1, 129.0, 128.2, 127.3, 121.8, 118.9 (q, 1JC-F = 322.1 Hz); 19F NMR (470 MHz, CDCl3, δ): −72.8. Spectral data are consistent with those previously reported.4
3’-Methyl-[1,1’-biphenyl]-4-yl trifluoromethanesulfonate (11a): Product 11a was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 3-tolylboronic acid (68.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and toluene (0.8 mL) followed by purification procedure A. Purification by flash column chromatography (Rf = 100% hexanes) provided 11a as a white solid (106.0 mg, 84% yield). 1H NMR (500 MHz, CDCl3, δ): 7.64 (d, J = 8.7 Hz, 2H); 7.38–7.31 (multiple peaks, 6H); 2.43 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 149.0, 142.0, 139.4, 138.8, 129.0, 128.9, 128.1, 127.6, 124.4, 121.7, 118.9 (q, 1JC-F = 322.0 Hz), 21.6; 19F NMR (470 MHz, CDCl3, δ): −72.8. HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C14H11F3O3S 316.0381; Found 316.0351.
4’-Methoxy-[1,1’-biphenyl]-4-yl trifluoromethanesulfonate (12a): Product 12a was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (91.2 mg, 0.6 mmol, 1.5 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.0120 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and dioxane (0.8 mL) followed by purification procedure B. The crude residue was purified by flash column chromatography (Rf = 0.41 in 1% Et3N, 3% benzene, 5% EtOAc, 91% hexanes) to yield 12a as a white solid (79.0 mg, 59% yield). 1H NMR (500 MHz, CDCl3, δ): 7.60 (d, J = 8.8 Hz, 2H), 7.49 (d, J = 8.8 Hz, 2H), 7.31 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 159.9, 148.6, 141.5, 131.9, 128.5, 128.4, 121.7, 119.0 (q, 1JCF = 321.2 Hz), 114.6, 55.5; 19F NMR (470 MHz, CDCl3, δ): −72.8. Spectral data are consistent with those previously reported.22
4’-Fluoro-[1,1’-biphenyl]-4-yl trifluoromethanesulfonate (15a): Product 15a was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 4-fluorophenylboronic acid (67.2 mg, 0.48 mmol, 1.2 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.0120 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and toluene (0.8 mL) followed by purification procedure B. The crude residue was purified by flash column chromatography (Rf = 0.37 in 1% Et3N, 5% benzene, 94% hexanes) to yield 15a as a colorless, low-melting solid (111.3 mg, 87% yield). 1H NMR (500 MHz, CDCl3, δ): 7.60 (d, J = 8.7 Hz, 2H), 7.52 (dd, 3JHH = 8.8 Hz, 3JHF = 5.3 Hz, 2H), 7.34 (d, J = 8.7 Hz, 2H), 7.16 (t, J = 8.8 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3, δ): 164.1, 162.1, 149.10, 140.91, 135.62 (d, 3JCF = 3.5 Hz), 128.95, 121.91, 119.14 (q, 1JCF = 320.8 Hz, CF3), 116.14 (d, 2JCF = 22.0 Hz); 19F NMR (470 MHz, CDCl3, δ): −72.8, −114.32; HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C13H8F4O3S 320.0130; Found 320.0111.
4’-(((Trifluoromethyl)sulfonyl)oxy)-[1,1’-biphenyl]-4-yl acetate (16a): Product 16a was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), (4-(methoxycarbonyl)phenyl)boronic acid (108.0 mg, 0.6 mmol, 1.5 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.0120 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and THF (0.8 mL, 0.5 M) followed by purification procedure B. The crude residue was purified by flash column chromatography (Rf = 0.38 in 1% Et3N, 10% EtOAc, 89% hexanes) to yield 16a as a white solid (98.0 mg, 68% yield). 1H NMR (500 MHz, CDCl3, δ): 8.13 (d, J = 8.2 Hz, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.63 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.8 Hz, 2H), 3.95 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 166.9, 149.6, 143.7, 140.7, 130.4, 129.9, 129.3, 127.3, 122.0, 119.0 (q, 1JCF = 322.0 Hz), 52.4; 19F NMR (470 MHz, CDCl3, δ): −72.7; HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C15H12F3O5S 361.0352; Found 361.0345.
4’-(Trifluoromethyl)-[1,1’-biphenyl]-4-yl trifluoromethanesulfonate (18a): Product 18a was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), (4-(trifluoromethyl)phenyl)boronic acid (91.2 mg, 0.48 mmol, 1.2 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.0120 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and toluene (0.8 mL) followed by purification procedure B. The crude residue was purified by flash column chromatography (Rf = 0.37 in 1% Et3N, 5% benzene, 94% hexanes) to yield 18a as a white solid (109.8 mg, 74% yield); m.p. 56–59 °C [uncorrected, measured against benzoic acid (108–112 °C)]; 1H NMR (500 MHz, CDCl3, δ): 7.73 (d, J = 8.1 Hz, 2H), 7.68–7.65 (multiple peaks, 4H), 7.39 (d, J = 8.8 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3, δ): 149.70, 142.97, 140.42, 130.40 (q, 2JCF = 32.9 Hz), 129.33, 127.74, 126.17 (q, 3JCF = 3.7 Hz), 124.29 (q, 1JCF = 272.5 Hz, ArCF3), 122.14, 119.05 (q, 1JCF = 320.0 Hz, SCF3); 19F NMR (470 MHz, CDCl3, δ): −62.6, −72.8; HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C14H8F6O3S 370.0098; Found 370.0087.
