

Abstract
DNA double-strand breaks (DSBs) are a highly toxic form of DNA damage produced by a number of carcinogens, drugs, and metabolic abnormalities. Involvement of DSBs in many pathologies has led to frequent measurements of these lesions, primarily via biodosimetry of S139-phosphorylated histone H2AX (γ-H2AX). However, γ-H2AX is also induced by some non-DSB conditions and abundantly formed in apoptosis, raising concerns about the overestimation of potential genotoxic agents and accuracy of DSB assessments. DSB-triggered γ-H2AX undergoes RNF168-mediated K13/K15 monoubiquitination, which is rarely analyzed in DSB/genotoxicity studies. Here we identified critical methodological factors that are necessary for the efficient detection of mono- (ub1) and diubiquitinated (ub2) γ-H2AX. Using optimized technical conditions, we found that γ-H2AX-ub1 was a predominant form of γ-H2AX in three primary human cell lines containing mechanistically distinct types of DSBs. Replication stress-associated DSBs also triggered extensive formation of γ-H2AX-ub1. For DSBs induced by oxidative damage or topoisomerase II, both γ-H2AX and γ-H2AX-ub1 showed dose-dependent increases whereas γ-H2AX-ub2 plateaued at low levels of breaks. Despite abundance of γ-H2AX, γ-H2AX-ub1,2 formation was blocked in apoptosis, which was associated with proteolytic cleavage of RNF168. Chromatin damage also caused only the production of γ-H2AX but not its ub1,2 forms. Our results revealed a major contribution of ubiquitinated forms to the overall γ-H2AX response and demonstrated the specificity of monoubiquitinated γ-H2AX as a biodosimeter of non-apoptotic DSBs.
Keywords: DNA double-strand break, histone H2AX, DNA damage, genotoxicity, RNF168
Graphical Abstract
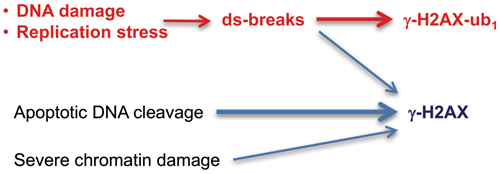
Introduction
DNA double-strand breaks (DSBs) are very dangerous genetic lesions that are frequently misrepaired resulting in the formation of deletions or chromosomal translocations (Povirk 2006; Wyman and Kanaar, 2006). If not timely repaired, DSBs act as strong inducers of cell death (Roos and Kaina, 2006). Ser139 phosphorylation of histone H2AX, which constitute approximately 10% of total histone H2A in human cells, plays a central role in orchestrating cellular responses to DSBs by acting as a platform for the recruitment of other components of DNA damage response (Scully and Xie, 2013). Initially detected as a novel histone H2AX species after ionizing radiation and named γ-H2AX (Rogakou et al., 1998), the discovery of H2AX-Ser139-phosphorylation has ushered in the era of extensive research on chromatin modifications that are critical for DSB repair (Lucas et al., 2011).
H2AX-S139 phosphorylation is principally performed by ATM kinase (Burma et al., 2001), which is activated by the DSB-sensing MRE11-RAD50-NBS1 complex (Lee and Paull, 2005; Falck et al., 2005). Kinase activity of ATM is further enhanced via its acetylation by KAT5, which is accumulated at the DSB sites through binding to H3K9me3 (Sun et al., 2007, 2009). The presence γ-H2AX is directly recognized by MDC1 via its tandem C-terminal BRCT repeat (Stucki et al., 2005; Lee et al., 2005). Chromatin-bound MDC1 initiates a protein ubiquitination cascade via recruitment of the E3 ligase RNF8 that ubiquitinates histone H1 (Thorslund et al., 2015). Ubiquitinated H1 in turn recruits another E3 ligase RNF168 that catalyzes monoubiquitination of Lys13/Lys15 in H2A and γ-H2AX, which is necessary for the subsequent K63-linked polyubiquitination of chromatin proteins (Mattiroli et al., 2012). These ubiquitination events are required for the activation of two main DSB repair pathways, namely, nonhomologous end-joining and homologous recombination. H2A/H2AX-K15 monoubiquitination is recognized and bound by 53BP1 (Fradet-Turcotte et al., 2013), which routes DSBs into repair via nonhomologous end-joining (Zimmermann and de Lange, 2014; Panier and Boulton, 2014). The K63-linked protein polyubiquitination is necessary for recruitment of the BRCA1-RAP80 complex and a subsequent activation of homology-based repair (Kim et al., 2007; Sobhian et al., 2007; Wang et al., 2007). In agreement with the biochemical evidence for RNF168 in regulation of DSB responses, inactivating mutations in this E3 ligase are the cause of the cancer-prone human syndrome RIDDLE (radiosensitivity, immunodeficiency, dysmorphic features and learning disabilities) (Stewart et al., 2007, 2009). Clinical characteristics of RIDDLE patients overlap with those in individuals with mutations in the upstream effectors such as ATM kinase and MRE11 (McKinnon, 2012).
Ionizing radiation and many cancer drugs have long been known to generate DSBs as the principal toxic lesions. However, DSBs can also play a major role in toxicity of drugs or carcinogens that directly do not produce strand breakage, which instead arise from normal or abnormal processing of the initial DNA modifications by other repair processes (Reynolds et al., 2009; Sawant et al., 2015). A large number of causes, including but not limited to defects in genome maintenance mechanisms, presence of activated oncogenes, chromatin abnormalities and metabolic deficiencies, can trigger replication stress, which results in the formation of variable amounts of toxic DSBs (Jeong et al., 2013; Macheret and Halazonetis, 2015). Growing recognition of a much wider importance of DSBs has led to increased interest in the formation and repair of these lesions. Measurements of γ-H2AX is the most widely used approach for the detection of DSBs in cells (26,600 hits in Google Scholar for the “γ H2AX assay” search at the time of writing). γ-H2AX is frequently described as a specific marker of DSBs although it can also be formed in the absence of DSBs. For example, heat shock induced well defined nuclear foci of γ-H2AX that were very similar to those formed by ionizing radiation but direct physical measurements and genetic approaches found no evidence for the production of DSBs (Hunt et al., 2007; Laszlo and Fleischer, 2009). Another frequent concern, especially in prolonged treatments or recovery studies, is apoptotic DNA cleavage, which triggers extensive formation of γ-H2AX (Rogakou et al., 2000). The apoptosis inducer staurosporine has long been used as a positive control by some suppliers of anti-γ-H2AX antibodies. The proposed use of γ-H2AX in high throughput screens for genotoxic agents (Khoury et al., 2013) can lead to many false positives if the apoptosis-induced responses are not filtered out.
