

Abstract
Introduction:
Inflammatory bowel disease (IBD) affects approximately 1/3 of patients with chronic granulomatous disease (CGD). Comprehensive investigation of the effect of allogeneic hematopoietic cell transplantation (HCT) on CGD IBD and the impact of IBD on transplant outcomes is lacking.
Methods:
We collected data retrospectively from 145 patients with CGD who had received allogeneic HCT at 26 Primary Immune Deficiency Treatment Consortium (PIDTC) centers between January 1, 2005 and June 30, 2016.
Results:
Forty-nine CGD patients with IBD and 96 patients without IBD underwent allogeneic HCT. Eighty-nine percent of patients with IBD and 93% of patients without IBD engrafted (p=0.476). Upper gastrointestinal acute GVHD occurred in 8.5% of patients with IBD and 3.5% of patients without IBD (p=0.246). Lower gastrointestinal acute GVHD occurred in 10.6% of patients with IBD and 11.8% of patients without IBD (p=0.845). The cumulative incidence of acute GVHD grades II-IV was 30% (CI 17–43%) in patients with IBD and 20% (CI 12–29%) in patients without IBD (p=0.09). Five-year overall survival was equivalent for patients with and without IBD: 80% [CI 66–89%] and 83% [CI 72–90%], respectively (p=0.689). All 33 surviving evaluable patients with a history of IBD experienced resolution of IBD by 2 years following allogeneic HCT.
Conclusions:
In this cohort allogeneic HCT was curative for CGD associated IBD. IBD should not contraindicate HCT, as it does not lead to an increased risk of mortality.
This study is registered at ClinicalTrials.gov NCT02082353.
Keywords: Allogeneic hematopoietic cell transplantation, Allogeneic bone marrow transplantation, Allogeneic hematopoietic stem cell transplantation, Chronic granulomatous disease, Primary immunodeficiency, Inflammatory bowel disease
Introduction
Chronic granulomatous disease (CGD) is a primary immune deficiency (PID) caused by pathogenic mutations in genes that encode components of the NADPH oxidase complex. Defects in the NADPH oxidase complex cripple normal neutrophil bactericidal and fungicidal function. Patients with CGD develop an array of life-threatening infections with specific organisms including Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, and Nocardia and Aspergillus species.
In addition to increased susceptibility to infection, patients with CGD exhibit a dysregulated inflammatory response to infection and can develop hyperinflammatory reactions requiring corticosteroids along with antimicrobial therapy. They also are prone to other autoinflammatory disease including colitis or inflammatory bowel disease (IBD), which affects approximately 1/3 of CGD patients.1,2 IBD in CGD patients usually presents during childhood, can affect any part of the gastrointestinal tract, and pathologically often resembles Crohn’s Disease.3 Rectal involvement is almost always observed.4 Peri-rectal or intra-abdominal abscesses and fistulas can lead to significant morbidity.
IBD is often difficult to treat in patients with CGD. Intermittent or chronic corticosteroid treatment is usually employed as first line therapy, but can result in significant steroid-associated morbidity. There is no standardized approach to treatment of CGD in steroid-refractory or steroid-dependent patients. Because tumor necrosis factor-alpha inhibitors can be associated with severe infectious complications in patients with CGD, they are often avoided.5 Other therapies have been used with mixed success.6–10
Allogeneic hematopoietic cell transplantation (HCT) is a curative treatment option for CGD. Historically, allogeneic HCT has been performed in CGD using myeloablative conditioning regimens.11,12 More recently, reduced toxicity/reduced intensity conditioning regimens have increased in use, and generally are associated with survival rates of greater than 80%.13–16 The number of transplants performed for CGD has increased in recent years. CGD was the third most common indication for allogeneic HCT among patients with primary immunodeficiencies reported to the Center for International Blood and Marrow Transplant Research (CIBMTR) from 2010–2016, behind only severe combined immune deficiency and hemophagocytic lymphohistiocytosis disorders.17
It is known that successful allogeneic HCT eliminates the risk of CGD-associated infections. Some reports have also suggested that IBD is cured with allogeneic HCT.11–13,18 However, to date no large-scale evaluations have been undertaken to test the hypothesis that allogeneic HCT resolves the IBD associated with CGD, or to assess the impact of pre-HCT IBD on survival and on the risk of developing gastrointestinal graft versus host disease (GVHD).
The Primary Immune Deficiency Treatment Consortium (PIDTC) consists of 44 centers in the U.S.A. and Canada focused on treatment approaches and outcomes for patients with PID disorders including severe combined immunodeficiency (SCID), Wiskott-Aldrich syndrome (WAS), and CGD. The PIDTC has been funded continuously by the NIH since 2009.19 We collected detailed information on the PIDTC CGD cohort regarding demographics, disease manifestations (including IBD and infections before and after transplant), and details of HCT treatment. We report here the long-term effect of allogeneic HCT on manifestations of CGD–associated IBD, and the effect of IBD on post-HCT survival and other outcomes.
Methods
The study was conducted by the PIDTC, a member of the Rare Diseases Clinical Research Network (RDCRN), in the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS). The PIDTC Protocol 6903 is an observational study registered at ClinicalTrials.gov NCT02082353. The data presented here were collected retrospectively and include 145 patients who underwent allogeneic HCT at 26 centers between January 1, 2005 through June 30, 2016 (Supplemental Table 1). The protocol was approved by the Institutional Review Boards of each participating center. Patient consent was not required as all data was collected using chart review and was deidentified.
Patients
A review panel consisting of immunology and HCT physicians confirmed eligibility for each CGD patient enrolled in PIDTC Protocol 6903. To be eligible, patients were required to have defective neutrophil function as assessed by either the dihydrorhodamine assay (DHR) or nitroblue tetrazolium oxidation test, plus a clinical history or family history consistent with CGD. In cases where either functional test results or patient or family history were equivocal, abnormal western or northern blot data, and/or pathogenic mutations identified by gene sequencing confirmed a diagnosis of CGD. Patient neutrophil function was classified as oxidase-null or oxidase-positive. Oxidase-null was defined as having a DHR assay Stimulation Index ≤ 2.5 or ferricytochrome C reduction assay with O2- < 2.3 nmoles/106 cells/h. Oxidase positive was defined as having results above these cut-offs. For patients who did not have a functional assay performed, those with no detectable protein or a genetic mutation very likely to be associated with absent oxidase production (e.g. null mutations) were also classified as oxidase-null.20
Patients were diagnosed with IBD clinically by their treating physician. No formal PIDTC definition for IBD was used. For patients with a history of IBD that was active or controlled with medications during the year prior to allogeneic HCT, information was collected regarding IBD treatment, physician-reported disease activity prior to allogeneic HCT, and presence or absence of IBD 2 years following allogeneic HCT. Data regarding serum albumin level, height, and weight prior to allogeneic HCT were also recorded. Infections that occurred within the year prior to allogeneic HCT were recorded, and any occurrence of surgical resections including lung, bowel, or liver were noted.
Transplant Procedures, Definitions, and Outcomes
Routine data regarding the first or only allogeneic HCT were collected including donor and recipient human leukocyte antigen (HLA) match, donor relation, graft source, conditioning regimen, and acute graft versus host disease (GVHD) prophylaxis. Patients who received a second allogeneic HCT were censored at the time of the second transplant.
