

SUMMARY
Despite recent successes of precision and immunotherapies there is a persisting need for novel targeted or multi-targeted approaches in complex diseases. Through a systems pharmacology approach including phenotypic screening, chemical and phosphoproteomics and RNA-Seq, we elucidated the targets and mechanisms underlying the differential anticancer activity of two structurally related multi-kinase inhibitors, foretinib and cabozantinib, in lung cancer cells. Biochemical and cellular target validation using probe molecules and RNA interference revealed a polypharmacology mechanism involving MEK1/2, FER and AURKB, which were each more potently inhibited by foretinib than cabozantinib. Based on this, we developed a synergistic combination of foretinib with barasertib, a more potent AURKB inhibitor, for MYC-amplified small cell lung cancer. This systems pharmacology approach showed that small structural changes of drugs can cumulatively, through multiple targets, result in pronounced anticancer activity differences and that detailed mechanistic understanding of polypharmacology can enable repurposing opportunities for cancers with unmet medical need.
Graphical Abstract
eTOC Blurb
Sumi et al. use a systems polypharmacology approach involving chemical proteomics, phosphoproteomics and RNA-Seq to elucidate the differential mechanism of action of two multi-targeted drugs with similar chemical structure. The understanding of the polypharmacology mechanism of foretinib allowed for the rational development of a synergistic drug combination.
INTRODUCTION
Many severe diseases, such as diabetes and cancer, are highly complex and involve various deregulated pathways. An attractive approach to developing new therapies is therefore based on targeting multiple disease-relevant pathways (Gentile et al., 2017). While this can be accomplished in many cases with drug combinations, it would be ideal to have a single drug with defined pharmacokinetic and pharmacodynamic properties capable of simultaneously targeting multiple disease-relevant targets and pathways, a concept known as “polypharmacology” (Anighoro et al., 2014; Hopkins, 2008; Knight et al., 2010). However, defining which combinations of targets confer vulnerability is often difficult, particularly in the absence of a known disease driver. Unbiased phenotypic screening using for example RNA interference or small molecule inhibitors can provide new starting points for identifying such vulnerabilities. Screening with small molecules is a particularly attractive strategy as it immediately infers “druggability” of the underlying target(s). Moreover, if the selected hit compound is a clinical drug/candidate, this approach entails the potential for fast and efficient repurposing. In addition, as targeted drugs can have widely varying selectivity profiles (Bantscheff et al., 2007; Davis et al., 2011; Knezevic et al., 2016; Ong et al., 2009; Zhang et al., 2009), the observed cellular activity can be the result of inhibition of the intended (“cognate”) or of non-canonical targets (“off-targets”). These on- or off-target effects can be readily distinguished by careful library design with multiple compounds for each individual cognate target (Ctortecka et al., 2018; Kuenzi et al., 2017). Considering that polypharmacology per definitionem involves multiple targets, it is important to elucidate off-target mechanisms that translate into cellular activity, which can lead to identification of new clinical opportunities (Kuenzi et al., 2017; Li et al., 2010). This can be achieved by applying systems pharmacology approaches involving, for instance, global proteomics and transcriptomics or a combination thereof (Lamb et al., 2006; Winter et al., 2012).
We here explore these concepts in lung cancer, the leading cause of cancer-related death in the US (Siegel et al., 2018). Through unbiased viability-based drug screening in a panel of non-small cell lung cancer (NSCLC) cell lines, we observed differential cellular activity of the multi-targeted clinical kinase inhibitors cabozantinib (XL184, 1) and foretinib (XL880, 2) across multiple cell lines with foretinib displaying markedly higher potency than cabozantinib. Foretinib and cabozantinib show high structural similarity and similar potency for their cognate targets MET and VEGFR-2 (Qian et al., 2009; Yakes et al., 2011; You et al., 2011) suggesting that foretinib’s mechanism of action (MoA) in these cells involves one or more unrecognized off-targets. In order to identify these targets, we applied an integrated systems pharmacology approach comprised of mass spectrometry (MS)-based chemical proteomics, global and tyrosine phosphoproteomics, as well as RNA-Seq-based transcriptomics. This combined strategy revealed a complex polypharmacology MoA for foretinib, which involves simultaneous inhibition of MEK1/2, FER and AURKB kinases, and led to the rational design of a synergistic drug combination with a more potent AURKB inhibitor in MYC-amplified small cell lung cancer (SCLC) cells, which depend on AURKB and constitute a highly unmet medical need. Notably, comparison of foretinib and cabozantinib also highlights that small changes in the chemical structures of closely related compounds can result in moderate differences in potency across multiple individual targets. This in turn can lead to pronounced differences in overall anticancer activity when amplified by differential polypharmacology mechanisms that involve several of these targets. We furthermore demonstrate that an unbiased and integrated systems pharmacology approach can elucidate these complex mechanisms and lead to new therapeutic opportunities.
RESULTS
Foretinib displays anticancer activity in NSCLC cells independent of its cognate targets
To identify new vulnerabilities in lung cancer, we performed a viability-based drug screen in a panel of NSCLC cell lines with a customized library of 240 compounds, which included diverse classes of targeted drugs. This screen revealed markedly different potency of the two related multi-kinase inhibitors foretinib and cabozantinib across various cell lines (Figure 1A,B). Foretinib and cabozantinib were developed as dual MET/VEGFR-2 inhibitors and display similarly potent biochemical activity of approximately 1 nM for both targets, as well as high structural similarity based on a trisubstituted quinoline scaffold (Qian et al., 2009; Yakes et al., 2011). The only structural differences featured by foretinib are a fluorine substituent on the central phenyl ring and a terminal N-propyl-morpholino moiety shown by X-ray co-crystallization of MET and foretinib to extend into the solvent space (pdb: 3LQ8) (Qian et al., 2009). Despite these similarities, foretinib displayed much higher cellular activity compared to cabozantinib in our screen, which was confirmed in 14 NSCLC cell lines (Figure 1C, Figure S1). PF-04217903 and motesanib, other potent inhibitors of MET and VEGFR-2, respectively, like cabozantinib showed little to no effects suggesting foretinib’s activity in these cells to be independent of its cognate targets. Consistently, although foretinib and cabozantinib both inhibited MET autophosphorylation in H1648 cells (Figure S2A), most NSCLC cell lines, including H1155 and A427 cells, did not display active MET signaling under basal conditions (Figure S2B). Additionally, in contrast to MET inhibitor-sensitive EBC1 cells (Figure S2C,D), inhibition of efflux pumps by verapamil did not sensitize these cells to cabozantinib suggesting that cabozantinib’s lack of efficacy was not simply due to drug efflux (Figure S2E). Foretinib’s activity was also not correlated with any specific oncogene driver (Figure 1B). However, foretinib, in contrast to cabozantinib, inhibited A427 cell growth also in a crystal violet clonogenic assay (Figure 1D). Finally, foretinib, but not cabozantinib, induced apoptosis in H1155 and A427 cells as demonstrated by immunoblot analysis of PARP1 and caspase 3 cleavage (Figure 2A), as well as live-cell imaging of caspase 3/7 induction (Figure 2B). Taken together, the multi-kinase inhibitor foretinib, but not its close structural analogue cabozantinib, displays anticancer activity across multiple NSCLC cell lines independently of its cognate targets.
Figure 1. Foretinib displays non-canonical anticancer activity in NSCLC cells.
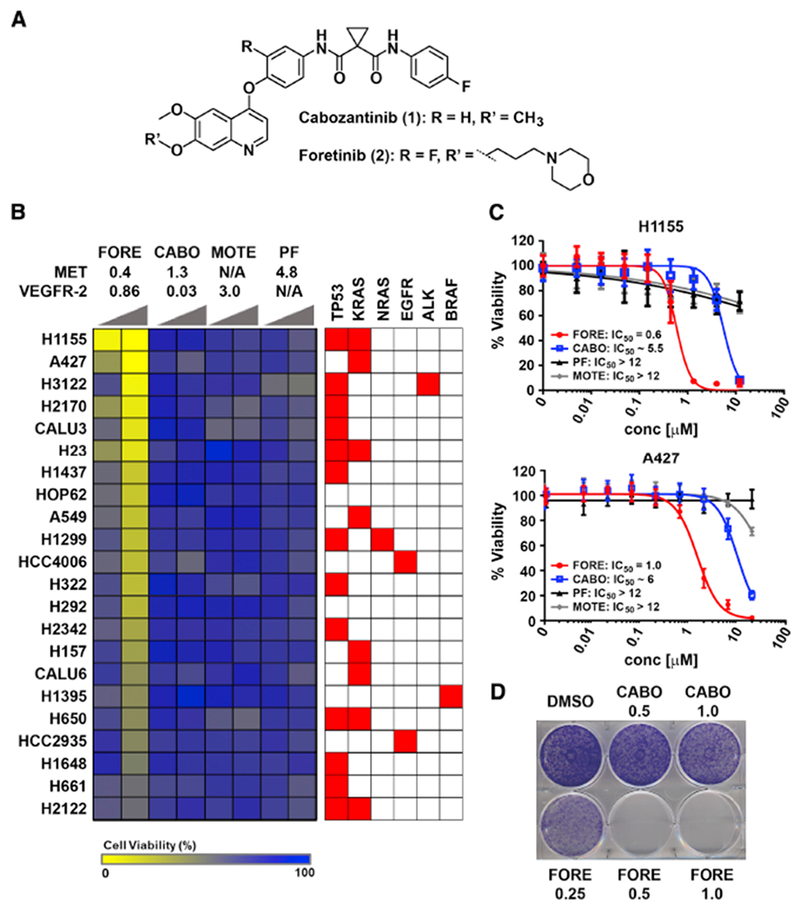
(A) Chemical structures of cabozantinib and foretinib.
(B) Viability-based cellular drug screening after 72 hours of treatment with foretinib (FORE), cabozantinib (CABO), motesanib (MOTE) and PF-04217903 (PF) in a total of 22 NSCLC cells with different genetic background. Displayed above the heatmap are the IC50 or Ki values (in nM) for MET and VEGFR-2 inhibition. Increasing wedges indicate concentrations of 0.5 μM and 2.5 μM. N/A: not applicable.
(C) Dose response curves of FORE, CABO, PF and MOTE for inhibition of viability of H1155 and A427 cells after 72 hours treatment and the respective IC50 values [μM], n = 3, Data are represented as mean ± SD.
(D) Representative clonogenic assay in A427 cells. Cells were treated with the indicated doses of FORE and CABO for 10 days. Data is representative of three independent experiments.
Figure 2. Foretinib induces apoptosis in NSCLC cells.
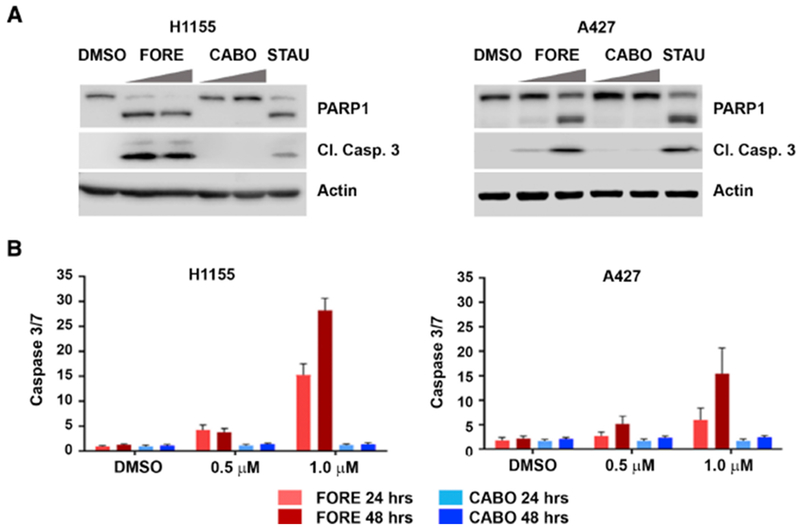
(A) Western blot for PARP1 and cleaved caspase 3 in H1155 (left) and A427 (right) cells. Cells were treated with 0.5 μM and 1 μM of FORE and CABO for 24 hours. Staurosporine (STAU; 50 nM) was used as positive control.
(B)Quantification of cleaved caspase 3/7 using fluorescence microscopy after 24 and 48 hours of treatment with FORE and CABO in H1155 (left) and A427 (right) cells, n = 3, Data are represented as mean ± SD.
Systems pharmacology approach reveals differential targets and pathways of foretinib and cabozantinib
To elucidate the MoA responsible for the differential cellular activity of foretinib and cabozantinib, we applied a systems pharmacology approach. We compared the target profiles of foretinib and cabozantinib, their network-wide signaling effects and gene expression changes upon drug treatment by a combination of chemical proteomics, phosphoproteomics and RNA-Seq (Figure 3). Data integration using Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) nominated specific targets and pathways for functional validation.
Figure 3. Systems pharmacology approach to interrogate the drug-modulated proteome and transcriptome.
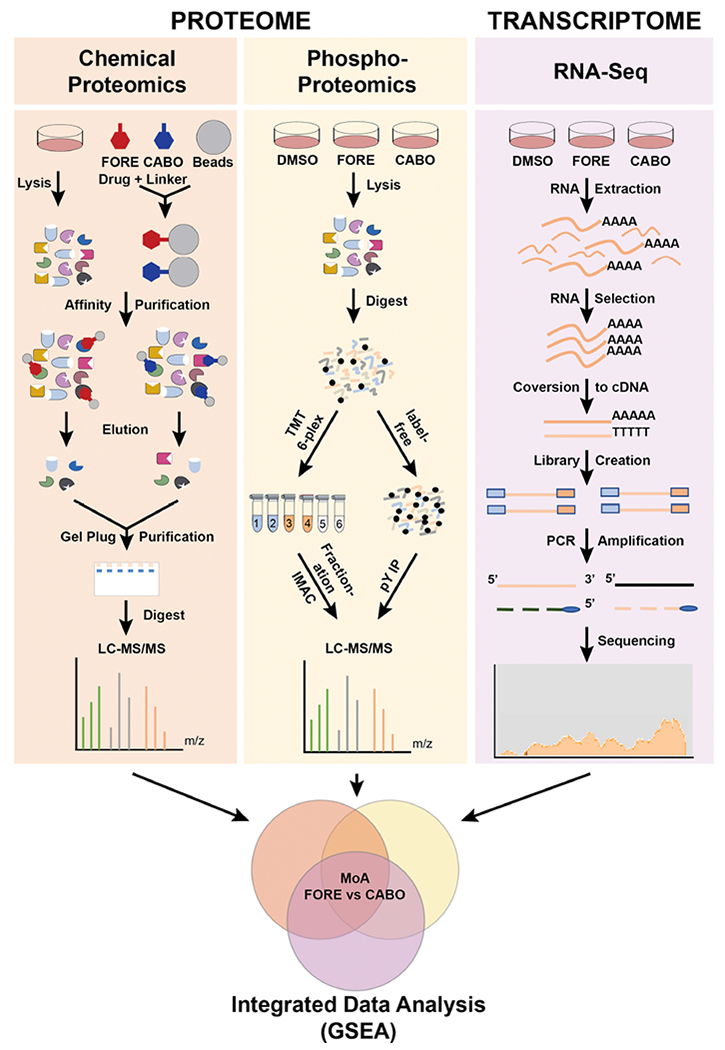
Scheme of the applied systems pharmacology approach including chemical proteomics, global and tyrosine phosphoproteomics and RNA-Seq performed in parallel. FORE: foretinib, CABO: cabozantinib.
