

SUMMARY
Long INterspersed Element-1 (L1) retrotransposition poses a threat to genome integrity and cells have evolved mechanisms to restrict retrotransposition. However, how cellular proteins facilitate L1 retrotransposition requires elucidation. Here, we demonstrate that single strand DNA breaks induced by the L1 endonuclease trigger the recruitment of poly(ADP-ribose) polymerase 2 (PARP2) to L1 integration sites and that PARP2 activation leads to the subsequent recruitment of the replication protein A (RPA) complex to facilitate retrotransposition. We further demonstrate that RPA directly binds activated PARP2 through poly(ADP-ribosylation) and can protect single strand L1 integration intermediates from APOBEC3-mediated cytidine deamination in vitro. Paradoxically, we provide evidence that RPA can guide APOBEC3A, and perhaps other APOBEC3 proteins, to sites of L1 integration. Thus, the interplay of L1-encoded and evolutionarily conserved cellular proteins is required for efficient retrotransposition; however, these interactions also may be exploited to restrict L1 retrotransposition in the human genome.
Keywords: Mobile genetic element, retrotransposon, LINE-1, integration, retrotransposition, poly(ADP-ribose) polymerase 2, replication protein A complex, cytidine deamination, APOBEC3 proteins
Graphical Abstract
INTRODUCTION
An average individual genome contains ~80–100 full-length Long INterspersed Element-1 (L1) sequences that can move to new locations (Brouha et al., 2003; Sassaman et al., 1997) by a process termed retrotransposition. Full-length retrotransposition-competent human L1s (RC-L1s) are ~6 kb and contain a 5’ untranslated region (UTR) harboring sense and anti-sense RNA polymerase II promoters, two open reading frames (ORF1 and ORF2), and a 3’ UTR that ends in a poly(A) tract (Richardson et al., 2015). ORF1 encodes a ~40 kDa protein (ORF1p) with RNA binding and nucleic acid chaperone activities (Hohjoh and Singer, 1996; Khazina et al., 2011; Martin and Bushman, 2001). ORF2 encodes a ~150 kDa protein (ORF2p) with endonuclease (EN) and reverse transcriptase (RT) activities (Doucet et al., 2010; Feng et al., 1996; Mathias et al., 1991; Moran et al., 1996).
L1 retrotransposition occurs by target-site primed reverse transcription (TPRT) (Cost et al., 2002; Feng et al., 1996; Luan et al., 1993). The L1 5’UTR sense strand promoter (Athanikar et al., 2004; Swergold, 1990) produces a full-length polyadenylated L1 transcript, which then is exported to the cytoplasm. ORF1p and ORF2p preferentially associate with their encoding transcript (Doucet et al., 2015; Esnault et al., 2000; Wei et al., 2001), leading to the formation of an L1 ribonucleoprotein particle (RNP) (Doucet et al., 2010; Hohjoh and Singer, 1996; Kulpa and Moran, 2005; Martin, 1991). Components of the L1 RNP gain nuclear access (Kubo et al., 2006), where L1 EN activity generates a single strand endonucleolytic nick at a degenerate consensus sequence (5’-TTTTT/AA-3’ and variants of that sequence) in genomic DNA, generating a 3’-hydroxyl group (Cost and Boeke, 1998; Feng et al., 1996; Flasch et al., 2019; Sultana et al., 2019). Annealing between the thymidine residues liberated by L1 EN cleavage and L1 mRNA poly(A) tail provides a primer/template complex that the L1 RT uses to reverse transcribe (−) strand L1 cDNA from L1 RNA (Doucet et al., 2015; Kulpa and Moran, 2006; Monot et al., 2013). Second strand genomic DNA cleavage, (+) strand L1 cDNA synthesis, and L1 cDNA ligation to genomic DNA require elucidation, but likely involve host-encoded proteins (Goodier, 2016; Pizarro and Cristofari, 2016).
Cells have evolved mechanisms to restrict L1 retrotransposition (Levin and Moran, 2011; Pizarro and Cristofari, 2016). For example, L1 expression can be repressed by transcriptional and post-transcriptional mechanisms and host factors involved in the innate immune response can destabilize L1 RNA, L1 cDNA, and/or the L1-encoded proteins (Levin and Moran, 2011; Richardson et al., 2015). Proteins involved in the DNA damage response and/or DNA repair also restrict L1 retrotransposition (Coufal et al., 2011; Gasior et al., 2008; Liu et al., 2018; Servant et al., 2017).
Proteins involved in DNA replication and/or DNA repair can facilitate L1 retrotransposition, but their mechanisms of action require elucidation (Benitez-Guijarro et al., 2018; Flasch et al., 2019; Mita et al., 2018; Sultana et al., 2019; Suzuki et al., 2009; Taylor et al., 2018; Taylor et al., 2013). Here, we conducted immunoprecipitation (IP)-coupled mass spectrometry analyses to identify L1 ORF2p interacting proteins. We demonstrate poly(ADP-ribose) (PAR) polymerase 2 (PARP2) is recruited to single strand DNA (ssDNA) breaks generated by L1 EN cleavage. The subsequent enzymatic activation of PARP2 leads to the synthesis of PAR, which promotes the direct recruitment of the replication protein A (RPA) complex to sites of L1 integration. RPA can decrease the vulnerability of ssDNA intermediates generated during TPRT to APOBEC3A (A3A)-mediated cytidine deamination in vitro. However, A3A can associate with RPA, which may guide it to L1 integration sites. Thus, interactions between L1 ORF2p, activated PARP2, and RPA can facilitate retrotransposition, whereas the association of RPA with APOBEC3 proteins may, in principle, restrict L1 retrotransposition.
RESULTS
Identification of L1 ORF2p interacting proteins
We conducted IP-coupled mass spectrometry analyses to identify ORF2p interacting proteins. We constructed an L1 expression vector (pTMF3) that contains a T7 gene10 epitope tag and three tandem copies of a FLAG epitope tag at the carboxyl termini of ORF1p and ORF2p, respectively (Figure S1A). ORF2p-3FLAG co-precipitated with ORF1p-T7 (Figure S1B) and the epitope tags did not significantly affect retrotransposition efficiency (Figure S1C).
Human ORF2p is expressed at lower levels than ORF1p (Doucet et al., 2010; McMillan and Singer, 1993; Taylor et al., 2013). Thus, we generated an ORF2p-3FLAG monocistronic expression vector (pTMO2F3) to increase the production of ORF2p in HEK293T cells (Figure 1A). Expression of pTMO2F3 led to an increase in γ-H2AX levels, a well-known marker for double stranded DNA breaks (Hustedt and Durocher, 2016), when compared to a negative control lacking both L1 EN and L1 RT activities (Figure S1D). HEK293T cells were next transfected with pTMO2F3, an anti-FLAG antibody was used to purify the ORF2p-3FLAG complex, and associated host proteins were identified using mass spectrometry (Figure 1B). We identified ORF2p-3FLAG associated proteins that were absent (Table S1A) or significantly reduced (Table S1B) in controls. Some proteins were known to associate with L1 RNPs (e.g., PABPC1, PCNA, HSPA8, UPF1, and MOV10) (Dai et al., 2012; Goodier et al., 2013; Taylor et al., 2013); others are involved in RNA metabolism and nuclear processes (Table S1A and S1B).
Figure 1. Identification of the L1 ORF2p-3FLAG protein complex.
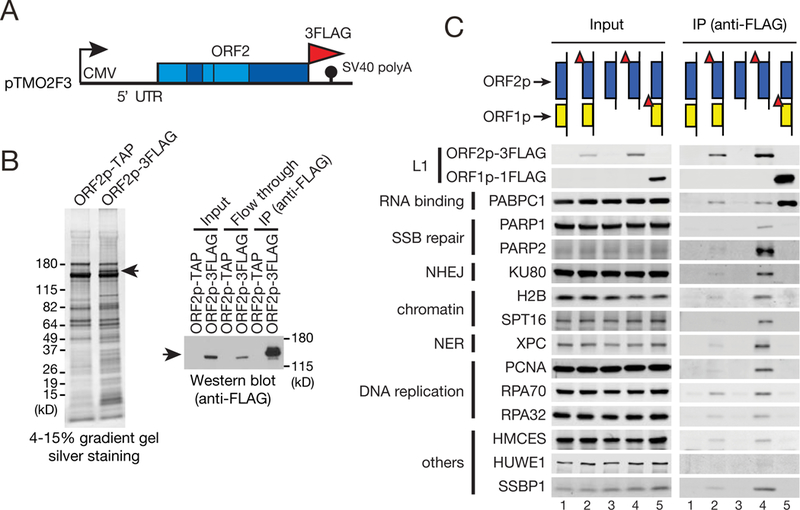
(A) The L1.3 ORF2p-3FLAG expression vector (pTMO2F3). The CMV promoter (black arrow) and 5’ UTR augment L1 expression. Also indicated are the FLAG epitope tag, SV40 polyadenylation signal, and the endonuclease (EN), Z, reverse transcriptase (RT), and cysteine-rich (C) domains.
(B) The ORF2p-3FLAG complex. HEK293T cells were transfected with pAD500 (ORF2p-TAP) or pTMO2F3 (ORF2p-3FLAG). Left: the ORF2p-3FLAG complex was purified using anti-FLAG antibody-conjugated beads, separated on a denaturing polyacrylamide gel, and visualized by silver staining. Right: ORF2p-3FLAG was detected by western blot. Input, the whole cell lysate used in the IP. Arrows, ORF2p-3FLAG.
(C) Validation of proteins in the ORF2p-3FLAG complex. ORF2p-3FLAG complexes were purified (see panel B) from HEK293T cells transfected with a full-length (pTMF3, lane 2) or monocistronic (pTMO2F3, lane 4) L1 expression constructs. The ORF1p-FLAG complex was purified from cells transfected with pJM101/L1.3FLAG (lane 5). The pJM101/L1.3 and pAD001 samples (lanes 1 and 3, respectively) served as negative controls. Input, western blot lanes of whole cell lysate used in the IP. Red triangles, FLAG tags; SSB, single-strand break; NHEJ, non-homologous end-joining; NER, nucleotide excision repair.
We conducted IP followed by western blot to validate a subset of the ORF2p-3FLAG interacting proteins (Figure 1C). ORF2p-3FLAG co-precipitated with PARP1, PARP2, KU80, H2B, SPT16, XPC, PCNA, RPA, HMCES, HUWE1, and SSBP1. Most of these proteins also associated at reduced levels with ORF2p-3FLAG expressed from a full-length RC-L1 (Figure 1C) (Doucet et al., 2010). ORF1p-FLAG did not or only exhibited subtle interactions with the ORF2p-3FLAG associated nuclear proteins (Figure 1C), suggesting that ORF1p is more abundant in cytoplasmic vs. nuclear L1 RNPs or might form unstable complexes with ORF2p associated nuclear proteins. Thus, in addition to the previously reported associations with PARP1, PCNA, RPA, and HMCES (Mita et al., 2018; Taylor et al., 2018; Taylor et al., 2013), ORF2p interacts with at least seven other nuclear proteins.
PARP1 and PARP2 are required for efficient L1 retrotransposition
L1 EN activity is required for initiating TPRT (Feng et al., 1996; Moran et al., 1996). Because ssDNA breaks are sensed and repaired by two PAR polymerase family members (PARP1 and PARP2) (Gibson and Kraus, 2012), we used established assays to test whether a PARP inhibitor (PARPi), oliparib, affects L1 retrotransposition (Figure 2A; STAR METHODS) (Gibson and Kraus, 2012; Moran et al., 1996; Ostertag et al., 2000; Wei et al., 2001). L1 retrotransposition efficiency decreased significantly upon increasing the PARPi concentration (Figures 2B and S2A-C) and controls, in which HeLa-JVM cells were transfected with a plasmid expressing a blasticidin resistant gene (pcDNA6), indicated that the reduction in retrotransposition efficiency was not due to PARPi toxicity (Figure 2B).
Figure 2. PARP activity is required for efficient L1 retrotransposition.
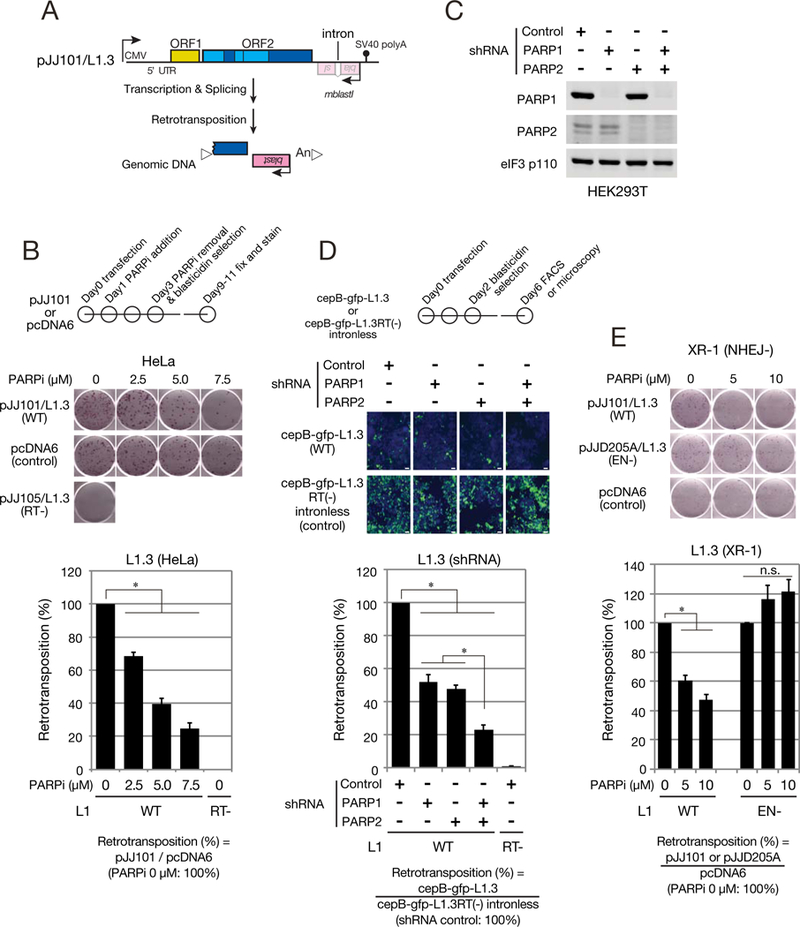
(A) The L1 retrotransposition assay. A backward copy of an mblastl retrotransposition indicator cassette (pink box) was inserted into the L1 3’ UTR. The cassette is interrupted by an intron in the L1 transcriptional orientation. Retrotransposition allows activation of the blasticidin deaminase gene, conferring blasticidin resistance to cells. White triangles, target-site duplications (TSDs) presumed to flank the retrotransposed L1. “An”, the L1 poly(A) tract.
(B) Retrotransposition in PARPi treated HeLa-JVM cells. Top: timeline of the assay. HeLa-JVM cells were transfected with either a WT L1 expression construct (pJJ 101/L1.3) or a construct containing an intronless blasticidin-resistant gene (pcDNA6). Middle: representative retrotransposition assay. An RT-deficient L1 (pJJ105/L1.3; D702A) served as a negative control. Bottom: retrotransposition efficiencies were calculated as described in STAR METHODS. X-axis, PARPi (olaparib) concentration and L1 constructs. Y-axis, normalized retrotransposition efficiencies. Assays conducted with the WT L1 in the absence of olaparib are set to 100%. Standard errors (black error bar lines) were calculated from three independent biological replicates. P-values were calculated using one-way analysis of variance (ANOVA) followed by a Bonferroni multiple comparison test. Asterisks (*), p-value of < 0.005.
(C) Western blots of PARP1 and PARP2 protein levels in shRNA-knockdown cells. The eukaryotic translation initiation factor 3 subunit protein (eIF3, p110) served as a loading control.
(D) Retrotransposition in PARP1 and/or PARP2 shRNA-knockdown cells. Top: timeline of the assay. HEK293T cells were transfected with WT L1 expression construct containing an enhanced green fluorescent protein-based retrotransposition indicator cassette (cepB-gfp-L1.3). Blasticidin selection (the selectable marker in the vector) began two days post-transfection. Middle: EGFP-positive cells (green) and nuclei (blue) after selection. White bars, 50 μm. Bottom: relative retrotransposition efficiencies were calculated as described in STAR METHODS X-axis, the shRNA knockdown cell lines and L1 constructs. Y-axis, the normalized retrotransposition efficiencies compared to the non-mammalian targeting shRNA control (set at 100%). An RT mutant (cepB-gfp-L1.3RT(−)) served as a negative control. Standard errors were calculated from seven independent biological replicates. P-values were calculated as in panel B.