HLA match was confirmed by the CGD protocol team based on review of HLA typing. HLA match was classified based on allele-level typing for 8 HLA alleles (HLA-A, B, C, and DR) for patients who received a bone marrow (BM) or peripheral blood stem cell (PBSC) graft, and based on allele level or antigen level typing for 6 (HLA-A, B, and DR) or 8 HLA (HLA-A, B, C, and DR) alleles for patients who received a cord blood graft, with the exception of 5 patients who received an unrelated donor PBSC or BM graft and had only antigen level typing. HLA match and relationship categories used for this study included matched sibling donor (MSD), matched other related donor (MORD), mismatched related donor (MMRD), matched unrelated donor (MUD), and mismatched unrelated donor (MMUD).
Conditioning regimen intensity was classified using CIBMTR workshop definitions with some exceptions.21 Exceptions included those patients who received a regimen based on reduced toxicity busulfan and fludarabine, and patients who received 10 mg/kg busulfan without an additional chemotherapeutic agent; these were classified together with patients who received reduced intensity conditioning regimens in a “reduced intensity/reduced toxicity” (RIC/RTC) conditioning regimen category. RIC/RTC regimens received by 107 patients included: busulfan plus fludarabine (n=42); busulfan plus low-dose total body irradiation (300 cGy) (n=26); fludarabine plus melphalan, with or without additional chemotherapeutic agents (n=22); or other regimens (n=17). MAC regimens consisted of busulfan and cyclophosphamide with or without additional chemotherapeutic agents (n=38). One patient who received 7mg/kg busulfan, but maintained a myeloablative exposure and also received cyclophosphamide was classified as having received a myeloablative conditioning (MAC) regimen.
Neutrophil recovery following allogeneic HCT was defined as the first instance of 3 consecutive measurements with an absolute neutrophil count above 500 cells/uL. Results of chimerism studies were collected. Graft failure was originally defined in the 6903 protocol as failure to achieve ≥5% donor cells in all lineages or in whole blood by 100 days post-HCT, or receipt of a second allogeneic HCT by day +100, and 9 patients met this definition of graft failure. Based on very recent data in a large number of female carriers of X-linked CGD, in whom a significant risk of infections was associated with <10% oxidase positive neutrophils,22 we amended the graft failure definition to include failure to achieve ≥10% donor cells in the myeloid lineage (as evidenced by chimerism analysis or by neutrophil oxidative burst testing); 3 additional patients were classified as graft failure. Two additional patients were included as having graft failure as each received a second allogeneic HCT at 112 and 117 days following the initial HCT.
Data regarding allogeneic HCT complications and survival were also collected. Acute GVHD was graded according to consensus criteria,23 and centers additionally clarified whether or not there were upper intestinal tract symptoms of persistent nausea or vomiting. Chronic GVHD was graded as limited or extensive.24
Statistics
Data are presented with medians and ranges or with categories and percentages where applicable. Numerical data were compared between patients with and without IBD using the Kruskal–Wallis test. For height and weight percentile Z-scores, data were compared using the Wilcoxon rank-sum test. Categorical data were compared between patients with and without IBD using the chi-squared test. Cumulative incidence curves were generated for acute GVHD grades I-IV, grades II-IV, and grades III-IV, and for chronic GVHD, treating death as a competing risk; cumulative incidence estimates were compared between patients with vs. without IBD prior to allogeneic HCT using Gray’s test. Kaplan-Meier survival curves were generated, and groups were compared using the log-rank test. Patients who underwent a second transplant were censored at the time of second transplant. Multivariate analyses were performed using Cox proportional hazards regression modeling. Covariates were selected using step-wise variable selection; the main effect of IBD was forced into all models. The following variables were considered in the variable selection: age at HCT, year of HCT, oxidase stratum, Karnofsky or Lansky performance score, donor type, HLA matching, conditioning intensity, and history of Aspergillus. The multivariate analysis of acute GVHD risk also included GVHD prophylaxis category. The proportional hazards assumption was assessed for all variables using graphical methods and time-dependent covariates. A p-value of <0.05 was considered statistically significant.
Results
Patients
Of a total of 145 patients, 49 (34%) with IBD and 96 (66%) without IBD underwent allogeneic HCT. Basic demographic data and pre-HCT complications are presented in Table 1. Patient age at the time of CGD diagnosis was similar between patients with and without IBD, with median ages of 1.5 years and 1 year, respectively (p=0.452). The distribution of genetic causes of CGD was similar between groups (p=0.172) with the majority of patients having X-linked CGD. Oxidase function, as defined above, was also comparable between groups (p=0.718). Stratifying for oxidase function, we found that approximately 35% percent of either oxidase-positive or oxidase-null patients had IBD. Patients with IBD were more likely to have received steroids within one year of HCT (p <0.001) and to have received immunosuppressive treatments other than steroids (p <0.001). Despite this, infection rates during the year prior to allogeneic HCT were similar in patients with and without IBD, and the percentages of patients who had undergone a previous lung or liver resection were also similar. As expected, patients with IBD had lower serum albumin levels than patients without IBD, and more patients with IBD had heights and weights below the 5th percentile for age (all p <0.05). A greater percentage of patients within the IBD group had Lansky or Karnofsky scores below 90 (p=0.007). Thirty-two (65%) of the patients with IBD had physician-reported control of their IBD prior to allogeneic HCT.
Table 1.
Demographics and co-morbidities in patients with and without IBD.
| IBD N=49 |
No IBD N=96 |
P | |
|---|---|---|---|
| Median Age (Years) at CGD Diagnosis (Range) | 1.5# (<1–16) | 1# (<1–18) | 0.452 |
| Median Age (Years) at Allogeneic HCT (Range) | 11 (1–26) | 5 (<1–21) | <0.001 |
| Genetic Mutation (Protein Deficiency) | |||
| CYBA (p22phox) | 2 (4.1%) | 6 (6.3%) | 0.172 |
| NCF4 (p40phox) | 1 (2.0%) | ||
| NCF1 (p47phox) | 2 (4.1%) | 15 (15.6%) | |
| NCF2 (p67phox) | 1 (2.0%) | 3 (3.1%) | |
| CYBB (gp91phox) | 40 (81.6%) | 66 (68.8%) | |
| Unknown | 3 (6.1%) | 6 (6.2%) | |
| Oxidase Function | |||
| Unable to Be Determined | 6 (12.2%) | 16 (16.7%) | 0.718 |
| Oxidase Null | 28 (57.1%) | 55 (57.3%) | |
| Oxidase Positive | 15 (30.6%) | 25 (26.0%) | |
| History of Infection(s) Within 1 Year Prior to Allogeneic HCT | 38 (77.6%) | 64 (66.7%) | 0.175 |
| Staphylococcus Aureus | 1 (2.0%) | 8 (8.3%) | 0.137 |
| Nocardia Species Infection | 1 (2.0%) | 4 (4.2%) | 0.507 |
| Aspergillus Species Infection | 7 (14.3%) | 17 (17.7%) | 0.600 |
| History of Pneumonia Within 1 Year Prior to Allogeneic HCT | 23 (46.9%) | 37 (38.5%) | 0.332 |
| History of Lung Resection | 3 (6.1%) | 8 (8.4%) | 0.623 |
| History of Liver Resection | 1 (2.0%) | 0 (0%) | 0.162 |
| History of Gastrointestinal Resection | 0 | 0 | N/A |
| Median Albumin g/dL (Range) | 3.5@ (1.7–4.4) | 3.8@ (2.5–5.0) | <0.001 |
| Weight <5th Percentile for Age | 17* (35.4%) | 14 (14.6%) | 0.004 |
| Height <5th Percentile for Age | 24∧ (51.1%) | 25 (26.0%) | 0.003 |
| Lansky or Karnofsky Score <90 Prior to Allogeneic HCT | 10+ (20.4%) | 5+ (5.2%) | 0.007 |
| Received IBD Therapy | 47− (97.9%) | ||
| IBD Controlled Prior to Allogeneic HCT | 32$ (65%) |
Age at diagnosis was reported for 24 IBD patients and 71 patients without IBD
Albumin data was reported for 48 IBD patients and 92 patients without IBD
Weight data was reported for 48 IBD patients
Height data was reported for 47 IBD patients
Lansky or Karnofsky was unknown for 1 IBD patient and 9 patients without IBD
IBD therapy was reported for 48 IBD patients
IBD status was reported for 48 IBD patients (and reported as unknown for 1 IBD Patient)
Transplant Procedures
Patients with IBD who underwent allogeneic HCT were older (median 11 years (range 1 year to 26 years)), as compared to those without IBD (median 5 years (range <1 year to 21 years)) (p<0.001) (Table 1). Patients undergoing allogeneic HCT received either RIC/RTC (n=108) or MAC (n=37) (Table 2) regimens. Over 90% of patients received serotherapy with either alemtuzumab or anti-thymocyte globulin. Bone marrow was the most common graft source in patients without IBD (63.5%), while mobilized peripheral blood stem cell (PBSC) grafts were the most common in patients with IBD (46.9%) (p<0.001). Just over half of patients in both groups received grafts from MUDs. GVHD prophylaxis varied greatly among patients (Supplemental Table 2). The most commonly used GVHD prophylaxis regimens were a calcineurin inhibitor plus mycophenolate mofetil (n=47), sirolimus monotherapy (n=28), a calcineurin inhibitor plus methotrexate (n=27), and a calcineurin inhibitor plus steroids (n=23); others (n=18) received different prophylactic regimens. There was a higher use of calcineurin inhibitors in combination with various other agents among patients without IBD (82% versus 59%) and a higher use of other therapies, predominantly sirolimus monotherapy, among patients with IBD (41% versus 16%) (p=0.003) (Table 2 and Supplemental Table 2).