Chemical proteomics, unbiased drug affinity chromatography followed by mass spectrometry (MS) (Rix and Superti-Furga, 2009), was performed to identify protein targets for both compounds. Based on the published X-ray co-crystal structure of foretinib in complex with MET (pdb: 3LQ8) (Qian et al., 2009), we synthesized immobilizable analogues of cabozantinib (i-cabozantinib, 10) and foretinib (i-foretinib, 18) by adding a linker to the solvent-exposed morpholino moiety of foretinib and the equivalent position of cabozantinib, for which no X-ray co-crystal structure has been reported (Figure S3A,B). Pulldowns with lysate from H1648 cells, which express high MET levels, and in vitro MET kinase assays indicated that both probes retained their ability to bind and inhibit MET (Figure S4A,B), suggesting i-foretinib and i-cabozantinib to be generally suitable probe molecules. Employing these probes for chemical proteomics in H1155 cells (Table S1), a total of 89 protein kinases were detected with a minimum of 2 unique peptides, 41 of which had normalized spectrum abundance factor (NSAF) values greater than 0.0006 for foretinib, a metric for relative protein abundance in the eluate (Zybailov et al., 2006). Foretinb and cabozantinib shared the majority of their targets (Figure 4A). This was particularly the case for more prominent kinase targets while differences were mostly discernible for kinases with low NSAF values indicating either low expression, binding affinity or both. Consistently, comparison with the publicly available LINCS (Library of Integrated Network-based Cellular Signatures) dataset (ID 20176) (Davis et al., 2011), which contains dissociation constants (Kd) for multiple foretinib kinase targets (cabozantinib is not included in LINCS), showed weak or no (grey circles) Kd values for foretinib-specific, but less prominent targets, such as PERK (E2AK3), STK33 and AMPKα1 (AAPK1) (Figure S4C). No individual kinase showed strong preferential binding to foretinib over cabozantinib and high NSAF value and this was similar in A427 cells (Figure S4D, Table S1). One exception was MAP4K5 (M4K5) (Kd = 3.5 nM), which was also prominently enriched using activity-based protein profiling (ABPP) (Barglow and Cravatt, 2007; Patricelli et al., 2011) of foretinib and cabozantinib in H1155 cells (Figure S4E, Table S1). However, siRNA-mediated silencing of MAP4K5 alone or in combination with cabozantinib did not reduce cell viability indicating that its inhibition is not responsible for the difference between foretinib and cabozantinib activity (Figure S4F).
Figure 4. Systems pharmacology reveals differential targets and pathways of foretinib and cabozantinib.
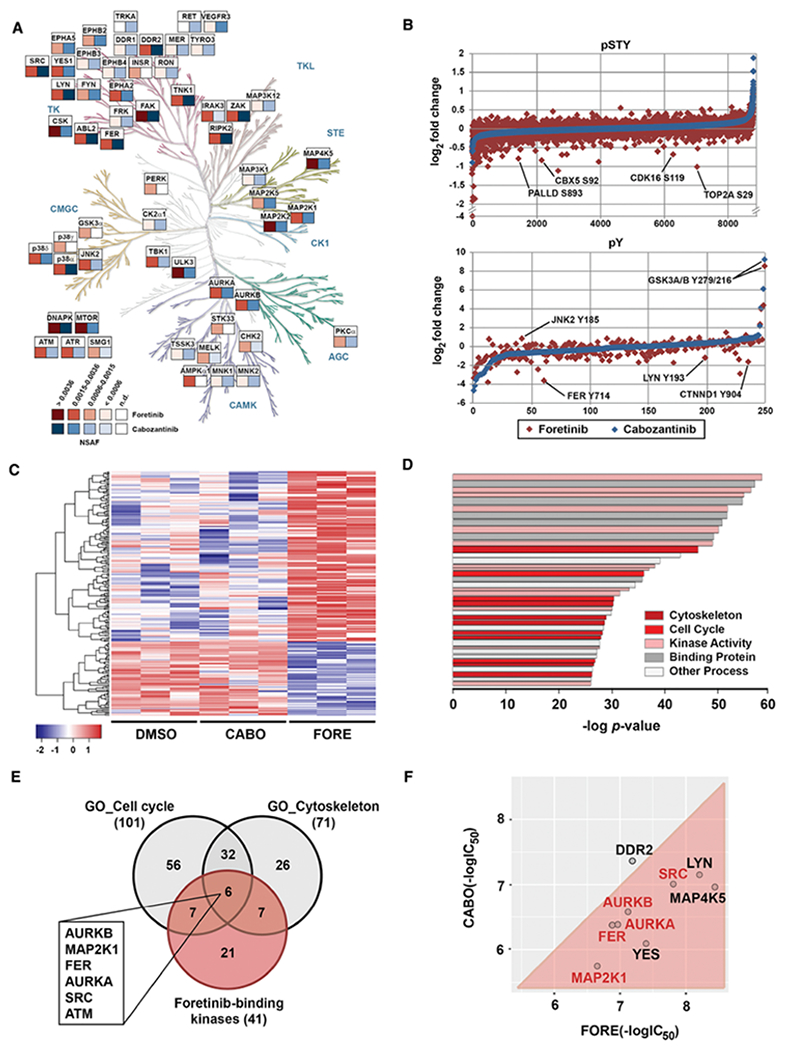
(A) Chemical proteomics comparison between foretinib (red) and cabozantinib (blue) kinase targets presenting NSAF > 0.0006 in H1155 cells (see also Figure S4C). Kinome phylogenetic tree adapted courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com).
(B) Global (pSTY) and phosphotyrosine (pY) proteomics data for H1155 cells after 3 hours treatment with 1 μM of foretinib (red) or cabozantinib (blue).
(C) Heatmap representation of RNA-Seq analysis in H1155 cells after 4 hours of treatment with DMSO, cabozantinib (CABO) or foretinib (FORE) at 1 μM. Displayed are the 231 genes that were differentially expressed.
(D) Pathway analysis using GSEA: top 40 pathways assigned with GSEA after combining the specific data. Colors classify pathways into clusters. Bar strength correlates with the number of genes in each pathway.
(E) Venn diagram analysis of foretinib-binding kinases and the most predominant differentially regulated pathways between foretinib and cabozantinib. Highlighted are foretinib-binding kinases that are shared by both pathways
(F) Comparison of in vitro kinase assay IC50 values for foretinib (FORE) and cabozantinib (CABO) for select kinase targets. Displayed are the negative log IC50 values (in M).
In parallel, we performed quantitative phosphoproteomics in H1155 cells upon treatment with foretinib and cabozantinib. Global phosphoproteomics was conducted using a 6-plex tandem mass tag (TMT) isobaric labeling approach followed by basic pH reversed-phase off-line fractionation and global enrichment of phosphoserine/threonine/tyrosine (pSTY) peptides by immobilized metal ion affinity chromatography (IMAC). Tyrosine phosphoproteomics was performed by immunoprecipitation of phosphotyrosine (pY) peptides followed by label-free quantitative LC-MS/MS. Both datasets showed even intensity distributions and good correlation between biological replicates. In the global phosphoproteomics dataset, 9,934 peptides were detected, of which 8,819 presented a PEP score < 0.05 (Table S2). In the pY dataset, a total of 484 peptides were detected, 250 of which had a PEP score < 0.05 (Table S3). Comparing foretinib and cabozantinib treatment showed multiple differential phosphorylation changes, such as for palladin (PALLD), heterochromatin protein 1 (HP1A-CBX5), DNA topoisomerase 2-alpha (TOP2A), tyrosine-protein kinase FER and δ-catenin (CTNND1) (Figure 4B). In contrast, the majority of proteins did not show any appreciable phosphorylation changes upon drug treatment or, as for GSK3α/β, any changes different from those induced by cabozantinib.
Finally, genome-wide gene expression changes in H1155 cells upon 4 hours of drug treatment with foretinib and cabozantinib were determined by RNA-Seq All samples reached more than 40,000,000 reads and average total alignment was 80%. A total of 15,165 genes were detected and 231 genes passed the specific selection criteria (see STAR Methods, Data analysis) (Table S4). Foretinib treatment resulted in a significantly different gene expression profile compared to DMSO and cabozantinib, which were much more similar to each other (Figure 4C).
The different data sets were integrated using Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005). Direct foretinib kinase targets with NSAF > 0.0006 were selected from the chemical proteomics data set. In addition, we selected signaling proteins that were differentially regulated by foretinib compared to cabozantinib in the phosphotyrosine (p-value < 0.1 or rescue criteria as outlined in STAR Methods, Data analysis; 108 proteins) and global (+/− 2 average standard deviations; 409 proteins) phosphoprotemics data sets. Finally, the 231 genes displaying differential expression when comparing foretinib treatment with DMSO and cabozantinib were selected from the RNA-Seq data. Subsequent combined GSEA analysis highlighted multiple pathways/terms, such as “Kinase activity” and “Binding” as expected (Table S5). In addition, several pathways representing biological processes like regulation of cell contact, “cytoskeleton” and the “cell cycle” were significantly enriched suggesting these to participate in the MoA of foretinib (Figure 4D). To identify which direct foretinib targets were involved in modulating these pathways, we determined which kinase targets were shared between the cytoskeleton and the cell cycle pathways (Figure 4E, Table S6,7). This analysis nominated six kinases, AURKB, FER, MAP2K1 (MEK1), AURKA, SRC and ATM, as potentially relevant foretinib targets. ATM displayed only a low NSAF and no associated Kd in LINCS and is likely an indirect foretinib binding protein via a different direct target. However, biochemical validation by in vitro kinase assays with foretinib and cabozantinib confirmed MEK1, SRC, FER and AURKA/B to be functional foretinib targets (Figure 4F), which, being more potently inhibited by foretinib than by cabozantinib, were selected for further investigation. In summary, an unbiased interrogation of targets, signaling pathways and gene expression profiles that are differentially modulated by foretinib and cabozantinib nominated several targets as mechanistically relevant candidates.
Foretinib kills NSCLC cells through a polypharmacology mechanism involving MEK1/2, FER and AURKB
We next asked if the selected targets were also inhibited in live cells by investigating changes in downstream signaling. Even though foretinib is a reasonably potent SRC, LYN and YES inhibitor, it unexpectedly did not decrease SRC autophosphorylation at Y419 in H1155 cells (data not shown). However, foretinib, in contrast to cabozantinib, decreased phosphorylation of pERK at T202/Y204, which are direct downstream substrate sites of MEK1/2, in both H1155 and A427 cells (Figure 5A, Figure S5A). Other MET and/or VEGFR inhibitors had no effect on pERK in these cells. In addition, foretinib decreased phosphorylation of histone H3 at S10 (Figure 5B), which is a bona fide downstream target of AURKB required for the binding of HP1A to chromatin (Fischle et al., 2005; Hirota et al., 2005). Considering that AURKB is a major regulator of the cell cycle and mitosis, we also investigated drug-induced cell cycle changes in H1155 and A427 cells (Figure 5C, Figure S5B). This analysis showed that foretinib, but not cabozantinib, caused a pronounced accumulation of cells in G2 phase consistent with inhibition of AURKB. Finally, since reliable antibodies for the FER phosphorylation site of interest were lacking, we performed pY immunoprecipitation and observed phosphorylation changes of FER in response to treatment with foretinib and the FER inhibitor GSK1838705A, but not cabozantinib or the pan-SRC inhibitor saracatinib (Figure 5D). These analyses suggest that foretinib inhibits MEK1/2, AURKB and FER signaling in NSCLC cells.
Figure 5. Foretinib kills NSCLC cells through a polypharmacology mechanism involving inhibition of MEK1/2, FER and AURKB.
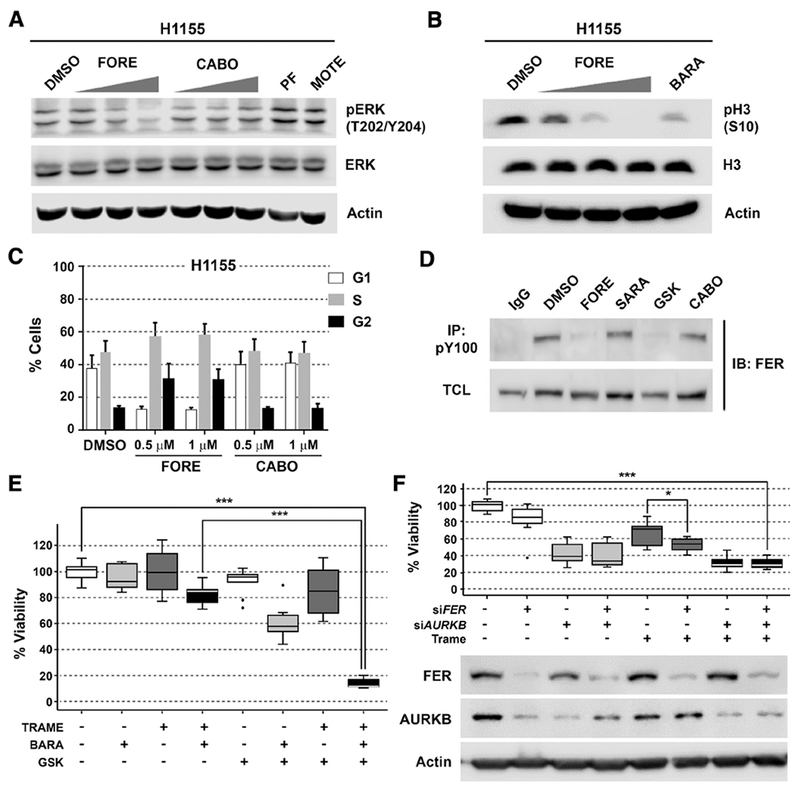
(A) Western blot of pERK (T202/Y204) after 1 hour treatment of H1155 cells with 0.1, 0.5 and 1 μM foretinib (FORE) or cabozantinib (CABO), 0.1 μM of PF-04217903 (PF-METi) or 0.1 μM of motesanib (MOTE - VEGFRi). Blots are representative of three independent experiments.
(B) Western blot of pH3 (S10) after 6 hours treatment of H1155 cells with 0.05, 0.25 and 0.5 μM of foretinib (FORE) and 0.05 μM of barasertib (AURKBi). Blots are representative of three independent experiments.
(C) Cell cycle analysis of H1155 cells after 6 hours of treatment with foretinib (FORE) and cabozantinib (CABO) at indicated concentrations, n = 3, Data are represented as mean + SD.
(D) Immunopreciptation of tyrosine phosphorylated FER in H1155 cells after 3 hours treatment with 1 μM each of foretinib (FORE), saracatinib (SARA – SRCi), GSK1838705A (GSK - FERi), cabozantinib (CABO) or DMSO control. TCL: total cell lysate. Blots are representative of three independent experiments.
(E) H1155 cell viability upon treatment with GSK1838705A (2.5 μM), barasertib (50 nM) and trametinib (35 nM) alone or in indicated combinations. Cell viability was determined by CellTiter-Glo assay after 72 hours of treatment and normalized to DMSO (first white bar), n = 3, Data are represented as mean ± SD. *** indicates p value < 0.001 as determined using Student’s t-test.
(F) RNA interference of FER and AURKB in combination with trametinib (35 mM) in H1155 cells. Cell viability was determined after 72 hours of treatment with DMSO or trametinib by trypan blue-based cell counting, n = 3, Data are represented as mean ± SD. Blots are representative of three independent experiments. * and *** indicate p values of < 0.05 and < 0.001, respectively, as determined using Student’s t-test.
