

SUMMARY
Evidence suggests that Polycomb (Pc) is present at chromatin loop anchors in Drosophila. Pc is recruited to DNA through interactions with the GAGA binding factors GAF and Pipsqueak (Psq). Using HiChIP in Drosophila cells, we find that the psq gene, which has diverse roles in development and tumorigenesis, encodes distinct isoforms with unanticipated roles in genome 3D architecture. The BR-C, ttk, and bab domain (BTB)-containing Psq isoform (PsqL) colocalizes genome-wide with known architectural proteins. Conversely, Psq lacking the BTB domain (PsqS) is consistently found at Pc loop anchors and at active enhancers, including those that respond to the hormone ecdysone. After stimulation by this hormone, chromatin 3D organization is altered to connect promoters and ecdysone-responsive enhancers bound by PsqS. Our findings link Psq variants lacking the BTB domain to Pc-bound active enhancers, thus shedding light into their molecular function in chromatin changes underlying the response to hormone stimulus.
Graphical Abstract
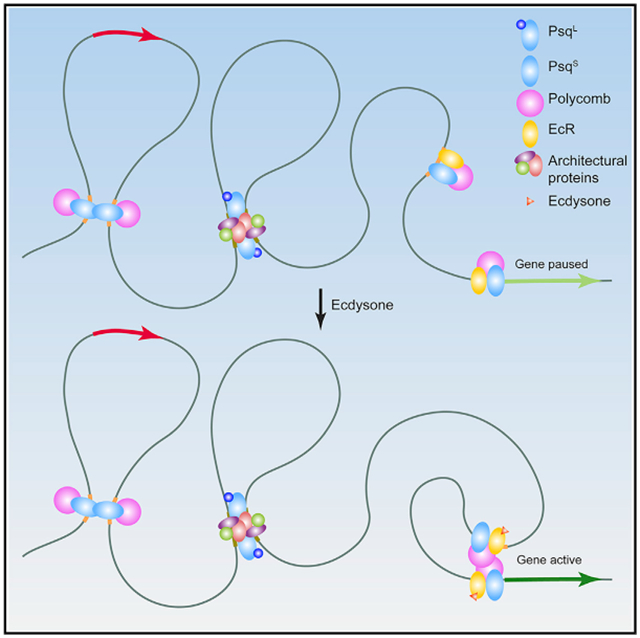
In Brief
Gutierrez-Perez et al. show that BTB domain-containing isoforms of Pipsqueak associate with architectural proteins, whereas Psq lacking BTB colocalizes with Polycomb. Induction of differentiation by the hormone 20-hydroxyecdysone results in recruitment of the ecdysone receptor and Psq lacking BTB to enhancers and establishment of interactions with promoters of activated genes.
INTRODUCTION
Genomes are organized in the three-dimensional (3D) nuclear space to ensure that processes such as transcription are fine-tuned in time and space (Rowley and Corces, 2016). The first experiments using Hi-C described the segregation of chromatin into A (active) and B (inactive) compartments that interact with other genomic regions in a similar transcriptional state (Lieberman-Aiden et al., 2009). More recently, experiments using high-resolution Hi-C data have found that the segregation of active and inactive chromatin scales to small compartmental domains of tens to hundreds of kilobases (kb) (Rao et al., 2014, 2017; Rowley et al., 2017). In addition, high-resolution Hi-C in mammalian cells has led to the discovery of thousands of point-to-point interactions representing CCCTC-binding factor (CTCF) loops (Rao et al., 2014). Drosophila cells lack loops anchored by CTCF (Rowley et al., 2017). Instead, Hi-C heatmaps in Drosophila cultured cells and embryos have shown the existence of two classes of loops formed by contacts between specific sites. The first class represents hundreds of point-to-point interactions present in early embryos and whose anchors are enriched in RNA polymerase II (Pol II) and the transcription factor Zelda (Ogiyama et al., 2018). The second class of loops was originally discovered in Kc167 cells and represents a few hundred point-to-point interactions whose anchors are enriched in other architectural proteins (Cubeñas-Potts et al., 2017). These loops are frequently located within B compartmental domains, and their anchors are enriched in Polycomb (Pc) (Eagen et al., 2017), a member of the Polycomb repressor complex 1 (PRC1) that mediates recognition and binding to the histone modification histone H3 lysine 27 trimethylation (H3K27me3) (Schwartz and Pirrotta, 2007). Pc and most components of PRC1 lack DNA binding activity and are recruited to Polycomb response elements (PREs) containing GAGA sequence motif (GAGA) consensus binding sites by sequence-specific transcription factors (Schwendemann and Lehmann, 2002; Lehmann et al., 1998; Huang et al., 2002; Huang and Chang, 2004; Farkas et al., 1994; Chaharbakhshi and Jemc, 2016). Deletion of PREs or GAGA motifs present at loop anchors results in loss of the corresponding loops and decreased Polycomb group (PcG)-mediated gene silencing during development (Ogiyama et al., 2018). However, the GAGA binding factor or factors that mediate point-to-point interactions leading to the formation of these loops remain undefined. Two sequence-specific transcription factors, Trithorax-like/GAF (Soeller et al., 1993) and Pipsqueak (Psq) (Lehmann et al., 1998) bind GAGA sequences. Interactions among GAF, Psq, and Pc have also been identified (Huang et al., 2002; Schwendemann and Lehmann, 2002; Horard et al., 2000), and these physical interactions are supported by genetic interactions among these genes (Ferres-Marco et al., 2006; Schwendemann and Lehmann, 2002; Huang et al., 2002).
The psq gene is a complex locus encoding two types of isoforms containing or lacking a BTB domain (Siegmund and Lehmann, 2002). We refer to the BR-C, ttk, and bab domain (BTB)-containing Psq isoforms as PsqL and the Psq isoforms lacking the BTB domain as PsqS. Both types of isoforms share a helix-turn-helix (HTH) DNA binding domain (Horowitz and Berg, 1996). The long PsqL isoforms, like GAF, contain a conserved BTB domain involved in protein-protein interactions. BTB domain-containing proteins have the ability to oligo- and multi-merize in solution with other BTB or non-BTB-containing proteins (Perez-Torrado et al., 2006; Stogios et al., 2005; Smaldone et al., 2016). This ability and their location in distinct nuclear substructures suggest that BTB-containing proteins may interact with distant proteins in the genome, altering chromatin structure (Gurudatta and Corces, 2009; Schoborg and Labrador, 2014; Van Bortle and Corces, 2013; Gómez-Díaz and Corces, 2014). Several Drosophila architectural proteins, such as CP190 and Mod(mdg4), contain BTB domains, and they colocalize in different combinations and levels of occupancy at architectural protein binding sites (APBSs) (Van Bortle et al., 2014). Several studies have suggested that interactions among GAF, Psq, and Pc involve the BTB domains of GAF and Psq (Horard et al., 2000; Schwendemann and Lehmann, 2002). Therefore, it has been assumed that the BTB-containing Psq is responsible for the recruitment of Pc to GAGA sequences. However, this notion is at odds with earlier findings showing that the presence of a BTB domain inhibits DNA binding (Bardwell and Treisman, 1994). Thus, the role of the BTB domain in the function of sequence-specific transcription factors and in the recruitment of PcG complexes remains unresolved. Given the expanding role of BTB domain-containing proteins and the PcG machinery in chromatin structure and cancer (Simon and Kingston, 2013), it is important to characterize how variants lacking the BTB domain act in transcriptional regulation and cell differentiation.
Here, we characterize the in vivo function of Psq isoforms containing or lacking the BTB domain, their differential chromatin binding, and their associated long-range chromatin contacts using chromatin immunoprecipitation sequencing (ChIP-seq) and chromosome conformation capture (3C) coupled with sequencing combined with chromatin immunoprecipitation (HiChIP). We found that PsqL colocalizes with Suppressor of Hairy wing (Su(Hw)) and other architectural proteins at sequences classically defined as insulators. In contrast to what was previously assumed, PsqS, rather than BTB-containing PsqL, colocalizes with GAF and Pc at enhancer elements and may therefore be responsible for the classical GAGA binding function assigned to the Psq protein. HiChIP analysis identifies two types of Pc-associated interactions. The first corresponds to Pc loops established by high-frequency point-to-point interactions between anchors containing PsqS. The second type corresponds to contacts between large, repressive Pc domains that form broad interactions similar to those mediated by B compartmental domains. To analyze the functional role of these interactions, we then examined changes in the 3D organization of chromatin during the ecdysone-inducible response and show that PsqS-bound enhancers undergo dramatic changes in their contacts with the promoters of ecdysone-induced genes. These findings suggest distinct roles for Psq isoforms containing or lacking the BTB domain in Pc function and 3D chromatin architecture in response to developmental cues elicited by ecdysone.
RESULTS
Psq Isoforms Have Distinct Roles
To analyze the role of the BTB domain in the function of Psq isoforms and their ability to bind to GAGA sites in vivo, we first examined isoform-specific functions by analyzing their effects on tumorigenesis. The psq gene encodes 12 annotated transcripts generated by alternative use of promoters (Figure S1A). Previous work using northern blot analyses concluded that only isoforms lacking the BTB domain are expressed in the larval imaginal tissues (Horowitz and Berg, 1996; Weber et al., 1995). Although the phenotypic classes of psq alleles were initially attributed to defects in the long or short transcripts encoding Psq isoforms containing (PsqL) or lacking (PsqS) the BTB domain, subsequent studies indicate an intricate relationship between Psq isoforms and complex roles in epigenetic silencing in conjunction with Pc (Ferres-Marco et al., 2006; Huang et al., 2002). Since PsqS is fully contained within the long PsqL isoform (Figure S1A), we tested their function in vivo using isoform-specific transgenes driven by the yeast Gal4 upstream activating sequence (UAS-PsqL and UAS-PsqS). Because mutations that drive Gal4-mediated overexpression of the psq gene (psqGS88A8) (Figure S1A) can cause tumorigenesis when combined with another oncogene (Ferres-Marco et al., 2006), we assayed each isoform for its ability to trigger tissue overgrowth and tumors when coexpressed with a Notch ligand Delta (UAS-Dl) transgene in the growing eye imaginal disc with the help of the ey-Gal4 driver (Figure S1B). Although PsqL overexpression efficiently drove tissue overgrowth/tumor, PsqS did not (Figures S1B and S1C). This observation supports the notion that tumor formation is driven by mis-expression of isoforms containing both the BTB and the Psq HTH domains. Because point mutations within a conserved amino acid in the HTH DNA binding region of psq reverse tumorigenesis by the gain-of-function psqGS88A8 mutation (Figure S1B), we hypothesize that the tumor-promoting role of PsqL may rely on its ability to interact with DNA. However, binding of the full-length Psq proteins to DNA remains undefined. Moreover, the PsqS isoform uses the HTH DNA binding domain, and in vitro studies have shown that Psq proteins containing this region can selectively bind GAGA sequences (Lehmann et al., 1998). Nevertheless, PsqS does not drive tissue overgrowth in the transgenic overexpression assay. We therefore hypothesize that the presence of the BTB domain might regulate the affinity, specificity, or ability of the HTH region to interact with DNA, consistent with earlier observations (Lehmann et al., 1998; Bardwell and Treisman, 1994). These results indicate that the presence or absence of the BTB domain in the Psq protein has functional consequences.
