

Abstract
Background:
Approximately 13% of African-American individuals carry two copies of the APOL1 risk alleles G1 or G2, which are associated with 1.5-2.5 fold increased risk of chronic kidney disease (CKD). There have been conflicting reports as to whether an association exists between APOL1 risk alleles and cardiovascular disease, independent of the effects of APOL1 on kidney disease. We sought to test the association of APOL1 G1/G2 alleles with coronary artery disease (CAD), peripheral artery disease (PAD), and stroke among African American individuals in the Million Veteran Program (MVP).
Methods:
We performed a time-to-event analysis of retrospective electronic health record (EHR) data using Cox proportional hazard and competing risks Fine and Gray sub-distribution hazard models. The primary exposure was APOL1 risk allele status. The primary outcome was incident CAD amongst individuals without CKD during the 12.5 year follow up period. Separately we analyzed the cross-sectional association of APOL1 risk allele status with lipid traits and 115 cardiovascular diseases using phenome-wide association.
Results:
Among 30,903 African American MVP participants, 3,941 (13%) carried the two APOL1 risk allele high-risk genotype. Individuals with normal kidney function at baseline with two risk alleles had slightly higher risk of developing CAD compared to those with no risk alleles (Hazard Ratio (HR): 1.11, 95% Confidence Interval (CI): 1.01-1.21, p=0.039). Similarly, modest associations were identified with incident stroke (HR: 1.20, 95% CI: 1.05-1.36, p=0.007) and PAD (HR: 1.15, 95% CI:1.01-1.29, p=0.031). When modeling both cardiovascular and renal outcomes, APOL1 was strongly associated with incident renal disease, while no significant association with the cardiovascular disease endpoints could be detected. Cardiovascular phenome-wide association analyses did not identify additional significant associations with cardiovascular disease subsets.
Conclusions:
APOL1 risk variants display a modest association with cardiovascular disease and this association is likely mediated by the known APOL1 association with CKD.
Keywords: Apolipoprotein L, genetics, cardiovascular disease, chronic kidney disease
Introduction
African-American individuals with kidney disease experience a 3-5 fold increased risk of cardiovascular disease (CVD) compared to Caucasians.1 While a subset of this risk is associated with differences in traditional risk factors and sociologic disparities, a significant excess risk for CVD in general and coronary artery disease (CAD) in particular remains even after accounting for these factors.2–4
Genome wide association analyses have revealed that a substantial fraction of the previously unexplained disparity in the incidence of end stage renal disease between African Americans and individuals of European ancestry can be explained by two alleles in apolipoprotein L1 (APOL1), a component of High Density Lipoprotein cholesterol (HDL-C). The APOL1 G1 allele consists of two missense variants in very high linkage disequilibrium (APOL1 p.S342G, rs73885319; APOL1 p.I384M; rs6091014) and the G2 allele is a two amino acid deletion (APOL1 p.delN388/Y389, rs60910145). As kidney disease confers greater risk for CVD in African Americans than individuals of European descent, recent reports have sought to evaluate if APOL1 risk alleles also confer increased risk of CVD. These studies have reported conflicting results (summarized in Supplemental Table 1). Despite initial reports of a 2-fold increased risk of CVD in APOL1 two-risk allele carriers in the Jackson Heart Study,5 subsequent analyses failed to identify any association of APOL1 risk allele status with CVD6–10 or reported modest associations11,12.
We sought to determine the association between APOL1 risk alleles and incident CAD, Peripheral Artery Disease (PAD) and stroke among 30,903 African American participants in the Million Veteran Program (MVP). We utilized the longitudinal medical records for MVP participants to temporally censor individuals upon developing kidney disease, thereby enabling us to adjust for the differing rates with which APOL1 variant carriers develop kidney disease. As APOL1 is a component of HDL-c, we also considered the association between APOL1 and lipid levels as a possible mechanistic link between APOL1 and atherosclerosis. Lastly, we used phenome-wide association to test the association between APOL1 and all cardiovascular conditions in an unbiased way.
Methods
Study population
The MVP is a Department of Veterans Affairs (VA) health care system observational cohort study and mega-scale biobank.13 The study was approved by the VA central institutional review board (IRB) and all participants provided informed consent. The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure without approval of the VA central IRB. Recruitment began in 2011 and is ongoing with more than 700,000 veterans accrued to date. Data was collected from participants using questionnaires and the VA electronic health record (EHR). A blood sample was collected for genomic studies at time of enrollment. For the present study, we included 30,903 African-American MVP participants who were directly genotyped for APOL1 risk variants and who had two or more clinical encounters at the VA healthcare system in the two years prior to the analytic cohort entry date of January 1, 2005, the start of our time-to-event analysis, suggesting that these individuals were active users of the VA healthcare system.
African American ancestry was defined using a combination of self-reported race and ethnicity and principal components of ancestry using previously described methods.14
Genotyping
The two APOL1 risk allele variants G1 (rs73885319 p.S342G; rs60910145 p.I384M) and G2 (rs71785313, a six base pair deletion that removes amino acids N388 and Y389) were directly genotyped on the Affymetrix Axiom Biobank Array platform on DNA that was extracted from whole blood (Supplemental Figure 1). Individuals who carried G1S342G alleles (the functional missense variant in the G1 allele) in the absence of the G1I384M variant were considered to be G1 allele carriers. Participants were defined as two risk allele carriers if they were homozygotes for G1/G1, homozygotes for G2/G2, or compound heterozygotes for G1/G2.
Study design, follow up and covariates
The study was designed as a time-to-event analysis of retrospective EHR data using a cohort of African American Veterans, 18 years and older, who had enrolled in MVP and used the VA as their primary source of care. Baseline covariates were assessed during the 730 days prior to cohort entry, using the closest value to January 1, 2005, for time varying covariates with multiple measurements.
