

Abstract
While considerable research has focused on studying individual-species, we now face the challenge of determining how interspecies interactions alter bacterial behaviours and pathogenesis. Pseudomonas aeruginosa and Staphylococcus aureus are often found to co-infect cystic-fibrosis patients. Curiously, their interaction is reported as competitive under laboratory conditions. Selecting appropriate methodologies is therefore critical to analyse multi-species communities. Herein, we demonstrated the major biases associated with qPCR quantification of bacterial populations and optimized a RNA-based qPCR able not only to quantify but also to characterize microbial interactions within dual-species biofilms composed by P. aeruginosa and S. aureus, as assessed by gene expression quantification. qPCR quantification was compared with flow-cytometry and culture-based quantification. Discrepancies between culture independent and culture dependent methods could be the result of the presence of viable but not-cultivable bacteria within the biofilm. Fluorescence microscopy confirmed this. A higher sensitivity to detect viable cells further highlights the potentialities of qPCR approach to quantify biofilm communities. By using bacterial RNA and an exogenous mRNA control, it was also possible to characterize bacterial transcriptomic profile, being this a major advantage of this method.
Subject terms: Biofilms, Microbial ecology
Introduction
The recognition that biofilm communities, which typically comprise multiple species1, with a spatiotemporal heterogeneous chemical, physiological and genetic composition2 poses a serious healthcare concern regarding the synergies that arise from the co-infecting species3,4. Hence, selecting appropriate methodologies that provides reliable quantitative measures of individual populations within the polymicrobial biofilms is crucial to predict the behaviour and dynamic of these communities.
Culture-based methods have long been used to detect microbial pathogens; however, since these methodologies require the growth of the cultures, it takes a long period of time to obtain results. Furthermore, culturing is not always an option since some bacterial species are difficult to growth in the laboratory5,6 and many species develop a viable but non-cultivable state7–9. Other limitations are the presence of cell-aggregates that strongly influence the outcome of bacterial culture quantification10,11. Alternatively, culture-independent assessments of microbial communities, particularly through quantitative real-time PCR (qPCR) or sequencing, have allowed a rapid screening and/or quantification of the specific microorganisms within the consortia12–16. The most frequently used qPCR method is based in the amplification of gDNA molecules. Unfortunately, DNA-based methodologies cannot unequivocally differentiate between live (including viable but non-cultivable cells (VBNC)) and dead cells17–20 and, therefore, are likely to overestimate the microbial communities19. Other specialized methods, such as propidium-monoazide (PMA)-qPCR21, are becoming more popular as the function and organization of the entire community depends on which members are alive or dead22. Nevertheless, PMA-based approaches have reportedly important limitations, including changes in cell membrane permeability (influencing the uptake of PMA dye) and false-positive signals23.
A less explored alternative is the utilization of RNA-based qPCR approaches for the detection of live members of a polymicrobial consortia. This principle was recent demonstrated by several independent studies which demonstrated that RNA-based quantification, was superior to DNA-based methods, in detecting live bacterial cells, providing a more relevant insight into the microbial community composition24,25. Of utmost importance, RNA-based qPCR approach has the significant potential of performing not only a quantification analysis, but also addressing specific microbial interactions, as detected by the expression of key virulence genes of interest.
In fact, infections caused by the presence of multispecies are often more virulent or recalcitrant to treatment than those caused by each independent species26. However, relatively little is known about the interspecies interactions that modulate the community dynamics in terms of composition and virulence; ultimately contributing to increased infection severity and chronicity27.
P. aeruginosa and S. aureus are versatile bacterial pathogens and common etiological agents in several polymicrobial infections, including wounds, otitis media and oral infections, and CF lung disease3. Of particular interest is the ecological interactions between P. aeruginosa and S. aureus, since microbial communities containing both of these pathogens can display enhanced virulence27. Despite routine administration of antibiotics, these infections are often highly resilient and tolerant to treatment28. This might explain why despite aggressive antibiotics treatment, patients with CF will eventually succumb to the chronic persistent infections29,30. Robust measures of these community changes (composition and/or abundance) are therefore urgent, since misinterpretation of these communities can impair antimicrobial treatment31–34.
Herein, the objective of this study was to evaluate an optimal RNA-based qPCR method to investigate the community dynamics and microbial interactions between P. aeruginosa and S. aureus. Thus, microbial composition of 24- and 48-h-old dual-species biofilms was assessed by RNA-based qPCR as well as changes in the transcriptomic profile of key virulence-related genes. The qPCR bacterial quantification was first fully optimized to polymicrobial samples and further compared with conventional culture and flow-cytometry techniques.
Results and Discussion
Quantification of dual-species biofilm communities: plate count versus flow cytometry
Dual-species biofilms were first characterized over the course of 24- and 48-h by enumerating P. aeruginosa and S. aureus through CFU assessment on selective media (PIA and MSA, respectively) and also by using flow-cytometry (Fig. 1). At both time points, P. aeruginosa was the prevalent species detected, being the differences between P. aeruginosa and S. aureus more relevant at 48-h of growth. Interestingly, these differences were more dramatic (~6 log10 CFU/mL) when considering culture quantification only, but significantly lower (~2 log10 cells/mL) when quantified by flow- cytometry. These results highlight that culture methods are not reliable to analyse biofilm population composition in certain conditions11. This has been justified by the fact that, with time, bacteria within biofilms can enter a VBNC state and, therefore, are not detectable by conventional CFU plating35.
Figure 1.
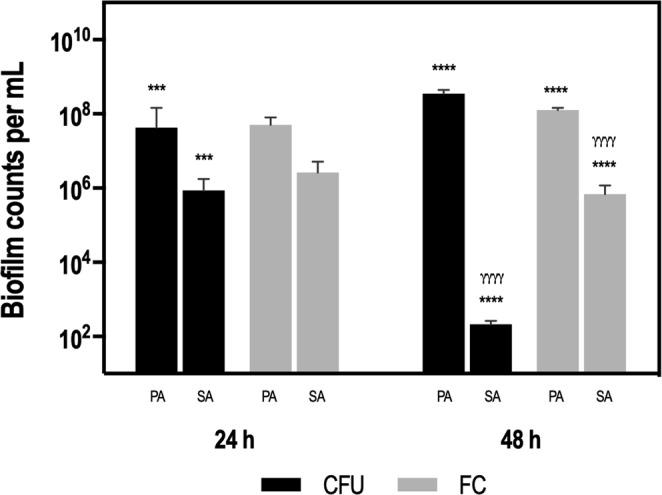
Quantification of P. aeruginosa and S. aureus populations in dual-species biofilms by plate count (CFU) and flow-cytometry (FC), following 24- and 48-h of growth. Biofilm counts are expressed as means ± SD of CFU per mL or cells per mL, respectively. For each condition three independent experiments were performed. Statistical significance was determined by performing an ANOVA followed by a Tukey’s multiple comparison test to compare: (*) significantly different P. aeruginosa counts versus S. aureus counts for each method; ***P < 0.001; ****P < 0.0001. (γ) significantly different P. aeruginosa counts between methods and significantly different S. aureus counts between methods; γγγγP < 0.0001. Abbreviations: PA = P. aeruginosa, SA = S. aureus.
In order to analyze and compare the biofilm structures, 24- and 48-h dual-species biofilms were directly examined using scanning electron microscopy (Supplementary Figure S1). The dual-species consortium showed, at 24-h, a non-contiguous layer of cells, that evolved, at 48-h, into mature biofilm producing a thick layer of co-aggregated cells surrounded by extracellular matrix. Both P. aeruginosa (road-shape) and S. aureus (coccus-shape) were distinguishable at 24-h but at 48-h, the dense structure of the biofilm did now allow to distinguish the bacterial species.