The presence of multiple signaling steps prior to the initiation of DSB repair offers cells the opportunity to integrate input from additional sensors and prevent unnecessary large-scale chromatin changes by non-DSB stressors. γ-H2AX ubiquitination by RNF168 is a relatively late step in DSB signaling, representing an advanced commitment by the cell. Despite the clearly established importance of H2A/H2AX-K13/K15 monoubiquitination in DSB repair, a vast majority of publications utilizing γ-H2AX westerns do not show its mono- (ub1) and diubiquitinated (ub2) products and in rare cases when shown, they appeared as minor species. This lack of attention to γ-H2AX-ub1,2 forms probably reflects a wide use of γ-H2AX prior to the discovery of its regulatory ubiquitination and is further reinforced by the absence or near absence of higher bands on westerns provided by all major suppliers of γ-H2AX antibodies. However, antibody vendors often use either UV (non-DSB damage at environmental doses) or the apoptosis inducer staurosporine for their positive samples. If γ-H2AX-ub1,2 are abundant but undetected products, then measurements of only γ-H2AX would give an incomplete assessment of the entire DSB response and may lead to inaccurate conclusions about DSB repair if γ-H2AX-ub1,2 and γ-H2AX display different decay kinetics.
In this work, we identified the necessary technical conditions for the efficient detection of γ-H2AX products and found that at low-moderate doses of mechanistically distinct DSB inducers, γ-H2AX-ub1 was the predominant form accounting up to 80–90% of total γ-H2AX in normal human cells. We also found that DSB-like signaling by false-positive stimuli was truncated prior to γ-H2AX monoubiquitination, identifying γ-H2AX-ub1 as a specific and dose-dependent marker of nonapoptotic DSBs.
Materials and Methods
Materials
Etoposide (C1383, 100 mM stock in DMSO, stored at −20°C), camptothecin (C9911, 10 mM stock in DMSO, −20°C) and hydroxyurea (H8627, freshly prepared 0.6 M stock in cell culture media) were from Sigma. AZ-20 (S7050, 10 mM stock in DMSO, −80°C) and VE821 (S8007, 10 mM stock in DMSO, −80°C) were from Selleckchem. DRB (10010302, 100 mM stock in DMSO, −20°C) was from Cayman Chemical. KU55933 (1685–5, 10 mM stock in DMSO, −20°C) was from Bio-Vision. Staurosporine (sc-3510A, 10 mM stock in DMSO, −20°C) and aphidicolin (sc-201535, 10 mM stock in DMSO, −20°C) were from Santa Cruz Biotechnology. Bleomycin sulfate (B4518, 1.32 mM stock in sterile H2O, −20°C) was from LKT Laboratories. Human recombinant TRAIL/APO2L (GF092, 0.5 μg/μL stock in sterile H2O, −20°C) was from Millipore.
Cells
IMR90, WI38, normal human neonatal keratinocytes (PCS-200–010), H460, A549, U2OS, mouse embryonic fibroblasts (MEFs, SCRC-1040), telomerase-immortalized human bronchial epithelial HBEC3 (CRL-4051), Daudi, HCT116, Ramos, Raji and CA46 were obtained from ATCC. RCC4 cells were gift from Dr. W. Kaelin. H460 and lymphoma lines Daudi, Ramos, Raji and CA46 were maintained in RPMI-1640 medium (Gibco, 11875–119). IMR90, WI38, MEFs, RCC4 and U2OS cells were maintained in DMEM medium (Gibco, 12430–062). A549 and HCT116 cells were grown in F12-K (ATCC, 30–2004) and DMEM/F12 medium (Gibco, 11320–033), respectively. All media were supplemented with 10% fetal bovine serum (Rocky Mountain Biologicals, FBS-BBT-5XM) and 1% Pen-Strep solution (100 Units/ml penicillin and 100 μg/ml streptomycin) (Gibco, 15140–122). Keratinocytes were grown in Dermal Cell Basal Medium (ATCC, PCS-200–300) supplemented with Keratinocyte Growth Kit (ATCC, PCS-200–040). HBEC3 cells were propagated in Airway Epithelial Cell Basal Medium (ATCC, PCS-300–030) supplemented with Bronchial Epithelial Growth Kit (ATCC, PCS-300–040). All cells were grown at 37°C in a 5% CO2 humidified atmosphere containing either 5%O2 (IMR90, WI38 and MEFs) or 20% O2 (all other cells).
Proteins extraction and quantitation
Attached cells were collected by scraping, combined with floating cells, washed twice with ice-cold Dulbecco’s Phosphate-Buffered Saline (DPBS) (Gibco, 21600–010) and centrifuged at 500xg for 5 min at 4°C. Total proteins were extracted using 2% SDS Lysis Buffer (2% SDS, 50 mM Tris-HCl pH 6.8, 10% glycerol) supplemented with Halt Protease and Phosphatase Inhibitor (ThermoFisher, 78443) and PMSF (Sigma-Aldrich, 93482). Cells were boiled in 2% SDS Lysis Buffer for 10 min, cooled down to room temperature and collected by centrifugation at 12000×g for 10 min at room temperature. Proteins concentration was measured using DC Protein Assay Kit I (Bio-Rad, 5000111) according to manufacturer protocol. Absorbance (A=750 nm) was read by the Endpoint method using SpectraMax M2 (Molecular Devices).