(E) Retrotransposition assays in NHEJ-deficient XR-1 cells. Retrotransposition assays were conducted as denoted in panel B. Top: XR-1 cells were transfected with a WT (pJJ101/L1.3) or EN-deficient (pJJD205A/L1.3) L1 expression construct. Bottom: the relative L1 retrotransposition efficiencies were calculated as described in STAR METHODS X-axis, the olaparib concentration and L1 constructs. Y-axis, the normalized retrotransposition efficiencies compared to the WT L1 in the absence of olaparib (set at 100%). Standard errors were calculated from three independent biological replicates. P-values were calculated as noted in panel B. Asterisks (*), p-value < 0.005.
See also Figure S2.
We next examined whether altering PARP1 and/or PARP2 expression affects L1 retrotransposition (Figure S2D). Ectopic overexpression of PARP1 and/or PARP2 did not affect L1 retrotransposition. By comparison, short hairpin RNA (shRNA)-mediated knockdown of PARP1 or PARP2 in HEK293T cells led to a ~50% reduction in L1 retrotransposition (Figures 2C and 2D).
Mouse homozygous knockouts of either Parp1 or Parp2 are viable, whereas double knockouts lead to embryonic lethality, suggesting Parp1 and Parp2 may be functionally redundant (Menissier de Murcia et al., 2003). Knockdown of both PARP1 and PARP2 in HEK293T cells led to an ~80% reduction in L1 retrotransposition (Figures 2D and S2E), but treating these cells with PARPi did not further reduce retrotransposition (Figure S2F). Controls revealed that the reduction in L1 retrotransposition is not due to alterations in steady state ORF2p levels or HEK293T survival rates (Figures S2G and S2H).
PARP activity is important for canonical L1 retrotransposition
L1 retrotransposition can occur by an alternative EN-independent pathway in cells that are p53-defective and contain mutations in genes critical for non-homologous end-joining (NHEJ) DNA repair, suggesting that L1 can exploit endogenous DNA lesions to initiate retrotransposition (Coufal et al., 2011; Kopera et al., 2011; Morrish et al., 2007; Morrish et al., 2002). Thus, we transfected a NHEJ-defective Chinese Hamster Ovary (CHO) cell line (Li et al., 1995) with a wild-type (WT) or EN-deficient L1 expression construct and observed efficient retrotransposition in the absence of the PARPi; however, increasing the PARPi concentration only led to a decrease in WT L1 retrotransposition (Figure 2E). Thus, PARP activity is important for canonical TPRT, but not EN-independent retrotransposition.
PARP2 specifically recognizes DNA breaks created by L1 EN
PARP1 and PARP2 have different DNA-binding domains and are independently recruited to DNA breaks (Mortusewicz et al., 2007). Thus, we examined whether L1-induced ssDNA breaks trigger the recruitment of PARP1 and/or PARP2 to the ORF2p-3FLAG complex. We prepared WT, EN-deficient, RT-deficient, EN/RT-deficient, or cysteine-rich (C) domain-deficient ORF2p-3FLAG protein complexes (Figures 3A and 3B). IP demonstrated that PARP1 and PABPC1 associated with the WT, EN-deficient, RT-deficient, and EN/RT-deficient ORF2p-3FLAG proteins, but only minimally associated with the C-domain-deficient protein (Figure 3B). In contrast, PARP2 only associated with WT and RT-deficient ORF2p (Figure 3B), indicating that L1 EN activity is critical for ORF2p-3FLAG/PARP2 association. SSBP1 and HMCES also exhibited a reduced ability to associate with the RT-deficient and EN/RT-deficient ORF2p-3FLAG protein (Figure S3A), suggesting their association with ORF2p-3FLAG may be facilitated by ssDNAs generated during TPRT.
Figure 3. PARP2 recognizes DNA breaks created by ORF2p.
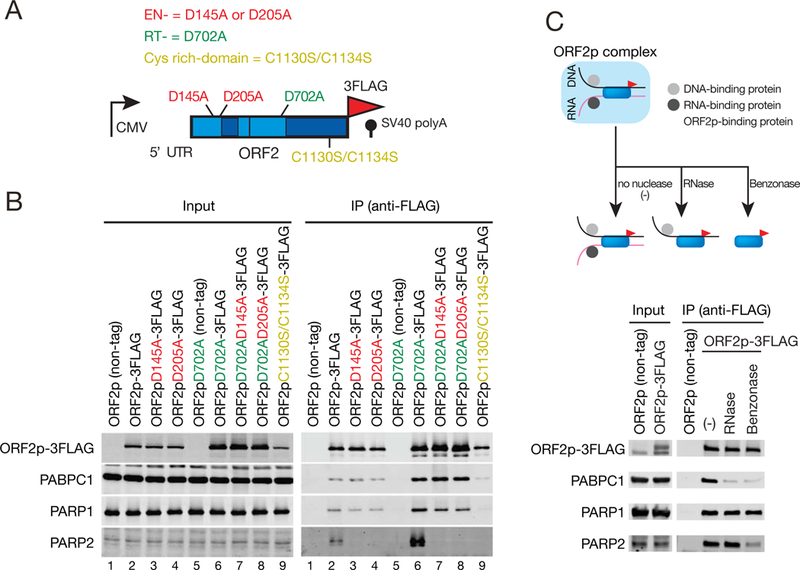
(A) L1 mutants used in the study. Indicated are mutations in the EN domain (red), RT domain (green), or C domain (gold).
(B) L1 EN activity is necessary for ORF2p-3FLAG/PARP2 association. HEK293T cells were transfected with: Lane 1, pAD001; Lane 2, pTMO2F3; Lane 3, pTMO2F3D145A; Lane 4, pTMO2F3D205A; Lane 5, pAD035; Lane 6, pTMO2F3D702A; Lane 7, pTMO2F3D145AD702A; Lane 8, pTMO2F3D205AD702A; or Lane 9, pTMO2F3C1130S/C1134S (see STAR METHODS). ORF2p-3FLAG complex preparation and western blots were conducted as described in Figure 1B. The ORF2p (non-tag) and ORF2pD702A (non-tag) samples served as negative controls.
(C) DNA is required for ORF2p-3FLAG/PARP2 association. ORF2p-3FLAG complexes were purified as in Figure 1B and were treated in either the absence or presence of RNase or benzonase. Western blots were used to detect the indicated proteins.
See also Figure S3.
We next tested whether L1 ORF2p-3FLAG directly interacts with PARP1 or PARP2 (Figure S3B). PARP1 interacted with an ORF2p-3FLAG derivative that lacks the EN domain; however, deleting the central regions of ORF2p (i.e., the Z or RT domains) led to reduced interactions; PARP2 did not associate with any of the ORF2p-3FLAG deletion derivatives (Figure S3B).
PARP2 may associate with ORF2p after L1 EN nicks target site DNA. Thus, we treated the ORF2p-3FLAG IP complex with RNase or benzonase—a nuclease that can degrade RNA and DNA (Figure 3C). Benzonase treatment reduced the ORF2p-3FLAG/PARP2, but not the ORF2p-3FLAG/PARP1, interaction (Figure 3C). Controls revealed that RNase or benzonase treatment abolished the PABPC1/ORF2p-3FALG interaction (Dai et al., 2012). Thus, protein-protein interactions may mediate the association of PARP1 with ORF2p-3FLAG, whereas DNA may mediate PARP2/ORF2p interactions. Notably, benzonase treatment also reduced interactions between ORF2p-3FLAG and KU80, H2B, PCNA, RPA, HMCES, and SSBP1 (Figure S3C), suggesting these proteins may interact with ORF2p-3FLAG in a DNA-dependent manner.
DNA substrates that mimic L1 EN cleavage events selectively activate PARP2
PARP1 and PARP2 activation lead to their auto-poly(ADP-ribosyl)ation and PARP2 is activated by DNA nicks that contain a 5’ phosphate group (Langelier et al., 2014). Because L1 EN generates a single strand nick that contains a 5’ phosphate and 3’ hydroxyl group (Feng et al., 1996), we tested whether various DNA substrates could lead to PARP2 enzymatic activation. We incubated recombinant PARP2 with double strand (ds) DNA substrates and used an antibody to detect newly synthesized PAR (Figures 4A and S4A). PARP2 was activated to different extents by each substrate, but the most intense PAR signals were obtained from reactions containing a 5’ phosphorylated nicked DNA substrate (Figure 4A). Controls revealed that PARP2 activation requires both DNA and nicotinamide adenine dinucleotide (NAD), was abolished in reactions containing a PARPi (Figure 4A), and that recombinant PARP1 was activated to similar extents by each DNA substrate (Figure S4B), which agrees with reports that PARP1 plays multiple roles in DNA repair (Langelier et al., 2014).
Figure 4. PARP2 promotes RPA recruitment to the ORF2p complex.
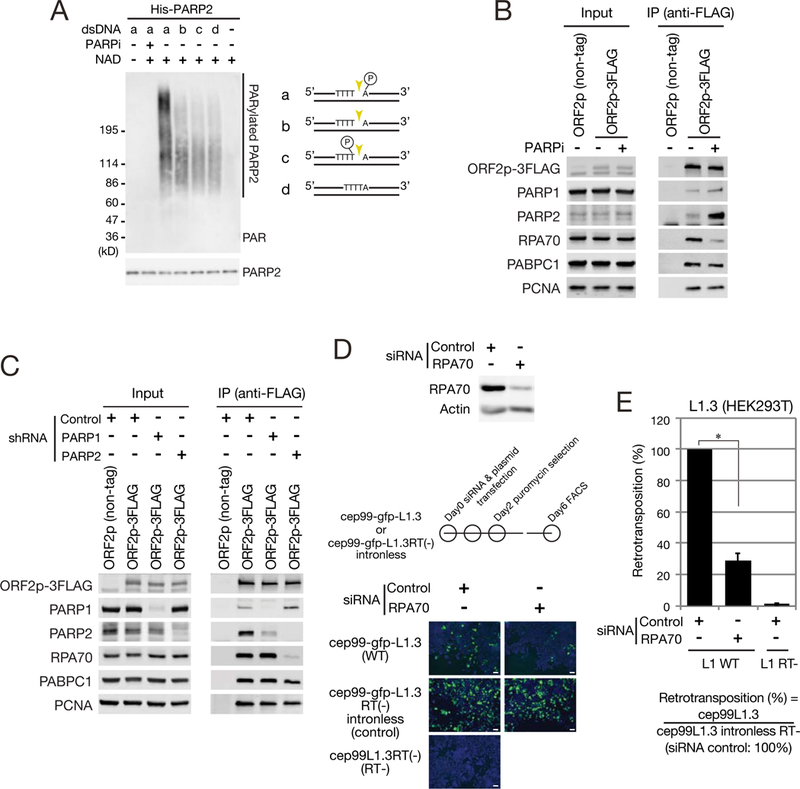
(A) A 5’ phosphate nicked DNA substrate selectively activates PARP2. Left: recombinant PARP2 was incubated with or without 20 μM NAD, 10 μM olaparib (PARPi), and 20 μM of dsDNA substrate. PARylated-PARP2 was separated on an SDS-PAGE gel and detected by western blot. Signals obtained with an anti-PARP2 antibody served as a loading control. Right: dsDNA substrates tested for PARP2 activation: (a) a 5’ phosphorylated nick; (b) an unphosphorylated nick; (c) a 3’ phosphorylated nick; and (d) a dsDNA lacking a nick. Gold triangles, the positions of the nick in the L1 ORF2p EN consensus cleavage sequence. Encircled “P”, the phosphate group.
(B) The ORF2p-3FLAG/RPA association is reduced upon treatment with a PARPi. HEK293T cells were transfected with pTMO2F3 and treated with 10 μM olaparib (PARPi) for ~12 hours prior to cell collection. DMSO served as a control (PARPi [-]). The ORF2p-3FLAG complexes were prepared as described in Figure 1B and analyzed by western blot.
(C) PARP2 can recruit RPA to the ORF2p complex. The ORF2p-3FLAG complexes in PARP1 or PARP2 shRNA knockdown HEK293T cells were purified as described in Figure 1B and analyzed by western blot.
(D) Retrotransposition in RPA siRNA-knockdown cells. Top: Whole cell lysates from siRNA transfected HEK293T cells were subjected to western blot. β-Actin served as a loading control. Middle: timeline of the retrotransposition assay. HEK293T cells were transfected with a WT L1 expression construct (cep99-gfp-L1.3) in the presence of a non-targeting siRNA (control) or a siRNA against RPA70. Puromycin selection (the selectable marker in the vector) began two days post-transfection. Bottom: EGFP-positive cells (green) and nuclei (blue) after selection. White bar, 50 μm.
(E) L1 retrotransposition efficiency in RPA70 siRNA-knockdown cells. Retrotransposition assays were conducted as indicated in panel D. Relative retrotransposition efficiencies were calculated as described in STAR METHODS. X-axis, the siRNAs and L1 constructs. Y-axis, the normalized retrotransposition efficiency compared to cells transfected with a non-targeting siRNA control (set at 100%). Standard errors were calculated from four independent biological replicates. P-values were calculated using an unpaired two-tailed t-test. Asterisks (*), p-value < 0.01.
PARP2 promotes RPA recruitment to the ORF2p complex
PARP activation at DNA breaks leads to the recruitment of DNA repair proteins (Gibson and Kraus, 2012). Our IP/mass spectrometry and IP-western blot analyses failed to detect known PARP2-interacting proteins (e.g., XRCC1, LIG3, or DNA pol P) in the ORF2p-3FLAG complex (Table S1) (Schreiber et al., 2002). Thus, PARP2 activation at sites of L1 EN cleavage may trigger the recruitment of a different set of host proteins. Control IP/western blot experiments revealed that PARPi did not alter ORF1p, ORF2p, or PABPC1 levels in the ORF2p-3FLAG complex (Figures 4B and S4C), suggesting that it did not interfere with cytoplasmic L1 RNP formation.
Hyper-poly(ADP-ribosyl)ation (i.e., PARylation) is proposed to decrease the ability of PARPs to bind DNA (Langelier et al., 2014). Thus, we next tested whether poly(ADP-ribose) polymerase activity is critical for OFR2p-3FLAG/nuclear protein interactions. In agreement with our findings that DNA mediates ORF2p/PARP2 interactions, whereas protein-protein contacts mediate ORF2p/PARP1 interactions (Figure 3C and S3B), IP/western blot experiments demonstrated an increase in ORF2p-3FLAG/PARP2 in the presence of PARPi (Figure 4B). However, it is possible that excessive PARylation inhibits antibody recognition of PARP2, leading to an underestimate of PARP2 protein levels in the absence of the PARPi (Figure S4D).
RPA is a heterotrimeric protein complex consisting of RPA70, RPA32, and RPA14 that binds ssDNA in eukaryotes (Chen and Wold, 2014). Intriguingly, RPA70 and RPA32 exhibited a decreased ability to associate with ORF2p-3FLAG in the presence of the PARPi (Figures 4B and S4E). Although PARP1 is required to recruit numerous repair proteins to DNA breaks, the ORF2p-3FLAG/RPA complex formation was only markedly reduced in the PARP2 knockdown cell line (Figure 4C). A reduction of ORF2p-3FLAG/PARP2 complexes, but not total PARP2 protein levels, also was observed in PARP1 knockdown cells (Figure 4C). The ORF2p-3FLAG/PCNA and ORF2p-3FLAG/PABPC1 complexes were not reduced in PARP1 or PARP2 knockdown cell lines (Figure 4C).
RPA facilitates L1 retrotransposition
We next used small interfering RNAs (siRNAs) to transiently knockdown RPA70 (Figure 4D). RPA70 knockdown led to a ~70% decrease in L1 retrotransposition efficiency when compared to controls (Figure 4E), but did not dramatically alter the cell cycle profile (Figure S4F), indicating that RPA facilitates L1 retrotransposition. Of note, RPA70 knockdown reduced L1 retrotransposition to a greater extent than PARP2 knockdown, suggesting RPA may also be recruited to L1 integrations sites by a PARP2-independent mechanism (e.g., autonomous binding to ssDNA) to facilitate L1 retrotransposition (Figure S4G).
Direct recognition of poly(ADP-ribose) by an OB-fold domain of RPA
The RPA heterotrimer has six oligonucleotide/oligosaccharide-binding (OB)-fold DNA binding domains (DBDs) that bind ssDNA (Figure 5A; DBD-F, A, B, C, D and E) and OB-fold domains in other proteins can directly interact with PAR (Zhang et al., 2014). To test whether RPA could bind directly to PAR, we expressed the three RPA subunits in bacteria, purified the heterotrimeric complex, and examined RPA/PAR interactions (Figure S5A; STAR METHODS). PAR bound to RPA, but not glutathione S-transferase (GST) (Figure S5A). RPA70 also co-precipitated with PAR in a PARP-activity dependent manner in vivo (Figure S5B).
Figure 5. OB-fold of RPA directly binds poly(ADP-ribose).