We next examined the genome-wide association of PsqL and PsqS with DNA by first raising two antibodies: a BTB isoform-specific antibody (aa 92–106, PsqL antibody) and an antibody that recognizes all Psq isoforms (aa 453–552, Psqtot antibody) (Figure S1A). The specificity of Psqtot antisera was demonstrated by labeling larval wing imaginal discs expressing an interference RNA transgene to silence the psq gene (Psq-IR) (Dietzl et al., 2007) using the MS1096-Gal4 driver (Figure S1D). Because the levels of the detected endogenous PsqL have little consistency (Figure S1D) with those of earlier analyses (Horowitz and Berg, 1996), we tested the PsqL-specific antisera in control larval imaginal discs expressing the UAS-PsqL transgene in a defined stripe of cells in the wing disc using dpp-Gal4 (Figure S1D). We additionally tested the specificity of the antibodies generated through immunoprecipitation (IP) (Figure S1E). PsqL antibody only immunoprecipitates a band of 150 kDa corresponding to the BTB-containing protein. Moreover, Psqtot detects both isoforms of Psq: the 75 kDa PsqS and the 150 kDa PsqL (Figure S1E). Having demonstrated the specificity of the two Psq antibodies, we then used them to study the genome-wide distribution of the different Psq isoforms to understand the functional role of the BTB domain.
BTB-Containing PsqL Colocalizes with Su(Hw)-Associated Architectural Proteins
To assess the impact of the BTB domain on DNA binding, we used the PsqL and Psqtot antibodies to examine the distribution of PsqL and PsqS by ChIP-seq in Drosophila Kc167 cells (Table S1), which express both Psq isoforms (Figure S1E). By combining both datasets, we detect 10,478 Psq peaks. Because of the specificity of the PsqL antibody, we assumed that peaks detected by the Psqtot antibody, but not detected by PsqL, could be used to identify PsqS binding sites (Figure 1A). Using this strategy, 6,386 (61%) peaks were identified as candidate binding sites of the PsqS isoform (Figure 1B). We detect 1,962 PsqL peaks that showed a ChIP-seq signal with the PsqL antibody and no signal with the Psqtot antibody (Figure S2A). The MD453–552 epitope used to prepare the Psqtot antibody, which is present in the amino-terminal part of PsqS and within the PsqL isoform (Figure S1A), could be occluded under native conditions by Psq-interacting proteins. If this is the case, the PsqL-specific peaks could represent complexes lacking PsqS, which could have more exposed epitopes recognized by the Psqtot antibody. In support of this, previous ChIP-seq experiments using the Organic ChIP-seq method and antibodies similar to Psqtot failed to detect these PsqL sites (Figure S2A; Kasinathan et al., 2014). We also identified a third category of peaks that were strongly enriched with both antibodies. These peaks either represent sites strongly bound by PsqL alone or by a combination of PsqL and PsqS (n = 2,130). When analyzing these peaks (as described later), we found similarities to both PsqL and PsqS binding sites; thus, we named the peaks detected by both antibodies PsqL&S.
Figure 1. PsqL Colocalizes with Su(Hw) and Its Associated BTB Domain Proteins in Kc167 Cells.

(A) Integrative genomics viewer (IGV) tracks showing peaks of PsqL(green) and PsqS (blue). Peaks in green alone detected by the PsqL antibody are classified as PsqL binding sites, while peaks with a signal in blue alone detected by the Psqtot antibodies, but not the PsqL antibodies, are classified as PsqS. Peaks with signals in both ChIP-seq datasets could be either PsqL alone or co-occupied by PsqL and PsqS.
(B) Pie chart showing the number of peaks occupied by each Psq isoform.
(C) Overlap between PsqL peaks and Su(Hw), GAGA, both, or neither motif.
(D) Cumulative fraction of PsqL peaks that overlap Su(Hw) motif locations called at varying q values by individual motif occurrences (fimo).
(E) Heatmap showing ChIP-seq signal for various proteins or histone modifications surrounding PsqS binding sites ± 2 kb. n = 1,962. The STARR-seq signal is from S2 cells, and the ChIP-seq signal is from Kc167 cells and is shown relative to immunoglobulin G (IgG).
(F) Western analysis of protein extracts from Kc167 cells containing input (left), IP of PsqL (middle), or IgG (right) using antibodies to Mod(mdg4)2.2 (top) or PsqL (bottom).
(G) Western analysis of protein extracts from Kc167 cells containing input (left), IP of Mod(mdg4)2.2 (middle), or IgG (right) using antibodies against PsqL (top) or Mod(mdg4)2.2 (bottom).
See also Figure S1.
We next performed motif analysis at PsqL-specific sites. These sites are highly enriched in the Su(Hw) binding motif. Approximately 91% of PsqL binding sites contain this motif, while only 1% exclusively contain the GAGA motif (Figure 1C). This observation is consistent with experiments suggesting that the BTB domain inhibits the interactions of the associated DNA binding domain with DNA (Lehmann et al., 1998). Only 4% of PsqS peaks overlap the Su(Hw) motif, indicating that the two Psq isoforms recognize different sites in the genome. We identified motif locations using q < 0.05; therefore, we tested whether the observed overlap could be explained by low-significance motifs. We found that 85% and 66% of PsqL sites overlap Su(Hw) motif positions called at q < 0.01 and q < 0.001, respectively (Figure 1D). The prevalent presence of Su(Hw) motifs at PsqL binding sites suggests an association between PsqL and architectural proteins, some of which also contain BTB domains, including CP190 and Mod(mdg4)2.2 (Gurudatta and Corces, 2009). To explore the possibility that PsqL colocalizes with architectural proteins, we compared its genomic distribution with that of 13 architectural proteins using published ChIP-seq data in Kc167 cells (see Key Resources Table). We plotted the signal of each feature across a 2-kb region surrounding the PsqL peak summits and identified distinct patterns of binding using k-means clustering. We found, as expected, that PsqL colocalizes with Su(Hw) (Figure 1E). There is also enrichment of BTB-containing proteins CP190 and Mod(mdg4)2.2 and weak enrichment of CTCF and Rad21 (Figure 1E). In contrast, PsqL does not colocalize with other architectural proteins, such as BEAF-32, Dref, Cap-H2, Chromator, Z4, Nup98, or L3mbt, suggesting that PsqL is not present at high-occupancy APBSs (Figure 1E). We also performed ChIP-seq for imitation SWI (ISWI) and found that PsqL sites are enriched for this ATP-dependent nucleosome-remodeling factor (Figure 1E). To test whether the colocalization between PsqL and Mod(mdg4)2.2 may result from direct interactions between these proteins through their BTB domains, we performed coimmunoprecipitation (coIP) experiments using antibodies for these two proteins. We detect Mod(mdg4)2.2 in material immunoprecipitated with the PsqL antibody (Figure 1F), as well as PsqL in material immunoprecipitated with the Mod(mdg4)2.2 antibody (Figure 1G), indicating that these two BTB-containing proteins interact with each other. Pc, GAF, and active or repressive histone modifications are largely absent from PsqL sites (Figure 1E). In agreement with this, PsqL is absent from the self-transcribing active regulatory region sequencing (STARR-seq) sites defining housekeeping (hkCP) and developmental (dCP) enhancers (Zabidi et al., 2015; Figure 1E).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti- MD92-106 | This Study | N/A |
| Anti- MD453-552 | This Study | N/A |
| Anti-Pc | (Li et al., 2015) | N/A |
| Anti-EcR | Hybridoma bank | Cat#AB_528209; RRID:AB_528209 |
| Anti-ISWI | Gift from John Tamkun | N/A |
| Deposited Data | ||
| MD92-106 ChIP-seq Kc167 Cells | This Study | GSE118047 |
| MD453-552 ChIP-seq Kc167 Cells | This Study | GSE118047 |
| MD92-106 20-HE ChIP-seq Kc167 Cells | This Study | GSE118047 |
| MD453-552 20-HE ChIP-seq Kc167 Cells | This Study | GSE118047 |
| ORGANIC Psq ChIP-seq S2 cells | (Kasinathan et al., 2014) | GSE45672 |
| EcR ChIP-seq Kc167 Cells | This Study | GSE118047 |
| EcR 20-HE ChIP-seq Kc167 Cells | This Study | GSE118047 |
| MD453-552 HiChIP Kc167 Cells | This Study | GSE118047 |
| MD453-552 20-HE HiChIP Kc167 Cells | This Study | GSE118047 |
| Pc HiChIP Kc167 Cells | This Study | GSE118047 |
| Pc 20-HE HiChIP Kc167 Cells | This Study | GSE118047 |
| Pc ChIP-seq Kc167 Cells | This Study; (Li et al., 2015) | GSE118047; GSE63518 |
| Pc 20-HE ChIP-seq Kc167 Cells | This Study | GSE118047 |
| GAF ChIP-seq Kc167 Cells | (Cubeñas-Potts et al., 2017) | GSE80702 |
| CBP ChIP-seq Kc167 Cells | (Li et al., 2015) | GSE63518 |