Western blotting – standard procedures
Typically, 20 μg of total proteins were used for each western blot analysis. Protein samples were prepared in a loading buffer (50 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 0.02% Bromophenol blue, 5% 2-mercaptoethanol) and unless indicated otherwise, separated by 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were electrotransferred onto Immun-Blot PVDF Membrane (Bio-Rad, 1620177) using semi-dry transfer system (PierceG2 Fast Blotter) or an overnight wet transfer procedure. In the semi-dry procedure, PVDF membranes and Western Blotting Filter Paper (ThermoFisher, 88600) were incubated for 10 min with 1-Step Transfer Buffer (ThermoFisher, 84731) supplemented with 12% ethanol followed by electrotransfer for 13 min at 25V/2.5A at room temperature. For the overnight wet transfer, proteins were electrotransferred onto PVDF membrane at 18V at 4°C using the Bjerrum Schafer-Nielsen Buffer (48 mM Tris, 40 mM glycine, pH 8.8). After the transfer (semi-dry or wet), PVDF membranes were washed three times for 5 min with 1x Tris-Buffered Saline/0.1% Tween 20 (TBST) buffer and blocked in 5% w/v nonfat dry milk (in 1xTBST) for 1 h at room temperature with shaking. The majority of γ-H2AX westerns were performed using rabbit polyclonal antibodies (Cell Signaling Technology Cat# 2577, 1:1000 dilution in 5% BSA), referred to as γ-H2AX Ab #1 in this work. Membranes were incubated with primary antibodies overnight at 4°C. Next day, membranes were washed three times for 5 min in 1xTBST buffer and incubated with secondary antibodies at room temperature for 1 h. Horseradish peroxidase-conjugated secondary antibodies were goat anti-mouse IgG (Millipore Cat# 12–349) and goat anti-rabbit IgG (Cell Signaling Technology Cat# 7074 and were used at 1:2000 dilution. Bands were revealed using Pierce ECL Western Blotting Substrate (ThermoFisher, 32106) or ECL Prime Western Blotting Detection Reagent (GE Healthcare, RPN2232) and detected using Blue Autoradiography & Western Blotting Film (USA Scientific, 1968–3810) and Agfa CP1000 Processor. Other primary antibodies were rabbit polyclonal anti-phospho-Chk1 (Ser317) (Cell Signaling, 2344, 1:1000 in 5% milk), rabbit polyclonal anti-PARP (Cell Signaling, 9542, 1:1000 in 5% milk), mouse monoclonal anti-RNF168 (Novus Biologicals, H00165918-M01, 1:500 in 5% milk), rabbit monoclonal anti-c-Myc (Cell Signaling, 13987, 1:1000 in 5% BSA), rabbit polyclonal anti-phospho RPA32 (S4/S8) (Bethyl, A300–245A, 1:1000 in 5% milk), rabbit monoclonal anti-L7A (Cell Signaling, 2415, 1:1000 in 5% milk) and mouse monoclonal anti-γ-tubulin (Sigma–Aldrich, T6557, 1:2000 in 5% milk).
Variations in western blotting for γ-H2AX
After separation on 12% SDS-PAGE, three different buffers were tested for electrotransfer: Bjerrum Schaffer-Nielsen Buffer (48 mM Tris, 40 mM glycine, pH 8.8), Towbin Buffer (25 mM Tris, 192 mM glycine, pH 8.3) and 1-Step Transfer Buffer. Bjerrum Schaffer-Nielsen and Towbin buffers were used for wet transfer overnight (18V, +4°C) whereas 1-Step Transfer Buffer was used for semi-dry transfer (25V/2.5A, 13 min at room temperature). The buffers were supplemented or not with 12% ethanol. After protein transfer, membranes were handled in the same way. A comparison of different γ-H2AX antibodies tested five antibodies: Ab #1 - rabbit polyclonal from Cell Signaling (Cat# 2577), Ab #2 - rabbit monoclonal from Cell Signaling (Cat# 9718), Ab #3 - rabbit polyclonal from Bethyl (Cat# A300–081A), Ab #4 - rabbit monoclonal from Abcam (Cat# ab81299) and Ab #5 - mouse monoclonal antibodies from Millipore (Cat# 05–636). Primary antibodies were used in 1:1000 dilution in 5% milk (Ab #3,4 and 5) or 5% BSA (Ab #1 and 2) and incubated with membranes overnight at +4°C. Secondary antibodies were used in 1:2000 dilution for 1 h at room temperature. ECL Prime Western Blotting Detection Reagent was used for signal detection.
Heat shock
Cells were seeded onto 100-mm dishes and allowed to attach overnight. Next day, all dishes were tightly sealed with parafilm and floated in 37, 43 or 45.5°C in water baths for 30 min. Cells were collected by scraping, washed twice with ice-cold DPBS and centrifuged at 500xg for 5 min at +4°C. Whole cell lysates were prepared by boiling in 2% SDS Lysis buffer as described above.
Colony formation
U2OS cells were seeded onto 6-well plates (200 cells per well) and next day were treated with the solvent or 10 μM KU55933 for 1 h before heat shock. Plates were parafilm-sealed and incubated at 37 or 45.5°C in water bath for 30 min followed by recovery for 3 days with or without 10 μM KU55933. After growth for additional growth in the regular medium for 4–5 days, colonies were fixed with methanol and stained with a Giemsa solution. Bleomycin-treated cells (1 h) were used as a positive control for ATMi activity.
Cell viability
Cytotoxic effects of hydroxyurea were evaluated with CellTiter-Glo Luminescent Cell Viability Assay (Promega, G7571). Lymphoma cells were seeded (4×103 cells/well) into black, 96-well optical bottom cell culture plates (ThermoFisher, 165305) and grown overnight. HU stock solutions were freshly prepared in RPMI1640 medium and sterile-filtered before use. Cells were incubated with HU for 72 h followed by the addition of assay reagents and measurements of luminescence at Cytation3 Imaging Reader (BioTek).
Pulsed field gel electrophoresis (PFGE)
A recently described procedure for the measurements of DSBs by PFGE was followed (Ortega-Atienza et al., 2016). Cells were seeded onto 150 mm dishes (4−5×106 cells) and the next day were subjected to treatments. After trypsinization and 2x washes in DPBS at room temperature, cells were resuspended in 0.5 ml of Cell Suspension Buffer (10 mM Tris, pH 7.2, 50 mM EDTA, 20 mM NaCl). After mixing with an equal volume of 2% UltraPure LMP agarose, solutions were immediately poured out into CHEF Mapper XA System Plug Molds (Bio-Rad) and allowed them to cool in refrigerator for 10 min. Solidified plugs were transferred to Proteinase K Reaction Buffer (10 mM Tris, pH 8.0, 100 mM EDTA, 1% N-lauroylsarcosine, 0.2% sodium deoxycholate, 1 mg/ml Proteinase K) and incubated overnight at room temperature with gentle mixing. Following 5xwashes for 1 h each in Wash Buffer (20 mM Tris, pH 8.0, 50 mM EDTA), plugs were stored at 4°C. Plugs were prepared with 4×105 (U2OS, H460) or 5×105 MEF cells. Bio-Rad CHEF MAPPER system was used PFGE. DNA was visualized by incubation for 1 h in 3xGelRed staining solution (Phoenix Research Products, RGB-4103) in H2O with 0.1 M NaCl. Gel Doc XP+ Imaging System (Bio-Rad) with Quantity One software were used for DNA quantitation.
siRNA knockdown
On-TARGETplus Non-Targeting Control Pool (Dharmacon, D-001810–10, 20 μM stock in sterile dH2O) and H2AX si-RNA-1 (Dharmacon, ZHIAA-000005, 20 μM stock in sterile dH2O) (CAACAAGAAGACGCGAAUCdTdT) were used for knockdowns of H2AX in IMR90 cells. A transfection procedure used 50 nM siRNA (final concentration) and 20 μL of Lipofectamine RNAiMAX (Invitrogen,13778150). After overnight growth (0.5×106 cells/100 mm dish), IMR90 were treated with the siRNA/Lipofectamine complex for 6 h twice at 24 h intervals. Following 24 h recovery after the last transfection, cells were seeded for experiments. Next day, cells were incubated with 0 and 100 μM etoposide for 1 h and collected for westerns.