Patients with kidney disease during the baseline period were excluded from the primary longitudinal analyses. Kidney disease was defined using the Kidney Disease: Improving Global Outcomes (KDIGO) definition of CKD, which defines CKD as patients having an estimated glomerular filtration rate (eGFR) < 60 ml/min/1.73 m2 on two separate measurements 90 days apart or on chronic renal replacement therapy (RRT), or have a history of kidney transplant15. RRT and kidney transplant were defined using International Classification of Disease 9th and 10th revisions (ICD-9-CM, ICD-10) diagnosis codes and Current Procedural Terminology (CPT) codes (Supplemental Table 2). Estimated glomerular filtration rate was calculated using the CKD Epidemiology Collaboration (CKD-EPI) equation16.
Individuals with prevalent CAD, PAD or stroke at the time of the analytic cohort start date of January 1, 2005 were excluded from each of the three analyses. These included all data available in the VA EHR or in the fee-for-service data documenting care paid for by the VA but provided outside of the VA.
Incident outcomes were collected from January 1, 2005, to July 1, 2017, from the national VA Corporate Data Warehouse, which aggregates data from all VA facilities. Patients were followed until they either experienced an outcome of interest, developed incident CKD/ESRD (as defined above), or until the date of their last encounter with the VA system prior to July 1, 2017.
The primary clinical endpoint, CAD, was defined based on ICD-9-CM, ICD-10 and CPT codes using the method described by Dewey and colleages17. CAD events were defined as individuals who had either an inpatient admission with a primary diagnosis or procedure code of CAD, a combination of CAD associated inpatient or outpatient encounters on two or more distinct days, or a coronary revascularization as of July 1, 2017 noted in the longitudinal VA EHR or in the fee-for-service data (Supplemental Table 2).
A similar combination of diagnosis and procedure codes was used to derive PAD definitions based on earlier work by Arya and colleagues18. In particular, PAD was defined as 1 primary inpatient diagnosis code; 2 diagnosis codes; 1 dx code and a procedure; 1 dx code and 2 visit to vascular surgery or 1 dx code and 2 ABI measurements in 14 months (Supplemental Table 2). Stroke was defined using the algorithm described by Tirschwell and Longstreth19.
For analyses of lipid traits, we extracted the maximum LDL cholesterol and triglyceride values and the minimum HDL cholesterol value present in the EHR to minimize the impact of lipid lowering medication. This approach was consistent with prior lipid genome-wide analyses that included longitudinal EHR data.20
All diagnosis codes for participants present in the VA EHR through July 1, 2017 were used to perform cardiovascular disease phenome wide association testing. ICD-10-CM codes were back mapped to ICD-9 using a combination of the Center for Medicare and Medicaid Services General Equivalency Mappings21 and manual curation prior to converting to phecodes22.
Power calculation
Previous studies estimated the odds ratio for CVD associated with carrying two APOL1 risk alleles in the range of 1.04 to 2.2, with a median of 1.24 (Supplemental Table 1). Given our sample size and study specific risk-allele frequencies, we had 80% power to detect a hazard ratio (HR) of 1.16 for the effect of two APOL1 risk alleles on CAD risk with a two-sided alpha of 0.05.
Statistical analyses
We calculated descriptive statistics for participant characteristics in the overall population and among those with and without APOL1 risk alleles. Tests of statistical significance for differences in population characteristics were conducted using the Wilcoxon-test for continuous measures and the Fisher exact test for count data.
A fully adjusted Cox proportional hazards model between APOL1 risk variants and incident CAD was the primary pre-specified analysis with stroke and PAD being secondary endpoints. Individuals with prevalent CAD or prevalent kidney disease (CKD or end stage renal disease (ESRD)) were excluded from these analyses. Individuals were marked as completing follow-up at the time of the clinical endpoint or upon developing kidney disease (CKD/ESRD), or censored at the time of their last encounter in the VA system prior to July 1, 2017. The proportional hazards model adjusted for baseline covariates of: age, sex, diabetes, systolic blood pressure, diastolic blood pressure, body mass index, LDL cholesterol, HDL cholesterol, triglycerides, the use of a statin, antihypertensive medication, aspirin, smoking status and baseline eGFR. In addition, the first 5 ancestry principal components were included in the proportional hazard model to further control for potential ancestry stratification. A p-value less than 0.05 was considered statistically significant.
Percent missing was computed for each covariate and missing data patterns were examined using cluster analysis of variables usually missing together. Observed patterns were suggestive of data being missing at random.
Multiple imputation for the missing values was performed using the aregImpute algorithm in the Hmisc package in R (version 3.2). Details of the aregImpute algorithm have been described elsewhere.23 The algorithm accounts for all aspects of uncertainty in the imputations by boot-strapping to approximate the process of drawing predicted values from a full Bayesian predictive distribution. Different bootstrap resamples are used for each of the multiple imputations. A flexible additive model is fit on a sample with replacement from the original data and this model is used to predict all of the original missing and non-missing values for the target variable, then the imputation models are run. In the imputation model, linearity is assumed for target variables (variables being imputed) while continuous predictors on the right-hand side of the model are transformed using restricted cubic splines with 5 knots. The algorithm uses predictive mean matching with weighted probability sampling of donors to fill-in the missing data.
Five imputations were performed, creating 5 complete datasets. The regression model (comprising all covariates included in the imputation model) was fit on each complete dataset and the regression coefficients were averaged over the multiple imputations. The distributions of measured and imputed values were highly comparable (Supplemental Figure 2) and the variance inflation due to the missing variables was modest (Supplemental Table 3).
Several sensitivity analyses were performed. First, a series of minimal models were created using the cohort described above with progressive adjustment for covariates including (1) a basic model adjusting for only age and sex and principle components and (2) further adjustment for cardiovascular disease covariates diabetes, SBP, DBP, BMI, LDL, HDL, TG, statin, anti-hypertensive medication, aspirin, tobacco use.
