

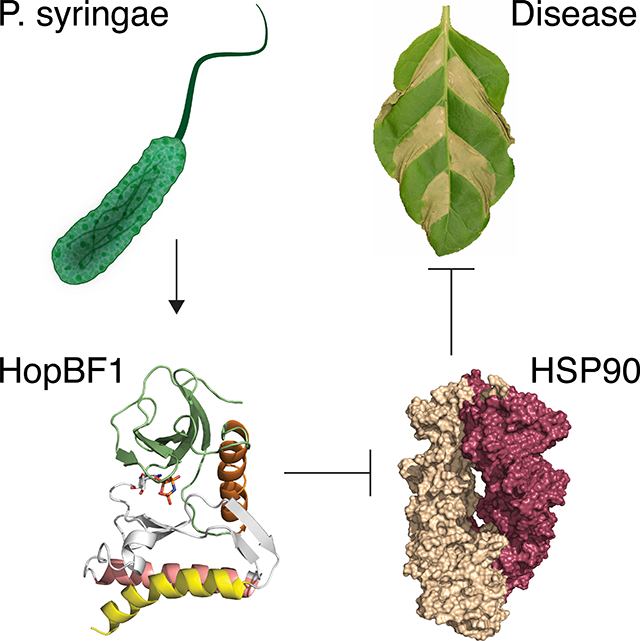
Summary
The molecular chaperone HSP90 facilitates the folding of several client proteins including innate immune receptors and protein kinases. HSP90 is an essential component of plant and animal immunity yet pathogenic strategies that directly target the chaperone have not been described. Here we identify the HopBF1 family of bacterial effectors as eukaryotic-specific HSP90 protein kinases. HopBF1 adopts a minimal protein kinase fold that is recognized by HSP90 as a host client. As a result, HopBF1 phosphorylates HSP90 to completely inhibit the chaperone’s ATPase activity. We demonstrate that phosphorylation of HSP90 prevents activation of immune receptors that trigger the hypersensitive response in plants. Consequently, HopBF1-dependent phosphorylation of HSP90 is sufficient to induce severe disease symptoms in plants infected with the bacterial pathogen, Pseudomonas syringae. Collectively, our results uncover a family of bacterial effector kinases with toxin-like properties and reveal a previously unrecognized betrayal mechanism by which bacterial pathogens modulate host immunity.
Keywords: HSP90, phosphorylation, chaperone, immunity, kinase
eTOCs
A bacterial effector operates through a “betrayal-like” mechanism by masquerading as an HSP90 client as a means to achieve specificity for its target. This minimal kinase then phosphorylates a critical active site residue to inactive an essential chaperone required for host innate immunity.
Graphical Abstract
Introduction
The millions of predicted amino acid sequences acquired in the post-genomic era have revealed remarkable diversity among protein families. This is especially noticeable in bacterial pathogens where the evolutionary arms race between the pathogen and its host is intense, and inter-kingdom horizontal gene transfer is common (Soucy et al., 2015). We have taken a bioinformatic approach in search of remote members of the protein kinase superfamily that are barely recognizable by their primary amino acid sequence, yet have maintained the kinase fold, and possibly catalytic activity. Bioinformatic methods combined with expert curation has enabled the identification of divergent kinase family members such as the Fam20 secretory pathway kinases (Tagliabracci et al., 2012; Tagliabracci et al., 2015), the CotH spore coat protein kinases (Nguyen et al., 2016) and the AMPylating pseudokinase SelO (Sreelatha et al., 2018). Using a similar approach, we identified the uncharacterized HopBF1 family of bacterial Type III secretion system (T3SS) effectors as remote homologs of classical protein kinases and of aminoglycoside phosphotransferases.
Several HopBF1 homologs are found in strains of the gram-negative plant pathogen Pseudomonas syringae (Baltrus et al., 2011; O’Brien et al., 2012), but also in a collection of phylogenetically scattered strains from gammaproteobacteria (e.g. genera Enterobacter, Klebsiella, Ewingella, Serratia), betaproteobacteria (e.g. genera Burkholderia, Ralstonia) as well as a handful of strains from alphaproteobacteria, Planctomycetes, Actinobacteria and Cyanobacteria (Figure S1A). Notably, some of these strains are animal and plant pathogens or symbionts, whereas others are free-living.
Evolution has provided bacterial pathogens with a diverse set of strategies to survive within the harsh environment of its host. To combat or evade host immunity, many Gram-negative pathogenic bacteria employ the T3SS, a needle like structure used to deliver virulence factors into host cells (Deng et al., 2017; O’Brien et al., 2012; Wessling et al., 2014). Bacterial effectors have evolved unique biochemical activities to disrupt the host immune response. While critical to a pathogen’s survival, the molecular functions of most effector proteins are unknown.
During an infection, the innate immune system of the host recognizes pathogen-derived molecules to stimulate a robust defense response (Jones et al., 2016). As a first line of protection, plants and animals use pattern recognition receptors to detect pathogen associated molecular patterns (PAMPs) and execute PAMP triggered immunity (PTI) (Ronald and Beutler, 2010). Moreover, the host cell can also employ an intracellular surveillance mechanism to detect PAMPs via a family of polymorphic intracellular nucleotide binding domain, leucine rich repeat proteins (NB-LRR) (Jones et al., 2016). These immune receptors require the molecular chaperone HSP90 to facilitate their activation (Hubert et al., 2003; Schulze-Lefert, 2004). HSP90 and its cochaperones maintain NB-LRRs in an inactive but signal competent form (Kadota and Shirasu, 2012). Despite the fact that HSP90 is an indispensable component of both plant (Schulze-Lefert, 2004; Shirasu, 2009) and animal (Mayor et al., 2007) immunity, pathogenic mechanisms that directly target HSP90 have not been described.
Here we show that HopBF1 adopts a minimal and atypical protein kinase fold that appears to be recognized by HSP90 as a host client. As a result, HopBF1 phosphorylates HSP90 resulting in the complete inactivation of the chaperone’s ATPase activity. Using the bacterial plant pathogen P. syringae as a model system, we demonstrate that HopBF1-dependent phosphorylation of HSP90 prevents activation of NB-LRRs. Accordingly, HopBF1 is sufficient to induce strong disease symptoms in plants infected with P. syringae. Our results uncover a novel betrayal mechanism of host-pathogen interaction.
Results
HopBF1 adopts a minimal and atypical protein kinase fold
Sequence logo analyses using 161 HopBF1 homologs suggest conservation of residues involved in protein kinase activity, including highly conserved glycine residues within the predicted β1-β2 ATP binding loop, and active site residues corresponding to D166 and D184 of the prototypical protein kinase A (PKA) (Figure 1A). However, some features differ; for example, there are no clear equivalents of the metal binding N171 and the predicted kinase domain appears to be missing much of the C-lobe (Figures S1B and S1C). In fact, the average length of the predicted kinase domain in the HopBF1 family is only ~183 amino acids, whereas a typical kinase domain encompasses approximately 265 amino acids on average.
Figure 1. HopBF1 adopts a minimal protein kinase fold (Related to Figure S1).

(A) Sequence logo analysis of 161 HopBF1 homologs depicting the conservation of predicted active site residues. The Gly-rich loop (β1-β2 loop) and the active site HRD (PKA, D166) and DFG (PKA, D184) are shown.
(B) Amino acid sequence of E. americana HopBF1 depicting the secondary structural elements. The N-lobe β-strands are in lime, the α1 (PKA αC equivalent) helix is in orange, the C-lobe β-strands and the α2 helix are in white, the α3 helix (PKA αE) is in salmon and the α4 helix (PKA αF) is in yellow. E. americana HopBF1 contains 203 amino acids.
(C) Ribbon representation of E. americana HopBF1. Color coding as in Figure 1B.
(D) Ribbon representation of PKA (PDB code 1ATP). Color coding as in Figure 1B. The GHI helical subdomain, which is missing in HopBF1, is in white.
(E) Superposition of the N-lobes of the Apo and AMP-PNP bound HopBF1 structures depicting the unfolded β1-strand in the Apo structure. The Apo structure is in yellow and the AMP-PNP bound structure is in teal.
(F) Enlarged image of the nucleotide-binding pocket showing the detailed molecular interactions involved in nucleotide binding. Active site residues are numbered and the PKA equivalent residues are shown in parentheses. The AMP-PNP molecule is also shown.
To gain insight into the molecular function of HopBF1, we solved crystal structures of HopBF1 from the opportunistic gram-negative human pathogen Ewingella americana (Hassan et al., 2012) in the nucleotide free (apo) and bound to the ATP analog, AMP-PNP forms (Figures 1B, 1C, and S1D; Table S1). E. americana HopBF1 exhibits a minimal protein kinase-like fold and can be superimposed onto PKA with a root mean square derivation (rmsd) of 3.2 Å over 150 Cα atoms (Figures 1C and 1D). As found in all members of the protein kinase superfamily, E. americana HopBF1 contains a β-strand rich N-lobe (Figure 1C, lime) containing the regulatory α-C helix (Figure 1C, orange) packing against the core N-lobe β-sheet. The C-lobe in canonical kinases is rich in α-helices; however, in E. americana HopBF1 there are more β-strands than α-helices within the C-lobe. The α-E and α-F helices (PKA equivalent) are present in the C-lobe, yet the GHI helical subdomain, which is typically involved in substrate recognition and allosteric regulation, is notably absent in E. americana HopBF1 (Figures 1C and 1D) (Taylor and Kornev, 2011). The apo and nucleotide bound structures of E. americana HopBF1 are highly similar with a rmsd of 0.86 Å for the Cα atoms (Figure S1D). The β1-strand in the apo structure is unfolded and forms a long loop that covers the ATP binding pocket (Figure 1E). Thus, HopBF1 adopts a minimal and atypical protein kinase-like fold.
The AMP-PNP nucleotide in the HopBF1 structure resides in a pocket between the N and C-lobes of the kinase (Figure 1F). As observed in all protein kinases, the Gly-rich loop (β1-β2 loop) extends over the nucleotide and stabilizes the phosphate groups (Figure 1F). The formation of an ion pair between a Lys in β3 (PKA K72) and a Glu in the α-C helix (PKA E91) is considered a hallmark of an activated protein kinase (Endicott et al., 2012). An equivalent ion pair is present in E. americana HopBF1; however, the Lys has migrated to the β1-β2 loop, analogous to the With No Lysine (WNK) kinases (Min et al., 2004). The catalytic “HRD” (D155) and the metal binding “DFG” (D170) are present in the C-lobe and positioned for catalysis. Although Mn2+ was present in our crystallization conditions, there is no clear electron density in the active site consistent with a divalent cation bound near the AMP-PNP nucleotide.
HopBF1 induces severe disease symptoms in plants
Because most HopBF1 homologs are found in strains of P. syringae, we used plants as a model host to investigate the role of HopBF1 during infection. To determine the impact of HopBF1 expression in planta, we generated mutants of P. syringae HopBF1 that we predicted would inactivate the kinase based on their location in the active site (Figure 1F). We transiently expressed wildtype (WT) or the predicted catalytically-inactive D154A and D169A (E. americana D155 and D170) P. syringae HopBF1 mutants by agroinfiltration in Nicotiana benthamiana and Nicotiana tabacum plants and observed robust necrosis and tissue collapse in leaves expressing WT but not the predicted inactive HopBF1 mutants (Figures 2A, 2B, S2A, and S2B). To test whether HopBF1 causes cell death during an infection, we expressed HopBF1 or the mutants in an effectorless derivative of P. syringae pv. tomato DC3000 (DC3000D28E) (Cunnac et al., 2011) and infected Arabidopsis thaliana and N. benthamiana leaves. Leaves inoculated with DC3000D28E expressing WT HopBF1 also displayed profound necrosis and tissue collapse that was not observed when the HopBF1 mutants were expressed in this strain (Figures 2C, 2D, S2C, and S2D). Thus, HopBF1 kinase activity is sufficient to cause severe disease symptoms in N. benthamiana, N. tabacum and A. thaliana.
Figure 2. HopBF1 induces severe disease symptoms in plants (Related to Figure S2).

(A) Agrobacterium-mediated expression of HopBF1 leads to leaf tissue collapse. Leaves of N. benthamiana plants were infiltrated with A. tumefaciens carrying HopBF1 or the predicted kinase inactive mutants D154A and D169A. PKA numbering is in parentheses. Photos were taken 7 days post infiltration.
(B) Agrobacterium-mediated expression of HopBF1 induces tissue collapse in N. tabacum. Sections of tobacco leaves were infiltrated with A. tumefaciens carrying HopBF1 or the predicted kinase inactive mutants D154A and D169A. YFP was used as a negative control. Tissue collapse in the infiltrated zones developed within 4 days.
(C) HopBF1 evokes leaf tissue collapse in Arabidopsis plants after infection with P. syringae. Three adult leaves were infiltrated with P. syringae pv. tomato DC3000 strain with 28 effectors deleted expressing HopBF1, the D169A mutant or mCherry as a control. Representative leaves were photographed 6 days post inoculation.
(D) HopBF1 evokes N. benthamiana leaf tissue collapse after infection with P. syringae. Leaves of N. benthamiana plants were infiltrated with P. syringae strain pv. tomato DC3000 with 28 effectors deleted expressing HopBF1, the D154A and D169A mutants or mCherry as a control. Representative leaves were photographed 7 days post inoculation.
HopBF1 phosphorylates eukaryotic HSP90
Bacterial effectors typically target evolutionarily conserved signaling pathways in eukaryotes (Alto and Orth, 2012). As such, Saccharomyces cerevisiae is commonly used as a proxy to identify substrates and pathways targeted by effectors. We expressed WT and predicted inactive P. syringae HopBF1 mutants under the control of a galactose inducible promoter in S. cerevisiae and observed a potent growth defect only when WT HopBF1 was expressed (Figure 3A and S3A). To identify potential HopBF1 substrates, we incubated recombinant P. syringae HopBF1 or the predicted inactive D154A mutant with a yeast extract and [γ32P]ATP. We observed a single 32P-labelled species that migrated during electrophoresis at ~80–90 kDa in extracts that were incubated with WT HopBF1 but not the predicted inactive mutant (Figure 3B).
Figure 3. HopBF1 phosphorylates eukaryotic HSP90 (Related to Figure S3).

(A) Spot assay depicting the growth of S. cerevisiae expressing Myc-tagged P. syringae HopBF1 or the predicted inactive D154A and D169A mutants.
(B) Time dependent incorporation of 32P from [γ32P]ATP into an unknown 80–90 kDa protein in a yeast extract catalyzed by recombinant P. syringae HopBF1 but not the D154A mutant. The Coomassie stained gel (upper), autoradiograph (middle) and HopBF1 immunoblot (lower) are shown.
(C) SDS PAGE analysis of anti-Flag immunoprecipitates from yeast extracts expressing empty vector or Flag-tagged P. syringae HopBF1 D169A. The immunoprecipitating proteins were eluted with Flag peptide, visualized by silver stain, and HopBF1 interacting proteins were identified by mass spectrometry.
(D) Protein immunoblotting of Flag-immunoprecipitates from yeast extracts expressing Flag-tagged P. syringae HopBF1 D169A. Cell extracts were also analyzed for HSP82/HSC82 and GAPDH as a loading control.