Results and Discussion
Critical methodological factors.
The amounts of ubiquitinated forms of γ-H2AX can potentially be affected by a cell background and/or a type of DSBs. The efficiency of detection of γ-H2AX forms may also vary, depending on technical variables during western blotting, such as the choice of antibodies, electrophoresis and/or transfer conditions. In our previous work on chromium-induced DSBs, we have readily detected mono- and diubiquitinated forms of γ-H2AX in transformed H460 and normal IMR90 human cells (DeLoughery et al., 2015). Carcinogenic chromium causes DSBs via a rather unusual processing of its DNA adducts by mismatch repair (Reynolds et al., 2009; Zecevic et al., 2009), leaving a possibility that high levels of γ-H2AX ubiquitination were a chromium-specific phenomenon. To evaluate the impact of technical variables during western blotting, we again tested H460 and IMR90 cells but treated them with more commonly used genotoxic stressors: radiomimetic bleomycin, dNTPs-depleting hydroxyurea and DNA polymerase inhibitor aphidicolin. Bleomycin is a potent and direct inducer of DSBs whereas hydroxyurea and aphidicolin are replication stressors that produce only a very few DSBs in cells with normal replication fork stability. Whole cell lysates prepared by boiling of cells in the lysis buffer containing 2% SDS, which rapidly and irreversibly inactivates deubiquitinating enzymes (Emmerich and Cohen, 2015), were tested throughout the entire project. Using the same antibodies as before (DeLoughery et al., 2015), we detected approximately equal amounts of γ-H2AX and γ-H2AX-ub1 forms in bleomycin-treated H460 cells irrespective of gel acrylamide percentage using semi-dry transfer with 12% ethanol-supplemented buffer (Fig. 1A, top panels). As expected, replication stressors produced only minimal amounts of γ-H2AX and its ubiquitinated forms. Using overnight wet transfer without ethanol, we found much lower levels of γ-H2AX-ub1 relative to nonubiquitinated γ-H2AX on three gels with different acrylamide percentage (Fig. 1A, bottom panels). For both semi-dry and wet transfer conditions, lower acrylamide percentage gels offered a modestly higher detection sensitivity for all γ-H2AX forms independent of their ubiquitination. The importance of ethanol in the semi-dry transfer buffer for the efficient detection of γ-H2AX-ub1 was also confirmed for DSBs induced by the topoisomerase II poison etoposide in IMR90 cells (Fig. 1B). The presence of ethanol in wet transfer buffers made them similarly sensitive to the semi-dry procedure (Fig. 1C). Ubiquitin is resistant to denaturation (Emmerich and Cohen, 2015), which diminishes its binding and retention by the membranes during transfer conditions. Positive effects of ethanol can be attributed to a more complete denaturation and stripping of SDS from γ-H2AX-conjugated ubiquitin which would increase its membrane binding.
Figure 1. Technical factors affecting detection of ubiquitinated forms of Ser139-phosphorylated histone H2AX (γ-H2AX) by western blotting.

Panels A-C used antibodies #1. (A) Impact of gel polyacrylamide percentage and transfer conditions to PVDF membranes. Treatments of H460 cells: 6 h with 3 mM hydroxyurea (HU), 6 or 16 h with 5 μM aphidicolin (Aphi) or 1 h with 5 μM bleomycin (Bleo). All γ-H2AX images had 5 s film exposures. (B) Ethanol effect in semi-dry transfer. IMR90 cells were treated with 40 μM etoposide (Eto) for 1 h. (C) Comparison of wet (pH 8.2 and pH 8.8) and semi-dry transfers using 12% ethanol-containing buffers (10 s film exposures for all γ-H2AX images). IMR90 cells were treated with 40 μM etoposide for 1 h. (D) Effectiveness of different antibodies using semi-dry transfer with 12% ethanol. Normal human keratinocytes were treated for 1 h with 30 μM etoposide or 0.5 μM bleomycin. Antibodies #1–3: 1:1000 dilutions and 5 s exposures, antibody #4: 1:1000 dilution and 60 s exposure. (E) Comparison of antibodies #1 and #5 (both at 1:1000 dilutions) in H460 cells treated with bleomycin for 1 h. (F) γ-H2AX blot (Ab#1) in control and H2AX-depleted IMR90 cells treated with 100 μM etoposide for 1h.
Next, we tested several commercial antibodies selected on the basis of their popularity (from citation statistics or a designation as “most popular with customers” on vendors’ webpages). We tested the selected set of five antibodies on whole cell lysates of normal human keratinocytes as these cells showed strong DSB-induced γ-H2AX ubiquitination, possibly reflecting their evolutionary selection for robust DNA damage responses. We found that in addition to the above used antibodies (Ab #1), which are rabbit polyclonal, two other antibodies detected similar (Ab #3, rabbit polyclonal) or even stronger (Ab #2, rabbit monoclonal) signals for γ-H2AX-ub1 in etoposide and bleomycin-treated keratinocytes (Fig. 1D). Blots with these three antibodies clearly showed that γ-H2AX-ub1 was the most abundant form of γ-H2AX. The 4th antibody (rabbit monoclonal) was distinctly less sensitive for the detection of mono- and diubiquitinated γ-H2AX despite longer exposure time. The 5th antibody (mouse monoclonal, supplier-recommended use for purified histones) did not produce satisfactory results in whole cell lysates from keratinocytes and was tested in bleomycin-treated H460 cells where it showed a less efficient detection of γ-H2AX-ub1 in comparison to the reference antibodies #1 (Fig. 1E). Finally, we confirmed H2AX specificity of γ-H2AX bands in our westerns by siRNA knockdown of this histone (Fig. 1F). In summary, a sensitive detection of γ-H2AX-ub1 requires the presence of ethanol in the transfer buffer (not tested but 2x concentration of more toxic methanol probably would work as well) and the use of appropriate antibodies. Although K13/K15 ubiquitination sites are relatively far from Ser139, the attachment of ubiquitin can change folding of γ-H2AX bound to the membrane, which could then alter the accessibility of some antibodies to the phospho-Ser139 epitope. This effect would more likely occur with monoclonal antibodies, which is consistent with our observations on the weakest γ-H2AX-ub detection by two out of three tested monoclonal antibodies. A lower sensitivity of monoclonal antibodies, including their complete inactivity, is a frequent problem in detection of ubiquitinated proteins (Emmerich and Cohen, 2015). γ-H2AX ubiquitination may also be associated with the formation of other posttranslational modifications in the vicinity of the phospho-Ser139 epitope, interfering with the binding efficiency of some antibodies. This effect would also be more pronounced for monoclonal antibodies.