The impact of RNA extraction variability in the utilization of 16S rRNA as a direct quantification tool
Molecular-based methodologies have greatly increased our understanding regarding ecological interactions occurring among members of polymicrobial communities, when compared to culture-based methods36. However, those techniques are not void of limitations and, especially when using RNA-based approaches, several controls and normalizations need to be considered37–41.
A key aspect in RNA-based qPCR quantification is the need to normalize RNA extraction efficiencies, since the quality and quantity of extracted RNA can affect downstream applications. While often underreported, RNA extraction efficiency varies38,39. To demonstrate this, we first assessed the variability of RNA extraction processes, followed an optimized protocol previously described39,42. Briefly, we split planktonic suspensions of either high or low cell concentrations of P. aeruginosa, in six aliquots from the same biological sample, to perform total RNA extraction. As observed in Fig. 2, a high variability of total RNA was obtained, especially when using high density populations. Total RNA concentrations ranged from 26 to 220 ng/μL in the 109 CFU/mL aliquots and 7 to 77 ng/μL in the 105 CFU/mL aliquots. Noteworthy, aliquots of the same biological sample yielded distinct RNA concentrations, with a higher inter-assay variability in high biomass samples (coefficient of variation of 0.66) compared to the low biomass (0.23) (Supplementary Table S1).
Figure 2.
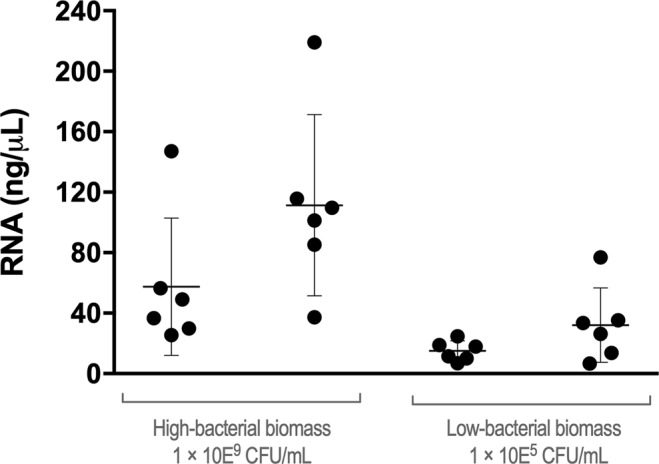
Total RNA extraction variability obtained from 109 CFU/mL and 105 CFU/mL of P. aeruginosa planktonic cultures. For both high- and low-bacterial concentrations, two independent biological samples were experienced, each one aliquoted in six technical replicates.
Next, we quantified 16S rRNA for each aliquot. Not surprising, the cycle threshold (Ct value) detection was highly variable, in a range of 3 to 5 cycles (Fig. 3). These results confirmed that quantifying 16S rRNA-target, by itself, is not a good strategy to quantify bacterial communities.
Figure 3.
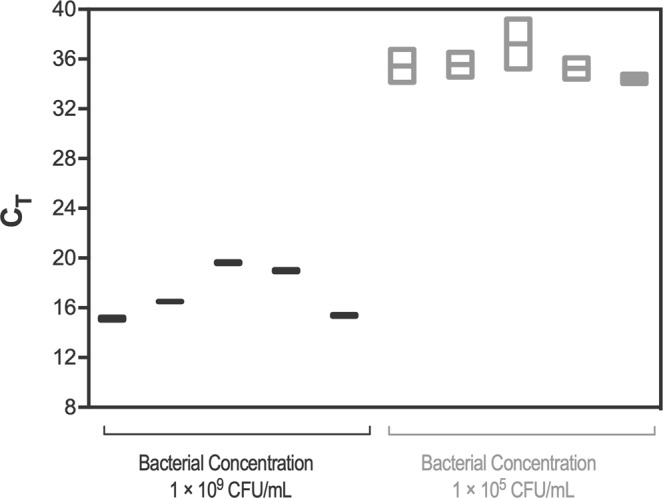
Variability in cycle threshold (Ct) values for 16S rRNA gene from 109 CFU/mL and 105 CFU/mL of P. aeruginosa planktonic samples corresponding to high- and low-bacterial concentrations. For each bacterial concentration five technical replicates from the same biological sample were used.
Impact of utilizing an external ref mRNA in bacterial quantification using 16 sRNA
When quantifying gene expression by qPCR, a normalization strategy is required and often it includes the quantification of a reference gene, also known as housekeeping gene43,44. Due to the variability of the experimental method39 the same principle needs to be applied when quantifying bacterial populations by qPCR. The most common alternative is to normalize the levels of the target mRNA to that of an exogenous mRNA spike-in transcript45–53. The exogenous mRNA control is characterized by: (i) not being originally found in the sample, (ii) its addition should be previous to the RNA extraction procedure and (iii) its initial concentration is known54. After laboratorial sample processing, the amount of recovered exogenous mRNA is determined by qPCR. Although exogenous ref mRNA approach has been previously used45–53, to the best of our knowledge, this study is the first evaluating polymicrobial biofilm samples.
In this study, commercial luciferase mRNA (ref mRNA) was preferred as the exogenous reference gene since the specific sequence is not found in any of the bacterial species tested, as assessed via NCBI GenBank database (www.ncbi.nlm.nih.gov). To quantify the ref mRNA, a standard curve was constructed, by plotting the Ct values against the known initial ref mRNA copy number (Supplementary Figure S2A). The standard curve generated covers a linear range along the selected five orders of magnitude with a correlation coefficient (R2) of 0.977, suggesting that all of the tested concentrations can be used in the subsequent analysis. Nevertheless, considering that the initial concentration of ref mRNA spiked into the samples may interfere with the mRNA quantification due to competition for qPCR reagents51, five aliquots derived from the same P. aeruginosa biological culture were amended with 1 × 107 to 1 × 1011 copies of ref mRNA, immediately after cell lysis (Supplementary Figure S2B). The significance of correlation between the total copies of ref mRNA added and total copies of ref mRNA extracted from each sample was calculated based on Pearson’s coefficient (r = 0.99, P < 0.05 for 109 CFU/mL samples and r = 0.90, P < 0.05 for 105 CFU/mL samples). Next, in order to test the sensitivity and accuracy of the qPCR assay, the 16S rRNA gene was normalized to the exogenous control. This was done either in the 109 and 105 CFU/mL samples. Figure 4 demonstrates that in almost all conditions tested, no significant variations were found in the relative gene expression determined. The exception was in the low biomass sample amended with 1 × 107 copies of ref mRNA. This suggests that at low amounts of genomic material, the efficiency drops below the linear range of the method55.
Figure 4.
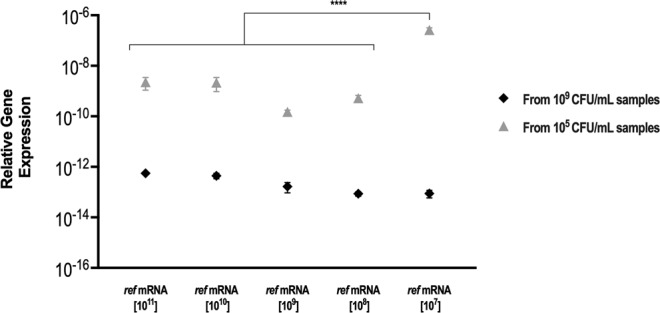
Effect of the ref mRNA normalization on the 16S rRNA expression levels. Relative gene expression of the 16S rRNA normalized to the ref mRNA, using the delta Ct method, in 109 CFU/mL and 105 CFU/mL samples adjusted to the initial copies of ref mRNA used. For each bacterial concentration five technical replicates were spiked with standard concentrations of ref mRNA prior to RNA extraction and subsequent reverse transcription. For each condition two independent biological samples were used. Statistical significance within each samples’ set was determined by performing one-way ANOVA analyses followed by a Tukey’s multiple comparison test (****P < 0.0001).