| ISWI ChIP-seq Kc167 Cells | This Study | GSE118047 |
| BEAF32 ChIP-seq Kc167 Cells | (Li et al., 2015; Yang et al., 2013) | GSE30740; GSE63518 |
| CAPH2 ChIP-seq Kc167 Cells | (Li et al., 2015; Van Bortle et al., 2014) | GSE54529, GSE63518 |
| Chromator ChIP-seq Kc167 Cells | (Li et al., 2015; Van Bortle et al., 2014) | GSE54529, GSE63518 |
| CP190 ChIP-seq Kc167 Cells | (Van Bortle et al., 2014; Yang et al., 2013) | GSE30740, GSE54529 |
| CTCF ChIP-seq Kc167 Cells | (Yang et al., 2013) | GSE30740 |
| DREF ChIP-seq Kc167 Cells | (Li et al., 2015; Yang et al., 2013) | GSE63518, GSE39664 |
| H3K27ac ChIP-seq Kc167 Cells | (Yang et al., 2013) | GSE36374 |
| H3K27me3 ChIP-seq Kc167 Cells | (Rowley et al., 2017) | GSE89244 |
| H3K4me1 ChIP-seq Kc167 Cells | (Li et al., 2015; Yang et al., 2013) | GSE36374, GSE63518 |
| H3K4me3 ChIP-seq Kc167 Cells | (Li et al., 2015) | GSE63518 |
| Hi-C Kc167 Cells | (Cubeñas-Potts et al., 2017) | GSE80702 |
| L3mbt ChIP-seq Kc167 Cells | (Li et al., 2015; Van Bortle and Corces, 2013) | GSE36393, GSE63518 |
| Mod(mdg4)2.2 ChIP-seq Kc167 Cells | (Van Bortle and Corces, 2013) | GSE36393 |
| Nup98 ChIP-seq Kc167 Cells | (Cubeñas-Potts et al., 2017) | GSE80702 |
| Rad21 ChIP-seq Kc167 Cells | (Li et al., 2015; Van Bortle et al., 2014) | GSE54529, GSE63518 |
| Su(Hw) ChIP-seq Kc167 Cells | (Yang et al., 2013) | GSE30740 |
| Z4 ChIP-seq Kc167 Cells | (Li et al., 2015) | GSE63518 |
| STARR-seq | (Shlyueva et al., 2014; Zabidi et al., 2015) | GSE47691, GSE57876 |
| Oligonucleotides | ||
| 5′- CGACGGCAATTACATACCAG –3′ | Psq-L&SFor | N/A |
| 5′- GTTCGTGCTCGTGGTCCT-3′ | Psq-L&S Rev | N/A |
| 5′- CAACAGCAACAACACCCAGA –3′ | Eip75B-RA For | N/A |
| 5′- CAGATCGGCACATGGCTTT –3′ | Eip75B-RA Rev | N/A |
| 5′- GCACAGGCGCATTGTGATA –3′ | Eip75B-RD For | N/A |
| 5′- GGCGAAGAACTCCCGATATT –3′ | Eip75B-RD Rev | N/A |
| 5′- GATCCATAAGTCCGCAGTCAAA –3′ | Vrille For | N/A |
| 5′- CTATGGAGATGGAATGATGGCC –3′ | Vrille Rev | N/A |
| 5′- GCGGCCTATCTGTCTACTATTG –3′ | CG44004 For | N/A |
| 5′- ACGGAGCAATCTACGAAACC –3′ | CG44004 Rev | N/A |
| Experimental Models: Cell Lines | ||
| D. melanogaster: Cell line Kc167 | Drosophila Genomics Resource Center (DGRC) | FlyBase: FBtc0000001 |
| Software and Algorithms | ||
| Juicebox | (Durand et al., 2016a) | http://aidenlab.org/juicebox/ |
| Juicer | (Durand et al., 2016b) | http://aidenlab.org/juicer/ |
| Chicago | (Cairns et al., 2016) | http://regulatorygenomicsgroup.org/chicago |
| ngsplot | (Shen et al., 2014) | https://github.com/shenlab-sinai/ngsplot |
| MANORM | (Shao et al., 2012) | http://bioinfo.sibs.ac.cn/shaolab/MAnorm/MAnorm.htm |
| Bowtie2 | (Langmead et al., 2009) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 | (Zhang et al., 2008) | https://github.com/taoliu/MACS |
Psq Isoforms Lacking the BTB Domain Colocalize with GAF and Pc at Enhancer Elements Containing GAGA Motifs
We next analyzed the enrichment of DNA binding motifs at ChIP-seq peaks for the PsqS isoform. The most significant motif corresponds to GAGA (Figure 2A), as described in native ChIP-seq experiments using an antibody recognizing all Psq isoforms (Kasinathan et al., 2014). Approximately 37% of PsqS peaks contain the GAGA motif within a 200 bp region surrounding the peak summit (Figure S2B). This enrichment is higher than expected for random genomic locations and is similar to that of ChIP-seq peaks for the GAF protein (Figure S2B). Stronger PsqS peaks are more likely to overlap the GAGA motif (Figure 2B, blue line). Because we saw a GAF ChIP-seq signal at PsqS binding sites, in line with enrichment of the GAGA motif (Figure 2C), we asked whether all GAF binding sites are also bound by PsqS. To test this hypothesis, we examined the Psq ChIP-seq signal across all detected GAF binding sites. We found that nearly half of GAF peaks show enrichment of Psq, most of which corresponds to the PsqS isoform (Figure S2C). Psq has been proposed to interact with GAF through the BTB domain (Schwendemann and Lehmann, 2002). However, the presence of PsqS, which lacks the BTB domain, at GAF sites suggests that the interaction may involve different domains of these proteins. The occurrence of PsqS co-incides with the strongest GAF signal, and PsqS is only present at GAF sites that are enriched for Pc (Figure S2C).
Figure 2. The PsqS Isoform Colocalizes with Pc and GAF at Active Enhancers in Kc167 Cells.

(A) Binding motif detected by multiple expectation maximization (EM) for motif elicitation designed to analyze ChIP-seq (MEME-ChIP) at PsqS peaks.
(B) Percentage of PsqS peaks that overlap GAGA motifs ranked by ChIP-seq signal intensity (blue). Regions upstream and downstream of PsqS summits were tested for comparison (gray).
(C) Example region showing PsqS binding sites as peaks with a Psqtot signal (blue), without a PsqL signal (green), and colocalizing with GAF, Pc, CBP, and H3K27ac (black).
(D) Heatmap showing ChIP-seq signal for various proteins or histone modifications surrounding PsqS binding sites ± 2 kb. n = 6,386. The STARR-seq signal is from S2 cells, and the ChIP-seq signal is from Kc167 cells and is shown relative to IgG.
(E) IGV track showing an example locus with ChIP-seq signal for PsqL and Psqtot overlapping with both Su(Hw) and GAF.
(F) ChIP-seq signal for various proteins and histone modifications in a 2-kb region surrounding sites enriched in PsqL and Psqtot. n = 2,130. The STARR-seq signal is from S2 cells, and the ChIP-seq signal is from Kc167 cells and is shown relative to IgG.
See also Figure S2.
We then examined the colocalization of PsqS binding sites with those of other transcription factors or histone modifications. As described earlier, we plotted the signal of each feature across a 2-kb region surrounding the PsqS peak summits and identified distinct patterns of binding using k-means clustering. We detect ChIP-seq signals corresponding to Pc at nearly all PsqS peaks, and vice versa (Figures 2D and S2D). We examined the relative binding strength of these two proteins and found a correlation between PsqS and Pc ChIP-seq signal (Figure S2E). This indicates that PsqS colocalizes with Pc genome-wide and overlaps with GAF (Figures 2D and S2D). In addition to its presence at narrow peaks lacking H3K27me3 (Figure 2C), Pc is distributed in broad domains termed Pc domains (Schuettengruber and Cavalli, 2009) that contain H3K27me3 signal. These domains correspond to cluster 1 with broad enrichment of Pc (Figure 2D), which may correspond to regions containing the classical PREs (Aranda et al., 2015). PsqS is present at these broad Pc domains, including several PcG-silenced Hox genes such as deformed (Dfd), Sex combs reduced (scr), and Antennapedia (Antp) (Figure S2F). These regions contain high levels of H3K27me3 and Pc, with most sites of PsqS and GAF colocalizing at the summits of Pc peaks (Figure S2F). We found that PsqS sites at repressive Pc domains represent a small fraction of the total, and most PsqS sites coincide with narrow Pc peaks containing active histone modifications (Figure 2D). For example, the sprouty (spry) and eIF5B genes are present in an active histone H3 lysine 27 acetylation (H3K27ac)-rich chromatin domain, where Pc and GAF colocalize at the summits of PsqS peaks (Figure 2C). PsqS sites overlapping with Pc and H3K27ac are also enriched in CREB-binding protein (CBP), which is typically found at enhancers (Figures 2C and 2D). We therefore tested whether PsqS is enriched at Pleiohomeotic (Pho)-occupied enhancers that were previously annotated in embryos (Erceg et al., 2017) and found great enrichment compared with random regions (Figure S2G). We also tested whether PsqS-bound regions correspond to enhancers by examining STARR-seq signal for hkCP and dCP enhancers (Zabidi et al., 2015). Cluster 2 is enriched for dCP enhancer signal, whereas cluster 3 is enriched for hkCP enhancer signal (Figure 2D). PsqS sites that overlap hkCP enhancers are enriched in histone H3 lysine 4 trimethylation (H3K4me3), whereas those overlapping dCP enhancers are enriched in histone H3 lysine 4 monomethylation (H3K4me1), which is consistent with previous results for these two types of enhancers (Cubeñas-Potts et al., 2017)(Figure S2H). Cluster 4 represents PsqS binding sites that lack histone modifications characteristic of enhancers but contain low levels of CBP and Pc (Figure 2D). These results reveal that the PsqS isoform is enriched at enhancer sites that contain the GAGA motif and suggest that PsqS binds to DNA through these sequences.