To determine the contributions of the identified targets to foretinib’s MoA, we applied a combined functional validation approach using selective small molecule probes and RNAi. The following tool compounds were utilized: GSK1838705A (FER inhibitor, Kd = 200 nM)(Davis et al., 2011), barasertib (selective AURKB inhibitor, IC50 = 0.37 nM)(Mortlock et al., 2007), and trametinib (selective MEK1/2 inhibitor, IC50 = 0.92 nM/1.8 nM)(Yamaguchi et al., 2011). Whereas single agent treatment with these probes showed no (H1155, Figure 5E) or only moderate (A427, Figure S5C) effects, pairwise and particularly triple combinations markedly decreased cell viability. This suggests that foretinib displays a polypharmacology MoA involving all three kinases. Consistently, combined inhibition of these three targets only minimally reduced cell viability of foretinib-insensitive H661 cells (Figure S6A). To verify that the effect of the combination with GSK1838705A in foretinib-sensitive H1155 cells is primarily due to inhibition of FER and not its other potent targets, IGF1R/INSR (Sabbatini et al., 2009), H1155 cells were treated with linsitinib (OSI-906), a selective IGF1R/INSR inhibitor with no potency against FER (Mulvihill et al., 2009). Indeed, combination of linsitinib with barasertib and trametinib did not show any additional effect on cell viability over the combination of barasertib with trametinib (Figure S6B). This indicates that inhibition of IGF1R/INSR is not responsible for the decrease in cell viability upon GSK1838795A treatment. As FAK has also been reported to be a weak off-target of GSK1838705A, we confirmed that FAK autophosphorylation was not affected by GSK1838705A (or foretinib) at the concentrations used (Figure S6C). In addition, to evaluate if only inhibition of AURKB or also AURKA is involved in the MoA of foretinib, H1155 cells were treated with alisertib (MLN8237), a potent and selective AURKA inhibitor (Gorgun et al., 2010). Notably, compared to the observed effects with AURKB inhibition, combining alisertib with FER and MEK1/2 inhibition resulted only in a minor decrease of viability (Figure S6D). Consistently, combined targeting of AURKB, FER and MEK1/2 using siRNA-mediated knockdown of AURKB and FER and the specific small molecule MEK inhibitor trametinib is more effective than each single treatment alone (Figure 5F, Figure S5D). To summarize, foretinib’s anticancer effects in NSCLC cells are the result of a polypharmacology mechanism involving MEK1/2, FER and AURKB.
Foretinib, but not cabozantinib, potently kills MYC-amplified SCLC cells
AURKB inhibition is known to confer strong sensitivity to small cell lung cancer (SCLC) cells with MYC amplification (den Hollander et al., 2010; Li et al., 2016; Sos et al., 2012), which occurs in approximately 5% of SCLC and is associated with poor survival (Iwakawa et al., 2013; Kim et al., 2006). However, no targeted therapies have been approved yet for SCLC and its treatment still relies on conventional chemotherapy, against which resistance rapidly develops (Skarlos et al., 1994; Waqar and Morgensztern, 2017). Based on our findings that foretinib is a moderately potent inhibitor of AURKB and that the polypharmacology mechanism of foretinib in NSCLC cells relies to large extent on AURKB inhibition, we hypothesized that MYC-amplified SCLC cells would also be sensitive to foretinib, but less so to cabozantinib as a weaker AURKB inhibitor. In addition, we hypothesized that MYC-amplified SCLC cells would be more sensitive to foretinib than non-MYC-amplified SCLC cells. Foretinib indeed showed significantly more potent inhibition of viability of MYC-amplified cells than of SCLC cells without MYC amplification (Figure 6A,B). In fact, MYC-amplified SCLC cell lines were on average slightly more sensitive to foretinib than NSCLC cell lines. Similar to NSCLC cells, however, foretinib was markedly more potent than cabozantinib in SCLC cell lines, particularly in those with MYC amplification. In addition, the VEGFR and MET inhibitors motesanib or PF-04217903, respectively, had no effect on SCLC cells indicating that foretinib’s effect on cell viability is due to inhibition of non-canonical foretinib targets, particularly AURKB. This was consistent with foretinib’s ability to inhibit phosphorylation of the AURKB downstream target H3 at S10 in 16HV SCLC cells as did the selective AURKB inhibitor barasertib (Figure 6C). Taken together, foretinib, in contrast to cabozantinib, potently inhibited viability of MYC-amplified SCLC cells, but not SCLC cells without MYC amplification.
Figure 6. Foretinib, but not cabozantinib, potently kills MYC-amplified SCLC cells.
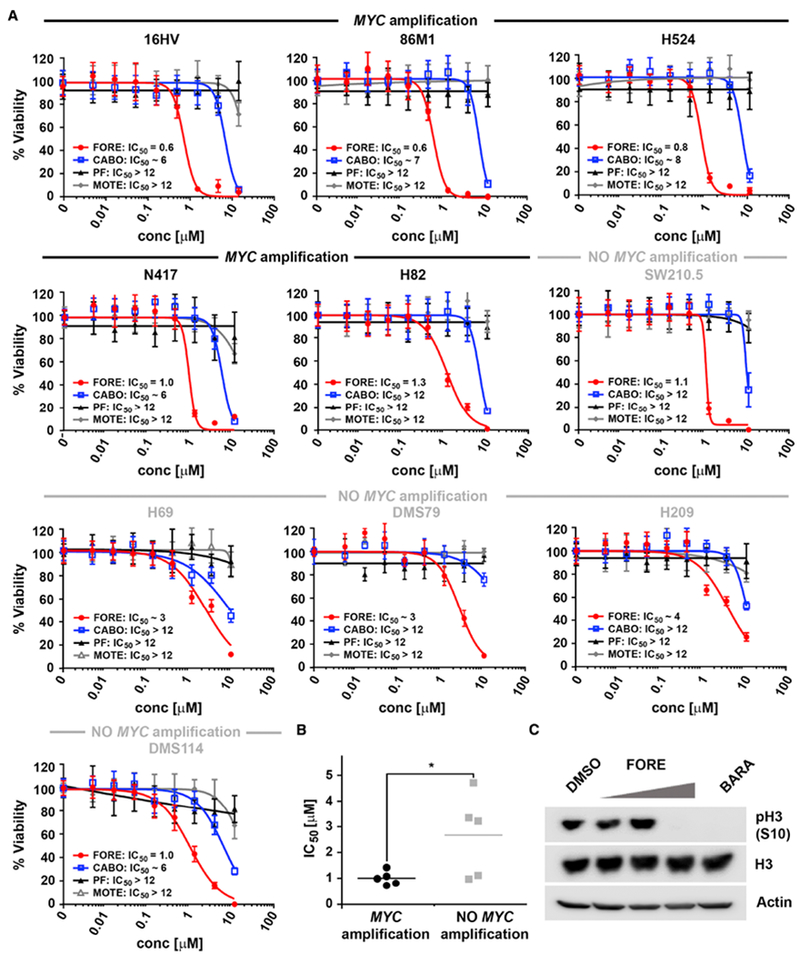
(A) Dose response curves of foretinib (FORE), cabozantinib (CABO), PF-04217903 (PF) and motesanib (MOTE) for inhibition of viability of various SCLC cell lines with indicated MYC amplification status after 72 hours treatment and the respective IC50 values [μM], n = 2 (H209, N417, DMS114, H69 and DMS79), n = 3 (16HV, 86M1, H524, H82, SW210.5), Data are represented as mean ± SD.
(B) IC50 comparison for inhibition of viability between MYC-amplified and non-MYC-amplified SCLC cells after 72 hours treatment with foretinib. * indicates p value < 0.05 as determined using Student’s t-test.
(C) Western blot of pH3 (S10) in 16HV cells after 6 hours treatment with 0.05, 0.25 and 0.5 μM foretinib (FORE) and 0.05 μM barasertib (BARA). Blots are representative of three independent experiments.
Foretinib synergizes with the potent AURKB inhibitor barasertib in MYC-amplified SCLC cells
Although foretinib is indeed a reasonably potent inhibitor of AURKB, in vitro kinase assays, as well as immunoblotting for H3 S10 phosphorylation (Figure 6C), indicated that barasertib, which has been optimized for AURKB inhibition, was yet several times more potent for this target. We therefore hypothesized that the combination of the potent and selective AURKB inhibitor barasertib with the polypharmacology-based compound foretinib would result in increased cell death of MYC-amplified SCLC cells. Indeed, addition of foretinib showed a pronounced shift of the barasertib dose-response curve in various MYC-amplified, but not non-MYC-amplified SCLC cells (Figure 7A, Figure S7A). Likewise, the addition of barasertib to foretinib dose-response curves strongly enhanced foretinib efficacy in MYC-amplified SCLC cells (Figure S7B). Interestingly, this was also apparent in H1155 cells, which like SCLC cells have neuroendocrine features, but not in A427 lung adenocarcinoma cells (Figure S7C,D). This increase in efficacy was statistically significant and applying the Bliss model of independence further indicated pronounced synergy in multiple MYC-amplified SCLC cell lines (Figure 7B). Finally, we investigated, whether the combination of foretinib with barasertib also increased the induction of apoptosis. Indeed, immunoblot analysis showed strong increases of PARP1 and caspase-3 cleavage upon combination treatment (Figure 7C). Importantly, this increased effect was only observed in MYC-amplified and not in non-MYC-amplified SCLC cells (Figure S7E). It is interesting to note, though, that the combination of foretinib with the BET inhibitor JQ1, which has been shown to exhibit single agent activity in MYC-amplified SCLC cells (Kaur et al., 2016), displayed minimal or no increase in activity (data not shown). In summary, combination of the polypharmacology exhibiting multi-kinase inhibitor foretinib with the more potent and selective AURKB inhibitor barasertib resulted in strong synergy in MYC-amplified, but not non-MYC-amplified SCLC cells.
Figure 7. Foretinib synergizes with the potent AURKB inhibitor barasertib in MYC-amplified SCLC cells.
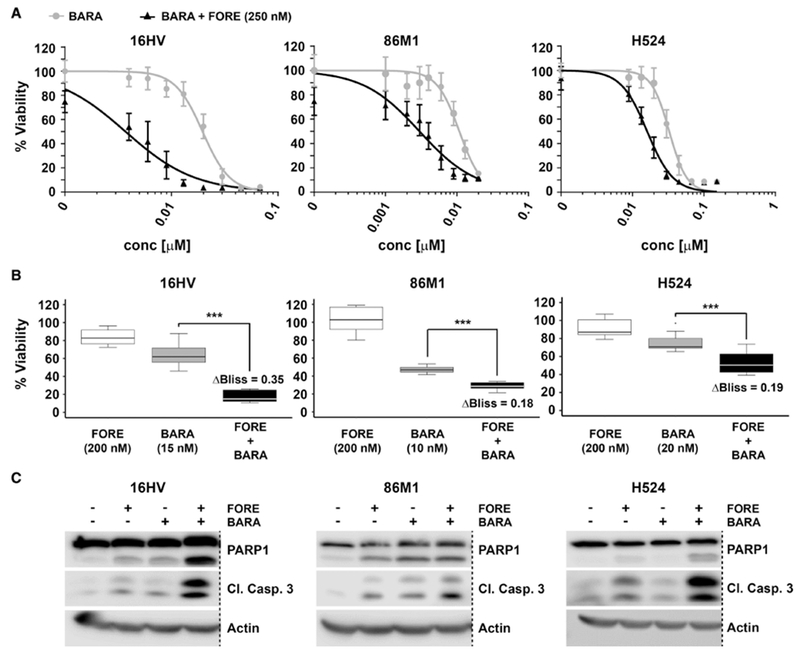
(A) Dose response curves for inhibition of viability of MYC-amplified SCLC cells after 72 hours treatment with barasertib (BARA) alone or in combination with 250 nM of foretinib (FORE), n = 3, Data are represented as mean ± SD.
(B) Analysis of synergistic effects on cell viability upon combination of foretinib (FORE) and barasertib (BARA) at the indicated concentrations. Synergy was determined using the Bliss model of independence. ΔBliss indicates the difference of observed inhibition of viability to the theoretically calculated additive effect. *** indicates p value < 0.001 as determined using Student’s t-test, n = 3, Data are represented as mean ± SD.
(C) Western blot analysis for PARP1 and caspase-3 cleavage in MYC-amplified SCLC cells after 24 hours of treatment with barasertib (concentrations as in (B)), foretinib (250 nM) or their combination. Dashed lines indicate deletion of additional lanes. Blots are representative of three independent experiments.
DISCUSSION
Recognizing that complex diseases require complex therapeutic solutions, the concept of polypharmacology has received significant interest over the last decade. However, our understanding of the optimal combination of targets for a disease and the design rules for drugs that engage these targets is still in its infancy. In this study, we present a systems pharmacology approach to elucidate the differential MoA of foretinib and cabozantinib, two dual MET/VEGFR inhibitors with similar chemical structures and target profiles. Despite their strong similarities, we observed that foretinib is more potent than cabozantinib in NSCLC cell lines. Foretinib has been shown to bind to MET within the ATP-binding site and an adjacent pocket causing the protein to adapt a “pseudo-inactive” form with the DFG motif in the “out” conformation stabilized by a stacking interaction of the DFG phenylalanine with foretinib’s fluorophenyl ring (Qian et al., 2009). Although it is generally expected that kinase inhibitors capable of inducing a DFG-out conformation are more selective (Liu and Gray, 2006), foretinib is known to be a multi-kinase inhibitor (Davis et al., 2011; Klaeger et al., 2017) consistent with our findings here. For cabozantinib, several additional targets like ROS1 and RET have also been found (Davare et al., 2013; Yakes et al., 2011), but a similarly comprehensive kinome-wide profiling has not been described. The discrepancy in cellular activity in lung cancer between these two related compounds may therefore be explained by differential target profiles and/or differential potency towards shared targets rooted in dissimilar binding modes. The latter was a particularly intriguing possibility considering the importance of the fluorophenyl moiety for foretinib binding and the fact that one of the major differences of cabozantinib is the lack of the respective fluorine. Interestingly, a recent study reported an induced dipole force, caused by a halogen substituted phenyl moiety within a kinase inhibitor and involving an alanine directly adjacent to the DFG motif, to lead to a pronounced outwards shift of the DFG motif in AURKA (Martin et al., 2012). Notably, also AURKB and several other kinases more potently inhibited by foretinib than cabozantinib (e.g. MAP4K5, SRC, YES, LYN) feature an alanine in this position suggesting that a similar phenomenon may contribute to the kinome-wide target differences between these two compounds. On the other hand, MET and DDR2 also feature such an ADFG motif, but are similarly inhibited by both drugs. However, as AURKB makes a major contribution to foretinib’s MoA, this feature appears to be relevant for its anticancer activity in these cells.
Although AURKB was indeed an important foretinib target in NSCLC cells, foretinib’s overall cellular activity was the result of complex polypharmacology involving multiple targets, particularly MEK1/2 and FER in addition to AURKB. This is best illustrated by the finding that single target inhibition showed only little effect whereas combined targeting was as effective as foretinib treatment itself. Notably, the more pronounced effects observed with siRNA-mediated gene silencing of AURKB compared to pharmacological inhibition are consistent with previous reports that describe more profound effects of AURKB knockdown due to loss of AURKB protein expression and subsequent disruption of the chromosomal passenger complex (Ditchfield et al., 2003; Weiss et al., 2007). While the mechanistic interplay between foretinib’s mechanistic targets here is largely unknown, we consistently observed that AURKB was downregulated upon FER knockdown in H1155 cells, which however was prevented by MEK inhibition with trametinib. As this was not similarly observed in A427 cells it is unlikely that it was the result of off-target silencing by siFER. Rather, it might indicate functional crosstalk and compensatory signaling between these targets that is inhibited by foretinib. The relevance of foretinib’s simultaneous targeting of MEK and the mitotic kinase AURKB is supported by reports that microtubule polymerization by MEK, independent of ERK, leads to defective mitotic spindle formation and polyploid cells (Cao et al., 2010), although MEK can also inhibit microtubule stabilization through ERK phosphorylation (Reszka et al., 1995). In addition, the BRAF/MEK/ERK pathway transcriptionally regulates AURKB through activation of the FOXM1 transcription factor (Bonet et al., 2012). Consistently, BI-847325, a dual MEK/AURKB inhibitor, can overcome BRAF inhibitor resistance in BRAF-mutant melanoma cells by decreasing ERK and histone H3 phosphorylation (Phadke et al., 2015). Thus, there may be multiple mechanisms underlying the crosstalk between foretinib’s main targets in these cells.