4’-Methyl-[1,1’-biphenyl]-4-yl trifluoromethanesulfonate (21a): Product 21a was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), 4-tolylboronic acid (68.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and toluene (0.8 mL) followed by purification procedure B. The crude residue was purified by preparatory TLC (Rf = 0.47 in 100% pentane) to yield 21a as a white solid (117.7 mg, 93% yield). 1H NMR (500 MHz, CDCl3, δ): 7.63 (d, J = 8.9 Hz, 2H), 7.46 (d, J = 8.1 Hz, 2H), 7.33 (d, J = 8.9 Hz, 2H), 7.27 (d, J = 8.1 Hz, 2H, overlaps with solvent), 2.41 (s, 3H, CH3); 13C{1H} NMR (125 MHz, CDCl3, δ): 148.9, 141.8, 138.2, 136.6, 129.9, 128.8, 127.2, 121.8, 119.0 (q, 1JCF = 323.1 Hz), 21.3; 19F NMR (470 MHz, CDCl3, δ): −72.8. Spectral data are consistent with those previously reported.54
4’-Vinyl-[1,1’-biphenyl]-4-yl trifluoromethanesulfonate (22a): Product 22a was prepared according to the general procedure using 4-chlorophenyl trifluoromethanesulfonate (1, 70 μL, 0.4 mmol, 1 equiv), (4-vinylphenyl)boronic acid (74.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and dioxane (0.8 mL), with a reaction time of 4 h at 60 °C, followed by purification procedure B. The crude residue was purified by flash column chromatography (Rf = 0.4 in 1% Et3N, 5% benzene, 94% hexanes) to yield 22a as a white solid (77.8 mg, 59% yield). BHT (1.0 mg) was added to the product following column chromatography in order to prevent polymerization during concentration and storage. The product mass reported above excludes the added mass of the BHT. 1H NMR (500 MHz, CDCl3, δ): 7.65 (d, J = 8.8 Hz, 2H), 7.53 (d, J = 8.6 Hz, 2H), 7.50 (d, J = 8.6 Hz, 2H), 7.34 (d, J = 8.8 Hz, 2H), 6.76 (dd, J = 17.8, 10.9 Hz, 1H), 5.82 (dd, J = 17.8, 0.6 Hz, 1H), 5.31 (dd, J = 10.9, 0.6 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3, δ): 149.1, 141.4, 138.7, 137.6, 136.3, 128.9, 127.5, 127.0, 121.9, 119.2 (q, 1JCF = 320.8 Hz), 114.8; 19F NMR (470 MHz, CDCl3, δ): −72.8; HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C15H12F3O3S 329.0454; Found 329.0450.
4’-Methoxy-[1,1’-biphenyl]-3-yl trifluoromethanesulfonate (23a): Product 23a was prepared according to the general procedure using 3-chlorophenyl trifluoromethanesulfonate (23, 70.5 μL, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (91.2 mg, 0.6 mmol, 1.5 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and toluene (1.6 mL) followed by purification procedure B. The crude residue was purified by flash column chromatography (Rf = 0.52 in 1% Et3N, 10% EtOAc, 89% hexanes) to yield 23a as a white solid (100.9 mg, 76% yield). 1H NMR (500 MHz, CDCl3, δ): 7.56 (ddd, J = 7.9, 1.6, 1.6 Hz, 1H), 7.53–7.46 (multiple peaks, 3H), 7.46 (dd, J = 1.6, 1.6 Hz, 1H), 7.20 (m, 1H), 7.00 (d, J = 8.9 Hz, 2H), 3.87 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 160.0, 150.2, 143.7, 131.6, 130.6, 128.4, 126.7, 119.6, 119.3, 119.0 (q, 1JCF = 320.9 Hz), 114.5, 55.41; 19F NMR (470 MHz, CDCl3, δ): −72.8. Spectral data are consistent with those previously reported.55
4’-Methoxy-[1,1’-biphenyl]-2-yl trifluoromethanesulfonate (24a): Product 24a was prepared according to the general procedure using 2-chlorophenyl trifluoromethanesulfonate (24, 69.5 μL, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure B. The crude residue was purified by flash column chromatography (Rf = 0.58 in 10% DCM, 20% benzene, 70% hexanes) to yield 24a as a pale yellow oil (80.1 mg, 60% yield). 1H NMR (300 MHz, CDCl3, δ): 7.50–7.32 (multiple peaks, 6H), 6.99 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 159.9, 147.1, 135.5, 132.1, 130.7, 128.7, 128.1, 122.2, 118.6 (q, 1JCF = 321.2 Hz), 114.2, 55.5 (two signals are coincidentally overlapping); 19F NMR (282 MHz, CDCl3, δ): −74.0; HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C14H11F3O4S 332.0330; Found 332.0307.