For further analysis, we selected the ref mRNA concentration of 1 × 108 copies of ref mRNA/μL, since under these conditions the best mRNA recovery was found, for either the low or high cell concentrations (Supplementary Table S2). Interestingly, the low mRNA recovery estimated confirms that somewhat random physical losses during RNA isolation will impair some quantification approaches24,37. Overall, these results support the importance of a ref mRNA normalization for proper quantification of bacterial populations using the RNA-based method.
Validation of the ref mRNA normalization in controlled dual-species communities
To assess the robustness of this approach, the relative expression of the 16S rRNA gene was analysed in three well defined dual-species planktonic samples, (P. aeruginosa/S. aureus ratio equal to 10, 1 and 0.1). The CT of each 16S rRNA target was normalized to the ref mRNA in order to determine the experimental ratios, calculated by applying the equation described in the section Quantitative Real Time-PCR of the Experimental procedures (Supplementary Table S3). As shown in Fig. 5, the experimental ratios obtained by applying the ref mRNA strategy were consistent with the theoretical ratios for all P. aeruginosa and S. aureus populations. Statistical analysis showed no significant differences between the theoretical and the experimental PA/SA ratios (P > 0.05).
Figure 5.
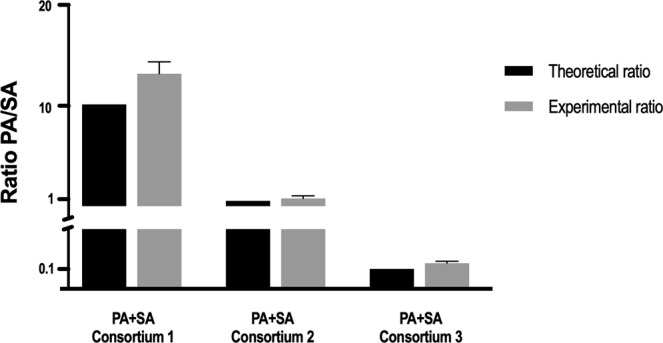
Comparison between the theoretical and the experimental qPCR PA/SA ratio of dual-species planktonic consortia (PA/SA ratios of 10, 1 and 0.1). For each condition two independent experiments were performed, each one with two technical replicates. Statistical significance was determined by performing independent ANOVA analyses followed by a Dunnett’s multiple comparison test.
Quantification of a dual-species biofilm communities by RNA-based qPCR
Following the experimental validation of the ref mRNA normalization strategy in the well-defined planktonic cultures, 24- and 48-h-old dual-species biofilms of P. aeruginosa and S. aureus were formed and quantified by qPCR (Supplementary Table S4). The same biofilms were also characterized by culture dependent (plate count) and culture independent (flow cytometry) methods (Supplementary Table S5). As shown in Fig. 6, the ratios of P. aeruginosa/S. aureus varied significantly accordingly with the method used, especially in the 48-h-old biofilm (P < 0.0001). The differences between culture-dependent and culture-independent methods were more significative than between both culture-independent methods. Interestingly, both qPCR and flow-cytometry methods have ascertained P. aeruginosa as the predominant species in the biofilm, but a significant population of S. aureus was also detected by these two methods in the 48-h-old consortium, in striking contrast with the CFU quantification. It is conceivable that after 48-h of co-culture with P. aeruginosa, a remarkable portion of S. aureus cells acquire a VBNC state. Indeed, as biofilms mature, often portions of bacteria within the community enter a VBNC state, in response to stressful conditions, such as starvation56 or antimicrobial treatments8,57,58, suggesting that this is an adaptive strategy for long-term survival under unfavourable conditions59. Although a series of physiological changes occur during the transition from cultivable to VBNC state, VBNC cells were found to maintain high levels of rRNA, similar to cultivable cells19,60, being qPCR RNA-based assays been commonly used to evaluate gene expression profile in cultures containing VBNC cells61–63. However, an important technical limitation of these studies is that transcription is evaluated as the global gene expression within the biofilm, not able to differentiate the physiological state of sub-populations. To determine the differences within such populations, the new generation of single-cells transcriptomic analysis64 should be used, but as far as we are aware, this has not been attempted in dual-species biofilms.
Figure 6.
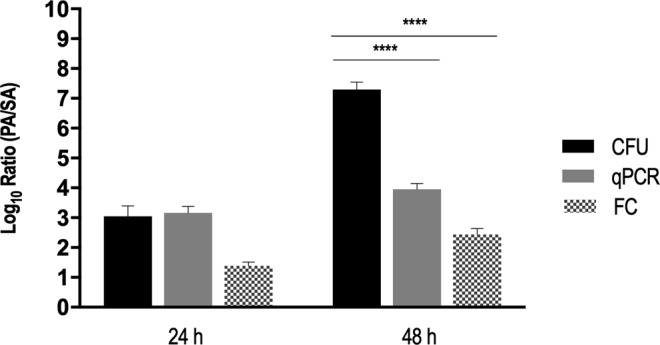
Relative quantification of 24-h and 48-h-old dual-species biofilms by plate count, qPCR and flow-cytometry. For each condition three independent experiments were performed. Statistical significance was determined by performing independent ANOVA analyses followed by a Dunnett’s multiple comparison test (****P < 0.0001).
We also performed a Live/Dead (L/D) viability assay on disrupted 48-h-old dual-species consortia, to inspect the overall biofilm-cell viability (Supplementary Figure S3). L/D staining showed that cocci-like bacterial cells were more abundant in the green than the red filter. Those results reinforce the previous conclusion that even though S. aureus lost its culture capability after 48-h of co-culture with P. aeruginosa, some bacteria remains in a viable state.
Applicability of the RNA- based qPCR quantification method to further study inter-species microbial interactions
A potential advantage of the RNA-based qPCR quantification approach to study dual-species biofilms, is its ability to not only discern between the relative proportions of each species, but also to determine if a specific gene is being induced or repressed. To achieve this, the gene of interest should be normalized to the reference gene (in this case, 16s RNA gene), while this reference gene is also normalized against the exogenous ref mRNA (in this case luciferase) in order to account for the relative quantification of each bacterial population.
As a demonstration of principle of this method potential, we quantified the differences in expression of key virulence genes of P. aeruginosa and S. aureus, between single- or dual-species biofilms. To inspect the impact of S. aureus on P. aeruginosa virulence potential, we analysed the expression of pqsA (Pseudomonas quinolone signal), rhlA (rhamnolipids) and mucA (alginate biosynthesis). Pseudomonas quinolone signal (PQS) is an important P. aeruginosa signalling molecule regulating the production of quorum-sensing (QS) dependent virulence factors, such as elastase, pyocyanin and rhamnolipids65. Rhamnolipids are biosurfactants produced by P. aeruginosa and several bacterial species acting as immune modulators and virulence factors; have antimicrobial activities and are involved in surface motility and in bacterial biofilm development66. Alginate is an important extracellular virulence factor and has been shown to impair host innate defences related to phagocytes67. Chronic P. aeruginosa infections are often associated with a mucoid phenotype due to the production of large quantities of the exopolysaccharide alginate68. The switch from nonmucoid to mucoid state is induced by the inactivation of mucA gene69. Notably, our results indicated that in 48-h-old dual-species biofilms, the expression levels of P. aeruginosa pqsA, rhlA and mucA were down-regulated, as compared with P. aeruginosa single-species (Fig. 7A). Interestingly, these differences were not detected in early (24-h-old) biofilms, suggesting a biofilm maturation-dependent effect.