Second, a mediation analysis was performed including all individuals regardless of prevalent kidney disease status and progressive adjustment for (1) a basic model of age, sex and principle components, (2) adjusting for cardiovascular disease risk factors (3) adjusting for cardiovascular disease risk factors and baseline eGFR and (4) adjusting for cardiovascular disease risk factors, eGFR and incident CKD.
Third, competing-risks analyses for the outcomes of interest (CAD, PAD and stroke) were performed using Fine and Gray sub-distribution hazard (SDH) models with CKD modeled as the competing event. The Fine and Gray method — based on cumulative incidence functions — fits a proportional hazards model for the sub-distribution hazard and effect estimates for the outcome of interest (in the presence of competing events) are presented as sub-distribution hazard ratios. Competing-risks analyses were conducted before and after performing multiple imputation for missing data using the MICE (multivariate imputation by chained equations) algorithm (mice package in R). Five imputations were performed, creating 5 complete datasets; the regression model was fit on each complete dataset and the regression coefficients were averaged over the multiple imputations.
We conducted a cardiovascular disease phenome-wide association study (PheWAS) to evaluate associations between APOL1 and CVD in an unbiased manner in individuals who had no evidence of kidney disease at baseline or during the follow up period. Broadly, the PheWAS included 115 cardiovascular conditions (observed in at least 40 individuals) including arrhythmias, cardiomyopathy, cerebrovascular disease, ischemic heart disease, vascular disease and valvular disease. The PheWAS models tested for association with APOL1 risk alleles in a recessive model adjusted for age and sex using the R PheWAS software package24adjusting for age, sex, and the first 5 principal components of ancestry. With a Bonferroni correction for 115 cardiovascular phenotype codes, a p-value of <4.3 × 10−4 was considered significant.
All analyses were conducted using R 3.2 in the VA GenISIS computational environment.13
Results
MVP Population characteristics
There were 30,903 African American MVP participants genotyped for APOL1 risk variants and active in the VA healthcare system prior to the cohort entry date in 2005 (Table 1). On average 12 years (± sd 0.7 years) of VA EHR follow up data were available. In the two years prior to the cohort entry date, the study participants had on average 34 encounters with the VA healthcare system.
Table 1.
Study population characteristics stratified by APOL1 risk allele status
| APOL1 risk alleles | ||||
|---|---|---|---|---|
| Baseline clinical characteristics | 0 | 1 | 2 | P* |
| N | 12510 (40%) | 14452 (47%) | 3941 (13%) | - |
| VA Clinical Encounters in baseline period, mean (sd) | 34 (46) | 33 (44) | 34 (47) | 0.73 |
| Follow up time, years, mean (sd) | 12 (0.7) | 12 (0.7) | 12 (0.8) | 0.93 |
| Age, n (%) | 52 (10) | 52 (10) | 52 (10) | 0.09 |
| Male, n (%) | 11096 (89%) | 12823 (89%) | 3477 (88%) | 0.13 |
| Diabetes, n (%) | 3078 (25%) | 3657 (25%) | 1011 (26%) | 0.07 |
| Hypertension, n (%) | 7577 (61%) | 8820 (61%) | 2475 (63%) | 0.004 |
| Tobacco use, n (%) | 9595 (77%) | 11105 (77%) | 2993 (76%) | 0.05 |
| Aspirin, n (%) | 1620 (13%) | 1810 (13%) | 472 (12%) | 0.04 |
| Statin, n (%) | 2934 (24%) | 3478 (24%) | 965 (25%) | 0.07 |
| Anti-hypertensive medication, n (%) | 6112 (49%) | 7249 (50%) | 2029 (52%) | 0.006 |
| Systolic BP mmHg, mean (sd) | 134 (17) | 134 (18) | 134 (17) | 0.64 |
| Diastolic BP mmHg, mean (sd) | 79 (11) | 79 (11) | 79 (11) | 0.06 |
| Body mass index, mean (sd) | 30 (6) | 30 (6) | 31 (6) | 0.03 |
| LDL-C mg/dL, mean (sd) | 112 (36) | 113 (37) | 113 (36) | 0.39 |
| HDL-C mg/dL, mean (sd) | 48 (16) | 48 (16) | 48 (15) | 0.84 |
| Triglyceride-C mg/dL, mean (sd) | 137 (125) | 137 (134) | 141 (139) | 0.02 |
| eGFR mL/min/1.73 m2, mean (sd) | 90 (21) | 90 (21) | 86 (24) | <0.001 |
| Prevalent Disease | ||||
| Prevalent CAD, n (%) | 1380 (11%) | 1640 (11%) | 461 (12%) | 0.07 |
| Prevalent Stroke, n (%) | 358 (3%) | 388 (3%) | 116 (3%) | 0.14 |
| Prevalent PAD, n (%) | 451 (4%) | 520 (4%) | 145 (4%) | 0.10 |
| Prevalent CKD, n (%) | 441 (4%) | 582 (4%) | 266 (7%) | <0.001 |
| Prevalent ESRD, n (%) | 44 (0%) | 53 (0%) | 88 (2%) | <0.001 |
| Incident Disease | ||||
| Follow up time, years, mean (sd) | 12 (0.7) | 12 (0.7) | 12 (0.8) | 0.93 |
| Incident CAD, n (%) | 1806 (14%) | 2155 (15%) | 604 (15%) | 0.06 |
| Incident Stroke, n (%) | 908 (7%) | 1075 (7%) | 319 (8%) | 0.01 |
| Incident PAD, n (%) | 1026 (8%) | 1216 (8%) | 364 (9%) | 0.02 |
| Incident CKD, n (%) | 4376 (35%) | 5167 (36%) | 1640 (42%) | <0.001 |
| Incident ESRD, n (%) | 388 (3%) | 518 (4%) | 347 (9%) | <0.001 |
P for comparison of 2 APOL1 risk alleles compared to 0 or 1 APOL1 risk alleles by Fisher exact test for proportions and Wilcoxon test for continuous variables. sd, standard deviation; BP, blood pressure; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; eGFR, estimated glomerular filtration rate; CAD, coronary artery disease; PAD, peripheral artery disease; CKD, chronic kidney disease; ESRD, end-stage renal disease
Among these 30,903 participants, 3941 (12.8%) carried two APOL1 risk alleles, 14,452 participants (46.7%) carried one APOL1 risk allele, and 12,510 participants (40.4%) carried no APOL1 risk alleles. This is identical to the proportion of two APOL1 risk allele carriers reported in the Atherosclerosis Risk in Communities study (12.8%)6, and highly comparable to the frequency of two APOL1 risk allele reported in other population based studies including the Dallas Heart Study (13.2%)25 and Multi-Ethnic Study of Atherosclerosis Study (12.1%)9. The G1 and G2 genotypes were in Hardy-Weinberg equilibrium. 88% of the study population was male. The mean age of the study population was 52 years as of the cohort entry date. Diabetes was present in 26% at baseline and an additional 30% developed diabetes in the follow up period. Individuals with two APOL1 risk alleles were observed to have a slightly higher prevalence of hypertension (63% vs 61%, p= 4×10−3). A greater proportion of individuals with two APOL1 risk variants had prevalent CKD relative to non-carriers (7% vs 4%, p= 1.4 × 10−16) and a greater proportion developed incident CKD over the follow up period (42% vs 35%, p=1.3 × 10−14, Table 1).