(E) Incorporation of 32P from [γ32P]ATP into S. cerevisiae HSP90 (HSP82) by recombinant P. syringae HopBF1 but not into the D154A or the D169A mutants. Bands were also excised from the gel and radioactive incorporation was quantified by scintillation counting revealing ~1:1 stoichiometry of phosphorylation.
(F) Incorporation of 32P from [γ32P]ATP into S. cerevisiae HSP90 (HSP82) by recombinant P. syringae, E. americana and S. rubidaea HopBF1. Reaction products were analyzed as in B.
(G) Incorporation of 32P from [γ32P]ATP into E. coli, P. syringae, H. sapiens, S. cerevisiae and T. aestivum HSP90 homologs by recombinant P. syringae HopBF1. Reaction products were analyzed as in B.
(H) Incorporation of 32P from [γ32P]ATP into the generic protein kinase substrates, osteopontin (OPN), myelin basic protein (MyBP), histone H4 and β-casein or the S. cerevisiae HSP90 chaperones, HSP82, HSC82 and the HSP70 chaperones, SSA1, SSA2, SSA3, SSC1 and KAR2 by recombinant P. syringae HopBF1. Reaction products were analyzed as in B.
Moreover, analysis of anti-Flag immunoprecipitates from yeast extracts expressing Flag-tagged HopBF1 D169A revealed the presence of a single coimmunoprecipitating protein, which also migrated as an ~80–90 kDa species during SDS PAGE (Figure 3C). Mass spectrometry identified tryptic peptides corresponding to HSP82 and HSC82, the yeast orthologs of human HSP90. Protein immunoblotting of Flag-immunoprecipitates confirmed HSP82/HSC82 as HopBF1 interacting proteins (Figure 3D).
HSP90 is a ubiquitous molecular chaperone that binds and hydrolyzes ATP while facilitating the maturation of a wide array of proteins referred to as clients. To test whether HSP90 is a substrate of HopBF1, we purified recombinant yeast HSP90 (HSP82) and performed in vitro kinase assays. P. syringae HopBF1, but not the D154A or the D169A mutants phosphorylated HSP82 in vitro and incorporated ~1 mol of phosphate per mol HSP82 (Figure 3E). The apparent Km for HSP82 was ~3 μM (Figure S3B), which is in the range of the ~17.5 μM concentration of HSP82 in yeast (Ghaemmaghami et al., 2003). HopBF1 homologs from P. syringae, E. americana, and Serratia rubidaea, but not from Burkholderia species phosphorylated HSP82 (Figures 3F and S3C). Moreover, P. syringae HopBF1 also phosphorylated Triticum aestivum (wheat) HSP90 and human HSP90β (Figure 3G). Interestingly, the prokaryotic HSP90 orthologs, htpG from E. coli and P. syringae were not phosphorylated by HopBF1 (Figure 3G). HopBF1 activity appeared to be highly specific for HSP90 because generic protein kinase substrates such as casein and myelin basic protein, as well as the yeast HSP70’s and Bip chaperones were not phosphorylated (Figure 3H). HopBF1 phosphorylated an ATP-binding deficient mutant of HSP82 and geldanamycin, an HSP90 inhibitor, did not inhibit the phosphorylation (Figure S3D). Thus, HopBF1 appears to be a eukaryotic-specific HSP90 kinase.
HopBF1-dependent phosphorylation of HSP90 inactivates ATPase activity and chaperone function
Mass spectrometry analysis of phosphorylated HSP82 revealed a phosphopeptide corresponding to the lid segment of the chaperone, which suggested that either Ser99 or Thr101 was phosphorylated (Figure 4A). Mutation of S99 to Ala or Glu completely abolished HopBF1-dependent phosphorylation of HSP82 (Figure 4B). In contrast, mutation of Thr101 to Ala had no effect on HSP82 phosphorylation by HopBF1. Ser99 is invariant in the HSP90 family and supports nucleotide binding by interacting with the β-phosphate of ATP (Figures 4C and 4D) (Ali et al., 2006; Verba et al., 2016) HopBF1 phosphorylation of HSP82 or the presence of a phosphomimetic Glu (S99E) reduced the chaperone’s ATPase activity to a similar extent as did geldanamycin, a potent HSP90 family inhibitor (Figure 4E).
Figure 4. HopBF1 inactivates HSP90 ATPase and chaperoning activity (Related to Figure S4).

(A) Representative MS/MS fragmentation spectra of a tryptic peptide (HSP82 87–102) depicting Ser99 phosphorylation of HSP82. Recombinant HSP82 was incubated with ATP and P. syringae HopBF1, separated by SDS PAGE and subjected to MS. Control experiments were performed in parallel using P. syringae HopBF1 D169A but no phosphopeptides were identified.
(B) Incorporation of 32P from [γ32P]ATP into S. cerevisiae HSP82 and the Thr101Ala mutant but not the Ser99Ala or the Ser99Glu mutants by recombinant P. syringae HopBF1. Reaction products were analyzed as in Figure 3B.
(C) Zoomed in view of the active site of HSP90, PDB: 2CG9 (Ali et al., 2006) depicting the interaction between the β-phosphate of ATP and the HopBF1 target Ser (Ser99 in yeast; Ser108 in human HSP90β).
(D) Multiple sequence alignment depicting the conservation of the HopBF1 target Ser in the HSP90 family of chaperones. HSP90 sequences from E. coli, P. syringae, S. cerevisiae, N. benthamiana, T. aestivum and H. sapiens are shown.
(E) ATPase activity of unphosphorylated (WT) and HopBF1-phosphorylated (p-WT) HSP82 was assayed by measuring free phosphate release from ATP using the Malachite Green method. The activities of the S99A and S99E mutants are also shown. The HSP90 inhibitor geldanamycin (Gel) was used as a control.
(F) Protein immunoblotting of HEK293A cell extracts expressing Flag-v-src with E. americana HopBF1 or the D170A mutant. Cells were also pretreated with geldanamycin. Extracts were analyzed for v-src activity as judged by global phospho-Tyr immunoblotting (α-pY; 4G10). Levels of Flag-v-src and HopBF1 are also shown. GAPDH is included as a loading control.
(G) Protein immunoblotting of HEK293A cell extracts expressing Flag-NLRP3 with E. americana HopBF1 or the D170A mutant. Cells were also pretreated with geldanamycin. Levels of Flag-NLRP3 and HopBF1 are also shown. Tubulin is included as a loading control.
(H) Spot assay depicting the growth of S. cerevisiae expressing Flag-tagged S. cerevisiae HSP82 (Flag-HSP82) with Myc-tagged P. syringae HopBF1 (Myc-HopBF1) or the hypomorphic E74A mutant. Images were taken 2 days after spotting (glucose) and 5 days after spotting (galactose, induced).
(I) Viability of HSP90 null yeast expressing empty vector, WT HSP82 or the Ser99Glu (S99E) phosphomimetic mutant deduced by a 5-fluoroorotic acid (5-FOA) shuffling assay (Nathan and Lindquist, 1995).
To test whether HSP90 chaperone-function was affected by HopBF1 phosphorylation, we co-expressed E. americana HopBF1 with two HSP90 clients in mammalian cells: the oncogenic tyrosine kinase, v-src (Xu and Lindquist, 1993), and the innate immune receptor, NLRP3 (Mayor et al., 2007). HopBF1, but not the inactive mutant, markedly reduced the levels of v-src, global phospho-tyrosine and NLRP3, but not GFP (Figures 4F, 4G, and S4A). Additionally, a phosphomimetic mutant of human HSP90β (S108E) could still interact with v-src, suggesting that the chaperone function but not the client interaction is perturbed by HopBF1 phosphorylation (Figure S4B). Collectively, these results suggest that the HopBF1 family of kinases phosphorylate and inactivate HSP90 ATPase activity and chaperone function.
HopBF1 phosphorylates and inactivates HSP90 in yeast
We used structure-guided mutagenesis to generate mutants of HopBF1 that we predicted would be involved in catalysis. Alanine substitutions of the ion pair Lys41, the catalytic Asp154, and the metal binding Asp169 in P. syringae HopBF1 prevented phosphorylation of HSP82 in vitro (Figures S4C and S4D). However, mutation of the ion pair residue E74A significantly reduced, but did not completely abolish, HopBF1 activity. Likewise, expression of HopBF1 E74A in yeast produced a less severe growth defect than that of WT HopBF1 (Figures 4H and S4D).
Because HSP90 is essential for yeast viability (Borkovich et al., 1989), we hypothesized that phosphorylation and inactivation of HSP90 was the mechanism by which HopBF1 induced a growth defect in yeast. Overexpression of HSP90 failed to suppress the growth defect caused by HopBF1, most likely because HopBF1 acts catalytically (Figure 4H). However, overexpression of HSP90 in yeast expressing the hypomorphic HopBF1 E74A mutant partially rescued the growth defect. Importantly, we also rescued viability of an HSP90 null yeast strain by expression of WT HSP90 but not an HSP90 S99E phosphomimetic mutant (Figure 4I, See methods). Collectively, these results suggest that the HopBF1-induced growth defect in yeast is a result of phosphorylation and inactivation of HSP90.
HopBF1 phosphorylates HSP90 during infection.
We expressed E. americana HopBF1 or the inactive mutant in an effectorless derivative of Yersinia pestis (YP37), a commonly used infection model system that delivers effectors into host cells through the T3SS. Following infection of HeLa cells, endogenous HSP90 was immunoprecipitated and analyzed by MS. We detected 14 modified phosphopeptides and 1 unmodified peptide corresponding to phosphorylated Ser108 of human HSP90β (Yeast Ser99 equivalent) when WT E. americana HopBF1 was translocated into HeLa cells (Figure S5A). In contrast, we failed to detect any phosphorylated peptides when the D170A mutant of HopBF1 was used in these experiments. By comparing the number of modified to unmodified peptides, we can infer that the stoichiometry of phosphorylation during infection is close to 93% (Figure S5A). Thus, HopBF1 phosphorylates HSP90 during bacterial infection.
To determine whether HSP90 is phosphorylated by P. syringae HopBF1 in planta, we co-expressed Myc-tagged HSP90 with P. syringae HopBF1 or the D169A mutant and analyzed anti-Myc immunoprecipitates from N. benthamiana leaf extracts by MS. Approximately 40% of the detected peptides containing S100 were phosphorylated when coexpressed with WT HopBF1; in contrast, only 7.7% were phosphorylated when coexpressed with the D169A mutant (Figure S5A).
To determine the impact of HSP90 phosphorylation during P. syringae infection, we infiltrated N. benthamiana leaves with DC3000D28E expressing HopBF1 and agroinfiltrated WT N. benthamiana HSP90 or the non-phosphorylatable S100F mutant (Figure S5B) and monitored disease symptoms. Interestingly, the S100F mutation in HSP90, which has very low ATPase activity (Hubert et al., 2009), was identified in a large-scale genetic screen for A. thaliana mutants with impaired resistance triggered by RPM1 (Resistance to Pseudomonas syringae pv maculicola 1) upon recognition of the P. syringae effector AvrRpm1 (Hubert et al., 2003). Leaves overexpressing HSP90 that were infiltrated with P. syringae HopBF1, but not the D169A mutant, displayed signs of tissue collapse as early as three-days post infiltration, which became more pronounced after 6 days (Figures 5A, S5C, and S5D). In contrast, leaves transiently expressing HSP90 S100F or YFP had significantly less disease symptoms after 3- and 6-days post-infection. The acceleration of disease symptom formation by HopBF1 in plants overexpressing HSP90 but not the Ser100 mutant suggests that elevated levels of Ser100 phosphorylated HSP90 may be toxic to plants. Taken together, our results suggest that HopBF1-dependent phosphorylation of HSP90 on Ser100 during infection causes tissue collapse and necrosis in planta.
Figure 5. HopBF1 targets plant HSP90 during P. syringae infection (Related to Figure S5).

(A) N. benthamiana leaves were infiltrated with P. syringae DC3000D28E expressing HopBF1 and appropriate A. tumefaciens strain to transiently express HSP90, HSP90 S100F or YFP as a control. Overexpression of HSP90 accelerated tissue necrotization triggered by bacteria expressing HopBF1 but not the S100F mutant or YFP. Photographs show representative leaves made 3 or 6 dpi as indicated. The experiment was performed 5 times with similar results.
(B) N. benthamiana leaves were co-infiltrated with appropriate Agrobacterium strains to express the autoactive variant of RPM1 (D505V) with or without HopBF1 or the D154A and D169A inactive mutants. Co-expression of WT HopBF1 but not the mutants strongly inhibited tissue necrotization. Photographs of the representative leaves were taken 5 days post infiltration.
(C) Model depicting the mechanism of HopBF1-dependent suppression of the HR in plants during P. syringae infection. T3SS modified from (Deng et al., 2017). We propose that HopBF1 has evolved to phosphorylate and inactive HSP90 to 1) prevent activation of NB-LRR proteins that trigger the HR in plants and 2) cause host cell death.
Plants protect themselves against pathogens by PAMP-triggered immunity (PTI) (Dangl et al., 2013; Dangl and McDowell, 2006). Bacterial plant pathogens utilize type III effectors to promote virulence by disrupting or suppressing PTI (Xin and He, 2013). Consequently, plants have evolved mechanisms to detect the presence of effectors, which results in the activation of “effector-triggered” immunity (ETI), often resulting in a form of programmed cell death termed the “hypersensitive response” (HR) (Cui et al., 2015). To test whether HopBF1 induces the HR, we inoculated N. benthamiana leaves with the virulent P. syringae B728a strain, which does not induce HR unless a recognized effector, such as HopQ1, is ectopically expressed in the strain (Giska et al., 2013). Bacteria expressing HopBF1 or the mutants multiplied to similar extent as a control strain and caused disease symptoms (Figure S5E). Thus, HopBF1 does not activate the HR in N. benthamiana.
In plants, HSP90 and its co-chaperones, SGT1 and RAR1 have well-documented roles in the host defense response to pathogens (Kadota and Shirasu, 2012). The current model suggests that many NB-LRR proteins, several of which are plant Resistance proteins (R-proteins), such as RPM1, are clients of the HSP90 complex (Hubert et al., 2003; Takahashi et al., 2003). Pathogen-induced activation of NB-LRR proteins leads to the HR and requires a functional HSP90 (Kadota and Shirasu, 2012). An autoactive mutant of RPM1 (D505V) recapitulates normal RPM1 activation and subsequent HR in the absence of pathogens (Gao et al., 2011). Because RPM1 requires HSP90 for its function, we reasoned that HopBF1 would inactivate HSP90 and prevent the HR in leaves expressing the autoactive RPM1 mutant. As expected, transient expression via agroinfiltration of RPM1 D505V in N. benthamiana resulted in widespread HR (Figure 5B, panel 1). However, co-expression of RPM1 D505V with HopBF1, but not the inactive D154A and D169A mutants, delayed and reduced HR development (Figure 5B, panels 2–4). Collectively, these results suggest that HopBF1 interferes with the activation of RPM1 D505V by phosphorylation and inactivation of HSP90. Because several R proteins require HSP90 for activation (Kadota and Shirasu, 2012), one consequence of HSP90 inhibition by HopBF1 is prevention of NB-LRR activation and the HR (Figure 5C). However, inactivation of HSP90 by HopBF1 will likely have pleiotropic effects across multiple signaling pathways, some of which may also contribute to P. syringae pathogenicity (See Discussion).