Figure 7.

Gene expression profile of (A) P. aeruginosa and (B) S. aureus virulence-related genes in 24- and 48-h-old dual-species biofilms, as compared to single-species biofilms. Transcripts were normalized to 16S rRNA and results were calculated as fold change relative to gene expression values obtained for single-species biofilms. Dashed lines represent a 2-fold cut-off value. For each condition at least two independent experiments were performed with three technical replicates. Statistical significance was determined by performing an unpaired two-tailed t test (***P < 0.001).
Additionally, the role of P. aeruginosa in the gene expression profile of select S. aureus QS- (agrB and sarA) and virulence-related genes (sodA and hld) was also assessed. Agr and Sar are two main QS systems in S. aureus that control the expression of several virulence genes and production of cell-wall-associated proteins70,71. The hld gene, codifying delta hemolysin, is positively controlled by agr system and plays a role in severe staphylococcal infections72. Superoxide dismutase is a metalloprotein, encoded by the sodA gene, that inactivates harmful superoxide radicals encountered during oxidative stress73. Under the tested conditions, the presence of P. aeruginosa promoted the downregulation of sodA, sarA, agrB and hld genes in 24-h dual-species biofilms, while for 48-h-old dual-species biofilms sodA and sarA, were up-regulated (Fig. 7B).
Major conclusions and study limitations
Our results underline the importance of selecting appropriate methodologies to analyse dual-species biofilm communities, since some techniques, if used alone, may not provide accurate insights into biofilm composition, resulting in misleading conclusions. Both culture-independent techniques used in this study provided a more rapid and sensitive assessment of the individual species present in the dual-species biofilm consortia, comparing with conventional culture, which underestimated the real abundance of S. aureus population. As each technique has its particularity, Table 1 summarise the advantages, disadvantages, and purposes of use of all methodologies applied in this study.
Table 1.
Comparison of the methodologies described in this study, conventional culture, flow-cytometry and qPCR. Advantages, disadvantages and purposes of use applicable to microbial communities’ analysis.
| Conventional Culture | Flow Cytometry | qPCR | |
|---|---|---|---|
| Advantages | ♦ Standardized method that allows the identification of the most abundant members78,79. | ♦ Allows a rapid detection of multi-species samples80–82. | ♦ Detection of live cells18,84. |
| ♦ High specificity and sensitivity to detect both viable and VBNC cells83. | ♦ Can be used for different applications: bacterial detection85 and to assess changes in the gene expression profile86. | ||
| Disadvantages | ♦ Time consuming analysis; | ♦ Require full optimization of the method to each species81. | ♦ optimization (primers specificity, RNA extraction)42,43,87. |
| ♦ Requires selective media87; | |||
| ♦ Not detection of cells in a viable but non-cultivable state7,35. | ♦ Limit of cell detection81. | ||
| Purposes of use | Screening method for bacterial enumeration. | Fast method for enumeration of a large number of samples. | Fast method for detection and quantification; |
| Can simultaneously analyse gene expression profile of key genes (e.g. virulence genes) in the tested samples. |
Of relevance, our experiments demonstrated the advantage of using a RNA-based qPCR quantification and highlighted how important is the use of an appropriate mRNA control molecule in the qPCR assay, in order to address experimental biases. Overall, this experiment confirmed that this qPCR method is able to assess specific gene expression profiling together with quantification of bacterial populations in dual-species biofilms. Despite the advantages of the qPCR assay optimized through this study, it must be recognized that potential bias can also be introduced. For instance, when gene transcript abundance is calculated by normalization against the ref mRNA, the assumption is that it reflects the behaviour among the whole RNA pool over the entire sample’s processing steps. One potential bias in normalization against the ref mRNA is that it cannot compensate the differences of RNA extraction and reverse transcription efficiencies between the targeted species. While the occurrence of this bias cannot be excluded under the tested conditions, it is believed that is safe to assume that it is not very significant, given the results achieved. The qPCR sensitivity to quantify species with very low cell numbers74 in certain scenarios (e.g. after exposure of an antimicrobial therapy targeting specifically a member of the polymicrobial community) may also limit its usefulness on the quantification of less prevalent species within the community.
Experimental Procedures
Bacterial strains and culture conditions
P. aeruginosa (strain UCBPP-PA14) and S. aureus (strain ATCC 25923) were used throughout this work. Both strains were stored at −80 ± 2 °C in Tryptic Soy Broth (TSB, Liofilchem) supplemented with 20% glycerol. Prior to each assay, bacteria were subcultured from frozen stock preparations onto plates of TSB supplemented with 2% (W/V) agar (TSA) and incubated aerobically at 37 °C for 24-h.
Planktonic growth
All assays were carried out by using a standardized bacterial inoculum. Briefly, a few colonies of each species were collected from the TSA plates and grown overnight in batches of TSB (Liofilchem) at 37 °C under agitation (120 rpm). Subsequently, cells were harvested by centrifugation (9000 g, 5 min) and washed in sterile saline solution (0.9% NaCl). The concentration of cellular suspensions was then adjusted in TSB by spectrophotometric measurement at 640 nm (calibrations were performed for each bacterial strain to relate the absorbance at 640 nm with the number of colony forming units, CFU) to obtain a final concentration of 1 × 105 CFU/mL or 1 × 109 CFU/mL. Unless otherwise stated, two independent assays with five to six replicates for each condition tested were performed. For the preparation of the dual-species planktonic suspension, overnight grown cultures of each species were adjusted to a final concentration of 1 × 109 CFU/mL and then mixed in three samples of various compositions (P. aeruginosa/S. aureus ratios of 10, 1 and 0.1). Two independent assays for each condition with two replicates were used. To perform the RNA extraction and subsequent reverse transcription, single- and dual-species planktonic suspension were centrifuged (7000 rpm, 10 min, 4 °C) and the pellet resuspended in RNA protect (Qiagen), as described below. Planktonic samples preparation is schematically represented in Fig. 8.
Figure 8.

Experimental workflow of the planktonic and biofilm samples preparation.
Biofilm formation
Dual-species biofilms encompassing P. aeruginosa and S. aureus were grown as previously described, with minor modifications4. Briefly, overnight cultures of each strain, grown in TSB at 37 °C and 120 rpm in aerobic conditions, were washed in sterile water and diluted in TSB to obtain 1 × 107 CFU/mL as final concentration. The dual-species cultures were then prepared by mixing the inoculum of each species in a 1:1 ratio. Each well of a 24-well polystyrene plate (Orange Scientific, Braine-l’Alleud, Belgium) was seeded with 1 mL of P. aeruginosa and S. aureus suspension and incubated at 37 °C on a horizontal shaker (120 rpm) for 24- and 48-h. Following biofilm growth, the planktonic fraction of the polystyrene plates was removed and the wells were washed twice with distilled sterile water. Biofilms cells were resuspended in 1 mL of 0.9% NaCl, detached by scraping and collected by centrifugation (7000 rpm, 10 min, 4 °C). Three biologic replicates of each condition were performed. To perform the RNA extraction and subsequent reverse transcription, a biofilm pooling (10 wells of a 24-well-plate) was resuspended in RNA protect (Qiagen). Biofilm samples preparation is schematically illustrated in Fig. 8.