4’-Methoxy-2-methyl-[1,1’-biphenyl]-4-yl trifluoromethanesulfonate (25a): Product 25a was prepared according to the general procedure using 4-chloro-2-methylphenyl trifluoromethanesulfonate (25, 74.7 μL, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL, 0.5 M) followed by purification procedure B. Purification by flash column chromatography (Rf = 0.57 in 10% DCM, 20% benzene, 70% hexanes) provided 25a as a pale yellow oil (80.5 mg, 58% yield). 1H NMR (500 MHz, CDCl3), δ (ppm): 7.48 (d, J = 8.8 Hz, 2H), 7.46 (d, J = 2.1 Hz, 1H), 7.41 (dd, J = 8.6, 2.4 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H, overlaps with solvent), 6.98 (d, J = 8.6 Hz, 2H), 3.86 (s, 3H), 2.43 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3), δ (ppm): 159.8, 147.6, 141.2, 132.1, 131.1, 130.5, 128.4, 125.9, 121.6, 118.8 (q, 1JC-F = 320 Hz), 114.5, 55.5, 16.7. 19F NMR (282 MHz, CDCl3), δ (ppm): −73.8. HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C15H13F3O4S 346.0487; Found 346.0461.
4’-Methoxy-2’-(trifluoromethyl)-[1,1’-biphenyl]-4-yl trifluoromethanesulfonate (26a): Product 26a was prepared according to the general procedure using 4-chloro-3-(trifluoromethyl) phenyl trifluoromethanesulfonate (26, 131.4 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and 1:1 dioxane/toluene (1.6 mL) followed by purification procedure A. The crude residue was purified by flash column chromatography (Rf = 0.41 in 3% EtOAc, 97% hexanes, 1% Et3N) to yield 26a as a cololrless oil (80.3 mg, 50% yield). 1H NMR (500 MHz, CDCl3, δ): 7.64 (d, J = 1.7 Hz, 1H), 7.49 (dd, J = 8.4, 1.7 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H, 7.24 (d, J = 8.2 Hz, 2H), 6.95 (d, J = 8.2 Hz, 2H), 3.86 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 159.9, 148.1, 142.0, 134.6, 130.9 (q, 2JCF = 31.3 Hz), 130.2, 124.3, 123.0 (q, 1JCF = 278.3 Hz), 119.7 (q, 3JCF = 5.5 Hz), 119.0 (q, 1JCF = 319.9 Hz), 113.7, 55.4 (two signals are coincidentally overlapping); 19F NMR (282 MHz, CDCl3, δ): −57.7, −72.7. HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C15H10F6O4S 400.0204; Found 400.0178.
4’-Methoxy-2-methyl-[1,1’-biphenyl]-4-yl trifluoromethanesulfonate (28a): Product 28a was prepared according to the general procedure using 3-chloro-4-methylphenyl trifluoromethanesulfonate (28, 109.9 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.0120 mmol, 3 mol %), degassed H2O (50 μL, 2.76 mmol, 6.9 equiv), and THF (0.8 mL) followed by purification procedure B. The crude residue was purified by flash column chromatography (Rf = 0.50 in 1% NEt3, 4% EtOAc, 10% benzene, 85% hexanes) to yield 28a as a colorless oil (98.8 mg, 71% yield). 1H NMR (500 MHz, CDCl3, δ): 7.48 (d, J = 8.7 Hz, 2H), 7.46 (d, J = 2.3 Hz, 1H), 7.40 (dd, J = 8.7, 2.3 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H, overlaps with solvent), 6.98 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H), 2.44 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 159.8, 147.6, 141.2, 132.1, 131.1, 130.5, 128.4, 125.9, 121.6, 118.8 (q, 1JCF = 320.1 Hz), 114.5, 55.5, 16.7; 19F NMR (282 MHz, CDCl3, δ): −73.8; HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C15H13F3O4S 346.0487; Found 346.0467.
4-Formyl-4’-methoxy-[1,1’-biphenyl]-2-yl trifluoromethanesulfonate (29a): Product 29a was prepared according to the general procedure using 2-chloro-5-formylphenyl trifluoromethanesulfonate (29, 115.4 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and dioxane (0.8 mL) followed by purification procedure A. The crude residue was purified by flash column chromatography (Rf = 0.36 in 15% EtOAc, 85% hexanes) to yield 29a as a yellow oil (83.7 mg, 58% yield). 1H NMR (500 MHz, CDCl3, δ); 10.0 (s, 1H), 7.94 (dd, J = 7.9, 1.3 Hz, 1H), 7.86 (d, J = 1.3 Hz, 1H), 7.65 (d, J = 7.9 Hz, 1H), 7.45 (d, J = 8.5 Hz, 2H), 7.02 (d, J = 8.5 Hz, 2H), 3.87 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 189.9, 160.6, 147.4, 141.3, 136.6, 132.7, 130.8, 129.6, 126.9, 123.0, 118.5 (q, 1JCF = 320.9 Hz), 114.4, 55.5; 19F NMR (282 MHz, CDCl3, δ): −73.8. Anal. Calcd for C15H11F3O5S: C, 50.00; H, 3.08. Found: C, 49.99, H, 3.06.