Abundance of γ-H2AX-ub1 in response to different DSBs.
Using polyclonal antibodies Ab #1, which gave a sensitive detection of both γ-H2AX and its ubiquitinated forms across different cell types with whole cell lysates, we examined the dose-response relationships for three mechanistically distinct DSB inducers in three primary human cells (Fig. 2A–C). Keratinocytes and two fibroblast lines all showed dose-dependent increases in the levels of γ-H2AX and its ub1 form after treatments with the radiomimetic bleomycin and the topoisomerase II inhibitor etoposide. Also in all cell lines, γ-H2AX-ub1 was clearly more abundant (1.4‒3-times) relative to the nonubiquitinated γ-H2AX. The prevalence of the ub1 form was particularly evident in keratinocytes (Fig. 2A). Treatments with the topoisomerase I inhibitor camptothecin, which causes DSBs in a replication-dependent manner (Fig. 2D), also stimulated the predominant formation of γ-H2AX-ub1 (Fig. 2A–C, left panels). Induction of the three γ-H2AX forms by camptothecin in all normal cells and in H460 carcinoma cells (Fig. 2E) showed weak or no concentration dependence, which could have resulted from a rapid activation of S-phase checkpoint inhibiting the replication-associated production of DSBs. The presence of similar amounts of DSBs after different doses of camptothecin was confirmed by PFGE (Fig. 2E). In addition to the ub1 form, all three DSB-producing agents also triggered the appearance of diubiquitinated γ-H2AX, however, its levels did not show dose-dependent increases (Fig. 2A–E). In summary, γ-H2AX-ub1 was the main form of Ser139-phosphorylated H2AX in normal human cells after treatments with low-moderate doses of three mechanistically distinct DSB inducers.
Figure 2. γ-H2AX ubiquitination in human cells in response to different DSB inducers.

(A-C) γ-H2AX westerns for primary human cells treated with bleomycin, etoposide or camptothecin for 1 h. Left panel in C: 0.5 μM bleomycin treatment. ub1/ub0 is the ratio of monoubiquitinated to nonubiquitinated γ-H2AX. (D) Effects of replication and transcription inhibition on γ-H2AX formation by camptothecin (0.2 μM for 1h following pretreatments with either 5 μM aphidicolin or 100 μM DRB for 30 min). (E) γ-H2AX western for H460 lung carcinoma cells treated with camptothecin for 1 h. (F) Detection of DSBs in camptothecin (CPT)-treated H460 cells by PFGE.
γ-H2AX ubiquitination during replication stress.
Elevated replication stress, especially in cells with a weakened stability of replication forks, can lead to a significant production of toxic DSBs (Jeong et al., 2013; Macheret and Halazonetis, 2015). For example, two established causes of replication-associated DSBs are mutations in BRCA2 (Lomonosov et al., 2003; Schlacher et al., 2011) and overexpression of oncogenes causing accelerated progression of cancer cells from G1 into S phase (Macheret and Halazonetis, 2018). Many cancer chemotherapeutic regimes include inhibitors of nucleotide synthesis, which exploit inefficiency of cancer cells in maintaining stability of stalled replication forks and prevention of their conversion into toxic DSBs. A majority of cancers do not have major defects in stability of forks, which makes it important to identify vulnerable cases and develop effective drugs for disabling forks viability mechanisms by pharmacological means. The apical replication stress-responsive kinase ATR and its downstream target CHK1 kinase are two biochemical targets that are under extensive investigation for cancer treatment alone and in combination with already approved chemotherapeutics (Karnitz and Zou, 2015; Manic et al., 2015). To investigate γ-H2AX ubiquitination responses of cancer cells to replication stress, we tested the effects of the ribonucleotide reductase inhibitor hydroxyurea alone and in combination with two selective ATR inhibitors: VE821 and AZ20. In U2OS osteosarcoma cells, which is one of the commonly used biological models in studies of DNA damage responses, hydroxyurea alone did not cause significant increases in any form of γ-H2AX (Fig. 3A). A combination of hydroxyurea with either ATR inhibitor led to an abundant formation of both γ-H2AX and its monoubiquitinated form. Similar results were also found in H460 lung carcinoma and Daudi lymphoma cells (Fig. 3A). Although both inhibitors showed similar suppression of ATR activity assessed by the loss of phospho-S317-CHK1, in all three cell lines AZ20 produced a moderately higher ratio of γ-H2AX-ub1 to γ-H2AX-ub0 relative to VE821. Burkitt’s lymphoma is characterized by overexpression of c-MYC, which is known to increase replication stress and susceptibility of cells to CHK1 inhibitors (Ferrao et al., 2012; Murga et al., 2011). In parallel testing of three Burkitt’s lymphoma lines, we found that hydroxyurea triggered a massive accumulation of γ-H2AX-ub1 in Ramos cells (Fig. 3B). The differences in hydroxyurea-induced γ-H2AX-ub1,2 were unrelated to c-MYC expression as all three Burkitt’s lymphoma lines had comparable protein levels of this oncogene (Fig. 3C). The readout of ATR activity, phospho-S317-CHK1, also was not predictive of γ-H2AX-ub1 responses in Burkitt’s lymphomas. The abundance of hydroxyurea-induced γ-H2AX-ub1 strongly correlated with the levels of S4/8-phosphorylated RPA32 (Fig. 3C), which is a biochemical marker of nuclease-processed stalled forks and DSBs in S-phase (Sartori et al., 2007). Ramos, Burkitt’s lymphoma line with the strongest γ-H2AX-ub1,2 formation, was also the most sensitive to cytotoxic effects of hydroxyurea (Fig. 3D). Thus, monitoring of γ-H2AX-ub forms can help identify cancer cells that are especially sensitive to a drug-induced depletion of dNTPs and replication stress.
Figure 3. Formation of γ-H2AX in cancer cells under replication stress.

Cells were treated with hydroxyurea (HU) for 3h. (A) Westerns for cells treated with HU alone or after 1 h pretreatment with 10μM VE821 (ATRi1) or 1μM AZ20 (ATRi2). p-CHK1: phospho-S317-CHK1. (B,C) Westerns for HU-treated Burkitt’s lymphoma lines. (D) Viability of Burkitt’s lymphoma cells treated with HU for 72 h. Means±SD, **− p<0.001, ***−p<0.0001.
Absence of γ-H2AX ubiquitination in heat-shocked cells.