RNA extraction
RNA was extracted using the RNeasy mini kit (Qiagen), as previously optimized42. Briefly, bacteria were first resuspended in 500 μL lysis buffer (provided in the kit). Then, 500 μL of phenol solution (AppliChem) was added, and the entire suspension was transferred to a safe lock tube (2 mL) with 0.4 g of acid-washed 150–212 mm silica beads (Sigma). The tube content was vortexed for 20 s previously to being placed in the FastPrepH cell disruptor (BIO 101, Thermo Electron Corporation, Thermo Scientific) at 6.5 m/s for 35 s. The samples were then cooled on ice and the beat- beading step repeated twice. Afterwards, Afterwards, 700 μL of 70% ethanol (Fisher Scientific) was added to the supernatants, followed by 1 μL of 1 × 108 luciferase control RNA transcripts (ref mRNA) (Promega). To achieve the final RNA fraction, 50 μL of RNase-free water was added to the RNA columns. To digest possible contaminating genomic DNA, RNA was treated with DNase I (Fermentas, Ontario, Canada) following manufacturer instructions. Briefly, the RNA samples were incubated at 37 °C for 30 min after the addition of 2 μL of DNase I and 5 μL of reaction buffer. Then, 5 μL of 25 mM EDTA was added to the mixture and incubated at 65 °C for 10 min to inactivate the DNase I enzyme.
RNA quality determination
RNA integrity and purity were assessed by gel electrophoresis and the total RNA concentration was spectrophotometrically determined using a NanoDrop 1000TM (Thermo Scientific). As indicator of protein contamination and polysaccharide, phenol, and/or chaotropic salt contamination, the absorbance ratio A260/A280 and A260/A230 were measured, respectively75. The RNA integrity was assessed by 23S/16S banding pattern visualization. Electrophoresis was carried out at 80 V for 60 min using a 1.5% agarose gel. The gel was stained with GelRed (Thermo Fisher Scientific) and visualized using a GelDoc2000 (Bio-Rad, Hercules, CA, US) (Supplementary Figure S4). RNA was stored at −80 °C for further use.
Determination of RNA recovery using an exogenous internal reference mRNA
Luciferase mRNA (Promega) was used as the exogenous internal reference (ref mRNA) to estimate the percentage of RNA loss during extraction. After the centrifugation of the lysed cells, 1 μL of a range of 1 × 1011 to 1 × 107 ref mRNA transcripts/μL were added to the aqueous phase. After RNA extraction, the amount of ref mRNA recovered from samples was measured by qPCR and the standard curve generated from serial dilutions of ref mRNA. The recovery fraction was calculated by dividing the determined levels by the initial number of ref mRNA transcripts added to the samples.
Quantitative Real Time-PCR (qPCR)
For the quantification of the 16S rRNA by qPCR, total RNA was reversely transcribed to complementary DNA (cDNA) using GRS cDNA Synthesis Kit (Grisp). Specifically, 100 ng of total RNA was reversely transcribed in 10 μL of reaction volume by using random primers. The samples were incubated at 65 °C for 5 min, 37 °C for 60 min, and 70 °C for 10 min. Primers, specific for 16S rRNA, housekeeping gene, and virulence-related genes of P. aeruginosa and S. aureus were designed (Supplementary Table S5). The qPCR reaction was performed by mixing together 5 µL of master mix SYBR Xpert Fast SYBR (Grisp), 2 µL of 1:100 diluted cDNA, 0.5 µL of forward and reverse primes (5 µM), and water up to a total volume of 10 µL. No reverse transcriptase (NRT) and a no template control (NTC) were included to verify the reaction mixtures were DNA and other contaminants-free. The efficiency of the primers used was measured by the dilution method38. To perform the qPCR run, a CFX 96 (Bio-Rad) was used with the following cycle parameter: 95 °C for 3 minutes, 40 cycles of 95 °C for 5 s, and 60 °C for 20 s. qPCR products were analysed by melting curves for unspecific products or primer dimer formation. The qPCR was applied to independently quantify the 16S rRNA of P. aeruginosa and S. aureus and the ref mRNA in each cocktailed mixed sample. The normalized gene expression was determined by using the delta Ct method (2∆Ct), a variation of the Livak method, where ∆Ct = Ct (ref mRNA) − Ct (16S rRNA). After normalization of the 16S rRNA for both species in each cocktail sample, the PA/SA ratio was calculated by applying the following equation:
Relative fold increase of pqsA, rhlA, mucA, sodA, sarA, agrB and hld genes, was determined by applying the Pfaffl equation76 using 16S rRNA as reference gene. At least two biologic replicates of each condition were used with three technical replicates.
Biofilm cultivability
The number of adhering bacteria was determined after biofilm cell detachment. To remove any aggregates, biofilm suspensions were first sonicated for 10 s at 30% amplitude11 (Cole Parmer Ultrasonic Processor, IL, USA) and then the culturable cell count was carried out. Briefly, the disrupted biofilms were serially diluted (1:10) in 0.9% NaCl, streaked onto different selective agar media and incubated at 37 °C for 18–24 h, for CFU counting. Pseudomonas isolation agar (PIA) and Mannitol Salt Agar (MSA) were the selective media used to discriminate P. aeruginosa and S. aureus, respectively. Values of cultivable biofilm cells were expressed as CFU/mL and represented as the average of three independent experiments.
Flow-cytometry
The total number of P. aeruginosa and S. aureus cells in 24- and 48 h-old mixed biofilms was determined using flow-cytometry. In brief, biofilm cells were collected by centrifugation (7000 rpm, 10 min, 4 °C) and suspended in 100 µL of a solution with 25 µg/mL of wheat germ agglutinin (WGA) conjugated with FITC (Molecular probes, ThermoFisher Scientific). This suspension was incubated at room temperature, in the dark, for 15 min. Thereafter, cells were washed twice with 0.9% NaCl, suspended in 1 mL of the same solution and sonicated for 10 s at 30% amplitude (Cole Parmer Ultrasonic Processor, IL, USA) to disrupt aggregates. The bacterial fluorescence analysis was carried out using an EC800 Sony flow cytometer (Sony Biotechnologies Inc., CA, USA) equipped with a 488 nm laser. Multiparametric analyses were performed on both scattering signals (FSC, SSC) and FL1 channel. All the detectors were set to logarithmic amplification. Samples were acquired with a flow rate of 10 μL/min and the analysis stopped when 80 000 events were detected. Three biologic replicates of each condition were used and each sample was analysed at least twice to ensure an accurate counting. Data analysis was performed using EC800 analysis software (Sony Biotechnologies).
Assessment of cell viability by Live/Dead staining
To analyse the viability of dual-species biofilm consortia 48-h-old biofilms were scraped, and the biofilm disrupted-cells were collected in 1 mL sterile distilled water. Afterwards, disrupted biofilm cells were stained in the dark, for 15 min, with SYTO® BC (2 μM) and Propidium Iodide (15 μM) (both from Invitrogen™, CA, USA) prepared in saline solution, in order to differentially label live and dead cells by microscopy. For microscopic observation, an Olympus BX51 microscope fitted with fluorescence illumination was used. The optical filter combination consisted of 470 to 490 nm in combination with 530 to 550 nm excitation filters.
Scanning electron microscopy
Scanning electron microscopy (SEM) was used to examine the dual-species biofilm as described before, with minor modifications77. Briefly, dual-species biofilms were formed on Thermanox® plastic coverslips (Thermo Scientific™; Rochester, NY, USA) placed in the bottom of the wells of 24-well microtiter plates (Orange Scientific, Belgium). Prior to SEM observation, discs were added to aluminum pin stubs with electrically conductive carbon adhesive tape (PELCO Tabs™). Samples were coated with 2 nm of Au for improved conductivity. The aluminum pin stub was then placed inside a Phenom Standard Sample Holder or Phenom Charge Reduction Sample Holder. The analysis was conducted at 5 kV with intensity image. The samples were characterized using a desktop Scanning Electron Microscope (SEM) (Phenom ProX, Netherlands). All results were acquired using the ProSuite software.