4-[4’-Methoxy-(1,1’-biphenyl)-4-carbonyl]phenyl trifluoromethanesulfonate (30a): Product 30a was prepared according to the general procedure using 4-(4-chlorobenzoyl)phenyl trifluoromethanesulfonate (30, 145.9 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and toluene (1.6 mL) followed by purification procedure A. The crude residue was purified by flash column chromatography (Rf = 0.38 in 10% EtOAc, 10% acetone, 80% hexanes) to yield 30a as a white solid (124.4 mg, 57% yield); m.p. 124–128 °C [uncorrected, measured against benzoic acid (109–112 °C)].). 1H NMR (500 MHz, CDCl3, δ): 7.93 (d, J = 8.8 Hz, 2H), 7.86 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.8 Hz, 2H), 7.42 (d, J = 8.8 Hz, 2H), 7.02 (d, J = 8.8 Hz, 2H), 3.89 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 194.4, 160.2, 152.0, 145.7, 138.1, 134.5, 132.22, 132.19, 130.9, 128.6, 126.7, 121.5, 118.9 (q, 1JCF = 333.3 Hz), 114.7, 55.6; 19F NMR (282 MHz, CDCl3, δ): −72.7. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C21H15F3O5S 437.0671; Found 437.0652.
6-(4-Methoxyphenyl)pyridin-3-yl trifluoromethanesulfonate (35a): Product 35a was prepared according to the general procedure using 2-chloropyridin-5-yl trifluoromethanesulf onate (104.6 mg, 0.4 mmol, 1 equiv), 4-methoxyphenylboronic acid (76.0 mg, 0.5 mmol, 1.25 equiv), KF (69.7 mg, 1.2 mmol, 3 equiv), 3 (8.5 mg, 0.012 mmol, 3 mol %), degassed H2O (25 μL, 1.4 mmol, 3.5 equiv), and toluene (1.6 mL, 0.25 M) followed by workup A. The crude residue was purified by flash column chromatography (Rf = 0.28 in 5% EtOAc, 95% hexanes) to yield 35a as a white solid (103.1 mg, 77% yield); m.p. 63–64°C [uncorrected, measured against benzoic acid (109–112 °C)]. 1H NMR (500 MHz, CDCl3, δ); 8.60 (d, J = 2.8 Hz, 1H), 7.95 (d, J = 8.7 Hz, 2H), 7.75 (d, J = 8.9 Hz, 1H), 7.64 (dd, J = 8.9, 2.8 Hz, 1H), 7.01 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3, δ): 161.3, 157.3, 145.4, 142.4, 130.3, 129.7, 128.6, 120.5, 119.0 (q, 1JCF = 321.5 Hz), 114.5, 55.5; 19F NMR (282 MHz, CDCl3, δ): −72.5. HRMS (EI/Q-TOF) m/z: [M]+ Calcd for C13H10F3NO4S 333.0283; Found 333.0263.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NSF CAREER (CHE-1848090) and Montana State University. The NSF REU program (CHE-1461218) is acknowledged for support for J.N.H. Calculations were performed on Comet at SDSC through XSEDE (CHE-170089), which is supported by NSF (ACI-1548562). NMR spectra were recorded on an instrument purchased with support from the NSF (NSF-MRI:DBI-1532078) and the Murdock Charitable Trust Foundation (2015066:MNL). X-ray crystallographic data were collected at the University of Montana X-ray diffraction core facility (NIH, CoBRE NIGMS P20GM103546) using an instrument principally supported by NSF (MRI CHE-1337908). We thank Daniel A. Decato for his assistance in solving crystal structures and the Cloninger lab at MSU for use of HPLC.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website.
NMR spectra, crystallographic details, computational details, energies and Cartesian coordinates of minimum energy calculated structures (PDF)
X-ray crystallographic data for 3 and 5 (CIF)
The authors declare no competing financial interest.
