

Abstract
Rfam is a database of non-coding RNA families in which each family is represented by a multiple sequence alignment, a consensus secondary structure, and a covariance model. Using a combination of manual and literature-based curation and a custom software pipeline, Rfam converts descriptions of RNA families found in the scientific literature into computational models that can be used to annotate RNAs belonging to those families in any DNA or RNA sequence. Valuable research outputs that are often locked up in figures and supplementary information files are encapsulated in Rfam entries and made accessible through the Rfam website. The data produced by Rfam have a broad application, from genome annotation to providing training sets for algorithm development. This unit provides an overview of how to search and navigate the Rfam website, and how to annotate sequences with RNA families. The Rfam database is freely available at http://rfam.org.
Keywords: Non-coding RNA, RNA family, Rfam, Infernal, genome annotation
INTRODUCTION
Non-coding RNA (ncRNAs) can be classified into families that have evolved from a common ancestor. Sequence family classification can be helpful by revealing information about the structure, function, and evolution of these RNAs. Rfam is a database of ncRNA families, represented by multiple sequence alignments and covariance models (CMs) used for homolog detection and sequence alignment (Kalvari et al., 2017). The purpose of the Rfam database is to allow users to explore non-coding RNA families (Basic Protocols 1 and 2) and to provide the means to identify ncRNAs in sequence datasets both online (Basic Protocol 3) and locally (Alternate Protocol). A wide range of data is available in the Rfam FTP archive, and a public instance of the MySQL database enables the user to form complex queries not supported by the website search (Support Protocol).
BASIC PROTOCOL 1: USING TEXT SEARCH TO EXPLORE ncRNA FAMILIES, CLANS, MOTIFS, AND ncRNA GENOME ANNOTATIONS
With over 2,600 families from 19 RNA types in release 13.0, Rfam contains a wealth of data that can be explored using a faceted text search. It enables browsing and searching RNA families, Rfam clans (groups of homologous RNA families), RNA motifs, genomes annotated with ncRNAs, and individual ncRNA sequences. The protocol below describes the key features of the Rfam text search functionality.
Necessary Resources
Any device with Internet access and an up-to-date Web browser, such as Chrome, Safari, or Firefox.
Browse RNA families
1. Begin at the Rfam home page (http://rfam.org) and find the search box located at the centre of the home page and at the top of every page (Figure 12.5.1).
Figure 12.5.1.

Search box on the Rfam home page.
2. Click the Families link in the search box (Figure 12.5.1). Alternatively, navigate to the browse page (http://rfam.org/browse) by clicking on the Browse tab at the top of any page and select the “Browse families” option.
3. Examine the search results. For each family a preview of its secondary structure, RNA type, the size of the Seed alignment (see Background information), and the number of annotated species are displayed (Figure 12.5.2).
Figure 12.5.2.
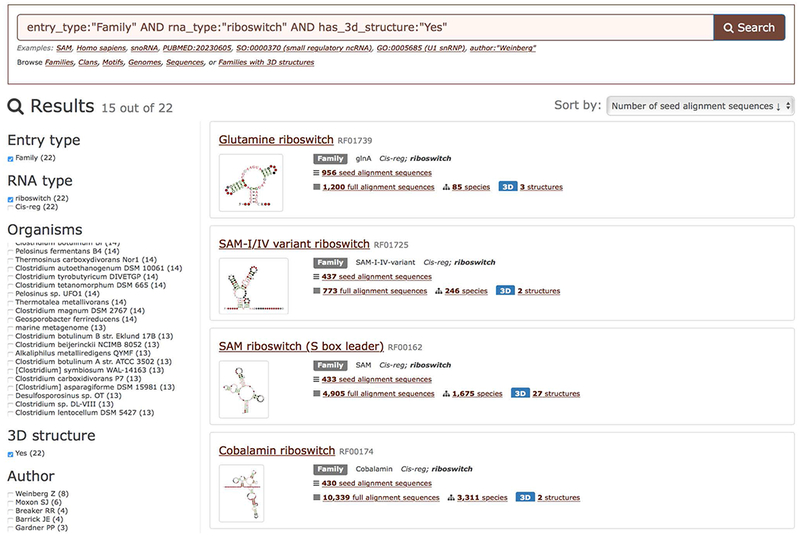
Example text search showing riboswitch families with a known 3D structure that are found in one or more Bacillus species. For each family the secondary structure, the number of annotated sequences, and the number of species where the family is found are displayed.
The search URL can be shared or bookmarked to repeat the search with the same parameters.
4. Filter the results page using facets. For instance, to focus on riboswitches, select “riboswitch” from the RNA type facet on the left of the search page (Figure 12.5.2). The results can also be filtered by organism and the availability of experimentally determined 3D structures.
5. Refine your query by adding keywords. For example, to find families identified in one or more Bacillus species, type “Bacillus” at the end of the query in the search box (Figure 12.5.2) and press Enter or click the Search button. It is possible to search for sequence accessions, organism names, and other types of keywords (see Rfam Help for the most up-to-date list at http://rfam.org/help).
6. Sort the results by various criteria. For example, in order to find families identified in the largest number of organisms or matching most sequences. The sorting options can be accessed using a dropdown menu at top right (Figure 12.5.2).
7. Visit a family page by clicking on a family name (for example, SAM riboswitch) to view a summary imported from Wikipedia and access family specific data such as sequences, alignments, secondary structures, phylogenetic trees, structures and motifs (if available), database cross-links, and curation details. Basic Protocol 2 provides more information about viewing families.
Browse RNA motifs and clans
Rfam organises related families into clans (Gardner et al., 2011). For example, the LSU clan (CL00112) groups together five families that describe different types of large ribosomal subunit RNAs, including bacterial, eukaryotic, and archaeal LSU families. RNA motifs in Rfam are based on the RMfam motif collection (Gardner & Eldai, 2015) and are defined as RNA sequences and/or secondary structures that can be found in a number of different RNA families. For example, UNCG tetraloop (RM00029) is a common 3D motif that is found in over 200 RNA families.
RNA clans and motifs can be searched using the same steps as described above for RNA families except that at step 2 Clans or Motifs should be selected instead of Families. Figure 12.5.3 shows example search results when browsing Clans and Motifs.
Figure 12.5.3.
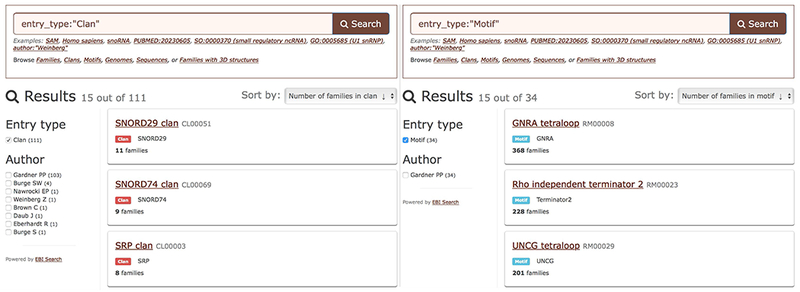
Browsing ncRNA clans (left) and motifs (right). For each clan the number of families is shown and the results can be sorted by the number of member families. For each motif the number of families where the motif occurs is shown and motifs can be sorted accordingly.
Browse non-coding RNAs in a specific genome
Starting with release 13.0, Rfam provides ncRNA annotations of a non-redundant set of complete genomes (Kalvari et al., 2017). The text search enables viewing RNA families found in a certain species or taxonomic group, comparing RNAs found in different genomes, and viewing annotations of individual ncRNA sequences found in a genome.
8. Begin at the Rfam home page (http://rfam.org) and find the search box located at the centre of the home page and at the top of every page (Figure 12.5.1).
9. Search for a specific genome. For example, type “human” or “Homo sapiens” in the search box. The search engine will retrieve various entries that relate to human (e.g. families, sequences or related genomes). Select “Genome” from the Entry type facet to focus on genome entries (Figure 12.5.4, left). Clicking a genome name will load a genome summary page (Figure 12.5.4, right) showing additional information, such as a short genome description, taxonomic identifier, and cross-links to related databases. From a genome summary page it is possible to browse families (step 3a) or individual ncRNA sequences (step 3b).
Figure 12.5.4.

Searching for the human genome (left) and viewing the human genome summary page (right).