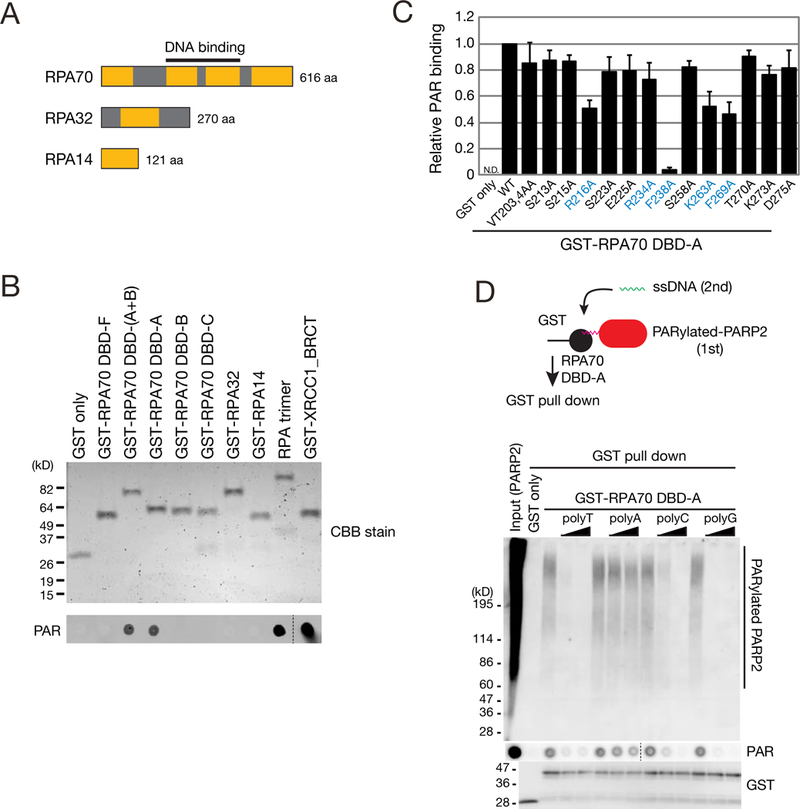
(A) The RPA heterotrimer consists of RPA70, RPA32, and RPA14 and possesses six OB-fold DNA binding domains (DBDs) (orange boxes, A-F).
(B) RPA70 DBD-A is sufficient to bind PAR. Top: The indicated N-terminal GST-tagged DBD proteins were purified from E. coli, subjected to SDS-PAGE, and visualized by CBB staining. Bottom: purified GST-DBD fusion proteins were analyzed for PAR binding assay (STAR METHODS). Recombinant GST-only and GST-XRCC1-BRCT domain proteins served as negative and positive controls, respectively (Li et al., 2013). The dotted black line indicates a spliced boundary between two different areas from the same membrane.
(C) PAR binding assays using RPA70 DBD-A missense mutations. The indicated GST-DBD fusion proteins were purified and assessed for PAR binding. PAR signal intensities were quantified and normalized to the negative control GST-only signals. X-axis, GST-fusion proteins. Y-axis, the level of PAR binding relative to the WT GST-RPA70-DBD-A fusion protein (set at 1.0). Standard errors were calculated from three independent biological replicates. Representative gel images are shown in Figure S5E.
(D) Single strand DNAs compete with PARylated PARP2 binding to RPA70 DBD-A. Top: the GST-RPA70 DBD-A was incubated with activated PARP2 in the absence or presence of ssDNA competitors followed by GST-pull down assays. Middle: PARP2-associated PAR/GST-RPA70-DBD-A complexes formed in the absence or presence (100 or 1,000 nM) of ssDNA competitors (polyT, TM154; polyA, TM155; polyC, TM156; polyG, TM157) were separated on an SDS-PAGE gel or spotted onto a nitrocellulose membrane. PAR was detected by western blot. The dotted black line indicates a spliced boundary between two different areas from the same membrane. Bottom: The GST-tagged fusion proteins were detected by western blot.
We next used GST fusion proteins containing different RPA OB-fold domains to determine which RPA OB-fold domains bind PAR. The RPA70 DBD-A domain was sufficient to bind PAR (Figure 5B). Moreover, GST/DBD-A fusion proteins from mouse, fish, frog, and yeast RPA70 homologs exhibited both ssDNA and PAR binding activity (Figures S5C and S5D), indicating PAR recognition by DBD-A domain is conserved among eukaryotes.
To identify DBD-A amino acid residues important for PAR binding, we generated GST-DBD-A fusion proteins containing alanine mutations in conserved amino acids (Figures 5C, S5C, and S5E). The R216A, K263A, F269A, and F238A DBD-A mutant proteins exhibited ~50% or less PAR binding when compared to the WT DBD-A control; these four amino acid residues directly contact ssDNA (Bochkarev et al., 1997).
The DBD-A domain likely recognizes both ssDNA and PAR. Thus, we performed DBD-A/PAR binding experiments in the absence or presence of homopolymeric ssDNA competitors followed by a GST-pull down assay (Figures 5D and S5F). The DBD-A domain efficiently pulled down PARP2-associated and recombinant PAR. These interactions were decreased upon adding increasing concentration of ssDNAs to the reactions, but the poly(A) oligonucleotide was the least effective competitor (Figures 5D and S5F), which agrees with previous studies (Kim et al., 1992). Why poly(A) is a less effective competitor requires further exploration, but may result, in part, from its ability to form higher order DNA structures (e.g., double helical or triplex structures) that impede RPA binding (Chakraborty et al., 2009; Tubbs et al., 2018).
RPA protects single strand DNA from cytidine deamination by APOBEC3A
The overexpression of A3A, a member of the APOBEC3 family of cytidine deaminase proteins, can inhibit L1 retrotransposition by deaminating transiently exposed ssDNAs during TPRT (Richardson et al., 2014). Thus, we tested whether RPA could counteract A3A-mediated deamination. Controls confirmed recombinant A3A (rA3A) could mediate C-to-U deamination in a single strand oligonucleotide substrate, whereas a rA3A mutant (C106S) lacking deaminase activity could not (Figure S6A) (Richardson et al., 2014). The addition of increasing amounts of RPA trimer (RPA70 WT) significantly inhibited A3A-mediated deamination, but a mutant RPA70 F238A trimer was less effective at inhibiting deamination (Figure 6A).
Figure 6. RPA protects single strand DNA from A3A-medited cytosine deamination.
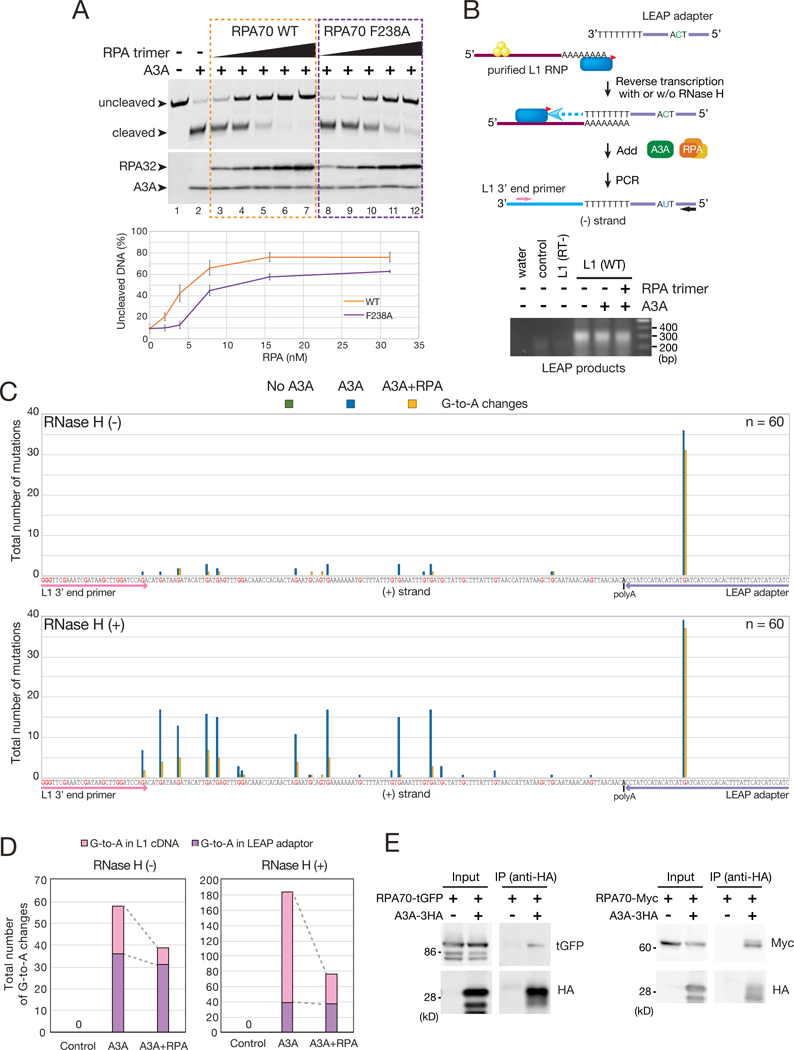
(A) RPA suppresses A3A-mediated deamination. Top: a single-strand LEAP adapter oligonucleotide (TM165) was incubated with His-A3A in the absence or presence of the WT RPA trimer or a mutant RPA trimer containing a F238A RPA70 subunit at concentrations ranging from 31.25 to 1.95 nM. Arrows indicate the uncleaved (no deamination) and cleaved (deamination) ssDNAs. A3A and RPA32 were detected by western blot. Bottom: quantification of non-deaminated ssDNA. X-axis, the RPA concentration. Y-axis, the percentage of uncleaved ssDNA. A reaction conducted in the absence of A3A and RPA (lane 1) was set to 100% uncleaved ssDNA. Error bars represent the standard errors calculated from three independent experiments.
(B) Rationale of the LEAP Assay. Top: The yellow circles and blue oval with the red triangle indicate ORF1p and ORF2p-3FLAG, respectively. The pink and black arrows indicate primers, which are specific to the engineered L1 expression vector (TM160) and the LEAP adapter (TM159), respectively. These primers were used to PCR amplify L1 cDNAs. Bottom: LEAP assays were conducted with purified L1 RNPs in the absence or presence of A3A and/or RPA. The representative image was obtained after PCR amplification of the LEAP products in the presence of RNase H. L1 RNPs isolated from cells transfected with pJM101/L1.3 (control) or pTMF3D702A (RT-) served as negative controls.
(C) Sequence analysis of LEAP products in the absence (top) or presence (bottom) of RNase H. Reactions were conducted in the absence of A3A (green), the presence of A3A (blue), or the presence of A3A and RPA (yellow). X-axis, the (+) strand L1 cDNA LEAP sequence. Guanine nucleotides are shown in red. Sequences underlined with pink and purple denote the L1 3’ end primer (TM160) and LEAP adapter (TM158), respectively. Outlined “A”, the position of the variably sized poly(A) tract. Y-axis, total number of G-to-A mutations. Non-A3A-mediated mutations are shown in Figure S6D.
(D) Distribution of A3A-mediated mutations in LEAP products in the absence or presence of RNase H. X-axis, LEAP reaction condition. Y-axis, total number of G-to-A mutations on the (+) strand L1 cDNA (pink) and LEAP adapter (purple).
(E) RPA association with A3A. HEK293T cells were transfected with A3A-3HA and either the RPA70-turboGFP (tGFP) or RPA70-Myc expression plasmids. A3A-3HA was immunoprecipitated using an anti-HA antibody; associated proteins were examined by western blot. HEK293T cells transfected with pcDNA3.1(+), instead of A3A-3HA, served as a negative control.
We next used the L1 Element Amplification Protocol (LEAP) (Figures 6B and S6B) to determine whether RPA could protect ssDNA L1 retrotransposition intermediates from C-to-U deamination in vitro (Richardson et al., 2014; Kulpa and Moran, 2006). LEAP cDNA products were detected from WT, but not RT-defective, L1 RNPs (Figure 6B; STAR METHODS) and LEAP products generated in the presence of A3A exhibited evidence of C-to-U deamination at the 5’-TCA-3’ consensus sequence on (−) strand L1 cDNA and occurred most frequently in the LEAP oligonucleotide primer adapter (Figures 6C, 6D, S6C, S6D, and Table S2; denoted as G-to-A mutations on the L1 (+) strand cDNA sequence) (Richardson et al., 2014).
The L1 (−) cDNA remains associated with L1 RNA during the LEAP reaction, which protects the L1 cDNA, but not the LEAP oligonucleotide primer, from A3A-mediated deamination (Richardson et al., 2014). Addition of RNase H to the LEAP reaction resulted in A3A-mediated deamination events throughout the L1 cDNA at the A3A consensus sequence (Figure 6C) (Richardson et al., 2014) The characterized LEAP products had variable sized poly(A) tracts and different C-to-U mutations patterns, indicating they were derived from independent L1 cDNA molecules (Table S2). A3A-mediated C-to-U deamination events were only modestly decreased in the LEAP oligonucleotide adapter in either the presence or absence of RNase H when RPA was included in the LEAP reaction (Figures 6C and 6D), which could be due to the small size of the LEAP oligonucleotide adapter (58 nucleotides) or lower RPA/substrate ratios used in LEAP reactions when compared to in vitro deamination reactions (Figure 6A; STAR METHODS). Regardless, we observed a significant reduction in A3A-mediated C-to-U deamination events throughout the L1 cDNA upon the inclusion of RPA in the reaction (Figures 6C and 6D). Because RNase H2 is required for efficient L1 retrotransposition (Benitez-Guijarro et al., 2018), we speculate that RPA may bind transiently exposed single strand L1 cDNAs that arise upon RNase H2 digestion during TPRT to protect them from C-to-U deamination.
Although A3A is a potent inhibitor of L1 retrotransposition, how it gains access to L1 TPRT ssDNA intermediates requires elucidation. Previous studies reported RPA interacts with activation-induced cytidine deaminase (AID) to mediate somatic hyper-mutation during immunoglobulin class switch recombination (Basu et al., 2008; Chaudhuri et al., 2004). IP experiments demonstrated, that analogous to the situation observed for AID, A3A could interact with RPA70 (Figure 6E).
DISCUSSION
Diverse nuclear proteins interact with L1 ORF2p
Previous studies revealed that a putative PCNA-interacting protein box within L1 ORF2p (Taylor et al., 2013), proteins involved in NHEJ (Suzuki et al., 2009), and an association between the L1-encoded proteins and DNA replication machinery facilitate retrotransposition (Flasch et al., 2019; Sultana et al., 2019). Here, we validated seven new interactions between ORF2p and nuclear proteins that participate in DNA repair and/or DNA replication (Figure 1). We also validated the interaction of ORF2p with SPT16, a subunit of the FACT complex that acts as a histone chaperone to reorganize nucleosomes during transcription, replication, and DNA repair (Ransom et al., 2010) and HMCES, a protein that binds oxidized derivatives of methyl-cytosine that arise during DNA demethylation (Figure 1 and Table S1) (Spruijt et al., 2013). In most cases, DNA enhanced the association between ORF2p and these proteins in an L1 EN and/or RT-dependent manner. (Figure 3 and S3). Thus, interactions between L1 RNPs and a diverse set of host proteins likely play critical roles in L1 retrotransposition.
PARP2 specifically recognizes DNA breaks generated by ORF2p during TPRT
L1 EN activity is critical for the initiation of TPRT (Cost et al., 2002; Feng et al., 1996). Overexpression of L1 ORF2p can lead to the formation of dsDNA breaks in cultured human cells (Gasior et al., 2006), whereas proteins involved in the DNA damage response may target TPRT intermediates to restrict L1 retrotransposition (Coufal et al., 2011; Gasior et al., 2008; Servant et al., 2017). However, how host proteins “sense” L1 EN breaks remains an open question. Studies using bicistronic L1 expression constructs did not detect significant ORF2p/PARP2 interactions (Taylor et al., 2018; Taylor et al., 2013). By comparison, expressing ORF2p from a monocistronic construct provided several lines of evidence that PARP2 specifically recognizes L1 EN single strand breaks generated during TPRT (Figure 7). First, EN-deficient ORF2p mutants could not interact with PARP2 (Figure 3). Second, dsDNA substrates mimicking an L1 EN cleavage site led to PARP2 auto-PARylation (Figure 4). Third, activated PARP2 directly interacted with RPA, which could localize it to presumptive TPRT intermediates (Figure 5). Finally, PARP2 or RPA knockdown led to a reduction in engineered L1 retrotransposition efficiency (Figures 2 and 4). Thus, we propose PARP2 PARylation acts as a post-translational modification to recruit RPA to L1 EN-induced breaks during TPRT to facilitate L1 retrotransposition.
Figure 7. Working model for how PARP2 and RPA facilitate L1 retrotransposition.