Importantly, each of the three targets, which were mainly responsible for foretinib’s activity in NSCLC cells, was individually slightly more potently inhibited by foretinib than by cabozantinib. These moderate differences would not have been sufficient to explain such pronounced activity differences between the two drugs on the cellular level, if this effect had been the result of inhibition of only a single target. Rather, as all three kinases, AURKB, FER and MEK1/2, play important roles in foretinib’s polypharmacology MoA, these individual differences cumulatively resulted in markedly distinct cellular potency. This is particularly noteworthy as foretinib and cabozantinib are very close structural analogues. This illustrates how even minor modifications in the chemical structure of a multi-targeted drug, such as the addition of a halogen substituent, can have broad and unexpected consequences that, while potentially undesirable, may in some cases reveal new vulnerabilities, which can be harnessed for therapeutic purposes. Although medicinal chemists utilize this concept already regarding lead optimization for single or a panel of related targets, a proteome-wide approach may uncover new opportunities for drug design and development. However, dissection of the underlying mechanisms can be challenging and requires an unbiased, systems-wide strategy that integrates identification of differential drug targets with the downstream signaling pathways modulated by these drugs. The systems approach described here, which integrated chemical proteomics, phosphoproteomics and RNA-Seq data using GSEA therefore was critical to reveal the specific targets responsible for the biological effect of foretinib in lung cancer cells, although other systems pharmacology approaches may be equally suited.
Though challenging, elucidation of the underlying MoA of a polypharmacology drug is critical for the rational design of drug combinations that enhance the overall cellular efficacy. This is important as the involved targets are most likely, as is the case for foretinib, targeted by chance and therefore not as potently inhibited as could be achieved by medicinal chemistry efforts specifically directed at these targets. Thus, to generate translational opportunities based on these mechanisms, it is necessary to either engage in such a challenging medicinal chemistry project (Hopkins, 2008), or develop a mechanistically-informed drug combination with enhanced cellular efficacy, such as illustrated here on the example of MYC-amplified SCLC, a recalcitrant cancer known to exhibit some sensitivity to AURKB inhibition (den Hollander et al., 2010; Li et al., 2016; Sos et al., 2012). Importantly, the combination of foretinib, its polypharmacology MoA based in part on moderate AURKB inhibition, with the highly potent AURKB inhibitor barasertib exhibited pronounced synergy and potently killed MYC-amplified SCLC cells at physiologically achievable concentrations. Interestingly, combination of foretinib with the bromodomain inhibitor JQ1, which also displays single drug activity in these cells (Kaur et al., 2016), did not produce similar synergy. This demonstrates that synergy is not always obtained upon combination of two individually active compounds, but requires a strong mechanistic foundation as shown here for foretinib and barasertib and the enhanced targeting of the functionally relevant target AURKB in these cells.
SIGNIFICANCE
Targeted drugs have become an important cornerstone in modern medicine. However, many patients do not benefit from these drugs due to, for instance, lack of actionable genetic aberrations that function as disease drivers. In such cases a feasible alternative may be based on network pharmacology, which targets multiple disease-relevant proteins at the same time and can be achieved, for example, through drug polypharmacology. However, the identification of novel and potentially actionable targets that cooperate with each other within a disease network is challenging. Here, we present an unbiased systems pharmacology approach, including phenotype-based drug screening, mass spectrometry-based chemical and phosphoproteomics and RNA-Seq-based gene expression profiling, that is capable of differentiating the mechanisms of action of two multi-targeted drugs with highly similar chemical structures and shared canonical targets, but markedly different cellular efficacy in lung cancer cells. The underlying polypharmacology mechanism involved simultaneous targeting of at least three kinases, which were not mutated, but cooperatively play important roles for cancer cell survival. Importantly, this analysis highlighted that small changes in the chemical structures of multi-targeted drugs can result in moderate potency differences across multiple individual targets. Cumulatively, this effect can lead to pronounced differences in overall cellular activity of these drugs when amplified by differential polypharmacology mechanisms that involve several of these targets, indicating new opportunities for drug design and development. Furthermore, the mechanistic understanding gained from this study allowed for the rational development of a synergistic drug combination in a recalcitrant cancer known to depend on the affected signaling pathway. In summary, such a comprehensive approach allows for the identification of drug targets and polypharmacology mechanisms potentially resulting in the repurposing of multi-targeted drugs and in the design of potent combination therapies for the treatment of complex diseases lacking directly actionable drivers.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Uwe Rix (uwe.rix@moffitt.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Cell lines were provided by the Moffitt Lung Cancer Center of Excellence Cell Line Core after testing negative for mycoplasma and authentication by short tandem repeat (STR) analysis. Cells were cultivated in RPMI-1640 media with 10% FBS (complete RPMI) and maintained in the incubator with 5% CO2 at 37°C.
METHOD DETAILS
Compounds
Foretinib (GSK1363089, EXEL2880, XL880; Chemietek), cabozantinib (XL184, BMS-907351; Chemietek), motesanib free base (AMG-706; Selleckchem), PF-04217903 (Selleckchem), barasertib (AZD1152-HQPA; Selleckchem), trametinib (GSK1120212; Chemietek), GSK1838705A (Selleckchem), PF-573228 (Selleckchem), linsitinib (OSI-906; Chemietek) and alisertib (MLN-8237; Selleckchem) were dissolved in DMSO (10 mM). Drug dilutions were made in DMSO as necessary.
Viability assays
Cells were generally plated with a density of 1,000 cells per well in a black, clear bottom 384 well tissue culture plate (Fisher). A549 cells were plated with a density of 500 cells per well. After 24 hours, cells were drug treated and incubated for an additional 72 hours. Inhibitors were diluted in complete RPMI media to the desired final concentration as described in each experiment. The viability of cells was measured with CellTiter-Glo Luminescent Cell Viability Assay reagent (Promega). After addition of CellTiter-Glo reagent, plates were shaken at 400 rpm for 2 minutes and placed in the dark for another 10 minutes. A M5 Spectramax plate reader (Molecular Devices) using 500 ms integration was used to collect the data. Data were analyzed using GraphPad Prism and R. Values were normalized to the DMSO control of each drug dilution.
Crystal violet assay
A427 cells were plated at a density of 3,000 cells per well in a 6-well plate. After 24 hours, cells were treated as indicated with complete RPMI media and were grown for an additional 9 days. Cells were fixed with 1 ml of methanol and stained with 0.1% crystal violet solution. Plates were imaged on a tabletop scanner. Crystal violet was extracted using methanol and quantified on a M5 Spectramax plate reader (Molecular Devices) using a readout of 540 nm. Data were analyzed using GraphPad Prism.
Cell cycle analysis
Cells were plated at a density of 4.5 × 105 cells per well in a 6-well dish. After 24 hours, cells were treated as indicated with complete RPMI media for 6 hours. Cells were harvested and incubated with Accutase (Innovative Cell Technologies) for 5 minutes to disrupt cell aggregation. Cells were fixed with ice-cold 70% ethanol until use. Cells were stained with DAPI (4’,6-diamidino-2-phenylindole; Sigma D9542)/0.1% Triton X-100/PBS solution at a final concentration of 1 μg/ml. Cells were analyzed using a FACSCanto II benchtop analyzer (BD Biosciences) at 340 to 380 nm. Data was analyzed using ModFitLT V3.2.1 and Excel.
Quantification of caspase 3/7 by fluorescence microscopy
Cell lines were plated at 1,000 cells per well in a black, clear bottom 384 well tissue culture plate (Corning). After 24 hours in the incubator, cells were treated as indicated with complete RPMI media containing 5 μM of Incucyte Caspase 3/7 reagent (Essen Bioscience #4440). Plates were placed in the IncuCyte and scanned every 2 hours for green fluorescence and in the phase contrast channel up to a total of 72 hours. Data was analyzed using the IncuCyte Zoom software, which determined the phase and green object confluence of each well. Green object confluence was normalized to the phase object confluence. Further analyses were performed using GraphPad Prism.
Cell lysate preparation for immunoblotting and immunoprecipitation
Cells were harvested, pelleted by centrifugation and lysed with lysisbuffer (0.20% NP40, 50 mM Tris pH 7.5, 5% Glycerol, 1.5 mM MgCl2 and 100 mM NaCl) plus Roche cOmplete protease inhibitor cocktail and phosphatase inhibitor cocktail 2 (Sigma, #P5726). Samples were centrifuged twice at 21,000 × g and 4°C (10 min, 20 min) and the protein concentration determined using a Bradford assay.
Immunoblotting
Total cell lysates were separated by SDS-PAGE and transferred to activated PVDF membranes using the TransBlot Turbo system (BioRad). Membranes were blocked and probed with primary and secondary antibodies according to standard techniques. Images were acquired using an Odyssey FC Imager (LI-COR) and quantified using Image J. Immunoblotting was performed with primary antibodies against actin (Sigma, A5441), pMET(T1234/1235) (D26)XP (Cell Signaling, #3077), MET (D1C2)XP (Cell Signaling #8198), p44/42MAPK(ERK1/2 - T202/204) (E10) (Cell Signaling, #9106), ERK1/2/MAP-Kinase (Sigma, #M5670), pY100 (Cell Signaling, #9411), FER (5D2) (Cell Signaling, #4268), pHistone H3(S10) (D2C8)XP (Cell Signaling, #3377), Histone H3 (D1H2)XP (Cell Signaling, #4499), cleaved caspase-3 (Asp175) (Cell Signaling, #9661), PARP1 (Cell Signaling, #9542), Aurora B/AIM-1 (BD Bioscience, #611082), pFAK(T397) (D20B1) (Cell Signaling, #8556), FAK (D2R2E) (Cell Signaling, #130095), MAP4K5 (KHS [E-5]) (Santa Cruz, sc-374070). Secondary antibodies were anti-rabbit and anti-mouse (GE Healthcare).
Immunoprecipitation
H1155 cells were plated at 4.5 × 106 cells per 100 mm dish/sample. After 24 hours, cells were treated as indicated for 3 hours. Lysates were prepared using the lysis buffer described in the cell lysates preparation method. For each sample, 50 μl of Protein A/G beads Plus – Agarose (Santa Cruz, #sc-2003) was incubated horizontally at 4°C with 2 μl of pY100 antibody (Cell Signaling, #9411) or normal mouse IgG (Santa Cruz, #sc-2025) for 4 hours. Beads were washed 3 times with 1 ml lysis buffer, with spins at 1,000 × g for 1 minute at 4°C between washes. A small portion of total cell lysate (TCL-100 μg) of each sample was kept aside to test for the presence of the protein of interest. The other 1 mg of protein of each sample was added to the coated beads and rotated at 4°C overnight. S ubsequently, beads were spun at 1,000 × g for 2 minutes at 4°C and washed 4 times with 1 ml l ysis buffer, again with spins at 1,000 × g for 1 minute at 4°C between washes. The elution was performed by adding 20 μl of 4× Laemmli buffer and denaturation at 90°C. Samples were spun at 14,000 × g for 1 minute and analyzed by SDS-PAGE, in comparison to the TCL samples. Immunoblotting was performed using the FER (5D2) (Cell Signaling, #4268) antibody.
RNA interference
siRNA SMART pool (Dharmacon) stocks were re-suspended at 20 μM with 1× siRNA buffer (1:5 Thermo Scientific 5× siRNA Buffer (B-002000-UB-100) with RNase-free water (B-002000-WB-100)) and shaken at 37 °C for 1 hour. siRNA stocks were diluted to the desired final concentration in Opti-MEM media (Fisher 31985062), depending on the cell line (See table below), and incubated for 5 minutes. siRNA was incubated for additional 20 minutes in the respective well with the lipofectamine mix (OptiMEM media plus lipofectamine mix) at 1:1 ratio. For the A427 cell line we used lipofectamine RNAiMax (Fisher 13-778-150) and for H1155 we used lipofectamine 2000 (Fisher 11668019). A427 and H1155 cells were plated at 1.5 × 105 cell per well, left to attach for 24 hours and treated with trametinib (35 nM) or DMSO. Cells were counted with trypan blue (Sigma) in triplicate after 72 hours of treatment. Data was analyzed using Microsoft Excel and R. Leftover samples were used to confirm knockdown by western blotting. All the samples received the same concentration of siRNA; non-target siRNA (siNT) was used to balance the amount of siRNA in all samples to match the concentration of the double knockdown sample.
| siRNA reagents (Dharmacon) | Final Concentration in cells |
|---|---|
| ON-TARGET plus Non-target Pool (D-001810-10-20) | 100 nM – H1155 40 nM – A427 |
| ON-TARGET plus Human MAP4K5 (11183) – SMARTPOOL (L-003589-00-0005) | 20 nM – H1155 |
| ON-TARGET plus Human AURKB (9212) – SMARTPOOL (L-003326-00-0005) |
80 nM – H1155 20 nM – A427 |
| ON-TARGET plus Human FER (2241) – SMARTPOOL (L-003129-00-0005) |
20 nM – H1155 20 nM – A427 |
Kinase assays
In vitro kinase inhibition assays were performed for two doses of foretinib and cabozantinib (0.05 μM and 0.25 μM); and IC50 values were determined using 10 μM ATP and a 3-fold drug dilution series. Assays were performed in duplicate using the Reaction Biology Kinase Hotspot platform or Eurofins platform. The Reaction Biology Kinase Hotspot platform was used to determine the IC50 for the Aurora kinases (A/B/C) and MAP4K5, as well the two dose inhibition assay for MAP4K5. The Eurofins platform was used to determine the two dose inhibition for MET, CSK, cSRC, FAK, FER, JNK2α2, LYN and YES, Aurora kinase B, DDR2, IGF1R, IR and MEK1. IC50’s for cSRC, DDR2, FER, LYN, MEK1 and YES were also determined on the Eurofins Kinase Profiler™ platform.
Chemical proteomics
Experiments were performed as previously described (Knezevic et al., 2016). Beads were incubated with 2.5 μl of i-foretinib or i-cabozantinib. Pre-treatment was performed by incubating cell lysates with 20 μM of foretinib and cabozantinib. Experiments were performed in duplicate (H1155) or triplicate (A427) using 5 mg protein per pulldown. SDS-PAGE, in-gel digestion with trypsin and LC-MS/MS analyses were performed as described previously. The samples were analyzed on a QExactive Plus hybrid orbitrap mass spectrometer (Thermo) coupled to a nanoflow liquid chromatograph (RSLC, Dionex). Data was searched with Mascot (Matrix Science) against the human SwissProt 2015 (Dec.) and SwissProt 2016 (Jul.) database for H1155 and A427, respectively. For both searches, two missed trypsin cleavages were allowed. Carbamidomethylation of cysteine and methionine oxidation were selected as variable modifications. Results were imported to Scaffold 4.0 and data were analyzed setting the minimum number of peptides to 2 and the protein and peptide threshold to 95% confidence.
