

Abstract
Ex vivo CRISPR gene editing in hematopoietic stem and progenitor cells has opened potential treatment modalities for numerous diseases. The current process uses electroporation, sometimes followed by virus transduction. While this complex manipulation has resulted in high levels of gene editing at some genetic loci, cellular toxicity was observed. We have developed a CRISPR nanoformulation based on colloidal gold nanoparticles with a unique loading design capable of cellular entry without the need for electroporation or viruses. This highly monodispersed nanoformulation avoids lysosomal entrapment and localizes to the nucleus in primary human blood progenitors without toxicity. Nanoformulation-mediated gene editing is efficient and sustained with different CRISPR nucleases at multiple loci of therapeutic interest. Engraftment kinetics of nanoformulation-treated primary cells in humanized mice are better relative to non-treated cells, with no differences in differentiation. Here we demonstrate nontoxic delivery of the entire CRISPR payload into primary human blood progenitors.
Keywords: gold nanoparticle, CRISPR, nanoformulation, homology directed repair, hematopoietic stem, progenitor cells
Retrovirus-mediated gene correction in hematopoietic stem and progenitor cells (HSPC) has demonstrated curative outcomes for various genetic, infectious and malignant disorders1,2,3,4,5. The use of gene-modified autologous, or “self”, HSPC eliminates the risk of graft-host immune responses, negating the need for immunosuppressive drugs required in allogeneic hematopoietic stem cell transplant. However, effective implementation of HSPC gene therapy faces several major challenges. Currently, limited quantities of therapeutic retrovirus vector can be produced at Good Manufacturing Practices (GMP) quality, creating a major bottleneck to widespread use. In addition to the challenges of manufacturing sufficient vector quantities, there is a known risk of genotoxicity associated with retrovirus vectors for gene transfer, evidenced by the development of malignancy due to insertional mutagenesis 6,7,8,9. All of these have inspired the development of non-viral means for genetic modification.
Gene editing has been proposed as a safer alternative to retrovirus-mediated gene transfer, made possible by the development of engineered nucleases such as clustered regularly interspaced short palindromic repeat (CRISPR)-Cas nucleases10. These programmable nucleases incorporate one or more RNA molecules to target specific sequences in the DNA for cutting. Of these, Cas9 nuclease is the most well studied. This nuclease complexes with two RNA molecules, a guide RNA (crRNA) and a tracer RNA (tracrRNA), to recognize a cognate protospacer adjacent motif (PAM) site consisting of an NGG sequence and makes a blunt-end DNA double strand break. This break is most commonly repaired by either non-homologous end joining (NHEJ) or homology-directed repair (HDR)11. For the latter to occur, an intact template sequence homologous to the cut site must be present. The sister chromatid can serve as a template, but synthetic template molecules provided in surplus can enhance HDR efficiency. The flanking regions of this template must match DNA flanking the cut site; however, new genetic code can be inserted within, permitting precise editing or addition of endogenous DNA to the genome when HDR occurs, whereas with NHEJ, insertions and/or deletions (indels) are the most likely outcome11. Recently, Cpf1 (or Cas12a), has also demonstrated utility in genome editing. Cpf1 recognizes a different protospacer adjacent motif (PAM) site (e.g. TTTN, where N can be either A, C, G or T), requires a single guide RNA and results in staggered cutting of the DNA with 5’ overhangs 12. The smaller size and staggered cutting of Cpf1 are postulated to enhance delivery and likelihood of HDR when template oligonucleotides are provided, but this has yet to be demonstrated.
For utility in HSPC gene therapy, a delivery platform including the designer nuclease of choice, with or without a DNA template, which performs efficiently and reliably without cytotoxicity would be ideal. Current state of the art for this approach in HSPC requires electroporation of engineered nuclease components as mRNA or ribonucleoprotein (RNP) complexes. If HDR is preferred, the most effective method has been electroporation followed by transduction with non-integrating virus vectors13, or simultaneous electroporation of defined concentrations of engineered nuclease components with chemically modified, single-stranded oligonucleotide (ssODN) template at specified cell concentrations14. Electroporation is known to induce toxicity, and there is no means to control the number of cells which take up each component of the payload or the concentrations of each component that are successfully delivered by electroporation15. Finally, where non-integrating viruses are used as templates, the system still depends on avaibility of GMP-grade viral particles. Thus, nanoparticle-based delivery is being actively pursued for the delivery of CRISPR components16.
Lipid-based, polymer-based, and gold nanoparticles (AuNP) carry great potential for the delivery of CRISPR components to cells17,18,19. While polymer and lipid nanoparticles represent “encapsulating” or “entrapping” delivery vehicles, the unique surface loading of AuNP facilitates precise modification and functionalization by different molecules, such as RNA, DNA, and proteins20. Because the surface area is known, controlled loading of payload components ensures uniformity of AuNP preparations, leading to more predictable delivery21. Finally, AuNP are considered relatively nontoxic compared to lipid and polymer nanocarriers22,23,24, which is critical for nonmalignant dividing somatic cells such as HSPC. Indeed, Lee et al. have demonstrated the utility of a polymer-encapsulated AuNP design in the delivery of CRISPR Cas9 and Cpf1 to non-dividing somatic tissues such as muscle and brain18,19, but these carriers have not demonstrated efficacy in HSPC or with accompanying oligonucleotide templates. Moreover, the combination of polymer encapsulation with a gold nanocore greatly increases overall nanoparticle size and alters the nanoformulation cytotoxicity profile.
We have designed a simple AuNP-based CRISPR nanoformulation (AuNP/CRISPR) with layer-by-layer conjugation of the CRISPR components (guide RNA and nuclease) on the surface of AuNP with or without a single-stranded DNA template to support HDR, hereby referred to as “homology directed repair template (HDT)”, which does not require polymer encapsulation (Fig. 1a and b, Supplementary Figure 1).
Figure 1. Layer by layer conjugation of CRISPR components onto gold nanoparticles.
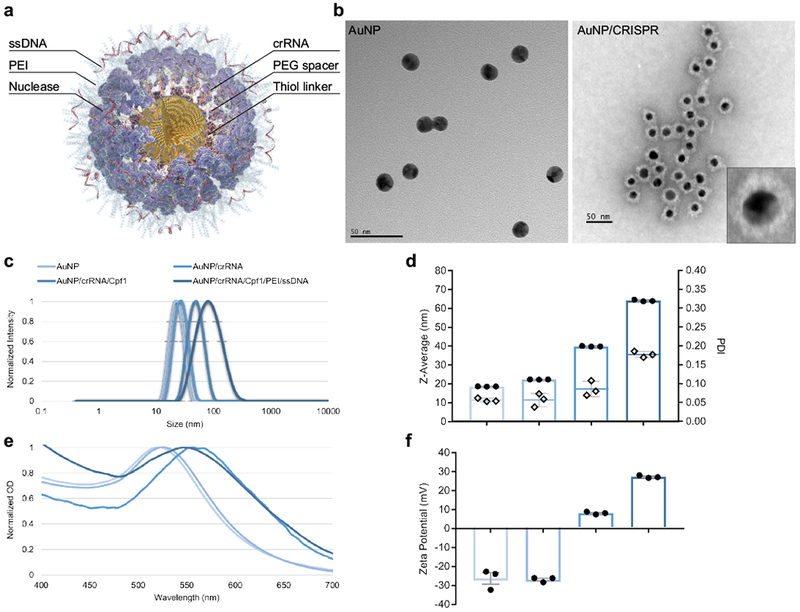
a, Schematic representation of AuNP/CRISPR nanoformulation. b, TEM images of AuNP and AuNP/CRISPR nanoformulations. Inset shows successful attachment of all cargo components on the surface. Experiment was repeated twice with similar results. c, dynamic light scattering characterization of AuNPs after each layering step. Sharp single peaks and shifts in size after adding each layer demonstrate precise attachment to the surface. Data have been normalized. Experiment was repeated twice with similar results. d, Average size (Z-Average, bar graph with dots plotted on the primary axis) and polydispersity index (PDI, diamonds plotted on the secondary axis) of AuNPs after each layering step. PDI values <0.2 show high monodipersity without aggregation. Data are means ± SEM (n=3 technical replicates). e, Red shifts in localized surface plasmon resonance (LSPR) of AuNPs after adding each component confirm cargo loading. Data have been normalized. Experiment was repeated twice with similar results. f, Zeta potential measurements after adding each layer changed from −26 mV for AuNPs to +27 mV for the final AuNP/CRISPR nanoformulation. Data are means ± SEM (n=3 technical replicates).