10a. Browse Rfam families found in a genome by clicking “Browse families” on the Genome summary page (Figure 12.5.4, right). The results are shown in Figure 12.5.5 and can be filtered by RNA type and other criteria using facets or sorted using the dropdown menu at the top right.
Figure 12.5.5.
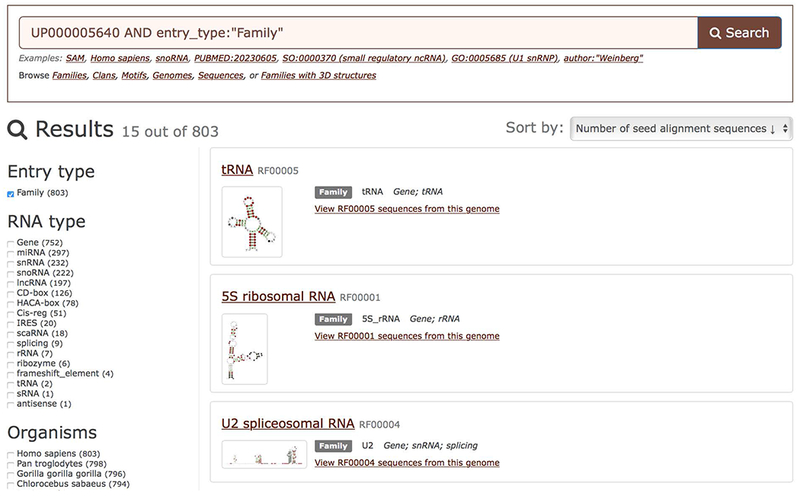
Searching for ncRNA families found in the human genome.
One can list all sequences from each family found in a specific genome. For example, to view all matches for the tRNA family (RF00005) found in the human genome click “View RF00005 sequences from this genome” (Figure 12.5.5).
10b. Browse RNA sequences found in a genome by clicking “Browse sequences” on the Genome summary page (Figure 12.5.4, right). The sequences can be filtered using the RNA type facet to focus on a specific RNA type, such as snoRNA (Figure 12.5.6).
Figure 12.5.6.
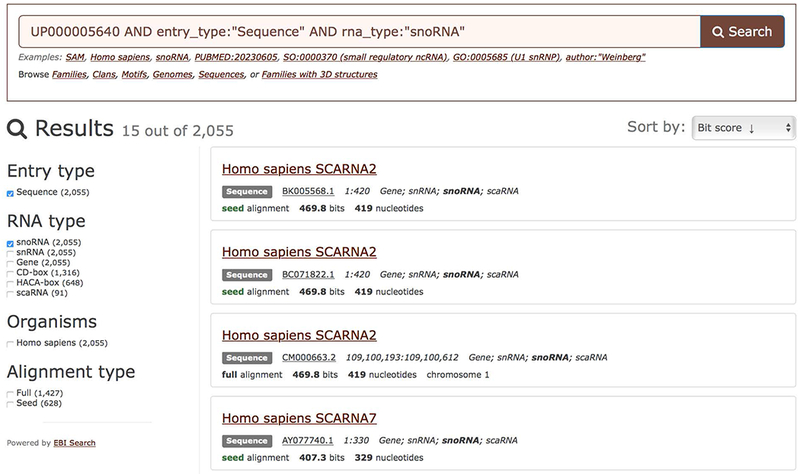
Browsing human snoRNA sequences. The results can be sorted by bit score or E-value (see Guidelines for Understanding Results for more information).
Each sequence has a summary page where the sequence can be downloaded in FASTA format. The summary page also contains links to the RNAcentral database (The RNAcentral Consortium, 2017) that provides links to other resources annotating the same sequence as well as publications and other information. Where possible, an embedded genome browser shows the genomic location of the sequence alongside the genes from Ensembl (Aken et al., 2017) (Figure 12.5.7).
Figure 12.5.7.
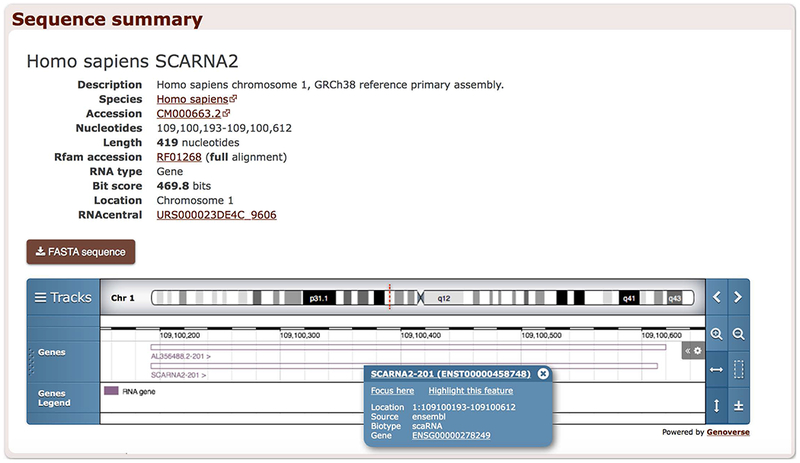
Sequence summary page of a human SCARNA2 sequence. The embedded genome browser shows the location of a small Cajal body-specific RNA 2 sequence (RF01268) on chromosome 1.
Advanced search examples
Rfam text search provides a flexible way of combining different search strategies using the Lucene search syntax. The following section provides several examples of using the query syntax (see Rfam Help at http://rfam.org/help for the most up-to-date information). Queries that are not supported by the online interface may be performed using the public MySQL database described in Support Protocol.
11. Search for species-specific information using NCBI Taxonomy identifiers (Federhen, 2012). For example, to search for all families found in human type the following in the search box:
taxonomy:”9606” entry_type:”family”
where 9606 is the NCBI taxonomy identifier for Homo sapiens. The currently available entry types include family, clan, motif, genome, and sequence (using the Entry type facet is equivalent to adding entry_type to the query).
12. Search using taxonomic classification. For example, type the following in the search box to find all Rfam families that have been identified in primates or mammals:
tax_string:”Primates” entry_type:”family”
tax_string:”Mammalia” entry_type:”family”
13. Use logic operators. Sometimes it is useful to exclude some entries which can be achieved using the NOT operator. For example, the following query lists all RNAs on human chromosome 1 (accession CM00063.2) except for rRNA and tRNA:
CM000663.2 not rna_type:”rRNA” not rna_type:”tRNA”
Another useful logic operator is OR. For example, the following query retrieves all Rfam families that are found either in Fungi or Archaea.
entry_type:”family” (tax_string:”Fungi” or tax_string:”Archaea”)
Note that without the OR operator and the parentheses, the query finds families that are found in both Fungi and Archaea:
entry_type:”family” tax_string:”Fungi” tax_string:”Archaea”
14. Use field-specific search. By default the search terms found in a query are matched against all the indexed data, and in some cases it may be desirable to search only a subset of metadata, such as family descriptions or species names.
Several queries above contain examples of field-specific searches, for instance, entry_type:”family” where entry_type is a field that is being searched and family is the query term. Other searchable fields include: description, scientific_name, common_name, tax_string, and others. The complete list of searchable fields is available in Rfam Help at http://rfam.org/help.
15. Use double quotes. Another way of performing a more specific query is to surround the search terms with double quotes. For example, the following query finds all entries where the terms “group” and “II” occur independently, including group II introns:
group II
However, this query is better expressed as follows (note the double quotes):
“group II”
because it finds only the entries where the words occur next to each other.
BASIC PROTOCOL 2: VIEWING Rfam FAMILY
The family page is the central point of information of any Rfam family. It is split into several tabs which offer specific information about the family, such as functional annotation, sequences, secondary structure, PDB structures, species distribution, Seed alignment, and covariance model details.
Necessary Resources
Any device with Internet access and an up-to-date Web browser, such as Chrome, Safari, or Firefox.
Explore the family Summary
1. Navigate to the SAM riboswitch family page (http://rfam.org/family/RF00162) directly or by searching Rfam for the accession number RF00162 (see Basic Protocol 1 for more information about Rfam text search).
The Summary tab is the starting point of a family page. The tab shows a Wikipedia article describing the family. Any user can contribute to these articles on Wikipedia, and the content is regularly synchronised with Rfam.