Activity-based protein profiling (ABPP)
The ATP-probe-based ABPP was performed according to the manufacturer’s instructions of Pierce™ Enrichment Kit with ATP Probe (ThermoFisher Scientific, # 88310). H1155 cells were lysed using a 10 M urea/Pierce IP lysis buffer containing protease and phosphatase inhibitors (1:100). Samples were sonicated for 1 minute and then incubated for 10 minutes on ice. Samples were centrifuged at 16,000 rpm at 4°C for 1 0 minutes. Buffer exchange to remove endogenous ATP was performed using Zeba spin desalting columns (Thermo Fisher Scientific), which were prepared following the manufacturer’s instructions, and protein concentration was determined using Coomassie protein assay reagent (Thermo Fisher Scientific). Additional protease and phosphatase inhibitors (1:100) were added to the lysates. Lysate was divided into 6 samples of 1 mg of protein (duplicates of DMSO, cabozantinib and foretinib treatment) and 10 ul of 1 M MnCl2 were added to each sample. 20 μM of DMSO, foretinib or cabozantinib was added to the respective tubes and rotated for 15 minutes at room temperature. Desthiobiotin-ATP probe was added to each sample at a final concentration of 5 μM and incubated for 15 minutes at room temperature. Reduction and alkylation were performed as described by the manufacturer. Trypsin digestion (20 μg/μl) was performed at 37°C overnight. The 50% high capacity streptavidin agarose resin slurry (50 ul) was added to each sample and rotated for 1 h at room temperature. After washes, peptide elution was performed by adding three times 100 μl elution buffer (50% ACN, 0.1% trifluoroacetic acid (TFA) to each sample. Samples were frozen and lyophilized. Samples were resuspended in 20 μl of 2% ACN in 0.1% TFA solution containig Thermo Scientific Pierce Retention Time Calibration Mixture (PRTC) in order to confirm correct operation of the LC-MS instrument.
A nanoflow ultra high performance liquid chromatograph (RSLC, Dionex) coupled to a nanoelectrospray benchtop Orbitrap mass spectrometer (QExactive Plus, Thermo) was used to acquire MS/MS peptide sequences. The samples were loaded onto a pre-column (2 cm × 100 μm ID packed with C18 PepMap100 reversed-phase resin, 5 μm particle size, 100 Å pore size) and washed for 8 minutes with aqueous 2% ACN and 0.04% TFA. The captured peptides were eluted onto the analytical column, (C18 PepMap100, 75 μm ID × 50 cm, 2 μm particle size, 100 Å pore size, Dionex). A 120-minute gradient was programmed as: 95% solvent A (2% ACN + 0.1% FA) for 8 minutes, solvent B (90% ACN + 0.1% FA) from 5% to 38.5% in 90 minutes, then solvent B from 50% to 90% B in 7 minutes and held at 90% for 5 minutes, followed by solvent B from 90% to 5% in 1 minute and re-equilibration for 10 minutes. The flow rate on the analytical column was 300 nl/min. Sixteen tandem mass spectra were collected in a data-dependent manner following each survey scan. 60 second dynamic exclusion was used for previously sampled peptide peaks. Each sample was injected twice (technical replicates).
Database searches and quantification were performed using MaxQuant 1.2.2.5. The fragment tolerance for MS/MS scans was set to 20 ppm. Desthiobiotin was set as a variable modification. The false discovery rate (FDR) was set to 0.05.
Phosphoproteomics
For each of the six samples (foretinib, cabozantinib and DMSO – in duplicate), eleven 150 mm dishes were plated with 1.5 × 107 H1155 cells. After 24 hours, plates were treated with 1 μM of foretinib or cabozantinib for 3 hours; DMSO was used as control. Cells were harvested, washed with phosphate-buffered saline (PBS) and pellets from each treatment were combined. Phosphoproteomic samples were prepared following the PTMScan Kit Protocol (Cell Signaling) and as previously described (Ctortecka et al., 2018). Peptide purification was performed using Sep-Pak® C18 columns (Waters). Eluates were collected in the same tube and then aliquoted for pY enrichment (20 mg per sample) or global phosphoproteomics (400 μg per sample). Samples were frozen at −80°0 overnight and lyophili zed for two days to ensure that there was no TFA left in the samples. Tyrosine phosphoproteomics was performed by immunoprecipitation of phosphotyrosine peptides using the Antibody-beads [PTMScan® Phospho-Tyrosine Rabbit mAb (P-Tyr-1000) Kit, Cell Signaling #8803] followed by a label-free quantitative LC-MS/MS analysis.
Peptides for global phosphoproteomics (pSTY) were labeled using TMT 6-plex reagents accordingly to TMT Mass Tagging Kits and Reagent Instruction (Thermo Scientific Mass Tagging Kit, #90064). Two channels were used for each type of sample (126 and 127 – DMSO1/2; 128 and 129 – foretinib1/2; 130 and 131 – cabozantinib1/2) and incubated for 1 hour at room temperature. Label efficiency was confirmed by injecting approximately 1 μg of peptides for LC-MS/MS; all samples showed more than 99.4% labelling efficiency by spectral counting. Reactions were quenched with 5% hydroxylamine for 15 minutes. Samples were combined and lyophilized overnight. Samples were re-dissolved with 350 μl of 20 mM of ammonium formate (pH 10.0), vortexed for 10 minutes and separated with basic pH reversed phase LC into 12 concatenated fractions using a Waters Xbridge C18 column (4.6 × 100 mm). Peptides were lyophilized and pSTY peptides were enriched using an IMAC enrichment kit (Sigma I1408). All samples were evaporated using a speed vacuum centrifuge and resuspended in 2% ACN / 0.1% FA, which contained 50 fmol/μl of PRTC (Thermo Scientific Pierce Retention Time Calibration Mixture, PIERCE) to confirm consistent performance of the analyses.
Data were acquired as described in ABPP methods. pY and IMAC data were queried using MaxQuant 1.2.2.5 and 1.5.2.8, respectively. The fragment tolerance for MS/MS scans was set to 20 ppm. Fixed modifications were carbamidomethylation of cysteine; variable modifications were set to oxidation of methionine, acetylation of the protein N-terminus and phosphorylation of serine, threonine and tyrosine. The false discovery rate (FDR) was set to 0.05. The intensities of label-free phosphopeptides with PEP scores < 0.05 were manually validated.
RNA-Seq
H1155 cells were plated at a density of 0.5 × 106 cells in a 35 mm dish/treatment. After 24 hours, cells were treated with 1 μM of foretinib or cabozantinib for 4 hours and then collected, pelleted and kept at −80°0 until all biological replicates were harvested. DMSO treatment was used as control. RNA was extracted following manufacturer instructions (RNeasy Mini Kit – Qiagen, #74104), with the addition of an extra DNase digestion step to increase the quality of the RNA. This digestion was performed following the method in appendix E: DNase digestion of RNA before RNA cleanup from the RNase-Free DNase Set (Qiagen, #79254). To 87.5 μl of RNA sample was added 10 μl Buffer RDD and 2.5 μl DNase I stock solution (provided by the Moffitt’s Molecular Genomics Core). The sample volumes were brought up to 100 μl with RNase-free water and incubated at room temperature for 10 minutes before proceeding with the cleanup protocol of the RNeasy Mini Kit (Qiagen). Additionally, ethanol precipitation was performed after RNA extraction. The volume of each sample was brought up to 200 μl and 1/10 volume (20 μl) of 5 M NaCl plus 2.5 volumes (500 μl) of absolute ethanol were added prior to thorough vortex. Samples were centrifuged at 1,000 × g for 10 minutes; the supernatant was discarded, and pellets were washed with 500 μl of freshly diluted 75% ethanol in RNase-free water. Samples were centrifuged again at 1,000 × g for 10 minutes, supernatant was discarded, and samples were left to air-dry. Samples were resuspended in RNase-free water and analyzed by Nanodrop to verify the 260/280 and 260/230 ratio > 1.8 and ~2.0. Before cDNA synthesis, RNA quality and quantity were verified by Agilent RNA Screen Tape System and Quant-iT Assays (Invitrogen). Quant-iT was used to quantify the RNA in the samples and 100 ng per sample were used to start the cDNA synthesis. RNA-Seq library generation was performed following the Encore Complete RNA-Seq Library System protocol (Nugen). The KAPA library amplification kit for Illumina platform (KAPABiosystem) was used as described in the technical data sheet. Beads were resuspended in 20 μl of elution buffer. RNA-Seq was performed on an Illumina NextSeq 500. All samples had a minimum of 40,000,000 reads. Paired-end reads were aligned using TopHat2 (Kim et al., 2013) and HTSeq (Anders et al., 2015) was used to count reads that were mapped to the genes. Reads were normalized using DESeq2 based on estimated library size. The average mapping rate was 80% across samples (range 75.5%-89.2%). Biological samples of each condition were run using 5 replicates. For each condition, the three replicates with the fewest missing data were retained for the further analysis.
Chemical synthesis
All reagents were purchased from commercial suppliers and used without further purification. 1H NMR spectra were recorded on an Agilent-Varian Mercury 400 MHz spectrometer with CDCl3, CD3OD, or DMSO-d6 as the solvent. 13C NMR spectra were recorded at 100 MHz. All coupling constants are measured in Hertz, and the chemical shifts(δH and δC) are reported in parts per million. High-resolution mass spectrometry was carried out on an Agilent 6210 LC–MS (ESI-TOF) system. Microwave reactions were performed in Biotage Initiator 8 machines. HPLC analysis was performed using a JASCO HPLC system equipped with a PU-2089 Plus quaternary gradient pump and a UV-2075 Plus UV–vis detector, using an Alltech Kromasil C-18 column (150 × 4.6 mm, 5 μm) and an Agilent Eclipse XDB-C18 column (150 × 4.6 mm, 5 μm). The purities of the final compounds that underwent biological assessment were >97% as measured by HPLC. Melting points were recorded on an Optimelt automated melting point system (Stanford Research Systems). Thin-layer chromatography was performed using SiO2 60 F254 plates (Fisher), with observation under UV when necessary. Anhydrous solvents (dimethylformamide, dichloromethane, methanol, and tetrahydrofuran) were used as purchased from Aldrich. Burdick and Jackson HPLC-grade solvents (methanol and water) were purchased from VWR for HPLC and high-resolution mass analysis. HPLC-grade TFA was purchased from Fisher. The SiO2 column purification of compounds were carried out using Biotage automated Isolera purification system using a Biotage SiO2 prepacked columns.
i-Cabozantinib (10)
tert-Butyl (3-((4-chloro-6-methoxyquinolin-7-yl)oxy)propyl)carbamate (4): In a 50 mL round bottom flask, 4-chloro-6-methoxyquinolin-7-ol (3) (1.17 g, 5.30 mmol) was dissolved in dry DMF (35.0 mL) and K2CO3 (7.32 g, 53 mmol) was added under argon atmosphere. The mixture was stirred at RT for 15 minutes followed by the addition of tert-butyl (3-bromopropyl)carbamate (1.30 g, 5.30 mmol). After stirring the reaction at RT. for 16 hours, crude NMR showed ~ 60% conversion to the product. Added more tert-butyl (3-bromopropyl)carbamate (0.5 equivalent, 650 mg, 2.65 mmol) and stirring was continued for an additional 8 hours. The reaction was repeated using the same procedure employing 1.0 g (4.77 mmol) of 4-chloro-6-methoxyquinolin-7-ol. The combined crude mixture from both reactions was filtered and the solid was washed with ~ 30 mL of EtOAc. The filtrate was washed with water (2 × 50 mL) and concentrated. The crude solid was sonicated with methanol (3 mL) and solvent was decanted. The solid was further rinsed with methanol (2 × 1.5 mL) and dried under vacuum to give the title compound as beige solid (2.92 g, 80%). Mp: 133-136 °C (dec.). 1H NMR (400 MHz, DMSO-d6): δ 8.60 (d, J = 4.8 Hz, 1H), 7.57 (d, J = 4.8 Hz, 1H), 7.43 (s, 1H), 7.37 (s, 1H), 6.93 (t, J = 5.3 Hz, 1H, disappeared on D2O shake), 4.17 (t, J = 6.2 Hz, 2H), 3.97 (s, 3H), 3.13 (m, 2H), 1.92 (m, 2H), 1.38 (s, 9H); HPLC-MS (ESI+): m/z 367.2 [100%, (M35Cl+H)+], 369.2 [40%, (M37Cl+H)+].
tert-Butyl (3-((6-methoxy-4-(4-nitrophenoxy)quinolin-7-yl)oxy)propyl)carbamate (5): The tert-Butyl (3-((4-chloro-6-methoxyquinolin-7-yl)oxy)propyl)carbamate (4) (2.46 g, 6.71 mmol) and p-nitrophenol (1.03 g, 7.38 mmol, 1.10 equiv.) were dissolved in a 20 mL microwave vial and dry DIPEA (14.0 mL) was added. The microwave vial was capped and the reaction mixture was stirred at 130 °C (oil bath temp.) overnight. After completion of the reaction (monitored by HPLC-MS), the DIPEA was evaporated using a V-10 system and the resulting thick brown oil was triturated using EtOAc/hexane to give the title compound as a white solid (2.1 g, 67%). Mp: 212 °C (dec). 1H NMR (400 MHz, DMSO-d6): δ 8.61 (d, J = 5.1 Hz, 1H), 8.34 (d, J = 9.2 Hz, 2H), 7.44 (d, J = 9.2 Hz, 2H), 7.43 (s, 1H), 7.37 (s, 1H), 6.94 (t, J = 6.2 Hz, 1H, peak intensity go down on D2O shake), 6.87 (d, J = 5.1 Hz, 1H), 4.17 (t, J = 6.4 Hz, 2H), 3.89 (s, 3H), 3.14 (m, 2H), 1.89-1.90 (m, 2H), 1.38 (s, 9H); HPLC-MS (ESI+): m/z 470.2 [100%, (M+H)+].
tert-Butyl (3-((4-(4-aminophenoxy)-6-methoxyquinolin-7-yl)oxy)propyl)carbamate (8): In a 50 mL round bottom flask, 10% Pd/C (455.61 mg, 0.2 equiv.) was added to dry MeOH (50 mL) and argon gas was bubbled through the mixture for 10 minutes. The tert-Butyl (3-((6-methoxy-4-(4-nitrophenoxy)quinolin-7-yl)oxy)propyl)carbamate (5) (2.01 g, 4.28 mmol) was added to the flask followed by hydrazine monohydrate (1.26 mL, 25.69 mmol, 6 equiv.). After stirring for 2 h at rt, HPLC-MS showed complete conversion of the starting material to the product. The reaction mixture was filtered through a short pad of celite and the filtrate was concentrated. The crude product was stirred in EtOAc/hexane (95:5,18 mL) at RT, filtered and dried under vacuum to give the title compound as a white solid (1.61 g, 86%). Mp: 205 °C (dec). 1H NMR (400 MHz, DMSO-d6): δ 8.42 (d, J = 5.2 Hz, 1H), 7.50 (s, 1H), 7.34 (s, 1H), 6.94 (brs, partially overlapped, 1H, 40% reduced on D2O shake), 6.92 (d, J = 8.6 Hz, 2H), 6.66 (d, J = 8.6 Hz, 2H), 6.36 (d, J = 5.2 Hz, 1H), 5.17 (s, 2H), 4.15 (t, J = 6.2 Hz, 2H), 3.93 (s, 3H), 3.14 (m, 2H), 1.92 (m 2H), 1.38 (s, 9H); HPLC-MS (ESI+): m/z 440.2 [100%, (M+H)+].