REFERENCES
- 1.Selected reviews: (a) Magano J; Dunetz JR Large-Scale Applications of Transition Metal-Catalyzed Couplings for the Synthesis of Pharmaceuticals. Chem. Rev 2011, 111, 2177–2250; [DOI] [PubMed] [Google Scholar]; (b) Miyaura N; Suzuki A Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev 1995, 95, 2457–2483; [Google Scholar]; (c) Seechurn CCCJ; Kitching MO; Colacot TJ; Snieckus V, Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed 2012, 51, 5062–5085; [DOI] [PubMed] [Google Scholar]; (d) Suzuki A Cross-Coupling Reactions Of Organoboranes: An Easy Way To Construct C—C Bonds (Nobel Lecture). Angew. Chem. Int. Ed 2011, 50, 6722–6737; [DOI] [PubMed] [Google Scholar]; (e) Kotha S; Lahiri K; Kashinath D Recent Applications of the Suzuki-Miyaura Cross-Coupling Reaction in Organic Synthesis. Tetrahedron 2002, 58, 9633–9695. [Google Scholar]
- 2.For a review see: Dobrounig P; Trobe M; Breinbauer R Sequential and Iterative Pd-Catalyzed Cross-Coupling Reactions in Organic Synthesis. Monatsh Chem 2017, 148, 3–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. (a).Mahatthananchai J; Dumas AM; Bode JW Catalytic Selective Synthesis. Angew. Chem. Int. Ed 2012, 51, 10954–10990; [DOI] [PubMed] [Google Scholar]; (b) Hartwig J Catalyst-Controlled Site-Selective Bond Activation. Acc. Chem. Res 2017, 50, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Littke AF; Dai C; Fu GC Versatile Catalysts for the Suzuki Cross-Coupling of Arylboronic Acids with Aryl and Vinyl Halides and Triflates under Mild Conditions. J. Am. Chem. Soc 2000, 122, 4020–4028. [Google Scholar]
- 5.For a related isolated example using P(iPr)(tBu)2, see Proutiere F; Lyngvi E; Aufiero M; Sanhueza IA; Schoenebeck F Combining the Reactivity Properties of PCy3 and PtBu3 into a Single Ligand, P(iPr)(tBu)2. Reaction via Mono- or Bisphosphine Palladium(0) Centers and Palladium(I) Dimer Formation. Organometallics 2014, 33, 6879–6884. [Google Scholar]
- 6.Other phosphine ligands provide varying selectivity (or predicted selectivity) in the reaction of 1 with 2; see Niemeyer ZL; Milo A; Hickey DP; Sigman MS Parameterization of Phosphine Ligands Reveals Mechanistic Pathways and Predicts Reaction Outcomes. Nature Chem 2016, 8, 610–617. [DOI] [PubMed] [Google Scholar]
- 7.Keaveney ST; Kundu G; Schoenebeck F Modular Functionalization of Arenes in a Triply Selective Sequence: Rapid C(sp(2)) and C(sp(3)) Coupling of C—Br, C—OTf, and C—Cl Bonds Enabled by a Single Palladium(I) Dimer. Angew. Chem. Int. Ed 2018, 57, 12573–12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Pd/PtBu3 system has been used for selective cross-coupling of aryl chlorides with triflate-containing arylboron reagents: Manabe K; Ohba M; Matsushima Y, A Repetitive One-Step Method for Oligoarene Synthesis Using Catalyst-Controlled Chemoselective Cross-Coupling. Org. Lett 2011, 13, 2436–2439. [DOI] [PubMed] [Google Scholar]
- 9.Selected reviews: (a) Marion N; Nolan SP Well-Defined N-Heterocyclic Carbenes-Palladium(II) Precatalysts for Cross-Coupling Reactions. Acc. Chem. Res 2008, 41, 1440–1449; [DOI] [PubMed] [Google Scholar]; (b) Shi SC; Nolan SP; Szostak M Well-Defined Palladium(II)-NHC Precatalysts for Cross-Coupling Reactions of Amides and Esters by Selective N—C/O—C Cleavage. Acc. Chem. Res 2018, 51, 2589–2599; [DOI] [PubMed] [Google Scholar]; (c) Hazari N; Melvin PR; Beromi MM Well-Defined Nickel and Palladium Precatalysts for Cross-Coupling. Nat. Rev. Chem 2017, 1, 0025; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Valente C; Calimsiz S; Hoi KH; Mallik D; Sayah M; Organ MG The Development of Bulky Palladium NHC Complexes for the Most-Challenging Cross-Coupling Reactions. Angew. Chem. Int. Ed 2012, 51, 3314–3332; [DOI] [PubMed] [Google Scholar]; (e) Fortman GC; Nolan SP, N-Heterocyclic Carbene (NHC) Ligands and Palladium in Homogeneous Cross-Coupling Catalysis: a Perfect Union. Chem. Soc. Rev 2011, 40, 5151–5169. [DOI] [PubMed] [Google Scholar]
- 10.For a recent example, see: Melvin PR; Nova A; Balcells D; Dai W; Hazari N; Hruszkewycz DP; Shah HP; Tudge MT Design of a Versatile and Improved Precatalyst Scaffold for Palladium-Catalyzed Cross-Coupling: (η3-1-tBu-indenyl)2(μ-Cl)2Pd2. ACS Catal 2015, 5, 3680–3688. [Google Scholar]
- 11.Organ MG; Abdel-Hadi M; Avola S; Hadei N; Nasielski J; O’Brien CJ; Valente C Biaryls Made Easy: PEPPSI and the Kumada–Tamao–Corriu Reaction. Chem. Eur. J 2007, 13, 150–157. [DOI] [PubMed] [Google Scholar]