If a family belongs to a clan, as is the case with the SAM riboswitch, a section in the Summary page will provide clan membership information.
All family pages have a header which includes the name of the family, Rfam accession, and a short description. On the right side of the header, three icons provide a summary of the number of sequences, the number of species, and 3D structures in that family (Figure 12.5.8). Clicking on these icons is a quick way to access the appropriate tabs where this information can be found.
Figure 12.5.8.
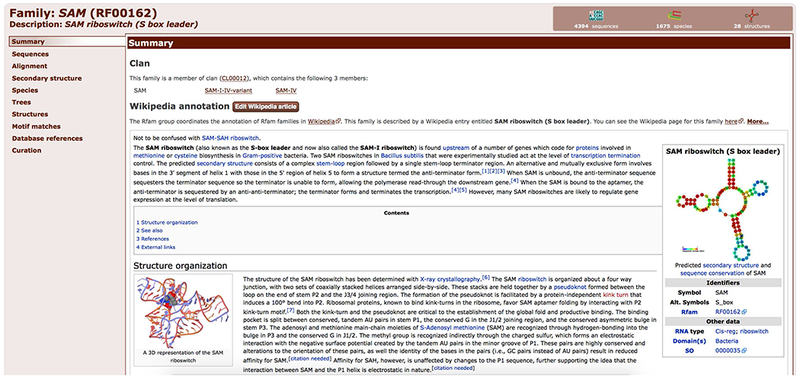
Summary tab for the SAM riboswitch family page (RF00162) showing a Wikipedia article about the family and a list of Rfam clans the family belongs to.
View and download Seed alignment
Seed alignments (described in Background Information) are available to view or download in several formats, including Stockholm, Pfam, and FASTA (gapped and ungapped).
1. Click on the Alignment tab.
2. Select a format from the dropdown menu under the Formatting options section. Select the “Download” option, and click “Generate” (Figure 12.5.9).
Figure 12.5.9.
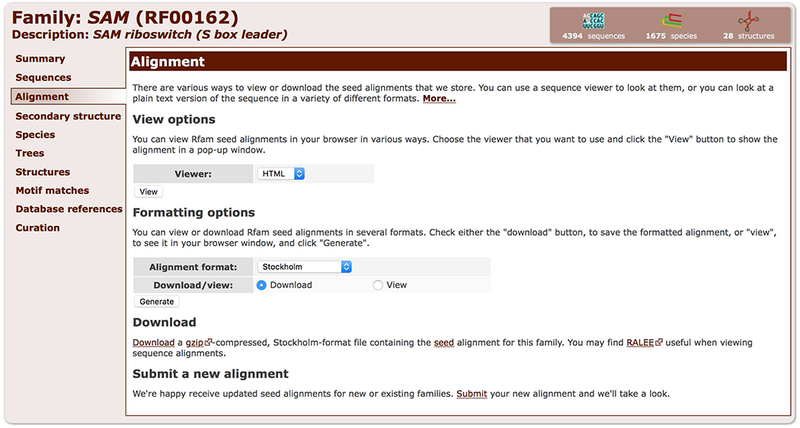
The Alignment tab enables viewing and downloading the Seed alignment in several formats.
The alignment can also be viewed as a text file in the browser, in any of these formats, by selecting the View option, and then clicking the Generate button. Additionally, a compressed version of the file in Stockholm format is also available under the Download section.
3. To view the Seed alignment, select HTML from the View options section and click View. This will open a pop-up window with the alignment (Figure 12.5.10).
Figure 12.5.10.

Viewing part of the Seed alignment for SAM riboswitch. The alignment is colored by secondary structure helical regions.
View and download ncRNA sequences
The Sequences tab provides a full list of sequence regions that belong to the SAM riboswitch family.
4. Click the Sequences tab and use the table to explore the first 300 sequences (highest bit scores) of the family (Figure 12.5.11). The table columns can be sorted, for example, by clicking on the “Bit score” column header.
Figure 12.5.11.
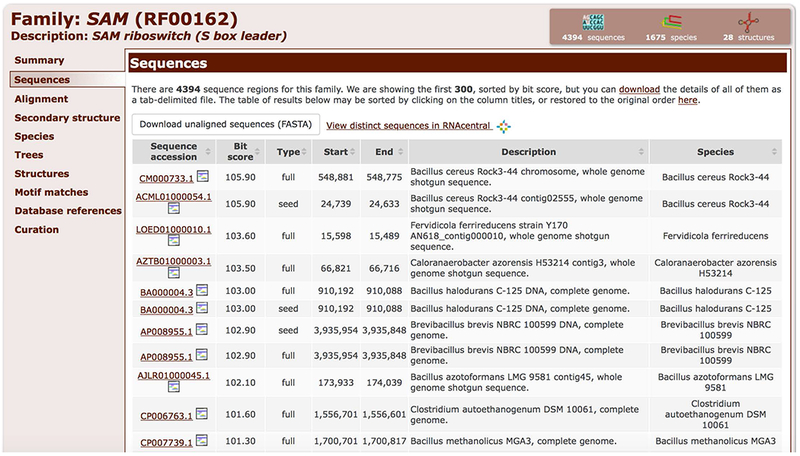
A list of sequence regions that belong to a family can be found in the Sequences tab.
5. Download all sequences by clicking on the “Download unaligned sequences (FASTA)” button on the top of the page (Figure 12.5.11).
The downloaded file is in gzipped ungapped FASTA format. The Infernal software can be used to align these sequences to the Rfam CM (see Alternate Protocol).
6. Click the “View distinct sequences in RNAcentral” link at the top of the page to find SAM riboswitch sequences in RNAcentral. The RNAcentral database integrates sequences and annotations from over 25 different resources (The RNAcentral Consortium, 2017) that can provide additional context for the sequence.
View structural information
7. Click on the “Secondary Structure” tab and explore the different representations of the consensus secondary structure.
The main visualization is generated with the R-scape software (Rivas, Clements, & Eddy, 2017), which annotates statistically significant covarying basepairs and highlights them in green in the secondary structure diagram (Figure 12.5.12).
Figure 12.5.12.
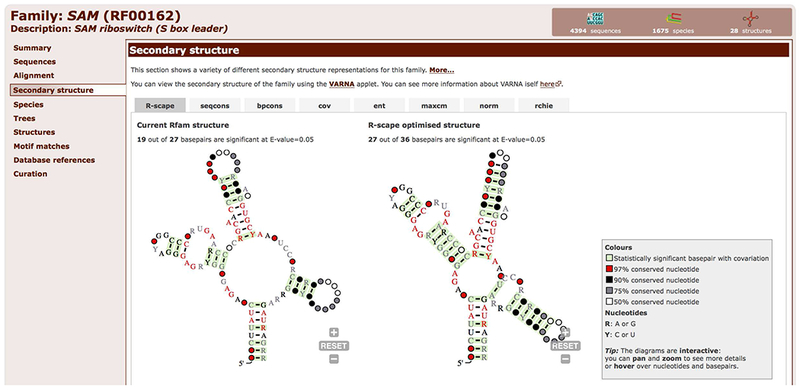
R-scape secondary structure visualisations for the SAM riboswitch (RF00162) shown in the Secondary structure tab. Two structures are shown: On the left, the R-scape analysis of the current secondary structure in the Rfam Seed alignment. On the right, an R-scape optimised structure predicted using the statistically significant covarying basepairs as folding constraints.
8. Explore other Secondary Structure tabs (Figure 12.5.13). They show several coloring schemes for sequence conservation (seqcons), base-pair conservation (bpcons), covariation (cov), sequence entropy (ent), scores from the covariance model’s consensus sequence (maxcm), and stem-loop colouring (norm).
Figure 12.5.13.