HopBF1 mimics an HSP90 client
Because HopBF1 and HSP90 appear to interact in near stoichiometric quantities (Figure 3C), we wondered whether HopBF1 was being recognized as a host HSP90 client. Interestingly, the most prevalent HSP90 clientele are members of the protein kinase superfamily (Taipale et al., 2010; Taipale et al., 2012). Therefore, to test whether HSP90 recognizes HopBF1 as a client, we expressed the catalytically inactive E. americana HopBF1 D170A in HEK293A cells and treated the cells with the HSP90 inhibitor geldanamycin. Inhibition of HSP90 decreased levels of HopBF1 D170A and the bona fide HSP90 client v-src, but not GFP (Figure 6A). Likewise, levels of WT HopBF1 were consistently lower than that of the D170A mutant, indicating that WT HopBF1 expression phenocopies geldanamycin treatment of the D170A mutant (Figure S6A).
Figure 6. HopBF1 mimics an HSP90 client (Related to Figure S6).

(A) Protein immunoblotting of HEK293A cell extracts depicting the levels of E. americana Flag-HopBF1 D170A (upper), the HSP90 kinase client Flag-v-src (positive control; middle) and GFP (negative control; lower). Tubulin and GAPDH are used as loading controls.
(B) Protein immunoblotting of anti-Flag immunoprecipitates from HEK293A cell extracts expressing Flag-tagged GFP (Flag-GFP), v-src (Flag v-src) or E. americana HopBF1 D170A (Flag HopBF1 D170A). Immunoprecipitates and cell extracts were analyzed for HSP90 and CDC37. Cell extracts were also analyzed for HSP90, CDC37 and GAPDH as a loading control.
(C) Protein immunoblotting of anti-Flag immunoprecipitates from HEK293A cell extracts expressing Flag-tagged GFP (Flag-GFP), v-src (Flag v-src) or E. americana HopBF1 D170A (Flag HopBF1 DA) following depletion of CDC37 using siRNA. Immunoprecipitates and cell extracts were also analyzed for HSP90 and CDC37.
(D) Spot assay depicting the growth of S. cerevisiae expressing Flag-tagged E. americana HopBF1, the inactive D170A mutant or mutations within the αC-β4 loop. The primary and secondary structure of the αC-β4 loop is shown above and the structure of this region is shown on right in ribbon representation highlighting the mutated residues.
(E) Protein immunoblotting of anti-Flag immunoprecipitates from HEK293A cell extracts expressing Flag-tagged E. americana HopBF1 D170A (Flag-HopBF1 D170A) and the V89D; D170A mutant (Flag HopBF1 V89D; D170A). Cells were also pretreated with the proteasome inhibitor MG132 to prevent degradation of unfolded proteins.
(F) Photographs depicting leaves of N. benthamiana plants infiltrated with A. tumefaciens carrying WT HopBF1 or the indicated mutants V88D and D169A or YFP as a control. Photos were taken 7 days post infiltration.
In eukaryotes, HSP90 works together with the co-chaperone CDC37 to facilitate the maturation of client kinases (Karnitz and Felts, 2007; Pearl, 2005). To test whether HopBF1 requires CDC37, we analyzed anti-Flag immunoprecipitates from HEK293A cell extracts expressing Flag-tagged-GFP, v-src and HopBF1 D170A. As expected, protein immunoblotting of anti-Flag immunoprecipitates revealed that HSP90 and CDC37 interact with v-src; however, we failed to detect CDC37 in HopBF1 immunoprecipitates (Figure 6B). Furthermore, deletion of CDC37 using small interfering RNA (siRNA) abolished v-src interaction with HSP90, but had no effect on HopBF1/HSP90 binding (Figure 6C). Thus, HopBF1 does not require CDC37 to interact with HSP90.
The αC- β4 loop in client kinases is involved in kinase recognition by HSP90 via conserved hydrophobic residues flanked by a proline (Citri et al., 2006; Xu et al., 2005). Several HopBF1 homologs contain a G-F-P-V-V motif in the αC- β4 loop (Figure 6D). Therefore, we generated acidic mutations in this region of HopBF1 to screen for mutants that lost the ability to induce a growth defect in yeast. Expression of E. americana HopBF1 V89D displayed a minor growth phenotype compared to WT HopBF1 (Figures 6D and S6B).
Steady state kinetic analysis of P. syringae V88D (V89D in E. americana HopBF1) revealed an ~10-fold increase in the Km and a ~150-fold decrease in Vmax for S. cerevisiae HSP82 (Figure S6C). Despite more than a 1000-fold decrease in catalytic efficiency (Kcat/Km), P. syringae HopBF1 V88D appeared to be well-folded when heterologously expressed and purified from E. coli. When expressed in mammalian cells, E. americana HopBF1 V89D was undetectable by protein immunoblotting (Figure 6E; compare lanes 1 and 3). Improperly folded clients of HSP90 are targeted to the proteasome for degradation (Connell et al., 2001; Ehrlich et al., 2009; Murata et al., 2001; Neckers, 2002; Theodoraki and Caplan, 2012; Zhang et al., 2008). Indeed, proteasomal inhibition using MG132 restored E. americana HopBF1 V89D protein levels, indicating that the V89D mutant is targeted to the proteasome for degradation. Importantly, E. americana HopBF1 V89D was unable to interact with HSP90 in co-immunoprecipitation experiments (Figure 6E; compare lanes 2 and 4). Thus, the αC-β4 loop of HopBF1 is necessary for HSP90 recognition.
To determine the impact of HopBF1 V88D expression in planta, we transiently expressed P. syringae HopBF1 or mutants by agroinfiltration in N. benthamiana plants. We observed robust necrosis and tissue collapse in leaves expressing WT HopBF1, but not the HSP90 interacting mutant V88D or the catalytic D169A mutant (Figures 6F and S6D). Collectively, these data strongly suggest that HopBF1 is mimicking an HSP90 client.
Discussion
We have discovered that the uncharacterized HopBF1 family of T3SS effectors target HSP90 to dampen the host defense response. HopBF1 is specific for eukaryotic HSP90, despite the fact that the target Ser is strictly conserved in prokaryotic homologs (Figure 5D & 6D). This is a clever mechanism that appears to have evolved to prevent phosphorylation and inactivation of the pathogen’s own HSP90.
HSP90 lies at the hub of plant and animal immunity and our results describe the first pathogenic mechanism to directly inactivate the chaperone. In animals, HSP90 binds the cochaperone SGT1 and this complex is essential for the immune response initiated by the NB-LRR immune sensors NOD½ and NLRP3 (da Silva Correia et al., 2007; Mayor et al., 2007). Likewise, plant HSP90 and SGT1 are also required for proper activation of NB-LRR proteins; however, unlike animal HSP90/SGT1, the plant complex also requires the CHORD domain containing protein RAR1 (Azevedo et al., 2002; Hubert et al., 2003; Shirasu, 2009; Takahashi et al., 2003). Plant HSP90 binds several NB-LRR proteins including RPM1 (Hubert et al., 2003), N (Liu et al., 2004), MLA1 and MLA6 (Bieri et al., 2004), and is required for NB-LRR function. For example, knockout or silencing of RAR1, SGT1, or HSP90 compromises resistance against various pathogens (Austin et al., 2002; Azevedo et al., 2002; Kadota and Shirasu, 2012; Shirasu et al., 1999; Takahashi et al., 2003).
In addition to dampening the host defense response by interfering with NB-LRR function, HopBF1 may also contribute to the necrotic stage of the P. syringae lifestyle. Unlike biotrophic and necrotrophic pathogens, which obtain nutrients from living and dead tissue, respectively, P. syringae is hemibiotrophic (Xin and He, 2013). During a successful disease cycle in a susceptible host, P. syringae multiplies aggressively but does not cause host cell death in the initial phase of infection. However, infected tissues show extensive necrosis during the late stages of infection (Xin et al., 2018). Notably, both agroinfiltration and natural delivery of HopBF1 during infection caused profound tissue collapse and necrosis in N. benthamiana, N. tabacum and A. thaliana plants (Figure 2). Thus, HopBF1 may have at least two roles during infection: 1) dampen ETI by interfering with NB-LRR activation and 2) inducing host cell death during the late stage of infection. Future work will be required to understand the temporal regulation of HopBF1 expression during P. syringae infection.
Recently, Harrison and coworkers (Hulin et al., 2018) studied a large set of P. syringae strains aiming to elucidate molecular features associated with pathogenicity on cherry (Prunus avium). Interestingly, they concluded that HopBF1 is the effector most significantly associated with pathogenicity on this host. Although only the HopBF1 proteins from P. syringae have been characterized to date as type III effectors, genomic neighborhoods of more distant HopBF1 family members suggest these proteins are delivered to target cells by diverse secretion systems. For example, the many Burkholderia HopBF1 genes are located in clusters of T3SS genes. Some other gammaproteobacterial HopBF1 proteins are found within clusters of Type VI secretion system genes while an actinobacterial HopBF1 homolog is next to a Type VII secretion system gene cluster. These observations suggest that the HopBF1 minimal kinase domain is a conserved and versatile molecular machine, delivered in very different ways to target cells.
Some homologs of the HopBF1 kinase are found in a remarkably diverse set of proteins, some of which contain multiple structural domains (Figure S1A). Several of these long HopBF1 homologs contain a domain structure reminiscent of the polymorphic toxins used by bacteria against hosts or during inter-bacterial conflicts (Zhang et al., 2012). Polymorphic toxins contain several ordered functional segments including: structural repeats for adhesion and/or presentation on target cells, a releasing protease domain, and a toxin domain. Numerous HopBF1 homologs, contain filamentous hemagglutinin and RHS (Recombination Hot Spot) repeats, and cysteine- and metallo-protease domains. A few of these long HopBF1 homologs contain two or three HopBF1-type kinase domains. The similarity of HopBF1 homologs to polymorphic toxins suggests that the HopBF1 kinase domain may also function as a toxin.
Structure homology searches using the DALI server (Holm and Rosenstrom, 2010), indicate that E. americana HopBF1 is similar to the Shigella flexneri effector OspG (Pruneda et al., 2014) and the Enterohemorrhagic Escherichia coli (EHEC) effector NleH1 (Grishin et al., 2014). However, OspG and NleH1 are not recognized by standard BLAST sequence analysis when using HopBF1 as a query in the search. Nevertheless, all three effectors adopt a similar three-dimensional fold. OspG and NleH1 are capable of undergoing autophosphorylation (Gao et al., 2009; Kim et al., 2005; Zhou et al., 2013); though their substrates have yet to be identified. OspG binds ubiquitin and ubiquitin loaded E2 enzymes, attenuates host NF-κβ signaling and negatively regulates the host immune response stimulated by S. flexneri, the causative agent of shigellosis in humans (Kim et al., 2005; Pruneda et al., 2014). Likewise, NleH1 also regulates host NF-κβ signaling and promotes bacterial colonization and diarrhea in piglets infected with the foodborne pathogen E. coli strain O157:H7 (Wan et al., 2011). Despite sharing similar structural folds to HopBF1, OspG and NleH1 do not phosphorylate HSP90 in vitro (not shown). Nonetheless, it appears as though these kinases diverged early in the evolution of the protein kinase superfamily and may possess the minimal architecture needed to catalyze kinase-dependent protein phosphorylation.
In canonical kinases, specificity is commonly achieved by the primary amino acid sequence of the substrate and interactions that occur within the C-lobe of the kinase domain (Miller and Turk, 2018). HopBF1 lacks the C-lobe GHI helical subdomain, yet is highly specific for eukaryotic HSP90, suggesting a unique mode of recognition. In fact, we were unable to phosphorylate peptides derived from the sequence surrounding the phospho-acceptor site of HSP90. HSP90 is not considered to be a general chaperone like the HSP70 family. Instead, HSP90 preferentially facilitates the folding of client proteins involved in signal transduction, most notably protein kinases (Taipale et al., 2012). Because of HSP90’s unique role in facilitating the maturation of protein kinases, HopBF1 appears to exploit the chaperone’s affinity for kinases to achieve specificity.
The transient nature of kinase-substrate interactions (de Oliveira et al., 2016), often prevents identification of protein kinase substrates by co-immunoprecipitation experiments. The ~1:1 stoichiometry of interaction between HopBF1 and HSP90 suggests that binding may not be a typical kinase-substrate interaction, but rather a chaperone-client interaction, or a combination of both. Notably, treatment of mammalian cells with geldanamycin significantly reduced HopBF1 levels, suggesting that HopBF1 is an HSP90 client (Figure 6A). HSP90 generally works with the co-chaperone CDC37 to aid the maturation of client kinases; (Hunter and Poon, 1997) (Verba and Agard, 2017); however, the HSP90 kinase clients FLT4 and STK11 do not require, or only weakly require, CDC37 for HSP90 interaction (Taipale et al., 2012).
Likewise, HopBF1 does not require CDC37 for its interaction with HSP90 (Figure 6B and 6C). Cryo EM structures of the HSP90/CDC37/CDK4 ternary complex indicate that CDC37 serves to help unwind client kinases, separating the N and C-lobes and stabilizing a partially unfolded and inactive state (Verba and Agard, 2017; Verba et al., 2016). Therefore, it appears that HopBF1 is not unfolded by HSP90 in a typical client:kinase interaction. This would explain why upon HSP90 binding, HopBF1 is activated and capable of phosphorylating HSP90.
HSP90 recognizes many client kinases via hydrophobic residues within the α-C-β−4 loop region of the kinase domain. Sequence alignments of HopBF1 homologs revealed conservation of aliphatic residues within the α-C-β−4 loop (Figure 6D). A single mutation in this region drastically lowered HopBF1 activity and, importantly, prevented interaction with HSP90 in cells. These results suggest that HSP90 recognizes HopBF1 via a prototypical chaperone-kinase surface and that HopBF1 has evolved to mimic a host kinase client to specifically target and inactivate HSP90.
In summary, we have identified a bacterial kinase family that catalytically targets and inactivates eukaryotic HSP90 chaperones. HSP90 is an essential component of both plant and animal immunity and facilitates the folding of innate immune receptors and many protein kinases. Therefore, we propose that HopBF1 is mimicking an HSP90 client, or being recognized as a “pseudoclient”, which results in the phosphorylation and inactivation of the chaperone in a “betrayal” mechanism. Our results highlight a novel mechanism of host-pathogen interaction and reiterate the importance of HSP90 in immunity.
STAR Methods Text
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vincent S. Tagliabracci (vincent.tagliabracci@utsouthwestern.edu). Plasmids, primers, recombinant protein, experimental strains and any other research reagents generated by the authors will be distributed upon request to other research investigators under a Material Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
DH5α chemically competent cells were used for molecular cloning. Rosetta (DE3) chemically competent cells were used for protein expression. Both of them were grown in LB medium cultured at 37°C. After induction, Ros etta (DE3) cells were transferred to room temperature. S. cerevisiae (BY4741) were grown in YPD or synthetic media. Y. pestis (YP37) was grown in LB medium cultured at room temperature. YP37 cells were transferred to 37°C for induction of the secretion system. HeLa and HEK293A cells were grown on Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum and 1% Penicillin/Streptomycin in a 37°C incubator under 5% CO 2, unless otherwise stated. Agrobacterium tumefaciens GV3101 used for transient Agrobacterium-mediated gene expression were grown in YEP medium cultured at 28°C . Pseudomonas syringae pv. tomato DC3000D28E or Pseudomonas syringae pv. syringae B728a were grown in LB medium cultured at 28°C. Nicotiana benthamiana or Nicotiana tabacum cv. Xanthi-nc were grown in soil under controlled environmental conditions (21°C, 16 hours of light, 8 hours of dar k). Arabidopsis thaliana (Col-0) plants were grown in Jiffy7 pots in controlled-environment chambers (Percival Scientific, Iowa, USA) at 22°C, 40% humidity, under 8 hours of light, 16 hours of dark conditions.