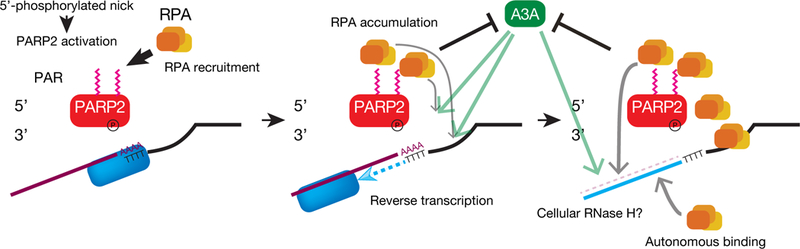
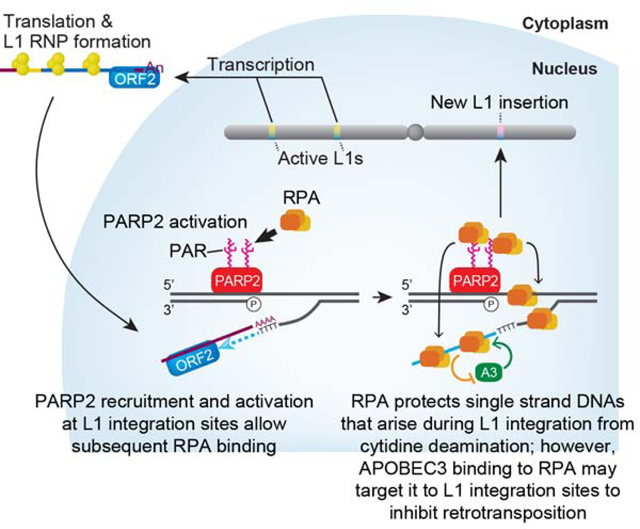
ORF2p EN activity creates a nick in genomic DNA to generate a 5’ phosphate and 3’ hydroxyl group to initiate TPRT. The 5’ phosphate group is recognized by PARP2, which leads to a local accumulation of PAR (magenta zigzags on PARP2). PARP2 PARylation recruits RPA to the L1 integration site. RPA may then bind to transiently exposed ssDNA regions (i.e., ssDNA on the DNA strand opposed to the L1 EN break and/or the newly synthesized L1 cDNA) that arise during TPRT (curved gray arrows) to protect ssDNAs from forming secondary structures and/or attack by APOBEC3 cytidine deaminase proteins. The association of APOBEC3 with RPA might allow it to localize to L1 TPRT intermediates.
PARP1 is also critical for L1 retrotransposition
PARP1 knockdown led to a reduction in engineered L1 retrotransposition efficiency. However, the association of L1 ORF2p with PARP1 is likely mediated by protein-protein interactions (Figure S3B). Like PARP2, PARP1 can undergo auto-PARylation and can PARylate other cellular proteins. The addition of a long PAR chain is predicted to impart a highly negative charge to a target protein, which could alter its conformation and/or affect its ability to bind nucleic acids (Gibson and Kraus, 2012). Because L1 ORF2p is PARylated in vivo and in vitro (TM and JVM, unpublished data), we speculate PARP1-mediated PARylation may modulate ORF2p enzymatic activities (e.g., by triggering ORF2p conformational changes that allow it to switch from endonuclease to reverse transcriptase activities) and/or alter ORF2p nucleic acid binding during TPRT. Alternatively, PARP1 PARylation could lead to the recruitment of host proteins to L1 integration sites. For example, PARP1 participates in the alternative-NHEJ (alt-NHEJ) pathway of DNA repair (Hustedt and Durocher, 2016). Because sequence microhomologies are often observed at genomic DNA/5’ L1 cDNA junctions (Gilbert et al., 2005; Kojima, 2010; Zingler et al., 2005), we speculate that PARylated PARP1 may facilitate L1 integration via alternative-NHEJ.
How is RPA recruited to TPRT intermediates?
PARP1 and PARP2 can indirectly recruit RPA to ssDNA through the regulation of a cellular nuclease activity (Bryant et al., 2009). We demonstrated that the RPA DBD-A domain could directly bind to PARylated PARP2, providing a pathway to directly recruit RPA to TPRT intermediates independently of ssDNA (Figure 4). Because the PAR used in our assays is heterogeneous in length, we cannot determine whether RPA binds PAR or ssDNA with a higher affinity. However, single strand oligonucleotides can outcompete PAR for RPA70-DBD-A binding (Figures 5 and S5). Thus, we propose that RPA may initially bind to activated PARP2 then transfer to ssDNAs to stabilize TPRT intermediates, which could, in principle, protect ssDNA intermediates generated during TPRT from nucleolytic degradation and/or prevent the formation of secondary structures that impair L1 cDNA synthesis and/or the completion of L1 integration (Figure 7).
Do APOBEC3 proteins associate with TPRT intermediates by interacting with RPA?
Although A3A is not expressed in endogenous cell types that accommodate L1 retrotransposition in vivo (Richardson et al., 2014), we provide proof of principle data that RPA may protect ssDNAs generated during TPRT from C-to-U deamination (Figure 6) in vitro and that A3A can associate with RPA (Figure 6E). Of note, RPA also attenuates DNA deamination induced by the ectopic expression of A3A and A3B in yeast cells during lagging strand DNA replication (Hoopes et al., 2016). Thus, these data raise the possibility that, analogous to AID (Basu et al., 2008; Chaudhuri et al., 2004), interactions between RPA and APOBEC3 proteins expressed in cells that support endogenous L1 retrotransposition in vivo (e.g., APOBEC3B) (Bogerd et al., 2006) may guide APOBEC3 proteins to ssDNA L1 integration intermediates. If so, such a scenario would provide an example of genetic conflict, whereby a host protein required for efficient retrotransposition subsequently becomes a target of a restriction factor to inhibit transposition (Figure 7).
STAR METHODS
CONTACTS FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact: Tomoichiro Miyoshi (miyoshi.tomoichiro.5e@kyoto-u.ac.jp).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture
HeLa-JVM (Moran et al., 1996) and HEK293T cells were grown at 37°C in 100% humidified incubators supplied with 5 or 7% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) high glucose medium (Gibco or Nacalai Tesque) supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U/mL penicillin, 100 μg/mL streptomycin, and 290 μg/mL L-glutamine (DMEM complete medium).
XR-1 Chinese Hamster Ovary (CHO) cells (Morrish et al., 2002) were grown at 37°C in 100%-humidified incubators supplied with 7% CO2 in DMEM low glucose medium (Gibco) supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 290 μg/mL L-glutamine, and 1X non-essential amino acids (Gibco) (DMEM low glucose medium).
Platinum-A (Plat-A) (Morita et al., 2000) (kindly gifted from Dr. T. Kitamura) cells were used and grown at 37°C in 100%-humidified incubators supplied with 5% CO2 in DMEM high glucose medium (Naclai Tesque) supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 290 μg/mL L-glutamine (DMEM complete medium).
The absence of Mycoplasma spp. was confirmed using a PCR-based assay (Sigma). STR-genotyping was used to validate the identity of HeLa-JVM and HEK293T cells. XR-1 cells were verified in the following publications (Morrish et al., 2007; Morrish et al., 2002).
METHOD DETAILS
Plasmids used in this study
Plasmids for mammalian transfection experiments were purified using a Midiprep Plasmid DNA kit (Qiagen) or GenElute HP Plasmid Midiprep Kit (Sigma). Plasmids for bacterial expression experiments were purified using a Wizard Plus SV Minipreps DNA Purification Systems (Promega) or GenElute Plasmid Miniprep Kit (Sigma). All L1-expressing plasmids contain a retrotransposition-competent “hot” L1 (L1.3; accession number L19088) subcloned into the pCEP4 episomal expression plasmid (Invitrogen) unless noted elsewhere. The amino acid residues of ORF2p were counted from the first methionine of the L1.3 ORF2p sequence.
pJM101/L1.3: was described previously (Sassaman et al., 1997). The full-length L1.3 is cloned into the pCEP4 plasmid. The cytomegalovirus immediate early (CMV) promoter and L1 5’ UTR augment L1.3 expression. The mneoI retrotransposition indicator cassette was inserted into the L1.3 3’ UTR as described previously (Moran et al., 1996).
pTMF3: is a derivative of pJM101/L1.3. A sequence encoding a T7 gene10 or three tandem copies of a FLAG epitope-tag was fused in-frame to the 3’ end of the L1.3 ORF1- or ORF2-coding regions, respectively.
pJM101/L1.3FLAG: was described previously (Moldovan and Moran, 2015). This plasmid is a derivative of pJM101/L1.3 and contains a single FLAG epitope tag sequence fused in frame to the 3’ end of the L1.3 ORF1-coding region.
pDK101: was described previously (Kulpa and Moran, 2005). This plasmid is a derivative of pJM101/L1.3 and contains a T7 gene10 epitope tag sequence fused in frame to the 3’ end of the L1.3 ORF1-coding region.
pTMO2F3: was described previously (Doucet et al., 2015). It contains a monocistronic L1.3 ORF2-coding sequence containing three tandem copies of a FLAG epitope-tag sequence fused in frame to the 3’ end of the L1.3 ORF2-coding region. A CMV promoter and the L1 5’ UTR augment L1.3 ORF2 expression. This plasmid does not contain a retrotransposition indicator cassette.
pAD001: was described previously (Doucet et al., 2015). It is similar to pTMO2F3, but lacks the FLAG epitope-tag sequence.
pAD035: is identical to pAD001, but contains the D702A mutation in the reverse transcriptase domain of ORF2p.
pAD500: was described previously (Doucet et al., 2010). It is similar to pTMO2F3, but contains a TAP epitope-tag sequence fused in frame to the 3’ end of the L1.3 ORF2-coding region.
pTMO2F3D145A: is identical to pTMO2F3, but contains the D145A mutation in the endonuclease domain of ORF2p (Wei et al., 2001).
pTMO2F3D205A: is identical to pTMO2F3, but contains the D205A mutation in the endonuclease domain of ORF2p (Wei et al., 2001).
pTMO2F3D702A: is identical to pTMO2F3, but contains the D702A mutation in the reverse transcriptase domain of ORF2p (Wei et al., 2001).
pTMO2F3D145AD702A: is identical to pTMO2F3, but contains the D145A and D702A mutations in the endonuclease and reverse transcriptase domains of ORF2p, respectively (Wei et al., 2001).
pTMO2F3D205AD702A: is identical to pTMO2F3, but contains the D205A and D702A mutations in the endonuclease and reverse transcriptase domains of ORF2p, respectively (Wei et al., 2001).
pTMO2F3C1130S/C1134S: is identical to pTMO2F3, but contains the C1130S and C1134S mutations in the C-terminus cysteine-rich domain of ORF2p (Moran et al., 1996; Wei et al., 2001).
pTMO2F3Z-C: is similar to pTMO2F3, but only contains the partial ORF2 sequence encoding K240 to the end of ORF2p.
pTMO2F3RT-C: is similar to pTMO2F3, but only contains the partial ORF2 sequence encoding Y481 to the end of ORF2p.
pTMO2F3pRT-C: is similar to pTMO2F3, but only contains the partial ORF2 sequence encoding T774 to the end of ORF2p.
pTMO2F3C: is similar to pTMO2F3, but only contains the partial ORF2 sequence encoding T1025 to the end of ORF2p.
pTMO2F3EN-pRT: is similar to pTMO2F3, but only contains the partial ORF2 sequence encoding the first methionine to E1024.
pTMO2F3EN-RT: is similar to pTMO2F3, but only contains the partial ORF2 sequence encoding the first methionine to L773.
pTMO2F3EN-Z: is similar to pTMO2F3, but only contains the partial ORF2 sequence encoding the first methionine to Q497.
pTMO2F3EN: is similar to pTMO2F3, but only contains the partial ORF2 sequence encoding the first methionine to R379.
pTMO2F3ΔZ: is similar to pTMO2F3, but lacks the ORF2 sequence encoding L380 to F480.
pTMO2F3ΔRT: is similar to pTMO2F3, but lacks the ORF2 sequence encoding S498 to L773.
pTMO2F3ΔpRT: is similar to pTMO2F3, but lacks the ORF2 sequence encoding T774 to E1024.
phrGFP-C (Agilent Technologies): contains the humanized renilla green fluorescent protein gene driven by the CMV promoter.
pJJ101/L1.3: was described previously (Kopera et al., 2011). It is similar to pJM101/L1.3 except that it contains the mblastI retrotransposition indicator cassette in the 3’ UTR of L1.3.
pJJ105/L1.3: was described previously (Kopera et al., 2011). It is identical to pJJ 101/L1.3, but contains the D702A mutation in the ORF2p reverse transcriptase domain.
pJJD205A/L1.3: was described previously (Kopera et al., 2011). It is identical to pJJ 101/L1.3, but contains the D205A mutation in the ORF2p endonuclease domain.
pcDNA6: is a derivative of pcDNA6/TR (Invitrogen). It contains the blasticidin S-deaminase (BSD) gene but lacks the TetR gene. Dr. John B. Moldovan (University of Michigan Medical School) generated this plasmid.
cep99-gfp-L1.3: contains a full-length L1.3 and an enhanced green fluorescent protein retrotransposition indicator cassette (mEGFPI) in the 3’UTR of the L1.3 (Ostertag et al., 2000). The L1 construct was cloned into a modified-pCEP4 vector that contains a puromycin resistant gene as a selectable marker. The L1 5’ UTR drives L1 expression.
cep99-gfp-L1.3RT(−): is identical to cep99-gfp-L1.3, but contains the D702A mutation in the ORF2p reverse transcriptase domain.
cep99-gfp-L1.3RT(−) intronless: is similar to cep99-gfp-L1.3RT(−), but lacks the intron sequence in the mEGFPI cassette, allowing cells with this plasmid to express EGFP without retrotransposition (Wissing et al., 2011).
cepB-gfp-L1.3: is similar to cep99-gfp-L1.3, but contains the blasticidin S-deaminase (BSD) gene from pcDNA6/TR as a selectable marker.
cepB-gfp-L1.3RT(−): is identical to cepB-gfp-L1.3, but contains the D702A mutation in the ORF2p reverse transcriptase domain.
cepB-gfp-L1.3RT(−) intronless: is similar to cepB-gfp-L1.3RT(−), but lacks the intron sequence in the mEGFPI cassette (Wissing et al., 2011).
cepZ-gfp-L1.3: is similar to cep99-gfp-L1.3, but contains the Zeocin resistant gene from pFRT//acZeo (Invitrogen) as a selectable marker.
cepZ-gfp-L1.3RT(−): is identical to cepZ-gfp-L1.3, but contains the D702A mutation in the ORF2p reverse transcriptase domain.
cepZ-gfp-L1.3RT(−) intronless: is similar to cepZ-gfp-L1.3RT(−), but lacks the intron sequence in the mEGFPI cassette (Wissing et al., 2011).
pLKO.1-TurboGFP: is a lentivirus vector (Sigma: SHC002) that expresses short hairpin (sh) RNA targeted to the TurboGFP gene. Due to sequence differences, this shRNA construct does not target the EGFP gene. Cells expressing this shRNA were used as a control for shRNA-based experiments described in this study.
pLKO.1-PARP1: (Sigma: SHCLNG-NM001618, clone#: TRCN0000356550) is a lentivirus vector that expresses shRNA targeted to the 3’ UTR of the human PARP1 gene.
pLKO.1-PARP2: (Sigma: SHCLNG-NM005484, clone#: TRCN0000235596) is a lentivirus vector that expresses shRNA targeted to the coding sequence of the human PARP2 gene.
psMD2.G: is a lentivirus envelope expression vector that was a gift from Didier Trono (Addgene plasmid # 12259).
psPAX2: is a lentivirus packaging vector that was a gift from Didier Trono (Addgene plasmid # 12260).
pMX-hyg: is a retrovirus vector that is a derivative of the pMX vector series described previously (Kitamura et al., 2003) (kindly gifted from Dr. T. Kitamura). It contains the hygromycin resistant gene and was used as a mock control for the pMX-hyg-RPA70myc plasmid.
pMX-hyg-RPA70myc: contains a Myc-tagged RPA70 coding sequence in pMX-hyg. The Myc tag sequence was inserted at the 3’ end of RPA70.
pGEX-2T: (GE Healthcare) is a bacterial expression vector that expresses glutathione S-transferase (GST). This plasmid was used to obtain GST alone as a negative control for both the in vitro DNA-binding and PAR-binding assays (Figures 5 and S5).
pGEX-2T-OBF: expresses GST-fused to the DBD-F domain (amino acids 1–150) of human RPA70.
pGEX-2T-OB(A+B): expresses GST-fused to the DBD-A+B domain (amino acids 151–433) of human RPA70.
pGEX-2T-OBA: expresses GST-fused to the DBD-A domain (amino acids 151–333) of human RPA70.