An AuNP core of 19 nm was synthesized using the citrate reduction method25. Synthesized nanoparticles were highly monodisperse with an observed polydispersity index (PDI) of 0.05 (Fig. 1c and d). The preparation and layer conjugation process can be found in Supplementary Figure 1. First, crRNA for Cpf1 or Cas9 synthesized with an 18-nucleotide oligo ethylene glycol (OEG) spacer and a terminal thiol linker (crRNA-18 spacer-SH) was attached to the surface of gold by semi covalent gold-thiol interaction (all sequence information can be found in Supplementary Table 1). Analysis of the published crystal structures of these Cas nucleases with crRNA and/or tracrRNA and double-stranded DNA suggested that adding a spacer-thiol linker to the crRNA would not have any effect on the recognition of the guide segment and nuclease activity26,27. The inclusion of the OEG spacer arm reduced electrostatic repulsion between crRNA strands to increase loading capacity on the AuNP surface. As shown in Fig. 1c, the AuNP core with crRNA resulted in a nanoparticle size of 22 nm with a PDI of 0.05. Nuclease proteins were then attached to the 5’ handle of surface-loaded crRNA by the natural affinity of nuclease to the 3D structure of crRNA. Nuclease attachment increased the size of nanoparticles to 40 nm with a PDI of 0.08 for Cpf1. This RNP-loaded AuNP served as a basis for comparison of nuclease activity without HDT present. For HDT loading, RNP-layered AuNP were further coated with branched low molecular weight (2000 g/mol by light scattering) polyethylenimine (PEI) to prepare the base for electrostatic conjugation of HDT in the outermost layer. This “fully loaded” AuNP demonstrated a size of 64 nm and remained highly monodispersed with an observed PDI of 0.17 (Fig. 1b–d). Uniform morphology without aggregation was inferred from transmission electron microscope images and by looking at fine localized surface plasmon resonance (LSPR) shifts after each attachment step (Fig. 1b and e). Zeta potential of the nanoformulation changed from −26 mV to +27 mV with complete layering (Fig. 1f). This positive charge of the final nanformulation likely prevented precipitation and aggregation over time, as these were not observed over a period of 48 hours following formulation.
This stable and monodisperse structure resulted from the adjustment of weight/weight (w/w) ratios between AuNP and CRISPR components. Analysis of different w/w ratios between AuNP and Cpf1 demonstrated that higher ratios of Cpf1 can trigger aggregation with an optimal w/w ratio of 0.6 (Supplementary Figure 2a and b). The loading capacity of Cpf1 was found to be 8.8 µg/mL in this ratio. In contrast, lower w/w ratio between AuNP and HDT lead to aggregation with an optimal w/w ratio of 1 (Supplementary Figure 2c and d).
We isolated primary HSPC from leukapheresis products on the basis of CD34 expression from granulocyte colony stimulating factor (G-CSF)-mobilized healthy adult volunteers. Cells were cultured in supportive media, and nanoformulations were added to culture at a concentration of 10 µg/mL. Potential toxicity in CD34+ cells was analyzed by both live-dead staining, and trypan blue dye exclusion assays after 24-hr and 48-hr incubations with AuNP/CRISPR nanoformulations (Supplementary Figure 3). AuNP/CRISPR-treated samples demonstrated >80% viability in both assays, with no variation between treated and untreated cells by trypan blue assay.
Although HSPCs are considered difficult to transfect, within 6 hours after treatment with AuNP/CRISPR carrying fluorescently labeled crRNA and HDT, confocal microscopy imaging showed uptake and nuclear localization of gene editing components (Fig. 2a–d).
Figure 2. AuNP/CRISPR can deliver CRISPR components to the nucleus of HSPCs.
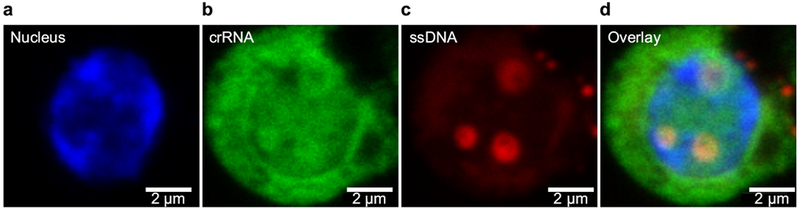
a, Nucleus of primary human CD34+ HSPC following addition of AuNP/CRISPR to the culture (blue, Hoechst). b, Fluorophore tagged crRNA (green, Alexa488). c, Fluorophore tagged ssDNA (Red, Alexa660). Visible small vesicles on the far left side of the image suggest passive uptake by endocytosis. d, Overlay of all three stains showed colocalization of crRNA and ssDNA. a-d, Images are at 60× magnification. Data are representative of a single human donor different from all other donors reported in this study. Experiment was repeated once as an independent biological replicate with a similar result.
To test the utility of AuNP/CRISPR for gene editing, we targeted two different genomic loci with demonstrated therapeutic value in HSPC: the chemokine receptor 5 (CCR5) gene on chromosome 3, and the gamma globin (γ-globin) gene promoter on chromosome 11. Disruption of CCR5 has been associated with resistance to human immunodeficiency virus (HIV) infection by eliminating attachment and entry of the virus through the expressed CCR5 co-receptor28. Targeting this disruption in HSPC renders future T cell progeny resistant to HIV infection. Alternatively, introduction of a specific deletion within the γ-globin promoter recapitulates a naturally-occurring phenomenon known as “hereditary persistence of fetal hemoglobin” (HPFH), which has been shown to be useful for the treatment of hemoglobinopathies such as sickle cell disease and β-thalassemia29,30
In silico off-target analysis of the human CCR5 target by CasOFFinder software demonstrated no homology with fewer than 3 bp mismatches for Cpf1 (Supplementary Table 2)31. We chose a target site encoding both Cpf1 and Cas9 PAM sites accessible with a single guide RNA to directly compare these two CRISPR nucleases (Supplementary Figure 4a and b). However, before testing we first wanted to optimize the HDT for Cpf1. Previous data have demonstrated cleavage of the non-target strand by the RuvC domain is a prerequisite for target strand cleavage by the Nuc domain26. Therefore, we tested HDTs designed for the DNA target and non-target strands. Our HDT was comprised of 40 bp homology arms flanking the Cpf1 cut site (17 bp downstream from the PAM) on each end, with 8 bp of NotI restriction enzyme cut site in the middle to disrupt CCR5 expression and enable HDR analysis. Using tracking of indels by decomposition (TIDE), we observed a total editing rate of 8.1% for the non-target strand and 7.8% for the target strand, with 7.3% HDR when HDT designed against the non-target strand was used, compared to 5.4% HDR when HDT designed against the target strand was used (Fig. 3a and b). These results were confirmed by T7EI and NotI restriction enzyme digestion assays (Fig. 3c), and were in agreement with previously published data by Yamano et al26.
Figure 3. Optimization of HDR conditions and optimal editing dosage.