APOL1 and CAD, Stroke, and PAD
There were 26,486 individuals who had no evidence of CKD or CAD at baseline and were eligible for the primary analysis. Amongst these individuals, those with two risk alleles had slightly higher risk of developing CAD compared to those with no risk alleles in fully adjusted Cox proportional hazard models (HR: 1.11 95% CI: 1.01-1.21, p=0.039, Figure 1 and 2, Supplemental Table 4). Similarly, weak associations were identified with incident stroke (HR: 1.20 95% CI: 1.05-1.36, p=0.007) and PAD (HR: 1.15 95% CI:1.01-1.29) among the 28,788 and 28,609 individuals without either CKD or those conditions at baseline, respectively. No evidence for association was observed among individuals with a single risk allele to either the primary or either of the secondary endpoints.
Figure 1. Association of APOL1 and incident CAD, Stroke and PAD.
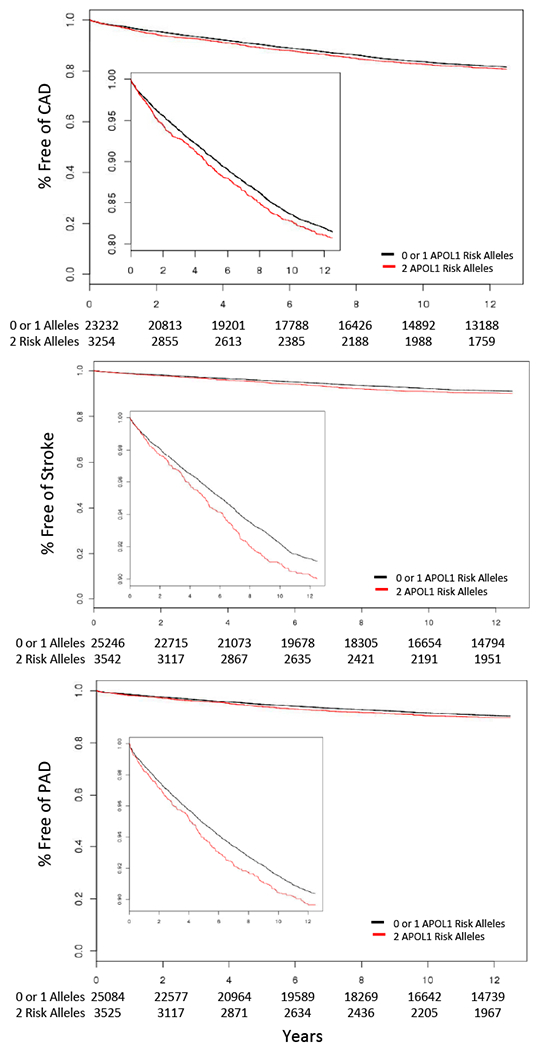
Kaplan Meier plots depicting disease free survival amongst individuals free of kidney disease at baseline over the 12.5 year follow up period stratified by APOL1 2 risk allele status. Data is censored at last VA clinical encounter or upon developing chronic kidney disease. Inset plots displays the same data on an expanded y-axis. Risk-sum tables for each phenotype are displayed below the x axis. CAD, coronary artery disease; PAD, peripheral artery disease.
Figure 2. Association of APOL1 and incident CAD, Stroke and PAD.
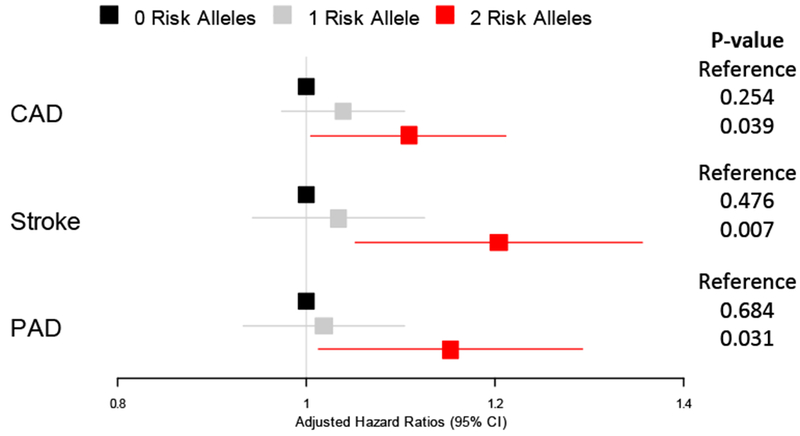
Forest plot representing hazard ratios of disease-free survival stratified by APOL1 risk allele carrier status in fully adjusted Cox proportional hazard models. CAD, coronary artery disease; PAD, peripheral artery disease.