pGEX-2T-OBB: expresses GST-fused to the DBD-B domain (amino acids 281–433) of human RPA70.
pGEX-2T-OBC: expresses GST-fused to the DBD-C domain (amino acids 424–616) of human RPA70.
pGEX-2T-RPA32: expresses GST-fused to full-length human RPA32.
pGEX-2T-RPA14: expresses GST-fused to full-length human RPA14.
pGEX-2T-XRCC1-BRCT: expresses GST-fused to the BRCT-domain (amino acids 274–432) of human XRCC1.
pGEX-2T-mOBA: expresses GST-fused to the DBD-A domain (amino acids 160–341) of Mus musculus RPA70. The RPA70 cDNA (MMM1013-202766360) was obtained from GE Healthcare Dharmacon.
pGEX-2T-dOBA: expresses GST-fused to the DBD-A domain (amino acids 145–319) of the Danio rerio RPA70 homolog. The RPA70 cDNA (MDR1734-202803987) was obtained from GE Healthcare Dharmacon.
pGEX-2T-xOBA: expresses the GST-fused DBD-A domain (amino acids 148–323) of the Xenopus laevis RPA70 homolog. The RPA70 cDNA (MXT1765-202786446) was obtained from GE Healthcare Dharmacon.
pGEX-2T-scOBA: expresses GST-fused to the DBD-A domain (amino acids 156–332) of the Saccharomyces cerevisiae RPA70 homolog. The RPA70 cDNA (YSC3867-202325048) was obtained from GE Healthcare Dharmacon.
pGEX-2T-OBA(VT203,204AA): is identical to pGEX-2T-OBA, but contains the V203A and T204A mutations in the DBD-A domain.
pGEX-2T-OBA(S213A): is identical to pGEX-2T-OBA, but contains the S213A mutation in the DBD-A domain.
pGEX-2T-OBA(S215A): is identical to pGEX-2T-OBA, but contains the S215A mutation in the DBD-A domain.
pGEX-2T-OBA(R216A): is identical to pGEX-2T-OBA, but contains the R216A mutation in the DBD-A domain.
pGEX-2T-OBA(S223A): is identical to pGEX-2T-OBA, but contains the S223A mutation in the DBD-A domain.
pGEX-2T-OBA(E225A): is identical to pGEX-2T-OBA, but contains the E225A mutation in the DBD-A domain.
pGEX-2T-OBA(R234A): is identical to pGEX-2T-OBA, but contains the R234A mutation in the DBD-A domain.
pGEX-2T-OBA(F238A): is identical to pGEX-2T-OBA, but contains the F238A mutation in the DBD-A domain.
pGEX-2T-OBA(S258A): is identical to pGEX-2T-OBA, but contains the S258A mutation in the DBD-A domain.
pGEX-2T-OBA(K263A): is identical to pGEX-2T-OBA, but contains the K263A mutation in the DBD-A domain.
pGEX-2T-OBA(F269A): is identical to pGEX-2T-OBA, but contains the F269A mutation in the DBD-A domain.
pGEX-2T-OBA(T270A): is identical to pGEX-2T-O, but contains the T270A mutation in the DBD-A domain.
pGEX-2T-OBA(K273A): is identical to pGEX-2T-OBA, but contains the K273A mutation in the DBD-A domain.
pGEX-2T-OBA(D275A): is identical to pGEX-2T-OBA, but contains the D275A mutation in the DBD-A domain.
hA3A-pET-28a: expresses human APOBEC3A protein containing an in-frame hexa-histidine-tag at its amino-terminus. The cDNA was cloned into a pET-28a(+) vector (Merck Millipore).
hA3AC106S-pET-28a: is identical to hA3A-pET-28a, but contains the C106S mutation in the hA3A coding sequence.
pK A3A: was described previously (Bogerd et al., 2006; Richardson et al., 2014). This plasmid contains three tandem in-frame copies of an HA epitope tag at the 3’ end of A3A.
RPA70 (RPA1) (NM 002945) Human Tagged ORF Clone (ORIGENE: RG202066): is a mammalian vector that expresses a human RPA70 protein containing an in-frame carboxyl-terminal turboGFP tag.
RPA70myc-pCMV6AC: is similar to the RPA70 (RPA1) (NM_002945) Human Tagged ORF Clone, but expresses a human RPA70 protein containing an in-frame carboxyl-terminal Myc-tag.
pCMV-3Tag-8 (Stratagene: 240203): is a C-terminal triple FLAG tagging vector.
PARP1/ pCMV-3Tag-9: is a pCMV-3Tag-8 derivative vector that expresses a human PARP1 containing an in-frame carboxyl-terminal triple FLAG tag.
PARP1E988K/ pCMV-3Tag-9: is identical to PARP1/ pCMV-3Tag-9, but contains the E988K mutation in the PARP catalytic domain.
3FLAG C1-PARP2: is a mammalian vector that expresses a human PARP2 containing an in-frame amino-terminal triple FLAG tag.
3FLAG C1-PARP2E558K: is identical to 3FLAG C1-PARP2, but contains the E558K mutation in the PARP catalytic domain.
Immunoprecipitation-coupled mass spectrometry analysis
To obtain ORF2p expressing cells, HEK293T cells were plated in a T175 flask (BD Biosciences) containing DMEM-complete medium at approximately 5×106 cells per flask. Two flasks were used for transfection with each plasmid DNA. The following day (day 0), the cells were transfected with 15 μg of pAD500 or pTMO2F3 DNA using 45 μL of the FuGENE6 transfection reagent (Promega) and 1.5 mL of Opti-MEM (Gibco) according to the manufacturer’s protocol. The following day (day 1), the transfection medium was replaced with fresh DMEM-complete medium. On days 2 through 5, the medium was replaced daily with DMEM-complete medium containing 200 μg/mL hygromycin B (Gibco). On day 5, cells were collected, washed with cold 1X PBS (Gibco), and flash frozen in dry ice ethanol bath.
The following steps were performed on ice or at 4°C unless noted otherwise. Five mL of Lysis300 buffer (20 mM Tris-HCl (pH 7.5), 2.5 mM MgCl2, 1 mM dithiothreitol (DTT), 300 mM KCl, 0.1% IGEPAL CA-630) containing 1X cOmplete EDTA-free protease inhibitor cocktail (Roche), and 0.2 mM phenylmethylsufonyl fluoride (PMSF) was added to the transfected cells and the cells were incubated for 1 hour with gentle rotation. The insoluble cell debris was removed by centrifugation at 12,000 rpm for 5 minutes and the supernatants were transferred into a new tube. Small aliquots from each sample were saved as the “Input” fraction in Figure 1. The remaining lysates were incubated with 100 of EZview Red ANTI-FLAG M2 Affinity Gel (Sigma) (50% slurry in 1X PBS) overnight with gentle rotation. The following day, the lysates were centrifuged at 3,000 rpm for ~2 to 3 minutes and the supernatants were saved as the “Flow through” fraction. After removing the supernatants, the beads were washed five times with 1 mL of the Lysis300 buffer. To elute the antibody-attached proteins, the beads were incubated twice with 100 μL of the Lysis300 buffer containing 200 μg/mL of 3X FLAG peptide (Sigma) for ~1 hour each time. The eluents were mixed together (~200 μL in total per sample) and 5 volumes of ice-cold acetone (1 mL) were added to the eluent followed by incubation at −20°C for 4–5 hours. After centrifugation at 12,000 rpm for 30 minutes, the supernatants were removed and the resultant pellets were dried at room temperature to allow evaporation of the solvents. The dried pellets then were resuspended in 50 μL of 1X Laemmli Sample Buffer (Bio-Rad) containing with 50 mM DTT and boiled at 100°C for 5 minutes. Fifteen microliters of the sample were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis and the proteins were visualized using the SilverQuest Silver Staining Kit (Invitrogen) according to the manufacturer’s protocol.
The proteomics core facility at the Fred Hutchinson Cancer Research Center performed mass spectrometry analyses as described previously (Moldovan and Moran, 2015). Briefly, an entire gel lane derived from each sample was excised into several gel fragments. After removing silver ions, these slices were incubated with sequencing-grade modified trypsin and the extracted peptides were subjected to liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) on an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). The detected MS/MS spectra were analyzed and searched against the human International Protein Index (IPI) database using the SEQUEST HT search engine and the Proteome Discoverer software (Thermo Fisher Scientific) with peptide sequence information that showed a ≤0.05 false discovery rate. Common laboratory contaminants (e.g., keratin) were filtered manually and excluded using the common Repository of Adventitious Proteins, cRAP database (UniProt). To identify the ORF2p-3FLAG interacting proteins, the proteins detected in the control (ORF2p-TAP: pAD500) were subtracted from the list obtained from the ORF2p-3FLAG (pTMO2F3) precipitate. The proteins that were found only with ORF2p-3FLAG, but not with the control sample, are shown in Table S1A. Several proteins that were significantly enriched in the ORF2p-3FLAG precipitate, but were also found in the negative control sample, are listed in Table S1B.
Immunoprecipitation and western blotting
To obtain the L1-expressing cells, HEK293T cells were plated in a T175 flask (BD Biosciences) containing DMEM-complete medium at approximately 5×106 cells per flask. A single flask was used for each sample preparation. On the following day (day 0), cells were transfected with 15 μg of a plasmid DNA using 45 μL of the FuGENE6 transfection reagent and 1.5 mL of Opti-MEM according to the manufacturer’s protocol. The following day (day 1), the medium was replaced with fresh DMEM-complete medium. From days 2 through 5, the medium was replaced daily with DMEM-complete medium containing 200 μg/mL hygromycin B. On day 5, the transfected cells were harvested, washed with cold 1X PBS, and stored at −80°C. When treated with a PARP inhibitor, transfected cells were incubated with DMEM-complete medium containing 10 μM of olaparib (AZD2281; Selleck Chemicals) overnight prior to cell collection on day 5.
The following steps were performed on ice or at 4°C unless noted otherwise. Two mL of Lysis300 buffer (20 mM Tris-HCl (pH 7.5), 2.5 mM MgCl2, 1 mM dithiothreitol (DTT), 300 mM KCl, 0.1% IGEPAL CA-630) containing 1X cOmplete EDTA-free protease inhibitor cocktail (Roche), and 0.2 mM phenylmethylsufonyl fluoride (PMSF) was added to the transfected cells and the cells were incubated for 1 hour with gentle rotation. The cell debris was removed by centrifugation at 12,000 rpm for 5 minutes and the supernatants were placed into tubes. Ten microliters of each sample were saved as the “Input” fraction. Prior to immunoprecipitation, antibody attached beads were prepared by incubating 2 μg of anti-FLAG M2 antibody (Sigma) with 20 μL of Dynabeads Protein G (Life Technologies) in 1X PBS containing 0.1% Triton X-100 and 0.5% Bovine Serum Albumin fraction V (Sigma) for 3–4 hours. The beads were washed twice with the Lysis300 buffer. The cleared cell lysates were incubated with the anti-FLAG antibody-attached magnetic beads or 20 μl of EZview Red ANTI-FLAG M2 Affinity Gel (Sigma) (50% slurry in 1X PBS) for 3–4 hours with gentle rotation. The beads then were washed four times with 200 μL of the Lysis300 buffer without the proteinase inhibitor. When treated with nucleases, before washing with the Lysis300 buffer, the beads were incubated with either 1.5 μL of RNase cocktail Enzyme Mix (RNase A: 0.5 U/μL; RNase T1: 20 U/μL) (Ambion) and 0.5 μL of RNase I (100 U/μL) (Ambion) or 1 μL of benzonase (25 U/μL) (Merck Millipore) in 100 μL of the Lysis50 buffer (20 mM Tris-HCl (pH 7.5), 2.5 mM MgCl2, 1 mM dithiothreitol (DTT), 50 mM KCl, 0.1% IGEPAL CA-630) containing 1X cOmplete EDTA-free protease inhibitor cocktail and 0.2 mM PMSF at 37 °C for 5 minutes. The beads were incubated with 30 μL of the Lysis300 buffer containing 200 μg/mL of 3X FLAG peptide for ~1 hour with gentle rotation. The beads were removed and then boiled in 15 μL of the SDS sample buffer (0.25 M Tris-HCl (pH 6.8), 8.2% SDS, 40% glycerol, 400 mM DTT, 0.02% bromophenol blue) at 100°C for 5 minutes to collect the eluents. Five microliters of the input fraction and 10 μL of the eluent were subjected to SDS-PAGE and western blotting.
To detect γ-H2AX, HEK293T cells were plated in a 6-well plate (IWAKI) containing DMEM-complete medium at approximately 2×105 cells per well. The following day (day 0), the growing cells were transfected with 1 μg of plasmid DNA (pCEP4, pTMO2F3, or pTMO2F3D145AD702A) using 3 μL of the FuGENE HD transfection reagent and 100 μL of Opti-MEM. The following day (day 1), the medium was replaced with fresh DMEM-complete medium containing 100 μg/mL hygromycin B. On day 3, the transfected cells were harvested, washed with cold 1X PBS, and stored at −80°C. To induce DNA breaks as a positive control for γ-H2AX accumulation, pCEP4-transfected cells were incubated with DMEM-complete medium containing 15 μM of etoposide (Sigma) for 4 hours prior to cell collection on day 3. Frozen cells were resuspended in ~20 to 30 μL of 1XPBS containing 1X cOmplete EDTA-free protease inhibitor cocktail and incubated with the same volume of the SDS sample buffer containing 0.4 U/μL benzonase without bromophenol blue for 30 minutes on ice. The resultant cell lysates were boiled for 5 minutes and the protein concentration was measured using the Bradford Protein Assay (Bio-Rad). Five micrograms of each sample was subjected to western blotting after boiling with the bromophenol blue-containing SDS sample buffer.
The protein levels of PARP1 and PARP2 were examined as follows. The control (non-mammalian shRNA targeting), PARP1, PARP2, and PARP1/PARP2 (double knockdown) shRNA knockdown cells growing in DMEM-complete media were washed with 1X PBS, collected, and stored at −80°C until lysing. The cells were lysed with Radio-Immunoprecipitation Assay (RIPA) buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% IGEPAL CA-630, 0.5% Sodium Deoxycholate, 0.1% SDS) containing 1X cOmplete EDTA-free protease inhibitor cocktail on ice for 30 minutes. The insoluble cell debris was cleared by centrifugation at 12,000 rpm for 5 minutes at 4°C. Protein concentrations of the resultant supernatants were measured using the Bradford Protein Assay (Bio-Rad). Approximately 30 μg of proteins were subjected to western blotting. To detect ORF2p-3FLAG in these shRNA knockdown cells, each cell line was transfected with pTMO2F3, cells were collected on day 5 post-transfection, whole cell lysates were prepared using RIPA buffer, and the lysates were subjected to western blotting as described above.
To detect RPA/poly(ADP-ribose), RPA70-Myc expressing HEK293T cells (see “Virus transduction”) were plated in 10 cm dishes (Fine Plus International) containing DMEM-complete medium at approximately 2×106 cells per dish. On day 2 post plating, cells were incubated with DMEM-complete medium containing 10 μM of olaparib or DMSO alone overnight. On the following day, the cells were harvested, washed with cold 1X PBS, and stored at −80°C. Five hundred microliters of NETN100 buffer (20 mM Tris-HCl (pH 8), 100 mM NaCl, 0.5 mM EDTA, 0.5% IGEPAL C-630) containing 1X cOmplete EDTA-free protease inhibitor cocktail (Roche) and 0.2 mM PMSF was added to the collected cells and the cells were incubated for 1 hour with gentle rotation. The cell debris was removed by centrifugation at 12,000 rpm for 5 minutes and the supernatants were placed into tubes. Ten microliters of each sample were saved as the “Input” fraction. Prior to immunoprecipitation, the antibody-attached beads were prepared by incubating 1 μL of anti-Myc antibody (CST) with 10 μL of Dynabeads Protein G (Life Technologies) in 1X PBS containing 0.1% Triton X-100 and 0.5% Bovine Serum Albumin fraction V (Sigma) for 3–4 hours. The beads were washed twice with the NETN100 buffer. The cleared cell lysates were incubated with the anti-Myc antibody-attached magnetic beads for 3–4 hours with gentle rotation. The beads then were washed four times with 200 μL of the NETN100 buffer without the proteinase inhibitor. The beads were boiled in 20 μL of the SDS sample buffer at 100°C for 5 minutes and then were removed from the sample. Five microliters of the input fraction and 10 μL of the eluent were subjected to SDS-PAGE and western blotting.