Heat shock is a ubiquitous environmental stress for the skin and the upper digestive and respiratory tracts in humans. Hyperthermia is also used in treatment of human malignancies (Arends et al., 2016; Van Driel et al., 2018). A combination of hyperthermia with DNA-damaging agents frequently results in synergistic cell killing, leading to mechanistic studies involving measurements of DNA damage responses. Exposure of cells to elevated temperatures is known to increase the levels of γ-H2AX, which was detected not only by westerns but also as well-defined nuclear foci that were morphologically indistinguishable from those induced by ionizing radiation (Kaneko et a., 2005; Takahashi et al., 2008). Although initially these results were taken as evidence of DSBs due to the perceived specificity of γ-H2AX for these lesions, subsequent investigations showed that heat shock was a false-positive DSB response likely related to activation of ATM kinase by chromatin alterations (Shiloh and Ziv, 2013). A study by a group of experienced investigators in the field of DNA damage (Hunt et al., 2007) using three different direct assays failed to detect the formation of DSBs in heat-shocked cells despite the appearance of nuclear foci of γ-H2AX and its sensor MDC1. Similar to radiation, heat shock triggered H2AX-S139 phosphorylation by ATM kinase, whose activation however was independent on the DSB-sensing MRN complex (Hunt et al., 2007). Induction of γ-H2AX by hyperthermia also did not vary in a panel of cells with variable thermotolerance and H2AX knockout produced no effect on survival of heat-shocked cells (Laszlo and Fleischer, 2009). Interestingly, hyperthermia did not cause the appearance of nuclear foci of 53BP1 (Hunt et al., 2007), which is a reader of DSB-induced H2A/H2AX-K15 ubiquitination (Fradet-Turcotte, 2013). We found that 30-min long exposure to 45.5°C induced a cell type-dependent formation of γ-H2AX but there was no even faint appearance of its ubiquitinated bands (Fig. 4A). We next examined the levels of DSBs using their direct measurements by PFGE in two cell lines (U2OS and MEFs) with the most extensive production of γ-H2AX. Neither cell line showed any changes in DSBs above the background after hyperthermia (Fig. 4B,C). As evidence of the PFGE assay sensitivity, both U2OS and MEFs gave clear increases in high-molecular weight DNA fragments after 30 min treatments with a low concentration of the DSBs-causing etoposide (Fig. 4D). Since ATM is responsible for H2AX-S139 phosphorylation in hyperthermia (Hunt et al., 2007), we also examined the impact of ATM inactivation on a long-term survival of cells subjected to mild or severe heat shock. Consistent with the absence of DSBs, we found that disabling of ATM with its kinase inhibitor KU55933 had no significant effect on the colony-forming ability of heat-shocked cells (Fig. 4E). As expected, the same inhibitor severely diminished (>3-fold) colony formation by cells treated with the radiomimetic bleomycin (Fig. 4F). Overall, our results further confirmed the absence of DSBs after hyperthermia and demonstrated that ubiquitinated forms of γ-H2AX are not produced in the heat shock-induced chromatin damage response. Our findings on the absence of γ-H2AX ubiquitination are also consistent with and provide a mechanistic explanation for the absence of 53BP1 foci in heat-shocked cells with γ-H2AX foci (Hunt et al., 2007).
Figure 4. Heat shock induces γ-H2AX but not its ubiquitination or DSB formation.

(A) Westerns for γ-H2AX (Ab#1) in cells exposed to high temperature for 30 min. (B) Representative PFGE images of DNA from control and heat-shocked cells. (C) Percentage of fragmented DNA (DSBs) as determined by PFGE (means±SD for three biological replicates). (D) Detection of DSBs by PFGE in cells treated with etoposide (30 μM for 30 min). (E) Clonogenic survival of heat-shocked U2OS cells grown without and with 10 μM KU55933 (ATMi). ATMi wad added 1 h before heat shock and was present during the first 3 days of growth. Data are means±SD for five biological replicates. (F) Suppression of clonogenic survival in bleomycin-treated U2OS cells by ATMi (3 μM bleomycin for 1 h). KU55933 was used as in panel E. Data are means±SD (n=3, ***p<0.001).
Apoptosis-induced γ-H2AX.
Irrespective of the type of the initial trigger, apoptosis induces nuclease-mediated DNA fragmentation, which leads to the formation of γ-H2AX (Rogakou et al., 2000). Historically, a majority of commercial suppliers of anti-phospho-S139-H2AX antibodies showed westerns with an apoptosis-inducing agent as a positive control. A classic apoptosis inducer staurosporine has been most commonly used for the formation of γ-H2AX in cells, which, is a current positive control for Ab #5 on its vendor’s webpage. γ-H2AX formation from apoptotic DSBs makes it difficult to interpret results with prolonged treatments with nongenotoxic stressors or chronic exposures with direct inducers of DSBs, as the late time points would almost unavoidably include populations of cells undergoing apoptosis and therefore, producing γ-H2AX.