Overall, these data indicate that while PsqS is present at sites bound by GAF, CBP, and Pc, PsqL colocalizes with Su(Hw), CP190, Mod(mdg4)2.2, and ISWI, suggesting different roles for the two isoforms. In addition, we examined PsqL&S peaks and found examples in which both GAF and Su(Hw) overlap these binding sites (Figure 2E). Analysis of ChIP-seq data at these undetermined Psq peaks found enrichment of GAF, Pc, CBP, and STARR-seq enhancers, which is similar to what we found for PsqS (Figure 2F). We also found enrichment for CP190 and Mod(mdg4)2.2, which is similar to what we found for PsqL (Figure 2F). We also detect a slight enrichment of Su(Hw), although not as strongly as at PsqL sites (compare Figure 2F with Figure S2H). Hence, PsqL&S peaks exhibit characteristics of both PsqL and PsqS binding sites and are thus likely bound by both.
PsqS Colocalizes with Pc at Chromatin Loops Distinct from Repressive Pc Domains
Because the PsqS isoform is responsible for GAGA binding, we wondered whether PsqS regulates Pc at chromatin loops. Computational annotation of strong point-to-point interactions in Drosophila Kc167 cells has identified 458 potential loops (Cubeñas-Potts et al., 2017). These structures have also been annotated visually, resulting in the identification of 120 loops (Eagen et al., 2017). We found that Pc overlaps 30% of loop anchors annotated computationally and 68% of those annotated visually. PsqS peaks overlap 21% and 53% of these two classes of loop anchors, respectively, whereas PsqL only overlaps 3% and 5%. These loop anchors have sharp peaks of PsqS and sharp peaks of Pc instead of the broad Pc signal representative of repressive Pc domains (Figures 3A and S3A). Although overlap of loops with Pc ChIP-seq has been reported (Eagen et al., 2017; Ogiyama et al., 2018), it is unclear whether Pc directly participates in the establishment of these loops. Therefore, we performed HiChIP using a Pc antibody (see Table S2 for quality controls and statistics of the Pc HiChIP libraries) and used these data to examine Pc-bound loops (Figure S3B, top right). These loops represent strong or frequent interactions compared with surrounding regions as seen by metaplot analysis of Hi-C data (Figure 3B, left) and are enriched in Pc HiChIP data (Figure 3B, right). To determine the chromatin state of Pc loop anchors identified by HiChIP, we plotted active and inactive marks around the Pc ChIP-seq summit found at loop anchors. We found high levels of H3K27me3 in the surrounding region but a dip precisely at the loop anchor (Figure 3C). Instead of H3K27me3, the Pc loop anchor precisely corresponds to a peak of H3K27ac (Figure 3C). Because there is high overlap between Pc and PsqS peaks in ChIP-seq data (Figure 2D), we examined whether loop anchors identified by Pc HiChIP are enriched in PsqS. We detect enrichment of the GAF, CBP, ISWI, and PsqS signal at these anchors (Figure S3C). To confirm that PsqS is directly associated with Pc loops, we then performed HiChIP using the Psqtot antibody (see Table S3 for quality controls and statistics of Psq HiChIP libraries) and found that Pc loops are enriched in Psq HiChIP signal (Figure S3B, bottom left, and Figure 3D). These data suggest that strong point-to-point Pc interactions detected by HiChIP represent loops anchored by sites of active chromatin cobound by PsqS, GAF, CBP, ISWI, and H3K27ac (Figure 3A).
Figure 3. Pc Loop Anchors Contain PsqS and Form Interactions Distinct from Those Involving Pc-Repressive Domains.

(A) Example locus showing Hi-C in Kc167 cells signals for loops associated with Pc (circles). Tracks showing ChIP-seq for H3K27ac, H3K27me3, Psqtot, and Pc are shown above and to the left. Genes are shown at the bottom.
(B) 2D metaplots of Hi-C (left) and Pc HiChIP (right) data centered on significant interactions called by Pc HiChIP. n = 206. The score indicates the enrichment of the center pixel compared with the top left corner.
(C) Average ChIP-seq profile for H3K27ac (orange) and H3K27me3 (pink) surrounding Pc loop anchors identified by Pc HiChIP. n = 206. The shaded area indicates SD.
(D) 2D metaplot of Psqtot HiChIP data centered on significant interactions called by Pc HiChIP. n = 206. The score indicates the enrichment of the center pixel compared with the top left corner.
(E) Hi-C interaction plot showing an example locus in which two distinct Pc domains interact more strongly with each other (black box) than with other inactive B compartmental domains (green box). Tracks showing H3K27ac, H3K27me3, Pc, and A or B compartmental domains are shown above and to the left.
(F) Zoomed-in area of the Pc domain shown in (E).
(G) Boxplot showing the distribution of average interaction signals occurring between Pc domains (Pc-Pc, n = 102), between Pc domains and other inactive B compartmental domains (Pc-B, n = 1,365), or between inactive B compartmental domains (B-B, n = 17,208). p < 0.001, Wilcoxon test for Pc-Pc versus Pc-B.
See also Figure S3.
To investigate chromatin organization in the context of Pc-repressed chromatin, we identified domains in which Pc was enriched in regions of a minimum length of 10 kb (see STAR Methods). We examined differences between large Pc domains and inactive B compartmental domains (Rowley et al., 2017). We found that although B compartmental domains are enriched in H3K27me3 versus H3K27ac, Pc domains contain higher levels of H3K27me3 than standard B domains (Figure S3D; see also Figure S3A). Pc HiChIP data enriched Pc domains, but not B compartmental domains, when compared with Hi-C (Figure S3E). In contrast, Psq HiChIP did not enrich these repressive Pc domains (Figure S3F), indicating that repressive Pc domain interactions are distinct from Psq/Pc looping interactions. By examining interactions from Hi-C data at Pc domains, we found that Pc domains interact more with each other than with other inactive B compartmental domains (Figure 3E, black versus green box). We examined more closely a Pc domain containing different levels of Pc and H3K27me3, and we observed a correlation between interaction frequency and levels of Pc and H3K27me3 (Figure 3F). This suggests that Pc domains associate with other Pc domains preferentially over other inactive chromatin, which is in agreement with studies in Drosophila embryos (Ogiyama et al., 2018). To test this genome-wide, we classified interactions as those with repressive Pc domains on both sides (Pc-Pc), those with a Pc domain on one side and a B compartmental domain on the other (Pc-B), or those with B compartmental domains on both sides (B-B). We found that Pc-Pc interactions are stronger than Pc-B interactions and B-B interactions (Figure 3G). Pc-B interactions were similar to B-B interactions, indicating that Pc domains are not prevented from interacting with other B compartmental domains but that they interact more frequently with other Pc domains (Figure 3G). Altogether, these results suggest the existence of two types of Pc-mediated chromatin organization: those resulting from interactions between broad repressive Pc domains, which are similar to compartmental interactions, and those resulting from point-to-point interactions associated with PsqS and visible as intense punctate signals in Hi-C heatmaps (Figure 3A).
A Role for Steroid Hormone 20-Hydroxyecdysone in Psq Chromatin Loops
Given that narrow peaks of Pc overlapping with PsqS are present at dCP enhancers defined by STARR-seq (Figure 2D, dCP), we wondered whether PsqS and Pc are involved in dCP transcriptional responses via enhancer-promoter interactions. To this end, we examined changes in Psq and Pc interactions during the response to the steroid hormone 20-hydroxyecdysone (20-HE) (D’Avino and Thummel, 2000). Treatment of Drosophila macrophage-like Kc167 cells with 20-HE triggers their differentiation, recapitulating events occurring during metamorphosis (Van Bortle et al., 2015). Visual inspection of ChIP-seq data indicates the presence of PsqS peaks at genes induced by 20-HE, suggesting that Psq may be involved in the response to this hormone. This hypothesis is supported by colocalization between PsqS and nucleoporin Nup98 (Figure S2H), which is involved in the ecdysone response (Pascual-Garcia et al., 2017). The expression of the ecdysone receptor (EcR) gene is induced by 20-HE and provides an autoregulatory loop that increases the level of receptor protein available for ligand binding and target-gene activation (Karim and Thummel, 1992; Riddiford et al., 2000). We therefore examined the genome-wide distribution of Psq, Pc, and EcR binding sites by ChIP-seq in Kc167 cells (Table S1). We identified 845 loci to which EcR binds under normal conditions, with strong enrichment of PsqS and PsqL (Figure 4A). EcR binding sites in cluster 1 overlap with sites containing PsqS or PsqL&S, which are enriched for GAF, Pc, CBP, hkCP, and dCP enhancers, and active histone modifications (Figure 4A and example in Figure S4A). EcR binding sites in cluster 2 overlap with the PsqL isoform, Su(Hw), CP190, and Mod(mdg4)2.2 (Figure 4A and example in Figure S4B).
Figure 4. Psq Colocalizes with EcR at Ecdysone-Induced Genes.

(A) ChIP-seq signal in Kc167 cells for various proteins and histone modifications in a 2-kb region surrounding EcR sites. n = 845. The ChIP-seq signal is shown relative to IgG.
(B) IGV tracks showing the ChIP-seq signal before and after 20-HE treatment for EcR (red), PsqL (green), Psqtot (blue), and Pc (black). The purple box highlights a region where binding is altered after ecdysone treatment.
(C) Fold expression changes for genes with overlapping Psq peaks that increase (pink) or are unchanged (gray) after ecdysone treatment. *p < 0.01, Wilcoxon test.
(D) Left: IGV tracks showing the ChIP-seq signal before and after 20-HE treatment for EcR (red), PsqL (green), and Psqtot (blue). Right: expression change of CG44004 after 20-HE treatment relative to the control (CTL) as measured by qPCR.
(E) Left: IGV tracks showing ChIP-seq signal before and after 20-HE treatment for EcR (red), PsqL (green), and Psqtot (blue). Right: expression change of Vrille after 20-HE treatment relative to the control (CTL) as measured by qPCR.
See also Figure S4.