In analyses that disaggregated the high-risk alleles by composition, G1/G1 homozygote risk allele carriers and G1/G2 compound heterozygote risk allele carriers both had increased risk of CAD (HR:1.13, 95% CI: 0.97-1.29, p=0.11, and HR: 1.16, 95% CI: 1.02-1.3, p=0.02, respectively). However, G2/G2 homozygote carriers have no association with CAD (HR: 0.89, 95% CI: 0.7-1.0, p=0.33).
To ensure that these findings were not an artifact of covariate selection, we performed several sensitivity analyses with progressive covariate adjustment which were consistent with the results of the fully adjusted models (Supplemental Table 5–7).
We also performed a mediation analysis including all individuals regardless of kidney disease status at baseline for our three CVD outcomes (Supplemental table 8–10). In this series of models, progressive adjustment for cardiovascular and kidney disease risk factors eliminated the association between APOL1 genotype and disease.
We further sought to delineate the renal and cardiovascular effects of APOL1 genotype using a competing risks Fine and Gray sub-distribution hazard model. Consistent with the cause specific models described above, when modeling both cardiovascular and renal outcomes, APOL1 was strongly associated with incident renal disease, while no significant association with the cardiovascular disease endpoints could be detected (Figure 3, Supplemental Figure 3,4, Supplemental Table 11).
Figure 3. Cumulative incidence function estimates from competing risks modeling of CAD and CKD/ESRD outcomes.
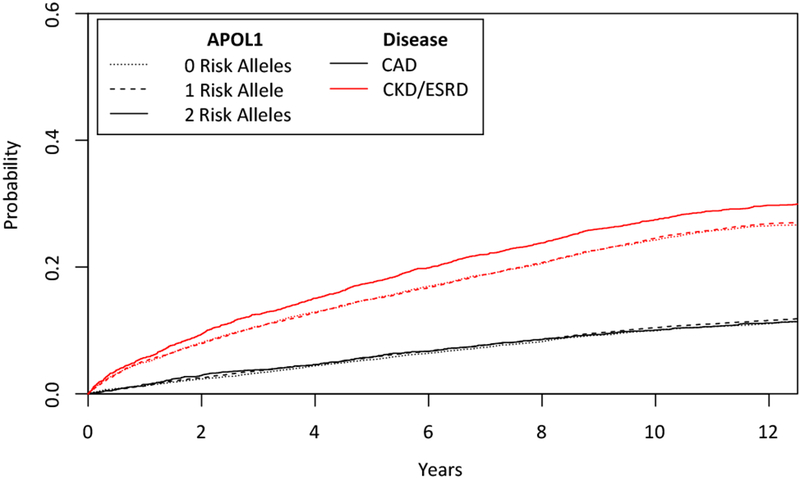
Test for equality across APOL1 risk allele groups CAD: p=0.56. CKD/ESRD p=5.1 × 10−4. CAD, coronary artery disease; PAD, peripheral artery disease; ESRD, end-stage renal disease
We performed stratified analyses by sex and baseline diabetes status to evaluate if there were subgroup differences in outcomes. Although both sub-group analyses were underpowered, the consistency of hazard ratios between subgroups suggested that there was no significant heterogeneity by sex or diabetes status (Supplemental Tables 12–13)
APOL1 and Lipids
APOL1 two risk allele status was associated with modestly increased LDL cholesterol (effect estimate 0.02 for 2 allele carrier status on log2 LDL, 95% CI 0.005-0.035, p=0.008) and increased HDL cholesterol (effect estimate 0.02 for 2 allele carrier status on log2 HDL, 95% CI 0.005-0.038, p= 0.012, Supplemental Table 14). No significant associations were observed between two APOL1 risk allele status and triglycerides (effect estimate 0.01 for 2 allele carrier status on log2 Triglycerides, 95% CI: −0.01, 0.03, p=0.56). Further stratifying the association with LDL cholesterol by APOL1 genotype revealed that the association with increased LDL cholesterol was only present in the G1/G1 and G1/G2 group (p=0.02 and p=0.03 respectively).
APOL1 and cardiovascular phenome-wide association study
We tested the associations of APOL1 two-risk allele status and 115 diverse cardiovascular conditions in 19,701 individuals with no evidence of kidney disease at baseline or during follow-up to evaluate potential APOL1 associations with additional CVD phenotypes in individuals without CKD (Figure 4, Supplemental Table 15). No conditions exceeded the Bonferroni-adjusted significance threshold. Of note, we did not find evidence for association of APOL1 and heart failure, angina or chronic venous insufficiency (Supplemental Table 16).
Figure 4. Cardiovascular phenome-wide association of APOL1 two risk allele status in individuals free of kidney disease.