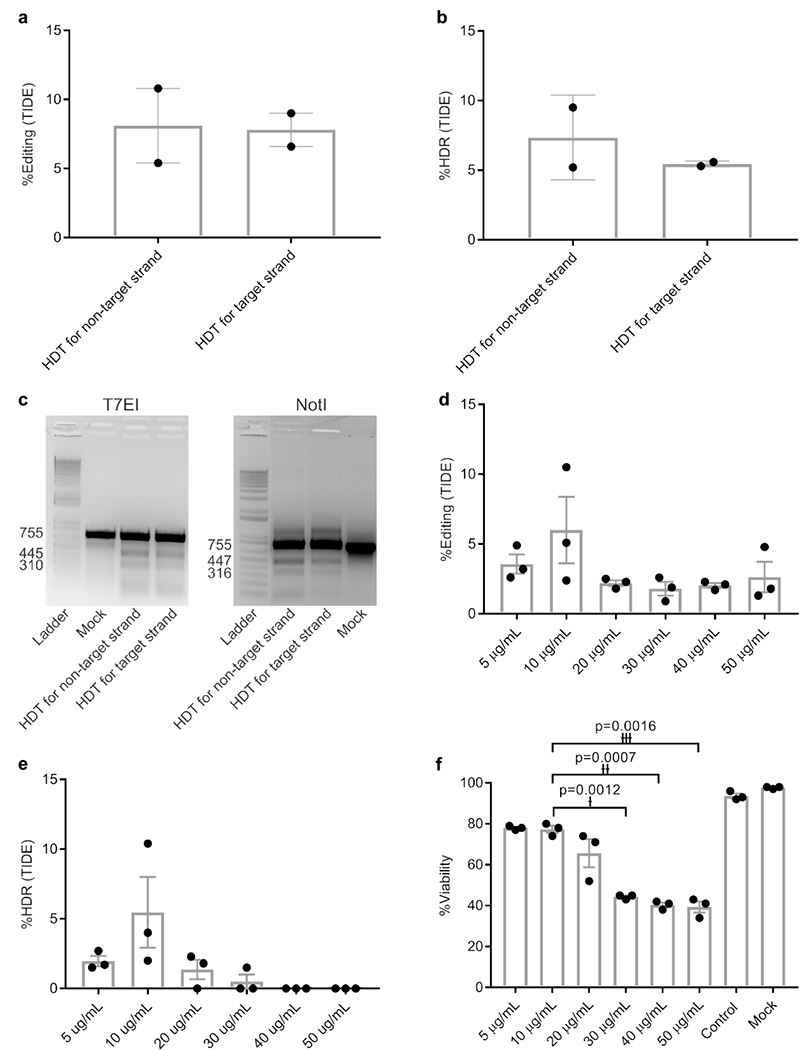
a and b, Total editing and HDR levels. Data are means ± SEM (n=2 independent experiments). c, T7EI and NotI restriction enzyme digestions showing the related digestion bands. Experiment has been repeated once with similar results. d and e, Effect of different AuNP/CRISPR nanoformulation concentrations on total editing and HDR in primary human HSPC. Data are means ± SEM (n=3 independent experiments). f, Toxicity in primary human CD34+ cells as a function of concentration. Data are means ± SEM (n=3 independent experiments). Statistical significance was determined by a two-sided t-test. †: t value=28.58, degrees of freedom (df)=2, effect size (d)=70.79; ††: t value=37, df=2, d=56.11; †††: t value=28.58, df=2, d=36.73. Data are representative of a single human donor different from all other donors reported in this study.
We next optimized the efficiency of HDR in primary HSPC by preparing AuNP/CRISPR-HDT in different concentrations (5–50 µg/mL) based on the amount of AuNP core in suspension. A concentration of 10 µg/mL demonstrated the highest total editing and HDR rate, with increasing concentrations demonstrating increased cytotoxicity and lower rates of HDR (Fig. 3d–f).
Typically, during clinical manipulation for ex vivo gene transfer, HSPC are cultured in serum-free media containing recombinant human growth factors on a layer of recombinant fibronectin fragment (retronectin). Retronectin stabilizes blood stem cells in culture by providing a mode of extracellular adhesion to the culture surface. Thus, we hypothesized that HSPC stabilization could impact AuNP/CRISPR uptake and activity. HSPC proliferation can also impact AuNP uptake and activity. The addition of serum to HSPC in culture has been shown to promote viability and proliferation, but can also drive differentiation and loss of stem cell function. It has also been shown that nanoparticles will form an albumin corona in the presence of serum albumin, which can alter stability and uptake of nanoparticles by cells.32 While the media used in HSPC culture includes bovine serum albumin, we wanted to test whether retronectin, human serum or human serum albumin (HSA) could impact AuNP-mediated HSPC gene editing. We tested nanoformulations that included HSA or pooled human A/B serum, as well as addition of nanoformulations to HSPC cultured on retronectin. No change in cytotoxicity was observed for any of the reagents tested (Supplementary Figure 5a), but all reagents reduced total editing and HDR rates (Supplementary Figure 5b and c). Thus, for all subsequent experiments, HDT (where included in the formulation) was designed against the non-target DNA strand, all formulations were added to HSPC in culture at a concentration of 10 µg/mL, and HSPC were cultured in serum-free, supportive media without retronectin or HSA.
We hypothesized that staggered cutting by Cpf1 would favor HDR moreso than Cas9 blunt-ended cuts. To test this hypothesis, we prepared AuNP/CRISPR nanoformulations targeting the CCR5 locus with and without HDT for both Cpf1 and Cas9. For comparison, we performed delivery side by side with electroporation at identical concentrations of each component. Notably, we did not include additional chemical modifications to the guide RNA, such as 2′ O-methyl ribonucleotide, 2′-deoxy-2′-fluoro-ribonucleotide, and phosphorothioates33, in any condition. TIDE analysis demonstrated a range of total editing between 2% and 25% without significance (Fig. 4a). However, we observed increased NotI restriction site incorporation, indicative of HDR in HSPC treated with Cpf1 or Cas9 delivered by our AuNP/CRISPR nanoformulation, compared to electroporation by both TIDE and next generation sequencing, with Cpf1 outperforming Cas9 (Fig. 4b and c). Cell viabilities for all samples were above 70%, but with higher viability observed in samples treated with AuNP, and in particular, significantly higher viability when Cas9 was delivered by AuNP rather than electroporation (Fig. 4d). HSPC fitness in these samples was analyzed by a colony-forming cell (CFC) assay with no observed differences in CFC potential or morphology (Fig. 4e and f). This standard CFC assay is representative of more short-term blood progenitors34; thus, as a crude measure of long-term repopulating capacity, colonies from the original assay were re-plated. No significant differences in number or type of secondary CFCs were observed relative to the mock (untreated) control sample, but the pattern of higher CFC numbers in AuNP-treated samples relative to electroporated samples was not observed (Supplementary Figure 6a and b).
Figure 4. AuNP/CRISPR nanoformulations carrying Cpf1 outperform Cas9 in terms of HDR.
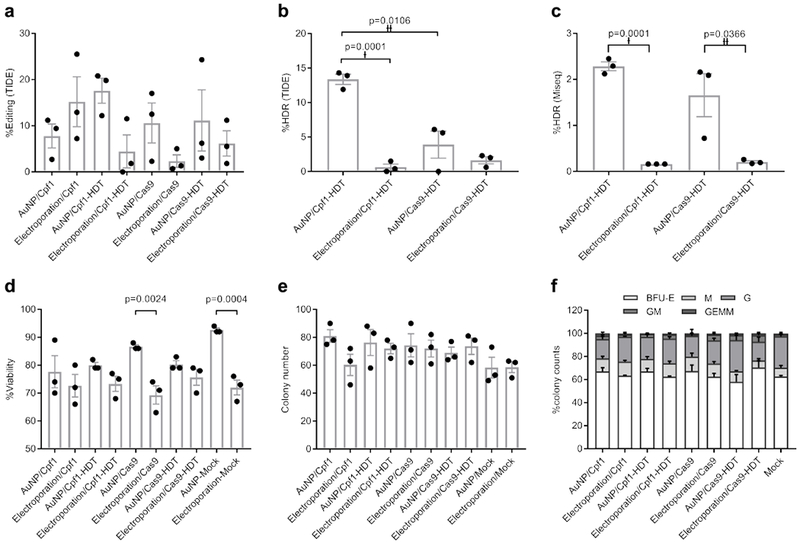
a, Total editing results by TIDE assay. Data are means ± SEM (n=3 independent experiments). b, HDR results by TIDE assay. Data are means ± SEM (n=3 independent experiments). Statistical significance was determined by a two-sided t-test. †: t value=14.68, df=4, d=16.63; ††: t value=4.526, df=4, d=9.05. c, MiSeq analysis to confirm TIDE assay results. Data are means ± SEM (n=3 independent experiments; sample source same as in a and b). Statistical significance was determined by a two-sided t-test. †: t value=22.32, df=4, d=36.32; ††: t value=3.088, df=4, d=5.31. d, Cell viability of CD34+ cells after treatment with CRISPR Cpf1 and Cas9 using AuNP/CRISPR and electroporation methods. Data are means ± SEM (n=3 independent experiments). Statistical significance was determined by one-way ANOVA (F value=5.966, df=20, d=55.86 (p=0.0024), 70.30 (p=0.0004)). e, CFC assay results showing total colony numbers. BFU-E: burst-forming unit-erythroid; M: macrophage; G: granulocyte; GM: granulocyte-macrophage; GEMM: granulocyte-erythrocyte-macrophage-monocyte. Data are means ± SEM (n=3 independent experiments). f, CFC assay results showing percentage of different colony morphologies. Data are means ± SEM (n=3 independent experiments). Data are representative of a single human donor different from all other donors reported in this study.
We tested the same hypothesis at the γ-globin promoter locus to affirm the Cpf1 preference for HDR. Here again we were able to identify both Cpf1 and Cas9 PAM sequences with an identical target cut site and no predicted off-target cutting (Supplementary Figure 7a and b; Supplementary Table 2). We designed HDT to insert a documented HPFH-associated, 13-bp deletion overlapping a repressor binding site in this promoter29. Obtained results in primary HSPC showed the same trend at this locus, with higher levels of HDR for Cpf1-containing AuNP/CRISPR nanoformulations as compared to Cas9-containing nanoformulations (Supplementary Figure 7c).