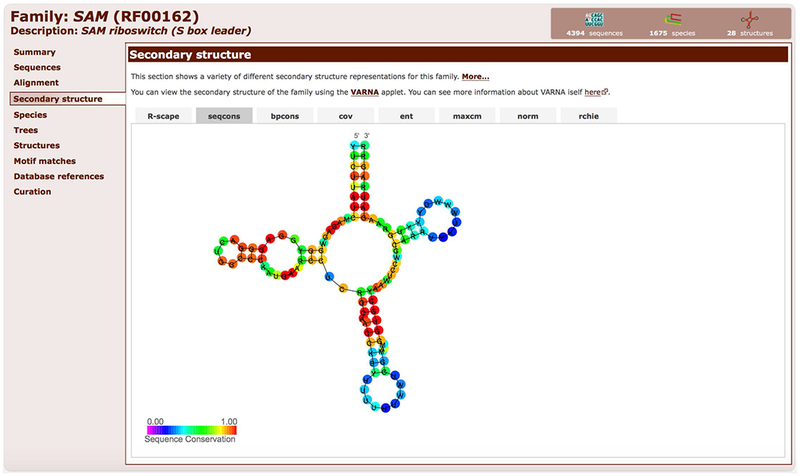
Secondary structure of the SAM riboswitch (RF00162) colored by sequence conservation (conserved nucleotides are red, variable nucleotides are blue).
9. Click on the last Secondary Structure tab, rchie.
This representation uses the R-chie package (Lai, Proctor, Zhu, & Meyer, 2012) to visualize the secondary structure as an arc diagram and highlight the basepairs in the sequence alignment (Figure 12.5.14). The alignment and the arcs are color coded to indicate covariation and identify invalid basepairs giving an insights into the alignment quality.
Figure 12.5.14.

R-chie visualisation of the Seed alignment and the consensus secondary structure of the SAM riboswitch (RF00162). Canonical basepairs are shown as blue arcs, and the green alignment columns indicate valid basepairs. This visualisation suggests that the Seed alignment is of reasonable quality.
10. Click on the Structures tab to access the 3D structures from the Protein Data Bank that match the Rfam family (Figure 12.5.15).
Figure 12.5.15.
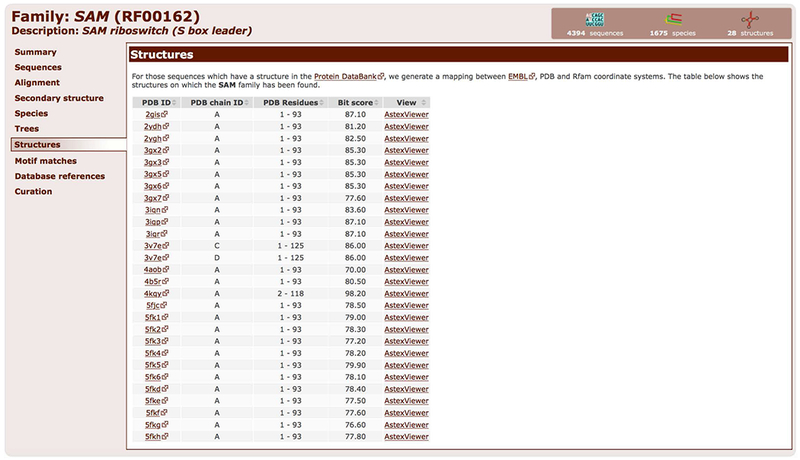
The Structures tab lists the 3D structures from the Protein Data Bank that match the SAM riboswitch Rfam family (RF00162).
The table can be sorted by clicking on the column titles. The PDB ID column contains links to Protein Data Bank where the 3D structures can be explored or downloaded.
Explore the distribution of the family across taxonomic groups
11. Click on the Species tab to see the species distribution of the family in a Sunburst or Tree diagram.
The sunburst and tree diagrams are generated using taxonomic lineages of all species where the RNA family was found (Figure 12.5.16).
Figure 12.5.16.
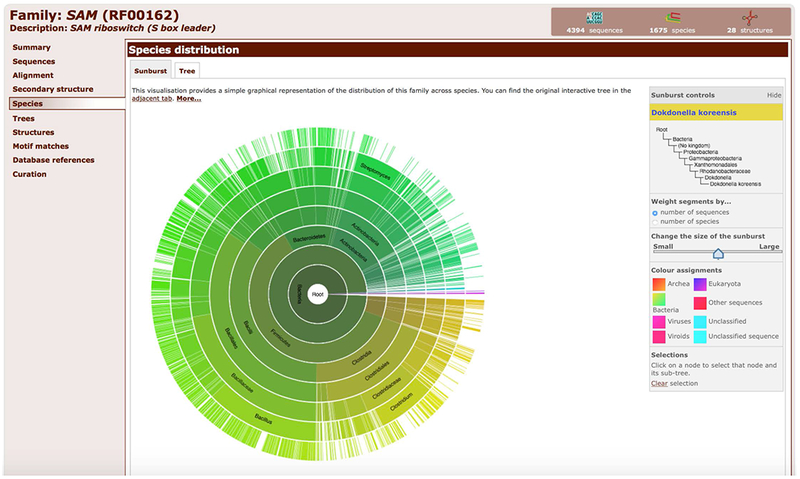
Sunburst representation of the taxonomic distribution of the SAM riboswitch family (RF00162).
12. Click on the Trees tab to see predicted phylogenetic trees for the alignment.
The trees are generated using maximum likelihood or neighbour-joining methods.
Examine detailed curation information
13. Click on the Curation tab to see detailed information about how the family was created, including Authors, Structure source, model parameters, and covariance model download (Figure 12.5.17).
Figure 12.5.17.
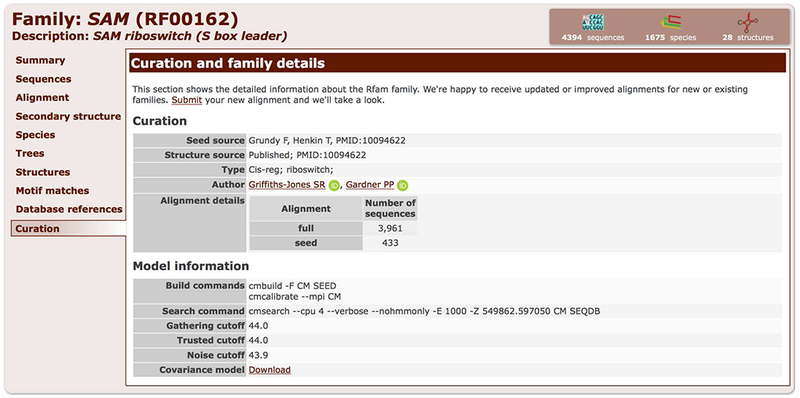
Curation tab of the SAM riboswitch family (RF00162) showing the source of the Seed alignment and the secondary structure, the authors of the family, and the parameters used to build the covariance model.
BASIC PROTOCOL 3: USING Rfam SEQUENCE SEARCH ONLINE
Necessary Resources
Hardware and software
Any device with Internet access and an up-to-date Web browser, such as Chrome, Safari, or Firefox.
Files
A FASTA file (supplementary file ‘sequence-search-example.fa’) containing nucleic acid sequences of interest.
The Rfam website enables annotation of a DNA or RNA sequence with RNA families using the Infernal cmscan program for single sequences or small datasets. See the Alternate Protocol 1 for running the searches locally.
Find non-coding RNAs in a single sequence
1. Begin at the Rfam home page (http://rfam.org) and locate the Sequence Search tab on the Rfam home page. Alternatively, navigate directly to http://rfam.org/search and choose “Sequence search”.
2. Paste a DNA or RNA sequence in the Sequence text box (see supplementary file ‘sequence-search-example.fa’) and click the Submit button.
3. Examine the search results once they become available (Figure 12.5.18).
Figure 12.5.18.
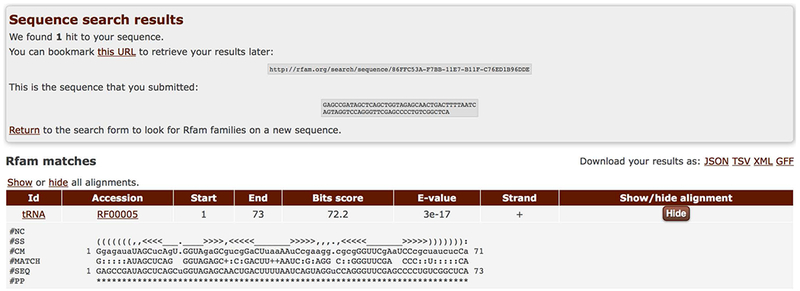
Sequence search results showing an alignment between the query sequence (the #SEQ line) matching the covariance model of the tRNA family (the #CM line). The secondary structure predicted for the query sequence is shown in the #SS line.