To detect the association of RPA with A3A, HEK293T cells were plated in 10 cm dishes (Fine Plus International) containing DMEM-complete medium at approximately 2×106 cells per dish. On the following day (day 0), cells were transfected with 2.5 μg of pcDNA3.1(+) and RPA70 (RPA1) (NM_002945) Human Tagged ORF Clone, pK_A3A and RPA70 (RPA1) (NM_002945) Human Tagged ORF Clone, pcDNA3.1(+) and RPA70myc-pCMV6AC, or pK_A3A and RPA70myc-pCMV6AC using 15 μL of the FuGENE HD transfection reagent and 0.5 mL of Opti-MEM according to the manufacturer’s protocol. The following day (day 1), the medium was replaced with fresh DMEM-complete medium. On day 2, the transfected cells were harvested, washed with cold 1X PBS, and stored at −80°C. Immunoprecipitation was performed as described in the RPA/poly(ADP-ribose) interaction except for antibodies used for immunoprecipitation. Briefly, 1 μL of anti-HA antibody (BioLegend) instead of anti-Myc antibody was used and incubated with 10 μL of Dynabeads Protein G prior to immunoprecipitation.
Western blotting was performed following standard protocols. Briefly, protein samples were separated on a SDS-PAGE gel, followed by transfer to a polyvinylidene difluoride (PVDF) membrane (Immobilon-FL PVDF for a fluorescence-based detection or Immobilon-P PVDF for a chemiluminescence-based detection (Merck Millipore)) with 10 mM CAPS (3-(cyclohexylamino)-1-propanesulfonic acid (pH 11)) using a Mini Trans-Blot Electrophoretic Transfer Cell tank (Bio-Rad) according to the manufacturer’s protocol. The resultant membranes were incubated with 1X PBS containing 3% nonfat skim milk (Kroger or Nacalai Tesque). After washing four times with 1X PBS, the membranes were incubated with the primary antibodies shown in the KEY RESOURCES TABLE in 1X PBS containing 0.1% Tween-20 at 4 °C for overnight. The membranes were washed four times with 1X PBS containing 0.1% Tween-20, and then were incubated with the secondary antibodies shown in the KEY RESOURCES TABLE in 1X PBS containing 0.1% Tween-20 and 0.01% SDS at room temperature for 1 to 2 hours. The membranes were washed four times with 1X PBS containing 0.1% Tween-20. The fluorescence signals were detected by the Odyssey CLx imaging system (LI-COR) and analyzed with Image Studio Software (LI-COR). The chemiluminescent signals were visualized by the LAS-3000mini CCD camera system (Fujifilm) with ECL Western Blotting Detection Reagents (GE Healthcare) or by Hyperfilm ECL (GE Healthcare) with SuperSignal West Pico Chemiluminescent Substrate (Pierce) or SuperSignal West Femto Maximum Sensitivity Substrate (Pierce).
L1 retrotransposition assay
L1 retrotransposition assays were performed as described previously with some modifications (Kopera et al., 2016; Moldovan and Moran, 2015; Moran et al., 1996; Morrish et al., 2002; Ostertag et al., 2000). To measure the retrotransposition efficiency with pJM101/L1.3 and pTMF3, HeLa-JVM cells were plated in 6-well plates (BD Biosciences) at ~1×104 cells per well in DMEM-complete medium. The following day (day 0), the growing cells were co-transfected with 0.5 μg of pJM101/L1.3 or pTMF3 and 0.5 μg of phrGFP-C using 3 μL of the FuGENE6 transfection reagent and 100 μL of Opti-MEM according to the manufacturer’s protocol. The following day (day 1), the medium was replaced with fresh DMEM-complete medium. On day 3 forward, the medium was replaced daily with DMEM-complete medium supplemented with 400 μg/mL G418 (Gibco) until there were no cells remaining in the negative control reactions (day 10 or 11). The G418-resistant foci were washed with 1X PBS, fixed with 1X PBS containing 2% paraformaldehyde and 0.4% glutaraldehyde, and visualized by a crystal violet staining. To determine transfection efficiencies, cells independently transfected with the same plasmid set were washed with 1X PBS, trypsinized by 0.25% Trypsin-EDTA (Gibco) and harvested to count GFP positive cells out of 10,000 cells as a transfection efficiency control using a flow cytometer, BD Accuri C6 (BD Biosciences) three days post-transfection (day 3). The percentage of GFP positive cells was used to normalize the numbers of G418-resistant foci to calculate “Retrotransposition (%)” in Figure S1C. Each transfection was carried out with at least two technical replicates. The representative images of stained-colonies are shown in Figure S1C.
To measure the L1 retrotransposition efficiency in the presence of a PARP inhibitor (olaparib dissolved in dimethyl sulfoxide (DMSO)), HeLa-JVM cells or CHO cells were plated in 6-well plates at ~1×104 or ~4×104 cells per well in DMEM-complete medium or DMEM-low glucose medium, respectively. The following day (day 0), the cells were transfected with 0.5 μg of pJJ 101/L1.3, pJJ105/L1.3, or pJJD205A/L1.3 using 1.5 μL of the FuGENE6 transfection reagent and 100 μL of Opti-MEM according to the manufacturer’s protocol. The following day (day 1), the medium was replaced with fresh medium containing the PARP inhibitor (PARPi) or DMSO alone as a control (0 μM). On day 3, the medium was replaced with fresh medium supplemented with 10 μg/mL blasticidin (Merck Millipore) without DMSO or PARPi. Every two or three days, the medium was changed until there were no cells remaining in the negative control reactions (day 9 through 11). The resultant blasticidin-resistant foci were washed with 1X PBS, fixed with 1X PBS containing 2% paraformaldehyde and 0.4% glutaraldehyde, and visualized by a crystal violet staining. Normalization of the retrotransposition efficiency was carried out using pcDNA6, which contains the blasticidin S-deaminase (BSD) gene. Briefly, HeLa-JVM cells or CHO cells, respectively, were plated in 6-well plates at ~1×103 or ~5×102 cells per well and transfected with 0.5 μg of pcDNA6 using 1.5 μL of the FuGENE6 transfection reagent and 100 μL of Opti-MEM. The olaparib treatment, the blasticidin selection, and the cell fixation were conducted in the same manner as the retrotransposition assays. The colony numbers obtained with pcDNA6 were used to normalize those with the L1 constructs to calculate “Retrotransposition (%)” in Figure 2. Each transfection was carried out with at least two technical replicates. The representative images of staining colonies were shown in Figure 2.
For retrotransposition assays using HEK293T cells with the PARP inhibitor, we used an EGFP-based retrotransposition assay (Kopera et al., 2016; Ostertag et al., 2000). HEK293T cells were plated in 6-well plates at ~2 to 5×104 cells per well in DMEM-complete medium. The following day (day 0), cells were transfected with either 0.5 μg of cep99-gfp-L1.3, cep99-gfp-L1.3RT(−) or cep99-gfp-L1.3RT(−) intronless, all of which contain a puromycin resistant gene on the pCEP4 vector backbone, using 1.5 μL of the FuGENE6 transfection reagent and 100 μL of Opti-MEM according to the manufacturer’s protocol. On the following day (day 1), the medium was replaced with fresh DMEM-complete medium containing DMSO alone as a control (0 μM) or the PARP inhibitor (PARPi) at the indicated concentrations. On day 3, the medium was replaced by DMEM-complete medium supplemented with 1 μg/mL puromycin (Gibco). On day 5, the medium was replaced with fresh DMEM-complete medium supplemented with 1 μg/mL puromycin. On days 6 or 7, puromycin resistant cells were washed with 1X PBS, trypsinized by 0.25% Trypsin-EDTA, and harvested to determine the percentage of EGFP positive cells out of 20,000 cells using a flow cytometer (BD Accuri C6). The percentages of EGFP-positive cells obtained in transfection experiments conducted with either the wild-type L1.3 or the RT mutant construct were normalized with the number of EGFP-positive cells obtained in transfection experiments conducted with the cep99-gfp-L1.3RT(−)intronless to calculate “Retrotransposition (%)” in Figure S2. Each transfection was carried out with at least two technical replicates.
For the assays using shRNA-knockdown HEK293T cells, the retrotransposition assays were carried out without the PARP inhibitor in the same manner as described above using cepB-or cepZ-based L1 episomal expression plasmids. Antibiotic selection (10 μg/ml blasticidin (Merck Millipore) for cepB plasmids or 100 μg/mL of zeocin (Gibco) for cepZ plasmids) began two days post-transfection (day 2).
For retrotransposition assays using siRNA, HEK293T cells were plated in 6-well plates (IWAKI) at ~1×105 cells per well in DMEM-complete medium. On the next day (~12 hours post plating), the cells were transfected with 25 nM of small interfering RNA (siRNA) (for a non-targeting control: ON-TARGETplus Non-targeting Pool, D-001810–10-0020; for RPA70: ON-TARGETplus RPA1 siRNA, J-015749–10-0020) using 7.5 μL of Lipofectamine RNAiMAX transfection reagent (Invitrogen) and 150 μL of Opti-MEM according to the manufacturer’s protocol. Approximately 24 hours post plating, the medium containing the siRNA transfection reagents was replaced with fresh medium DMEM-complete medium and the cells were then transfected with 0.5 μg of cep99-gfp-L1.3, cep99-gfp-L1.3RT(−), or cep99-gfp-L1.3RT(−)intronless using 1.5 μL of the FuGENE HD transfection reagent and 100 μL of Opti-MEM. On the following day (day 1), the medium was replaced with fresh DMEM-complete medium. On the following day (day 2), the medium was replaced by DMEM-complete medium supplemented with 1 μg/mL puromycin (Gibco). On day 5, the medium was replaced with fresh DMEM-complete medium supplemented with 1 μg/mL puromycin. On days 6 or 7, puromycin resistant cells were washed with 1X PBS, trypsinized by 0.25% Trypsin-EDTA, and harvested to determine the percentage of EGFP positive cells out of 20,000 cells using a flow cytometer (BD FACSCalibur). The percentages of EGFP-positive cells obtained in transfection experiments conducted with either the wild-type L1.3 or the RT mutant construct were normalized with the number of EGFP-positive cells obtained in transfection experiments conducted with the cep99-gfp-L1.3RT(−)intronless to calculate “Retrotransposition (%)” in Figures 4 and S4. Each transfection was carried out with at least two technical replicates.
For retrotransposition assays using the PARP1 or PARP2 expression vectors were plated in 6-well plates (Greiner) at ~5×104 cells per well in DMEM-complete medium. The following day (day 0), cells were transfected simultaneously with either 0.5 μg of cep99-gfp-L1.3 or cep99-gfp-L1.3RT(−)intronless and either 0.25 μg of pCMV-3Tag-9, PARP1/pCMV-3Tag-9, PARP1E988K/pCMV-3Tag-9, 3FLAG C1-PARP2, or 3FLAG C1-PARP2E558K using 2.25 μL of the FuGENE HD transfection reagent and 100 μL of Opti-MEM according to the manufacturer’s protocol. On the following day (day 1), the medium was replaced with fresh DMEM-complete medium. On day 2, the medium was replaced by DMEM-complete medium supplemented with 1 μg/mL puromycin (Gibco). On day 5, the medium was replaced with fresh DMEM-complete medium supplemented with 1 μg/mL puromycin. On days 6, puromycin resistant cells were washed with 1X PBS, trypsinized by 0.25% Trypsin-EDTA, and harvested to determine the percentage of EGFP positive cells out of 20,000 cells using a flow cytometer (BD FACSCalibur). The percentages of EGFP-positive cells obtained in transfection experiments conducted with the wild-type L1.3 construct were normalized with the number of EGFP-positive cells obtained in transfection experiments conducted with the cep99-gfp-L1.3RT(−)intronless to calculate “Retrotransposition (%)” in Figure S2. Each transfection was carried out with at least two technical replicates.
To examine cell cycle progression and RPA70 protein levels, HEK293T cells were independently plated in 6-well plates and transfected with siRNAs as noted above. The cells in DMEM-complete media were washed with 1X PBS and collected 2 and 4 days post transfection. The cells were fixed by treating them with ice-cold 70% ethanol at −20°C overnight, washed two times with 1X PBS, incubated with 100 μg/mL RNase A (Nacalai Tesque), stained with 50 μg/mL propidium iodide (PI) (Nacalai Tesque), and subjected to flow cytometer analysis using BD FACSCalibur (BD Biosciences). To measure protein levels, cell lysates prepared using RIPA buffer were subjected to SDS-PAGE and western blotting as described above.
To obtain images of EGFP positive cells, the retrotransposition assays were independently performed in the same manner described above. On day 6 or 7 post-transfection, transfected cells were washed with 1X PBS and fixed with 4% paraformaldehyde for 10 minutes at room temperature. After washing with water three times, the cells were permeabilized with the KCM buffer (10 mM Tris-HCl (pH 7.5), 20 mM NaCl, 120 mM KCl, 0.1% Triton X-100) for 10 minutes at room temperature. The cells then were washed three times with 1X PBST (1X PBS containing 0.1% Tween-20). In the second wash, 0.1 μg/mL of 4’, 6-diamidino-2-phenylindole (DAPI) was added to the wash buffer to stain the nuclei. The images of EGFP positive cells were observed and captured by a BZ-X710 all-in-one fluorescence microscopy (KEYENCE) with a 10X object lens (Nikon). Representative images are indicated for each experiment.
Trypan blue exclusion assay
To measure cell viability, the control (non-mammalian shRNA targeting), PARP1, PARP2, and PARP1/PARP2 (double knockdown) shRNA knockdown cells were transfected with either cepB-gfp-L1.3 (Figure 2D) or cepZ-gfp-L1.3 (Figure S2E) and then were subjected to a trypan blue exclusion assay. Briefly, the cells were trypsinized using 0.25% Trypsin-EDTA and resuspended in an equal volume of 0.4% trypan blue solution (Gibco). A Countess automated cell counter (Invitrogen) was then used to quantify the number of trypan blue negative (i.e., viable) cells.
Virus transduction
To produce virus particles, HEK293T cells were plated on 10 cm dishes (BD Biosciences) at ~2×106 cells per dish in DMEM-complete medium. The following day (day 0), the cells were co-transfected with 6 μg of an shRNA plasmid, 3.75 μg of psPAX2, and 1.25 μg of pMD2.G using 30 of the FuGENE HD transfection reagent (Promega) and 500 of Opti-MEM according to the manufacturer’s protocol. On the following day (day 1), the medium containing the transfection reagents was replaced with fresh DMEM-complete medium. On the following day (day 2) the virus-containing media was collected, filtered through a 0.45 μm filter (Pall), and mixed with 8 μg/mL polybrene (Merck Millipore). For virus transduction, HEK293T cells were plated in a 6-well plate at 2–3×105 cells per well in DMEM-complete medium. The following day (day 1), the medium was replaced with the virus-containing medium prepared above and incubated for ~24 hours. On day 2, the medium was replaced with fresh DMEM-complete medium. On day 3, the medium was replaced by DMEM-complete medium supplemented with 1 μg/mL puromycin. This medium then was changed daily until the uninfected cells died (typically 6–7 days post transduction). To obtain double knockdown cells, single knockdown cells were subsequently transduced with a second virus using the protocol described above. The transduced cells were maintained in DMEM-complete medium containing puromycin (1 μg/mL) and used for subsequent analyses.
To produce virus particles containing a RPA70-Myc expressing construct, Plat-A retroviral packaging cells were plated on a 10-cm dish at 2–3×106 cells per dish in DMEM-complete medium (Morita et al., 2000). The following day (day 0), cells were transfected with 5 μg of pMX-hyg or pMX-hyg-RPA70myc plasmid using 15 μL of the FuGENE HD transfection reagent and 500 μL of Opti-MEM. Preparation of the virus supernatant and transduction of HEK293T cells were carried out in the same manner described above except that 100 μg/mL of hygromycin B was used for selection instead of puromycin.
Purification of recombinant proteins
All recombinant proteins except for the Histidine-tagged APOBEC3A protein (His-A3A) were produced in the E. coli BL21(DE3) strain using bacterial expression plasmids. For induction of His-PARP1, His-PARP2, and all GST-tagged proteins, 0.5 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) was added to the culture of the transformed cells growing at 37°C in 2X YT medium (16 g tryptone, 5 g NaCl and 10 g yeast extracts per liter) containing 10 μg/mL kanamycin for PARPs or in LB medium (10 g tryptone, 10 g NaCl and 5 g yeast extracts per liter) containing 100 μg/mL ampicillin for GST fusion proteins. For induction of the human RPA trimer, 0.5 mM IPTG was added to culture of the transformed cells growing at 37°C in TB (10 g tryptone and 5 g NaCl per liter) containing 100 μg/mL ampicillin. After 2–4 hours of induction, the cells were collected, washed with 1X PBS, and stored at −80°C until cell lysis. His-A3A expression plasmids were transformed into the E. coli C43 strain (Lucigen). Induction was conducted in LB medium containing 10 μg/mL kanamycin at 37°C by adding 1 mM IPTG for 5–6 hours using a previously established protocol (Richardson et al., 2014).