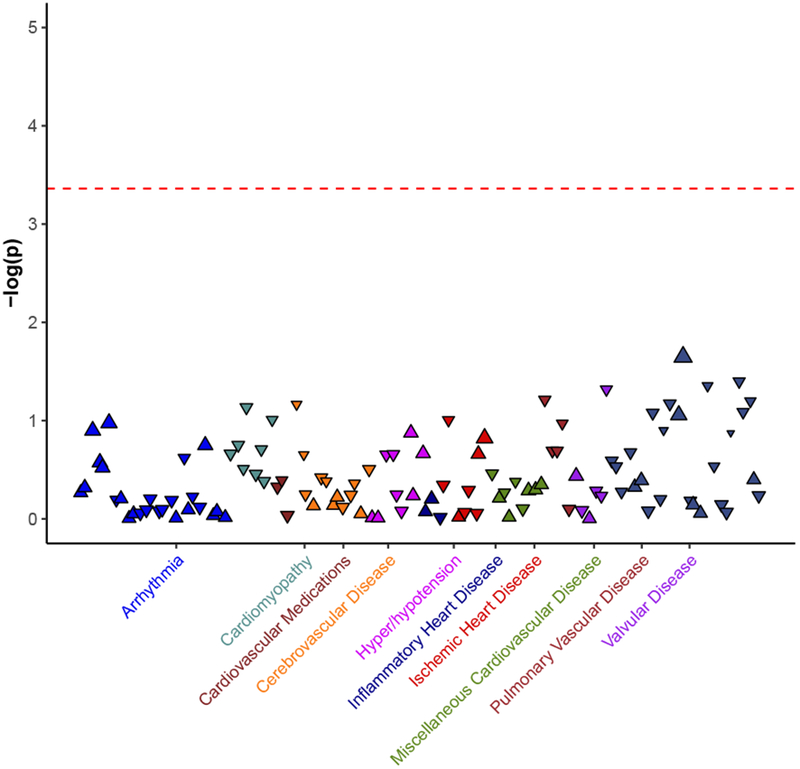
Phenome-wide association did not identify any significant cardiovascular disease associations with APOL1. Dashed horizontal line represents experiment wide significant p-value adjusted for 115 cardiovascular phenotypes (p<4 × 10−4). Each triangle represents a subset of CVD disease as defined by ICD-9 codes, colored by cardiovascular disease category.
Discussion
We evaluated the associations between APOL1 risk alleles and heart and vascular disease in a sample of approximately 30,000 African American veterans. Overall, we found a modest association between the APOL1 2 risk allele genotype and CAD, PAD and stroke in individuals without CKD. From these results we infer that APOL1 does not represent a strong independent risk factor for cardiovascular disease.
Interestingly, analyses stratified by risk allele suggested that the G1 and G2 alleles have differential effects on CAD risk, with G1 having an increased risk in the homozygous state and G2 having a neutral or potentially protective effect. The LDL cholesterol profile is consistent with this allelic heterogeneity, and only G1 homozygous allele status being associated with increased LDL cholesterol levels (Supplemental Table 14). Consistent with several prior reports5,26,27 but in contrast to others28, APOL1 risk allele status was associated with higher HDL cholesterol in fully adjusted models (Supplemental Table 3). Although numerous studies have found an association between low levels of HDL cholesterol with increased CVD risk, recently the emphasis has shifted from circulating levels to functionality of HDL particles as a better predictor of CVD29–31. Whether APOL1 risk variants affect HDL particle function remains to be determined.
The impact of APOL1 on cardiovascular traits beyond CAD has been evaluated by a number of investigators5,9,32,33. Cardiovascular PheWAS provides a method for exploring this diversity in an unbiased fashion. Although diverse associations have been reported for APOL1 and cardiovascular disease traits, we do not detect such associations in our unbiased PheWAS analysis. It is possible that differential markers of sub-clinical atherosclerosis that have been reported (i.e. carotid artery atherosclerosis) are not well captured by the ICD based PheWAS phenotyping approach and may require more specific phenotyping methods.
Considering our study in the context of the ongoing controversy, conflicting reports in the published literature on the association between APOL1 and CVD (Supplemental Table 1) may be ascribed to (1) demographic differences between studies with different proportions of CKD and different methods for adjusting for it; (2) genotypic differences with varying proportion of G1 and G2 in a given study; (3) differential association models used (recessive vs additive and adjustments for covariates); (4) differential composite outcomes for CVD in general and the inclusion of heart failure as part of the composite outcomes in particular - which is especially difficult to adjudicate in the setting of concomitant CKD. Based upon our findings, we suggest that in retrospect, many of the published studies lacked statistical power to detect what we find is an extremely modest increase in CAD risk.
The strengths of this study include a sample size that is >3 fold larger than previous investigations with >10 fold more CHD events observed compared to the largest previous investigations deriving kidney disease and cardiovascular disease endpoints from the VA EHR to censor individuals upon developing kidney disease, the use of a single rather than composite endpoints, and the ability to interrogate numerous heart and vascular disease endpoints in an unbiased fashion using PheWAS. Limitations of this study include the significant underrepresentation of female participants, an older population raising the possibility of survival bias, reliance on EHR diagnostic codes to determine endpoints and the possibility that outcomes in VA healthcare system may not be entirely representative of African American outcomes outside the VA.
Our study suggests several new directions for the field. Although G1 and G2 appear to have concordant effects on kidney disease risk, they may have distinct effects on the cardiovascular system and lipid profile that could be better evaluated in a model system. Although controlling these CVD risk factors are an important component of cardiovascular and renal health for all individuals, it may be of particular importance in reducing disease burden in individuals with APOL1 risk alleles.
Supplementary Material
Clinical Perspective.
1) What is new?
African-American individuals with kidney disease have increased risk of cardiovascular disease compared to Caucasians with kidney disease
Recent reports have sought to evaluate if APOL1 risk alleles, that cause kidney disease in African Americans, also confer increased risk of CVD and reached conflicting conclusions.
In this study, of 30,903 African American Veterans cared for in the US VA System, the authors found a modest increase in risk for coronary disease, stroke and peripheral artery disease in kidney disease free individuals, however this association was not found when simultaneously modeling kidney disease.
2) What are the clinical implications?
This study suggests that the impact of APOL1 high risk variants amongst African Americans without kidney disease on cardiovascular disease is modest.
It may have clinical implications for screening and counseling African American patients to manage modifiable cardiovascular risk factors that are also associated with kidney disease, such as hypertension and diabetes regardless of APOL1 genotype status.
Acknowledgements
We greatly appreciate the participation of the Million Veterans Program participants and the support of the Million Veterans Program staff.