Rfam search will report the Rfam identifier and accession of the matching model, start and end positions of the match, the strand, and the corresponding scores (bit score and E-value). Click on the Show button to view the alignment and secondary structuThe Rfam distribution from: Rfam cm filere (Figure 12.5.18). The results can be downloaded in several formats including JSON and GFF, and the search URL can be bookmarked for future reference.
Find non-coding RNAs in multiple sequences using batch search
4. Navigate to http://rfam.org/search and select the “Batch search” option (Figure 12.5.19).
Figure 12.5.19.
Batch sequence search interface.
5. Upload a FASTA file with RNA or DNA sequences (see sample file batch-search-example.fasta). Enter your email address to be notified when the results are ready, and start the search by clicking the “Submit” button.
Note that the online sequence search cannot process large datasets (FASTA files with over 100,000 lines or more than 200,000 nucleotides per sequence). See the Alternate Protocol for more information about using Infernal locally to annotate sequence datasets of arbitrary size.
6. Examine the results once they become available (Figure 12.5.20).
Figure 12.5.20.
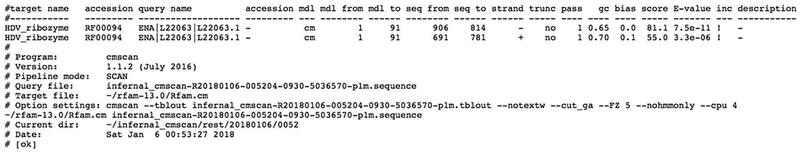
Batch search results in a tabular format showing ncRNA families found in Hepatitis delta virus genotype III (L22063.1).
ALTERNATE PROTOCOL 1: USING Rfam AND Infernal FOR RNA SEQUENCE ANALYSIS OF NUCLEOTIDE SEQUENCE DATASETS
Rfam covariance models are available to be downloaded and used locally along with the software package, Infernal (Nawrocki & Eddy, 2013), for various RNA sequence analysis tasks, such as identifying ncRNA homologs in sequence datasets and creating multiple alignments of ncRNAs. Infernal is computationally expensive, and searches in sequence datasets on the order of gigabases or larger, and alignment of thousands of sequences, can take a long time (Nawrocki et al., 2015). Additionally, Infernal programs must be run on the command-line, making this protocol more advanced than the Basic Protocols above.
Necessary Resources
Hardware
Any Unix/Linux/Mac laptop, workstation, or compute farm
Software
Infernal (open source BSD license)
Files
The Rfam distribution from:
Rfam cm file: ftp://ftp.ebi.ac.uk/pub/databases/Rfam/CURRENT/Rfam.cm.gz
Claninfo file: ftp://ftp.ebi.ac.uk/pub/databases/Rfam/CURRENT/Rfam.clanin
FASTA file of all hits to an example family (RF01831): ftp://ftp.ebi.ac.uk/pub/databases/Rfam/CURRENT/fasta_files/RF01831.fa.gz
A FASTA file (see supplementary file my.fa) containing some nucleic acid sequences of interest
1. Download and install Infernal as described in the Infernal user’s guide (http://eddylab.org/infernal). After installing Infernal, ensure that the executable programs (in the src and easel/minapps subdirectories) are in the user’s PATH. Also, download the Rfam files listed above and unpack those that end with a .gz suffix using gunzip.
Identify all non-coding RNAs from Rfam families in a nucleotide sequence dataset using Infernal’s cmscan program
2. Run the cmpress program on Rfam.cm to prepare it for use with the cmscan program:
cmpress Rfam.cm
This command should produce four additional files Rfam.cm.i1f, Rfam.cm.i1i, Rfam.cm.i1m and Rfam.cm.i1p. For more information on the various file formats see Infernal’s User’s Guide (Internet Resources)
3. Use the cmscan program to identify all subsequences that match any Rfam family with a score above the gathering cutoff (GA) selected by the Rfam curators:
cmscan --nohmmonly --rfam --cut_ga --fmt 2 --oclan --oskip --clanin Rfam.clanin -o my.cmscan.out --tblout my.cmscan.tblout Rfam.cm my.fa
This command may take up to several hours to complete. There are two output files from cmscan when the above command is used.
The file my.cmscan.tblout is a tabular output file that lists information on each hit above the GA cutoff for all families (see the Guidelines for Understanding Results section), one line per hit. The set of hits in the tblout file will not include any hits that overlap in the same sequence between models in the same clan (a set of homologous models, for example LSU_rRNA_archaea, LSU_rRNA_bacteria and LSU_rRNA_eukarya). This lack of overlapping hits is one potential advantage of using cmscan on the command line instead of via the Rfam website single sequence search or batch search, which does not remove any overlaps.
The my.cmscan.out file includes more information, such as alignments of all hits to the query models, including overlaps that may have been removed in the tblout file.
Both output files include all hits for the first sequence in the supplementary file ‘my.fa’, then all hits to the second sequence, and so on. You can find explanations of the format and information in these files in the Infernal user’s guide that is included in the Infernal distribution and available on the website (http://eddylab.org/infernal).
Identify examples of a single RNA family in a sequence dataset using cmsearch
4. Fetch the model of interest from Rfam.cm or use your own. Here, the THF Riboswitch family (RF01831) is used as an example.
cmfetch Rfam.cm RF01831 > RF01831.cm
Alternatively, several models can be fetched from Rfam.cm using the -f option and an input file that lists the desired accessions or names, or non-Rfam models created using cmbuild can be used. The Infernal user’s guide tutorial has examples of these alternatives.
5. Use the cmsearch program to search a sequence dataset for all RF01831 hits above the GA cutoff to RF01831.
cmsearch --nohmmonly --rfam --cut_ga -o my.cmsearch.out --tblout my.cmsearch.tblout RF01831.cm my.fa
This command searches with only one model and should complete in less than one minute. As with the cmscan example above, cmsearch creates two output files, my.cmsearch.tblout and my.cmsearch.out, which are in very similar formats to the cmscan output files. A difference between these files and the cmscan files is that the hits are not sorted by the target sequence, but rather by the query Rfam model (all hits to model 1, then all hits to model 2, etc.).
6. Optionally, fetch the high-scoring hits from the target sequence file (my.fa) using the esl-sfetch utility program that is included with Infernal. This requires a preliminary step of indexing the sequence file:
esl-sfetch --index my.fa
The command for fetching the hit subsequences requires piping output from the Unix program grep into the Unix program awk and the output of awk into the esl-sfetch program, as follows:
grep -v \^# my.cmsearch.tblout | awk ‘{ printf (“%s/%d-%d %d %d %s\n”, $1, $8, $9, $8, $9, $1); }’ | esl-sfetch -Cf my.fa - > my.RF01831.fa
The file my.RF01831.fa will include the hit subsequences listed in my.cmsearch.tblout.
Align all Rfam homologs for a particular RNA family to create the full alignment:
7. Use cmalign to create a sequence and secondary structure based multiple alignment in Stockholm format:
cmalign RF01831.cm RF01831.fa > RF01831.stk
8. Optionally, reformat the file to aligned fasta (afa) format using esl-reformat:
esl-reformat afa RF01831.stk > RF01831.afa
Prepare a sequence dataset to map short reads against
In order to identify non-coding RNAs in a short read library using Rfam it is recommended to first prepare a sequence dataset of all Rfam hits in the target genome or other dataset and then map reads against that. In this example, suppose that ‘my.fa’ contains the five complete genomes that the short reads derive from. The following step will create the dataset of all Rfam hits in ‘my.fa’. It builds on two earlier steps: in step 3, all Rfam hits were output to the file ‘my.cmscan.tblout’, and in step 6 the ‘my.fa’ file was indexed so that esl-sfetch could be used to fetch subsequences from it.
9. Using the .tblout file created in step 3, use esl-sfetch to fetch all subsequences that were a hit to an Rfam model, again using ‘awk’ and ‘grep’ as in step 6.
grep -v \^# my.cmscan.tblout | awk ‘{ printf (“%s.%s/%d-%d %d %d %s\n”, $2, $4, $8, $9, $8, $9, $4); }’ | esl-sfetch -Cf my.fa - > my.rfam.fa
The my.rfam.fa file can now be used to map reads against to identify non-coding RNAs from families in Rfam. Do not use Infernal (cmsearch or cmscan) to map the reads directly, as Infernal is not designed for short reads and will not give good results. Use a dedicated read mapping program instead. Note that the fields (e.g. $2, $4) used in this command differ from those used in the similar command in step 6. That is because the order and meaning of fields in the cmscan and cmsearch .tblout files differ.