1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxylic acid (7): The Cyclopropane-1,1-dicarboxylic acid (6) (2 g, 15.37 mmol) was dissolved in dry THF (20 mL) in a round bottom flask and the mixture was cooled to 0 °C (ice-water bath). Triethylamine (1.12 mL, 15.37 mmol) was slowly added (~over 2 min.) to the mixture and stirring was continued for 30 min. at the same temperature. Thionyl chloride (1.12 mL, 15. 37 mmol) was added and stirring was continued for another 30 min. The p-fluoroaniline (1.48 mL, 15.37 mmol) was dissolved in THF (20 mL) and added to the reaction mixture via a dropping funnel (over 20 min.). The reaction was stirred at 0 °C until the reaction went to comp letion (2 hours), diluted with EtOAc (~30 mL) and washed with brine (2 × 20 mL). The organic layer was separated and dried (Na2SO4). The crude solid product obtained was triturated using EtOAc/hexane (1:3) to give the title compound as an off-white solid (2.43 g, 67%). Mp: 166-172 °C reported 167-170 °C (Literature (Exelixis, WO2005/30140, p=195). 1H NMR (400 MHz, DMSO-d6): δ 13.09 (s, 1H, disappeared on D2O shake), 10.57 (s, 1H), 7.66 – 7.57 (m, 2H), 7.17 – 7.11 (m, 2H), 1.40 (s, 4H); 19F NMR (400 MHz, DMSO-d6): δ −119.02 – −119.22 (m); HPLC-MS (ESI+): m/z 222.1 [100%, (M-H)+], 246.1 [100%, (M+Na)+], 224.2 [80%, (M+H)+], 469.2 [35%, (2M+Na)+].
tert-butyl (3-((4-(4-(1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxamido)phenoxy)-6-methoxyquinolin-7-yl)oxy)propyl)carbamate-bisTFA (9): The 1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxylic acid (7) (60 mg, 0.26 mmol) was dissolved in dry DMF (0.8 mL), cooled to 0 °C, followed by the addition of DIPEA (103.3 μL, 0.59 mmol, 2.2 equiv.), HOBt (79.91 mg, 0.59 mmol, 2.2 eq.) and EDCI ( 113.37 mg, 0.59 mmol, 2.2 eq.). After stirring the reaction mixture for 40 minutes, tert-Butyl (3-((4-(4-aminophenoxy)-6-methoxyquinolin-7-yl)oxy)propyl)carbamate (8) (118.15 mg, 0.26 mmol, 1 eq.) was added and the reaction was first warmed to RT and then heated at 70 °C (oil bath) for 12 h. The crude reaction mixture was diluted with water (~3 mL) and extracted with EtOAc (2 × 5 mL). The organic layer was dried (Na2SO4), evaporated and the crude product was purified using SiO2 chromatography (eluents: MeOH:DCM , 5:95). The resulting material was further purified using preparative HPLC (Method: Gradient methanol-water with 0.1% TFA, 17 minutes run, flow rate 20 mL/min). The title compound was obtained (in the form of bis-TFA salt) as a thick oil (39 mg, 23%,) which partially solidified after drying under high-vacuum overnight. 1H NMR (400 MHz, DMSO-d6): δ 10.32 (s, 1H, disappeared on D2O shake), 10.02 (s, 1H, disappeared on D2O shake), 8.77 (d, J = 6.4 Hz, 1H), 7.85 (d, J = 9.0 Hz, 2H), 7.73 (s, 1H), 7.68 – 7.60 (m, 2H), 7.50 (s, 1H), 7.37 (d, J = 9.0 Hz, 2H), 7.21–7.12 (m, 2H), 6.97 (t, J = 6.0 Hz, 1H, disappeared on D2O shake), 6.79 (d, J = 6.4 Hz, 1H), 4.23 (t, J = 6.0 Hz, 2H), 4.03 (s, 3H), 3.18–3.11 (m, 2H), 2.04–1.92 (m, 2H), 1.55–1.44 (m, 4H), 1.38 (s, 9H); 19F NMR (400 MHz, DMSO-d6): δ −74.08 (s), −118.95 – −119.07 (m); HPLC–MS (ESI+): m/z 645.3 [100%, (M+H)+].
N-(4-((7-(3-aminopropoxy)-6-methoxyquinolin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide.bisTFA (10): The tert-butyl (3-((4-(4-(1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxamido)phenoxy)-6-methoxyquinolin-7-yl)oxy)propyl)carbamate-bisTFA (9) (18 mg, 0.027 mmol) was dissolved in dry DCM (0.8 mL) and TFA (0.2 mL) was added. The reaction mixture was stirred at RT for 2 h at which point TLC showed complete consumption of the starting material. The solvent was evaporated and the residual TFA was removed by evaporating the mixture with EtOAc (~ 4 mL) and DCM (~ 4 mL). Using a 4 mL vial, the crude compound was partitioned between 0.5 mL of 1N NaHCO3 (aq.) and EtOAc (2 × 2 mL). The aqueous layer was re-extracted with DCM (2 × 2 mL) and the combined organic layers were dried (Na2SO4), evaporated and lyophilized. The title compound was obtained as a white solid (11.7 mg, 77%) Mp: 203 °C (dec). HPLC 99% [tR = 10.3 min, Gradient: MeOH-water (with 0.1% TFA) 40-95%, 1mL/min, 20 min]; 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H, disappeared on D2O shake), 10.04 (s, 1H, disappeared on D2O shake), 8.61 (s, 1H), 7.84 (brs, 2H, disappeared on D2O shake), 7.81 (d, J = 9.1 Hz, 2H), 7.64 (dd, J = 9.1, 5.1 Hz, 3H), 7.50 (s, 1H), 7.28 (d, J = 9.0 Hz, 2H), 7.18–7.13 (m, 2H), 6.60 (s, 1H), 4.29 (t, J = 5.9 Hz, 2H), 3.99 (s, 3H), 3.05-3.00 (m, 2H), 2.17–2.11 (m, 2H), 1.48 (d, J = 3.9 Hz, 4H); 19F NMR (400 MHz, DMSO-d6): δ −73.64 (brs), −118.97 – −119.09 (m); HPLC–MS (ESI+): m/z 545.3 [95%, (M+H)+], 273.2 [100%, (M+2H)2+]; LC-MS 545.2 [100%, (M+H)+]; HRMS (ESI+) m/z calculated for C30H29FN4O5 (M+H)+ 545.2194, found 545.2193.
i-Foretinib (18)
N-(3-fluoro-4-hydroxyphenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (11): The 1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxylic acid (7), which was synthesized as described for the synthesis of i-cabozantinib, (100 mg, 0.45 mmol) was dissolved in dry THF (1 mL) and 1 drop of dry DMF was added at RT. The mixture was cooled to 0 °C; and oxalyl chloride (38.4 μL, 0.45 mmol) was slowly added via a syringe. The mixture was stirred for 2 h at the same temperature and 3-fluoro-4-hydroxyaniline (56.9 mg, 68.3 mmol, 1.20 equiv.) and pyridine (0.5 ml, 500 mmol) were added in THF (1 mL). The reaction was stirred at 0 °C for 1 h, diluted with EtOAc (~5 mL) and the organic layer was washed with water (~5 mL). The organic layer was separated and stirred with 1M HCl (3 mL) for 30 minutes at rt. The organic layer was separated and the aqueous layer was extracted with EtOAc (5 mL). The combined organics were dried (Na2SO4), evaporated and the solid obtained was triturated from EtOAc/hexane (1:3) to give the title compound as a beige solid (72 mg, 48%). Mp: 187 °C (dec). 1H NMR (400 MHz, DMSO-d6): δ 10.05 (s, 1H, disappeared on D2O shake), 9.90 (s, 1H, disappeared on D2O shake), 9.58 (s, 1H, disappeared on D2O shake), 7.61 (dd, J = 8.9, 5.1 Hz, 2H), 7.52 (dd, J = 13.5, 2.0 Hz, 1H), 7.14 (t, J = 8.9 Hz, 3H), 6.93–6.80 (m, 1H), 1.43 (brs, 4H); 19F NMR (376 MHz, DMSO-d6): δ −119.02 – −119.22 (m), −135.34 (dd, J = 13.3, 10.2 Hz, 1H); HPLC–MS (ESI+): m/z 333.1 [90%, (M+H)+], 355.1 [100%, (M+Na)+], 687.2 [80%, (2M+Na)+].
tert-Butyl 4-(3-chloropropyl)piperazine-1-carboxylate (13): In a 5 mL microwave reaction vial, 1-boc-piperazine (12) (186 mg, 1 mmol) and 1-bromo-3-chloropropane (197.8 μL, 2 mmol) were dissolved in dry DMF (1 mL) and K2CO3 (276 mg, 2 mmol) was added under argon. The reaction mixture was heated at 80 °C (oil bath) for 1 h at which point TLC showed complete consumption of the starting material piperazine. The reaction mixture was filtered and concentrated to obtain a thick brown oil. The crude was rinsed with hexane (5 mL × 4) to give the title compound as a light yellow oil (217 mg, 87%) which partially solidified overnight at 4 °C. Reaction was repeated with 1-boc-piperazine (12) (1.86 g, 10 mmol) using the same procedure described here to obtain the title compound (2.2 g, 84%). 1H NMR (400 MHz, CDCl3): δ 3.60 (t, J = 6.5 Hz, 2H), 3.43 – 3.41 (t, J = 5.2 Hz, 4H), 2.48 (t, J = 7.0 Hz, 2H), 2.38 – 2.36 (J = 5.2 Hz, 4H), 1.94 (p, J = 6.8 Hz, 2H), 1.46 (s, 9H); HPLC–MS (ESI+): m/z 263.2 [100%, (M35Cl+H)+], 265.2 [40%, (M37Cl+H)+].
terf-Butyl 4-(3-((4-chloro-6-methoxyquinolin-7-yl)oxy)propyl)piperazine-1-carboxylate (14): The 4-chloro-6-methoxyquinolin-7-ol (1.0 g, 4.77 mmol) was dissolved in dry DMF (10 mL) in a 20 ml microwave vial and K2CO3 (1.32 g, 9.54 mmol) was added at r.t. The mixture was stirred under argon for 10 minutes followed by the addition of tert-Butyl 4-(3-chloropropyl)piperazine-1-carboxylate (13) (1.5 g, 5.72 mmol). The reaction mixture was heated at 80 °C (oil bath) for 6 h at which point HPLC-MS showed complete consumption of the starting material. The crude mixture was filtered and the solid was washed with EtOAc (~ 5 mL). The organic filtrate was combined and evaporated. The crude solid was dissolved in EtOAc (~100 mL) and the organic layer was washed with water (50 mL × 2) and brine (50 mL) and dried (Na2SO4). After evaporation of the organic layer, the crude mixture was triturated from EtOAc/hexane to give the title compound as light green solid (1.78 g, 86%). Mp: 116-119 °C. 1H NMR (400 MHz, MeOD): δ 8.51 (d, J = 4.0 Hz, 1H), 7.49 (d, J = 4.0 Hz, 1H), 7.46 (s, 1H), 7.38 (s, 1H), 4.24 (t, J = 5.2 Hz, 2H), 4.01 (s, 3H), 3.45 (broad t, J = 3.6 Hz, 4H), 2.63 (t, J = 5.6 Hz, 3H), 2.48 (t, J = 4.4 Hz, 4H), 2.14 – 2.09 (m, 2H), 1.45 (s, 9H); HPLC–MS (ESI+): m/z 438.2 [40%, (M37Cl+H)+], 436.2 [100%, (M35Cl+H)+].
tert-Butyl4-(3-((4-(2-fluoro-4-(1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxamido) phenoxy)-6-methoxyquinolin-7-yl)oxy)propyl)piperazine-1-carboxylate (15): The N-(3-fluoro-4-hydroxyphenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (11) (381 mg, 1.15 mmol) and tert-Butyl 4-(3-((4-chloro-6-methoxyquinolin-7-yl)oxy)propyl)piperazine-1-carboxylate (14) (500 mg, 1.15 mmol) were dissolved in DIPEA (1.15 mL) in a 5 ml microwave vial and the reaction mixture was stirred at 130 °C (oil bath) for 3 h. The TLC showed ~50% conversion of the starting material to the product. However, undesired side products also started to form as indicated by TLC and HPLC-MS. At this stage, MeOH (1-2 mL) was added and the solvent mixture was evaporated using Biotage V-10 system. The crude compound was purified using SiO2 column chromatography (eluting with EtOAc: Hexane, 0:10 to 10:0 followed by MeOH:DCM, 0:10 to 1.2:8.8). The title compound was obtained as a gray solid (207 mg, 25%). Notably, doing the same reaction at elevated temperature or in a microwave reactor resulted in the formation of side products (indicated by TLC). Mp: 178 °C (dec). 1H NMR (400 MHz, DMSO-d6): δ 10.39 (s, 1H, disappeared on D2O shake), 10.01 (s, 1H, disappeared on on D2O shake), 8.46 (d, J = 5.1 Hz, 1H), 7.90 (dd, J = 13.2, 2.0 Hz, 1H), 7.64 (dd, J = 9.0, 5.1 Hz, 2H), 7.52 (s, 2H), 7.43-7.38 (m, 2H), 7.15 (t, J = 9.0 Hz, 2H), 6.41 (d, J = 5.0 Hz, 1H), 4.19 (t, J = 6.0 Hz, 2H), 3.95 (s, 3H), 3.61 (brs, 2H), 3.17 – 3. 11 (m, 4H), 2.35 (brs, 4H), 1.99 – 1.96 (m, 2H), 1.48 (brs, 4H), 1.40 (s, 9H); 19F NMR (376 MHz, DMSO-d6) δ −118.88 – −119.30 (m), −128.81 (dd, J = 13.3, 10.2 Hz); HPLC–MS (ESI+): m/z 732.3 [100%, (M+H)+].