METHOD DETAILS
Identification of HopBF1 by bioinformatics
Similarity of HopBF1 to kinases was recognized by analysing a collection of gammaproteobacterial effector protein sequences using the FFAS system for remote sequence similarity recognition (Xu et al., 2014) and verified using HHpred and Phyre2 servers for protein structure prediction (Kelley et al., 2015; Zimmermann et al., 2018). Significance of similarity was assessed using standard significance thresholds specific for each method.
An exhaustive search for homologues of the HopBF1-like kinase domain was performed using Jackhmmer (Finn et al., 2015). Sequences were aligned by MAFFT (Katoh and Standley, 2013) and the alignment curated manually. Sequence logos were created with Weblogo (Crooks et al., 2004).
Phylogenetic trees were built for selected HopBF1-like kinase domains after removing redundancy at the 95% sequence identity level, using the PhyML Maximum Likelihood method (Dereeper et al., 2008) with an Approximate Likelihood Ratio Test for branch support, and visualized with iToL (Letunic and Bork, 2016).
Genomic neighborhoods were investigated using the Integrated Microbial Genomes system (Chen et al., 2017).
Generation of plasmids and strains
The P. syringae HopBF1 and htpg coding sequences were amplified by PCR using P. syringae pv. syringae FF5 genomic DNA (gDNA) as a template. The Ewingella americana, and Serratia rubidaea, Burkholderia sp. B26, and Burkholderia sp. MSMB1072 coding sequences were synthesized as gBlocks (Integrative DNA Technologies) and used as a template in PCR reactions. Coding sequences for E. coli htpg (BL21), S. cerevisiae (BY4741) hsc82 and hsp82, ssa1, ssa2. ssa3, ssc1 and kar2, were all amplified from gDNA. The human HSP90β coding sequence was amplified from the Ultimate™ ORF Lite human cDNA collection (Life Technologies). N. benthamiana and Triticum aestivum HSP90 coding sequences were amplified from cDNA.
HopBF1 coding sequences were cloned into a modified pET28a bacterial expression plasmid containing an N-terminal 6XHis-Sumo tag (ppSumo) or pMMB67HE (a gift from the Orth Lab). Single amino acid mutations were introduced via Quick Change site directed mutagenesis. E. coli htpg, S. cerevisiae hsc82, hsp82, ssa1, ssa2, ssa3, ssc1, kar2, N. benthamiana HSP90 and T. aestivum HSP90 were cloned into pET28a bacterial expression vector. HopBF1 coding sequences were also cloned into a galactose-driven yeast expression vector pESC-Leu (Agilent Technologies) to generate Myc and Flag-tagged HopBF1. Similarly, the S. cerevisiae hsp82 coding sequence was amplified with an N-terminal Flag-tag and was cloned into the p423-GPD vector (Mumberg et al., 1995) for overexpression in yeast. Yeast expression plasmids were introduced into BY4741 via lithium acetate-mediated transformation and were selected on agar plates with the appropriate auxotrophic medium. Single colonies were picked and expression was verified by SDS PAGE and immunoblotting of yeast protein lysates.
To test the viability of HSP90 mutations of Ser 99, we generated plasmid shuffling strains following a previously described strategy (Nathan and Lindquist, 1995). We obtained an Hsc82 knockout yeast strain (ΔHSC82::KANMX) derived from BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0), and the pRS315 and pRS316 yeast vectors as kind gifts from Amanda Casey and Kim Orth. The Hsp82 coding sequence (including 1000 bp upstream of the initiator ATG) was cloned into pRS316 and pRS315 from yeast genomic DNA. Further, an S99E mutant was generated from the WT Hsp82 pRS315 vector using site-directed mutagenesis. The Hsp82 pRS316 plasmid was then introduced into the ΔHSC82::KANMX strain via lithium acetate-mediated transformation and single colonies were selected using SD agar without uracil. Subsequently, the S. cerevisiae HSP82 gene was replaced with a hygromycin cassette using a PCR based modification (Longtine et al., 1998). Briefly, a hygromycin resistance cassette with homologous sequences flanking the C-terminus of the HSP82 gene was amplified by PCR. The product of the reaction was then introduced by lithium acetate-mediated transformation and recovered overnight in 2 mL of SD media without uracil. Recombinants were selected by plating 100 μL of the transformation on SD agar without uracil supplemented with 200 μg/mL hygromycin. Successful recombination was verified by colony PCR using primers flanking the endogenous locus and the inserted cassette. Lastly, the pRS315 empty vector, WT Hsp82 pRS315, and S99E Hsp82 pRS315 were introduced to the double knockout strain by lithium acetate mediated transformation and single colonies were selected using SD agar without uracil and leucine.
For mammalian cell expression, the WT and mutant HopBF1 coding sequences were cloned into the pFLAG-CMV vector with N-terminal Flag tags. The Rous sarcoma virus v-src coding sequence was amplified from vectors received as a generous gift from Jenna Jewell and cloned into a pcDNA based vector (pCCF) with an N-terminal Flag tag. The EGFP coding sequence was amplified from pEGFP-C1 and cloned into the pFLAG-CMV vector with an N-terminal Flag tag. A vector coding for Flag-NLRP3 was received as a kind gift from Dr. James Chen. The Hsp90β coding sequence was amplified and cloned into the pCDNA 4.0 vector with an N-terminal V5 tag. All amino acid mutations were generated via site-directed mutagenesis.
Sequences encoding HopBF1 (KEZ70135.1) from Pseudomonas syringae pv. syringae FF5 or full-length complementary DNA (cDNA) encoding N. benthamiana Hsp90 (AY368905) were PCR amplified and cloned into pENTR/D-TOPO vector. P. syringae HopBF1 and N. benthamiana Hsp90 variants were generated by site-directed mutagenesis. To create constructs expressing HopBF1 variants fused to 2xSTREP-FLAG or Hsp90 protein variants N-terminally tagged with 10xMyc, the entry clones were LR recombined with appropriate Gateway destination vectors from the pGWB series (Golisz et al., 2013; Nakagawa et al., 2007).
To express HopBF1 variants in P. syringae, the sequences were PCR amplified with primers adding BglII and XhoI restriction sites to the opposite ends of the products and cloned into pJET 1.2 (Thermo Fisher Scientific). Next, the sequences were cut with the restriction enzymes and cloned under the control of Tac promoter in pBBR1MCS2-pTac, the modified broad-host-range vector pBBR1MCS-2 (Giska et al., 2013). The constructs were electroporated into P. syringae pv. tomato DC3000D28E or Pseudomonas syringae pv. syringae B728a.
To generate the Yersinia YP37 strains expressing HopBF1, triparental matings were performed between an E. coli strain (DH5α) harboring pMMB67HE vectors, an E. coli strain (DH5α) containing the pRK2013 plasmid (a kind gift from Dr. Kim Orth), and YP37 at room temperature overnight on LB agar. Transconjugants were selected on LB agar supplemented with chloramphenicol, kanamycin and ampicillin.
Protein purification
Rosetta (BL21 derivative) cells were transformed with ppSumo-HopBF1 plasmid and grown at 37°C in LB media under kanamycin selection until the OD600 reached approximately 0.6 – 1.0 absorbance units. Cultures were induced with final concentrations of 0.4μM IPTG and incubated with shaking overnight at room temperature.
The following buffers were used for Ni-NTA purification of HopBF1: Resuspension Buffer: 50mM Tris-HCl (pH 7.5), 300mM NaCl, 1mM DTT, 1mM PMSF. Wash Buffer: 50mM Tris-HCl (pH 7.5), 300mM NaCl, 1mM DTT, 20mM Imidazole (pH 8). Elution Buffer: 50mM Tris-HCl (pH 7.5), 300mM NaCl, 1mM DTT, 300mM Imidazole (pH 8). For purification of HSP90 and homologs, 1mM EDTA was added to all buffers.
Overnight cultures were pelleted by centrifugation at 5000 × g for 15 minutes at 4°C. Cells were re-suspended in 50mL of the appropriate resuspension buffer. The solution was split into two 25mL fractions and sonicated individually on ice. The sonication protocol uses 15 second pulses with 30 second rest times for a total “on” process time of 2 minutes and 30 seconds. The resulting lysate was spun at 20,000 × g for 30 minutes at 4°C. The supernatant was collected and incubated with 1mL of packed Ni-NTA resin (Bio-Rad His-Pur 88222) (washed with 1× TBS) at 4°C with nutating for 30 min – 1hour. The beads were then washed with 25mL of the appropriate wash buffer. Finally, proteins were eluted using 10mL of corresponding elution buffer.
λ-phosphatase and ULP treatment
The elution was warmed to room temperature and treated with 1 μL of λ-phosphatase (400,000 Units/mL New England Biolabs P0753S) and 1mM MnCl2 for 1–2 hours at room temperature. In some cases, the elution’s were treated with 20μL of 2mg/mL 6X His ULP-protease (purified as above) at 4°C overnight to remove the 6x-His-Sumo tag.
MonoQ anion exchange
Ni-NTA purified proteins were buffer exchanged to an anion exchange buffer containing 50mM Tris-HCl (pH 7.5), 50mM NaCl, and 1mM DTT. The protein was then subjected to anion exchange chromatography on a MonoQ column. Initially, the column was equilibrated with Buffer A1 containing 50mM Tris-HCl (pH 7.5) and 1mM DTT. Finally, the protein was eluted using Buffer B1 containing 50mM Tris-HCl (pH 7.5), 1M NaCl, and 1mM DTT. The gradient target concentration was 50% Buffer B1 and fraction sizes were set to 500 μL for optimal separation. Fractions corresponding to HopBF1 were collected and concentrated to 2 mL for use in the Superdex 200 gel filtration column.
Gel filtration
The concentrated proteins were loaded onto a Superdex 200 gel filtration column. The column was pre-equilibrated with a buffer containing 50mM Tris-HCl (pH 7.5), 150mM NaCl, and 1mM DTT. Fraction sizes were 1.5mL. Fractions containing the protein were concentrated to 2mg/mL and frozen in liquid N2 to be used in enzyme assays. For structural studies, E. americana HopBF1 was buffer exchanged and concentrated to 11mg/mL (see below).
Production of E. americana HopBF1 anti-sera
E. americana HopBF1 was purified as a 6X-His-Sumo fusion protein as above. The protein was used to inoculate rabbits for generation of rabbit anti-E. americana HopBF1 anti-serum (Cocalico Biologicals).
In vitro kinase assays
Kinase reactions were carried out in a buffer solution containing 50mM Tris-HCl (pH 7.5), 1mM DTT, 5mM MnCl2, 100μM [γ−32P]ATP, specific activity ~1000cpm/pmol. For standard reactions, 4μg of substrate was included with 26.7ng of HopBF1 in a final volume of 20μL. Reactions were incubated at room temperature for 5–15 minutes and stopped with 3μL of 500mM EDTA followed by the addition of 6μL of 5× SDS-loading dye containing 5% β-mercaptoethanol. Reaction products were loaded onto a poly-acrylamide gel for electrophoresis and visualized by Coomassie staining and autoradiography. Assays were typically performed a minimum of three times.
Kinase assays in yeast extracts were carried out in a similar manner with 40μg total yeast extract and 1μM staurosporine included. Proteins were resolved by SDS-PAGE and either stained with Coomassie or transferred to a nitrocellulose membrane and analyzed for HopBF1 by protein immunoblotting using anti-HopBF1 anti serum.
Kinetic analysis
For kinetic analysis, S. cerevisiae HSP82 concentrations were varied as indicated and incubated with 60nM P. syringae HopBF1 in a final volume of 20μL in a buffer containing 50mM Tris-HCl (pH 7.5), 1mM DTT, 5mM MnCl2, 100μM [γ−32P]ATP, specific activity ~5000cpm/pmol. Reactions were incubated at room temperature for 15 minutes and terminated with 3μL of 500mM EDTA followed by the addition of 6μL of 5× SDS-loading buffer containing 5% β-mercaptoethanol. Reaction products were resolved by SDS-PAGE and visualized by Coomassie staining. Radioactive gel bands corresponding to HSP82 were excised and incorporated radioactivity was quantified by scintillation counting (Budget-solve 111167; Beckmann scintillation counter). Assays were performed in technical duplicates in three independent experiments.
Crystallization and X-ray data collection
Recombinant E. americana HopBF1 was purified as described above and concentrated to 11 mg/mL in 10mM Tris-HCl pH = 7.5, 50mM NaCl, 1mM TCEP. Native apo crystals of HopBF1 were grown at 4°C by the hanging drop vap or diffusion method using a 1:1 ratio of protein/reservoir solution containing 1.4M ammonium sulfate, 100mM sodium citrate (pH 5.6) and 200mM sodium potassium tartrate, and were flash-frozen in 1.4M ammonium sulfate, 100mM sodium citrate (pH 5.6), 200mM sodium potassium tartrate, 50mM NaCl, and 25% ethylene glycol. Apo HopBF1 crystals exhibited the symmetry of space group R32 with cell dimensions of a = 134.95 Å, c = 124.49 Å, contained one molecule per asymmetric unit, and diffracted to a minimum Bragg spacing (dmin) of 2.50 Å when exposed to synchrotron radiation.
To obtain a heavy atom derivative, crystals were soaked in 1.4M ammonium sulfate, 100mM sodium citrate (pH 5.6), 200mM sodium potassium tartrate, 50mM NaCl and 10mM 2-chloromercuri-4-nitrophenol for 15 minutes, and were back-soaked by transferring to 1.4 M ammonium sulfate, 100mM sodium citrate (pH 5.6), 200mM sodium potassium tartrate, 50mM NaCl and 25% ethylene glycol. Nucleotide bound crystals were obtained by incubating the protein with 1mM AMPNP and 5mM MnCl2 prior to crystallization screening. Nucleotide bound crystals were grown at 4°C by the hanging drop vapor diffusion method using a 1:1 ratio of protein/reservoir solution containing 200 mM sodium citrate (pH 5.6) and 19% (w/v) PEG3000. Crystals were flash frozen in 22% (w/v) PEG3000, 200mM sodium citrate (pH 5.6) and 25% ethylene glycol. Nucleotide bound HopBF1 crystals exhibited the symmetry of space group R32 with cell dimensions of a = 76.19 Å, c = 351.36 Å, contained two molecules per asymmetric unit, and diffracted to a minimum Bragg spacing (dmin) of 1.90 Å when exposed to synchrotron radiation.
Diffraction data were collected at 100 K at the Advanced Photon Source beamline 19-ID. Native data for the nucleotide bound HopBF1 were collected near the Se K-edge, and native apo and mercury-derivative data were collected at the Hg K-edge. Data were indexed, integrated, and scaled using the HKL-3000 program package (Minor et al., 2006). Analysis of the self-Patterson function calculated with the nucleotide bound HopBF1 data revealed a significant off-origin peak at approximately (1/3, 2/3, 1/6) and 55% the height of the origin peak, indicating pseudo-translational symmetry. Data collection statistics are provided in Table S1.