Bulk CD34+ HSPC are known to contain a mixture of cell types including long-term repopulating stem cells (LT-HSC), multipotent progenitor cells (MPP), and other, more committed progenitor cells such as common myeloid progenitors (CMP), granulocyte-monocyte progenitors (GMP), and megakaryocyte-erythrocyte progenitors (MEP).35 To determine the extent of gene editing following nanoformulation treatment in these fractions, we sorted each subtype by fluorescence-activated cell sorting (FACS) from a single donor and compared AuNP/CRISPR-HDT activity to both mock-treated cells of the same phenotype and bulk CD34+ cells from the same donor (Supplementary Figure 8a). We observed reduced cell viability after AuNP treatment in the sorted populations, likely owing to the stress of FACS on HSPC (Supplementary Figure 8b). Interestingly, we observed slightly increased viability and CFC numbers in AuNP/CRISPR-HDT-treated LT-HSC and MPP fractions, the fractions believed to be responsible for the majority of hematopoietic reconstitution after transplant, relative to mock-treated counterparts (Supplementary Figure 8c). Highest levels of total gene editing were observed in the LT-HSC fraction, but bulk (unsorted) CD34+ cells demonstrated the highest levels of HDR, suggesting that sorting stress could compromise HDR efficiency or that other cell phenotypes in the bulk CD34+ cell population which were not sorted, owing to their lack of documented contribution to hematopoiesis, are more amenable to HDR (Supplementary Figure 8d).
We next wanted to determine whether ex vivo nanoformulation-treated HSPC engrafted following reinfusion into a myelosuppressed host. Primary human CD34+ HSPC were treated with AuNP/CRISPR-HDT nanoformulations ex vivo and infused into sub-lethally irradiated immunodeficient (Il2r gamma–/–) mice at 106 cells/per mouse. Mice were followed for 22 weeks, with maximum engraftment observed at 8 weeks following transplant and stable engraftment established around week 16 after transplant (Fig. 5a). Mouse weights were stable over time (Supplementary Figure 9). To our surprise, HSPC treated with AuNP/CRISPR-HDT or AuNP alone engrafted at higher levels than mock (untreated) cells, but with similar kinetics (Fig. 5a and b). This correlated with our observation that both LT-HSC and MPP cells treated with AuNP/CRISPR-HDT displayed higher CFC numbers and viability in vitro. Among different blood cell lineages, reconstitution of B cells reached peak at 10 weeks after transplant and then started to level-off through week 22 (Fig. 5c). Initial monocyte engraftment was high but decreased over the first 8 weeks and stabilized (Fig. 5d). We observed low levels of T cells until week 16, which then increased for all the study groups (Fig. 5e). No significant differences in the proportion of B cells, monocytes or T cells were observed relative to the ex vivo HSPC treatment administered.
Figure 5. AuNP treatment enhanced HSPC engraftment in neonatal immune-deficient mice.
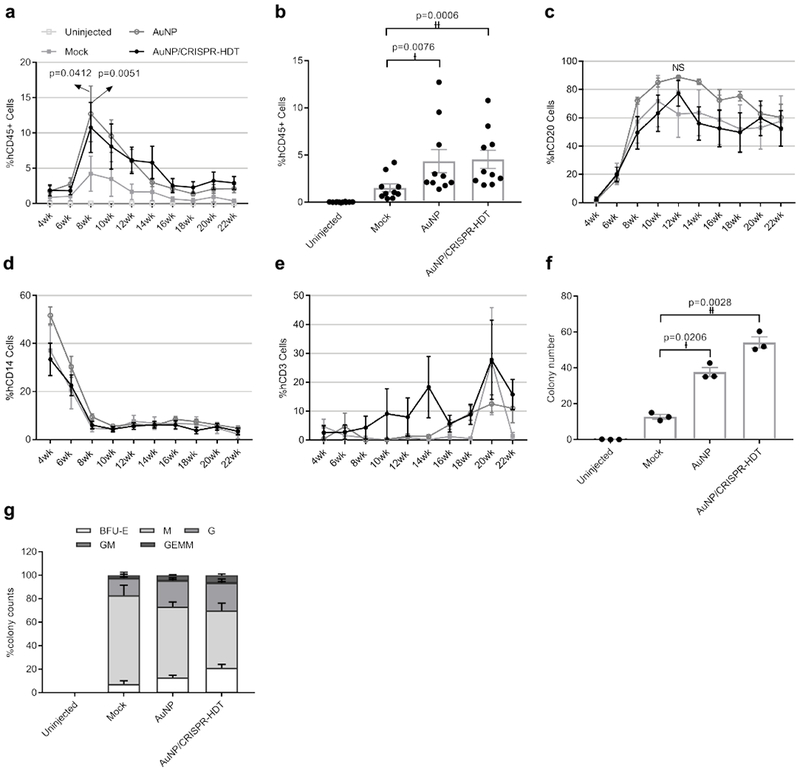
a,b, Engraftment measured as percentage of human CD45-expressing cells in peripheral blood of mice. Data are means ± SEM (n=10 mice receiving AuNP/CRISPR-HDT-treated HSPC, n=10 mice receiving AuNP-treated HSPC, n=5 Mock-treated mice, n=4 un-injected mice). Statistical significance was determined by two-way ANOVA in panel a (F value=11.51, df=244, d=2.10 (p=0.0412), 2.69 (p=0.0051)), and two-sided t-test in panel b (†: t value=3.42, df=9, d=2.39; ††: t value=5.168, df=9, d=2.94). c, Human CD20+ B cell engraftment kinetics in peripheral blood. Data are means ± SEM (n=10 mice receiving AuNP/CRISPR-HDT-treated HSPC, n=10 mice receiving AuNP-treated HSPC, n=5 Mock-treated mice, n=4 un-injected mice). NS (not significant p=0.5093). Statistical significance was determined by two-way ANOVA (F value=7.028, df=215, d=32.25) d, Human CD14+ monocyte engraftment kinetics in peripheral blood. Data are means ± s.e (n=10 mice receiving AuNP/CRISPR-HDT-treated HSPC, n=10 mice receiving AuNP-treated HSPC, n=5 Mock-treated mice, n=4 un-injected mice). e, Human CD3+ T cell engraftment kinetics in peripheral blood. f and g, CFC assay showing the total colony numbers and the frequency of different morphologies for bone marrow samples. BFU-E: burst-forming unit-erythroid; M: macrophage; G: granulocyte; GM: granulocyte-macrophage; GEMM: granulocyte-erythrocyte-macrophage-monocyte. (n=10 mice receiving AuNP/CRISPR-HDT-treated HSPC, n=10 mice receiving AuNP-treated HSPC, n=5 Mock-treated mice, n=4 un-injected mice). Data are represented as means of each study group ± SEM for three independent experiments. Statistical significance was determined by a two-sided t-test. †: t value=6.861, df=2, d=18.81; ††: t value=18.88, df=2, d=21.61. Data are representative of a single human donor different from all other donors reported in this study.
Mice were sacrificed after 22 weeks and bone marrow, spleen, thymus, and peripheral blood samples were retrieved. Flow cytometry analysis of these samples showed that AuNP and AuNP/CRISPR-HDT treated groups were associated with higher levels of engraftment (Supplementary Figure 10a–d). The frequency of multipotent CD34+ cells was higher in bone marrow of AuNP-treated animals (Supplementary Figure 10a, b and d), and the frequency of CD20-expressing cells was higher in the spleen, thymus and peripheral blood (Supplementary Figure 10b, c and d). A human-specific CFC assay of bone marrow cells correlated with engraftment results, with AuNP- and AuNP/CRISPR-HDT-treated groups demonstrating significantly higher colony numbers compared to the mock treated group (Fig. 5f). This was closely related with a greater diversity of colony types observed in these groups, suggestive of higher MPP numbers (Fig. 5g). These results aligned with pre-transplant CFC assay results, suggesting a positive effect of AuNP treatment in ex vivo cultured HSPC (Supplementary Figure 11a and b). Colony morphologies for all the treated samples are shown in Supplementary Figure 12.
We observed 9.8% total gene editing and 9.3% HDR by TIDE analysis in HSPC at the time of transplant (Fig. 6a, Supplementary Figure 13). Stable levels of total gene editing (~5%) were observed in peripheral blood cells (Fig. 6b). Interestingly, levels of NotI restriction enzyme incorporation were consistently lower than 1% across all time points (Fig. 6c). Necropsy samples from different tissues showed that HDR was comparably low in blood, bone marrow and spleen (Fig. 6d and e), supporting observations in sorted HSPC fractions.