An alternative method for identifying ncRNAs from Rfam families in sequencing read datasets is to first map your reads against the complete target genome or dataset, and then look for overlaps between those mapped reads and Rfam hits found using cmscan (or cmsearch).
10. To do this, first convert the my.cmscan .tblout file created in step 3 to GFF format:
grep -v ^\# my.cmscan.tblout | awk ‘ { printf(“%s\tinfernal\t%s\t%s\t%s\t%s\t%s\t.\n”, $4, $2, $10, $11, $17, $12); }’ > my.cmscan.gff
The my.cmscan.gff file can then be used as input to a program for identifying overlaps between two datasets, such as the bedtools intersect program (Quinlan, 2014).
A similar command could be used to convert the cmsearch .tblout output from step 5 to GFF format:
grep -v ^\# my.cmsearch.tblout | awk ‘ { printf (“%s\tinfernal\t%s\t%s\t%s\t%s\t%s\t.\n”, $1, $3, $8, $9, $15, $10); }’ > my.cmsearch.gffSUPPORT PROTOCOL 1: USING Rfam PUBLIC MySQL DATABASE TO FORM COMPLEX QUERIES
Necessary Resources
Hardware
A computer that can connect to a MySQL database
Software
A MySQL database management application like MySQL Workbench or Sequel Pro.
Files
The Rfam database dumps from: ftp://ftp.ebi.ac.uk/pub/databases/Rfam/CURRENT/database_files
Use public Rfam MySQL database
The Rfam web interface enables searching and browsing the data (see Basic Protocol 1) but some types of searches may not be supported. Users can use a MySQL database management application such as MySQL Workbench to connect to the Rfam public MySQL database and perform SQL queries to retrieve different types of data.
1. Connect to the public Rfam database
mysql --user rfamro --host mysql-rfam-public.ebi.ac.uk --port 4497 --database Rfam
Further information on how to establish a connection to the database is available in Rfam Help at http://rfam.org/help.
2. Execute the following query to select all high-scoring snoRNA regions found in Mammals:
SELECT fr.rfam_acc, fr.rfamseq_acc, fr.seq_start, fr.seq_end, fr.bit_score
FROM full_region fr, rfamseq rf, taxonomy tx, family f
WHERE
rf.ncbi_id = tx.ncbi_id
AND f.rfam_acc = fr.rfam_acc
AND fr.rfamseq_acc = rf.rfamseq_acc
AND tx.tax_string LIKE ‘%Mammalia%’
AND f.type LIKE ‘%snoRNA%’
AND is_significant = 1;
The first several lines of the output are shown in Figure 12.5.21. The results can be saved locally and used for further analysis. For example, sequence accessions and start/stop coordinates (labelled respectively rfamseq_acc, seq_start, and seq_end in Figure 12.5.21) can be used to download the corresponding sequence fragments from a sequence archive, such as ENA, using a REST API.
Figure 12.5.21.
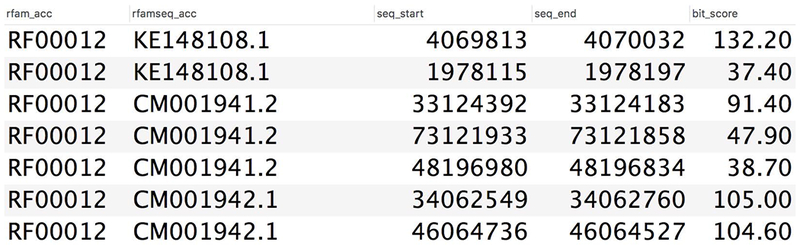
Example output of an SQL query showing Rfam accessions (rfam_acc), sequence accessions (rfamseq_acc), the start and stop coordinates of the ncRNAs relative to the sequence accessions (seq_start and seq_end), and bit score (bit_score, see Background information for more details about bit scores).
3. Execute the following query to get all high-scoring ncRNAs found in the human genome.
SELECT fr.rfam_acc, fr.rfamseq_acc, fr.seq_start, fr.seq_end, fr.bit_score
FROM full_region fr, genseq gs, genome g
WHERE
fr.rfamseq_acc=gs.rfamseq_acc
AND g.upid = gs.upid
AND g.scientific_name = ‘homo sapiens’
AND fr.is_significant = 1;
The output is in a similar format to that shown in Figure 12.5.21 above.
Create a local MySQL database for any available Rfam release
It is also possible to restore a specific version of the Rfam database on any computer using MySQL backup files available in the FTP archive for each release under database_files (see Internet Resources section). The CURRENT directory always contains the files from the most recent release, but previous versions are also available.
4. Create a new directory and download all files from the database_files directory on the FTP archive:
mkdir rfam_database_files && cd rfam_database_files wget ftp://ftp.ebi.ac.uk/pub/databases/Rfam/CURRENT/database_files/* .
The files with .sql extension contain SQL statements used to create each table, and the tabular files (.txt) contain the data stored in the tables.
5. Concatenate all .sql files in a single file:
cat *.sql > rfamdb.sql
6. Connect to a local MySQL server:
mysql --user username --host localhost -p
-p option will prompt the user for a password
7. Disable MySQL variable foreign_key_checks to allow the tables to be created in an arbitrary order:
set foreign_key_checks=OFF;
8. Create a new schema and switch to it:
create schema rfam_local;
use rfam_local;
Create all tables using the concatenated .sql file from step 2 and exit the MySQL server when the process is complete:
source rfamdb.sql;
exit;
Populate the tables using mysqlimport client and the data in the .txt dumps located in the rfam_database_files:
gunzip *.gz
mysqlimport --user username --host localhost --password --local rfam_local *.txt
11. The database can now be used to perform SQL queries as described above.
GUIDELINES FOR UNDERSTANDING RESULTS
Infernal bit scores, E-values, and Rfam GA cutoff scores
The Rfam sequence and batch searches in the Basic Protocol 3 section as well as cmsearch and cmscan searches in the Alternate Protocol section output a bit score and E-value for all hits found in the target sequence or database. The bit score (S) is a log-odds score, the logarithm, base 2, of the ratio of the probability that the sequence was generated from the covariance model (CM) versus from a null model. The E-value (E) is the expected number of hits at or above score S in a search of a random database of the same size (Z).
Rfam curators have defined a specific score for each family, called the gathering threshold, or GA cutoff, based on examination of hits in a large database called RFAMSEQ such that all hits with scores above the GA cutoff are believed to be true homologs. The GA cutoffs serve as family-specific thresholds for users when doing Rfam searches. The sequence and batch searches on the Rfam website automatically enforce the GA cutoffs, returning only hits that have scores exceeding them, as do the cmscan and cmsearch examples in Alternate Protocol 1 due to the use of the --cut_ga option. As explained in the Critical Parameters and Troubleshooting section, users may want to use different, less strict, thresholds when doing searches in some instances.
The role of database size in interpreting Infernal results
An Infernal E-value can be very helpful in interpreting search results because it is an estimate of the statistical significance of a hit, of how likely it is that the hit is occurring by chance as opposed to due to sequence homology. The database size is integral to E-values. For example, imagine you find a hit of 20 bits with an E-value of 0.01 in a database of size 2000 nucleotides (2Kb). The same hit in a database with size 2,000,000 (2Mb) will still be 20 bits but will have an E-value of 10. The E-value has increased by 1000-fold because the database size has increased by 1000-fold.
Importantly, database size is determined differently for different types of searches. For single sequence and batch searches through the website as well as with cmscan on the command line, the database size is defined as the length of the current (single) target sequence multiplied by two (because both strands are searched) multiplied by the number of query CM models. For website and cmscan searches using Rfam.cm the number of query models is the number of models in Rfam (e.g. 2,686 in Rfam 13.0).
Alternatively, for cmsearch on the command line the database size is the total number of nucleotides in the entire sequence file being searched, and does not depend on the number of models in the query CM file. Because of these differences, E-values for the same hit (same subsequence to the same model with the same bit score) may be different depending on how the search was performed because the database size will be different. Which program was used (website/batch/cmscan or cmsearch), the number of models in the CM file, and the total length of sequence that the hit occurs in can all affect the E-value because they can all affect the database size.