N-(3-Fluoro-4-((6-methoxy-7-(3-(piperazin-1-yl)propoxy)quinolin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (16): tert-Butyl4-(3-((4-(2-fluoro-4-(1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxamido) phenoxy)-6-methoxyquinolin-7-yl)oxy)propyl)piperazine-1-carboxylate (15) (100 mg, 0.136 mmol) was dissolved in 20% TFA in DCM (1 mL) and stirred at RT for 2 h. The TLC and HPLC-MS showed complete consumption of the starting material. The solvent was evaporated and residual TFA was removed by evaporating the reaction mixture with DCM (2 mL × 4) and EtOAc (2 mL × 4). The crude product was rinsed with DCM (2 mL) and hexane (2 mL) and dried under high vacuum to give the title compound (tris TFA salt) as a gummy oil which (partly solidified after storage in a refrigerator overnight) (106 mg, 123%). 1H NMR (400 MHz, DMSO-d6): 10.47 (s, 1H, 50% disappeared on D2O shake), 9.98 (s, 1H, 40% disappeared on D2O shake), 8.96 (s, 1H, disappeared on D2O shake), 8.67 (d, J = 5.4 Hz, 1H), 7.95 (dd, J = 13.2, 1.7 Hz, 1H), 7.67 (s, 1H), 7.64 (dd, J = 8.9, 5.1 Hz, 2H), 7.59 – 7.55 (m, 1H), 7.54 (s, 1H), 7.48 (t, J = 8.9 Hz, 1H), 7.16 (t, J = 8.9 Hz, 2H), 6.71 (brs, 1H), 4.29 (t, J = 5.8 Hz, 2H), 4.01 (s, 3H), 3.66 – 3.56 (m, 2H), 3.30 (brs, 4H), 3.15-3.12 (m, 2H), 2.18 (brs, 2H), 1.53 – 1.42 (m, 4H), 1.26 (t, J = 6.3 Hz, 4H); 19F NMR (376 MHz, DMSO-d6) δ −73.99 (s), −118.91 – −119.01 (m), −128.69 (dd, J = 11.4, 8.3 Hz); HPLC–MS (ESI+): m/z 632.3 [100%, (M+H)+], 316.7 [100%, (M+2H)2+].
tert-Butyl(3-(4-(3-((4-(2-fluoro-4-(1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxamido)phenoxy)-6-methoxyquinolin-7-yl)oxy)propyl)piperazin-1-yl)propyl)carbamate (17): The N-(3-Fluoro-4-((6-methoxy-7-(3-(piperazin-1-yl)propoxy)quinolin-4-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (16) (104 mg, 0.13) was dissolved in dry DMF (0.2 mL) under inert conditions, powdered and dry K2CO3 (55.79 mg, 0.40 mmol) was added to the mixture. After stirring at RT for 15 minutes, tert-butyl (3-bromopropyl)carbamate (41.66 mg, 0.17 mmol, 1.3 equiv.) was added to the reaction mixture and heated at 80 °C for 12 h. The HPLC-MS and TLC showed complete consumption of the starting material. The crude mixture was concentrated, MeOH (5 mL) was added to the crude mixture before filtration. The filtrate was concentrated under reduced pressure and purified using Biotage Isolera purification system (SiO2, eluting with MeOH:DCM, 0:10 to 1.5: 8.5). The title compound (in the form of bis TFA salt) was obtained as a light yellow solid (67 mg, 63%). Mp: 188.2 °C (dec). 1H NMR (400 MHz, DMSO-d6): 10.39 (s, 1H, disappeared on D2O shake), 10.01 (s, 1H, disappeared on D2O shake), 8.47 (d, J = 5.0 Hz, 1H), 7.90 (d, J = 13.4 Hz, 1H), 7.64 (dd, J = 8.9, 5.0 Hz, 2H), 7.53–7.49 (m, 2H), 7.43 – 7.39 (m, 2H), 7.16 (t, J = 8.8 Hz, 2H), 6.82 (brs, 1H, disappeared on D2O shake), 6.41 (d, J = 5.2 Hz, 1H), 4.19 (brt, J = 5.8 Hz, 2H), 3.95 (s, 3H), 3.108–3.03 (broad m, 9H), 2.95–2.90 (m, 3H), 2.29 – 2.23 (m, 4H), 2.02–1.96 (m, 2H), 1.47 (broad s, 4H), 1.37 (s, 9H); HPLC-MS (ESI+): m/z 789.4 [30%, (M+H)+], 395.2 [100%, (M+2H)2+].
N-(4-((7-(3-(4-(3-aminopropyl)piperazin-1-yl)propoxy)-6-methoxyquinolin-4-yl)oxy)-3-fluorophenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide.TrisTFA (18): The tert-Butyl(3-(4-(3-((4-(2-fluoro-4-(1-((4-fluorophenyl)carbamoyl)cyclopropane-1-carboxamido)phenoxy)-6-methoxyquinolin-7-yl)oxy)propyl)piperazin-1-yl)propyl)carbamate (17) (64 mg, 0.08 mmol) was dissolved in dry DCM (2 mL) and TFA (0.2 mL) was added to the mixture. After stirring the reaction mixture at RT for 2 h, TLC showed complete conversion of the starting material to the product. The solvent was evaporated and residual TFA was removed by evaporating the product with DCM (2 mL × 4) and MeOH (2 mL × 4). The crude product was loaded on a short plug of celite and low polar impurities were removed by eluting with DCM (10 mL). The product was eluted with MeOH:DCM (2:8), concentrated, and rinsed with DCM (~2 mL × 3) and lyophilized for 24 h. The title compound (tris TFA salt) was obtained as a light yellow gummy solid (20 mg, 36%). Mp: 227 °C (dec). HPLC: 98% [tR = 14.5 min, gradient MeOH-water (with 0.1% TFA), 5-95% over 20 min.]; 1H NMR (400 MHz, CD3OD): δ 8.73 (dd, J = 6.7, 2.1 Hz, 1H), 7.96 (dd, J = 12.7, 2.1 Hz, 1H), 7.87-7.85 (m, 1H), 7.59 – 7.54 (m, 3H), 7.51-7.47 (m, 2H), 7.07 (t, J =, 8.4 Hz, 2H), 6.99 (d, J = 6.0 Hz, 1H), 4.52 – 4.32 (m, 3H), 4.22-4.14 (m, 1H), 4.11 (s, 3H), 3.98 – 3.88 (m, 3H), 3.58 – 3.39 (m, 3H), 3.14 (t, J = 6.8 Hz, 2H), 3.06 (t, J = 7.2 Hz, 2H), 2.78 (t, J = 6.8 Hz, 1H), 2.72-2.65 (m, 1H), 2.47-2.42 (m, 1H), 2.38 – 2.27 (m, 1H), 2.01-1.89 (m, 1H), 1.66 (s, 4H); 19F NMR (376 MHz, DMSO-d6): δ −73.91 (s), −118.85 – −119.10 (m), −128.62 – −128.91 (m); HPLC-MS (ESI+): m/z 689.3 [30%, (M+H)+], 345.3 [100%, (M+2H)2+]; LC-MS [100%, (M+H)+], 345.2 [55%, (M+2H)2+]; HRMS (ESI+) m/z calculated for C37H42F2N6O2 (M+H)+ 689.3257, found 689.3255.
QUANTIFICATION AND STATISTICAL ANALYSIS
Most data were obtained from three different experiments and presented as the mean ± standard deviation (SD). Details of each exact number of replicates are provided in the figure legends. Data were analyzed using GraphPad Prism and R. Statistical analyses were performed using Student’s t-test and synergy was determined using the Bliss model of independence. All statistical details are included in the figure legends. Additional details for each experiment type are described in the Methods Details section of the STAR Methods.
Based on the scatterplot densities, a log2 abundance of 7.5 was selected as cutoff for the analysis of RNA-Seq data to avoid abundance noise. The selection criteria were: 1) either treatment group average must be > 7.5; 2) t-test < 0.01; 3) | fold change|≥ 1.5; 4) Hellinger distance > 0.25. Genes that were significantly regulated in at least 1 of the 3 comparisons – fore vs DMSO + cabo, fore vs DMSO or fore vs cabo – were selected for further analysis.
All MaxQuant data were first filtered for peptides with PEP score < 0.05. In addition, reverse and contaminant peptides and peptides with no intensity were excluded. Data were then normalized using IRON (iterative rank-order normalization) (Welsh et al. 2013). Our analyses further on mainly focused on the comparison between foretinib versus cabozantinib treatment.
In the ABPP analysis, first the technical duplicates were averaged, and then averaged between biological duplicate. Peptides with | fold change | ≥ 1.5 and p-value < 0.05 were kept for further analysis.
For the pY proteomics, after averaging between replicates, peptides with p-value < 0.1 were used for further analysis. In addition, to overcome the missingness between duplicates in the dataset, we manually rescued peptides that 1) each treatment had 3 values and the difference between them were less than 50%; 2) one treatment had 3 or 4 values and the other only 2, but the ratio between the two values was less than 1.5 and the difference between treatment were less than 50%; 3) one treatment had 3 or 2 values and the other treatment none. The cutoff for the global proteomics was +/− 2 average standard deviations.
Kinases specifically binding to foretinib over cabozantinib in chemical proteomics were prioritized using foretinib’s NSAF (normalized spectrum abundance factor) > 0.0006 as additional criteria. We also compared the fold change between the two treatments, taking into consideration the available Kd values from the HMS (Harvard Medical School) LINCS (Library of Integrated Network-Based Cellular Signatures) dataset.
Integrated data analysis was performed by entering the gene names into the GSEA database (Broad Institute) and querying canonical pathways and gene ontology (GO) gene sets, which included GO biological process, GO cellular component and GO molecular function. Data were exported to Excel, analyzed and visualized in R.
DATA AND CODE AVAILABILITY
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) with the dataset identifiers PXD012961 (Drug Pulldowns), PXD012962 (Tyrosine Phosphorylation), PXD012963 (IMAC Phosphoproteomics) and PXD012965 (ABPP) (Vizcaino et al., 2016). RNA-Seq data have been deposited in the GEO database with the dataset identifier GSE126850.
Supplementary Material
Table S1 – related to Figure 4 and Figure S4: Chemical proteomics data set of H1155 and A427 cells. Data was searched by Mascot and analyzed in Scaffold with a peptide threshold at 95% and a minimum of 2 exclusive spectra counts. Protein kinases are highlighted in orange. Amp: ampicillin control, Cabo: cabozantinib, Fore: foretinib, PT: pre-treatment. Fore-NSAF > 0.005 was used as selection criteria. - ABPP data set. Displayed are the kinases identified by activity-based protein profiling in H1155 cells. Data was filtered for PEP < 0.05 and data was IRON normalized. Rows with all zero values, contaminant and reverse peptides were removed. p-value compares Fore (F) to Cabo (C).
Table S2 – related to Figure 4: pSTY phosphoproteomics data set. Global phosphoproteomics data was filtered for PEP < 0.05 and data was IRON normalized. Rows with all zero values, contaminants and reverse sequences were removed.
Table S3 – related to Figure 4: pY phosphoproteomics data set. Phosphotyrosine data was filtered for PEP < 0.05 and data was IRON normalized. Rows with all zero values, contaminant and reverse peptides were removed.
Table S4 – related to Figure 4: RNA-Seq data set. Paired-end reads were aligned using TopHat2 and HTSeq was used to count reads that were mapped to the genes. Genes that were significantly regulated accordingly to our selection criteria have a value 1 in the criteria column.
Table S5 – related to Figure 4: Integrated data analysis. Pathway analysis was performed by entering the gene names into the GSEA database and querying canonical pathways and gene ontology (GO) gene sets, which included GO biological process, GO cellular component and GO molecular function.
Table S6 - related to Figure 4: GO_Cytoskeleton: Kinases including in the GO_Cytoskeleton pathway from GSEA and which were used for further analysis.
Table S7 – related to Figure 4: GO_Cell Cycle: Kinases including in the GO_Cell Cycle pathway from GSEA and which were used for further analysis.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal ant-β-Actin | Sigma | Cat#A5441; RRID:AB_476744 |
| Rabbit monoclonal anti-Phospho MET(Tyr1234/1235) (D26)XP | Cell Signaling | Cat #3077; RRID:AB_2143884 |
| Rabbit monoclonal anti-MET (D1C2)XP | Cell Signaling | Cat #8198; RRID:AB_10858224 |
| Rabbit polyclonal anti-Phospho AKT(Ser473) | Cell Signaling | Cat #9271; RRID:AB_329825 |
| Rabbit polyclonal anti-Akt | Cell Signaling | Cat #9272; RRID:AB_329827 |
| Mouse monoclocal anti-Phospho p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (E10) | Cell Signaling | Cat #9106; RRID:AB_331768 |
| Rabbit polyclonal anti-MAP kinase (ERK-1, ERK-2) | Sigma | Cat #M5670; RRID:AB_477216 |
| Rabbit monoclonal anti-Phospho Histone H3 (Ser10) (D2C8) XP | Cell Signaling | Cat #3377; RRID:AB_1549592 |
| Rabbit monoclonal Histone H3 (D1H2) XP | Cell Signaling | Cat #4499; RRID:AB_10544537 |
| Rabbit polyclonal anti-Cleaved Caspase-3 (Asp175) | Cell Signaling | Cat#9661; RRID:AB_2341188 |
| Rabbit polyclonal anti-PARP1 | Cell Signaling | Cat #9542; RRID:AB_2160739 |
| Mouse monoclonal anti-FER (5D2) | Cell Signaling | Cat#4268; RRID:AB_2278286 |
| Mouse monoclonal anti-Phospho Tyrosine (P-Tyr-100) | Cell Signaling | Cat#9411; RRID:AB_331228 |
| Mouse monoclonal anti-AIM-1 | BD Bioscience | Cat #611082; RRID:AB_2227708 |
| Rabbit monoclonal anti-Phospho FAK(Tyr397) (D20B1) | Cell Signaling | Cat #8556; RRID:AB_10891442 |
| Rabbit monoclonal anti-FAK (D2R2E) | Cell Signaling | Cat #13009 |
| Mouse IgG, HRP-linked whole Ab | GE Healthcare | Cat# NA931; RRID:AB_772210 |
| Rabbit IgG, HRP-linked whole Ab | GE Healthcare | Cat# NA934; RRID:AB_2722659 |
| Normal mouse IgG | Santa Cruz | Cat#sc-2025; RRID:AB_737182 |
| PTMScan® Phospho-Tyrosine Rabbit mAb (P-Tyr-1000) | Cell Signaling | Cat#8803 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMSO used in proteomics | Acros Organics | Cat#167852500; CAS: 67-68-5 |
| DMSO used in drug dilutions | Acros Organics | Cat#348445000; CAS: 67-68-5 |
| DMSO used in cell culture | Sigma | D2438; CAS: 67-68-5 |
| Foretinib | Chemietek | Cat#CT-FORE, CT-XL880; CAS: 849217-64-7 |
| XL184 (CABOZANTINIB) (FREE BASE) | Chemietek | Cat#CT-XL184; CAS: 849217-68-1 |
| GSK1120212 (TRAMETINIB) | Chemietek | Cat#CT-GSK212; CAS: 871700-17-3 |
| Motesanib (AMG-706) | Selleckchem | Cat#S5793; CAS: 453562-69-1 |
| PF-04217903 | Selleckchem | Cat#S1094; CAS: 956905-27-4 |
| AZD1152-HQPA | Selleckchem | Cat#S1147; CAS: 722544-51-6 |
| GSK1838705A | Selleckchem | Cat#S2703, CAS: 1116235-97-2 |
| PF-573228 | Selleckchem | Cat#S2013; CAS: 869288-64-2 |
| OSI-906 (LINSITINIB) | Chemietek | Cat#CT-O906; CAS: 867160-71-2 |
| Alisertib (MLN8237) | Selleckchem | Cat#S1133; CAS: 1028486-01-2 |
| i-foretinib | This Paper | N/A |
| i-cabozantinib | This Paper | N/A |
| Critical Commercial Assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | G7573 |
| Crystal violet solution | Sigma | HT90132 |
| Caspase 3/7 reagent | Essen Bioscience | 4440 |
| Thermo Scientific™ Pierce™ Coomassie (Bradford) Protein Assay | ThermoFisher Scientific | Cat# PI23200 |
| RNeasy Mini Kit | Quiagen | Cat#74104 |
| IMAC enrichment Kit | Sigma | Cat#I1408 |
| Pierce™ Enrichment Kit ATP Probe | ThermoFisher Scientific | Cat#88310 |
| PTMScan Kit | Cell Signaling | https://media.cellsignal.com/pdf/8803.pdf |
| TMT Mass Tagging Kits and Reagent | ThermoFisher Scientific | Cat#90064 |
| Encore Complete RNA-Seq Library System | Nugen | N/A |
| KAPA library amplification kit for Illumina platform | KAPABiosystem | |
| Eurofin Kinase Profiler™ platform | Eurofins Pharma Discovery Services | https://www.eurofinsdiscoveryservices.com/cms/cms-content/services/in-vitro-assays/kinases/kinase-profiler/ |
| Reaction Biology Kinase Hotspot platform | Reaction Biology Corp | www.reactionbiology.com/webapps/site/KinaseAssay.aspx |
| Deposited Data | ||
| X-ray co-crystal structure of the MET kinase domain bound to XL880 (foretinib) | (Qian et al., 2009) | PDB ID: 3LQ8 |
| Chemical Proteomics in H1155 and A427 cells (Drug Pulldowns) | ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) | PXD012961 |
| Activity-based protein profiling (ABPP) | ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) | PXD012965 |
| Global phosphoproteomics (IMAC Phosphoproteomics) | ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) | PXD012963 |
| Tyrosine phosphoproteomics (Tyrosine Phosphorylation) | ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) | PXD012962 |
| RNA-Seq data | GEO database | GSE126850 |
| Experimental Models: Cell Lines | ||
| H1155 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| A427 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H3122 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H2170 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| CALU3 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H23 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H1437 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| HOP62 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| A549 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H1299 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| HCC4006 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H322 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H292 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H2342 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H157 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| CALU6 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H1395 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H650 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| HCC2935 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H1648 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H661 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H2122 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H1944 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H460 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H358 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| 16HV | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| 86M1 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H524 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| N417 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H82 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| SW210.5 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H69 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| DMS79 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| H209 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| DMS114 | Moffitt Lung Cancer Center of Excellence Cell Line Core | N/A |
| Oligonucleotides | ||
| ON-TARGET plus Non-target Pool | Dharmacon | D-001810-10-20 |
| SMARTPOOL: ON-TARGET plus Human MAP4K5 (11183) | Dharmacon | L-003589-00-0005 |
| SMARTPOOL: ON-TARGET plus Human AURKB (9212) | Dharmacon | L-003326-00-0005 |
| SMARTPOOL: ON-TARGET plus Human FER (2241) | Dharmacon | L-003129-00-0005 |
| Software and Algorithms | ||
| Prism 7.03 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| R program | The R foundation | https://cran.r-project.org/mirrors.html |
| Image J - Version1.48 | NIH.gov | https://imagej.nih.gov/ij/download.html |
| M5 Spectramax plate reader - SoftMaxPro Software 6.2.1 | Molecular Devices | https://www.moleculardevices.com/products/microplate-readers/acquisition-and-analysis-software/softmax-pro-software |
| ModFitLT V3.2.1 | BD Biosciences | http://www.bdbiosciences.com/ca/c/modfit-lt-v33-for-pc/p/346033 |
| Incucyte Zoom Software 2016B | Essen Bioscience | https://www.essenbioscience.com/en/products/software/ |
| Odyssey FC Imaging System - ImageStudio Lite Software 5.2.5 | LI-COR | https://www.licor.com/bio/products/software/image_studio/is_products_fc.html#fc |
| Mascot 2.4 | Matrix Science | http://www.matrixscience.com/help/apr2012.html |
| MaxQuant 1.2.2.5 and 1.5.2.8 | Max-Planck-Institute of Biochemistry | http://coxdocs.org/doku.php?id=maxquant:start |
| Swiss-Prot | UniProt | https://www.uniprot.org/downloads#uniprotkblink |
| Scaffold 4.0 | Proteome software | http://www.proteomesoftware.com/pdf/scaffold_brochure.pdf?v=fa4ef332 |
| Skyline 4.1 | MacCoss Lab Software | https://skyline.ms/project/home/software/Skyline/begin.view |
| Htseq-count | Stanford University and EMBL Heidelberg | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4287950/ |
| DESeq2 | Biocondutor |
https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html https://www.ncbi.nlm.nih.gov/pubmed/?term=25516281 https://www.ncbi.nlm.nih.gov/pubmed/?term=20979621 |
| Gene Set Enrichment Analysis (GSEA) | Broad Institute |
http://software.broadinstitute.org/gsea/msigdb/index.jsp https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1239896/ |
| TopHat2 | Johns Hopkins University | https://www.ncbi.nlm.nih.gov/pubmed/23618408 |
| Other | ||
| LINCS (Library of Integrated Network-Based Cellular Signatures) dataset | Harvard Medical School and NIH LINCS Program | http://lincs.hms.harvard.edu/db/datasets/20176/results?items_per_page=250&sort=&search=# |
Highlights.