Figure 6. Persistent editing levels after engraftment.

a, TIDE assay results for total editing and HDR levels before engraftment. b, Tracking of total editing levels in peripheral blood samples collected after transplant. Data are means ± SEM (n=10 mice). c, Tracking of HDR levels after engraftment. Data are means ± SEM (n=10 mice). d, Total editing levels in peripheral blood, bone marrow and spleen at necropsy. Data are means ± SEM (n=10 mice). e, HDR levels in peripheral blood, bone marrow, and spleen at necropsy. Total cells, without separation of human cells from murine cells were used to assess gene editing. Data are means ± SEM (n=10 mice).
CRISPR gene editing is a promising approach for genetic screening to identify unknown genes and understand gene function, and to correct defective genes in congenital or acquired genetic diseases 36. CRISPR technology is moving rapidly from basic science to clinical application, however current state of the art for delivery of CRISPR components in HSPC requires electroporation, possibly with adeno-associated virus (AAV) transduction, which is far more complex than retrovirus-mediated gene transfer. Despite all achieved experience from RNA, DNA and protein delivery, there is no generalizable, simple approach for CRISPR delivery which is both effective and safe, suggesting that various cell types and tissues may require different delivery strategies.
In this study we used gold material to develop a widely applicable CRISPR delivery system. Our multilayered CRISPR nanoformulation packaged all required gene editing components with or without a DNA repair template on a single gold nanoparticle core with little impact on monodispersity. Stringent characterization at each component loading step was critical to optimizing this design. Nanoparticles remained in a non-aggregated state and successfully penetrated into hard-to-transfect CD34+ hematopoietic cells. Data from other cell types has shown that AuNP/CRISPR nanoformulations are internalized through endocytosis inside small vesicles which then burst and release into the cytoplasm. A PEI-induced proton sponge effect could be facilitating escape from HSPC lysosomes 37. Additionally, PEI has been shown to play an active role in nuclear trafficking of the nanoformulation which in addition to nuclear localization signals on nuclease proteins could facilitate payload delivery38. The CCR5 and γ-globin promoter loci targeted here were very unique, encoding PAM sites for Cpf1 and Cas9 with the same guide recognition site, enabling unbiased comparison of these two nuclease platforms with our nanoformulation. Importantly, 10 µg/mL AuNP/CRISPR concentrations produced up to 17.6 % total editing with13.4% HDR at the CCR5 locus and 12.1% total editing with 8.8% HDR at the γ-globin promoter locus when Cpf1 nuclease was delivered. Total editing and HDR results were comparable to or higher than electroporation-mediated delivery, suggesting a HSPC biology more amenable to CRISPR gene editing when AuNPs are the delivery mode. Higher levels of HDR observed with Cpf1 in the nanoformulation suggest that staggered nuclease cutting may favor HDR, at least at these therapeutically-relevant loci 12,39.
Colony assay and xenoengraftment data demonstrate that AuNP/CRISPR-HDT treatment did not have any adverse effect on HSPC fitness following ex vivo treatment, and suggest that repopulating potential may even be increased. However, additional donors will need to be evaluated to confirm this observation. One possible explanation for this phenomenon could be the described anti-inflammatory and analgesic effects of gold, which have been exploited in the treatment of rheumatoid arthritis for decades 40. Although total editing and HDR levels decreased after initial hematopoietic repopulation following transplant, levels then stabilized over the remaining course of study. This phenomenon has been reported before with 5- to 20-fold decrease following transplantation and suggests a preference for delivery of gene editing machinery to non-repopulating CD34+ cells41. Our gene editing data in sorted CD34+ HSPC subpopulations as compared to unsorted CD34+ cells supports this interpretation. One solution to this problem would be to refine the cell pool for editing even further42,43. Editing levels could also be increased by screening multiple different target sites in the loci of interest, or incorporating guide modifications or nuclease fusions with increased activity44,45.
Our AuNP/CRISPR nanoformulation addresses several limitations to the current state of the art for HSPC gene editing such as the extent of ex vivo manipulation, electroporation-associated toxicity, and delivery of the RNP complex which has been shown to reduce off target cutting.16,46,47,48 Moreover, our nanoformulation could fairly simply be adapted to other gene editing platforms such as adenine and cytosine base editors for DNA and CRISPR-Cas13 for RNA editing. Currently, these two approaches rely on electroporation due to the low packaging limit and documented immunogenicity of AAV vectors.49 Finally, AuNP/CRISPR-HDT could have utility in other blood cell types. Chimeric antigen receptor (CAR) T cell products currently require CRISPR delivery by electroporation along with AAV vectors carrying the long CAR template.50 If AuNPs can be adapated to carry larger HDT payloads, they could make an attractive delivery alternative in this cell type.
We demonstrate AuNP/CRISPR nanoformulations that efficiently and safely deliver CRISPR gene editing machinery to HSPCs. Our study expands the available toolkit for CRISPR delivery, and speculates that AuNP/CRISPR could be a potent delivery vehicle in different cells or tissues, paving the way for the clinical translation of CRISPR gene editing technology.
METHODS
Synthesis and characterization of nanoformulations
Gold nanoparticles (AuNPs) were synthesized by Turkevich’s method with slight modification 25, 51. 0.25 mM Chloroauric acid solution (Sigma-Aldrich, USA) was brought to the boiling point and reduced by adding 3.33 % sodium citrate solution (Sigma-Aldrich) and stirred vigorously under reflux system for 10 min. Synthesized nanoparticles were washed three times by centrifuging at 17000 x g for 15 min and re-dispersed in ultra pure water (Invitrogen, USA).
All custom oligonucleotides used in this study were purchased from Integrated DNA Technologies (IDT, USA). Cas9 and Cpf1 enzymes were purchased from Aldevron (USA). crRNAs with an 18 oligo ethylene glycol (OEG) spacer-thiol modification on the 3’ end for AsCpf1 and 5’ end for SpCas9 were used (all the sequence information can be found in Supplementary Table 1). crRNA and tracrRNA duplex (gRNA) for Cas9 nuclease were made by mixing equimolar concentrations in duplex buffer, incubating at 95°C for 5 min and cooling on the bench top. AuNPs in 10 µg/mL concentration were added to crRNA or gRNA solution in AuNP/crRNA w/w ratio of 0.5. Citrate buffer (pH 3.0) was added to 10 mM and the resulting solution was mixed for 5 min. Prepared AuNP/crRNA nanoconjugates were centrifuged down and re-dispersed in 154 mM sodium chloride (NaCl) (Sigma-Aldrich). Then, nuclease was added in AuNP/Cpf1 or AuNP/Cas9 w/w ratio of 0.6, and mixed by pipetting the solution up and down and incubating for 15 min. Following that, nanoparticles were centrifuged at 16000 g for 15 min and redispersed in NaCl solution. Polyethyleneimine (PEI) of 2000 MW (Polysciences, USA) was added in 0.005% concentration, mixed thoroughly and after 10 min incubation nanoparticles were centrifuged at 15000 g for 15 min and redispersed in NaCl solution. In the final step, homology-directed repair template (HDT) was added in the AuNP/HDT w/w ratio of 2 and after 10 min incubation nanoparticles were centrifuged and redispersed in NaCl solution.
Size and shape of the prepared nanoformulations were characterized by transmission electron microscope (TEM) (JEOL JEM 1400, USA). Samples were negatively stained first by glow-discharging carbon-coated grid, using the PELCO easiGlow Glow Discharge system (Ted Pella Inc., USA). A volume of 2 µL of the sample was dropped on the grid and after 30s it was blotted off, washed and stained in 0.75% uranyl formate solution (Polysciences). Finally, grids were dried inside a desiccator overnight and imaged by TEM 52.
Hydrodynamic size and polydispersity index of the nanoformulation was characterized on a Zetasizer Nano S device (Malvern Analytical, UK). Measurements were carried out in triplicate and data were analyzed using Zetasizer software (version 7.13). Results were reported as mean ± standard error of the mean (SEM). Low volume disposable cuvettes (ZEN0040) (Malvern) were used for measurements.
Zeta potential of the nanoformulation was characterized on a Zetasizer Nano ZS (Malvern, UK). Disposable Folded Capillary Zeta Cells (Malvern, UK) were used for the measurements and data were analyzed using Zetasizer software (version 7.13). Results were reported as mean ± SEM
Layer by layer conjugation of the clustered regularly-interspaced palindromic repeat (CRISPR) components was characterized by measuring shifts in the localized surface plasmon resonance (LSPR) of gold nanoparticles using a nanodrop device (Thermo Fisher Scientific, USA).