Table 2.
Transplant procedures in patients with and without IBD.
| IBD N=49 |
No IBD N=96 |
P | |
|---|---|---|---|
| Conditioning Regimen Intensity | |||
| Myeloablative | 9 (18.4%) | 28 (29.2%) | 0.158 |
| Reduced Toxicity/Reduced Intensity | 40 (81.6%) | 68 (70.8%) | |
| Conditioning Regimen Serotherapy | |||
| Anti-Thymocyte Globulin | 19 (38.8%) | 40∧ (42.6%) |
0.908 |
| Alemtuzumab | 26 (53.1%) | 47∧ (50.0%) |
|
| None | 4 (8.2%) | 7A (7.4%) | |
| Donor and Recipient Relation and HLA Match | 0.689 | ||
| Matched Sibling Donor | 8 (16.3%) | 22 (22.9%) | |
| Matched Other Related Donor | 4 (8.2%) | 4 (4.2%) | |
| Mismatched Related Donor& | 2 (4.1%) | 5 (5.2%) | |
| Matched Unrelated Donor | 29 (59.2%) | 50 (52.1%) | |
| Mismatched Unrelated Donor% | 6 (12.2%) | 15 (15.6%) | |
| Graft Source | <0.001 | ||
| Bone Marrow | 20 (40.8%) | 61 (63.5%) | |
| Peripheral Blood Stem Cell | 23 (46.9%) | 15 (15.6%) | |
| Cord Blood | 6 (12.2%) | 17 (17.7%) | |
| Bone Marrow and Cord Blood | 0 | 3 (3.1%) | |
| Graft Manipulation to Achieve T-cell Depletion | 0 | 2* (2.1%) | 0.454 |
| Acute GVHD Prophylaxis | 0.003 | ||
| Calcineurin Inhibitor +/− 1 or More Additional Agents | 29 (59.2%) | 79 (82.3%) | |
| Other | 20 (40.8%) | 15 (15.6%) | |
| None | 0 | 2 (2.1%) | |
Serotherapy was reported for 94 patients without IBD
Three patients received a 7/8 HLA matched graft, 1 patient received a 6/8 HLA matched graft, 1 patient received a 5/8 HLA matched graft, and 2 patients received haploidentical grafts
Two patients received a 4/6 HLA matched graft, 3 patients received a 5/8 HLA matched graft, 5 patients received a 6/8 HLA matched graft, 11 patients received a 7/8 HLA matched graft
Graft manipulation was reported for 95 patients without IBD
Neutrophil Recovery and Donor Chimerism
Neutrophil recovery occurred before Day +100 in 89% of patients with IBD and 93% of patients without IBD (p=0.476). Fourteen patients (10%) developed graft failure by Day +100 (including patients who underwent a second allogeneic HCT within 4 months of the initial HCT). Ten of these patients underwent a second allogeneic HCT at a median of 112 days following the first HCT (range 39–134 days). The median myeloid donor chimerism through 5 years post-HCT was 100% in patients with and without IBD (range 0–100%) (Figure 1A). The majority of patients maintained stable donor myeloid chimerism of greater than 90% over time (Figure 1A). Median T-cell donor chimerism increased over time, and ranged from 62–82% for the IBD group whereas it was greater than 80–100% at all time points for patients without IBD (Figure 1B). The observed differences were statistically significant only at the 1-year timepoint.
Figure 1.
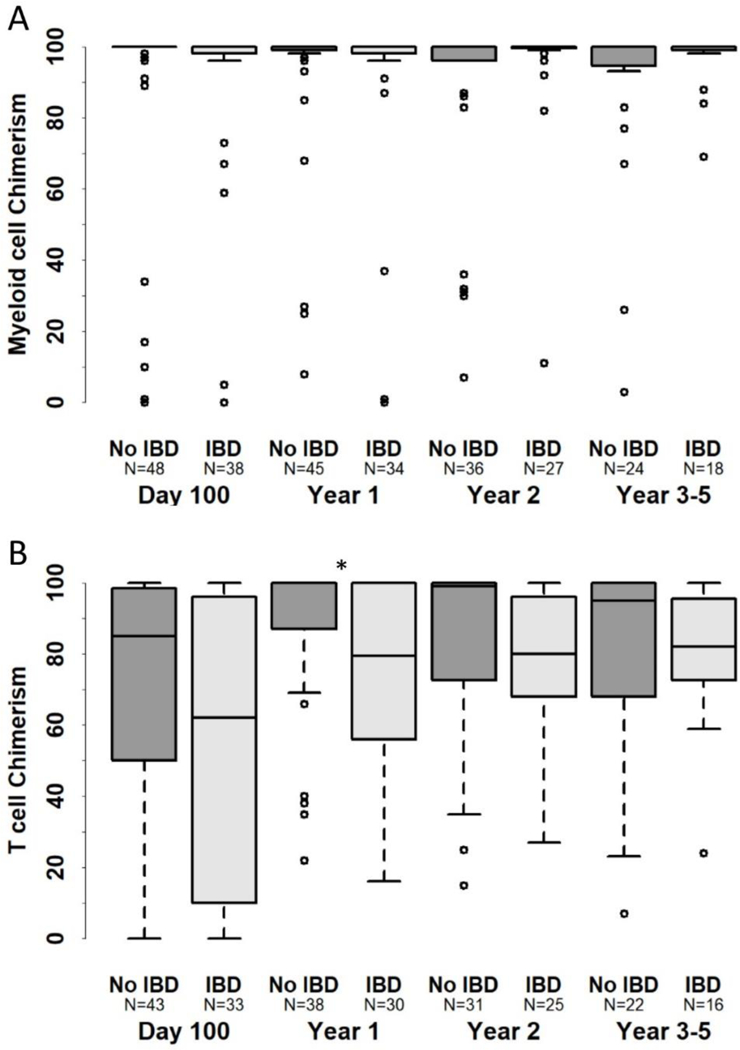
Myeloid (A) and T-cell (B) donor chimerism in patients with and without IBD.