- 12.Isolated examples of unexpected or divergent selectivity using NHC ligands have been reported in some unoptimized Pd-catalyzed cross couplings. (a) Ji JG; Li T; Bunnelle WH Selective Amination of Polyhalopyridines Catalyzed by a Palladium-Xantphos Complex. Org. Lett 2003, 5, 4611–4614; [DOI] [PubMed] [Google Scholar]; (b) Stroup BW; Szklennik PV; Forster CJ; Serrano-Wu MH Chemoselective Amination of 5-Bromo-2-chloro-3-fluoropyridine. Org. Lett 2007, 9, 2039–2042; [DOI] [PubMed] [Google Scholar]; (c) Keylor MH; Niemeyer ZL; Sigman MS; Tan KL Inverting Conventional Chemoselectivity in Pd-Catalyzed Amine Arylations with Multiply Halogenated Pyridines. J. Am. Chem. Soc 2017, 139, 10613–10616. [DOI] [PubMed] [Google Scholar]
- 13.For an example of an NHC ligand providing the opposite stereoselectivity as a phosphine ligand in a Ni-catalyzed cross coupling, see (a) Harris MR; Hanna LE; Greene MA; Moore CE; Jarvo ER Retention or Inversion in Stereospecific Nickel-Catalyzed Cross-Coupling of Benzylic Carbamates with Arylboronic Esters: Control of Absolute Stereochemistry with an Achiral Catalyst. J. Am. Chem. Soc 2013, 135, 3303–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang S-Q; Taylor BLH; Ji C-L; Gao Y; Harris MR; Hanna LE; Jarvo ER; Houk KN; Hong X Mechanism and Origins of Ligand-Controlled Stereoselectivity of Ni-Catalyzed Suzuki−Miyaura Coupling with Benzylic Esters: A Computational Study. J. Am. Chem. Soc 2017, 139, 12994–13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For an example of an NHC ligand providing the opposite regioselectivity as a phosphine ligand in a Cu-catalyzed aminoboration see Sakae R; Hirano K; Miura M Ligand-Controlled Regiodivergent Cu-Catalyzed Aminoboration of Unactivated Terminal Alkenes. J. Am. Chem. Soc 2015, 137, 6460–6463. [DOI] [PubMed] [Google Scholar]
- 15. A few substrate/arylboronic acid combinations that were evaluated led to poor conversions or to products that were challenging to isolate due to decomposition. In most of these cases, selectivity (analyzed by GC) appeared as expected based on ligand. However, yield and selectivity are poor for some strongly electronically biased substrates (in particular, those containing —CHO substituents). See the Supporting Information for details.
- 16. With some substrate/arylboronic acid combinations, we found that better results could be obtained by modifying the reaction conditions (solvent, equivalents of H2O, temperature) from the optimized conditions in Table 1. See the Supporting Information for details.
- 17.Suzuki-Miyaura couplings were recently shown to be favored over Heck couplings in the presence of water or another hydroxide source: Molloy JJ; Seath CP; West MJ; McLaughlin C; Fazakerley NJ; Kennedy AR; Nelson DJ; Watson AJB Interrogating Pd(II) Anion Metathesis Using a Bifunctional Chemical Probe: A Transmetalation Switch. J. Am. Chem. Soc 2018, 140, 126–130. [DOI] [PubMed] [Google Scholar]
- 18.Schoenebeck F; Houk KN Ligand-Controlled Regioselectivity in Palladium-Catalyzed Cross Coupling Reactions. J. Am. Chem. Soc 2010, 132, 2496–2497. [DOI] [PubMed] [Google Scholar]
- 19.Bickelhaupt FM; Houk KN Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anionic Pd species in cross coupling have been proposed. Selected examples: (a) Amatore C; Jutand A, Anionic Pd(0) and Pd(II) Intermediates in Palladium-Catalyzed Heck and Cross-Coupling Reactions. Acc. Chem. Res 2000, 33, 314—321; [DOI] [PubMed] [Google Scholar]; (b) Kozuch S; Shaik S; Jutand A; Amatore C Active Anionic Zero-Valent Palladium Catalysts: Characterization by Density Functional Calculations. Chem. Eur. J 2004, 10, 3072–3080; [DOI] [PubMed] [Google Scholar]; (c) Roy AH; Hartwig JF Oxidative Addition of Aryl Sulfonates to Palladium(0) Complexes of Mono- and Bidentate Phosphines. Mild Addition of Aryl Tosylates and the Effects of Anions on Rate and Mechanism. Organometallics 2004, 23, 194—202. [Google Scholar]; (d) Guest D; da Silva VHM; Batista APD; Roe SM; Braga AAC; Navarro O (N-Heterocyclic Carbene)-Palladate Complexes in Anionic Mizoroki-Heck Coupling Cycles: A Combined Experimental and Computational Study. Organometallics 2015, 34, 2463–2470. [Google Scholar]
- 21. Similar selectivity is expected with a Pd–fluoride rather than Pd–hydroxide species, but DFT calculations on transition structures for oxidative addition at C—Cl using [Pd(F)—SIMes]− fail to converge at the level of theory used for geometry optimizations.
- 22.Proutiere F; Schoenebeck F Solvent Effect on Palladium-Catalyzed Cross-Coupling Reactions and Implications on the Active Catalytic Species. Angew. Chem. Int. Ed 2011, 50, 8192–8195. [DOI] [PubMed] [Google Scholar]
- 23. Oxidative addition of aryl triflates at anionic [PdLX]– (L = P(o-tol)3, X = Br] has been proposed even in nonpolar solvents such as toluene; see reference 20c.