Isolation and culture of CD34+ cells
Primary human CD34+ cells were isolated from consenting healthy adult donors mobilized with granulocyte colony stimulating factor (G-CSF; Filgrastim, Amgen, USA) by the Fred Hutch Core Center of Excellence in Hematology Hematopoietic Cell Procurement and Processing Services Resource under a protocol approved by the Fred Hutch Institutional Review Board (IRB) (protocol No. 985.03) and in accordance with the Declaration of Helsinki and the Belmont Report. The Fred Hutch IRB determined this study to be “Not Human Subjects Research” since blood products were de-identified prior to purchase and no member of the study team had interactions with donors, access to identifiable private information or the ability to link study specimens or results to donors. Whole leukapheresis products were obtained and CD34-expressing cells were purified by immunomagnetic bead-based separation on a CliniMACS™ Prodigy device using previously published protocols 53. Resulting CD34+ cells were cultured in StemSpan Serum-Free Expansion Medium version II (SFEM II; Stem Cell Technologies, Canada) or Iscove’s Modified Dulbecco’s Medium (IMDM; Invitrogen) containing 10% fetal bovine serum (FBS; Gibco, USA), and 100 ng/mL each of recombinant human stem cell factor (SCF), Flt-3 ligand (Flt3) and thrombopoietin (TPO), all from CellGenix (Germany). Incubation conditions were 37°C, 85% relative humidity, 5% CO2 and normoxia.
To isolate long-term hematopoietic stem cell (LT-HSC), multipotent progenitor (MPP) and common myeloid progenitor/ granulocyte-monocyte progenitor/megakaryocyte-erythrocyte progenitor (CMP/GMP/MEP) subfractions, bulk CD34+ cells were suspended in a buffer containing sterile Dulbecco’s phosphate buffered saline (D-PBS) (Gibco), 1% FBS (Atlas Biologicals, USA) and 1mM ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich), and stained with antibodies including anti-human CD34 conjugated to brilliant violet 421 (BV421; Clone 581), CD38 conjugated to PerCP/Cy5.5 (Clone HIT2), CD45RA conjugated to allophycocyanin (APC; Clone 5H9), CD90 conjugated to phycoerythrin (PE; Clone 5E10), CD49f conjugated to PE/Cy7 (Clone GoH3). Anti-human CD90 was from BD Biosciences (USA), all other antibodies were from BioLegend (USA). All antibodies were used for staining following the manufacturer’s specifications. Labeled cells were washed and kept on ice for processing on a Sony MA900 Cell Sorter device (Sony, Japan) in the same buffer used for staining, but sorted into 5mL tubes containing 1mL SFEM II media. All sorted fractions were acquired on a fluorescence activate cell sorter (FACS) Canto II device (BD Biosciences) and analyzed by FlowJo software version 10.1 (Tree Star, USA) to conform >99% purity following sorting. An unsorted, but antibody-labeled fraction of bulk CD34+ cells was retained as a control. Sorted cells were washed in SFEM II media and cultured as above.
In vitro gene editing studies
CD34+ cells and sorted subfractions were thawed and pre-stimulated overnight in SFEM II media containing SCF, Flt3 and TPO. Following overnight culture, cells were seeded in a 96 well plate at 1×106/mL and treated with AuNP/CRISPR nanoformulations at 10 µg/mL concentration of AuNPs. All in vitro experiments were carried out in triplicate. After 48 h incubation, cells were washed with D-PBS (Gibco) and harvested for genomic DNA (gDNA) extraction and gene editing analysis.
Electroporation of the CRISPR components was also carried out for comparison. To do so, 49 pmol crRNA or gRNA was mixed with the same amount of Cpf1 or Cas9 nucleases (8.5 pmol) as present in nanoformulations tested in parallel and incubated for 15 min. Cells were dispersed in electroporation buffer and mixed with ribonucleoprotein (RNP) complex. The mixture was added to 1 mm electroporation cuvettes and electroporated under 125 V and 5 ms pulse duration using BTX electroporator device (Harvard Apparatus, USA). After electroporation, cells were cultured as above, washed after 24 h and cultured again for another 24 h incubation. After 48 h total incubation, cells were washed with D-PBS and harvested for gDNA extraction and gene editing analysis.
Cell viability analysis
Cell viability after treatment with AuNP/CRISPR nanoformulations and electroporation was analyzed at different time points using a Countess II FL Automated Cell Counter (ThermoFisher Scientific). 10 µL of trypan blue stain (0.4%) (Invitrogen) was mixed with 10 µL of cell suspension, and 10 µL of the mixture was applied to a disposable cell counting chamber slide and inserted into the device. Percent cell viability of each sample was recorded and reported as mean ± SEM
In order to confirm the results, cell viability was also analyzed using the LIVE/DEAD® assay kit (Invitrogen) which uses Calcein AM (retained by live cells) and ethidium homodimer (EthD-1) (taken up by dead cells). Cells were washed in D-PBS and sedimented by centrifugation. An aliquot of the cell suspension was transferred to a sterile, 22 mm square coverslip and cells were allowed to settle to the surface at 37°C in a covered 35 mm petri dish. Calcein AM (2 μM) and ethidium homodimer-1 (EthD-1) (4 μM) working solutions were prepared. A 150 μL volume of the combined LIVE/DEAD® assay reagents were added to the coverslip such that all cells were covered. Coverslips were incubated in a covered dish for 30 min at room temperature, after which 10 μL of D-PBS was added to a clean microscope slide and each coverslip was inverted and mounted onto an individual microscope slide. Labeled cells were imaged by fluorescence microscope (Nikon Ti Live, Japan) using excitation and emission values of 494/517 nm for Calcein AM, and 528/617 nm for EthD-1. Live and dead cells were counted using the cellomics vHCS software (v1.6.3.0, Thermo Fisher Scientific). Images were processed using ImageJ software (V 1.5i, National Institutes of Health, USA).
Colony Forming Cell (CFC) Assay
For CFC assays, cells were plated in methylcellulose (H4230: Stem Cell Technologies) containing recombinant human growth factors according to the manufacturer’s specifications and incubated for a period of 14 days. Resulting colonies were counted and scored for morphology under 4x magnification on a stereo microscope (ZEISS Stemi 508; ZEISS, Germany) to determine the number of colony-forming cells for every 500 cells plated.
For CFC replating, all colonies from a single 35mm plate for a given sample were collected into an excess volume of sterile D-PBS and washed by vortexing followed by centrifugation and supernatant aspiration to remove residual methylcellulose. Cell pellets were resuspended in SFEM II media as a single cell solution, and a small fraction of the homogenous solution equivalent to 5% of the total volume was plated as a new CFC assay.
Genome editing detection by T7 Endonuclease I
To analyze the total gene editing percentage, gDNA was extracted using the PureLink® Genomic DNA Mini Kit (Thermo Fisher Scientific) following the manufacturer’s protocol and amplified by polymerase chain reaction (PCR).
The genomic region flanking the CRISPR target site (755 bp) was PCR amplified (all the sequence information can be found in Supplementary Table 1), and products were purified using the PureLink™ PCR Purification Kit (Thermo Fisher Scientific) following the manufacturer’s protocol. A volume equivalent to 200 ng of the purified PCR products was mixed with 2 µL 10x NEBuffer 2 (New England BioLabs, USA) and ultrapure water to a final volume of 19 µL. Mixtures were subjected to a re-annealing process to enable heteroduplex formation with the following thermocycle conditions: 95°C for 5 min, ramping down from 95°C to 85°C at −2°C/s, then rampling down from 85°C to 25°C at −0.1°C/s, and a final 4°C hold. After re-annealing, products were treated with 1 µL of T7EI nuclease (New England BioLabs) and incubated for 15 min at 37°C. After incubation digested products were purified with the PureLink™ PCR Purification Kit and analyzed by electrophoresis in 2% agarose gel. Gels were imaged with a Gel Doc gel imaging system (Bio-Rad, USA). Quantification was determined by relative band intensities. Indel percentage was determined by the formula in Equation 1 below:
| Equation 1: |
NotI restriction enzyme digestion
Genomic region flanking the CRISPR target site (755 bp) was PCR amplified and products were purified using the PureLink™ PCR Purification Kit (Thermo Fisher Scientific) following the manufacturer’s protocol. A total of 1000 ng of the purified PCR products were mixed with 5 µL CutSmart® Buffer (New England BioLabs), 1 µL of NotI enzyme (New England BioLabs) and ultrapure water to a final volume of 50 µL. After incubation for 15 min at 37°C, digested products were purified using the PureLink™ PCR Purification Kit and analyzed by electrophoresis in 2% agarose gel. Gels were imaged with a Gel Doc gel imaging system (Bio-Rad, USA). Quantification was determined by relative band intensities. Gene insertion percentage was determined by Equation 1 above.