*Indicates p<0.05.
Acute GVHD
The frequency and staging of gastrointestinal acute GVHD was similar between patients with and without IBD (Table 3A). The Day +180 cumulative incidences of acute GVHD grades I-IV were also similar, 38% (95% confidence interval [CI] 24–52%) in patients with IBD and 33% in patients without IBD (CI 23–43%) (p=0.372) (Figure 2A). A trend toward increased grades II-IV acute GVHD in patients with IBD was suggested: the cumulative incidence of acute GVHD grades II-IV was 30% (CI 17–43%) in patients with IBD and 20% (CI 12–29%) in patients without IBD (p=0.09) (Figure 2B). The cumulative incidence of acute GVHD grades III-IV was 11% (CI 4–21%) in patients with IBD and 5% (CI 2–11%) in patients without IBD (p=0.19) (Figure 2C). The occurrence of acute GVHD was similar between patients reported to have controlled or uncontrolled IBD (Table 3B and Figure 3A-C).
Table 3.
Incidence of upper and lower gastrointestinal acute GVHD in evaluable patients with and without IBD prior to allogeneic HCT (A) and in patients with controlled or uncontrolled IBD at the time of allogeneic HCT (B).
| Table 3A: Baseline IBD | ||||
|---|---|---|---|---|
| Variables | Total N (%) |
No N (%) |
Yes N (%) |
P Value |
| Acute GVHD Upper Intestinal Tract (Nausea/Vomiting) | ||||
| No | 125 (94.7) | 82 (96.5) | 43 (91.5) | |
| Yes | 7 (5.3) | 3 (3.5) | 4 (8.5) | 0.246 |
| Acute GVHD Lower Intestinal Tract (Stage 1–4) | ||||
| No | 117 (88.6) | 75 (88.2) | 42 (89.4) | |
| Yes | 15 (11.4) | 10 (11.8) | 5 (10.6) | 0.845 |
| Table 3B: IBD Status (Controlled or Not) | |||||
|---|---|---|---|---|---|
| Variables | Total N (%) |
IBD Controlled with Meds N* (%) |
IBD Not Controlled with Meds N* (%) |
No IBD N (%) |
P Value |
| Acute GVHD Upper Intestinal Tract (Nausea/Vomiting) | |||||
| No | 124 (94.7) | 29 (93.5) | 13 (86.7) | 82 (96.5) | |
| Yes | 7 (5.3) | 2 (6.5) | 2 (13.3) | 3 (3.5) | 0.220 |
| Acute GVHD Lower Intestinal Tract (Stage 1–4) | |||||
| No | 117 (89.3) | 27 (87.1) | 15 (100.0) | 75 (88.2) | |
| Yes | 14 (10.7) | 4 (12.9) | 0 (0.0) | 10 (11.8) | 0.405 |
One IBD patient was reported to have unknown status of control at the time of allogeneic HCT, and so was excluded from analyses
Figure 2.
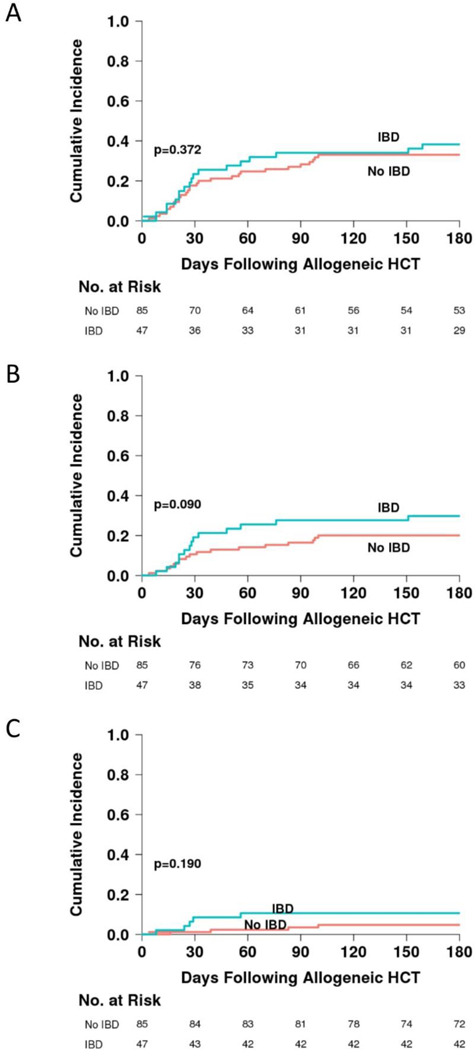
Cumulative incidence of acute GVHD grades I-IV (A), grades II-IV (B), and grades III-IV (C) in patients with and without IBD prior to allogeneic HCT.
Figure 3.
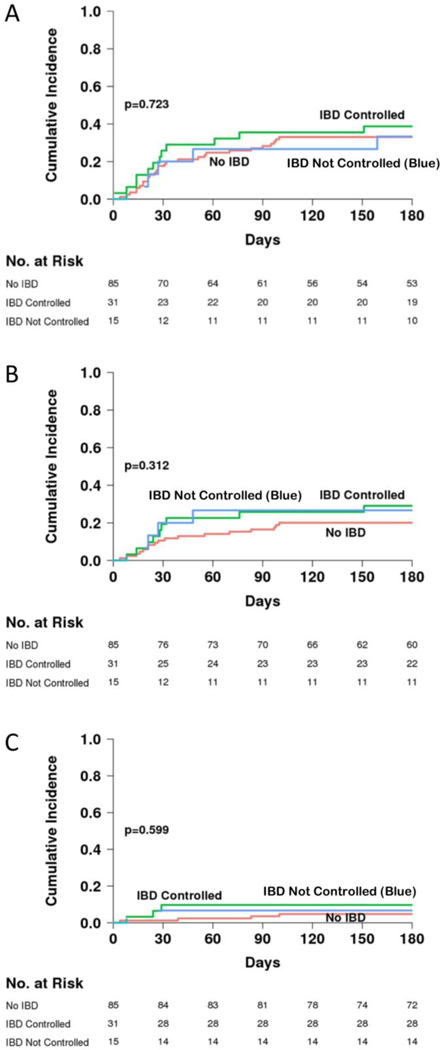
Cumulative incidence of acute GVHD grades I-IV (A), grades II-IV (B), and grades III-IV (C) in patients without IBD, in patients with IBD that was reported to be controlled, and patients with IBD that was reported to be uncontrolled prior to allogeneic HCT.
We performed multivariate analyses to assess the risks for acute GVHD grades II-IV. As expected, donor and patient HLA mismatch conferred a highly significant increased risk of grades II-IV acute GVHD (hazard ratio (HR) 4.03 (1.99–8.17), p=0.0001)) (Table 4A). IBD may have also influenced the risk of acute GVHD grades II-IV (HR 1.98 (1.00–3.94) but the effect did not quite reach statistical significance (p=0.051) (Table 4A). The risk of acute GVHD grades II-IV was similar between patients reported to have controlled or uncontrolled IBD (HR 1.89 and 1.86, respectively, Table 4B).
Table 4.