We hypothesize that the assembly of multiple simultaneous interactions of PsqS, EcR, and Pc at ecdysone-responsive genes might correlate with their transcriptional response. We thus performed ChIP-seq for PsqL, Psqtot, Pc, and EcR after treatment with 0.5 μM 20-HE for 3 h (Wood et al., 2011). The well-studied ecdysone-inducible Eip75B gene encodes different mRNA variants with different hormone sensitivity. EcR binding significantly increases at a region within the locus after 20-HE treatment (Figure 4B, purple box) accompanied by enhanced signal corresponding to PsqS (Figure 4B). The increase in EcR and PsqS occurs upstream of Eip75B-RA, which is the isoform with the highest response to the hormone (Figure S4C). We then used MAnorm (Shao et al., 2012) to compare PsqS peaks obtained before and after genome-wide treatment with the hormone. This analysis identified 35 peaks in which PsqS binding increases after 20-HE treatment (PsqS-inducible peaks) (Figure 4B). We found a similar number of peaks, 45 and 50, corresponding to PsqL and PsqL&S sites, respectively, that increase after ecdysone treatment. Genes that overlap with hormone-inducible PsqS peaks (not PsqL-containing peaks) were more likely to have increased expression after 20-HE treatment than genes that overlap with unaltered peaks (Figure 4C). We saw no difference for genes that overlap any other category of Psq peaks (Figure 4C). Because the total amount of Psq mRNA in the cell does not change significantly after 20-HE treatment (Figure S4D), we envision that hormone treatment facilitates Psq binding or recruitment to these ecdysone-induced genes. Some ecdysone-induced genes, such as CG44004 or Vrille (Figures 4D and 4E), do not show changes in PsqS, and 11 of 35 PsqS-inducible peaks are not close to known ecdysone-induced genes. This raises the possibility that PsqS might regulate the ecdysone response through 3D chromatin changes and these peaks may represent distal regulatory elements. Therefore, we next studied Psq-directed changes in chromatin architecture in response to ecdysone using HiChIP.
Ecdysone Induces the Establishment of Enhancer-Promoter Interactions Bound by PsqS and Pc
Enhancers regulate gene expression through long-range chromatin interactions with promoters. We thus asked whether Psq is involved in these interactions. First, we identified 180,058 active enhancer-promoter interactions using previously published H3K27ac HiChIP data (Rowley et al., 2017) in which one interaction anchor overlaps a promoter while the other overlaps a STARR-seq enhancer. Next, we examined which Psq isoform is most enriched at these enhancer-promoter interaction anchors. Of 94,483 enhancer-promoter interactions that are bound by Psq, we found that only 3% correspond to the PsqL isoform, whereas 76% correspond to the PsqS isoform and 21% correspond to PsqL&S (Figure 5A). This is consistent with our finding that PsqS binds to elements with enhancer potential as determined by STARR-seq (Figure 2D) and supports the conclusion that enhancer elements bound by the GAGA-motif-binding PsqS protein may participate in long-range interactions with target promoters.
Figure 5. PsqS-Bound Enhancer-Promoter Interactions Are Altered during the Ecdysone Response.

(A) Pie graph showing the relative binding of PsqS (purple), PsqL (green), and PsqL&S (pink) at enhancer-promoter interaction anchors determined by H3K27ac HiChIP in Kc167 cells. n = 94,483 interactions.
(B) 2D metaplot of Psq HiChIP signal around enhancer-promoter interactions determined by H3K27ac HiChIP found in (A). En, enhancers; Prm, promoters.
(C) Number of STARR-seq ecdysone enhancers (left) compared with random loci (right) that overlap PsqS (purple), PsqL (green), and PsqL&S (pink).
(D) Psq HiChIP data in a 1-Mb region of chromosome 2R showing changes in the interaction profile after ecdysone treatment (bottom left) compared with the control (top right). ChIP-seq tracks showing PsqS and EcR before (CTL) and after 20-HE treatment. The STARR-seq signal after ecdysone treatment is also shown.
(E) 2D metaplot of Psq HiChIP data before (top right) and after (bottom left) ecdysone treatment. Regions between STARR-seq ecdysone enhancers and nearby differentially expressed genes were scaled to equally sized bins. n = 86.
See also Figure S5.
We analyzed the presence of PsqS in enhancer-promoter interaction anchors using PsqS HiChIP data and found enrichment of interaction signals at enhancer-promoter contact points compared with surrounding regions (Figure 5B). Interactions detected using Hi-C data are also enriched at enhancer-promoter contacts identified using Psq HiChIP (Figure S5A). We therefore hypothesized that ecdysone treatment may alter gene expression through changes to a subset of these interactions. Using STARR-seq data obtained in ecdysone-treated cells to select for enhancers involved in the ecdysone response (Shlyueva et al., 2014), we tested the overlap of each Psq isoform with ecdysone-induced enhancers and found that these enhancers are enriched in PsqS (Figure 5C). Thus, although half of EcR binding sites are co-occupied by PsqL (Figure 4A), EcR sites overlapping PsqS are more likely to be functionally relevant ecdysone-induced enhancers. However, there is no change in Psq, Pc, or EcR at ecdysone enhancers upon ecdysone treatment (Figure S5B). Because of this overlap between ecdysone-induced enhancers and PsqS, and to obtain 1-kb resolution data necessary to accurately observe changes in 3D chromatin organization, we performed HiChIP for PsqS in cells treated with 20-HE (see Table S4 for quality control and statistics). We then compared this information to HiChIP data obtained in untreated cells. Example loci show the existence of many sites with a higher interaction signal after 20-HE treatment (Figure 5D, bottom left) compared with the control (Figure 5D, top right). These sites with an increased signal correspond to ecdysone enhancers identified by STARR-seq signal after ecdysone treatment (Figure 5D). We then tested whether ecdysone treatment changes chromatin interactions, specifically between ecdysone enhancers and ecdysone-regulated genes. We took ecdysone enhancers within 50 kb of genes differentially expressed after ecdysone treatment and performed a 2D metaplot analysis with the Psq HiChIP data. In control cells, there is little to no signal connecting ecdysone enhancers to transcription start sites (TSSs) of ecdysone-induced genes (Figure 5E, top right). After 20-HE treatment, the HiChIP signal is stronger between these sequences (Figure 5E, bottom left). This enrichment cannot be explained by different IP efficiencies before and after ecdysone, because the average ChIP-seq signal of PsqS is the same under both conditions at these interaction anchors (Figure S5C; see also Figure 5D). Of the 180,058 potential enhancer-promoter interactions examined in Figure 5A, 7,417 of them are occupied by EcR on at least one anchor. We then examined enhancer-promoter contacts containing EcR for changes in interaction frequency after ecdysone treatment using Hi-C and Psq HiChIP data. We found little to no HiChIP signal at these sites in the control and increased interactions upon ecdysone treatment (Figures S5D–S5G). This indicates that ecdysone treatment leads to changes in chromatin 3D organization between EcR-bound regions. However, no difference in Pc signal was observed at these interaction anchors before and after ecdysone treatment (Figure S5H). Thus, ecdysone treatment triggers chromatin interactions at PsqS-bound enhancers without changes in Pc. Because we found that ecdysone treatment induces enhancer-promoter interactions bound by PsqS and Pc, we analyzed possible changes in the intense puncta representing Pc loops in control cells (Eagen et al., 2017) (see Figure 3A). We found no change to these pre-existing Pc/PsqS loops, indicating that changes in 3D organization are specific to ecdysone enhancers without changes to Pc/PsqS loops manifested by strong puncta in Hi-C data (Figure S5I). This provides evidence that ecdysone regulates gene expression by changing the 3D organization of chromatin to increase the frequency of interactions between enhancers and promoters bound by PsqS and Pc.
DISCUSSION
The BTB domain of human PLZF, Bcl-6, and Drosophila Psq have been shown to contribute to the oncogenic roles of these proteins (Ferres-Marco et al., 2006, and citations therein). Most BTB-containing transcription factors also encode isoforms that lack the BTB domain (Ko et al., 2006) and the role of these short isoforms is uncertain. Here we show that different isoforms of Psq appear to play different roles in nuclear function, which may explain their opposing roles in tumorigenesis ascribed to the gene. We show that the BTB-containing PsqL isoform colocalizes with a specific class of architectural proteins that includes Su(Hw), CP190, and Mod(mdg4)2.2. In contrast, the PsqS isoform, which lacks the BTB domain, colocalizes with GAF and Pc at dCP enhancers and is mainly associated with active chromatin states. Therefore, PsqS appears to contribute to enhancer function, whereas PsqL is an architectural protein that binds to sequences that have insulator function. How these two isoforms display different genomic distributions while sharing the same DNA binding domain is unclear. However, based on previous findings (Lehmann et al., 1998), we can speculate that the conformation adopted by the protein in the presence of the BTB-interaction domain might inhibit its direct binding to DNA. In addition, the two isoforms coincide in regions in which both Pc and architectural proteins are found. This may explain the reported involvement of PsqL in the recruitment of PcG proteins to chromatin (Huang et al., 2002), where it might act with the help of other architectural proteins. In addition to its canonical role, Pc is found, together with PsqS, ISWI, GAF, and CBP, in regions containing H3K27ac and previously characterized experimentally as hkCP or dCP enhancers. These findings, suggesting an association of Pc with active enhancers, agree with previous observations showing that PRC1 can be recruited to active genes by the cohesin complex, where it affects phosphorylation of Pol II and Spt5 occupancy (Schaaf et al., 2013; Pherson et al., 2017).
H3K27me3 is present in the genome of Kc167 cells at very high levels in Pc-repressed domains such as Hox genes. The rest of the genome containing silenced genes in Kc167 cells has low but significant levels of H3K27me3 that represent B compartment sequences (Rowley et al., 2017). Pc HiChIP analysis provides insights into the dual role of Pc in regulating chromatin organization. Classical Pc-repressed domains interact with each other and with other B compartments with a frequency that correlates with the amount of H3K27me3 present in these compartments. Distinct from these interactions, Pc also forms punctate point-to-point contacts. Two types of loops, defined as puncta of an intense signal in Hi-C heatmaps, have been identified when analyzing changes in 3D organization during Drosophila embryonic development (Ogiyama et al., 2018). These loops were classified as active loops containing H3K27ac, Zelda, and Pol II at their anchors or as Pc loops bound by GAF. Zelda loops are absent from Kc167 cells (Ogiyama et al., 2018). Like Pc loop anchors observed in embryos, loops represented by puncta in Hi-C heatmaps of Kc167 cells are located within regions enriched in H3K27me3 (Cubeñas-Potts et al., 2017; Eagen et al., 2017). However, we found that the center of these sites in Kc167 cells is depleted of H3K27me3 and enriched in H3K27ac. The exact roles of H3K27ac, Pc, PsqS, and GAF found at these loop anchors are unknown, but we speculate that maintaining a localized active chromatin state may be important for the binding of these proteins and the establishment of these loops. These results suggest a dual and context-dependent function of regulatory elements and agree with previous studies showing that dCP enhancers can act as PREs, and vice versa, during Drosophila embryogenesis (Erceg et al., 2017).