Phase determination and structure refinement
Phases for the apo HopBF1 were obtained from a single-wavelength anomalous dispersion experiment using a mercury derivatized protein crystal with data collected at the mercury K-edge to a dmin of 2.90 Å. Two mercury sites were located using the program SHELXD (Schneider and Sheldrick, 2002) and phases were refined with the program Mlphare (Otwinowski, 1991), resulting in an over-all figure-of-merit of 0.19 for data between 42.65 and 2.90 Å. Phases were further improved by density modification in the program Parrot (Cowtan, 2010) resulting in a figure-of-merit of 0.61. An initial model containing 79% of all HopBF1 residues was automatically generated in the program Buccaneer (Cowtan, 2006).
As the mercury derivatized and native apo crystals were isomorphous, all further calculations for the apo structure were performed versus the native data. Additional residues for HopBF1 were manually modeled in the program Coot (Emsley et al., 2010). Positional and isotropic atomic displacement parameter (ADP) as well as TLS ADP refinement was performed to a resolution of 2.47 Å using the program Phenix (Afonine et al., 2010) with a random 10% of all data set aside for an Rfree calculation. The current model contains one HopBF1 monomer; included are residues 25 – 200 and 25 water molecules. The Rwork is 0.206, and the Rfree is 0.221. A Ramachandran plot generated with Molprobity (Chen et al., 2010) indicated that 95.4% of all protein residues are in the most favored regions and 0.6% (one residue) in disallowed regions.
Phases for the nucleotide bound HopBF1 were obtained by the molecular replacement method in the program Phaser (McCoy et al., 2007) using the coordinates for the native HopBF1 monomer. Model building and refinement were performed to a resolution of 1.90 Å using a similar protocol to the native structure. Two HopBF1 molecules were located in the asymmetric unit, two molecules of AMP-PNP and 232 waters. The Rwork is 0.208, and the Rfree is 0.240; the presence of pseudo-translational symmetry in this lattice is likely the cause of the higher than expected Rfree for this model. A Ramachandran plot generated with Molprobity (Chen et al., 2010) indicates that 98.6% of all protein residues are in the most favored regions and none in disallowed regions. Phasing and model refinement statistics for all structures are provided in Table S1.
Growth inhibition assays
Yeast cultures were grown overnight in the appropriate SD dropout media supplemented with 2% glucose for plasmid maintenance. The following day, cultures were normalized to 1 OD600 in sterile PBS and diluted serially to 10−3 OD600. Ten μL of each dilution were then plated on the appropriate SD dropout agar using either 2% glucose or 2% galactose and 1% raffinose. Spotted plates were then grown in a 30°C incubator for 2–5 days.
To verify protein expression, yeast cultures were grown overnight in the appropriate SD dropout media supplemented with 2% glucose. The overnight cultures were then diluted in the appropriate SD dropout media with 2% galactose and 1% raffinose. The induced and uninduced media (1 OD600) were pelleted and resuspended in yeast lysis buffer (4% 5N NaOH and 0.5% β-mercaptoethanol). The cells were incubated for 30 minutes on ice and then neutralized with 5N HCl. SDS loading buffer containing 1% β-mercaptoethanol was added to the samples, boiled, and resolved by SDS-PAGE. Proteins were then transferred to a nitrocellulose membrane, and immunoblotted with anti-Flag M2 (F3165, Sigma), anti-GAPDH (MA5–15738 Thermo Fisher Scientific), anti-c-Myc 9E10 (sc-40, Santa Cruz Biotechnology), and anti-HSP90 (sc-13119, Santa Cruz Biotechnologies). Results shown are representative of at least three independent experiments.
Plasmid shuffling experiments of HSP90 Ser99 mutants
Yeast cultures were streaked from glycerol stocks onto leucine and uracil SD dropout agar and grown for 2–3 days at 30°C. Plates were t hen re-streaked onto SD agar without leucine and uracil and agar supplemented with or without 5-fluororotic acid (1mg/mL). 5-FOA produces a toxic by-product and is used to negatively select against yeast containing URA vectors. WT genes were encoded on URA vectors and mutations tested were placed on a separate (non-URA) vector. As a result, lethal mutations were unable to support growth. The plates were allowed to grow for 2–3 days at 30°C. Results shown are representative of at least three independent experiments.
Immunoprecipitation of HopBF1
BY4741 was transformed with either pESC-Leu empty plasmid or pESC-Leu P. syringae D169A Flag-HopBF1. Cells were grown overnight in SD media without leucine containing 2% glucose. To induce protein expression, cells were washed three times with PBS and diluted into SD containing 2% galactose and 1% raffinose for overnight induction. Cells were pelleted the following morning and resuspended in approximately an equal volume of ice-cold yeast immunoprecipitation buffer (50mM Na-HEPES, 200mM NaOAc, 1mM EDTA, 1mM EGTA, 5mM MgOAc, 5% glycerol, 0.25% NP-40, 3mM DTT, 1mM PMSF, Roche protease inhibitor cocktail, and pH=7.5). Cleared cell lysates were obtained by glass bead beating followed by two subsequent spins at 3000 × g (2 min. at 4°C) and 20,000 × g (10 min. at 4°C). Lysates were then incubated on an orbital shaker for 3 hours at 4°C with anti-Flag M2 agarose affinity resin (Sigma A2220) that had been pre-blocked with 1% BSA in immunoprecipitation buffer. After 3 hours, the agarose was pelleted and washed four times with ice-cold yeast immunoprecipitation buffer without protease inhibitors. Immunoprecipitated proteins were then eluted using 3X-Flag peptide diluted in yeast immunoprecipitation buffer and SDS loading buffer containing 1% -mercaptoethanol was added to the samples and boiled. Samples were resolved by SDS-PAGE and analyzed by silver staining or immunoblotting with anti-Flag M2 (F3165, Sigma), anti-Hsc82/Hsp82 (a kind gift from Jill L. Johnson), and anti-GAPDH (MA5–15738 Thermo Fisher Scientific). Individual bands of silver stained-gel were cut for detection by mass spectrometry.
For immunoprecipitation from HEK293A cells, individual wells of a 6-well dish were transfected with pFlag-EGFP, pCCF-v-src, pFlag-E. americana (or mutants) plasmids using PolyJet transfection reagent (Signagen Laboratories) per manufacturer’s instructions. Fresh medium was added 5 hours after transfections. Cells were harvested the following day and lysed in ice-cold HENG buffer (50mM HEPES-KOH [pH 7.9], 150mM NaCl, 20mM Na2MoO4, 2mM EDTA, 5% glycerol, 1mM DTT, 0.5% Triton X-100) supplemented with a protease inhibitor cocktail (Roche). Whole cell lysates were then normalized by the total amount of protein. Anti-Flag M2 agarose affinity resin (Sigma) was added to the lysates and incubated for 3 hours at 4°C on an orbital shaker. The resin was pelleted by centrifugation and washed four times with ice-cold HENG buffer. Immunoprecipitated proteins were then eluted using 3X Flag peptide diluted in HENG buffer, filtered to remove contaminating resin (Millipore, Durapore-PVDF 22 μm; UFC30GV00 ) then concentrated (Millipore, Amicon Ultra-0.5mL 10Kda UFC501096). SDS loading buffer containing 1% β-mercaptoethanol was added to the samples and boiled. Samples were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with anti-Flag M2 (F3165, Sigma), anti-HSP90 (sc-13119, Santa Cruz Biotechnologies), anti-CDC37 (MA3–029, Thermo Fisher Scientific) and anti-GAPDH (CB1001, EMD Millipore) antibodies. Results shown are representative of at least three independent experiments.
HopBF1 inhibition of HSP90 chaperone activity in HEK293A cells
Individual wells of a 6 well-dish were transfected with pre-mixed pFlag (empty plasmid), pFlag-E. americana HopBF1 or the D170A E. americana HopBF1 mutant and pFlag-GFP, pCCF-v-src, or pcDNA-Flag-NLRP3 plasmids using PolyJet transfection reagent per manufacturer’s instructions. Fresh media with either 1μM geldanamycin (Fisher Scientific) or DMSO (as a vehicle control) was added 5 hours after transfection. Cells were harvested the following day and lysed in ice-cold lysis buffer (50mM Tris-HCl [pH 7.5], 150mM NaCl, 1mM EDTA, 1mM DTT, 1% Triton X-100) supplemented with a protease inhibitor cocktail (Roche). Lysates were cleared by two centrifugations at 3000 × g (2 min. at 4°C) and 20,000 × g (10 min. at 4°C). Cleared lysates were then diluted in SDS loading buffer containing 1% β-mercaptoethanol and boiled. Samples were resolved by SDS-PAGE, transferred to a nitrocellulose membrane and immunoblotted with anti-Flag M2 (F3165, Sigma), anti-pY 4G10 (a gift from the Dixon lab), anti-Tubulin (sc-9104, Santa Cruz Biotechnology) and anti-GAPDH (CB1001, EMD Millipore) antibodies. Results shown are representative of at least three independent experiments.
Client binding of S108E HSP90β in HEK293A cells
Individual wells of a 6 well-dish were transfected with pre-mixed pFlag-GFP, pCCF-v-src, pCCF-E. americana -HopBF1 D170A and pcDNA-V5-HSP90β or the S108E mutant plasmids using PolyJet transfection reagent per manufacturer’s instructions. Fresh media was added 5 hours after transfection. Cells were harvested the following day and lysed in ice-cold HENG buffer supplemented with a protease inhibitor cocktail (Roche). Whole cell lysates were then normalized by the total amount of protein. Anti-Flag M2 agarose affinity resin (Sigma) was added to the lysates and incubated for 3 hours at 4°C on an orbital shaker. The resin was pelleted by centrifugation and washed four times with ice-cold HENG buffer. Immunoprecipitated proteins were then eluted using 3X Flag peptide diluted in HENG buffer, filtered to remove contaminating resin (Millipore, Durapore-PVDF 22 μm; UFC30GV00) then concentrated (Millipore, Amicon Ultra-0.5mL 10Kda UFC501096). SDS loading buffer containing 1% β-mercaptoethanol was added to the samples and boiled. Samples were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with anti-Flag M2 (F3165, Sigma), anti-V5 (R960, Thermo Fisher Scientific), and anti-GAPDH (CB1001, EMD Millipore) antibodies. Results shown are representative of at least three independent experiments.
Depletion of CDC37 in HEK293A cells
Individual wells of a 6 well-dish were transfected with human CDC37 siRNA (L-003231–00-0005, Dharmacon) or a non-targeting control (D-001810–01-05, Dharmacon) using the Dharmafect1 transfection reagent (Dharmacon) per manufacturer’s instructions. Following 24 hours, cells were transfected with pFlag-GFP, pCCF-v-src, or Flag-E. americana HopBF1 plasmids using PolyJet transfection reagent. Fresh media was added 5 hours after transfections. Cells were harvested the following day and lysed in ice-cold HENG buffer supplemented with a protease inhibitor cocktail (Roche). Whole cell lysates were then normalized by the total amount of protein. Anti-Flag M2 agarose affinity resin (Sigma) was added to the lysates and incubated for 3 hours at 4°C on an orbital shaker. The resin was pelleted and washed four times with ice-cold HENG buffer. Immunoprecipitated proteins were then eluted using 3X Flag peptide diluted in HENG buffer, filtered to remove contaminating resin (Millipore, Durapore-PVDF 22 μm; UFC30GV00) then concentrated (Millipore, Amicon Ultra-0.5mL 10Kda UFC501096). SDS loading buffer containing 1% β-mercaptoethanol was added to the samples and boiled. Samples were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with anti-Flag M2 (F3165, Sigma), anti-HSP90 (sc-13119, Santa Cruz Biotechnologies), anti-CDC37 (MA3–029, Thermo Fisher Scientific) antibodies. Results shown are representative of three independent experiments.
Geldanamycin inhibition of HSP90 in HEK293A cells
Individual wells of a 6 well-dish were transfected with pFlag- E. americana HopBF1 (or indicated mutants), pFlag-GFP or pCCF-v-src plasmids using PolyJet transfection reagent per manufacturer’s instructions. Fresh media with either 1μM geldanamycin (Fisher Scientific) or DMSO (as a vehicle control) was added 5 hours after transfection. Cells were harvested the following day and lysed in ice-cold lysis buffer (50mM Tris-HCl [pH 7.5], 150mM NaCl, 1mM EDTA, 1mM DTT, 1% Triton X-100) supplemented with a protease inhibitor cocktail (Roche). Lysates were cleared by two subsequent centrifugations at 3000 × g (2 min. at 4°C) and 20, 000 × g (10 min. at 4°C). Cleared lysates were then diluted in SDS loading buffer containing 1% β-mercaptoethanol and boiled. Samples were resolved by SDS-PAGE, transferred to a nitrocellulose membrane and immunoblotted with anti-Flag M2 (F3165, Sigma), anti-Tubulin (sc-9104, Santa Cruz Biotechnology) and anti-GAPDH (CB1001, EMD Millipore) antibodies. Results shown are representative of at least three independent experiments.
Binding of the αC-β4 HopBF1 mutant in HEK293A cells
Individual wells of a 6 well-dish were transfected with pFlag- E. americana HopBF1 D170A or the V89D/D170A E. americana HopBF1 mutant plasmids using PolyJet transfection reagent per manufacturer’s instructions. Fresh media with either 10 μM MG-132 (Sigma) or DMSO (as a vehicle control) was added 5 hours after transfection. Cells were harvested the following day and lysed in ice-cold HENG buffer supplemented with a protease inhibitor cocktail (Roche). Whole cell lysates were then normalized by the total amount of protein. Anti-Flag M2 agarose affinity resin (Sigma) was added to the lysates and incubated for 3 hours at 4°C on an orbital shaker. The resin was pelleted and washed four times with ice-cold HENG buffer. Immunoprecipitated proteins were then eluted using 3X Flag peptide diluted in HENG buffer, filtered to remove contaminating resin (Millipore, Durapore-PVDF 22 μm; UFC30GV00 ) then concentrated (Millipore, Amicon Ultra-0.5mL 10Kda UFC501096). SDS loading buffer containing 1% β-mercaptoethanol was added to the samples and boiled. Samples were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with anti-Flag M2 (F3165, Sigma), anti-Tubulin (sc-9104, Santa Cruz Biotechnology), anti-GFP JL-8 (NC9777966, Thermo Fisher Scientific), and anti-HSP90 (sc-13119, Santa Cruz Biotechnologies) antibodies. Results shown are representative of at least three independent experiments.
Infection of mammalian cells with Y. pestis
Y. pestis (YP37) strains expressing pMMB67HE E. Americana HopBF1 or the D170A mutant were grown overnight in LB media at room temperature with the appropriate antibiotics. The next day, 800μL of the overnight cultures were added to 7 mL of LB media supplemented with 20mM sodium oxalate and 200μL MgCl2 for 1 hour at 37°C. After, IPTG was added to a final concentration of 0.4μM and incubated for 1 hour at 37°C. HeLa cells were washed three times with antibiotic-free DMEM supplemented with 10% FBS and infected at a multiplicity of infection of 10. Cells were incubated at 37°C for 2–3 hours and washed 3 times with 1X PBS. Cells were then lysed in 50mM Tris-HCl (pH 7.5), 150mM NaCl, 1mM EDTA, and 1% Triton X-100. Lysates were spun at 15,000 × g for 10min and supernatants were incubated with 200ng of anti-HSP90 antibody (sc-13119 Santa Cruz Biotechnology) overnight at 4°C. 20 μL of a 50% Protein A bead slurry (20333 Thermo Scientific) was added to the cleared lysates and incubated at 4°C for 1–2 hours. Beads were spun at 1000 × g for 1min and washed twice with 1mL of lysis buffer. Samples were diluted in SDS loading buffer with 1% β-mercaptoethanol and resolved by SDS-PAGE. Proteins were either stained with Coomassie or transferred to a nitrocellulose membrane and immunoblotted with anti-HSP90 (sc-13119, Santa Cruz Biotechnologies). Individual bands of Coomassie stained-gel were cut for detection by mass spectrometry. Results shown are representative of two independent experiments.