Multivariate analysis of risk of acute GVHD grades II-IV including patients with or without IBD (A, N=132 evaluable patients) or including patients stratified by control or not of IBD (B, N=131 evaluable patients).
| 4A Baseline IBD | |||
|---|---|---|---|
| Parameter | N (N Event) | HR (%95 CI) | P-value |
| Baseline IBD status | |||
| No (reference) | 85 (17) | 1.00 | |
| Yes | 47 (16) | 1.98 (1.00, 3.94) | 0.051 |
| Donor type (combined) | |||
| Matched donor (MUD, MORD or MSD) (reference) | 108 (20) | 1.00 | |
| Mismatched donor (MMUD or MMRD) | 24 (13) | 4.03 (1.99, 8.17) | 0.0001 |
| 4B IBD Status (Controlled or Not) | |||
|---|---|---|---|
| Parameter | N (N Event) | HR (%95 CI) | P-value |
| IBD status at Treatment | |||
| No (reference) | 85 (17) | 1.00 | |
| IBD Controlled | 31* (10) | 1.89 (0.86, 4.15) | 0.112 |
| IBD Uncontrolled | 15* (5) | 1.86 (0.69, 5.05) | 0.223 |
| Donor type (combined) | |||
| Matched donor (MUD, MORD or MSD) (reference) | 107(19) | 1.00 | |
| Mismatched donor (MMUD or MMRD) | 24 (13) | 4.16 (2.04, 8.51) | <0.0001 |
One patient with IBD was reported to have unknown control at the time of HCT and was excluded
MUD, Matched Unrelated Donor
MORD, Matched Other Related Donor
MSD, Matched Sibling Donor
MMUD, Mismatched Unrelated Donor
MMRD, Mismatched Related Donor
“N” is the number of subjects in the category of the variable; the “N event” is defined by acute GVHD grades II-IV.
The “HR” is for the estimate of acute GVHD grades II-IV.
For each “parameter”, the alternative category is compared with the reference group.
“P-value” provides the overall significance of the variable in the model.
Chronic GVHD
The 3-year cumulative incidence of chronic GVHD was 18% (8–31%) in patients with IBD. The 3-year cumulative incidence of chronic GVHD was 27% (17–37%) in patients without IBD (p=0.285).
Survival
Twenty-three patients died, with the primary causes of death reported to be related to organ toxicities (n=10), infections (n=8), GVHD (n=3), graft failure (n=1), and unknown (n=1). The probability of overall survival for all patients was 84% (CI 76–89%) at 2 years, and 82% (CI 74–88%) at 5 years, with a median follow-up of 3.5 years in surviving patients.
Survival was similar for patients with and without IBD. The probability of 5-year overall survival was 80% (CI 66–89%) for patients with IBD, and 83% (CI 72–90%) for patients without IBD (p=0.689) (Figure 4A). There was no difference in survival between patients reported to have controlled or uncontrolled IBD (Figure 4B). The probability of 5-year overall survival for patients who received MAC was 75% (CI 57–86%), and for patients who received RIC/RTC 84% (CI 75–91%) (Figure 4C) (p=0.130). Survival was equivalent for patients transplanted with HLA matched and mismatched donors in this study (Figure 4D). In multivariate analyses, no covariates were associated with a statistically significant adverse impact on survival, including IBD (p=0.689) (Table 5).
Figure 4.
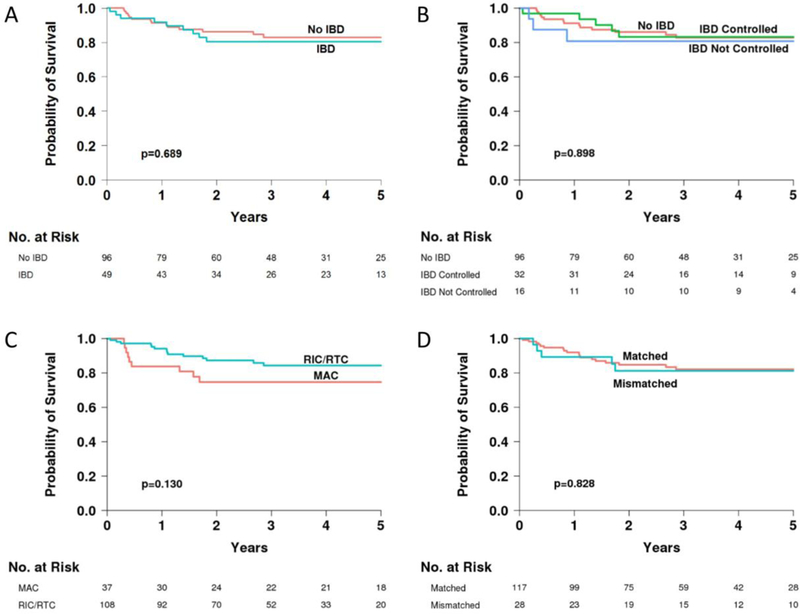
Five-year overall survival in patients with and without IBD (A), further divided into controlled or uncontrolled IBD (B), in patients who received myeloablative versus reduced intensity/reduced toxicity conditioning regimens (C), and in patients stratified by donor HLA match (D).
Table 5.
Multivariate analysis of risk of death (N=145).
| Parameter | N (N Event) | HR (%95 CI) | P-value |
|---|---|---|---|
| Baseline IBD status | |||
| No (reference) | 96(14) | 1.00 | |
| Yes | 49 (9) | 1.25 (0.54, 2.90) | 0.604 |
| Conditioning intensity | |||
| RIC/RTC (reference) | 108 (14) | 1.00 | |
| MAC | 37 (9) | 1.94 (0.82, 4.61) | 0.132 |
| Donor type (combined) | |||
| Matched donor (MUD, MORD or MSD) (reference) | 117 (18) | 1.00 | |
| Mismatched donor (MMUD or MMRD) | 28 (5) | 0.96 (0.35, 2.67) | 0.942 |
Note: No other predictors were significant in either stepwise selection or backward elimination. IBD, donor HLA match and relation, and conditioning intensity were forced into the model included here.
“N” is the number of subjects in the category of the variable; the “N event” is defined by death.
The “HR” is for the estimate of death.
For each “parameter”, the categories are compared with the reference group.
Resolution of IBD Following Allogeneic HCT
Of 49 patients who had suffered from IBD at baseline, 33 were alive and available for evaluation of IBD status 2 years after allogeneic HCT. All (100%) surviving and available patients experienced resolution of IBD (CI 89–100%). Sixteen of these patients (49%) were reported to have resolution by 100 days following HCT, and 10 additional patients (30%) were reported to have resolution at 1 year.
We examined the available 2-year neutrophil oxidative burst results and/or chimerism studies for these patients that were known to have resolution of IBD. All patients who had an oxidative burst reported at 2 years (N=20) had normal function in 83–100% of neutrophils except for 1 patient who had normal burst function in 12% of neutrophils. Seven patients who did not have an oxidative burst reported had engraftment studies performed on myeloid lineage cells which revealed 92% or greater donor chimerism. An additional 6 patients had engraftment studies performed on whole blood, peripheral blood mononuclear cell, or other sample which revealed 97% or greater donor chimerism. T-cell donor chimerism at 2 years was reported for 25 patients and ranged from 27–100%, with a median T-cell chimerism of 80%.
Growth Following Allogeneic HCT
Prior to transplant, more patients in the IBD group had heights and weights below the 5th percentile for age, and the median Z-score for height in patients with IBD was lower (−1.7) compared to patients without IBD (−1.0) (p=0.032) (Figure 5A). The median Z-score for weight also trended to be lower prior to transplant in patients with IBD compared to patients without IBD (−1.0 versus −0.5, p=0.059) (Figure 5B). By 3–5 years following allogeneic HCT, both groups had experienced increases in height and weight and made gains in Z scores, but patients with IBD maintained smaller heights and lower weights over time compared to patients without IBD (p<0.05) (Figure 5).