The late appearance or presence of γ-H2AX is often taken as evidence of some indirect mechanisms of DSB formation or existence of repair-resistant DSBs, respectively. However, these responses could largely or completely result from apoptotic DNA breaks. Thus, it would be very important to have a biochemical marker that can differentiate DNA damage-derived DSBs from those originating from apoptotic DNA cleavage. Therefore, we next examined γ-H2AX formation and its ubiquitination in cells undergoing apoptosis. Using staurosporine, a commonly employed chemical inducer of apoptosis, we assessed γ-H2AX formation in several human cell lines after 3 and 6 h treatments. Five out of six tested lines showed evidence of apoptosis as seen by the production of cleaved PARP (Fig. 5A). The same five cell lines also produced strong, time-dependent increases in γ-H2AX levels, which mirrored the appearance of cleaved PARP. Strikingly, essentially all staurosporine-induced γ-H2AX was present in its nonubiquitinated form irrespectively of its amount or time of staurosporine treatments. The same cell lines showed extensive accumulation of ubiquitinated γ-H2AX (mostly in the ub1 form) in response to etoposide-induced DSBs (Fig. 5B). A parallel run of H460 and A549 samples with staurosporine and etoposide treatments further illustrated the dramatic differences in γ-H2AX ubiquitination between DNA damage-derived and apoptotic DSBs (Fig. 5B). Analysis of staurosporine-treated samples under different gel/transfer conditions also found the absence of significant amounts of ubiquitinated γ-H2AX (Fig. 5C). Impaired γ-H2AX ubiquitination in response to staurosporine-induced apoptotic DNA fragmentation could have resulted from some unusual cellular phenotype caused by inactivation of multiple kinases by this inhibitor. To avoid similar potential complications with chemical stressors, we next examined a biologically specific apoptosis. We treated cells with TRAIL, which is a physiologically produced cytokine that triggers a caspase-mediated apoptosis via activation of death receptors DR4 and DR5. Cancer cells are typically more sensitive to TRAIL-induced apoptosis than normal cells (Fulda, 2015; de Miguel et al., 2016). We found that TRAIL-treated human cancer cells displayed very strong increases in γ-H2AX but the presence of the ub1 form was undetectable with the exception of a very faint band in H460 cells (Fig. 6A). Normal human keratinocytes showed a weaker PARP cleavage and a more moderate but still very significant accumulation of γ-H2AX, which was also induced exclusively in its nonubiquitinated form. A virtual absence of γ-H2AX-ub forms in TRAIL-treated cells was also observed in westerns using another polyclonal γ-H2AX antibody (Fig. 6B). This antibody (Ab #3) efficiently recognized ubiquitinated γ-H2AX in cells with nonapoptotic DSBs (Fig. 1D). To explore potential reasons for the lack of the ub1,2 forms of γ-H2AX in apoptosis, we looked at the protein levels of the E3 ligase RNF168 that is responsible for the DSB-induced K13/K15 ubiquitination of H2A/H2AX (Mattiroli et al., 2012). We found that TRAIL-triggered apoptosis was associated with cleavage of RNF168 in human cells of different histological origin (Fig. 6C). Thus, RNF168 joins a list of other key DSB repair proteins, such as DNAPK (Song et al., 1996), RAD51 (Huang et al., 1999) and BRCA1 (Zhan et al., 2002), that are cleaved during apoptosis, which prevents repair of apoptotic DNA breaks and survival of cells with severely damaged genome. Many cancer cells have resistance to apoptotic stimuli due to their altered balance of pro- and anti-apoptotic factors, which in some cases may result in a less effective truncation of the DSB response and the appearance of weak H2AX-ub1 signals (as in H460 cells). We note that no increases in residual H2AX-ub1 bands were observed during apoptosis in immortalized but not transformed HBEC3 cells (Fig. 5A) or primary keratinocytes (Fig. 6A).
Figure 5. γ-H2AX production in staurosporine-induced apoptosis.

(A) γ-H2AX and PARP westerns for cells treated with 1 μM staurosporine (Stn) for 3 or 6 h (cl.- cleaved). (B) Formation of γ-H2AX and its ubiquitinated forms in cells treated with 30 μM etoposide (Eto) for 1 h or as indicated. H460 and A549 panels also include 6-h staurosporine samples for comparison. (C) γ-H2AX westerns on the same samples as in panel A except that 10% gels and wet transfer were used.
Figure 6. γ-H2AX formation in TRAIL-induced apoptosis.

(A) γ-H2AX formation and PARP cleavage in cells treated with 0.1 μg/ml TRAIL for 3 or 6 h (cl. – cleaved). Semi-dry transfer (buffer with 12% ethanol) from 12% gel and polyclonal antibodies Ab #1 for γ-H2AX were used. (B) Samples from panel A were run on 12% gel, proteins were wet transferred overnight (pH 8.8 buffer with 12% ethanol) and membranes were probed with γ-H2AX polyclonal antibodies Ab #3. (C) Cleavage of RNF168 in cells undergoing TRAIL-induced apoptosis (0.1 μg/ml TRAIL for 3 or 6 h).
CONCLUSIONS
Our investigation showed that the effectiveness of detection of γ-H2AX-ub1 and γ-H2AX-ub2 forms was strongly influenced by western blotting conditions, especially by the choice of antibodies and the presence of ethanol in the transfer buffer. Under the optimized detection conditions, monoubiquitinated γ-H2AX was the predominant species in response to three mechanistically distinct inducers of DSBs in normal human cells. Ubiquitinated γ-H2AX was also extensively formed in cells with a poor tolerance of pharmacologically-induced replication stress. Unlike γ-H2AX, mono- and diubiquitinated γ-H2AX were not produced by the false-positive DSB stressor hyperthermia (nuclear/chromatin damage) and during apoptotic DNA fragmentation. Thus, analysis of ubiquitinated forms is important for the assessment of the overall formation of γ-H2AX, which offers a further advantage in specificity for nonapoptotic DSBs and discrimination of DSB-unrelated signaling. Quantitation of γ-H2AX-ub1 and γ-H2AX-ub2 forms can be critical in chronic treatments when even a small number of apoptotic cells containing large amounts of γ-H2AX could grossly inflate estimates for DNA damage-derived DSBs.
Acknowledgements.
This work was supported by grants ES008786 and ES028072 from the National Institute of Environmental Health Sciences.
Abbreviations:
- DSB
DNA double-strand break
- PFGE
pulsed-field gel electrophoresis
- ub1
monoubiquitinated
- ub2
diubiquitinated
References
- Arends TJ, Nativ O, Maffezzini M, de Cobelli O, Canepa G, Verweij F, Moskovitz B, van der Heijden AG, Witjes JA, 2016. Results of a randomised controlled trial comparing intravesical chemohyperthermia with mitomycin C versus Bacillus Calmette-Guérin for adjuvant treatment of patients with intermediate- and high-risk non-muscle-invasive bladder cancer. Eur. Urol 69, 1046–1052. [DOI] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ, 2001. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem 276, 42462–42467. [DOI] [PubMed] [Google Scholar]
- DeLoughery Z, Luczak MW, Ortega-Atienza S, Zhitkovich A, 2015. DNA double-strand breaks by Cr(VI) are targeted to euchromatin and cause ATR-dependent phosphorylation of histone H2AX and its ubiquitination. Toxicol Sci 143, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Driel WJ, Koole SN, Sonke GS, 2018. Hyperthermic Intraperitoneal Chemotherapy in Ovarian Cancer. N. Engl. J. Med 378, 1363–1364. [DOI] [PubMed] [Google Scholar]
- Emmerich CH, Cohen P, 2015. Optimising methods for the preservation, capture and identification of ubiquitin chains and ubiquitylated proteins by immunoblotting. Biochem. Biophys. Res. Commun 466, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP, 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434, 605–611. [DOI] [PubMed] [Google Scholar]