P. syringae secretion assays
P. syringae strains were incubated overnight at 28°C on a shake r in 5 mL of King’s broth media (KB) with the appropriate antibiotics. The following day, the bacterial cultures were diluted to OD600= 0.2 to and grown to early log-phase in 10 mL of KB (with the appropriate antibiotics for plasmid maintenance) at 28°C on a shaker. Samples were then pelleted and washed twice with PBS buffer. After washing, the cell pellets were resuspended in 10mL of minimal media supplemented with 10mM fructose. Cells were grown overnight at 24°C on a shaker. The next day, the bacterial cultures were pelleted by centrifugation. The supernatants were collected and filtered through 0.22 μm filters (SLGP033RS, Thermo Fisher Scientific) and 50μg of BSA (BP1600, Thermo Fisher Scientific) was added to facilitate protein precipitation. 0.5 OD600 of the cell pellets was resuspended in 100μL of 2X SDS loading buffer containing 1% β-mercaptoethanol and boiled. Proteins in the supernatants were precipitated for at least 1 hour at 4°C with the addition of Trichloroacetic acid (T0699, Millipore Sigma) to a final concentration of 20%. After, the precipitated proteins were pelleted by centrifugation at 20,000 × g for 15 min at 4°C then washed twice with 500μL of −20°C acetone (179124, Millipore Sigma). The washed pellets were resuspended in 100μL of 2X SDS loading buffer containing 1% β-mercaptoethanol and boiled. Proteins from both the cell pellets and supernatants were resolved by SDS-PAGE and analyzed by Ponceau-S (P3504, Millipore Sigma) staining and immunoblotting with anti-HopBF1, anti-GroEL (ADI-SPS-875-D Enzo Life Sciences), and anti-mCherry (ab125096 abcam) antibodies. Results shown are representative of two independent experiments.
Preparation of malachite green reagent
Malachite oxalate (13.5mg) was dissolved in 30mL of milli Q H2O followed by addition of 10mL of 4.2% (w/v) ammonium molybdate in 4M HCl as described (Tagliabracci et al., 2007). This solution was rotated on a nutator at 4°C for at least 45 minutes then filtered before use. The Malachite assay buffer consisted of 100mM Tris-HCl (pH 7.5), 6mM MgCl2, and 20mM KCl.
Malachite green assay
HSP90 activity was analyzed essentially as described (Rowlands et al., 2004). Briefly, 10μL of HSP90 (2mg/mL) and 2μL of ATP (100mM) was added to 188μL of Malachite assay buffer for a final concentration of 0.1mg/mL HSP90 and 1mM ATP. Reactions were incubated at 37°C for 1–3 hours. Reactions were stopped with 800μL of Malachite green reagent followed by addition of 100μL of 34% (w/v) sodium citrate. Resulting solutions were measured for absorbance at 620nm to monitor free phosphate release from ATP. Assays were performed in technical duplicates in three independent experiments.
Transient expression in plant cells
The appropriate constructs were electroporated into Agrobacterium tumefaciens (GV3101) cells. Subsequently, A. tumefaciens carrying the gene of interest on a binary vector were grown overnight in liquid medium (28°C at 225 rpm). The cells were harvested by centrifugation at 4000 × g for 7 minutes and resuspended in agroinfiltration buffer containing 10mM MES (pH 6.5), 10mM MgCl2 and 150μM acetosyringone (Romeis et al., 2001). The concentration of bacterial suspension was measured spectrophotometrically (OD600), adjusted to a final concentration (OD600 of 0.6 – 0.8), and incubated at room temperature for 2 – 3 hours. The bacterial suspension was then infiltrated using a needleless syringe, into leaves of N. benthamiana or N. tabacum cv. Xanthi-nc, that were grown in soil under controlled environmental conditions (21°C, 16 hours of light, 8 hours of dark). Results shown are representative of at least three independent experiments.
P. syringae inoculation
To assess HopBF1-dependent cell death, fully expanded leaves of A. thaliana (Col-0) or N. benthamiana were infiltrated with P. syringae pv. tomato DC3000D28E expressing WT HopBF1, the D169A mutant or mCherry as a control. Bacteria for inoculation were cultured overnight (18 h) on LB medium at 28°C with vigorous shaking. Following centrifugation at 3,500 × g for 10 minutes, the pellet was washed once and resuspended in sterile 10mM MgCl2. The bacterial suspension was adjusted to OD600 = 0.2 (that corresponds to approximately 108 colony forming units [cfu]/ml) (Giska et al., 2013). For avirulence assays, P. syringae pv. syringae B728a expressing WT HopBF1, kinase inactive variants or mCherry as a control were grown overnight in liquid LB medium (28°C at 200 rpm). Following centrifugation at 3,500 × g for 10 minutes, the pellet was washed once and resuspended in sterile milli-Q H2O and supplemented with Silwet L-77 (0.02%). N. benthamiana plants were dip inoculated by inverting whole plants into bacterial suspensions (culture density 106 [cfu]/ml) and gently agitated for 30 seconds. Following inoculation, plants were placed immediately under a plastic dome to maintain high humidity levels for 24 hours. Development of symptoms was assessed within 7 days. At the indicated time points, three 1 cm-diameter leaf disks were cut from infiltrated zones, surface-sterilized with 70% ethanol for 1 min, rinsed with sterile water for 1 min and ground in 300μl 10mM MgCl2. Serial dilutions were plated on LB agar plates for bacteria enumeration. Data are reported as means ± SD (Zembek et al., 2018). To analyse the effect of HSP90 on the HopBF1-dependent phenotype, cultures of Agrobacterium expressing HSP90 variants or YFP as a control were suspended in agroinfiltration buffer containing 10mM MES (pH 6.5), 10mM MgCl2 and 150μM acetosyringone and upon 2 hours incubation, a suspension of P. syringae pv. tomato DC3000D28E expressing HopBF1 or the D169A mutant was added to obtain a final OD600 = 0.7 and 0.2 for A. tumefaciens and P. syringae, respectively. The suspensions were then used to infiltrate fully expanded N. benthamiana leaves. To check whether HopBF1 is able to prevent autoactive RPM1-mediated HR response, A. tumefaciens expressing the autoactive variant of RPM1 (D505V) with or without HopBF1 or the D154A and D169A inactive mutants were co-infiltrated into fully expanded leaves of N. benthamiana plants. The final concentration of bacterial suspension was adjusted to OD600 = 0.7 for each strain. Results shown are representative of at least three independent experiments.
Quantification of disease symptoms in N. benthamiana and Arabidopsis plants
Images were analyzed with ImageJ software to manually measure the percentage of necrotic area per leaf. Necrotic areas were calculated with the following formula: [(Total leaf area − Healthy area)/Total leaf area)] × 100. The mean values of percentage-necrotic area for each sample were compared with the mCherry control sample. Analysis of variance was performed using the GraphPad Prism 8.1 statistics software. All experiments were conducted independently three times.
Mass spectrometric analysis of HopBF1 and HSP90
Myc-tagged N. benthamiana HSP90 was purified using Myc-Trap®_MA Chromotek beads according to manufacturer’s protocol with some modifications. N. benthamiana leaves were collected 48 h after agroinfiltration (ca. 2g), ground in liquid nitrogen and thawed in HENG buffer supplemented with protease (BioShop) and phosphatase (Sigma) inhibitor cocktails. Leaf extracts were incubated overnight with the beads at 4°C on a rotator. The samples were eluted with 2X Myc peptide, digested with Lys-C Protease and subjected to liquid chromatography-mass spectrometry analyses.
Recombinant HSP82 was incubated with ATP and either HopBF1 WT or HopBF1 D169A as described above, before being separated via SDS-PAGE and stained with Coomassie blue. Protein gel bands of interest were reduced with DTT and alkylated with iodoacetamide prior to overnight enzymatic digestion with trypsin at 37°C. Tryptic peptides were de-salted via solid phase extraction (SPE) prior to LC-MS/MS analysis. Experiments were performed on a Thermo Scientific EASY-nLC 1200 liquid chromatography system coupled to a Thermo Scientific Orbitrap Fusion Lumos mass spectrometer. MS1 spectra were acquired in the Orbitrap mass analyzer (resolution 120,000) and precursor ions were subjected to high-energy collision-induced dissociation (HCD) for fragmentation. MS2 spectra of fragment ions were collected in the ion trap. MS/MS spectral data were then searched using the Mascot search engine (Matrix Science) for peptide identification. Carbamidomethylation of cysteine residues (+57.021 Da) was set as a static modification, and oxidation of methionine (+15.995 Da) and phosphorylation of serine/tyrosine/threonine (+79.97 Da) were set as dynamic modifications. Precursor and product ion tolerances of 15 ppm and 0.6 Da, respectively, were used for all searches. The Ser99 phospho site on HSP82 was initially identified from the Mascot search and manually verified from the fragmentation spectra.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of HopBF1 and mutant kinase activity against HSP90 is represented as the mean of three independent experiments (n = 3). A student’s t-test was used to calculate a p value using GraphPad Prism 7. Error bars represent the standard error of the mean (SEM). Percentages of plant cell death were analyzed by one-way ANOVA analysis and Tukey’s multiple comparisons test using GraphPad Prism 8.
DATA AND CODE AVAILABILITY
The accession numbers for the coordinates and structure factors reported in this paper are PDB ID 6PWD for apo HopBF1, and PDB ID 6PWG for AMP-PNP bound HopBF1.
Table S1. Data collection and refinement statistics, E. americana HopBF1 structures. (Related to Figure 1)
ADDITIONAL RESOURCES
Table S2. List of Primers used in these studies (Related to Key Resource Table)
Supplementary Material
Figure S1. HopBF1-like kinase domains are present in a number of bacterial strains from diverse taxa and share sequence features typical of protein kinases (Related to Figure 1).
(A)Phylogenetic tree (Maximum Likelihood) for selected HopBF1 homologs built from a manually corrected MAFFT multiple sequence alignment of HopBF1 type kinase domains, collected using JackHMMer. Coloring is by taxonomy: Red, alphaproteobacteria; green, betaproteobacteria; yellow, gammaproteobacteria; cyan, Cyanobacteria; brown, Actinobacteria. Red squares denote proteins containing repeated (2 or 3) HopBF1 type kinase-like domains. Grey bars indicate protein sequence lengths (maximum: 5315 residues). For some branches, dominant genus shown (e.g. Pseudomonas). Black circles mark branches with bootstrap support better than 0.9.
(B, C)Sequence logos (Weblogos) for 161 HopBF1 homologs (B) and for 3,998 homologs of typical protein kinases (C) (Pfam domain PF00069). The two logos are aligned together by Mafft and filtered as to retain only columns matching human PKA sequence. Numbering in both logos corresponds to PKA. The height of the amino acid stack is proportional to the sequence conservation at that position.
(D) Superposition of the Apo and AMP-PNP bound HopBF1 structures. The Apo structure is in yellow and the AMP-PNP structure is in teal.
Figure S2. HopBF1 and its mutants are expressed in planta and secreted by. P syringae (Related to Figure 2).
(A-B) Protein immunoblots of leaf extracts showing HopBF1 levels in N. benthamiana (A) and N. tabacum (B). (C) Protein immunoblots of supernatants from DC300028E P. syringae depicting the levels of HopBF1 and mutants. (D-E). Quantification of tissue collapse and necrosis in N. benthamiana (D) and Arabidopsis (E) plants following P. syringae DC300028E infection with HopBF1, the inactive D154A, D169A mutants and mCherry. Bars represent the average percentage of necrotic leaf area with standard deviations.
Figure S3. HopBF1 is an HSP90 protein kinase (Related to Figure 3).
(A) Protein immunoblotting of yeast extracts depicting the levels of P. syringae Myc-tagged HopBF1 or the D154A and D169A mutants. GAPDH is shown as a loading control.
(B) Kinetic analysis depicting the concentration dependence of S. cerevisiae HSP82 on the rate of phosphate incorporation into S. cerevisiae HSP82 by E. americana HopBF1. (Inset) Km for S. cerevisiae HSP82 is indicated. Reaction products were resolved by SDS PAGE and radioactive gel bands were excised and 32P incorporation was quantified by scintillation counting.
(C) Incorporation of 32P from [γ32P]ATP into S. cerevisiae HSP90 (HSP82) by recombinant P. syringae HopBF1 or the Burkholderia orthologs. The reaction products were separated by SDS-PAGE and analyzed by Coomassie staining (upper) and autoradiography (lower).
(D) Incorporation of 32P from [γ32P]ATP into S. cerevisiae HSP90 (HSP82) or the ATPase inactive HSP82 D79N mutant by recombinant P. syringae HopBF1. HSP82 was also pretreated with geldanamycin. The reaction products were analyzed as in C.
Figure S4. HopBF1 phosphorylates HSP90 resulting in the inhibition of ATPase and chaperoning activity (Related to Figure 4).
(A) Protein immunoblotting of HEK293A cell extracts expressing Flag-GFP with E. americana HopBF1 or the D170A mutant. Cells were also pretreated with Geldanamycin. Levels of Flag-GFP and HopBF1 are also shown. GAPDH is included as a loading control. This is a control for the experiments present in Figures 4F and 4G.
(B) Protein immunoblotting of anti-Flag immunoprecipitates from HEK293A cells expressing Flag-GFP, Flag-v-src, Flag-E. americana HopBF1 D170A, V5-tagged human HSP90β or the S108E phosphomimetic HSP90β mutant. Extracts were also analyzed for expression. GAPDH is included as a loading control.
(C) Enlarged image of the HopBF1 nucleotide-binding pocket showing the molecular interactions involved in nucleotide binding. Active site residues are numbered and the PKA equivalent residues are shown in parentheses. The AMP-PNP molecule is also shown.
(D) Kinase activity of P. syringae HopBF1 and active site mutants using S. cerevisiae HSP90 (HSP82) as substrate. Reaction products were resolved by SDS PAGE and radioactive gel bands were excised and 32P incorporation was quantified by scintillation counting.
(E) Protein immunoblotting of yeast extracts depicting the levels of Flag-tagged HSP82 and P. syringae Myc-tagged HopBF1 or the E74A mutant before and after induction with galactose. GAPDH is shown as a loading control.
Figure S5. HopBF1 phosphorylates and inactivates HSP90 during infection (Related to Figure 5).
(A) Table depicting the percent of peptides modified on H. sapiens HSP90 Ser108 and N. benthamiana HSP90 Ser100. HeLa cells were infected with an effectorless Y. pestis strain expressing either E. americana HopBF1 or the D170A mutant. Endogenous HSP90 was immunoprecipitated (lower) and subjected to mass spectrometry analysis. P. syringae HopBF1 or the D169A mutant and HSP90 were also co-agroinfiltrated into N. benthamiana leaves. Myc-tagged HSP90 was immunoprecipitated and subjected to mass spectrometry (lower rows).