Figure 5.
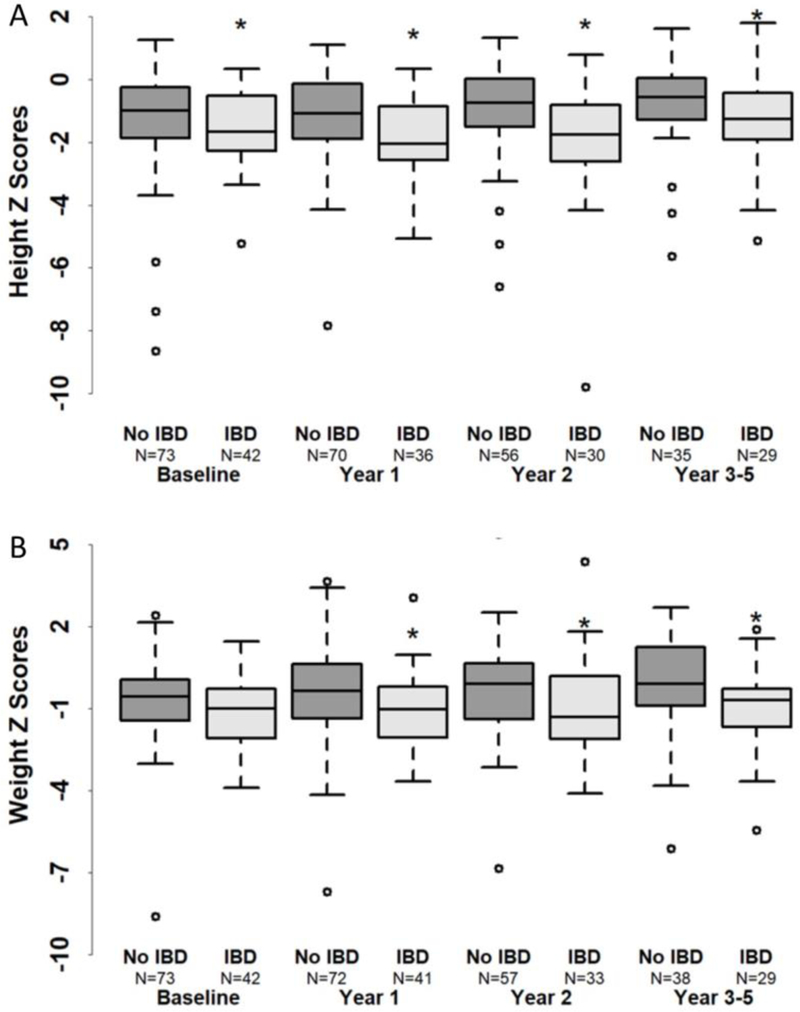
Height z score (A) and weight z score (B) in patients with and without IBD.
*Indicates p<0.05.
Discussion
Our data suggest that allogeneic HCT offers an attractive option for cure of CGD-associated IBD. Allogeneic HCT has been reported to result in resolution of colitis in small numbers of patients, 11,12,25 but systematic study of the impact of allogeneic HCT on the clinical course of colitis in a large group of patients has previously been lacking. To the best of our knowledge, this is the first such report in a large cohort of CGD patients. We show that successful allogeneic HCT indeed resulted in resolution of colitis in patients with CGD-related IBD. All evaluable patients were reported to experience resolution by 2 years following HCT. There are often confounding factors that make evaluation of IBD resolution difficult following transplant, including acute GVHD of the GI tract and possible effects from conditioning regimens and GVHD prophylaxis. Nevertheless, half of patients with IBD were reported to experience resolution of their IBD by Day +100, and three quarters of patients experienced resolution by 1 year.
We observed high levels of myeloid engraftment in all patients with IBD who were surviving at 2 years. A recent analysis of X-linked CGD carriers suggested that a %DHR+ value of <10% is strongly associated with risk of infections, suggesting that values of >10% myeloid chimerism are needed for protection against infection.22 However, such a threshold was not observed in 2 studies of patients or carriers with autoimmunity or inflammatory complications.22,26 It is concerning that levels of myeloid chimerism >10% may still be permissive for ongoing inflammatory or autoimmune manifestations despite being protective against infections.
Because all but one IBD patient had greater than 80% donor myeloid chimerism at 2 years following allogeneic HCT, we were unable to estimate a minimum donor neutrophil chimerism threshold needed for resolution of IBD. However, it is notable that IBD resolved in a patient with normal oxidative burst activity in 12% of neutrophils at 2 years following HCT, suggesting that this level of donor neutrophil chimerism can be curative.
Interestingly, in the T-cell compartment, substantial levels of mixed chimerism were observed for a majority of patients in this study. Approximately half of patients possessed donor chimerism within the T-cell compartment of <80%. These findings suggest that robust T-cell donor chimerism is not needed for resolution of CGD-associated IBD and should not be a goal of therapy for allogeneic HCT.
We observed that the age at HCT was higher in the IBD group. Older age at time of HCT suggests that patients being transplanted with IBD were referred later either due to an attempt to manage the disease with other modalities first, or due to a hesitation to consider HCT in a patient experiencing colitis. Later HCT may also reflect the evolution of colitis in CGD, i.e. patients with CGD often experience presentation of IBD at older ages. Despite older age at HCT, outcomes were not worse in this group.
Earlier reports suggested that CGD patients with active infection, inflammation, or end organ damage had an increased risk of post-HCT complications, such as severe acute GVHD and decreased survival. 11,12,25 We did not observe any clearly significant increased risk of acute GVHD in our cohort, although the analysis of the effect of IBD on grades II-IV acute GVHD may have been limited by the small sample size. Notably, we did not find an increased incidence of gastrointestinal acute GVHD. We speculate that a lack of T-cell involvement in the pathophysiology of CGD-associated IBD may account for the lack of the overtly increased risk of GVHD one would expect in the presence of active inflammation in the GI tract. Another explanation may be the fact that most patients were treated with serotherapy, which may have helped to attenuate active inflammation pre-HCT. In addition, most of the patients were reported to have their IBD under control at the time of HCT. However, the evaluation of the impact of IBD control was limited in our current study because no systematic assessment of IBD severity was performed; the assessment of IBD control or not at the time of allogeneic HCT was made on a clinical basis. Additional experience and study will be required to definitively determine if IBD has an impact on risk for acute GVHD. Importantly, the presence of IBD did not affect mortality. This is a reassuring finding for clinicians and patients as they weigh the risks and benefits of allogeneic HCT in the presence of IBD, and may encourage patients to proceed with HCT earlier even when colitis is present. Additional prospective studies in larger numbers of patients that can uniformly assess IBD severity at the time of allogeneic HCT are needed to more completely assess the risk of severe active IBD on transplant survival.
It is notable that patients with a history of IBD were smaller at the time of allogeneic HCT and maintained smaller heights and weights following transplant. Moving earlier to allogeneic HCT in patients with CGD IBD before significant declines in growth have occurred may have a beneficial effect on ultimate height achievement, and should be taken in to consideration.