Sources of Funding
This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration, and was supported by award CSP#G002, I01-01BX03340 (PIs: Wilson/Cho), I01-BX002641 (PIs: Tsao/Chang), and I01-BX003360 (PI: Hung). SMD is additionally supported by IK2-CX001780. This publication does not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
Conflict of Interest Disclosures
None
References
- 1.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019; 139:e56–e528. [DOI] [PubMed] [Google Scholar]
- 2.Safford MM, Brown TM, Muntner PM, Durant RW, Glasser S, Halanych JH, Shikany JM, Prineas RJ, Samdarshi T, Bittner VA, Lewis CE, Gamboa C, Cushman M, Howard V, Howard G. Association of Race and Sex With Risk of Incident Acute Coronary Heart Disease Events. JAMA. 2012; 308:1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tarver-Carr ME, Powe NR, Eberhardt MS, LaVeist TA, Kington RS, Coresh J, Brancati FL. Excess risk of chronic kidney disease among African-American versus white subjects in the United States: a population-based study of potential explanatory factors. J Am Soc Nephrol. 2002;13:2363–70. [DOI] [PubMed] [Google Scholar]
- 4.Weiner DE, Tighiouart H, Amin MG, Stark PC, MacLeod B, Griffith JL, Salem DN, Levey AS, Sarnak MJ. Chronic kidney disease as a risk factor for cardiovascular disease and all-cause mortality: a pooled analysis of community-based studies. J Am Soc Nephrol. 2004;15:1307–15. [DOI] [PubMed] [Google Scholar]
- 5.Ito K, Bick AG, Flannick J, Friedman DJ, Genovese G, Parfenov MG, Depalma SR, Gupta N, Gabriel SB, Taylor HA, Fox ER, Newton-Cheh C, Kathiresan S, Hirschhorn JN, Altshuler DM, Pollak MR, Wilson JG, Seidman JG, Seidman C. Increased burden of cardiovascular disease in carriers of APOL1 genetic variants. Circ Res. 2014;114:845–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grams ME, Rebholz CM, Chen Y, Rawlings AM, Estrella MM, Selvin E, Appel LJ, Tin A, Coresh J. Race, APOL1 Risk, and eGFR Decline in the General Population. J Am Soc Nephrol. 2016;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freedman BI, Rocco MV, Bates JT, Chonchol M, Hawfield AT, Lash JP, Papademetriou V, Sedor JR, Servilla K, Kimmel PL, Wall BM, Pajewski NM. APOL1 Renal-Risk Variants Do Not Associate With Incident Cardiovascular Disease or Mortality in the Systolic Blood Pressure Intervention Trial. 2017;2;713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen TK, Appel LJ, Grams ME, Tin A, Choi MJ, Lipkowitz MS, Winkler CA, Estrella MM. APOL1 Risk Variants and Cardiovascular Disease: Results From the AASK (African American Study of Kidney Disease and Hypertension). Arterioscler Thromb Vasc Biol. 2017; 37; 1765–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen TK, Katz R, Estrella MM, Gutierrez OM, Kramer H, Post WS, Shlipak MG, Wassel CL, Peralta CA. Association between APOL1 genotypes and risk of cardiovascular disease in MESA (Multi-Ethnic Study of Atherosclerosis). J Am Heart Assoc. 2017;6;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Pun PH, Kwee L, Craig D, Haynes C, Chryst-Ladd M, Svetkey LP, Patel UD, Hauser ER, Pollak MR, Kraus WE, Shah SH. Apolipoprotein L1 Genetic Variants Are Associated with Chronic Kidney Disease but Not with Cardiovascular Disease in a Population Referred for Cardiac Catheterization. CardioRenal Med. 2017;7:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukamal KJ, Tremaglio J, Friedman DJ, Ix JH, Kuller LH, Tracy RP, Pollak MR. APOL1 Genotype, Kidney and Cardiovascular Disease, and Death in Older Adults. Arterioscler Thromb Vasc Biol. 2016;36:398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutiérrez OM, Irvin MR, Chaudhary NS, Cushman M, Zakai NA, David VA, Limou S, Pamir N, Reiner AP, Naik RP, Sale MM, Safford MM, Hyacinth HI, Judd SE, Kopp JB, Winkler CA. APOL1 Nephropathy Risk Variants and Incident Cardiovascular Disease Events in Community-Dwelling Black Adults. Circ Genomic Precis Med. 2018;11: e002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, Whitbourne S, Deen J, Shannon C, Humphries D, Guarino P, Aslan M, Anderson D, LaFleur R, Hammond T, Schaa K, Moser J, Huang G, Muralidhar S, Przygodzki R, O’Leary TJ. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol. 2016;70:214–223. [DOI] [PubMed] [Google Scholar]
- 14.Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, Gagnon DR, DuVall SL, Li J, Peloso GM, Chaffin M, Small AM, Huang J, Tang H, Lynch JA, Ho Y-L, Liu DJ, Emdin CA, Li AH, Huffman JE, Lee JS, Natarajan P, Chowdhury R, Saleheen D, Vujkovic M, Baras A, Pyarajan S, Angelantonio E, Neale BM, Naheed A, Khera AV, Danesh J, Chang K-M, Abecasis G, Willer C, Dewey FE, Carey DJ, Concato J, Michael Gaziano J, Oamp CJ, Tsao PS, Kathiresan S, Rader DJ, F Wilson PW, Assimes TL. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat Genet. 2018; 50, 1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kliger AS, Foley RN, Goldfarb DS, Goldstein SL, Johansen K, Singh A, Szczech L. KDOQI US Commentary on the 2012 KDIGO Clinical Practice Guideline for Anemia in CKD. Am J Kidney Dis. 2013;62:849–859. [DOI] [PubMed] [Google Scholar]