(B) Incorporation of 32P from [γ32P]ATP into N. benthamiana HSP90 or the S100A and S100F mutants by recombinant P. syringae HopBF1. Reaction products were separated by SDS-PAGE and analyzed by Coomassie staining (upper) and autoradiography (lower).
(C) Protein immunoblotting of N. benthamiana leaf extracts depicting the levels of Myc-tagged HSP90 and Flag-tagged YFP. Leaves were infiltrated with P. syringae DC3000D28E expressing HopBF1 and appropriate A. tumefaciens strain to transiently express HSP90, HSP90 S100F or YFP as a control. The Coomassie stained membrane is included as a loading control.
(D) Photographs depicting N. benthamiana leaves following infiltration with P. syringae DC3000D28E expressing HopBF1 D169A and appropriate A. tumefaciens strain to transiently express HSP90, HSP90 S100F or YFP as a control. In contrast to leaves infected with HopBF1 WT, no signs of tissue necrotization were observed. Photographs show representative leaves made 6 dpi. The experiment was performed 5 times with similar results.
(E) Whole plants were dip-inoculated with a P. syringae pv. syringae B728a ectopically producing WT HopBF1, kinase inactive variants or mCherry as a control. At the indicated time points, bacteria were isolated from the inoculated leaves, spread on plates and counted. All the strains evoked strong disease symptoms and the plants eventually died. The photographs showing representative plants were made 7 days post inoculation.
(F) Protein immunoblotting depicting the levels of mCherry, HopBF1 and HopBF1 mutants in the P. syringae B728A strain in the cell pellet (lower) or the supernatant (upper) following induction of the T3SS. GroEL and the Ponceau membrane are also shown.
Figure S6. HSP90 recognizes HopBF1 as a client via the αC-β4 loop (Related to Figure 6).
(A) Protein immunoblotting of HEK293A cell extracts depicting the levels of WT E. americana Flag-HopBF1 or the D170A mutant. Tubulin is shown as a loading control. Note that WT HopBF1 protein levels are lower than the mutant, indicating that HopBF1, which phosphorylates and inactivates HSP90, phenocopies geldanamycin treatment of the D170A mutant.
(B) Protein immunoblotting of yeast extracts depicting the levels of E. americana Flag-tagged HopBF1 or the indicated mutants. HSP82 is also shown.
(C) Kinetic analysis depicting the concentration dependence of S. cerevisiae HSP82 on the rate of phosphate incorporation into S. cerevisiae HSP82 by P. syringae HopBF1 (left) or the V88D mutant (right, note the difference in numbering due to the different species). The Km for S. cerevisiae HSP82 and Vmax are shown in the inset. Reaction products were resolved by SDS PAGE and radioactive gel bands were excised and 32P incorporation was quantified by scintillation counting. As judged by Kcat/Km (catalytic efficiency), WT HopBF1 is more than 1000-fold more efficient than the V88D mutant.
(D) Protein immunoblotting of N. benthamiana extracts depicting the levels of Flag-tagged HopBF1 or indicated mutants and Flag-tagged YFP. Leaves were infiltrated with A. tumefaciens carrying Flag-tagged HopBF1 or the mutants and photos were taken 7 days post infiltration.
Highlights.
The bacterial effector HopBF1 adopts a minimal protein kinase fold
HopBF1 phosphorylates and inactivates eukaryotic HSP90
HopBF1 mimics an HSP90 client to achieve specificity
HopBF1 is sufficient to induce disease symptoms in plants
Acknowledgments
We thank Drs. Kim Orth, Neal Alto, Melanie Cobb, Greg Taylor, Dor Salomon and members of the Tagliabracci laboratory for insightful discussions. We also thank Dr. Farid El Kasmi for providing RPM1 constructs. Results shown in this report are derived from work performed at the Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. The equipment used was sponsored in part by the Centre for Preclinical Research and Technology (CePT), a project co-sponsored by European Regional Development Fund and Innovative Economy, The National Cohesion Strategy of Poland. This work was supported by NIH Grants R00DK099254 (V.S.T.), T32DK007257-37 (A.S.) and T32GM008203-29 (V.A.L.), Welch Foundation Grants I-1911(V.S.T) and a CPRIT grant RP170674 (V.S.T) and the Polish National Science Centre grants 2014/15/B/NZ1/03559 (K.P.) and 2017/25/B/NZ1/01883 (M.K.). V.S.T. is the Michael L. Rosenberg Scholar in Medical Research, Cancer Prevention Research Institute of Texas Scholar (RR150033) and Searle Scholar.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Mustyakimov M, Grosse-Kunstleve RW, Moriarty NW, Langan P, and Adams PD (2010). Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 66, 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, and Pearl LH (2006). Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440, 1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alto NM, and Orth K (2012). Subversion of cell signaling by pathogens. Cold Spring Harb Perspect Biol 4, a006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J Mol Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Austin MJ, Muskett P, Kahn K, Feys BJ, Jones JD, and Parker JE (2002). Regulatory role of SGT1 in early R gene-mediated plant defenses. Science 295, 2077–2080. [DOI] [PubMed] [Google Scholar]
- Azevedo C, Sadanandom A, Kitagawa K, Freialdenhoven A, Shirasu K, and Schulze-Lefert P (2002). The RAR1 interactor SGT1, an essential component of R gene-triggered disease resistance. Science 295, 2073–2076. [DOI] [PubMed] [Google Scholar]
- Baltrus DA, Nishimura MT, Romanchuk A, Chang JH, Mukhtar MS, Cherkis K, Roach J, Grant SR, Jones CD, and Dangl JL (2011). Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog 7, e1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieri S, Mauch S, Shen QH, Peart J, Devoto A, Casais C, Ceron F, Schulze S, Steinbiss HH, Shirasu K, et al. (2004). RAR1 positively controls steady state levels of barley MLA resistance proteins and enables sufficient MLA6 accumulation for effective resistance. Plant Cell 16, 3480–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich KA, Farrelly FW, Finkelstein DB, Taulien J, and Lindquist S (1989). hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol Cell Biol 9, 3919–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Natale DA, Finn RD, Huang H, Zhang J, Wu CH, and Mazumder R (2011). Representative proteomes: a stable, scalable and unbiased proteome set for sequence analysis and functional annotation. PLoS One 6, e18910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen IA, Markowitz VM, Chu K, Palaniappan K, Szeto E, Pillay M, Ratner A, Huang J, Andersen E, Huntemann M, et al. (2017). IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res 45, D507–D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Harari D, Shohat G, Ramakrishnan P, Gan J, Lavi S, Eisenstein M, Kimchi A, Wallach D, Pietrokovski S, et al. (2006). Hsp90 recognizes a common surface on client kinases. J Biol Chem 281, 14361–14369. [DOI] [PubMed] [Google Scholar]
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, and Patterson C (2001). The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol 3, 93–96. [DOI] [PubMed] [Google Scholar]
- Cowtan K (2006). The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr 62, 1002–1011. [DOI] [PubMed] [Google Scholar]
- Cowtan K (2010). Recent developments in classical density modification. Acta Crystallogr D Biol Crystallogr 66, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Tsuda K, and Parker JE (2015). Effector-triggered immunity: from pathogen perception to robust defense. Annu Rev Plant Biol 66, 487–511. [DOI] [PubMed] [Google Scholar]
- Cunnac S, Chakravarthy S, Kvitko BH, Russell AB, Martin GB, and Collmer A (2011). Genetic disassembly and combinatorial reassembly identify a minimal functional repertoire of type III effectors in Pseudomonas syringae. Proc Natl Acad Sci U S A 108, 2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Correia J, Miranda Y, Leonard N, and Ulevitch R (2007). SGT1 is essential for Nod1 activation. Proc Natl Acad Sci U S A 104, 6764–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangl JL, Horvath DM, and Staskawicz BJ (2013). Pivoting the plant immune system from dissection to deployment. Science 341, 746–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangl JL, and McDowell JM (2006). Two modes of pathogen recognition by plants. Proc Natl Acad Sci U S A 103, 8575–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira PS, Ferraz FA, Pena DA, Pramio DT, Morais FA, and Schechtman D (2016). Revisiting protein kinase-substrate interactions: Toward therapeutic development. Sci Signal 9, re3. [DOI] [PubMed] [Google Scholar]
- Deng W, Marshall NC, Rowland JL, McCoy JM, Worrall LJ, Santos AS, Strynadka NCJ, and Finlay BB (2017). Assembly, structure, function and regulation of type III secretion systems. Nat Rev Microbiol 15, 323–337. [DOI] [PubMed] [Google Scholar]
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36, W465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich ES, Wang T, Luo K, Xiao Z, Niewiadomska AM, Martinez T, Xu W, Neckers L, and Yu XF (2009). Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc Natl Acad Sci U S A 106, 20330–20335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott JA, Noble ME, and Johnson LN (2012). The structural basis for control of eukaryotic protein kinases. Annu Rev Biochem 81, 587–613. [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, and Eddy SR (2015). HMMER web server: 2015 update. Nucleic Acids Res 43, W30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44, D279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Wan F, Mateo K, Callegari E, Wang D, Deng W, Puente J, Li F, Chaussee MS, Finlay BB, et al. (2009). Bacterial effector binding to ribosomal protein s3 subverts NF-kappaB function. PLoS Pathog 5, e1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Chung EH, Eitas TK, and Dangl JL (2011). Plant intracellular innate immune receptor Resistance to Pseudomonas syringae pv. maculicola 1 (RPM1) is activated at, and functions on, the plasma membrane. Proc Natl Acad Sci U S A 108, 7619–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, and Weissman JS (2003). Global analysis of protein expression in yeast. Nature 425, 737–741. [DOI] [PubMed] [Google Scholar]
- Giska F, Lichocka M, Piechocki M, Dadlez M, Schmelzer E, Hennig J, and Krzymowska M (2013). Phosphorylation of HopQ1, a type III effector from Pseudomonas syringae, creates a binding site for host 14–3-3 proteins. Plant Physiol 161, 2049–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golisz A, Sikorski PJ, Kruszka K, and Kufel J (2013). Arabidopsis thaliana LSM proteins function in mRNA splicing and degradation. Nucleic Acids Res 41, 6232–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin AM, Cherney M, Anderson DH, Phanse S, Babu M, and Cygler M (2014). NleH defines a new family of bacterial effector kinases. Structure 22, 250–259. [DOI] [PubMed] [Google Scholar]
- Hassan S, Amer S, Mittal C, and Sharma R (2012). Ewingella americana: an emerging true pathogen. Case Rep Infect Dis 2012, 730720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, and Rosenstrom P (2010). Dali server: conservation mapping in 3D. Nucleic Acids Res 38, W545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert DA, He Y, McNulty BC, Tornero P, and Dangl JL (2009). Specific Arabidopsis HSP90.2 alleles recapitulate RAR1 cochaperone function in plant NB-LRR disease resistance protein regulation. Proc Natl Acad Sci U S A 106, 9556–9563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert DA, Tornero P, Belkhadir Y, Krishna P, Takahashi A, Shirasu K, and Dangl JL (2003). Cytosolic HSP90 associates with and modulates the Arabidopsis RPM1 disease resistance protein. EMBO J 22, 5679–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulin MT, Armitage AD, Vicente JG, Holub EB, Baxter L, Bates HJ, Mansfield JW, Jackson RW, and Harrison RJ (2018). Comparative genomics of Pseudomonas syringae reveals convergent gene gain and loss associated with specialization onto cherry (Prunus avium). New Phytol 219, 672–696. [DOI] [PubMed] [Google Scholar]
- Hunter T, and Poon RY (1997). Cdc37: a protein kinase chaperone? Trends Cell Biol 7, 157–161. [DOI] [PubMed] [Google Scholar]
- Jones JD, Vance RE, and Dangl JL (2016). Intracellular innate immune surveillance devices in plants and animals. Science 354. [DOI] [PubMed] [Google Scholar]
- Kadota Y, and Shirasu K (2012). The HSP90 complex of plants. Biochim Biophys Acta 1823, 689–697. [DOI] [PubMed] [Google Scholar]
- Karnitz LM, and Felts SJ (2007). Cdc37 regulation of the kinome: when to hold ‘em and when to fold ‘em. Sci STKE 2007, pe22. [DOI] [PubMed] [Google Scholar]
- Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Lenzen G, Page AL, Legrain P, Sansonetti PJ, and Parsot C (2005). The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc Natl Acad Sci U S A 102, 14046–14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, and Bork P (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44, W242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Burch-Smith T, Schiff M, Feng S, and Dinesh-Kumar SP (2004). Molecular chaperone Hsp90 associates with resistance protein N and its signaling proteins SGT1 and Rar1 to modulate an innate immune response in plants. J Biol Chem 279, 2101–2108. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Madej T, Lanczycki CJ, Zhang D, Thiessen PA, Geer RC, Marchler-Bauer A, and Bryant SH (2014). MMDB and VAST+: tracking structural similarities between macromolecular complexes. Nucleic Acids Res 42, D297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Mayor A, Martinon F, De Smedt T, Petrilli V, and Tschopp J (2007). A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol 8, 497–503. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CJ, and Turk BE (2018). Homing in: Mechanisms of Substrate Targeting by Protein Kinases. Trends Biochem Sci 43, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min X, Lee BH, Cobb MH, and Goldsmith EJ (2004). Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure 12, 1303–1311. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M (2006). HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Mumberg D, Muller R, and Funk M (1995). Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122. [DOI] [PubMed] [Google Scholar]
- Murata S, Minami Y, Minami M, Chiba T, and Tanaka K (2001). CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep 2, 1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Suzuki T, Murata S, Nakamura S, Hino T, Maeo K, Tabata R, Kawai T, Tanaka K, Niwa Y, et al. (2007). Improved Gateway binary vectors: high-performance vectors for creation of fusion constructs in transgenic analysis of plants. Biosci Biotechnol Biochem 71, 2095–2100. [DOI] [PubMed] [Google Scholar]
- Nathan DF, and Lindquist S (1995). Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol Cell Biol 15, 3917–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L (2002). Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med 8, S55–61. [DOI] [PubMed] [Google Scholar]
- Nguyen KB, Sreelatha A, Durrant ES, Lopez-Garrido J, Muszewska A, Dudkiewicz M, Grynberg M, Yee S, Pogliano K, Tomchick DR, et al. (2016). Phosphorylation of spore coat proteins by a family of atypical protein kinases. Proc Natl Acad Sci U S A 113, E3482–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien HE, Thakur S, Gong Y, Fung P, Zhang J, Yuan L, Wang PW, Yong C, Scortichini M, and Guttman DS (2012). Extensive remodeling of the Pseudomonas syringae pv. avellanae type III secretome associated with two independent host shifts onto hazelnut. BMC Microbiol 12, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z (1991). Maximum likelihood refinement of heavy atom paramaters. Paper presented at: CCP4 Study Weekend (Daresbury Laboratory: Science & Engineering Research Council). [Google Scholar]