In conclusion, the PIDTC Protocol 6903 transplant cohort is the largest described within the North American CGD population to date. Our results confirm that allogeneic HCT is curative for CGD colitis, and importantly, that colitis itself is not a contraindication to HCT as it does not lead to an increased risk of mortality. Although in this observational study we did show benefit of HCT, with resolution of CGD IBD, a comparable analysis of patients who were managed medically would be desirable, to more fully evaluate the benefits of HCT as compared to other treatments. For this purpose, the PIDTC is continuing to collect data on the outcomes of recipients of HCT and of non-HCT treatments for CGD on Protocol 6903. This report and our future evaluations should facilitate counseling of patients with CGD and their families and foster an improved understanding of treatment options for individual patients. Additionally, we believe that prospective multicenter studies evaluating novel reduced intensity/reduced toxicity allogenic HCT approaches for CGD patients with therapy-dependent/refractory IBD are warranted. Prospective studies would allow detailed uniform IBD assessments to be performed prior to allogeneic HCT to characterize the extent and activity of IBD immediately before transplant. Meaningful examinations of the impact of predetermined IBD severity status groupings and exploratory biomarker levels on outcomes can then be performed. Future efforts by the PIDTC and other groups to accomplish such trials will help optimize care for patients with CGD-associated IBD.
Supplementary Material
Key Points.
CGD-associated inflammatory bowel disease (IBD) resolves after allogeneic hematopoietic cell transplant (HCT).
Five-year survival following allogeneic HCT was greater than 80% in this PIDTC CGD cohort, and baseline IBD did not adversely affect survival.
Acknowledgement
The PIDTC is supported by the Division of Allergy, Immunology and Transplantation, National Institute of Allergy and Infectious Diseases (NIAID); and the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH), Bethesda, MD; Public Health Service grant/cooperative agreements U54-AI082973 (PI: MJ Cowan), U54-NS064808 and U01-TR001263 (PI: JP Krischer) and R13-AI094943 (PI: MJ Cowan).
The PIDTC is a part of the Rare Diseases Clinical Research Network (RDCRN) of ORDR, NCATS. The content and opinions expressed are solely the responsibility of the authors and do not represent the official policy or position of the NIAID, ORDR, NCATS, NIH, or any other agency of the US Government.
Support was also provided by the Division of Intramural Research, NIAID, NIH (EMK, LDN, HLM, PG and DG).
We thank Elizabeth Dunn and Tara Bani-Hashemi for project management and assistance. We thank all of the study coordinators for collection of clinical data from PIDTC sites. We thank all of the clinical care teams who provided care for patients.
LMG, an employee of DAIT, NIAID, the funding source, also participated as a PIDTC study team member in the interpretation of data, writing of the manuscript, and decision to submit the manuscript for publication.
Footnotes
Conflict-of-interest disclosure
The authors declare no competing financial interests.
Contributor Information
Rebecca A. Marsh, Department of Pediatrics, University of Cincinnati, and Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH.
Jennifer W. Leiding, Division of Allergy and Immunology, Department of Pediatrics, Johns Hopkins-All Children’s Hospital, University of South Florida, St. Petersburg, FL.
Brent R. Logan, Division of Biostatistics, Medical College of Wisconsin, Milwaukee, WI.
Linda M. Griffith, Division of Allergy, Immunology, and Transplantation, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD
Danielle Arnold, Allergy and Immunology, The Children’s Hospital of Philadelphia, Philadelphia, PA.
Elie Haddad, CHU Sainte-Justine, Immunology-Rheumatology Division, Department of Pediatrics, University of Montreal, QC, Canada.
E. Liana Falcone, Division of Immunity and Viral Infections, Institut de Recherches Cliniques de Montréal, Montréal, QC, Canada; and Department of Medicine, Université de Montréal, Montréal, QC, Canada.
Ziyan Yin, Division of Biostatistics, Medical College of Wisconsin, Milwaukee, WI.
Kadam Patel, Division of Biostatistics, Medical College of Wisconsin, Milwaukee, WI.
Erin Arbuckle, Department of Pediatrics, Duke University, Durham, NC.
Jack J. Bleesing, Department of Pediatrics, University of Cincinnati, and Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH
Kathleen E. Sullivan, Allergy and Immunology, The Children’s Hospital of Philadelphia, Philadelphia, PA
Jennifer Heimall, Allergy and Immunology, The Children’s Hospital of Philadelphia, Philadelphia, PA.
Lauri M. Burroughs, Fred Hutchinson Cancer Research Center, Seattle Children’s Hospital, and the University of Washington School of Medicine, Seattle, WA
Suzanne Skoda-Smith, Seattle Children’s Hospital, Seattle, WA.
Shanmuganathan Chandrakasan, Division of Bone Marrow Transplant, Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta, Emory University School of Medicine, Atlanta, Georgia.
Lolie Yu, Division of Hematology/Oncology and Hematopoietic Stem Cell Transplantation, The Center for Cancer and Blood Disorders, Children’s Hospital/Louisiana State University Medical Center, New Orleans, LA.
Benjamin R. Oshrine, Cancer and Blood Disorders Institute, Johns Hopkins All Children’s Hospital, St. Petersburg, FL
Geoffrey Cuvelier, Manitoba Blood and Marrow Transplant Program, CancerCare Manitoba, University of Manitoba, Winnipeg, MB, Canada.
Monica Thakar, Fred Hutchinson Cancer Research Center, Seattle Children’s Hospital, and the University of Washington School of Medicine, Seattle, WA.
Karin Chen, Division of Allergy and Immunology, Department of Pediatrics, University of Utah School of Medicine, Salt Lake City, UT, United States.
Pierre Teira, CHU Sainte-Justine, Hematology-Oncology Division, Department of Pediatrics, University of Montreal, QC, Canada.
Shalini Shenoy, Division of Pediatric Hematology/Oncology/Transplant, Washington University School of Medicine, St. Louis, MO.
Rachel Phelan, Pediatric Blood and Marrow Transplant Program, Division of Hematology, Oncology, and Blood Marrow Transplantation, Medical College of Wisconsin, Milwaukee, WI.
Lisa R. Forbes, Department of Pediatrics, Baylor College of Medicine, Houston, TX, United States and Section of Allergy, Immunology and Retrovirology, Texas Children’s Hospital William T. Shearer Center Human Immunobiology, Houston, TX, United States.
Deepak Chellapandian, Blood and Marrow Transplant Program, Johns Hopkins All Children’s Hospital, St. Petersburg, Florida.
Blachy J. Dávila Saldaña, Division of Blood and Marrow Transplantation, Children’s National Medical Center, Washington, District of Columbia, and Department of Pediatrics, The George Washington University, Washington, District of Columbia.
Ami J. Shah, Division of Stem Cell Transplantation and Regenerative Medicine, Lucille Packard Children’s Hospital, Stanford School of Medicine, Palo Alto, California
Katja Weinacht, Division of Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Stanford School of Medicine, Stanford, CA.
Avni Joshi, Division of Pediatric Allergy and Immunology, Mayo Clinic, Rochester, MN.
Farid Boulad, Department of Pediatrics, BMT Service, Memorial Sloan Kettering Cancer Center, New York.
Troy Quigg, Texas Transplant Institute, Methodist Children’s Hospital, San Antonio, TX.
Christopher C. Dvorak, Pediatric Allergy, Immunology, and Blood and Marrow Transplant Division, University of California, San Francisco Benioff Children’s Hospital, San Francisco, CA
Debi Grossman, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
Troy Torgerson, Department of Pediatrics, Divisions of Immunology/Rheumatology University of Washington and Seattle Children’s Hospital, Seattle, Washington.
Pamela Graham, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
Vinod Prasad, Division of Pediatric Blood and Marrow Transplant, Duke University Medical Center, Durham, North Carolina.