Tracking indels by decomposition (TIDE) assay
Genomic region flanking the CRISPR target site (755 bp) was PCR amplified (all the sequence information can be found in Supplementary Table 1). and products were purified using the PureLink™ PCR Purification Kit following the manufacturer’s protocol. Sanger sequencing was carried out by mixing 20 ng of DNA sample with 4 µL of BigDye® Terminator (Thermo Fisher Scientific), and ultrapure water to a final volume of 10 µL. After sequencing samples were analyzed on a 3730xl DNA Analyzer (Applied Biosystems, USA). Obtained sequences were processed using TIDE software (https://tide.nki.nl/) and results were reported as percent gene editing 54.
MiSeq analysis
A first PCR was carried out on the genomic region flanking the CRISPR target site (755 bp) (all the sequence information can be found in Supplementary Table 1) and products were purified using the PureLink™ PCR Purification Kit (Thermo Fisher Scientific) following the manufacturer’s protocol. A second PCR was carried out using primers with MiSeq adapter sequences on the genomic region flanking the CRISPR target site (157 bp) and products were purified using the PureLink™ PCR Purification Kit as above. Specific bands were checked by electrophoresing 5 µL of the sample in 2% agarose gel. Following electrophoresis, indexing of the DNA was carried out using the Nextera Index kit (96 indexes) (Illumina, USA) with 8 cycles and indexed products were purified using the PureLink™ PCR Purification Kit. Finally, the prepared library was diluted to 4 nM, pooled and analyzed by Illumina HiSeq 2500 (Illumina). Sequencing reads were analyzed using a publicly accessibly bioinformatics pipeline designed in-house (GitHub: FredHutch_CGT_Gene_Edit_1). Paired high-throughput sequencing reads (MiSeq) were combined with paired-end read merger (PEAR)55. Combined reads were then filtered with a custom python script. Reads without perfect primer sequences were discarded. Primer sequences were trimmed from the reads and then identical sequences were grouped together. A Needleman-Wunsch aligner from the emboss suite was used to align the sequence reads to the reference amplicon 56, 57. The options used with this aligner were: -gapopen 10.0, -gapextend 0.5, and -aformat3 sam. The custom python script then reads the Concise Idiosyncratic Gap Alignment Report (CIGAR) string from the Sequence Alignment Map (SAM) output and uses this information to identify and quantify insertions and deletions. Each aligned sequence was also compared to the reference amplicon to identify substitution mutations. Any mutation found in only one read was removed from the analysis. A table containing mutation sequences, read count, and frequency for each mutation was then output for further analysis. In each sequencing run, a control sample consisting of untreated cells from the same donor or a syngeneic, untransplanted animal determined the average frequency of mutation classes (insertion, deletion, substitution, insertion and substitution, etc.), and was used to perform a one-tailed binomial t-test on each mutation from the corresponding mutation class. Mutations from experimental samples were retained if they demonstrated a p-value < 0.05. All sequence data are available for download through the National Center for Biotechnology Information (BioProject ID: PRJNA529681).
In vivo engraftment studies in mice
All experiments involving live animals were conducted at the Fred Hutch. The Fred Hutch is an Association for the Assessment and Accreditation of Laboratory Animal Care International (AALAC)-accredited research institution. All live animal studies performed were conducted in accordance with the Office of Laboratory Animal Welfare (OLAW) Public Health Assurance (PHS) policy, United States Department of Agriculture (USDA) Animal Welfare Act and Regulations, the Guide for the Care and Use of Laboratory Animals and the Fred Hutch Institutional Animal Care and Use Committee (IACUC) policies (IACUC protocol No. 1864).
NOD.Cg-PrkdcscidIl2rgtm1Wjl/Szj (Il2r gamma–/–) mice were obtained from The Jackson Laboratory (USA) and bred in-house in pathogen-free housing conditions. Neonates (1–3 days old) received 175 cGy total body irradiation from a Cesium irradiator followed 3–4 hours later by a single, intrahepatic injection of 1 × 106 primary human CD34+ hematopoietic cells resuspended in 30 μL of phosphate-buffered saline (PBS; Invitrogen) containing 1% heparin (APP Pharmaceuticals, Puerto Rico). Four weeks post-engraftment blood was collected by retro-orbital puncture to determine the level of human blood cells by flow cytometry. Blood was collected every two weeks for the duration of follow-up. White blood cells were isolated and stained with anti-human CD45-PerCP antibody (Clone 2D1), CD3-FITC (Clone UCHT1), CD4-V450 (Clone RPA-T4), CD20-PE (Clone 2H7), and CD14-APC (Clone M5E2) (all from BD Biosciences) as previously reported58. Stained cells were acquired on a FACS Canto II (BD Biosciences) and analyzed using FlowJo software version 10.1 (Tree Star).
Confocal microscopy imaging
In order to track intracellular biodistribution, Cpf1 crRNA, and HDT template were fluorescently tagged by Alexa 488, and Alexa 660 fluorophores on the 5’ end, respectively (IDT). AuNP/CRISPR nanoformulations were prepared and incubated with primary human CD34+ cells for 6 h. At the end of incubation cells were washed and dispersed in FluoroBrite™ Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco) inside a FluoroDish. Two drops of NucBlue™ Live ReadyProbes™ Reagent (Ex/Em 360/460 nm) (Invitrogen) were added to the cells and incubated for 30 min at room temperature. Finally, cells were imaged on a ZEISS LSM 780 Confocal and Multi-Photon with Airyscan microscope. Images were analyzed using ZEN Lite software (ZEISS). Imaging was carried out using a 60x objective after background adjustments.
Statistical analysis
All data are reported as means ± SEM Statistical analysis was performed using the two-sided Student’s t-test or analysis of variance (ANOVA, one-way/two-way) where applicable, using GraphPad Prism software, version 7.03 for Windows, (GraphPad Software, USA). In all cases confidence intervals were 95%, and effect sizes were calculated as Cohen’s d (mean difference divided by the pooled standard deviation), where mean standard deviation was used when individual standard deviations were unequal. No adjustments were made for multiple comparisons in any statistical test. A p-value <0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank healthy donors who submitted to mobilization and leukapheresis collection. We thank the laboratory of Dr. Miqin Zhang at the University of Washington for access to and use of Nanosizer equipment. We extend a special thanks to H. Crawford for assistance in manuscript preparation, and J. Chen and C. Ironside for excellent support in animal studies. This work was primarily supported by funds to J.E.A. from the Fred Hutch including Development and Evergreen awards, and the Hartwell Foundation. This research was also funded in part through a pilot study award to R.S. from the NIDDK Cooperative Center of Excellence in Hematology Grant U54 DK106829. All shared resources used in this study were supported by the NIH/NCI Cancer Center Support Grant P30 CA015704. H.-P.K. is a Markey Molecular Medicine Investigator, the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Fred Hutch Endowed Chair for Cell and Gene Therapy. J.E.A. and R.S. are co-inventors on U.S. Patent WO2018226762A1 entitled “Genomic Safe Harbors for Genetic Therapies in Human Stem Cells and Engineered Nanoparticles to Provide Targeted Genetic Therapies.”
Footnotes
Competing Financial Interests
H.-P.K., O.H. and J.E.A. received licensing revenue from Rocket Pharmaceuticals for research unrelated to this manuscript. K.G.H. is employed by and holds equity in Nohla Therapeutics. The other authors declare no competing financial interests associated with this work.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Sequence data are available for download through the National Center for Biotechnology Information (BioProject ID: PRJNA529681).
ADDITIONAL INFORMATION
Supplementary information accompanies this paper, including two tables and thirteen figures.