Statistical analysis
All assays were carried out in triplicate and repeated at least twice, and the results are presented as means ± SDs. The Pearson’s correlation coefficient was calculated to determine the association between two variables. All P-values were based on two-tailed tests of significance, and confidence interval of 95% was used in all the analysis performed. Statistical analysis was performed using Graph Pad Prism 7.0.
Supplementary information
Acknowledgements
This study was supported by the Portuguese Foundation for Science and Technology (FCT) under the scope of the strategic funding of UID/BIO/04469/2019 unit and BioTecNorte operation (NORTE-01-0145-FEDER-000004) funded by the European Regional Development Fund under the scope of Norte2020 - Programa Operacional Regional do Norte. Grant of APM (SFRH/BD/132165/2017).
Author Contributions
N.C. and M.O.P. conceptualized and designed the study. A.P.M. and A.F. performed the experiments. All authors contributed in data interpretation. A.P.M. wrote the first draft of the paper. N.C., A.F. and M.O.P. reviewed the manuscript. All authors read and approved the final manuscript.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-50094-3.
References
- 1.Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the Natural environment to infectious diseases. Nat. Rev. Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 2.Stewart PS, Franklin MJ. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- 3.Peters BM, et al. Polymicrobial interactions: Impact on pathogenesis and human disease. Clin. Microbiol. Rev. 2012;25:193–213. doi: 10.1128/CMR.00013-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magalhães AP, Lopes SP, Pereira MO. Insights into Cystic Fibrosis Polymicrobial Consortia: The Role of Species Interactions in Biofilm Development, Phenotype, and Response to In-Use Antibiotics. Front. Microbiol. 2017;7:1–11. doi: 10.3389/fmicb.2016.02146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ammann TW, Bostanci N, Belibasakis GN, Thurnheer T. Validation of a quantitative real-time PCR assay and comparison with fluorescence microscopy and selective agar plate counting for species-specific quantification of an in vitro subgingival biofilm model. J. Periodontal Res. 2013;48:517–526. doi: 10.1111/jre.12034. [DOI] [PubMed] [Google Scholar]
- 6.Cereija TB, Castro J, Alves P, Cerca N. Influence of anaerobic conditions on vaginal microbiota recovery from bacterial vaginosis patients. Sex. Transm. Infect. 2013;89:307–307. doi: 10.1136/sextrans-2013-051100. [DOI] [PubMed] [Google Scholar]
- 7.Carvalhais V, et al. Tetracycline and rifampicin induced a viable but nonculturable state in Staphylococcus epidermidis biofilms. Future Microbiol. 2018;13:27–36. doi: 10.2217/fmb-2017-0107. [DOI] [PubMed] [Google Scholar]
- 8.Pasquaroli S, et al. Antibiotic pressure can induce the viable but non-culturable state in Staphylococcus aureus growing in biofilms. J. Antimicrob. Chemother. 2013;68:1812–1817. doi: 10.1093/jac/dkt086. [DOI] [PubMed] [Google Scholar]
- 9.Li L, et al. The importance of the viable but non-culturable state in human bacterial pathogens. Front. Microbiol. 2014;5:1–1. doi: 10.3389/fmicb.2014.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fux CA, Costerton JW, Stewart PS, Stoodley P. Survival strategies of infectious biofilms. Trends Microbiol. 2005;13:34–40. doi: 10.1016/j.tim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Freitas AI, Vasconcelos C, Vilanova M, Cerca N. Optimization of an automatic counting system for the quantification of Staphylococcus epidermidis cells in biofilms. J. Basic Microbiol. 2014;54:750–757. doi: 10.1002/jobm.201200603. [DOI] [PubMed] [Google Scholar]
- 12.Grattepanche F, Lacroix C, Audet P, Lapointe G. Quantification by real-time PCR of Lactococcus lactis subsp. cremoris in milk fermented by a mixed culture. Appl. Microbiol. Biotechnol. 2005;66:414–21. doi: 10.1007/s00253-004-1705-4. [DOI] [PubMed] [Google Scholar]
- 13.Smits THM, Devenoges C, Szynalski K, Maillard J, Holliger C. Development of a real-time PCR method for quantification of the three genera Dehalobacter, Dehalococcoides, and Desulfitobacterium in microbial communities. J. Microbiol. Methods. 2004;57:369–78. doi: 10.1016/j.mimet.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Winded C, Anneser B, Griebler C, Meckenstock RU, Lueders T. Depth-resolved quantification of anaerobic toluene degraders and aquifer microbial community patterns in distinct redox zones of a tar oil contaminant plume. Appl. Environ. Microbiol. 2008;74:792–801. doi: 10.1128/AEM.01951-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vuong J, et al. Development of Real-Time PCR Methods for the Detection of Bacterial Meningitis Pathogens without DNA Extraction. PLoS One. 2016;11:e0147765. doi: 10.1371/journal.pone.0147765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zemanick ET, et al. Reliability of quantitative real-time PCR for bacterial detection in cystic fibrosis airway specimens. PLoS One. 2010;5:e15101. doi: 10.1371/journal.pone.0015101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aellen S, Que Y-AA, Guignard B, Haenni M, Moreillon P. Detection of live and antibiotic-killed bacteria by quantitative real-time PCR of specific fragments of rRNA. Antimicrob. Agents Chemother. 2006;50:1913–1920. doi: 10.1128/AAC.00869-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keer JT, Birch L. Molecular methods for the assessment of bacterial viability. J. Microbiol. Methods. 2003;53:175–83. doi: 10.1016/S0167-7012(03)00025-3. [DOI] [PubMed] [Google Scholar]
- 19.Lahtinen SJ, et al. Degradation of 16S rRNA and attributes of viability of viable but nonculturable probiotic bacteria. Lett. Appl. Microbiol. 2008;46:693–698. doi: 10.1111/j.1472-765X.2008.02374.x. [DOI] [PubMed] [Google Scholar]
- 20.Rogers GB, et al. The exclusion of dead bacterial cells is essential for accurate molecular analysis of clinical samples. Clin. Microbiol. Infect. 2010;16:1656–1658. doi: 10.1111/j.1469-0691.2010.03189.x. [DOI] [PubMed] [Google Scholar]
- 21.Tavernier S, Coenye T. Quantification of Pseudomonas aeruginosa in multispecies biofilms using PMA-qPCR. PeerJ. 2015;3:e787. doi: 10.7717/peerj.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emerson JB, et al. Schrödinger’s microbes: Tools for distinguishing the living from the dead in microbial ecosystems. Microbiome. 2017;5:86. doi: 10.1186/s40168-017-0285-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gensberger ET, et al. Evaluation of quantitative PCR combined with PMA treatment for molecular assessment of microbial water quality. Water Res. 2014;67:367–376. doi: 10.1016/j.watres.2014.09.022. [DOI] [PubMed] [Google Scholar]
- 24.Li R, et al. Comparison of DNA-, PMA-, and RNA-based 16S rRNA Illumina sequencing for detection of live bacteria in water. Sci. Rep. 2017;7:5752. doi: 10.1038/s41598-017-02516-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomez-Silvan, C. et al. A comparison of methods used to unveil the genetic and metabolic pool in the built environment. Microbiome6 (2018). [DOI] [PMC free article] [PubMed]