- 16.Levey AS, Stevens LA. Estimating GFR using the CKD Epidemiology Collaboration (CKD-EPI) creatinine equation: more accurate GFR estimates, lower CKD prevalence estimates, and better risk predictions. Am J Kidney Dis. 2010;55:622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, McCarthy S, Van Hout CV., Bruse S, Dansky HM, Leader JB, Murray MF, Ritchie MD, Kirchner HL, Habegger L, Lopez A, Penn J, Zhao A, Shao W, Stahl N, Murphy AJ, Hamon S, Bouzelmat A, Zhang R, Shumel B, Pordy R, Gipe D, Herman GA, Sheu WHH, Lee I-T, Liang K-W, Guo X, Rotter JI, Chen Y-DI, Kraus WE, Shah SH, Damrauer S, Small A, Rader DJ, Wulff AB, Nordestgaard BG, Tybjærg-Hansen A, van den Hoek AM, Princen HMG, Ledbetter DH, Carey DJ, Overton JD, Reid JG, Sasiela WJ, Banerjee P, Shuldiner AR, Borecki IB, Teslovich TM, Yancopoulos GD, Mellis SJ, Gromada J, Baras A. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N Engl J Med. 2017;377:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arya S, Binney Z, Khakharia A, Brewster LP, Goodney P, Patzer R, Hockenberry J, Wilson PWF. Race and Socioeconomic Status Independently Affect Risk of Major Amputation in Peripheral Artery Disease. J Am Heart Assoc. 2018; 7: e007425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tirschwell DL, Longstreth WT. Validating administrative data in stroke research. Stroke. 2002; 33: 2465–70. [DOI] [PubMed] [Google Scholar]
- 20.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang H-Y, Demirkan A, Den Hertog HM, Do R, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkilä K, Hyppönen E, Isaacs A, Jackson AU, Johansson Å, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikäinen L-P, Magnusson PKE, Mangino M, Mihailov E, Montasser ME, Müller-Nurasyid M, Nolte IM, O’Connell JR, Palmer CD, Perola M, Petersen A-K, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney ASF, Döring A, Elliott P, Epstein SE, Eyjolfsson GI, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen A-L, Hayward C, Hernandez D, Hicks AA, Holm H, Hung Y-J, Illig T, Jones MR, Kaleebu P, Kastelein JJP, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimäki T, Lin SY, Lindström J, Loos RJF, Mach F, McArdle WL, Meisinger C, Mitchell BD, Müller G, Nagaraja R, Narisu N, Nieminen TVM, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stančáková A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YI, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrières J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Järvelin MR, Jula A, Kähönen M, Kaprio J, Kesäniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, März W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njølstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PEH, Sheu WH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BHR, Ordovas JM, Boerwinkle E, Palmer CNA, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Kathiresan S, Mohlke KL, Ingelsson E, Abecasis GR; Global Lipids Genetics Consortium. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013; 45:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.U.S. Centers for Medicare Medicaid Services. 2017 ICD-10-CM and GEMs. 2016. Available at: https://www.cms.gov/medicare/coding/icd10/2017-icd-10-cm-and-gems.html [PubMed]
- 22.Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, Field JR, Pulley JM, Ramirez AH, Bowton E, Basford MA, Carrell DS, Peissig PL, Kho AN, Pacheco JA, Rasmussen LV, Crosslin DR, Crane PK, Pathak J, Bielinski SJ, Pendergrass SA, Xu H, Hindorff LA, Li R, Manolio TA, Chute CG, Chisholm RL, Larson EB, Jarvik GP, Brilliant MH, McCarty CA, Kullo IJ, Haines JL, Crawford DC, Masys DR, Roden DM. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31:1102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrell FE. Regression Modeling Strategies : With Applications to Linear Models, Logistic Regression, and Survival Analysis. Springer; New York; 2001. [Google Scholar]
- 24.Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics. 2014;30:2375–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman DJ, Kozlitina J, Genovese G, Jog P, Pollak MR. Population-Based Risk Assessment of APOL1 on Renal Disease. J Am Soc Nephrol. 2011;22:2098–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gutierrez OM, Judd SE, Irvin MR, Zhi D, Limdi N, Palmer ND, Rich SS, Sale MM, Freedman BI. APOL1 nephropathy risk variants are associated with altered high-density lipoprotein profiles in African Americans. Nephrol Dial Transplant. 2016;31:602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bentley AR, Divers J, Shriner D, Doumatey AP, Gutiérrez OM, Adeyemo AA, Freedman BI, Rotimi CN. APOL1 G1 genotype modifies the association between HDLC and kidney function in African Americans. BMC Genomics. 2015;16:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozlitina J, Zhou H, Brown PN, Rohm RJ, Pan Y, Ayanoglu G, Du X, Rimmer E, Reilly DF, Roddy TP, Cully DF, Vogt TF, Blom D, Hoek M. Plasma Levels of Risk-Variant APOL1 Do Not Associate with Renal Disease in a Population-Based Cohort. J Am Soc Nephrol. 2016;27:3204–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol Efflux Capacity, High-Density Lipoprotein Function, and Atherosclerosis. N Engl J Med. 2011;364:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, Shaul PW. HDL Cholesterol Efflux Capacity and Incident Cardiovascular Events. N Engl J Med. 2014;371:2383–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saleheen D, Scott R, Javad S, Zhao W, Rodrigues A, Picataggi A, Lukmanova D, Mucksavage ML, Luben R, Billheimer J, Kastelein JJP, Boekholdt SM, Khaw K-T, Wareham N, Rader DJ. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case-control study. Lancet Diabetes Endocrinol. 2015;3:507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLean NO, Robinson TW, Freedman BI. APOL1 Gene Kidney Risk Variants and Cardiovascular Disease: Getting to the Heart of the Matter. Am J Kidney Dis. 2017;70:281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franceschini N, Kopp JB, Barac A, Martin LW, Li Y, Qian H, Reiner AP, Pollak M, Wallace RB, Rosamond WD, Winkler CA. Association of APOL1 With Heart Failure With Preserved Ejection Fraction in Postmenopausal African American Women. JAMA Cardiol. 2018;3:712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.