Small structural change in multi-kinase drugs can cause strong cellular difference
Functional proteomics and transcriptomics provide systems view of drug effects
Systems pharmacology can elucidate differential mechanisms of related compounds
Understanding polypharmacology can lead to repurposing of multi-targeted drugs
ACKNOWLEDGMENTS
This work was supported by the NIH/NCI R01 CA181746 (to U.R.), the Moffitt NIH/NCI SPORE in Lung Cancer P50 CA119997 (to E.B.H.), the NIH/NCI R50 CA211447 (to H.R.L.), Circle of Hope for Cancer Research (to N.J.S.), the Austrian Marshall Plan Scholarships (to C.C.) and the H. Lee Moffitt Cancer Center and Research Institute. We would like to thank Dr. Forest M. White, Dr. Nicholas J. Lawrence, Dr. Keiran S. Smalley and Dr. W. Douglas Cress for their constructive feedback. We further wish to acknowledge the Moffitt Lung Cancer Center of Excellence and the Moffitt Chemical Biology (Chemistry Unit), Proteomics and Metabolomics, Molecular Genomics, Cancer Informatics, Flow Cytometry and Analytical Microscopy Core Facilities. Moffitt Core Facilities are supported by the National Cancer Institute (Award No. P30-CA076292) as a Cancer Center Support Grant. The Proteomics and Metabolomics Core is also supported by the Moffitt Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anighoro A, Bajorath J, and Rastelli G (2014). Polypharmacology: challenges and opportunities in drug discovery. J Med Chem 57, 7874–7887. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, et al. (2007). Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol 25, 1035–1044. [DOI] [PubMed] [Google Scholar]
- Barglow KT, and Cravatt BF (2007). Activity-based protein profiling for the functional annotation of enzymes. Nat Methods 4, 822–827. [DOI] [PubMed] [Google Scholar]
- Bonet C, Giuliano S, Ohanna M, Bille K, Allegra M, Lacour JP, Bahadoran P, Rocchi S, Ballotti R, and Bertolotto C (2012). Aurora B is regulated by the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling pathway and is a valuable potential target in melanoma cells. J Biol Chem 287, 29887–29898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao JN, Shafee N, Vickery L, Kaluz S, Ru N, and Stanbridge EJ (2010). Mitogen-activated protein/extracellular signal-regulated kinase kinase 1act/tubulin interaction is an important determinant of mitotic stability in cultured HT1080 human fibrosarcoma cells. Cancer Res 70, 6004–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ctortecka C, Palve V, Kuenzi BM, Fang B, Sumi NJ, Izumi V, Novakova S, Kinose F, Remsing Rix LL, Haura EB, et al. (2018). Functional Proteomics and Deep Network Interrogation Reveal a Complex Mechanism of Action of Midostaurin in Lung Cancer Cells. Mol Cell Proteomics 17, 2434–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Saborowski A, Eide CA, Tognon C, Smith RL, Elferich J, Agarwal A, Tyner JW, Shinde UP, Lowe SW, et al. (2013). Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc Natl Acad Sci U S A 110, 19519–19524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, and Zarrinkar PP (2011). Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, Kremer M, Graf N, Scheerer M, Hall MA, et al. (2010). Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 116, 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, and Taylor SS (2003). Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol 161, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, and Allis CD (2005). Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116–1122. [DOI] [PubMed] [Google Scholar]
- Gentile C, Martorana A, Lauria A, and Bonsignore R (2017). Kinase Inhibitors in Multitargeted Cancer Therapy. Curr Med Chem 24, 1671–1686. [DOI] [PubMed] [Google Scholar]
- Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, Ikeda H, Bianchi G, Hu Y, Cirstea D, et al. (2010). A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood 115, 5202–5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Lipp JJ, Toh BH, and Peters JM (2005). Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 438, 1176–1180. [DOI] [PubMed] [Google Scholar]
- Hopkins AL (2008). Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 4, 682–690. [DOI] [PubMed] [Google Scholar]
- Iwakawa R, Takenaka M, Kohno T, Shimada Y, Totoki Y, Shibata T, Tsuta K, Nishikawa R, Noguchi M, Sato-Otsubo A, et al. (2013). Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosomes Cancer 52, 802–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur G, Reinhart RA, Monks A, Evans D, Morris J, Polley E, and Teicher BA (2016). Bromodomain and hedgehog pathway targets in small cell lung cancer. Cancer Lett 371, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Girard L, Giacomini CP, Wang P, Hernandez-Boussard T, Tibshirani R, Minna JD, and Pollack JR (2006). Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 25, 130–138. [DOI] [PubMed] [Google Scholar]
- Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA, Reinecke M, Ruprecht B, Petzoldt S, Meng C, et al. (2017). The target landscape of clinical kinase drugs. Science 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevic CE, Wright G, Remsing Rix LL, Kim W, Kuenzi BM, Luo Y, Watters JM, Koomen JM, Haura EB, Monteiro AN, et al. (2016). Proteome-wide Profiling of Clinical PARP Inhibitors Reveals Compound-Specific Secondary Targets. Cell Chem Biol 23, 1490–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Lin H, and Shokat KM (2010). Targeting the cancer kinome through polypharmacology. Nat Rev Cancer 10, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenzi BM, Remsing Rix LL, Stewart PA, Fang B, Kinose F, Bryant AT, Boyle TA, Koomen JM, Haura EB, and Rix U (2017). Polypharmacology-based ceritinib repurposing using integrated functional proteomics. Nat Chem Biol 13, 1222–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. (2006). The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Li J, Fang B, Kinose F, Bai Y, Kim JY, Chen YA, Rix U, Koomen JM, and Haura EB (2016). Target Identification in Small Cell Lung Cancer via Integrated Phenotypic Screening and Activity-Based Protein Profiling. Mol Cancer Ther 15, 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Rix U, Fang B, Bai Y, Edwards A, Colinge J, Bennett KL, Gao J, Song L, Eschrich S, et al. (2010). A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol 6, 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, and Gray NS (2006). Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2, 358–364. [DOI] [PubMed] [Google Scholar]
- Martin MP, Zhu JY, Lawrence HR, Pireddu R, Luo Y, Alam R, Ozcan S, Sebti SM, Lawrence NJ, and Schonbrunn E (2012). A novel mechanism by which small molecule inhibitors induce the DFG flip in Aurora A. ACS Chem Biol 7, 698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortlock AA, Foote KM, Heron NM, Jung FH, Pasquet G, Lohmann JJ, Warin N, Renaud F, De Savi C, Roberts NJ, et al. (2007). Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J Med Chem 50, 2213–2224. [DOI] [PubMed] [Google Scholar]
- Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, Buck E, Foreman K, Landfair D, O’Connor M, Pirritt C, Sun Y, Yao Y, et al. (2009). Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem 1, 1153–1171. [DOI] [PubMed] [Google Scholar]
- Ong SE, Schenone M, Margolin AA, Li X, Do K, Doud MK, Mani DR, Kuai L, Wang X, Wood JL, et al. (2009). Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc Natl Acad Sci U S A 106, 4617–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, et al. (2011). In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol 18, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phadke MS, Sini P, and Smalley KS (2015). The Novel ATP-Competitive MEK/Aurora Kinase Inhibitor BI-847325 Overcomes Acquired BRAF Inhibitor Resistance through Suppression of Mcl-1 and MEK Expression. Mol Cancer Ther 14, 1354–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian F, Engst S, Yamaguchi K, Yu P, Won KA, Mock L, Lou T, Tan J, Li C, Tam D, et al. (2009). Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res 69, 8009–8016. [DOI] [PubMed] [Google Scholar]
- Reszka AA, Seger R, Diltz CD, Krebs EG, and Fischer EH (1995). Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc Natl Acad Sci U S A 92, 8881–8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rix U, and Superti-Furga G (2009). Target profiling of small molecules by chemical proteomics. Nat Chem Biol 5, 616–624. [DOI] [PubMed] [Google Scholar]
- Sabbatini P, Korenchuk S, Rowand JL, Groy A, Liu Q, Leperi D, Atkins C, Dumble M, Yang J, Anderson K, et al. (2009). GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol Cancer Ther 8, 2811–2820. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2018). Cancer statistics, 2018. CA Cancer J Clin 68, 7–30. [DOI] [PubMed] [Google Scholar]
- Skarlos DV, Samantas E, Kosmidis P, Fountzilas G, Angelidou M, Palamidas P, Mylonakis N, Provata A, Papadakis E, Klouvas G, et al. (1994). Randomized comparison of etoposide-cisplatin vs. etoposide-carboplatin and irradiation in small-cell lung cancer. A Hellenic Co-operative Oncology Group study. Ann Oncol 5, 601–607. [DOI] [PubMed] [Google Scholar]
- Sos ML, Dietlein F, Peifer M, Schottle J, Balke-Want H, Muller C, Koker M, Richters A, Heynck S, Malchers F, et al. (2012). A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc Natl Acad Sci U S A 109, 17034–17039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, D447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waqar SN, and Morgensztern D (2017). Treatment advances in small cell lung cancer (SCLC). Pharmacol Ther 180, 16–23. [DOI] [PubMed] [Google Scholar]
- Weiss WA, Taylor SS, and Shokat KM (2007). Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat Chem Biol 3, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Rix U, Carlson SM, Gleixner KV, Grebien F, Gridling M, Muller AC, Breitwieser FP, Bilban M, Colinge J, et al. (2012). Systems-pharmacology dissection of a drug synergy in imatinib-resistant CML. Nat Chem Biol 8, 905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, et al. (2011). Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther 10, 2298–2308. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, and Sakai T (2011). Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol 39, 23–31. [DOI] [PubMed] [Google Scholar]
- You WK, Sennino B, Williamson CW, Falcon B, Hashizume H, Yao LC, Aftab DT, and McDonald DM (2011). VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res 71, 4758–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, and Gray NS (2009). Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 9, 28–39. [DOI] [PubMed] [Google Scholar]
- Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, and Washburn MP (2006). Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res 5, 2339–2347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 – related to Figure 4 and Figure S4: Chemical proteomics data set of H1155 and A427 cells. Data was searched by Mascot and analyzed in Scaffold with a peptide threshold at 95% and a minimum of 2 exclusive spectra counts. Protein kinases are highlighted in orange. Amp: ampicillin control, Cabo: cabozantinib, Fore: foretinib, PT: pre-treatment. Fore-NSAF > 0.005 was used as selection criteria. - ABPP data set. Displayed are the kinases identified by activity-based protein profiling in H1155 cells. Data was filtered for PEP < 0.05 and data was IRON normalized. Rows with all zero values, contaminant and reverse peptides were removed. p-value compares Fore (F) to Cabo (C).
Table S2 – related to Figure 4: pSTY phosphoproteomics data set. Global phosphoproteomics data was filtered for PEP < 0.05 and data was IRON normalized. Rows with all zero values, contaminants and reverse sequences were removed.
Table S3 – related to Figure 4: pY phosphoproteomics data set. Phosphotyrosine data was filtered for PEP < 0.05 and data was IRON normalized. Rows with all zero values, contaminant and reverse peptides were removed.
Table S4 – related to Figure 4: RNA-Seq data set. Paired-end reads were aligned using TopHat2 and HTSeq was used to count reads that were mapped to the genes. Genes that were significantly regulated accordingly to our selection criteria have a value 1 in the criteria column.
Table S5 – related to Figure 4: Integrated data analysis. Pathway analysis was performed by entering the gene names into the GSEA database and querying canonical pathways and gene ontology (GO) gene sets, which included GO biological process, GO cellular component and GO molecular function.
Table S6 - related to Figure 4: GO_Cytoskeleton: Kinases including in the GO_Cytoskeleton pathway from GSEA and which were used for further analysis.
Table S7 – related to Figure 4: GO_Cell Cycle: Kinases including in the GO_Cell Cycle pathway from GSEA and which were used for further analysis.
Data Availability Statement
The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) with the dataset identifiers PXD012961 (Drug Pulldowns), PXD012962 (Tyrosine Phosphorylation), PXD012963 (IMAC Phosphoproteomics) and PXD012965 (ABPP) (Vizcaino et al., 2016). RNA-Seq data have been deposited in the GEO database with the dataset identifier GSE126850.