- 24. It is logical to assume that, like other anionic bisligated complexes, [Pd(SIPr)X]− would favor reaction at C—OTf over C—Cl. However, we are unable to evaluate this computationally, as all attempts to calculate a transition structure for oxidative addition of 1 at C—Cl with [Pd(SIPr)X]− (X = F or OH) failed to converge.
- 25.For a recent review, see Almond-Thynne J; Blakemore DC; Pryde DC; Spivey AC Site-Selective Suzuki–Miyaura Coupling of Heteroaryl Halides – Understanding the Trends for Pharmaceutically Important Classes. Chem. Sci 2017, 8, 40–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maes BUW; Verbeeck S; Verhelst T; Ekomie A; von Wolff N; Lefèvre G; Mitchell EA; Jutand A Oxidative Addition of Haloheteroarenes to Palladium(0): Concerted versus SNAr-Type Mechanism. Chem. Eur. J 2015, 21, 7858–7865. [DOI] [PubMed] [Google Scholar]
- 27.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09 Wallingford, CT: Gaussian, Inc.; 2009. [Google Scholar]
- 28.Zhao Y; Truhlar DG A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions J. Chem. Phys 2006, 125, 194101. [DOI] [PubMed] [Google Scholar]
- 29. (a).Dolg M; Wedig U; Stoll H; Preuss H Energy-Adjusted ab initio Pseudopotentials for the First Row Transition Elements. J. Chem. Phys 1987, 86, 866–872; [Google Scholar]; (b) Andrae D; Haussermann U; Dolg M; Stoll H Preuss H Energy-Adjusted ab initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar]
- 30.Ribeiro RF; Marenich AV; Cramer CJ; Truhlar DG Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. J. Phys. Chem. B 2011, 115, 14556–14562. [DOI] [PubMed] [Google Scholar]
- 31. (a).Gonzalez C; Schlegel HB An Improved Algorithm for Reaction Path Following. J. Chem. Phys 1989, 90, 2154–2161; [Google Scholar]; (b) Gonzalez C; Schlegel HB Reaction Path Following in Mass-Weighted Internal Coordinates. J. Phys. Chem 1990, 94, 5523–5527; [Google Scholar]; (c) Fukui K The Path of Chemical Reactions - the IRC Approach. Acc. Chem. Res 1981, 14, 363–368. [Google Scholar]
- 32.Cossi M; Rega N; Scalmani G; Barone V Energies, Structures, and Electronic Properties of Molecules in Solution with the CPCM Solvation Model. J. Comput. Chem 2003, 24, 669–681. [DOI] [PubMed] [Google Scholar]
- 33.CYLview, 1.0b; Legault CY, Université de Sherbrooke, 2009. (http://www.cylview.org)
- 34.Goossen LJ; Rodríguez N; Linder C Decarboxylative Biaryl Synthesis from Aromatic Carboxylates and Aryl Triflates. J. Am. Chem. Soc 2008, 130, 15248–15249. [DOI] [PubMed] [Google Scholar]
- 35.Dyke AM; Gill DM; Harvey JN Hester AJ; Lloyd-Jones GC; Paz Muñoz M; Shepperson IR Decoupling Deprotonation from Metalation: Thia-Fries Rearrangement. Angew. Chem. Int. Ed 2008, 47, 5067–5070. [DOI] [PubMed] [Google Scholar]
- 36.Dawson MI; Xia Z; Liu G; Fontana JA; Farhana L; Patel BB; Arumugarajah S; Bhuiyan M; Zhang Z-K; Han Y-H; Stallcup WB; Fukushi J; Mustelin T; Tautz L; Su Y; Harris DL; Waleh N; Hobbs PD; Jong L; Chao W; Schiff LJ; Sani BP An Adamantyl-Substituted Retinoid-Derived Molecule That Inhibits Cancer Cell Growth and Angiogenesis by Inducing Apoptosis and Binds to Small Heterodimer Partner Nuclear Receptor: Effects of Modifying Its Carboxylate Group on Apoptosis, Proliferation, and Protein-Tyrosine Phosphatase Activity. J. Med. Chem 2007, 50, 2622–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yingqian L; Dan W; Liting Y; Zhongbang L; Xiang N; Liu Y; Xiaobing W; Zilong S; Gaoxiang Z; Benzophenone Hydrazone Sulfonylurea Compound and Its Preparation Method and Use CN Patent 10472528, December 20, 2013. [Google Scholar]
- 38.Wu J; Liu Y; Lu C; Shen Q Palladium-Catalyzed Difluoromethylthiolation of Heteroaryl Bromides, Iodides, Triflates and Aryl Iodides. Chem. Sci 2016, 7, 3757–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He Q; Wang L; Liang Y; Zhang Z; Wnuk SF Transition-Metal-Free Cross-Coupling of Aryl Halides with Arylstannanes. J. Org. Chem 2016, 81, 9422–9427. [DOI] [PubMed] [Google Scholar]