- 26.Burmølle M, et al. Enhanced biofilm formation and increased resistance to antimicrobial agents and bacterial invasion are caused by synergistic interactions in multispecies biofilms. Appl. Environ. Microbiol. 2006;72:3916–23. doi: 10.1128/AEM.03022-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeLeon S, et al. Synergistic interactions of Pseudomonas aeruginosa and Staphylococcus aureus in an In vitro wound model. Infect. Immun. 2014;82:4718–4728. doi: 10.1128/IAI.02198-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stressmann FA, et al. Long-term cultivation-independent microbial diversity analysis demonstrates that bacterial communities infecting the adult cystic fibrosis lung show stability and resilience. Thorax. 2012;67:867–73. doi: 10.1136/thoraxjnl-2011-200932. [DOI] [PubMed] [Google Scholar]
- 29.Cuthbertson L, et al. Respiratory microbiota resistance and resilience to pulmonary exacerbation and subsequent antimicrobial intervention. ISME J. 2016;10:1081–1091. doi: 10.1038/ismej.2015.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cigana C, et al. Staphylococcus aureus Impacts Pseudomonas aeruginosa Chronic Respiratory Disease in Murine Models. J. Infect. Dis. 2018;217:933–942. doi: 10.1093/infdis/jix621. [DOI] [PubMed] [Google Scholar]
- 31.Lopes SP, Azevedo NF, Pereira MO. Developing a model for cystic fibrosis sociomicrobiology based on antibiotic and environmental stress. Int. J. Med. Microbiol. 2017;307:460–470. doi: 10.1016/j.ijmm.2017.09.018. [DOI] [PubMed] [Google Scholar]
- 32.Tavernier, S. et al. Community composition determines activity of antibiotics against multispecies biofilms. Antimicrob. Agents Chemother. 61 (2017). [DOI] [PMC free article] [PubMed]
- 33.Zhao J, Murray S, Lipuma JJ. Modeling the impact of antibiotic exposure on human microbiota. Sci. Rep. 2014;4:4345. doi: 10.1038/srep04345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung C-Y, Wang J-C, Chuang H-S. Simultaneous and quantitative monitoring of co-cultured Pseudomonas aeruginosa and Staphylococcus aureus with antibiotics on a diffusometric platform. Sci. Rep. 2017;7:46336. doi: 10.1038/srep46336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oliver JD. The viable but nonculturable state in bacteria. J. Microbiol. 2005;43(Spec No):93–100. [PubMed] [Google Scholar]
- 36.Bittar F, et al. Molecular detection of multiple emerging pathogens in sputa from cystic fibrosis patients. PLoS One. 2008;3:e2908. doi: 10.1371/journal.pone.0002908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.França A, Bento JC, Cerca N. Variability of RNA quality extracted from biofilms of foodborne pathogens using different kits impacts mRNA quantification by qPCR. Curr. Microbiol. 2012;65:54–59. doi: 10.1007/s00284-012-0124-5. [DOI] [PubMed] [Google Scholar]
- 38.França A, Melo LD, Cerca N. Comparison of RNA extraction methods from biofilm samples of Staphylococcus epidermidis. BMC Res. Notes. 2011;4:572. doi: 10.1186/1756-0500-4-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sousa C, França A, Cerca N. Assessing and reducing sources of gene expression variability in Staphylococcus epidermidis biofilms. Biotechniques. 2014;57:295–301. doi: 10.2144/000114238. [DOI] [PubMed] [Google Scholar]
- 40.Sieber MW, et al. Substantial performance discrepancies among commercially available kits for reverse transcription quantitative polymerase chain reaction: A systematic comparative investigator-driven approach. Anal. Biochem. 2010;401:303–311. doi: 10.1016/j.ab.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 41.Rump LV, Asamoah B, Gonzalez-Escalona N. Comparison of commercial RNA extraction kits for preparation of DNA-free total RNA from Salmonella cells. BMC Res. Notes. 2010;3:211. doi: 10.1186/1756-0500-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.França A, Freitas AI, Henriques AF, Cerca N. Optimizing a qPCR gene expression quantification assay for S. epidermidis biofilms: a comparison between commercial kits and a customized protocol. PLoS One. 2012;7:e37480. doi: 10.1371/journal.pone.0037480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bustin SA, et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 44.Huggett J, Dheda K, Bustin S, Zumla A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005;6:279–284. doi: 10.1038/sj.gene.6364190. [DOI] [PubMed] [Google Scholar]
- 45.Lequarre AS, Traverso JM, Marchandise J, Donnay I. Poly(A) RNA is reduced by half during bovine oocyte maturation but increases when meiotic arrest is maintained with CDK inhibitors. Biol. Reprod. 2004;71:425–31. doi: 10.1095/biolreprod.103.026724. [DOI] [PubMed] [Google Scholar]
- 46.Vigneault C, McGraw S, Massicotte L, Sirard M-A. Transcription factor expression patterns in bovine in vitro-derived embryos prior to maternal-zygotic transition. Biol. Reprod. 2004;70:1701–9. doi: 10.1095/biolreprod.103.022970. [DOI] [PubMed] [Google Scholar]
- 47.Bower NI, Moser RJ, Hill JR, Lehnert SA. Universal reference method for real-time PCR gene expression analysis of preimplantation embryos. Biotechniques. 2007;42:199–206. doi: 10.2144/000112314. [DOI] [PubMed] [Google Scholar]
- 48.Bettegowda A, Patel OV, Ireland JJ, Smith GW. Quantitative analysis of messenger RNA abundance for ribosomal protein L-15, cyclophilin-A, phosphoglycerokinase, beta-glucuronidase, glyceraldehyde 3-phosphate dehydrogenase, beta-actin, and histone H2A during bovine oocyte maturation and early embryogenesis in vitro. Mol. Reprod. Dev. 2006;73:267–78. doi: 10.1002/mrd.20333. [DOI] [PubMed] [Google Scholar]
- 49.Baker PJ, O’Shaughnessy PJ. Expression of prostaglandin D synthetase during development in the mouse testis. Reproduction. 2001;122:553–9. doi: 10.1530/rep.0.1220553. [DOI] [PubMed] [Google Scholar]
- 50.Smith RD, Brown B, Ikonomi P, Schechter AN. Exogenous reference RNA for normalization of real-time quantitative PCR. Biotechniques. 2003;34:88–91. doi: 10.2144/03341st05. [DOI] [PubMed] [Google Scholar]
- 51.Johnson DR, Lee PKH, Holmes VF, Alvarez-Cohen L. An internal reference technique for accurately quantifying specific mRNAs by real-time PCR with application to the tceA reductive dehalogenase gene. Appl. Environ. Microbiol. 2005;71:3866–3871. doi: 10.1128/AEM.71.7.3866-3871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, et al. Transcription level of messenger RNA per gene copy determined with dual-spike-in strategy. Anal. Biochem. 2009;394:202–208. doi: 10.1016/j.ab.2009.07.043. [DOI] [PubMed] [Google Scholar]
- 53.Revilla-Fernández S, et al. The use of endogenous and exogenous reference RNAs for qualitative and quantitative detection of PRRSV in porcine semen. J. Virol. Methods. 2005;126:21–30. doi: 10.1016/j.jviromet.2005.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnston S, Gallaher Z, Czaja K. Exogenous reference gene normalization for real-time reverse transcription-polymerase chain reaction analysis under dynamic endogenous transcription. Neural Regen. Res. 2012;7:1064–1072. doi: 10.3969/j.issn.1673-5374.2012.14.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levesque-Sergerie J-P, Duquette M, Thibault C, Delbecchi L, Bissonnette N. Detection limits of several commercial reverse transcriptase enzymes: impact on the low- and high-abundance transcript levels assessed by quantitative RT-PCR. BMC Mol. Biol. 2007;8:93. doi: 10.1186/1471-2199-8-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dwidjosiswojo Z, Richard J, Moritz MM, Dopp E, Wingender J. Influence of copper ions on the viability and cytotoxicity of Pseudomonas aeruginosa under conditions relevant to drinking water environments. Int. J. Hyg. Environ. Health. 2011;214:485–492. doi: 10.1016/j.ijheh.