REFERENCES
- 1.Hacein-Bey-Abina S, Pai SY, Gaspar HB, Armant M, Berry CC, Blanche S, et al. A modified gamma-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med 2014, 371(15): 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cicalese MP, Ferrua F, Castagnaro L, Pajno R, Barzaghi F, Giannelli S, et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood 2016, 128(1): 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sessa M, Lorioli L, Fumagalli F, Acquati S, Redaelli D, Baldoli C, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase ½ trial. Lancet 2016, 388(10043): 476–487. [DOI] [PubMed] [Google Scholar]
- 4.Hacein-Bey Abina S, Gaspar HB, Blondeau J, Caccavelli L, Charrier S, Buckland K, et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. Jama 2015, 313(15): 1550–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science 2018, 359(6372). [DOI] [PubMed] [Google Scholar]
- 6.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302(5644): 415–419. [DOI] [PubMed] [Google Scholar]
- 7.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 2003, 348(3): 255–256. [DOI] [PubMed] [Google Scholar]
- 8.Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med 2006, 12(4): 401–409. [DOI] [PubMed] [Google Scholar]
- 9.Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 2010, 16(2): 198–204. [DOI] [PubMed] [Google Scholar]
- 10.Cornu TI, Mussolino C, Cathomen T. Refining strategies to translate genome editing to the clinic. Nat Med 2017, 23(4): 415–423. [DOI] [PubMed] [Google Scholar]
- 11.Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nature reviews Molecular cell biology 2017, 18(8): 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163(3): 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 2016, 539(7629): 384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Ravin SS, Li L, Wu X, Choi U, Allen C, Koontz S, et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Science translational medicine 2017, 9(372). [DOI] [PubMed] [Google Scholar]
- 15.Lefesvre P, Attema J, van Bekkum D. A comparison of efficacy and toxicity between electroporation and adenoviral gene transfer. BMC molecular biology 2002, 3: 12–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li L, He ZY, Wei XW, Gao GP, Wei YQ. Challenges in CRISPR/CAS9 delivery: Potential roles of nonviral vectors. Hum Gene Ther 2015, 26(7): 452–462. [DOI] [PubMed] [Google Scholar]
- 17.Finn JD, Smith AR, Patel MC, Shaw L, Youniss MR, van Heteren J, et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Reports 2018, 22(9): 2227–2235. [DOI] [PubMed] [Google Scholar]
- 18.Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering 2017, 1(11): 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee B, Lee K, Panda S, Gonzales-Rojas R, Chong A, Bugay V, et al. Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nature Biomedical Engineering 2018, 2(7): 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science 2006, 312(5776): 1027–1030. [DOI] [PubMed] [Google Scholar]
- 21.Ding Y, Jiang Z, Saha K, Kim CS, Kim ST, Landis RF, et al. Gold nanoparticles for nucleic acid delivery. Molecular Therapy 2014, 22(6): 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan Y, Neuss S, Leifert A, Fischler M, Wen F, Simon U, et al. Size-dependent cytotoxicity of gold nanoparticles. Small 2007, 3(11): 1941–1949. [DOI] [PubMed] [Google Scholar]
- 23.Alkilany AM, Murphy CJ. Toxicity and cellular uptake of gold nanoparticles: what we have learned so far? J Nanopart Res 2010, 12(7): 2313–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewinski N, Colvin V, Drezek R. Cytotoxicity of nanoparticles. Small 2008, 4(1): 26–49. [DOI] [PubMed] [Google Scholar]
- 25.Turkevich J, Stevenson PC, Hillier J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discussions of the Faraday Society 1951, 11(0): 55–75. [Google Scholar]
- 26.Yamano T, Nishimasu H, Zetsche B, Hirano H, Slaymaker IM, Li Y, et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell 2016, 165(4): 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee K, Mackley VA, Rao A, Chong AT, Dewitt MA, Corn JE, et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife 2017, 6: e25312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopalco L CCR5: From natural resistance to a new anti-HIV strategy. Viruses 2010, 2(2): 574–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akinsheye I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastiani P, et al. Fetal hemoglobin in sickle cell anemia. Blood 2011, 118(1): 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, et al. A genome-editing strategy to treat beta-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 2016, 22(9): 987–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bae S, Park J, Kim JS. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30(10): 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mariam J, Sivakami S, Dongre PM. Albumin corona on nanoparticles - a strategic approach in drug delivery. Drug Deliv 2016, 23(8): 2668–2676. [DOI] [PubMed] [Google Scholar]
- 33.Yin H, Song CQ, Suresh S, Wu Q, Walsh S, Rhym LH, et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat Biotechnol 2017, 35(12): 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basic Cell Culture Protocols, 4th edn Humana Press, 2013. [Google Scholar]
- 35.Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351(6269): aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong X, Chen M, Lim WA, Zhao D, Qi LS. CRISPR/Cas9 for human genome engineering and disease research. Annu Rev Genomics Hum Genet 2016, 17(1): 131–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benjaminsen RV, Mattebjerg MA, Henriksen JR, Moghimi SM, Andresen TL. The possible “proton sponge “ effect of polyethylenimine (PEI) does not include change in lysosomal pH. Molecular therapy : the journal of the American Society of Gene Therapy 2013, 21(1): 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reza S, Ilyas O, Gurkan O, Kezban U. Functionalized gold nanoparticles manifested as potent carriers for nucleolar targeting. Nanotechnology 2017, 28(2): 025103. [DOI] [PubMed] [Google Scholar]
- 39.Nakade S, Yamamoto T, Sakuma T. Cas9, Cpf1 and C2c½/3―What’s next? Bioengineered 2017, 8(3): 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Araújo RF, de Araújo AA, Pessoa JB, Freire Neto FP, da Silva GR, Leitão Oliveira ALCS, et al. Anti-inflammatory, analgesic and anti-tumor properties of gold nanoparticles. Pharmacological Reports 2017, 69(1): 119–129. [DOI] [PubMed] [Google Scholar]
- 41.Xu L, Yang H, Gao Y, Chen Z, Xie L, Liu Y, et al. CRISPR/Cas9-mediated CCR5 ablation in human hematopoietic stem/progenitor cells confers HIV-1 resistance in vivo. Molecular therapy : the journal of the American Society of Gene Therapy 2017, 25(8): 1782–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radtke S, Adair JE, Giese MA, Chan YY, Norgaard ZK, Enstrom M, et al. A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Science translational medicine 2017, 9(414): doi: 10.1126/scitranslmed.aan1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masiuk KE, Brown D, Laborada J, Hollis RP, Urbinati F, Kohn DB. Improving gene therapy efficiency through the enrichment of human hematopoietic stem cells. Molecular therapy : the journal of the American Society of Gene Therapy 2017, 25(9): 2163–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Charpentier M, Khedher AHY, Menoret S, Brion A, Lamribet K, Dardillac E, et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat Commun 2018, 9(1): 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McMahon MA, Prakash TP, Cleveland DW, Bennett CF, Rahdar M. Chemically modified Cpf1-CRISPR RNAs mediate efficient genome editing in mammalian cells. Molecular therapy : the journal of the American Society of Gene Therapy 2018, 26(5): 1228–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kimberland ML, Hou W, Alfonso-Pecchio A, Wilson S, Rao Y, Zhang S, et al. Strategies for controlling CRISPR/Cas9 off-target effects and biological variations in mammalian genome editing experiments. J Biotechnol 2018, 284: 91–101. [DOI] [PubMed] [Google Scholar]
- 47.Glass Z, Lee M, Li Y, Xu Q. Engineering the delivery system for CRISPR-based genome editing. Trends Biotechnol 2018, 36(2): 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lino CA, Harper JC, Carney JP, Timlin JA. Delivering CRISPR: a review of the challenges and approaches. Drug Deliv 2018, 25(1): 1234–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 2018, 19(12): 770–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543(7643): 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
REFERENCES TO METHODS
- 51.Shahbazi R, Asik E, Kahraman N, Turk M, Ozpolat B, Ulubayram K. Modified gold-based siRNA nanotherapeutics for targeted therapy of triple-negative breast cancer. Nanomedicine (London, England) 2017, 12(16): 1961–1973. [DOI] [PubMed] [Google Scholar]
- 52.Booth DS, Avila-Sakar A, Cheng Y. Visualizing Proteins and Macromolecular Complexes by Negative Stain EM: from Grid Preparation to Image Acquisition. J Vis Exp 2011(58): 3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adair JE, Waters T, Haworth KG, Kubek SP, Trobridge GD, Hocum JD, et al. Semi-automated closed system manufacturing of lentivirus gene-modified haematopoietic stem cells for gene therapy. Nat Commun 2016, 7: 13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research 2014, 42(22): e168–e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30(5): 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. Journal of Molecular Biology 1970, 48(3): 443–453. [DOI] [PubMed] [Google Scholar]
- 57.Kruskal JB. An overview of sequence comparison In: Sankoff D, Kruskal JB (eds). Time warps, string edits, and macromolecules: The theory and practice of sequence comparison. Addison-Wesley: Reading, MA, 1983, pp 1–44. [Google Scholar]
- 58.Haworth KG, Ironside C, Norgaard ZK, Obenza WM, Adair JE, Kiem HP. In vivo murine-matured human CD3(+) cells as a preclinical model for T cell-based immunotherapies. Mol Ther Methods Clin Dev 2017, 6: 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.