- 40.Chadwick J; Amewu RK; Marti F; Bousejra-El Garah F; Sharma R; Berry NG; Stocks PA; Burrell-Saward H; Wittlin S; Rottmann M; Brun R; Taramelli D; Parapini S; Ward SA; O’Neill PM Antimalarial Mannoxanes: Hybrid Antimalarial Drugs with Outstanding Oral Activity Profiles and A Potential Dual Mechanism of Action. Chem. Med. Chem 2011, 6, 1357–1361. [DOI] [PubMed] [Google Scholar]
- 41.Rangwala SM; O’Brien ML; Tortorella V; Longo A; Loiodice F; Noonan DJ; Feller DR Stereoselective Effects of Chiral Clofibric Acid Analogs on Rat Peroxisome Proliferator-Activated Receptor α (rPPARα) Activation and Peroxisomal Fatty Acid β-Oxidation. Chirality 1997, 9, 37–47. [DOI] [PubMed] [Google Scholar]
- 42.Antelo Miguez JM; Adrio LA; Sousa-Pedrares A; Vila JM; Hii KK A Practical and General Synthesis of Unsymmetrical Terphenyls. J. Org. Chem 2007, 72, 7771–7774. [DOI] [PubMed] [Google Scholar]
- 43.Klement I; Rottländer M; Tucker CE; Majid TN; Knochel P Preparation of Polyfunctional Aryl and Alkenyl Zinc Halides From Functionalized Unsaturated Organolithiums and their Reactivity in Cross-Coupling and Conjugated Addition Reactions. Tetrahedron 1996, 52, 7201–7220. [Google Scholar]
- 44.Yuan Y; Bian Y Efficient Homocoupling Reactions of Halide Compounds Catalyzed by Manganese (II) Chloride. Appl. Organometal. Chem 2008, 22, 15–18. [Google Scholar]
- 45.Malapit CA; Ichiishi N; Sanford MS Pd-Catalyzed Decarbonylative Cross-Couplings of Aroyl Chlorides. Org. Lett 2017, 19, 4142–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu P; Morandi B Nickel-Catalyzed Cyanation of Aryl Chlorides and Triflates Using Butyronitrile: Merging Retro-hydrocyanation with Cross-Coupling. Angew. Chem. Int. Ed 2017, 56, 15693–15697. [DOI] [PubMed] [Google Scholar]
- 47.Paul D; Rudra S; Rahman P; Khatua S; Pradhan M; Nath Chatterjee P Synthesis and Characterization of Pd-γ-Fe2O3 Nanocomposite and its Application as a Magnetically Recyclable Catalyst in Ligand-Free Suzuki-Miyaura Reaction in Water. J. Organomet. Chem 2018, 871, 96–102. [Google Scholar]
- 48.Gurung SK; Thapa S; Kafle A; Dickie DA; Giri R Copper-Catalyzed Suzuki–Miyaura Coupling of Arylboronate Esters: Transmetalation with (PN)CuF and Identification of Intermediates. Org. Lett 2014, 16, 1264–1267. [DOI] [PubMed] [Google Scholar]
- 49.Yang C; Edsall R; Harris HA; Zhang X; Manas ES; Mewshaw RE ERβ Ligands. Part 2: Synthesis and Structure–Activity Relationships of a Series of 4-Hydroxy-biphenyl-carbaldehyde Oxime Derivatives. Bioorg. Med. Chem 2004, 12, 2553–2570. [DOI] [PubMed] [Google Scholar]
- 50.Hamdi J; Blanco AA; Diehl B; Wiley JB; Trudell ML Room-Temperature Aqueous Suzuki-Miyaura Cross-Coupling Reactions Catalyzed via a Recyclable Palladium@Halloysite Nanocomposite. Org. Lett 2019, 21, 3471–3475. [DOI] [PubMed] [Google Scholar]
- 51.Xue D; Jia Z-H; Zhao C-J; Zhang Y-Y; Wang C; Xiao J Direct Arylation of N-Heteroarenes with Aryldiazonium Salts by Photoredox Catalysis in Water. Chem Eur. J 2014, 20, 2960–2965. [DOI] [PubMed] [Google Scholar]
- 52.Piller FM; Appukkuttan P; Gavryushin A; Helm M; Knochel P Convenient Preparation of Polyfunctional Aryl Magnesium Reagents by a Direct Magnesium Insertion in the Presence of LiCl. Angew. Chem. Int. Ed 2008, 47, 6802–6806. [DOI] [PubMed] [Google Scholar]
- 53.Thapa S; Gurung SK; Dickie DA; Giri R Copper-Catalyzed Coupling of Triaryl- and Trialkylindium Reagents with Aryl Iodides and Bromides through Consecutive Transmetalations. Angew. Chem. Int. Ed 2014, 53, 11620–11624. [DOI] [PubMed] [Google Scholar]
- 54.Saa JM; Martorell G Palladium-Catalyzed Cross-Coupling Synthesis of Hindered Biaryls and Terphenyls. Cocatalysis by Copper(I) Salts. J. Org. Chem 1993, 58, 1963–1966. [Google Scholar]
- 55.Joseph JT; Sajith AM; Ningegowda RC; Nagaraj A; Rangappa KS; Shashikanth S Aryl/Heteroaryl Pentafluorobenzenesulfonates (ArOPFBs): New Electrophilic Coupling Partners for Room Temperature Suzuki–Miyaura Cross-Coupling Reactions. Tetrahedron Lett 2015, 56, 5106–5111. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.