Analysis of the distribution of sites containing Pc and PsqS in the genome also uncovered enrichment of these proteins around ecdysone-inducible genes, although most EcR, Pc, and PsqS peaks do not change significantly after ecdysone treatment. This is consistent with previous observations indicating that EcR does not change at most enhancers induced by ecdysone (Shlyueva et al., 2014). Results from PsqS HiChIP experiments in control and ecdysone-treated cells suggest that hormone treatment leads to the establishment of new ecdysone-induced enhancer-promoter interactions without changes to pre-established Pc/PsqS loops. Early genes directly activated by ecdysone are paused before induction, and their expression is regulated at the level of Pol II release from promoter-proximal pausing (Ivaldi et al., 2007; Mazina et al., 2015). This suggests that activation of early gene expression by ecdysone requires the establishment of new enhancer-promoter interactions. The possible involvement of PsqS and Pc in the establishment of these interaction networks in the Drosophila embryo will be an interesting topic for future analyses.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Maria Dominguez (m.dominguez@umh.es, Phone: +34 965 91 9390).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila Cell Lines
Kc167 cells derived from a Drosophila melanogaster female embryo at the dorsal closure stage were obtained from the Drosophila Genomics Resource Center. Cells were grown at 25°C in Hyclone SFX insect culture media (GE Healthcare).
Drosophila Husbandry and Strains
Flies were reared in vials containing cornmeal medium. Flies were collected under CO2-induced anesthesia and housed at a density of 30 female flies per vial. All flies were kept in a humidified, temperature-controlled incubator with 12 h on/off light cycle at 25°C.A detailed description of the Drosophila stocks and transgenic flies used in this study can be found at http://flybase.org/ (MS1096-Gal4 and Dpp-Gal4) or at VDRC RNAi stock (Psq-IR), https://stockcenter.vdrc.at/control/main. Strain UAS-Psq-L was produced in the laboratory of Dr. Maria Dominguez.
METHOD DETAILS
Immunofluorescence and microscopy analysis
Third instar wing imaginal discs were fixed and stained using standard procedures and the following primary antibodies. Psqtot antibody (Rabbit 1:200) against the common epitope for both isoforms encompassing residues 453-552 of the long Psq isoform; PsqL antibody (Rabbit 1:200) recognizing the epitope encompassing residues 92-106. This epitope was designed in the laboratory of Dr. María Domínguez and synthetized by SDIX using SDIX Genomic Antibody Technology® and Eurogentec. Images were captured on a Leica TCS-NT Confocal microscope.
Cell culture, transfections and western analysis
Kc167 cells (DGRC cat. no. 1) were maintained in SFX medium supplemented with 10% inactivated fetal bovine serum (Invitrogen, ref. #10108-165) and penicillin/streptomycin stock of antibiotics (Sigma P4333-100ML) at 25°C without CO2. Ecdysone treatment was done by incubation with 0.5 μM 20-Hidroxyecdysone (20-HE) (Sigma H5142-10MG) in culture medium for 3 hr; vehicle control with ethanol was performed in parallel. Western analysis was performed using standard procedures. PVDF membranes were incubated with one of the following primary antibodies: polyclonal rabbit a-Psqtot (1:2000), polyclonal rabbit a-PsqL (1:2000), a-Actin (Sigma A2066, 1:500), rat a-Mod(mdg4)2.2 (1:2000), rabbit a-CP190 (1:2000). Proteins were detected using the chemiluminescent substrate ECL (Pierce, 32209), LAS-100 detector (FujiFilm) and Imagen Reader LAS-1000 software (FujiFilm). Transient transfection experiments were done in 6 well plates with 8 × 105 cells per well in 2 mL of medium and 1 μg of total DNA per well. The amount of each plasmid was adjusted to obtain equimolar concentrations. Cells were transfected using Cellfectin II Reagent (Invitrogen 10362-100). dsRNA was generated using the Megascript T7 High Yield Transcription Kit (Ambion NC. 1404051). Primers used for the RNAi KD recognizing all isoforms of Psq are For 5′-TAATACGACTCACGCTGCCCTGCTTA-3′; Rev 5′- TAATACGACTCACAAGGCTCA CAATG-3′).
qPCR
Total RNA was isolated from Kc167 cells using the RNeasy Mini Kit (Cat. 74106, QIAGEN) and treated with DNaseI to eliminate the remaining DNA from samples according to the manufacturer’s protocol. RNA (1 μg) was reverse-transcribed using SuperScript III Reverse Transcriptase and Oligo(dT) primers (Cat. 18418020, Invitrogen). Quantitative PCR reactions were performed using Power SYBR Green PCR Master Mix (Applied Biosystems), 10 ng of template cDNA, and 222 nM gene-specific primers in a 7500 Real-time PCR system (Applied Biosystems). Three separate samples were collected for each condition and triplicate measurements were conducted. Primers were designed using the Primer Quest online tool (https://www.idtdna.com/site/account/login?returnurl=%2FPrimerQuest%2FHome%2FIndex). Data is presented as mean ± standard error of the mean. Statistical analyses were performed using the two tailed Student’s t test.
Co-immunoprecipitation assays
To detect interaction between Pipsqueak and Mod2.2, Kc167 cells were grown in 10 cm plates. One plate with 5x106 cells in 10 mL medium was used for each condition. Co-IPs between PsqLand Mod2.2 were done using antibodies against the corresponding proteins and 1 μg of rabbit anti-Mod(mdg4)2.2, rabbit PsqL and rabbit Psq total. After electrophoresis and transfer, membranes were incubated with the following primary antibodies: rabbit polyclonal Psqtot antibody (1:2000), rabbit polyclonal PsqL antibody (1:2000), α-actin (Sigma, A2066,1:500), and rat a-Mod(mdg4)2.2 (1:2000). After overnight incubation at 4°C, membranes were incubated for 1 hr at room temperature with secondary antibodies: HRP-conjugated rabbit a-IgG (Sigma, A9169,1:10000), HRP-conjugated mouse a-IgG (Jackson, 115-035-062,1:5000) or HRP-conjugated Rat a-IgG (Jackson, 712-035-153), and diluted in PBS with 0.1% Tween-20 and 3% BSA. Proteins were detected using the ECL chemiluminescent substrate (Pierce, 32209), LAS-100 detector (Fujifilm) and LAS-1000 Image Reader software (FujiFilm).
ChIP-seq
Chromatin immunoprecipitation was performed as described (Bushey et al., 2009), with 5 μl of primary antibody. To generate sequencing libraries, ChIP DNA was prepared for adaptor ligation by end repair (End-It DNA End Repair Kit, Epicenter ER0720) and addition of ‘A’ base to 3’ ends (Klenow 3′-5′ exo, NEB M0212S). Illumina adaptors (Illumina PE-102-1001) were titrated based on the prepared DNA ChIP sample concentration and ligated with T4 ligase (NEB M0202S). Ligated ChIP samples were amplified by PCR using Illumina primers and Phusion DNA polymerase (NEB F-530L), and size selected for 200-300 bp by gel extraction. Two ChIP biological replicates were sequenced at the HudsonAlpha Institute for Biotechnology using an Illumina HiSeq 2500 instrument. To ensure antibody specificity during ChIP-seq, western blots were performed using the exact same nuclei isolation procedure. Cells were crosslinked for 10 minutes in 1% formaldehyde followed by inactivation in 125 M glycine for 5 min and by two washes in cold PBS. Afterward, samples were incubated in cell lysis buffer (5 mM PIPES pH 8, 85 mM KCl, 0.5% NP40, with protease inhibitors) for 15 min on ice. Nuclei were then collected via centrifugation and incubated in 200 μl nuclei lysis buffer (50 mM Tris HCl pH 8, 10 mM EDTA, 1% SDS, with protease inhibitors) for 20 min on ice. Samples were then diluted with 100 μl cold IP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 Tris HCl pH8, 167 mM NaCl, with protease inhibitors). This was followed by sonication for 28 cycles of 30 / 60 s on / off at 4°C. Chromatin was isolated by centrifugation for 10 min at max speed and keeping the supernatant. To de-crosslink and denature proteins in preparation for western blot, samples were incubated at 99°C for 10 min in 2X SDS loading buffer.
HiChIP Library Preparation
HiChIP samples were prepared as described in Rowley et al. (2017) but using Pc or PsqS antibodies. Kc167 cells were crosslinked in 1% formaldehyde for 10 min at room temperature and stopped in 0.2 M glycine for 5 min. Cells were pelleted and nuclei were isolated in 500 μl cold Hi-C lysis buffer (10 mM Tris-HCl pH8, 10 mM NaCl, 0.2% Igepal CA-630, and 1x Protease Inhibitors (Roche 11873580001) and incubated on ice for 1 h followed by centrifugation at 2500 rcf. for 5 min at 4°C. Nuclei were resuspended in 100 μl 0.5% SDS, and incubated for 5 min at 65°C. We then added 290 μl of H2O and 50 μl of 10% Triton X-100, with an incubation for 15 min at 37°C. Chromatin was digested with 50 μl of 10x DpnII buffer and 200 u of DpnII (NEB R0543) overnight at 37°C with rotation. The next day, DpnII was inactivated at 65°C for 20 min, and each sample was divided into two reactions and allowed to cool to room temperature. Biotin fill-in was done with 22.5 μl of water, 1.5 μl each of 10 mM dTTP, dATP, and dGTP, 15 μl of 1 mM biotin-16-dCTP (Jena Bioscience JBS-NU-809-BIO16), and 8 μl of 5 u/μl DNA polymerase I Large (Klenow) fragment (NEB M210) at 37°C for 1.5 h. Afterward chromatin was ligated for 4 h at room temperature with the addition of 663 μl H20,120 μl 10x NEB T4 DNA Ligase buffer, 100 μl 10% Triton X-100, 12 μl 10 mg/ml BSA, and 5 μl 400 u/μl T4 DNA Ligase (NEB M0202).