To manually set the database size used in the E-value calculation to <X> megabases when running cmsearch or cmscan on the command line, use the -Z <X> option. It makes sense to do this if, for example, a large sequence file has been split up into many smaller files, and searches have been performed in parallel on a compute cluster, with the results combined. In that scenario, if <X> is set as the total number of models used times the total number of nucleotides in all sequence files times two (for both strands), then the combined results should have appropriate E-values. That is, the expectation is that in the collection of all hits between all sequences and models there will be about 1 hit with an E-value of 1 or below by chance (not due to homology), about 10 with an E-value of 10 or below by chance, etc.
COMMENTARY
Background Information
Non-coding RNAs
Non-coding RNAs (ncRNAs) are transcribed from the genome sequence but are not translated into protein products. NcRNAs are responsible for a wide array of functions (Cech & Steitz, 2014). Some ncRNAs are highly abundant and essential for normal cell function in all organisms, such as, ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs) that are responsible for translating mRNAs into proteins. Several types of small ncRNAs perform regulatory functions, for example, microRNA (miRNA) and piwi-interacting RNA (piRNA) act as key regulators of gene expression in higher eukaryotes. Several types of ncRNAs, including riboswitches (McCown, Corbino, Stav, Sherlock, & Breaker, 2017) and RNA thermometers (Kortmann & Narberhaus, 2012), are found in bacteria where they play roles in gene expression, virulence, and stress response.
Related ncRNAs may have relatively weak sequence conservation, and homologs are difficult to find by sequence similarity alone. However, they often show conserved base-paired secondary structure. Therefore, it is necessary to look for similarities in both sequence and structure. Computational methods using the combination of RNA sequence and secondary structure have been successfully used to discover large numbers of RNAs (Weinberg, Lünse, & Corbino, 2017).
Constructing Rfam families
Construction of each RNA family begins with a manually curated alignment of representative sequences called a Seed alignment (Figure 12.5.22). The Seed alignment may include a consensus secondary structure based on the literature or predicted using RNAalifold (Bernhart, Hofacker, Will, Gruber, & Stadler, 2008) or similar software. The cmbuild program from the Infernal software package is then used to build a covariance model (CM) from the Seed alignment.
Figure 12.5.22.
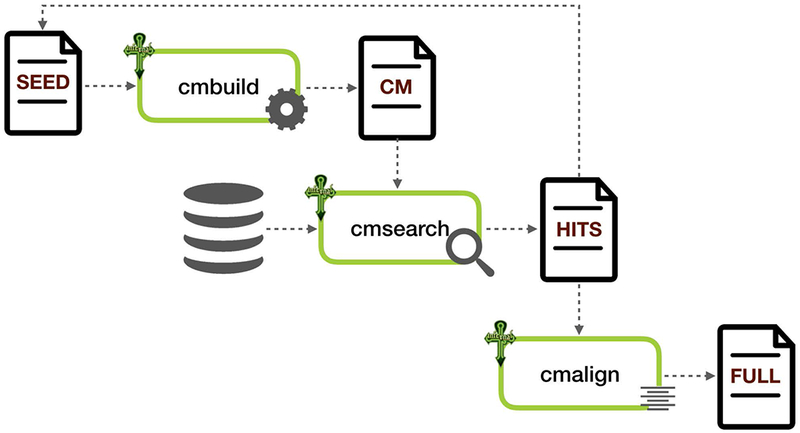
Building an RNA family using Infernal. The Seed alignment is a starting point used to build a covariance model (CM) which is then used to search for more hits in a large sequence database. The hits may be added to the Seed alignment, if necessary. The Full alignment is an alignment of all sequences in a family. Cmbuild, cmsearch, and cmalign are Infernal programs used for building CMs, searching sequence database, and aligning sequences to the CMs, respectively.
The CM is then used to search for related sequences in a sequence library called RFAMSEQ composed of non-redundant complete genomes (Kalvari et al., 2017) using Infernal’s cmsearch program. Each hit to the model is given a score, and the Rfam curator chooses a family-specific threshold score called the gathering threshold, or GA cutoff, that separates believed homologs from non-homologous sequences. The sequences above the GA cutoff are aligned to the CM using the cmalign program and are part of the Full alignment. In order to make the Seed alignment more diverse, the curator may add some sequences found in the Full to the Seed alignment and repeat the process of searching for new sequences. Further information about constructing RNA families can be found in Unit 12.13 (Barquist, Burge, & Gardner, 2016).
Multiple sequence alignments of ncRNA families have a number of uses. The protocols presented here demonstrate that alignments are the starting point for covariance model–based homolog detection, but they can also be used to learn about evolutionary relationships within a sequence family. Alignments are also useful for training and testing new ncRNA alignment and detection algorithms.
Critical Parameters / Troubleshooting
Finding hits with scores below the GA thresholds
It is possible that the online single sequence and batch search described in the Basic Protocol 3, as well as the cmscan and cmsearch programs described in the Alternate Protocol, will return search results with zero hits. This indicates that no hits to any of the query models with scores above the Rfam GA cutoffs have been found in the target sequence or sequences. As described above, the GA cutoffs are set as a score above the highest scoring false positive observed in a search in the large RFAMSEQ database, and are meant to be fairly strict, such that false positive hits in subsequent searches with the models are minimized. However, some true homologous RNA regions will have scores that are below the GA cutoff, and these may be the top scoring hits in other databases, especially ones that are considerably smaller than RFAMSEQ, such as a single bacterial or archaeal genome. To find such hits, the Alternate Protocol must be followed, but using options different than those specified above. In both the cmsearch and cmscan examples above, the --cut_ga option is used to enforce the GA cutoffs, but if this option is not used then all hits with E-values below 10 will be reported. Alternatively, the --cut_ga option can be replaced with -E <Y> to report all hits with an E-value of <Y> or below. As explained above in Guidelines for Understanding Results, E-values are dependent on the database size, and it may desirable to additionally use the -Z <X> option as well to specify the database size.
Another way to potentially increase the sensitivity of cmsearch and cmscan is to change the internal filter thresholds used by the programs. These thresholds control how much of the sequence database survives Infernal’s relatively fast, sequence-based filters and is evaluated using the more expensive sequence- and structure-based CM scoring algorithms. The Basic Protocol website and batch search as well as the Alternate Protocol cmsearch and cmscan examples above use the strictest possible filtering level (enforced with the --rfam option to cmsearch and cmscan). This option can be removed for less stringent filtering, but will result in slower searches. The level of filtering will be selected automatically based on database search size if --rfam is removed, or can be set to the various preset levels of filtering with the --rfam, --mid, --nohmm, or --max options, listed in order of most to least strict. With --nohmm and --max search times may be prohibitively slow, and it is recommended to test a search on a fraction of the database first to estimate the time required for the full database of interest.
Search time and further reference
Even with the strictest filters (--rfam), the cmsearch and cmscan programs are computationally expensive and require roughly 15 minutes per Mb of sequence (if all Rfam families are used as queries) when run on a single CPU (Nawrocki et. al, 2015). Typically, cmscan is 2 to 5 times slower than cmsearch, depending on the number and lengths of the sequences in the target database, due to differences in its implementation.
Infernal includes additional programs and command-line options not discussed here that are described in the Infernal user’s guide. The guide also includes more details on the filtering thresholds, E-values, and a tutorial with additional examples.
Pseudogenes
A serious limitation of Infernal is its inability to distinguish RNA pseudogenes from functional RNA genes. Infernal’s scoring system is designed to recognize homology by sequence similarity, and some RNA pseudogenes are similar enough to the functional RNA genes they derive from to score above Rfam GA thresholds and/or with statistically significant E-values. Signals, such as frameshift mutations, or loss of start and stop codons, which are helpful to identify protein coding gene pseudogenes are of no help in the context of RNA genes.