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 57.Rodrigues Maria E., Lopes Susana P., Pereira Cláudia R., Azevedo Nuno F., Lourenço Anália, Henriques Mariana, Pereira Maria O. Polymicrobial Ventilator-Associated Pneumonia: Fighting In Vitro Candida albicans-Pseudomonas aeruginosa Biofilms with Antifungal-Antibacterial Combination Therapy. PLOS ONE. 2017;12(1):e0170433. doi: 10.1371/journal.pone.0170433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gião MS, Azevedo NF, Wilks SA, Vieira MJ, Keevil CW. Interaction of Legionella pneumophila and Helicobacter pylori with bacterial species isolated from drinking water biofilms. BMC Microbiol. 2011;11:57. doi: 10.1186/1471-2180-11-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ducret A, Chabalier M, Dukan S. Characterization and resuscitation of ‘non-culturable’ cells of Legionella pneumophila. BMC Microbiol. 2014;14:3. doi: 10.1186/1471-2180-14-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramamurthy T, Ghosh A, Pazhani GP, Shinoda S. Current Perspectives on Viable but Non-Culturable (VBNC) Pathogenic Bacteria. Front. Public Heal. 2014;2:103. doi: 10.3389/fpubh.2014.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Toole D., Hunter R., Allen T., Zekarias B., Lehmann J., Kim K. S., Grab D., Corbeil L. B. Effect of Histophilus somni on Heart and Brain Microvascular Endothelial Cells. Veterinary Pathology. 2017;54(4):629–639. doi: 10.1177/0300985817691581. [DOI] [PubMed] [Google Scholar]
- 62.Dinu LD, Bach S. Induction of viable but nonculturable Escherichia coli O157:H7 in the phyllosphere of lettuce: A food safety risk factors. Appl. Environ. Microbiol. 2011;77:8295–8302. doi: 10.1128/AEM.05020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carvalhais V, et al. Dormancy within Staphylococcus epidermidis biofilms: a transcriptomic analysis by RNA-seq. Appl. Microbiol. Biotechnol. 2014;98:2585–96. doi: 10.1007/s00253-014-5548-3. [DOI] [PubMed] [Google Scholar]
- 64.Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018;50:96. doi: 10.1038/s12276-018-0071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Déziel E, et al. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: Multiple quorum sensingregulated genes are modulated without affecting IasRI, rhIRI or the production of N-acyl-L-homoserine lactones. Mol. Microbiol. 2005;55:998–1014. doi: 10.1111/j.1365-2958.2004.04448.x. [DOI] [PubMed] [Google Scholar]
- 66.Abdel-Mawgoud AM, Lépine F, Déziel E. Rhamnolipids: diversity of structures, microbial origins and roles. Appl. Microbiol. Biotechnol. 2010;86:1323–36. doi: 10.1007/s00253-010-2498-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei Q, Ma LZ. Biofilm matrix and its regulation in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2013;14:20983–1005. doi: 10.3390/ijms141020983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang L, et al. Bacterial adaptation during chronic infection revealed by independent component analysis of transcriptomic data. BMC Microbiol. 2011;11:184. doi: 10.1186/1471-2180-11-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martin DW, et al. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc. Natl. Acad. Sci. USA. 1993;90:8377–81. doi: 10.1073/pnas.90.18.8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bronner S, Monteil H, Prévost G. Regulation of virulence determinants in Staphylococcus aureus: Complexity and applications. FEMS Microbiol. Rev. 2004;28:183–200. doi: 10.1016/j.femsre.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 71.Dunman PM, et al. Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol. 2001;183:7341–7353. doi: 10.1128/JB.183.24.7341-7353.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kong C, Neoh H, Nathan S. Targeting Staphylococcus aureus Toxins: A Potential form of Anti-Virulence Therapy. Toxins (Basel) 2016;8:72. doi: 10.3390/toxins8030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Karavolos MH, Horsburgh M, Ingham E, Foster SJ. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology. 2003;149:2749–2758. doi: 10.1099/mic.0.26353-0. [DOI] [PubMed] [Google Scholar]
- 74.Kralik P, Ricchi M. A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions. Parameters, and Everything. Front. Microbiol. 2017;8:108. doi: 10.3389/fmicb.2017.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tavares L, Alves PM, Ferreira RB, Santos CN. Comparison of different methods for DNA-free RNA isolation from SK-N-MC neuroblastoma. BMC Res. Notes. 2011;4:3. doi: 10.1186/1756-0500-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zago CE, et al. Dynamics of Biofilm Formation and the Interaction between Candida albicans and Methicillin-Susceptible (MSSA) and –Resistant Staphylococcus aureus (MRSA) PLoS One. 2015;10:e0123206. doi: 10.1371/journal.pone.0123206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tunney MM, et al. Use of culture and molecular analysis to determine the effect of antibiotic treatment on microbial community diversity and abundance during exacerbation in patients with cystic fibrosis. Thorax. 2011;66:579–84. doi: 10.1136/thx.2010.137281. [DOI] [PubMed] [Google Scholar]
- 79.Burns JL, Rolain J-M. Culture-based diagnostic microbiology in cystic fibrosis: Can we simplify the complexity? J. Cyst. Fibros. 2014;13:1–9. doi: 10.1016/j.jcf.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 80.Liu Z, et al. Ecological Stability Properties of Microbial Communities Assessed by Flow Cytometry. mSphere. 2018;3:e00564–17. doi: 10.1128/mSphere.00564-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rüger M, Bensch G, Tüngler R, Reichl U. A flow cytometric method for viability assessment of Staphylococcus aureus and Burkholderia cepacia in mixed culture. Cytom. Part A. 2012;81 A:1055–1066. doi: 10.1002/cyto.a.22219. [DOI] [PubMed] [Google Scholar]
- 82.Rüger M, Ackermann M, Reichl U. Species-specific viability analysis of Pseudomonas aeruginosa, Burkholderia cepacia and Staphylococcus aureus in mixed culture by flow cytometry. BMC Microbiol. 2014;14:56. doi: 10.1186/1471-2180-14-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taimur Khan MM, Pyle BH, Camper AK. Specific and rapid enumeration of viable but nonculturable and viable-culturable gram-negative bacteria by using flow cytometry. Appl. Environ. Microbiol. 2010;76:5088–5096. doi: 10.1128/AEM.02932-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cangelosi GA, Weigel KM, Lefthand-Begay C, Meschke JS. Molecular detection of viable bacterial pathogens in water by ratiometric pre-rRNA analysis. Appl. Environ. Microbiol. 2010;76:960–2. doi: 10.1128/AEM.01810-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blazewicz SJ, Barnard RL, Daly RA, Firestone MK. Evaluating rRNA as an indicator of microbial activity in environmental communities: Limitations and uses. ISME J. 2013;7:2061–2068. doi: 10.1038/ismej.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Castro Joana, Machado Daniela, Cerca Nuno. Unveiling the role of Gardnerella vaginalis in polymicrobial Bacterial Vaginosis biofilms: the impact of other vaginal pathogens living as neighbors. The ISME Journal. 2019;13(5):1306–1317. doi: 10.1038/s41396-018-0337-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lopes SP, Azevedo NF, Pereira MO. Quantitative assessment of individual populations within polymicrobial biofilms. Sci. Rep. 2018;8:9494. doi: 10.1038/s41598-018-27497-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.