Following chromatin ligation, nuclei were pelleted and resuspended in 200 μl cold Nuclei Lysis Buffer (50 mM Tris-HCl pH 9,10 mM EDTA, 1% SDS, and 1x Protease Inhibitors) and incubated on ice for 20 min. Afterward we added 100 ml cold IP Dilution Buffer (0.01% SDS, 1.1% Triton X-100,1.2 mM EDTA, 16.7 Tris-HCl pH 8,16.7 mM NaCl, and 1x Protease Inhibitors) and sonicated. DNA for downstream libraries prepared with the standard protocol was sonicated to approximately 300 bp fragments, while DNA for libraries prepared with Tn5 transposase were sonicated to approximately 1 kb fragments. Cell debris was pelleted, and the supernatant was transferred into a new 1.5 mL tube for immunoprecipitation. To remove nonspecific IP, each sample was precleared before immunoprecipitation by taking 10 μl Protein A and 10 μl Protein G magnetic beads, washing 3x in 0.5% BSA in 1x PBS, followed by incubation with 10 μl pre-immune rabbit serum in 500 μl 0.5% BSA/PBS for 4 h at 4°C with rotation. Beads were then washed with 1 mL 0.5% BSA/PBS for 2 min at room temperature, followed by 2 washes in 1 mL IP Dilution Buffer, and resuspension in 300 μl cold IP Dilution Buffer. Beads with each antibody were prepared in the same manner. Before the IP, chromatin was diluted 5-fold with cold IP Dilution Buffer and incubated with pre-cleared beads for 1-2 h at 4°C with rotation. The unbound portion was then transferred to antibody-coated beads and incubated overnight at 4°C with rotation. After IP, samples were washed 3x with Low Salt Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA pH 8, 20 mM Tris-HCl pH 8,150 mM NaCl), 2x with High Salt Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA pH 8, 20 mM Tris-HCl pH 8, 500 mM NaCl), 2x with LiCl Buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 0.25 M LiCl, 1% Igepal CA-630, 1% DOC), and 1x with TE buffer. DNA was eluted 2x using IP elution buffer (0.1 M NaHCO3, 1% SDS) for 10 min at room temperature, followed by 5 min at 37°C and transferring to a new tube, combining eluates. For crosslink reversal, we added 20 μl 5 M NaCl, 8 μl 0.5 M EDTA and 16 μl 1 M Tris-HCl pH8, incubating 1.5 h at 68°C. Afterward we added 8 μl proteinase K and incubated at 50°C for 2 h. DNA was then precipitated in ethanol with sodium acetate, resuspending in 300 μl 10 mM Tris-Cl pH 8.5. To enrich for ligation events, we prepared Streptavidin beads by washing in 400 μl TWB (5 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1 M NaCl, 0.05% Tween 20) and resuspending in 300 μl of 2x Binding Buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 2 M NaCl). Beads were added to the sample and incubated at room temperature for 15 min with rotation. Samples were then washed 2x in TWB and the standard Hi-C library preparation was followed for some replicates (Rao et al., 2014) while the Tn5 library prep was followed for others (Mumbach et al., 2016).
Data Processing
Analysis of ChIP-seq data
Sequences were mapped to the dm6 genome with Bowtie 0.12.3 (Langmead et al., 2009) using default settings. Statistics for each ChIP-seq experiment can be found in Table S1. Peaks were then called with MACS2 with IgG as a control and a p value of 1e-3. Peaks detected by the PsqL and Psqtot antibodies were first combined into one list and overlapping summits +− 200 bp were merged into one peak. This total peak list was then categorized into PsqL sites such that read counts were greater than 99.5% of random regions for PsqL (FDR < 0.005) but less than that for PsqS. The remaining peaks were then split into PsqS peaks, i.e., those with signal only in PsqS (FDR < 0.005), and peaks with signal in both PsqLand PsqSto determine PsqL&S sites. Published ORGANIC ChIP-seq was mapped similarly to dm6 and then filtered for reads ≤ 50 bp in size as was done previously (Kasinathan et al., 2014). Changes in ChIP-seq occupancy after ecdysone treatment were measured by MAnorm using the parameters recommended for transcription factor binding (Shao et al., 2012). Motifs for Psq were identified by MEME-ChIP using default settings (Machanick and Bailey, 2011) and genomic coordinates were identified via fimo with a q-value < 0.05. Overlaps between peaks and motifs were evaluated using bedtools.
Analysis of Hi-C and HiChIP Data
Sequenced reads were mapped to the Drosophila dm6 genome, further processed to remove duplicates and self-ligations using the Juicer pipeline, and visualized using Juicebox (Durand et al., 2016a, 2016b). Statistics for each library can be found in Tables S2 and S3. Pc loops were identified using Chicago (Cairns et al., 2016) at 5 kb resolution using the recommended parameters. Due to the intensity of Pc loops, several bins surrounding the midpoint of the loop were called significant (Rao et al., 2014), thus only the pixel with the highest signal compared to the surrounding interactions was called. Enhancer-promoter interactions were called in H3K27ac HiChIP data (Rowley et al., 2017) using the Chicago package to identify statistically significant interactions (Cairns et al., 2016). These interactions were filtered to obtain those connecting STARR-seq enhancers to promoters. These interaction anchors were then overlapped with Psq binding sites to identify Psq-bound enhancer-promoter interactions. 2D metaplots were obtained by taking the median distance normalized Hi-C or HiChIP signal for the area surrounding either point-to-point interactions or across scaled regions. Scores for point-to-point interaction metaplots were calculated similar to APA (Rao et al., 2014) but instead of sums of interactions, which are affected by the variance, we took the median signal at the interaction point compared to the top right corner. Pc domains were identified by extracting 1 kb bins with more than 3-fold Pc ChIP-seq signal compared to IgG. Regions with more than 10 bins of enriched Pc with each bin within at least a 4 kb distance were kept. Compartmental interactions surrounding Pc domains in Figure 3E were visualized by the Pearson correlation matrix of the Hi-C data. Interaction strength between Pc domains and other features were calculated from the average distance normalized signal between these domains and other features.
QUANTIFICATION AND STATISTICAL ANALYSIS
Hi-C and HiChIP data represent the signal obtained from a population of cells, n = 20 million or n = 100 million respectively. Mapping statistics for HiChIP data were generated using Juicer (Durand et al., 2016b) and are displayed in Tables S2–S4. Metaplots of Hi-C or HiChIP data represent the median signal in each bin surrounding a central point or the median signal across bins scaled for features of non-uniform sizes, e.g., genes. Central point enrichment in Hi-C or HiChIP metaplots, termed the APA score (Rao et al., 2014), was calculated by dividing the signal in the center bin by the average signal across a 3x3 square in the upper right corner. The results of this test are labeled in the figures where applicable. Profiles of sequencing data across genomic loci represent the average or median signal as described on each figure and were generated using ngs.plot (Shen et al., 2014) or by using custom scripts. Correlations were evaluated using a Pearson correlation test and are displayed in the figures and in the figure legends. Boxplots were generated in R and the center line represents the median, the box represents the interquartile range (IQR) of quartiles Q1-Q3, and lower and upper whiskers represent the Q1 − 1.5*IQR or Q3 + 1.5*IQR. Significant differences were evaluated by the Wilcoxon Rank Sum test in R, the results of which are described in the figure legends.
DATA AND CODE AVAILABILITY
ChIP-seq, and HIChIP data are available from NCBI’s Gene Expression Omnibus (GEO). The accession number for all the datasets reported in this paper is GEO: GSE118047. Custom scripts were used to generate 2D metaplots of Hi-C and HiChIP data. These scripts are available without restrictions upon request.
Supplementary Material
Highlights.