While relatively rare in bacterial and archaeal sequences, RNA pseudogenes are more prevalent in eukaryotic sequences, especially in large vertebrate genomes. For example, some SINE repeat elements are derived from SRP RNA and transfer RNA genes and can number in the millions in vertebrate genomes. These elements will often receive high Infernal scores to the models of the genes they derive from, and currently, there are no effective computational tools for distinguishing them from real genes. For this reason, when interpreting Infernal/Rfam results for eukaryotic sequences from genomes in which RNA pseudogenes are common, extra care must be taken to distinguish hits to functional RNAs from hits to pseudogenes. One method for validation of real RNAs is to require high sequence identity to a previously annotated functional RNA sequence from a closely related organism, when possible.
Supplementary Material
Significance Statement.
Rfam (http://rfam.org) is a database of non-coding RNA families in which each family is represented by a multiple sequence alignment, a consensus secondary structure, and a covariance model. Using a combination of manual and literature-based curation and a custom software pipeline, Rfam converts descriptions of RNA families found in the scientific literature into computational models that can be used to annotate RNAs belonging to these families in any DNA or RNA sequence. Valuable research outputs that are often locked up in figures and supplementary files are encapsulated in Rfam entries and made accessible through the Rfam website. This unit provides an overview of how to navigate the Rfam website, and how to annotate sequences with Rfam families using the Infernal software.
ACKNOWLEDGEMENT
We would like to thank Sam Griffiths-Jones (University of Manchester) who wrote the original version of this protocol in 2005, as well as all previous Rfam team members. We also thank Sean Eddy (Harvard University) for developing the Infernal software and Elena Rivas (Harvard University) for developing R-Scape. This work was supported by Biotechnology and Biological Sciences Research Council (BBSRC) [BB/M011690/1] and the Intramural Research Program of the NIH National Library of Medicine.
APPENDIX: Rfam ALIGNMENT FORMAT
Rfam Seed and Full alignments (see Commentary) are distributed in blocked Stockholm format on the Rfam FTP archive (see Internet Resources). Stockholm format is described at http://sonnhammer.sbc.su.se/Stockholm.html.
Alignment lines take the form:
<EMBL accession>/<start>-<end> <aligned residues>
Stockholm format also allows annotation of the alignment, sequences, and residues to be embedded in the alignment by use of special tags (see Table 12.5.1):
#=GF <feature> <per file annotation>
#=GC <feature> <per column annotation>
#=GS <sequence name> <feature> <per sequence annotation>
#=GR <sequence name> <feature> <per residue annotation>
Table 12.5.1.
Annotation features found in Rfam Seed alignments
| Field name | Field tag | Description |
|---|---|---|
| Accession number | AC | Stable accession number for each Rfam family. It is of the form RFxxxxx, where x is a digit. |
| Identifier | ID | Short and meaningful name for the family. It is not necessarily stable between releases. |
| Description | DE | A one-line description of the family. |
| Author | AU | Author of the entry |
| Seed source | SE | Source of the Seed alignment. May be author, PubMed ID, literature reference, or alignment method. |
| Secondary structure | SS | Source of the secondary structure mark-up. May be Predicted specifying the software used or Published showing the PubMed ID of the publication where the structure was obtained. |
| Gathering cutoff | GA | Bit score cutoff chosen to determine whether a match is a real member of the family. |
| Trusted cutoff | TC | Bit score of the lowest scoring match above the GA threshold. |
| Noise cutoff | NC | Bit score of the highest scoring match below the GA threshold. |
| Type | TP | RNA type (controlled vocabulary) must be one
of the following: - Gene - antisense - antitoxin - CRISPR - microRNA - lncRNA - rRNA - ribozyme - sRNA - tRNA - snRNA -snoRNA - CD-box - HACA-box - scaRNA - splicing - Intron - Cis-regulatory element - IRES - frameshift element - leader - riboswitch - thermoregulator. |
| Build method | BM | Infernal command line parameters used to construct the family. |
| Database cross references | DR | GO: Gene Ontology terms SO: Sequence Ontology terms Optional: miRBase identifier, snOPY identifier, database URL, or other cross reference. |
| Comment | CC | Free text comment. |
| WK | Link to a Wikipedia article describing the family. | |
| Sequence count | SQ | Number of sequences in the alignment. |
| CL | Rfam clan (if a family is a member of a clan). |
The full and up-to-date list is available in the USERMAN file from the Rfam FTP archive (see Internet Resources).
Note that the nested relationship of these brackets does not allow the annotation of pseudoknots. These are marked-up in some alignments using capital letters in place of open brackets, and lowercase letters in place of closed brackets. The brackets <>, (), {}, and [] are all valid base pairs and are used in this order from inside to outside. Unpaired bases can be represented by the symbols shown in Table 12.5.2. Presently the Seed alignments are marked up with all base pairs as <>, and all gaps as “.”. The SS_cons line for the Full alignments is generated automatically by the Infernal software.
Table 12.5.2.
Symbols representing unpaired bases in the SS-cons line
| Symbol | Meaning |
|---|---|
| . | 5’ or 3’ terminal unpaired |
| , | Single strand between helices |
| - | Hairpin loop |
| — | Single stranded bulge |
| : | Insert state |
| ~ | Local insert state |
Footnotes
INTERNET RESOURCES
Rfam database: http://rfam.org
Rfam help and documentation: http://rfam.org/help
Rfam FTP archive: http://ftp.ebi.ac.uk/pub/databases/Rfam/
Infernal homepage and User’s Guide: http://eddylab.org/infernal
LITERATURE CITED
- Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, … Flicek P (2017). Ensembl 2017. Nucleic Acids Research, 45(D1), D635–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barquist L, Burge SW, & Gardner PP (2016). Studying RNA Homology and Conservation with Infernal: From Single Sequences to RNA Families. Current Protocols in Bioinformatics / Editorial Board, Andreas D. Baxevanis … [et Al.], 54, 12.13.1–12.13.25. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes building RNA families using Infernal and introduces related tools and workflows.
- Bernhart SH, Hofacker IL, Will S, Gruber AR, & Stadler PF (2008). RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics, 9, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech TR, & Steitz JA (2014). The Noncoding RNA Revolution—Trashing Old Rules to Forge New Ones. Cell, 157(1), 77–94. [DOI] [PubMed] [Google Scholar]
- Federhen S (2012). The NCBI Taxonomy database. Nucleic Acids Research, 40(Database issue), D136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, … Bateman A (2011). Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Research, 39(suppl_1), D141–D145. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the usage of Wikipedia for creating family descriptions and the introduction of Rfam clans.
- Gardner PP, & Eldai H (2015). Annotating RNA motifs in sequences and alignments. Nucleic Acids Research, 43(2), 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, & Eddy SR (2003). Rfam: an RNA family database. Nucleic Acids Research, 31(1), 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the first version of Rfam.
- Kalvari I, Argasinska J, Quinones-Olvera N, Nawrocki EP, Rivas E, Eddy SR, … Petrov AI (2017). Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Research. 10.1093/nar/gkx1038 [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes Rfam release 13.0 that introduced genome-centric sequence database
- Kortmann J, & Narberhaus F (2012). Bacterial RNA thermometers: molecular zippers and switches. Nature Reviews . Microbiology, 10(4), 255–265. [DOI] [PubMed] [Google Scholar]
- Lai D, Proctor JR, Zhu JYA, & Meyer IM (2012). R-CHIE: a web server and R package for visualizing RNA secondary structures. Nucleic Acids Research, 40(12), e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCown PJ, Corbino KA, Stav S, Sherlock ME, & Breaker RR (2017). Riboswitch diversity and distribution. RNA , 23(7), 995–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, Eddy SR, … Finn RD (2015). Rfam 12.0: updates to the RNA families database. Nucleic Acids Research, 43(Database issue), D130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes Rfam release 12.0 including the addition of RNA motifs to Rfam and migration to Infernal 1.1.
- Nawrocki EP, & Eddy SR (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics , 29(22), 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes using Infernal for annotating genomes with non-coding RNAs using the Rfam database.
- Quinlan AR (2014). BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Current Protocols in Bioinformatics / Editorial Board, Andreas D . Baxevanis … [et al. ], 47, 11.12.1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas E, Clements J, & Eddy SR (2017). A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nature Methods, 14(1), 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The RNAcentral Consortium. (2017). RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic Acids Research, 45(D1), D128–D134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Lünse CE, & Corbino KA (2017). Detection of 224 candidate structured RNAs by comparative analysis of specific subsets of intergenic regions. Nucleic Acids. Retrieved from https://academic.oup.com/nar/article/4080188 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.