For purification of His-tagged proteins, IPTG-treated cells were lysed in His lysis buffer (10 mM Tris-HCl (pH 8), 100 μg/mL Lysozyme, 500 mM NaCl, 1% IGEPAL CA-630, 0.2 mM PMSF, 1X cOmplete EDTA-free protease inhibitor cocktail) and sonicated using a Branson Sonifier 185 system (Branson) with condition of power 1–2 (duty 50%) for 2–10 minutes. The cell debris and insoluble proteins were removed by centrifugation at 12,000 rpm for 5 minutes and the supernatants were transferred into new tubes. The cleared cell lysates were incubated with 50 μL of Ni-NTA Agarose (Invitrogen) or TALON Metal Affinity Resin (BD Biosciences) (50% slurry in 1X PBS) for 2–3 hours at 4°C. After washing five times with the His wash buffer (10 mM Tris-HCl (pH 8), 500 mM NaCl, 5 mM imidazole (35 mM imidazole for His-A3A)), the beads were incubated with the His elution buffer (10 mM Tris-HCl (pH 8), 500 mM NaCl, 500 mM imidazole) for 10 minutes to release His-tagged proteins from the beads. The eluate was subjected to dialysis using Slide-A-Lyzer MINI Dialysis Device 20k MWCO (Thermo Fisher Scientific) in TBST (20 mM Tris-HCl (pH 8), 150 mM NaCl, 0.05% Tween 20) for overnight at 4°C.
For purification of GST-tagged proteins, IPTG-treated cells were lysed with the GST lysis buffer (20 mM Tris-HCl (pH 8), 100 μg/mL Lysozyme, 150 mM NaCl, 1% IGEPAL CA-630, 1 mM EDTA, 1 mM DTT) containing 1X cOmplete EDTA-free protease inhibitor cocktail and sonicated using a Branson Sonifier 185 system with condition of power 1–2 (duty 50%) for 2–3 minutes. The cell debris and insoluble proteins were removed by centrifugation at 12,000 rpm for 5 minutes. The cleared cell lysates were incubated with 50 μL of Glutathione sepharose 4B (GE Healthcare) or Glutathione agarose (Pierce) (50% slurry in 1X PBS) for 2–3 hours at 4°C. After washing five times with the GST lysis buffer, the sepharose then was incubated with TBST containing 10 mM glutathione for 10 minutes to release GST fusion proteins from sepharose. The dialysis was carried out in the same manner described above.
Purified proteins were subjected to SDS-PAGE and visualized by Coomassie Brilliant Blue staining. The images were captured using the Odyssey CLx imaging system and analyzed with Image Studio Software to calculate protein amounts using a dilution series of BSA (New England Biolabs or TaKaRa Bio) as a standard.
In vitro poly(ADP-ribosyl)ation
Activation of recombinant PARP1 or PARP2 was performed as previously reported with minor modifications (Langelier et al., 2014). Briefly, purified His-PARP1 (~8 nM) or His-PARP2 (~80 nM) was incubated in 100 μL of the PARP activation buffer (20 mM Tris-HCl (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 250 μM DTT, 20 jM nicotinamide adenine dinucleotide (NAD)) for 1 minute at 30°C with or without 20 μM dsDNAs. To prepare dsDNAs 20 micromoles of each single strand DNA were mixed with the complementary strand in the DNA annealing buffer (20 mM Tris-HCl (pH 7.5), 25 mM NaCl) followed by heat denaturation at 95°C for 3 minutes on an aluminum heat block. The heat block was covered with aluminum foil and left at room temperature for overnight, allowing them to anneal.
The following oligonucleotide DNAs were used in the assay (Table S3):
TM54: 5’-gtatcctcgtagtgcagatgTTTT-3’ (no phosphorylation)
TM55: 5’P-Atcggactgattcggtagatctg-3’ (phosphorylation at the 5’ end)
TM56: 5’-Atcggactgattcggtagatctg-3’ (no phosphorylation)
TM57: 5’-gtatcctcgtagtgcagatgTTTTAtcggactgattcggtagatctg-3’
TM58: 5’-cagatctaccgaatcagtccgaTAAAAcatctgcactacgaggatac-3’
TM60: 5’-gtatcctcgtagtgcagatgTTTT-3’P (phosphorylation at the 3’ end)
The 5’ and 3’ ends of the oligonucleotides contain either a hydroxyl or phosphate group. The dsDNAs in Figure 4 consist of the following oligonucleotide combinations: (a: TM54, TM55, and TM58), (b: TM54, TM56, and TM58), (c: TM60, TM56, and TM58), and (d: TM57 and TM58). The underlined capital letters indicate the L1 EN consensus sequence. After the reaction, PARylated products were boiled in the SDS sample buffer and separated by an SDS-PAGE gel followed by western blotting using anti-PAR antibody (Trevigen, 4336-APC-050).
PAR binding assays
Monitoring of PAR-binding to recombinant proteins was carried out using a previously established protocol with minor modifications (Zhang et al., 2014). Briefly, purified proteins (~20–60 ng) were directly blotted on a nitrocellulose membrane (LI-COR or Bio-Rad). The membrane was dried at room temperature, washed twice with TBST (20 mM Tris-HCl (pH 8), 150 mM NaCl, 0.05% Tween 20) buffer, and incubated with 1 nM poly(ADP-ribose) (PAR) polymer (Trevigen) in TBST for overnight at 4°C. After washing with TBST four times, the membrane was incubated with 1X PBS containing 3% nonfat skim milk and subjected to western blotting using anti-PAR antibody (Trevigen, 4335-MC-100). The images were captured using an Odyssey CLx imaging system and analyzed with Image Studio Software to calculate protein amounts.
Membrane stripping was performed using a NewBlot IR Stripping Buffer (Li-COR) according to the manufacturer’s protocol. Briefly, a membrane was washed more than two times with NewBlot IR Stripping Buffer at room temperature for ~20 minutes followed by TBST washing. After checking that no detectable signals remained on the membrane, the same membrane was subjected to western blotting using anti-GST antibody (CST). The obtained signals were measured by an Odyssey CLx imaging system and quantified with Image Studio Software. The PAR signals obtained were normalized with each GST amount to calculate relative PAR-bindings of GST-fused DBD-A mutants in Figures 5C and S5E.
GST-pull down assays
GST-pull down assay was carried using a previously established protocol with minor modifications (Zhang et al., 2014). Briefly, purified GST fusion proteins (20 μM for PAR polymer, 200 μM for PARylated PARP2) were incubated with 10 μL of glutathione sepharose 4B (50% slurry in 1X PBS) and 1 nM PAR polymer or 5 μL of PARylated PARP2 prepared in the above section labeled “In vitro poly(ADP-ribosyl)ation” in 200 μL of the 1X PBST buffer for 2–3 hours at 4°C. The poly(T)50 (TM154), poly(A)50 (TM155), poly(C)50 (TM156), or poly(G)50 (TM157) oligomers (10, 100 or 1,000 nM) also were added to the reactions as a competitor (Table S3). After washing the GST beads with 1X PBST three times, protein-bound PAR polymer or PARylated PARP2 was released from beads by adding 30 μL of 1X PBST containing 10 mM glutathione for 10 minutes at room temperature, or heated with 30 μL of the SDS buffer (150 mM Tris-HCl (pH 7.5), 10 mM EDTA, 10% SDS) for 10 minutes at 80°C, respectively. The resultant eluates were directly blotted on a nitrocellulose membrane followed by western blotting using anti-PAR antibody (Trevigen, 4336-APC-050) described above. The DBD-A bound PARylated PARP2 also was subjected to SDS-PAGE and western blotting using the same antibody.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA assays using an IRDye oligonucleotide were performed according to the protocol provided on the LI-COR web site with modification (https://www.licor.com/documents/1mqrtw8bbwd31jgcpk5r). Briefly, 10 to 75 ng of each protein was incubated in 20 μL of the EMSA buffer (10 mM Tris-HCl (pH 7.5), 50 mM KCl, 3.5 mM DTT, 0.25% Tween 20, 5 nM TM326) for 20 minutes at 25°C. Integrated DNA Technologies (IDT) synthesized high-performance liquid chromatography (HPLC) TM326, which is a poly(T)30 oligonucleotide labeled with IRDye800 at the 5’ end (Table S3). Three microliters of 10X loading dye (250 mM Tris-HCl (pH 7.5), 0.2% Orange G, 40% glycerol) was added to the reaction and 10 μL were subjected to electrophoresis in a 5% acrylamide/TBE gel (45 mM Tris-HCl (pH 7.5), 45 mM boric acid, 2 mM EDTA, 5% acrylamide/bisacrylamide (37.5:1), 2.5% glycerol). After running the 5% acrylamide/TBE gel, the signals of single strand DNAs were detected using an Odyssey CLx imaging system and analyzed with Image Studio Software.
A3A deamination assay
A3A-mediated C-to-U deamination activity was measured using a previously established protocol with minor modifications (Richardson et al., 2014). Briefly, the LEAP adaptor oligonucleotide TM165: 5’-IRD800-GATGGATGATGAATAAAGTGTGGGATGATCATGATGTATGGATAGGTTTTTTTTTTTT-3’ (Table S3), where the underlining indicates an A3A deamination consensus sequence, labeled with IRDye800 fluorescein at the 5’ end (5 nM) (IDT) was incubated with 15.625 to 125 nM (Figure S6A) or 125 nM (Figure 6) His-A3A proteins in A3A deamination buffer (40 mM Tris-HCl (pH 8.0), 40 mM KCl, 50 mM NaCl, 5 mM EDTA, 1 mM DTT, 8% glycerol) at 37°C for 2 hours in the absence or presence of RPA (1.95 to 31.25 nM). The reactions then were inactivated by heat treatment (90°C for 5 minutes). After cooling on ice, the deaminated single strand DNAs were treated with 0.425 U/μL of uracil DNA glycosylase (New England Biolabs) in a buffer containing 20 mM Tris-HCl (pH 8.0) and 1 mM DTT at 37°C for 1 hour, and subsequently were treated with 150 mM NaOH at 37°C for 30 minutes. The reaction mixtures then were heated to 95°C for 5 minutes and chilled on ice. The resultant products were denatured in TBE/urea buffer (89 mM Tris-borate, 2 mM EDTA, 10 M urea) at 70°C for 4 minutes and separated into 7 M-urea containing 15% polyacrylamide gels with 1X TBE (89 mM Tris-borate, 2 mM EDTA) buffer. The single strand DNA signals were captured by the Odyssey CLx imaging system and analyzed with Image Studio Software.
L1 RNP preparation and LEAP assay
To obtain L1 expressing cells, HEK293T cells were plated in a 10-cm dish (Fine Plus International) containing DMEM-complete medium at approximately 2×106 cells per dish. A single tissue culture dish was used for each sample preparation. On the following day (day 0), the cells were transfected with 5 jg of plasmid DNA using 15 μL of the FuGENE HD transfection reagent and 500 μL of Opti-MEM according to the manufacturer’s protocol. The following day (day 1), the medium was replaced with fresh DMEM-complete medium. From day 2 onward, the medium was replaced daily with DMEM-complete medium containing 200 μg/mL hygromycin B. On day 4, the transfected cells were harvested by centrifugation, washed with cold 1X PBS, and stored at −80°C.
L1 RNP preparation was conducted as described above (see “Immunoprecipitation and western blotting”), using 500 μL or 30 μL of Lysis300 buffer for cell lysis or 3X FLAG peptide elution, respectively. Aliquots (2.5 μL of the whole cell extracts and 3.5 μL of the purified L1 RNP (“Input” and “IP” respectively in Figure S6B) were subjected to western blotting or RNA preparation for RT-PCR. RNAs were extracted using RNeasy Mini Kit (QIAGEN) and eluted in 50 μL of RNase free water. An aliquot (2.5 μL) of the purified RNAs were used to synthesize cDNA with 0.175 U/μL Avian Myeloblastosis Virus Reverse Transcriptase XL (TaKaRa Bio) and 0.5 μM LEAP adaptor (TM158: 5’-GATGGATGATGAATAAAGTGTGGGATGATCATGATGTATGGATAGGTTTTTTTTTTTT-3’) (Table S3) in a total reaction volume of 10 μL (10 mM Tris-HCl (pH 8.3), 50 mM KCl, 5 mM MgCl2, 1 U/μL RNase inhibitor (TaKaRa Bio), 1 mM dNTPs) by incubation at 42°C for 30 minutes according to the manufacturer’s protocol. The reaction was inactivated by heat treatment (95°C for 5 minutes). Two microliters of each cDNA was amplified using 0.025 U/μL Ex Taq DNA polymerase (TaKaRa Bio) and 0.2 μM of each primer, TM159: (5’-GATGGATGATGAATAAAGTG-3’) and TM160: (5’-GGGTTCGAAATCGATAAGCTTGGATCCAGAC-3’) for L1 cDNA or TM159 and TM182: (5’-GGAGGTGATAGCAATGCTTTCGTG-3’) for ACTB cDNA (Table S3) in a reaction volume of 25 μL using the following PCR conditions: incubation at 95°C for 3 minutes followed by 30 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds with an additional extension period of 7 minutes at 72 °C. The final PCR products were separated into 2% agarose gels with 1X TAE buffer (40 mM Tris, 20 mM acetic acid, 1 mM EDTA) and visualized with ethidium bromide.
For the LEAP assay, 1.5 μL of the L1 RNP was incubated with LEAP buffer (50 mM Tris-HCl (pH 8.0), 50 mM KCl, 5 mM MgCh, 10 mM DTT, 0.05% Tween 20, 0.4 U/μL RNasin (Promega), 200 μM dNTPs, 400 nM LEAP adaptor (TM158)) in the absence or presence of 0.3 U/μL RNase H (TaKaRa Bio) in a reaction volume of 20 μL at 37°C for 60 minutes. Midway through the reaction (at the 30 minutes time point), recombinant A3A (28.13 nM) and RPA trimer (56.25 nM) were added to the LEAP reaction. The reaction was inactivated by heat treatment (95°C for 5 minutes). A sample was prepared by immunoprecipitation using the full-length L1.3 without a FLAG tag expressing cells as a negative control. An aliquot (1.5 μL) of the resultant cDNA or water then was incubated to amplify the RT products using Pfu Turbo Cx Hotstart DNA polymerase (Agilent Technologies) according to the manufacturer’s protocol using the following PCR conditions: incubation at 95°C for 3 minutes followed by 35 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds with an additional extension period of 7 minutes at 72 °C. The final PCR products were separated on 2% agarose gels with 1X TAE buffer and visualized with ethidium bromide. The obtained LEAP products were cut out from the gels, purified using QIAquick Gel Extraction Kit (Qiagen), and cloned into ZERO Blunt PCR cloning vector (Invitrogen). Each plasmid was purified and sequenced by ABI PRISM 3100 Genetic Analyzer (Applied Biosystems) using M13 forward: (5’-GTAAAACGACGGCCAGTGA-3’) and M13 reverse: (5’-AGGAAACAGCTATGACCAT-3’) primers (Table S3).
QUANTIFICATION AND STATISTICAL ANALYSIS
An unpaired two-tailed t-test was used for comparison of the two samples in Figure 4E. As described in the Figure Legends, one-way analysis of variance (ANOVA) followed by a Bonferroni multiple comparison test was used for comparison of the three or four samples in Figures 2B, 2D, 2E, S2C, S2D, S2E, and S2F. Statistical analyses were conducted using Excel software (Microsoft).
As described in the Figure Legends, gel images were analyzed and the signal intensities were measured using Image Studio software (LI-COR) in Figures 5C, S5E, and 6A.
DATA AND SOFTWARE AVAILABILITY
All datasets are present in this study or supplemental tables.
Supplementary Material
Table S1A (tab 1) indicates proteins specifically detected in the ORF2p-3FLAG immunoprecipitation reactions. Table S1B (tab 2) indicates proteins relatively enriched in the ORF2p-3FLAG immunoprecipitation reactions compared to those detected in the ORF2p-TAP control sample. Column A: the UniProt protein accession number. Column B: the names of the proteins; blue highlighting indicates ORF2p-3FLAG/protein interactions that were confirmed by western blot experiments. Column C: the number of amino acids (# AAs) in the protein. Column D: the predicted molecular weight (MW [kDa]) of the protein. Columns E and H: Score, the sum of the scores (Xcorr values) of the individual peptides calculated by SEQUEST search algorithm (Eng et al., 1994). Columns F and I: Coverage, the percentage of amino acid coverage (%) of identified peptides to the entire protein sequence. Columns G and J: Unique Peptides, the number of distinct peptides identified. Column K: the value of subtraction of score (2) (ORF2p-TAP) from score (1) (ORF2p-3FLAG). Candidates were eliminated if the score in column K was less than 4.0. Column L: whether the protein listed in Columns A and B was detected in (Taylor et al., 2013). Column M: whether the protein listed in Columns A and B was detected in (Goodier et al., 2013). Column N: whether the protein listed in Columns A and B was detected in (Moldovan and Moran, 2015). Column O: whether the protein listed in Columns A and B was detected in (Peddigari et al., 2013). The notation “Y” indicates that association of the listed protein with ORF1p and/or ORF2p in the previous studies. The notation “n.s.” means that the detection level was below the statistical threshold determined in the study.