PsqL and PsqS variants play distinct roles in gene expression and tumorigenesis
PsqS colocalizes with GAF, CBP, ISWI, H3K27ac, and Pc at enhancers
PsqS and Pc form loops, whereas Pc domains mediate compartmental interactions
PsqS and Pc-bound enhancers respond to ecdysone, altering 3D chromatin interactions
ACKNOWLEDGMENTS
We thank I. Oliveira, M.C. Martinez-Moratalla, and L. Mira for technical assistance; I. Gutierrez-Garcia and J. Bolivar for the UAS-PsqL transgenic line; the Bloomington Drosophila Stock Center (NIH P40OD018537) for fly stocks; and the Vienna Drosophila Resource Center (VDRC, https://stockcenter.vdrc.at/control/main) for transgenic RNAi fly stocks. We also thank the Genomic Services Lab at the HudsonAlpha Institute for Biotechnology, and especially Drs. Angela Jones and Braden Boone, for help in performing Illumina sequencing of samples. I.G.-P. was supported by a predoctoral FPI fellowship from the Spanish Ministry of Economy and Competitiveness (MINECO) (BES-2010-030958). M.J.R. was supported by NIH Pathway to Independence Award NIGMS K99GM127671. This work was funded by the Fundación Botín Grant, the Generalitat Valenciana (PROMETEO/2017/146), the Fundación Cientifica Asociación Española Contra el Cáncer (AECC) (CICPF16001DOMí), the Spanish Ministry of Economy and Competitiveness (MINECO) (BFU2015-64239-R), and cofinanced by the European Regional Development Fund (ERDF) and the Spanish State Research Agency through the “Severo Ochoa” Program for Centers of Excellence in R&D (SEV-2013-0317) to M.D. This work was also supported by Pathway to Independence Award K99/R00 GM127671 (to M.J.R.) and U.S. Public Health Service Award R01 GM035463 (to V.G.C.) from the NIH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.096.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aranda S, Mas G, and Di Croce L (2015). Regulation of gene transcription by Polycomb proteins. Sci. Adv 1, e1500737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell VJ, Treisman R (1994). The POZ domain: a conserved protein-protein interaction motif. Genes Dev. 8, 1664–1677. [DOI] [PubMed] [Google Scholar]
- Bushey AM, Ramos E, and Corces VG (2009). Three subclasses of a Drosophila insulator show distinct and cell type-specific genomic distributions. Genes Dev. 23, 1338–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J, Freire-Pritchett P, Wingett SW, Várnai C, Dimond A, Plagnol V, Zerbino D, Schoenfelder S, Javierre BM, Osborne C, et al. (2016). CHiCAGO: robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol. 17, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaharbakhshi E, and Jemc JC (2016). Broad-complex, tramtrack, and bric-à-brac (BTB) proteins: Critical regulators of development. Genesis 54, 505–518. [DOI] [PubMed] [Google Scholar]
- Cubeñas-Potts C, Rowley MJ, Lyu X, Li G, Lei EP, and Corces VG (2017). Different enhancer classes in Drosophila bind distinct architectural proteins and mediate unique chromatin interactions and 3D architecture. Nucleic Acids Res. 45, 1714–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Avino PP, and Thummel CS (2000). The ecdysone regulatory pathway controls wing morphogenesis and integrin expression during Drosophila metamorphosis. Dev. Biol 220, 211–224. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, Couto A, Marra V, Keleman, and Dickson BJ (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156. [DOI] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016a). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016b). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagen KP, Aiden EL, and Kornberg RD (2017). Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc. Natl. Acad. Sci. USA 114, 8764–8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erceg J, Pakozdi T, Marco-Ferreres R, Ghavi-Helm Y, Girardot C, Bracken AP, and Furlong EE (2017). Dual functionality of cis-regulatory elements as developmental enhancers and Polycomb response elements. Genes Dev. 31, 590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas G, Gausz J, Galloni M, Reuter G, Gyurkovics H, and Karch F (1994). The Trithorax-like gene encodes the Drosophila GAGA factor. Nature 371, 806–808. [DOI] [PubMed] [Google Scholar]
- Ferres-Marco D, Gutierrez-Garcia I, Vallejo DM, Bolivar J, Gutierrez-Aviño FJ, and Dominguez M (2006). Epigenetic silencers and Notch collaborate to promote malignant tumours by Rb silencing. Nature 439, 430–436. [DOI] [PubMed] [Google Scholar]
- Gómez-Díaz E, and Corces VG (2014). Architectural proteins: regulators of 3D genome organization in cell fate. Trends Cell Biol. 24, 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurudatta BV, and Corces VG (2009). Chromatin insulators: lessons from the fly. Brief. Funct. Genomics Proteomics 8, 276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horard B, Tatout C, Poux S, and Pirrotta V (2000). Structure of a polycomb response element and in vitro binding of polycomb group complexes containing GAGA factor. Mol. Cell. Biol 20, 3187–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz H, and Berg CA (1996). The Drosophila pipsqueak gene encodes a nuclear BTB-domain-containing protein required early in oogenesis. Development 122, 1859–1871. [DOI] [PubMed] [Google Scholar]
- Huang DH, and Chang YL (2004). Isolation and characterization of CHRASCH, a polycomb-containing silencing complex. Methods Enzymol. 377, 267–282. [DOI] [PubMed] [Google Scholar]
- Huang DH, Chang YL, Yang CC, Pan IC, and King B (2002). pipsqueak encodes a factor essential for sequence-specific targeting of a polycomb group protein complex. Mol. Cell. Biol 22, 6261–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivaldi MS, Karam CS, and Corces VG (2007). Phosphorylation of histone H3 at Ser10 facilitates RNA polymerase II release from promoter-proximal pausing in Drosophila. Genes Dev. 21, 2818–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim FD, and Thummel CS (1992). Temporal coordination of regulatory gene expression by the steroid hormone ecdysone. EMBO J. 11, 4083–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinathan S, Orsi GA, Zentner GE, Ahmad K, and Henikoff S (2014). High-resolution mapping of transcription factor binding sites on native chromatin. Nat. Methods 11 , 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JH, Son W, Bae GY, Kang JH, Oh W, and Yoo OJ (2006). A new hepatocytic isoform of PLZF lacking the BTB domain interacts with ATP7B, the Wilson disease protein, and positively regulates ERK signal transduction. J. Cell. Biochem 99, 719–734. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Siegmund T, Lintermann KG, and Korge G (1998). The pipsqueak protein of Drosophila melanogaster binds to GAGA sequences through a novel DNA-binding domain. J. Biol. Chem 273, 28504–28509. [DOI] [PubMed] [Google Scholar]
- Li L, Lyu X, Hou C, Takenaka N, Nguyen HQ, Ong CT, Cubeñas-Potts C, Hu M, Lei EP, Bosco G, et al. (2015). Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol. Cell 58, 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, and Bailey TL (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazina MY, Nikolenko JV, Fursova NA, Nedil’ko PN, Krasnov AN, and Vorobyeva NE (2015). Early-late genes of the ecdysone cascade as models for transcriptional studies. Cell Cycle 14, 3593–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiyama Y, Schuettengruber B, Papadopoulos GL, Chang J-M, and Cavalli G (2018). Polycomb-Dependent Chromatin Looping Contributes to Gene Silencing during Drosophila Development. Mol. Cell 71, 73–88.e5. [DOI] [PubMed] [Google Scholar]
- Pascual-Garcia P, Debo B, Aleman JR, Talamas JA, Lan Y, Nguyen NH, Won KJ, and Capelson M (2017). Metazoan Nuclear Pores Provide a Scaffold for Poised Genes and Mediate Induced Enhancer-Promoter Contacts. Mol. Cell 66, 63–76.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Torrado R, Yamada D, and Defossez PA (2006). Born to bind: the BTB protein-protein interaction domain. BioEssays 28, 1194–1202. [DOI] [PubMed] [Google Scholar]
- Pherson M, Misulovin Z, Gause M, Mihindukulasuriya K, Swain A, and Dorsett D (2017). Polycomb repressive complex 1 modifies transcription of active genes. Sci. Adv 3, e1700944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang S-C, Glenn St. Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon K-R, Sanborn AL, Johnstone SE, Bochkov ID, Huang X, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddiford LM, Cherbas P, and Truman JW (2000). Ecdysone receptors and their biological actions. Vitam. Horm 60, 1–73. [DOI] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2016). The three-dimensional genome: principles and roles of long-distance interactions. Curr. Opin. Cell Biol. 40, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, and Corces VG (2017). Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol. Cell 67, 837–852.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CA, Misulovin Z, Gause M, Koenig A, Gohara DW, Watson A, and Dorsett D (2013). Cohesin and polycomb proteins functionally interact to control transcription at silenced and active genes. PLoS Genet. 9, e1003560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoborg T, and Labrador M (2014). Expanding the roles of chromatin insulators in nuclear architecture, chromatin organization and genome function. Cell. Mol. Life Sci. 71, 4089–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, and Cavalli G (2009). Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 136, 3531–3542. [DOI] [PubMed] [Google Scholar]
- Schwartz YB, and Pirrotta V (2007). Polycomb silencing mechanisms and the management of genomic programmes. Nat. Rev. Genet 8, 9–22. [DOI] [PubMed] [Google Scholar]
- Schwendemann A, and Lehmann M (2002). Pipsqueak and GAGA factor act in concert as partners at homeotic and many other loci. Proc. Natl. Acad. Sci. USA 99, 12883–12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Yuan GC, Orkin SH, and Waxman DJ (2012). MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 13, R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stelzer C, Gerlach D, Yáñez-Cuna JO, Rath M, Boryń LM, Arnold CD, and Stark A (2014). Hormone-responsive enhancer-activity maps reveal predictive motifs, indirect repression, and targeting of closed chromatin. Mol. Cell 54, 180–192. [DOI] [PubMed] [Google Scholar]
- Siegmund T, and Lehmann M (2002). The Drosophila Pipsqueak protein defines a new family of helix-turn-helix DNA-binding proteins. Dev. Genes Evol. 212, 152–157. [DOI] [PubMed] [Google Scholar]
- Simon JA, and Kingston RE (2013). Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 49, 808–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaldone G, Pirone L, Pedone E, Marlovits T, Vitagliano L, and Ciccarelli L (2016). The BTB domains of the potassium channel tetramerization domain proteins prevalently assume pentameric states. FEBS Lett. 590, 1663–1671. [DOI] [PubMed] [Google Scholar]
- Soeller WC, Oh CE, and Kornberg TB (1993). Isolation of cDNAs encoding the Drosophila GAGA transcription factor. Mol. Cell. Biol 13, 7961–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, and Prive GG (2005). Sequence and structural analysis of BTB domain proteins. Genome Biol. 6, R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bortle K, and Corces VG (2013). The role of chromatin insulators in nuclear architecture and genome function. Curr. Opin. Genet. Dev 23, 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bortle K, Nichols MH, Li L, Ong CT, Takenaka N, Qin ZS, and Corces VG (2014). Insulator function and topological domain border strength scale with architectural protein occupancy. Genome Biol. 15, R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bortle K, Nichols MH, Ramos E, and Corces VG (2015). Integrated tRNA, transcript, and protein profiles in response to steroid hormone signaling. RNA 21, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber U, Siegel V, and Mlodzik M (1995). pipsqueak encodes a novel nuclear protein required downstream of seven-up for the development of photoreceptor R3 and R4. EMBO J. 14, 6247–6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AM, Van Bortle K, Ramos E, Takenaka N, Rohrbaugh M, Jones BC, Jones KC, and Corces VG (2011). Regulation of chromatin organization and inducible gene expression by a Drosophila insulator. Mol. Cell 44, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Sung E, Donlin-Asp PG, and Corces VG (2013). A subset of Drosophila Myc sites remain associated with mitotic chromosomes colocalized with insulator proteins. Nat. Commun 4, 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabidi MA, Arnold CD, Schernhuber K, Pagani M, Rath M, Frank O, and Stark A (2015). Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation. Nature 518, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq, and HIChIP data are available from NCBI’s Gene Expression Omnibus (GEO). The accession number for all the datasets reported in this paper is GEO: GSE118047. Custom scripts were used to generate 2D metaplots of Hi-C and HiChIP data. These scripts are available without restrictions upon request.