- Ferrao PT, Bukczynska EP, Johnstone RW, McArthur GA, 2012. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene 31, 1661–1672. [DOI] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, Durocher D, 2013. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, 2015. Targeting extrinsic apoptosis in cancer: Challenges and opportunities. Semin. Cell. Dev. Biol 39, 20–25. [DOI] [PubMed] [Google Scholar]
- Huang Y, Nakada S, Ishiko T, Utsugisawa T, Datta R, Kharbanda S, Yoshida K, Talanian RV, Weichselbaum R, Yuan ZM, 1999. Role for caspase-mediated cleavage of Rad51 in induction of apoptosis by DNA damage. Mol. Cell. Biol 19, 2986–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt CR, Pandita RK, Laszlo A, Higashikubo R, Agarwal M, Kitamura T, Gupta A, Rief N, Horikoshi N, Baskaran R, Lee JH, Löbrich M, Paull TT, Roti JL, Pandita TK, 2007. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res. 67, 3010–3017. [DOI] [PubMed] [Google Scholar]
- Jeong YT, Rossi M, Cermak L, Saraf A, Florens L, Washburn MP, Sung P, Schildkraut CL, Pagano M, 2013. FBH1 promotes DNA double-strand breakage and apoptosis in response to DNA replication stress. J. Cell. Biol 200, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Igarashi K, Kataoka K, Miura M, 2005. Heat shock induces phosphorylation of histone H2AX in mammalian cells. Biochem. Biophys. Res. Commun 328, 1101–1106. [DOI] [PubMed] [Google Scholar]
- Karnitz LM, Zou L, 2015. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res 21, 4780–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury L, Zalko D, Audebert M, 2013. Validation of high-throughput genotoxicity assay screening using γH2AX in-cell western assay on HepG2 cells. Environ. Mol. Mutagen 54, 737–746. [DOI] [PubMed] [Google Scholar]
- Kim H, Chen J, Yu X, 2007. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 316, 1202–1205. [DOI] [PubMed] [Google Scholar]
- Laszlo A, Fleischer I, 2009. The heat-induced gamma-H2AX response does not play a role in hyperthermic cell killing. Int. J. Hyperthermia 25,199–209. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT, 2005. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554. [DOI] [PubMed] [Google Scholar]
- Lee MS, Edwards RA, Thede GL, Glover JN, 2005. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J. Biol. Chem 280, 32053–32056. [DOI] [PubMed] [Google Scholar]
- Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR, 2003. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 17, 3017–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J, Lukas C, Bartek J, 2011. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell. Biol 13, 1161–1169. [DOI] [PubMed] [Google Scholar]
- Manic G, Obrist F, Sistigu A, Vitale I, 2015. Trial Watch: Targeting ATM-CHK2 and ATR CHK1 pathways for anticancer therapy. Mol. Cell. Oncol 2, e1012976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, Halazonetis TD, 2015. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol 10, 425–448. [DOI] [PubMed] [Google Scholar]
- Macheret M, Halazonetis TD, 2018. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK, 2012. RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 150, 1182–1195. [DOI] [PubMed] [Google Scholar]
- McKinnon PJ, 2012. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu. Rev. Pathol 7, 303–321. [DOI] [PubMed] [Google Scholar]
- de Miguel D, Lemke J, Anel A, Walczak H, Martinez-Lostao L, 2016. Onto better TRAILs for cancer treatment. Cell Death Differ. 23, 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montaña MF, Artista L, Schleker T, Guerra C, Garcia E, Barbacid M, Hidalgo M, Amati B, Fernandez-Capetillo O, 2011. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol 18,1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Atienza S, Wong VC, DeLoughery Z, Luczak MW, Zhitkovich A, 2016. ATM and KAT5 safeguard replicating chromatin against formaldehyde damage. Nucleic Acids Res. 44, 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S, Boulton SJ, 2014. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell. Biol 15, 7–18. [DOI] [PubMed] [Google Scholar]
- Povirk LF, 2006. Biochemical mechanisms of chromosomal translocations resulting from DNA double-stranded breaks. DNA Repair 5, 1199–1212. [DOI] [PubMed] [Google Scholar]
- Reynolds MF, Peterson-Roth EC, Bespalov IA, Johnston T, Gurel VM, Menard HL, Zhitkovich A, 2009. Rapid DNA double-strand breaks resulting from processing of Cr-DNA cross-links by both MutS dimers. Cancer Res. 69, 1071–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM, 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM, 2000. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem, 275, 9390–9395. [DOI] [PubMed] [Google Scholar]
- Roos WP, Kaina B (2006) DNA damage-induced cell death by apoptosis. Trends Mol. Med, 12, 440–450. [DOI] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J and Jackson SP (2007) Human CtIP promotes DNA end resection. Nature 450, 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawant A, Kothandapani A, Sobol RW, Zhitkovich A, Patrick SM, 2015. Role of mismatch repair proteins in the processing of cisplatin interstrand cross-links. DNA Repair 35, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M, 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Xie A, 2013. Double strand break repair functions of histone H2AX. Mutat. Res 750, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y, 2013. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell. Biol 14, 197–210. [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA, 2007. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316, 1198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q, Lees-Miller SP, Kumar S, Zhang Z, Chan DW, Smith GC, Jackson SP, Alnemri ES, Litwack G, Khanna KK, Lavin MF, 1996. DNA-dependent protein kinase catalytic subunit: a target for an ICE-like protease in apoptosis. EMBO J. 15, 3238–3246. [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Stankovic T, Byrd PJ, Wechsler T, Miller ES, Huissoon A, Drayson MT, West SC, Elledge SJ, Taylor AM, 2007. RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling. Proc. Natl. Acad. Sci. U.S.A 104, 16910–16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, Oldreive C, Wildenhain J, Tagliaferro A, Pelletier L, Taubenheim N, Durandy A, Byrd PJ, Stankovic T, Taylor AM, Durocher D, 2009. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434. [DOI] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. (2005) MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226. [DOI] [PubMed] [Google Scholar]
- Sun Y, Xu Y, Roy K, Price BD, 2007. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell. Biol 27, 8502–8509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD, 2009. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell. Biol 11, 1376–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Mori E, Somakos GI, Ohnishi K, Ohnishi T, 2008. Heat induces gammaH2AX foci formation in mammalian cells. Mutat. Res 656, 88–92. [DOI] [PubMed] [Google Scholar]
- Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker-Jensen S, Mailand N, 2015. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 527, 389–393. [DOI] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ, 2007. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316, 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman C, Kanaar R, 2006. DNA double-strand break repair: all’s well that ends well. Annu. Rev. Genet 40, 363–383. [DOI] [PubMed] [Google Scholar]
- Zecevic A, Menard H, Gurel V, Hagan E, DeCaro R, Zhitkovich A, 2009. WRN helicase promotes repair of DNA double-strand breaks caused by aberrant mismatch repair of chromium-DNA adducts. Cell Cycle, 8, 2769–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Q, Jin S, Ng B, Plisket J, Shangary S, Rathi A, Brown KD, Baskaran R, 2002. Caspase-3 mediated cleavage of BRCA1 during UV-induced apoptosis. Oncogene 21, 5335–5345. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, de Lange T, 2014. 53BP1: pro choice in DNA repair. Trends Cell. Biol 24, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]