Alan Knutsen, Pediatric Allergy and Immunology, Saint Louis University, Cardinal Glennon Children’s Medical Center, St. Louis, MO.
Hey Chong, UPMC Children’s Hospital of Pittsburgh, Pittsburgh, PA.
Holly Miller, Center for Cancer and Blood Disorders, Phoenix Children’s Hospital, Phoenix, AZ.
M. Teresa de la Morena, Department of Pediatrics/Immunology, University of Washington and Seattle Children’s Hospital, Seattle, WA.
Kenneth DeSantes, American Family Children’s Hospital, University of Wisconsin, Madison, WI.
Morton J. Cowan, Pediatric Allergy, Immunology, and Blood and Marrow Transplant Division, University of California, San Francisco Benioff Children’s Hospital, San Francisco, CA
Luigi D. Notarangelo, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD
Donald B. Kohn, Pediatrics, David Geffen School of Medicine at University of California, Los Angeles, Los Angeles, CA
Elizabeth Stenger, Aflac Center and Blood Disorders Center, Children’s Healthcare of Atlanta, Emory University, Atlanta, GA.
Sung-Yun Pai, Hematology-Oncology, Boston Children’s Hospital, Boston, MA.
John M. Routes, Division of Allergy and Immunology, Department of Pediatrics, Medical College of Wisconsin, Milwaukee, WI
Jennifer M. Puck, Pediatric Allergy, Immunology, and Blood and Marrow Transplant Division, University of California, San Francisco Benioff Children’s Hospital, San Francisco, CA
Neena Kapoor, Blood and Marrow Transplant Program, Division of Hematology, Oncology and Blood and Marrow Transplantation, Children’s Hospital Los Angeles, Keck School of Medicine, University of Southern California, Los Angeles, CA.
Michael A. Pulsiphe, Blood and Marrow Transplant Program, Division of Hematology, Oncology and Blood and Marrow Transplantation, Children’s Hospital Los Angeles, Keck School of Medicine, University of Southern California, Los Angeles, CA
Harry L. Malech, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
Suhag Parikh, Division of Pediatric Blood and Marrow Transplant, Duke University, Durham, NC.
Elizabeth M. Kang, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD.
References
- 1.Marciano BE, Rosenzweig SD, Kleiner DE, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004;114(2):462–468. [DOI] [PubMed] [Google Scholar]
- 2.Jones LB, McGrogan P, Flood TJ, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol. 2008;152(2):211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marks DJ, Miyagi K, Rahman FZ, Novelli M, Bloom SL, Segal AW. Inflammatory bowel disease in CGD reproduces the clinicopathological features of Crohn’s disease. Am J Gastroenterol. 2009; 104(1): 117–124. [DOI] [PubMed] [Google Scholar]
- 4.Khangura SK, Kamal N, Ho N, et al. Gastrointestinal Features of Chronic Granulomatous Disease Found During Endoscopy. Clin Gastroenterol Hepatol. 2016;14(3):395–402 e395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uzel G, Orange JS, Poliak N, Marciano BE, Heller T, Holland SM. Complications of tumor necrosis factor-alpha blockade in chronic granulomatous disease-related colitis. Clin Infect Dis. 2010;51(12):1429–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell N, Chapdelaine H. Treatment of chronic granulomatous disease-associated fistulising colitis with vedolizumab. J Allergy Clin Immunol Pract. 2017;5(6):1748–1749. [DOI] [PubMed] [Google Scholar]
- 7.de Luca A, Smeekens SP, Casagrande A, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A. 2014;111(9):3526–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van de Veerdonk FL, Netea MG, Dinarello CA, van der Meer JW. Anakinra for the inflammatory complications of chronic granulomatous disease. Neth J Med. 2011;69(2):95. [PubMed] [Google Scholar]
- 9.Hahn KJ, Ho N, Yockey L, et al. Treatment With Anakinra, a Recombinant IL-1 Receptor Antagonist, Unlikely to Induce Lasting Remission in Patients With CGD Colitis. Am J Gastroenterol. 2015;110(6):938–939. [DOI] [PubMed] [Google Scholar]
- 10.Angelino G, De Angelis P, Faraci S, et al. Inflammatory bowel disease in chronic granulomatous disease: An emerging problem over a twenty years’ experience. Pediatr Allergy Immunol. 2017;28(8):801–809. [DOI] [PubMed] [Google Scholar]
- 11.Soncini E, Slatter MA, Jones LB, et al. Unrelated donor and HLA-identical sibling haematopoietic stem cell transplantation cure chronic granulomatous disease with good long-term outcome and growth. Br J Haematol. 2009;145(1):73–83. [DOI] [PubMed] [Google Scholar]
- 12.Seger RA, Gungor T, Belohradsky BH, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985–2000. Blood. 2002;100(13):4344–4350. [DOI] [PubMed] [Google Scholar]
- 13.Gungor T, Teira P, Slatter M, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383(9915):436–448. [DOI] [PubMed] [Google Scholar]
- 14.Khandelwal P, Bleesing JJ, Davies SM, Marsh RA. A Single-Center Experience Comparing Alemtuzumab, Fludarabine, and Melphalan Reduced-Intensity Conditioning with Myeloablative Busulfan, Cyclophosphamide, and Antithymocyte Globulin for Chronic Granulomatous Disease. Biol Blood Marrow Transplant. 2016;22(11):2011–2018. [DOI] [PubMed] [Google Scholar]
- 15.Parta M, Kelly C, Kwatemaa N, et al. Allogeneic Reduced-Intensity Hematopoietic Stem Cell Transplantation for Chronic Granulomatous Disease: a Single-Center Prospective Trial. J Clin Immunol. 2017;37(6):548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morillo-Gutierrez B, Beier R, Rao K, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood. 2016;128(3):440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marsh R, Hebert KM, Keesler D, et al. Practice Pattern Changes and Improvements in Hematopoietic Cell Transplantation for Primary Immunodeficiencies. J Allergy Clin Immunol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauck F, Koletzko S, Walz C, et al. Diagnostic and Treatment Options for Severe IBD in Female X-CGD Carriers with Non-random X-inactivation. J Crohns Colitis. 2016;10(1):112–115. [DOI] [PubMed] [Google Scholar]
- 19.Griffith LM, Cowan MJ, Notarangelo LD, et al. Primary Immune Deficiency Treatment Consortium (PIDTC) update. J Allergy Clin Immunol. 2016;138(2):375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363(27):2600–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bacigalupo A, Ballen K, Rizzo D, et al. Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant. 2009;15(12):1628–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marciano BE, Zerbe CS, Falcone EL, et al. X-linked carriers of chronic granulomatous disease: Illness, lyonization, and stability. J Allergy Clin Immunol. 2018;141(1):365–371. [DOI] [PubMed] [Google Scholar]
- 23.Przepiorka D, Weisdorf D, Martin P, et al. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant. 1995;15(6):825–828. [PubMed] [Google Scholar]
- 24.Shulman HM, Sullivan KM, Weiden PL, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980;69(2):204–217. [DOI] [PubMed] [Google Scholar]
- 25.Seger RA. Hematopoietic stem cell transplantation for chronic granulomatous disease. Immunol Allergy Clin North Am. 2010;30(2):195–208. [DOI] [PubMed] [Google Scholar]
- 26.Battersby AC, Braggins H, Pearce MS, et al. Inflammatory and autoimmune manifestations in X-linked carriers of chronic granulomatous disease in the United Kingdom. J Allergy Clin Immunol. 2017;140(2):628–630 e626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.