Sixty LEAP products were sequenced from each reaction condition. Column 1: the name of the clone. Column 2: the number of G-to-A mutations in the (+) strand L1 cDNA product. Column 3: the sequence of the LEAP product. Red characters indicate G-to-A mutations. Green characters indicate other nucleotide changes. The poly(A) tract lengths of several LEAP products could not been determined to base pair resolution because of the difficulty in sequencing a long poly(A) tract.
ACKNOWLEGEMENTS
We thank Drs. F. Ishikawa, M. Wold, F. Hanaoka, X. Yu, T. Kitamura and D. Trono for reagents, Moran lab members and Dr. M. Miyoshi for discussions and critical reading of the manuscript, the Fred Hutchinson Cancer Research Center Proteomics Core for mass spectrometry assistance, the University of Michigan DNA Sequencing Core for technical support, and N. Leff for editorial assistance. T.M. was supported by a Grant-in-Aid for Young Scientists (B: 16K18471) and Scientific Research (C: 18K06180) from the Japan Society for the Promotion of Science, the Takeda Science Foundation, and the Kanae Foundation. J.V.M. was supported by a National Institutes of Health grant (GM060518) and previous funding from the Howard Hughes Medical Institute.
Footnotes
COMPETING INTEREST STATEMENT
J.V.M. is an inventor on patent US6150160, is a paid consultant for Gilead Sciences and a privately held company founded by Flagship Pioneering, and is on the American Society of Human Genetics Board of Directors. The other authors do not have competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Armougom F, Moretti S, Poirot O, Audic S, Dumas P, Schaeli B, Keduas V, and Notredame C (2006). Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res 34, W604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athanikar JN, Badge RM, and Moran JV (2004). A YY1-binding site is required for accurate human LINE-1 transcription initiation. Nucleic Acids Res 32, 3846–3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu U, Wang Y, and Alt FW (2008). Evolution of phosphorylation-dependent regulation of activation-induced cytidine deaminase. Mol Cell 32, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitez-Guijarro M, Lopez-Ruiz C, Tarnauskaite Z, Murina O, Mian Mohammad M, Williams TC, Fluteau A, Sanchez L, Vilar-Astasio R, Garcia-Canadas M, et al. (2018). RNase H2, mutated in Aicardi-Goutieres syndrome, promotes LINE-1 retrotransposition. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkarev A, Pfuetzner RA, Edwards AM, and Frappier L (1997). Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature 385, 176–181. [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O’Shea KS, Moran JV, and Cullen BR (2006). Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci U S A 103, 8780–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, and Kazazian HH Jr. (2003). Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci U S A 100, 5280–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, and Helleday T (2009). PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 28, 2601–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Sharma S, Maiti PK, and Krishnan Y (2009). The poly dA helix: a new structural motif for high performance DNA-based molecular switches. Nucleic Acids Res 37, 2810–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Khuong C, and Alt FW (2004). Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature 430, 992–998. [DOI] [PubMed] [Google Scholar]
- Chen R, and Wold MS (2014). Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 36, 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost GJ, and Boeke JD (1998). Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure. Biochemistry 37, 18081–18093. [DOI] [PubMed] [Google Scholar]
- Cost GJ, Feng Q, Jacquier A, and Boeke JD (2002). Human L1 element target-primed reverse transcription in vitro. EMBO J 21, 5899–5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Marchetto MC, Muotri AR, Mu Y, Carson CT, Macia A, Moran JV, and Gage FH (2011). Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc Natl Acad Sci U S A 108, 20382–20387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Taylor MS, O’Donnell KA, and Boeke JD (2012). Poly(A) binding protein C1 is essential for efficient L1 retrotransposition and affects L1 RNP formation. Mol Cell Biol 32, 4323–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet AJ, Hulme AE, Sahinovic E, Kulpa DA, Moldovan JB, Kopera HC, Athanikar JN, Hasnaoui M, Bucheton A, Moran JV, et al. (2010). Characterization of LINE-1 ribonucleoprotein particles. PLoS Genet 6, e1001150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet AJ, Wilusz JE, Miyoshi T, Liu Y, and Moran JV (2015). A 3’ Poly(A) Tract Is Required for LINE-1 Retrotransposition. Mol Cell 60, 728–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Esnault C, Maestre J, and Heidmann T (2000). Human LINE retrotransposons generate processed pseudogenes. Nat Genet 24, 363–367. [DOI] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH Jr., and Boeke JD (1996). Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87, 905–916. [DOI] [PubMed] [Google Scholar]
- Flasch DA, Macia A, Sanchez L, Ljungman M, Heras SR, Garcia-Perez JL, Wilson TE, and Moran JV (2019). Genome-wide de novo L1 Retrotransposition Connects Endonuclease Activity with Replication. Cell 177, 837–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior SL, Roy-Engel AM, and Deininger PL (2008). ERCC1/XPF limits L1 retrotransposition. DNA Repair (Amst) 7, 983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior SL, Wakeman TP, Xu B, and Deininger PL (2006). The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol 357, 1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, and Kraus WL (2012). New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13, 411–424. [DOI] [PubMed] [Google Scholar]
- Gilbert N, Lutz S, Morrish TA, and Moran JV (2005). Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol Cell Biol 25, 7780–7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL (2016). Restricting retrotransposons: a review. Mob DNA 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Cheung LE, and Kazazian HH Jr. (2013). Mapping the LINE1 ORF1 protein interactome reveals associated inhibitors of human retrotransposition. Nucleic Acids Res 41, 7401–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohjoh H, and Singer MF (1996). Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J 15, 630–639. [PMC free article] [PubMed] [Google Scholar]
- Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, and Roberts SA (2016). APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging Strand Template during DNA Replication. Cell Rep 14, 1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, and Durocher D (2016). The control of DNA repair by the cell cycle. Nat Cell Biol 19, 1–9. [DOI] [PubMed] [Google Scholar]
- Khazina E, Truffault V, Buttner R, Schmidt S, Coles M, and Weichenrieder O (2011). Trimeric structure and flexibility of the L1ORF1 protein in human L1 retrotransposition. Nat Struct Mol Biol 18, 1006–1014. [DOI] [PubMed] [Google Scholar]
- Kim C, Snyder RO, and Wold MS (1992). Binding properties of replication protein A from human and yeast cells. Mol Cell Biol 12, 3050–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, and Kumagai H (2003). Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp Hematol 31, 1007–1014. [PubMed] [Google Scholar]
- Kojima KK (2010). Different integration site structures between L1 protein-mediated retrotransposition in cis and retrotransposition in trans. Mob DNA 1, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopera HC, Larson PA, Moldovan JB, Richardson SR, Liu Y, and Moran JV (2016). LINE-1 Cultured Cell Retrotransposition Assay. Methods Mol Biol 1400, 139–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopera HC, Moldovan JB, Morrish TA, Garcia-Perez JL, and Moran JV (2011). Similarities between long interspersed element-1 (LINE-1) reverse transcriptase and telomerase. Proc Natl Acad Sci U S A 108, 20345–20350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo S, Seleme MC, Soifer HS, Perez JL, Moran JV, Kazazian HH Jr., and Kasahara N (2006). L1 retrotransposition in nondividing and primary human somatic cells. Proc Natl Acad Sci U S A 103, 8036–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulpa DA, and Moran JV (2005). Ribonucleoprotein particle formation is necessary but not sufficient for LINE-1 retrotransposition. Hum Mol Genet 14, 3237–3248. [DOI] [PubMed] [Google Scholar]
- Kulpa DA, and Moran JV (2006). Cis-preferential LINE-1 reverse transcriptase activity in ribonucleoprotein particles. Nat Struct Mol Biol 13, 655–660. [DOI] [PubMed] [Google Scholar]
- Langelier MF, Riccio AA, and Pascal JM (2014). PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res 42, 7762–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HL, and Moran JV (2011). Dynamic interactions between transposable elements and their hosts. Nat Rev Genet 12, 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Lu LY, Yang CY, Wang S, and Yu X (2013). The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev 27, 1752–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Otevrel T, Gao Y, Cheng HL, Seed B, Stamato TD, Taccioli GE, and Alt FW (1995). The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell 83, 1079–1089. [DOI] [PubMed] [Google Scholar]
- Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC, and Wysocka J (2018). Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 553, 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, and Eickbush TH (1993). Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell 72, 595–605. [DOI] [PubMed] [Google Scholar]
- Martin SL (1991). Ribonucleoprotein particles with LINE-1 RNA in mouse embryonal carcinoma cells. Mol Cell Biol 11, 4804–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SL, and Bushman FD (2001). Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Mol Cell Biol 21, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias SL, Scott AF, Kazazian HH Jr., Boeke JD, and Gabriel A (1991). Reverse transcriptase encoded by a human transposable element. Science 254, 1808–1810. [DOI] [PubMed] [Google Scholar]
- McMillan JP, and Singer MF (1993). Translation of the human LINE-1 element, L1 Hs. Proc Natl Acad Sci U S A 90, 11533–11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, LeMeur M, et al. (2003). Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J 22, 2255–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita P, Wudzinska A, Sun X, Andrade J, Nayak S, Kahler DJ, Badri S, LaCava J, Ueberheide B, Yun CY, et al. (2018). LINE-1 protein localization and functional dynamics during the cell cycle. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan JB, and Moran JV (2015). The Zinc-Finger Antiviral Protein ZAP Inhibits LINE and Alu Retrotransposition. pLoS Genet 11, e1005121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monot C, Kuciak M, Viollet S, Mir AA, Gabus C, Darlix JL, and Cristofari G (2013). The specificity and flexibility of l1 reverse transcription priming at imperfect T-tracts. PLoS Genet 9, e1003499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, and Kazazian HH Jr. (1996). High frequency retrotransposition in cultured mammalian cells. Cell 87, 917–927. [DOI] [PubMed] [Google Scholar]
- Morita S, Kojima T, and Kitamura T (2000). Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 7, 1063–1066. [DOI] [PubMed] [Google Scholar]
- Morrish TA, Garcia-Perez JL, Stamato TD, Taccioli GE, Sekiguchi J, and Moran JV (2007). Endonuclease-independent LINE-1 retrotransposition at mammalian telomeres. Nature 446, 208–212. [DOI] [PubMed] [Google Scholar]
- Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, and Moran JV (2002). DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet 31, 159–165. [DOI] [PubMed] [Google Scholar]
- Mortusewicz O, Ame JC, Schreiber V, and Leonhardt H (2007). Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res 35, 7665–7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, and Kazazian HH Jr. (2000). Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res 28, 1418–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peddigari S, Li PW, Rabe JL, and Martin SL (2013). hnRNPL and nucleolin bind LINE-1 RNA and function as host factors to modulate retrotransposition. Nucleic Acids Res 41, 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro JG, and Cristofari G (2016). Post-Transcriptional Control of LINE-1 Retrotransposition by Cellular Host Factors in Somatic Cells. Front Cell Dev Biol 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom M, Dennehey BK, and Tyler JK (2010). Chaperoning histones during DNA replication and repair. Cell 140, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SR, Doucet AJ, Kopera HC, Moldovan JB, Garcia-Perez JL, and Moran JV (2015). The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes. Microbiol Spectr 3, MDNA3–0061-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SR, Narvaiza I, Planegger RA, Weitzman MD, and Moran JV (2014). APOBEC3A deaminates transiently exposed single-strand DNA during LINE-1 retrotransposition. Elife 3, e02008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassaman DM, Dombroski BA, Moran JV, Kimberland ML, Naas TP, DeBerardinis RJ, Gabriel A, Swergold GD, and Kazazian HH Jr. (1997). Many human L1 elements are capable of retrotransposition. Nat Genet 16, 37–43. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, Menissier-de Murcia J, and de Murcia G (2002). Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem 277, 23028–23036. [DOI] [PubMed] [Google Scholar]
- Servant G, Streva VA, Derbes RS, Wijetunge MI, Neeland M, White TB, Belancio VP, Roy-Engel AM, and Deininger PL (2017). The Nucleotide Excision Repair Pathway Limits L1 Retrotransposition. Genetics 205, 139–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, Munzel M, Wagner M, Muller M, Khan F, et al. (2013). Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152, 1146–1159. [DOI] [PubMed] [Google Scholar]
- Sultana T, van Essen D, Siol O, Bailly-Bechet M, Philippe C, Zine El Aabidine A, Pioger L, Nigumann P, Saccani S, Andrau JC, et al. (2019). The Landscape of L1 Retrotransposons in the Human Genome Is Shaped by Pre-insertion Sequence Biases and Post-insertion Selection. Mol Cell 74, 555–570. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Yamaguchi K, Kajikawa M, Ichiyanagi K, Adachi N, Koyama H, Takeda S, and Okada N (2009). Genetic evidence that the non-homologous end-joining repair pathway is involved in LINE retrotransposition. PLoS Genet 5, e1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swergold GD (1990). Identification, characterization, and cell specificity of a human LINE-1 promoter. Mol Cell Biol 10, 6718–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MS, Altukhov I, Molloy KR, Mita P, Jiang H, Adney EM, Wudzinska A, Badri S, Ischenko D, Eng G, et al. (2018). Dissection of affinity captured LINE-1 macromolecular complexes. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MS, LaCava J, Mita P, Molloy KR, Huang CR, Li D, Adney EM, Jiang H, Burns KH, Chait BT, et al. (2013). Affinity proteomics reveals human host factors implicated in discrete stages of LINE-1 retrotransposition. Cell 155, 1034–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, Sridharan S, van Wietmarschen N, Maman Y, Callen E, Stanlie A, Wu W, Wu X, Day A, Wong N, et al. (2018). Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 174, 1127–1142 e1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD, and Moran JV (2001). Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol 21, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissing S, Montano M, Garcia-Perez JL, Moran JV, and Greene WC (2011). Endogenous APOBEC3B restricts LINE-1 retrotransposition in transformed cells and human embryonic stem cells. J Biol Chem 286, 36427–36437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Chen Y, Li M, and Yu X (2014). The oligonucleotide/oligosaccharide-binding fold motif is a poly(ADP-ribose)-binding domain that mediates DNA damage response. Proc Natl Acad Sci U S A 111, 7278–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingler N, Willhoeft U, Brose HP, Schoder V, Jahns T, Hanschmann KM, Morrish TA, Lower J, and Schumann GG (2005). Analysis of 5’ junctions of human LINE-1 and Alu retrotransposons suggests an alternative model for 5’-end attachment requiring microhomology-mediated end-joining. Genome Res 15, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1A (tab 1) indicates proteins specifically detected in the ORF2p-3FLAG immunoprecipitation reactions. Table S1B (tab 2) indicates proteins relatively enriched in the ORF2p-3FLAG immunoprecipitation reactions compared to those detected in the ORF2p-TAP control sample. Column A: the UniProt protein accession number. Column B: the names of the proteins; blue highlighting indicates ORF2p-3FLAG/protein interactions that were confirmed by western blot experiments. Column C: the number of amino acids (# AAs) in the protein. Column D: the predicted molecular weight (MW [kDa]) of the protein. Columns E and H: Score, the sum of the scores (Xcorr values) of the individual peptides calculated by SEQUEST search algorithm (Eng et al., 1994). Columns F and I: Coverage, the percentage of amino acid coverage (%) of identified peptides to the entire protein sequence. Columns G and J: Unique Peptides, the number of distinct peptides identified. Column K: the value of subtraction of score (2) (ORF2p-TAP) from score (1) (ORF2p-3FLAG). Candidates were eliminated if the score in column K was less than 4.0. Column L: whether the protein listed in Columns A and B was detected in (Taylor et al., 2013). Column M: whether the protein listed in Columns A and B was detected in (Goodier et al., 2013). Column N: whether the protein listed in Columns A and B was detected in (Moldovan and Moran, 2015). Column O: whether the protein listed in Columns A and B was detected in (Peddigari et al., 2013). The notation “Y” indicates that association of the listed protein with ORF1p and/or ORF2p in the previous studies. The notation “n.s.” means that the detection level was below the statistical threshold determined in the study.
Sixty LEAP products were sequenced from each reaction condition. Column 1: the name of the clone. Column 2: the number of G-to-A mutations in the (+) strand L1 cDNA product. Column 3: the sequence of the LEAP product. Red characters indicate G-to-A mutations. Green characters indicate other nucleotide changes. The poly(A) tract lengths of several LEAP products could not been determined to base pair resolution because of the difficulty in sequencing a long poly(A) tract.
Data Availability Statement
All datasets are present in this study or supplemental tables.