- Pearl LH (2005). Hsp90 and Cdc37 -- a chaperone cancer conspiracy. Curr Opin Genet Dev 15, 55–61. [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, and Cottrell JS (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- Piechocki M, Giska F, Koczyk G, Grynberg M, and Krzymowska M (2018). An Engineered Distant Homolog of Pseudomonas syringae TTSS Effector From Physcomitrella patens Can Act as a Bacterial Virulence Factor. Front Microbiol 9, 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda JN, Smith FD, Daurie A, Swaney DL, Villen J, Scott JD, Stadnyk AW, Le Trong I, Stenkamp RE, Klevit RE, et al. (2014). E2~Ub conjugates regulate the kinase activity of Shigella effector OspG during pathogenesis. EMBO J 33, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeis T, Ludwig AA, Martin R, and Jones JD (2001). Calcium-dependent protein kinases play an essential role in a plant defence response. Embo J 20, 5556–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald PC, and Beutler B (2010). Plant and animal sensors of conserved microbial signatures. Science 330, 1061–1064. [DOI] [PubMed] [Google Scholar]
- Rowlands MG, Newbatt YM, Prodromou C, Pearl LH, Workman P, and Aherne W (2004). High-throughput screening assay for inhibitors of heat-shock protein 90 ATPase activity. Anal Biochem 327, 176–183. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, and Maniatis T (1989). Molecular cloning : a laboratory manual (Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory; ). [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TR, and Sheldrick GM (2002). Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr 58, 1772–1779. [DOI] [PubMed] [Google Scholar]
- Schulze-Lefert P (2004). Plant immunity: the origami of receptor activation. Curr Biol 14, R22–24. [PubMed] [Google Scholar]
- Shirasu K (2009). The HSP90-SGT1 chaperone complex for NLR immune sensors. Annu Rev Plant Biol 60, 139–164. [DOI] [PubMed] [Google Scholar]
- Shirasu K, Lahaye T, Tan MW, Zhou F, Azevedo C, and Schulze-Lefert P (1999). A novel class of eukaryotic zinc-binding proteins is required for disease resistance signaling in barley and development in C. elegans. Cell 99, 355–366. [DOI] [PubMed] [Google Scholar]
- Soucy SM, Huang J, and Gogarten JP (2015). Horizontal gene transfer: building the web of life. Nat Rev Genet 16, 472–482. [DOI] [PubMed] [Google Scholar]
- Sreelatha A, Yee SS, Lopez VA, Park BC, Kinch LN, Pilch S, Servage KA, Zhang J, Jiou J, Karasiewicz-Urbanska M, et al. (2018). Protein AMPylation by an Evolutionarily Conserved Pseudokinase. Cell 175, 809–821 e819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Engel JL, Wen J, Wiley SE, Worby CA, Kinch LN, Xiao J, Grishin NV, and Dixon JE (2012). Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 336, 1150–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Turnbull J, Wang W, Girard JM, Zhao X, Skurat AV, Delgado-Escueta AV, Minassian BA, Depaoli-Roach AA, and Roach PJ (2007). Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci U S A 104, 19262–19266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Wiley SE, Guo X, Kinch LN, Durrant E, Wen J, Xiao J, Cui J, Nguyen KB, Engel JL, et al. (2015). A Single Kinase Generates the Majority of the Secreted Phosphoproteome. Cell 161, 1619–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF, and Lindquist S (2010). HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11, 515–528. [DOI] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, and Lindquist S (2012). Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Casais C, Ichimura K, and Shirasu K (2003). HSP90 interacts with RAR1 and SGT1 and is essential for RPS2-mediated disease resistance in Arabidopsis. Proc Natl Acad Sci U S A 100, 11777–11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, and Kornev AP (2011). Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci 36, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoraki MA, and Caplan AJ (2012). Quality control and fate determination of Hsp90 client proteins. Biochim Biophys Acta 1823, 683–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verba KA, and Agard DA (2017). How Hsp90 and Cdc37 Lubricate Kinase Molecular Switches. Trends Biochem Sci 42, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verba KA, Wang RY, Arakawa A, Liu Y, Shirouzu M, Yokoyama S, and Agard DA (2016). Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinatzer BA, Teitzel GM, Lee MW, Jelenska J, Hotton S, Fairfax K, Jenrette J, and Greenberg JT (2006). The type III effector repertoire of Pseudomonas syringae pv. syringae B728a and its role in survival and disease on host and non-host plants. Mol Microbiol 62, 26–44. [DOI] [PubMed] [Google Scholar]
- Wan F, Weaver A, Gao X, Bern M, Hardwidge PR, and Lenardo MJ (2011). IKKbeta phosphorylation regulates RPS3 nuclear translocation and NF-kappaB function during infection with Escherichia coli strain O157:H7. Nat Immunol 12, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessling R, Epple P, Altmann S, He Y, Yang L, Henz SR, McDonald N, Wiley K, Bader KC, Glasser C, et al. (2014). Convergent targeting of a common host protein-network by pathogen effectors from three kingdoms of life. Cell Host Microbe 16, 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin XF, and He SY (2013). Pseudomonas syringae pv. tomato DC3000: a model pathogen for probing disease susceptibility and hormone signaling in plants. Annu Rev Phytopathol 51, 473–498. [DOI] [PubMed] [Google Scholar]
- Xin XF, Kvitko B, and He SY (2018). Pseudomonas syringae: what it takes to be a pathogen. Nat Rev Microbiol 16, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Jaroszewski L, Li Z, and Godzik A (2014). FFAS-3D: improving fold recognition by including optimized structural features and template re-ranking. Bioinformatics 30, 660–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yuan X, Xiang Z, Mimnaugh E, Marcu M, and Neckers L (2005). Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat Struct Mol Biol 12, 120–126. [DOI] [PubMed] [Google Scholar]
- Xu Y, and Lindquist S (1993). Heat-shock protein hsp90 governs the activity of pp60v-src kinase. Proc Natl Acad Sci U S A 90, 7074–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zembek P, Danilecka A, Hoser R, Eschen-Lippold L, Benicka M, Grech-Baran M, Rymaszewski W, Barymow-Filoniuk I, Morgiewicz K, Kwiatkowski J, et al. (2018). Two Strategies of Pseudomonas syringae to Avoid Recognition of the HopQ1 Effector in Nicotiana Species. Front Plant Sci 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, de Souza RF, Anantharaman V, Iyer LM, and Aravind L (2012). Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Boter M, Li K, Kadota Y, Panaretou B, Prodromou C, Shirasu K, and Pearl LH (2008). Structural and functional coupling of Hsp90- and Sgt1-centred multi-protein complexes. EMBO J 27, 2789–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Dong N, Hu L, and Shao F (2013). The Shigella type three secretion system effector OspG directly and specifically binds to host ubiquitin for activation. PLoS One 8, e57558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann L, Stephens A, Nam SZ, Rau D, Kubler J, Lozajic M, Gabler F, Soding J, Lupas AN, and Alva V (2017). A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J Mol Biol. [DOI] [PubMed] [Google Scholar]
- Zimmermann L, Stephens A, Nam SZ, Rau D, Kubler J, Lozajic M, Gabler F, Soding J, Lupas AN, and Alva V (2018). A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J Mol Biol 430, 2237–2243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. HopBF1-like kinase domains are present in a number of bacterial strains from diverse taxa and share sequence features typical of protein kinases (Related to Figure 1).
(A)Phylogenetic tree (Maximum Likelihood) for selected HopBF1 homologs built from a manually corrected MAFFT multiple sequence alignment of HopBF1 type kinase domains, collected using JackHMMer. Coloring is by taxonomy: Red, alphaproteobacteria; green, betaproteobacteria; yellow, gammaproteobacteria; cyan, Cyanobacteria; brown, Actinobacteria. Red squares denote proteins containing repeated (2 or 3) HopBF1 type kinase-like domains. Grey bars indicate protein sequence lengths (maximum: 5315 residues). For some branches, dominant genus shown (e.g. Pseudomonas). Black circles mark branches with bootstrap support better than 0.9.
(B, C)Sequence logos (Weblogos) for 161 HopBF1 homologs (B) and for 3,998 homologs of typical protein kinases (C) (Pfam domain PF00069). The two logos are aligned together by Mafft and filtered as to retain only columns matching human PKA sequence. Numbering in both logos corresponds to PKA. The height of the amino acid stack is proportional to the sequence conservation at that position.
(D) Superposition of the Apo and AMP-PNP bound HopBF1 structures. The Apo structure is in yellow and the AMP-PNP structure is in teal.
Figure S2. HopBF1 and its mutants are expressed in planta and secreted by. P syringae (Related to Figure 2).
(A-B) Protein immunoblots of leaf extracts showing HopBF1 levels in N. benthamiana (A) and N. tabacum (B). (C) Protein immunoblots of supernatants from DC300028E P. syringae depicting the levels of HopBF1 and mutants. (D-E). Quantification of tissue collapse and necrosis in N. benthamiana (D) and Arabidopsis (E) plants following P. syringae DC300028E infection with HopBF1, the inactive D154A, D169A mutants and mCherry. Bars represent the average percentage of necrotic leaf area with standard deviations.
Figure S3. HopBF1 is an HSP90 protein kinase (Related to Figure 3).
(A) Protein immunoblotting of yeast extracts depicting the levels of P. syringae Myc-tagged HopBF1 or the D154A and D169A mutants. GAPDH is shown as a loading control.
(B) Kinetic analysis depicting the concentration dependence of S. cerevisiae HSP82 on the rate of phosphate incorporation into S. cerevisiae HSP82 by E. americana HopBF1. (Inset) Km for S. cerevisiae HSP82 is indicated. Reaction products were resolved by SDS PAGE and radioactive gel bands were excised and 32P incorporation was quantified by scintillation counting.
(C) Incorporation of 32P from [γ32P]ATP into S. cerevisiae HSP90 (HSP82) by recombinant P. syringae HopBF1 or the Burkholderia orthologs. The reaction products were separated by SDS-PAGE and analyzed by Coomassie staining (upper) and autoradiography (lower).
(D) Incorporation of 32P from [γ32P]ATP into S. cerevisiae HSP90 (HSP82) or the ATPase inactive HSP82 D79N mutant by recombinant P. syringae HopBF1. HSP82 was also pretreated with geldanamycin. The reaction products were analyzed as in C.
Figure S4. HopBF1 phosphorylates HSP90 resulting in the inhibition of ATPase and chaperoning activity (Related to Figure 4).
(A) Protein immunoblotting of HEK293A cell extracts expressing Flag-GFP with E. americana HopBF1 or the D170A mutant. Cells were also pretreated with Geldanamycin. Levels of Flag-GFP and HopBF1 are also shown. GAPDH is included as a loading control. This is a control for the experiments present in Figures 4F and 4G.
(B) Protein immunoblotting of anti-Flag immunoprecipitates from HEK293A cells expressing Flag-GFP, Flag-v-src, Flag-E. americana HopBF1 D170A, V5-tagged human HSP90β or the S108E phosphomimetic HSP90β mutant. Extracts were also analyzed for expression. GAPDH is included as a loading control.
(C) Enlarged image of the HopBF1 nucleotide-binding pocket showing the molecular interactions involved in nucleotide binding. Active site residues are numbered and the PKA equivalent residues are shown in parentheses. The AMP-PNP molecule is also shown.
(D) Kinase activity of P. syringae HopBF1 and active site mutants using S. cerevisiae HSP90 (HSP82) as substrate. Reaction products were resolved by SDS PAGE and radioactive gel bands were excised and 32P incorporation was quantified by scintillation counting.
(E) Protein immunoblotting of yeast extracts depicting the levels of Flag-tagged HSP82 and P. syringae Myc-tagged HopBF1 or the E74A mutant before and after induction with galactose. GAPDH is shown as a loading control.
Figure S5. HopBF1 phosphorylates and inactivates HSP90 during infection (Related to Figure 5).
(A) Table depicting the percent of peptides modified on H. sapiens HSP90 Ser108 and N. benthamiana HSP90 Ser100. HeLa cells were infected with an effectorless Y. pestis strain expressing either E. americana HopBF1 or the D170A mutant. Endogenous HSP90 was immunoprecipitated (lower) and subjected to mass spectrometry analysis. P. syringae HopBF1 or the D169A mutant and HSP90 were also co-agroinfiltrated into N. benthamiana leaves. Myc-tagged HSP90 was immunoprecipitated and subjected to mass spectrometry (lower rows).
(B) Incorporation of 32P from [γ32P]ATP into N. benthamiana HSP90 or the S100A and S100F mutants by recombinant P. syringae HopBF1. Reaction products were separated by SDS-PAGE and analyzed by Coomassie staining (upper) and autoradiography (lower).
(C) Protein immunoblotting of N. benthamiana leaf extracts depicting the levels of Myc-tagged HSP90 and Flag-tagged YFP. Leaves were infiltrated with P. syringae DC3000D28E expressing HopBF1 and appropriate A. tumefaciens strain to transiently express HSP90, HSP90 S100F or YFP as a control. The Coomassie stained membrane is included as a loading control.
(D) Photographs depicting N. benthamiana leaves following infiltration with P. syringae DC3000D28E expressing HopBF1 D169A and appropriate A. tumefaciens strain to transiently express HSP90, HSP90 S100F or YFP as a control. In contrast to leaves infected with HopBF1 WT, no signs of tissue necrotization were observed. Photographs show representative leaves made 6 dpi. The experiment was performed 5 times with similar results.
(E) Whole plants were dip-inoculated with a P. syringae pv. syringae B728a ectopically producing WT HopBF1, kinase inactive variants or mCherry as a control. At the indicated time points, bacteria were isolated from the inoculated leaves, spread on plates and counted. All the strains evoked strong disease symptoms and the plants eventually died. The photographs showing representative plants were made 7 days post inoculation.
(F) Protein immunoblotting depicting the levels of mCherry, HopBF1 and HopBF1 mutants in the P. syringae B728A strain in the cell pellet (lower) or the supernatant (upper) following induction of the T3SS. GroEL and the Ponceau membrane are also shown.
Figure S6. HSP90 recognizes HopBF1 as a client via the αC-β4 loop (Related to Figure 6).
(A) Protein immunoblotting of HEK293A cell extracts depicting the levels of WT E. americana Flag-HopBF1 or the D170A mutant. Tubulin is shown as a loading control. Note that WT HopBF1 protein levels are lower than the mutant, indicating that HopBF1, which phosphorylates and inactivates HSP90, phenocopies geldanamycin treatment of the D170A mutant.
(B) Protein immunoblotting of yeast extracts depicting the levels of E. americana Flag-tagged HopBF1 or the indicated mutants. HSP82 is also shown.
(C) Kinetic analysis depicting the concentration dependence of S. cerevisiae HSP82 on the rate of phosphate incorporation into S. cerevisiae HSP82 by P. syringae HopBF1 (left) or the V88D mutant (right, note the difference in numbering due to the different species). The Km for S. cerevisiae HSP82 and Vmax are shown in the inset. Reaction products were resolved by SDS PAGE and radioactive gel bands were excised and 32P incorporation was quantified by scintillation counting. As judged by Kcat/Km (catalytic efficiency), WT HopBF1 is more than 1000-fold more efficient than the V88D mutant.
(D) Protein immunoblotting of N. benthamiana extracts depicting the levels of Flag-tagged HopBF1 or indicated mutants and Flag-tagged YFP. Leaves were infiltrated with A. tumefaciens carrying Flag-tagged HopBF1 or the mutants and photos were taken 7 days post infiltration.
Data Availability Statement
The accession numbers for the coordinates and structure factors reported in this paper are PDB ID 6PWD for apo HopBF1, and PDB ID 6PWG for AMP-PNP bound HopBF1.
Table S1. Data collection and refinement statistics, E. americana HopBF1 structures. (Related to Figure 1)