

Abstract
The imprinted delta like 1 homolog (DLK1) - thyroxine deiodinase type III (DIO3) locus regulates development and growth. Its imprinting regulation involves two differentially methylated regions (DMRs), intergenic-DMR (IG-DMR) and maternally expressed gene 3-DMR (Meg3-DMR). In mice, a maternal deletion of the IG-DMR leads to LOI in the locus, proving that the IG-DMR is a cis-acting imprinting control region of the locus. However, the Meg3-DMR overlaps with the promoter, exon 1 and intron 1 of the Meg3 gene. Because deletion of the Meg3-DMR inactivates the Meg3 gene, their roles in imprinting regulation of Meg3-DMR mice is unknown. Therefore, we generated two mouse models: Meg3Δ(1-4) and Meg3Δ(2-4), respectively targeting exons 1-4 and exons 2-4 of the Meg3 gene. A maternal deletion of Meg3Δ(1-4) caused embryonic death and LOI in both embryos and placentas, but did not affect methylation status of the IG-DMR. In contrast, mice carrying a maternal deletion of Meg3Δ(2-4) were born normally and did not have LOI. These data indicate that it is the Meg3-DMR, not the Meg3 gene, which regulates imprinting of the Dlk1-Dio3 locus.
Keywords: DLK1-DIO3 locus, genomic imprinting, IG-DMR, imprinting control region, MEG3, MEG3-DMR
Introduction
Genomic imprinting is an epigenetic phenomenon in which expression of imprinted genes is parental-origin-dependent (1). An imprinted locus is formed by a cluster of imprinted genes in which their expression is tightly regulated by imprinting mechanisms. The delta like 1 homolog (DLK1) - thyroxine deiodinase type III (DIO3) locus is located on chromosome 14 in humans and on distal chromosome 12 in mice (2–4). Balanced gene expression in the DLK1-DIO3 locus plays a pivotal role in development and growth. Loss of imprinting (LOI) in this locus in humans leads to Kagami-Ogata syndrome (5, 6) or Temple syndrome (7). In mice, LOI of the locus causes developmental defects and premature death (8, 9). The locus is also essential in maintaining pluripotentiality of induced pluripotent stem cells (10).
The cis-elements controlling genomic imprinting are differentially methylated regions (DMRs) (11). Two DMRs, intergenic DMR (IG-DMR) and maternally expressed gene 3-DMR (Meg3-DMR), are thought to play important roles regulating imprinting of the DLK1-DIO3 locus (2, 6). Kagami et al reported that a patient carrying a microdeletion overlapping with the IG-DMR showed LOI in the locus (6). Similarly, Lin et al demonstrated LOI in mice carrying a maternal deletion of the IG-DMR (ΔIG-DMR/+)(Fig. S3) (12, 13). These data proved that the IG-DMR is an imprinting control region (ICR). The MEG3-DMR starts approximately 1.5 kb upstream of the MEG3 gene and extends into the MEG3 intron 1. We previously generated a Meg3-5kb mouse model, in which a 5-kb genomic DNA was targeted including a small portion of the Meg3 promoter and exons 1 through 5 of the Meg3 gene (Fig. S3) (14). The maternal deletion of the targeted region resulted in LOI in the locus (14), indicating that the targeted region is necessary for the imprinting regulation of the Dlk1-Dio3 locus. In agreement, Kagami et al reported that a patient with a microdeletion containing the MEG3-DMR had LOI in DLK1-DIO3 locus (6). Recently, Sanli et al made deletions in the Meg3 promoter or intron 1 in embryonic stem cells using CRISPR-Cas9 technique and found that these deletions caused LOI (15). These data support the concept that the MEG3-DMR plays a role in imprinting regulation. However, a contradictory finding was reported by Takahashi et al, who generated a mouse model carrying an approximately 10-kb deletion including Meg3 exons 1 through 5 (designated as Meg3-10kb)(Fig. S3) (16). Mice carrying a maternal deletion of Meg3-10kb did not demonstrate LOI in the Dlk1-Dio3 locus, although Meg3 expression was completely abolished. IG-DMR methylation was not affected by the Meg3-10kb deletion (16). These data imply that the Meg3-DMR does not play a significant role in imprinting regulation. Furthermore, because the Meg3-DMR overlaps with the Meg3 gene promoter, any deletion in this region inactivates both Meg3-DMR and the Meg3 gene. Therefore, it is still unknown as to whether the effects on imprinting in the Dlk1-Dio3 locus were caused by lack of maternal Meg3-DMR, or Meg3 expression.
To address this key unanswered question, we created two mouse models, one with a deletion of exons 1 through 4 of the Meg3 gene (Meg3Δ(1-4)) and the other with a deletion of exons 2 through 4 of the gene (Meg3Δ(2-4)). We show that LOI was only observed in embryos with a maternal Meg3Δ(1-4) deletion (Meg3Δ(1-4)/+), not in those with a maternal deletion of Meg3Δ(2-4). In addition, the maternal allele of the IG-DMR remains unmethylated in Meg3Δ(1-4)/+ embryos. Our data demonstrate that it is the Meg3-DMR, not the Meg3 gene, that regulates imprinting, likely by regulating expression of maternally expressed genes.
Materials and Methods
Mice:
All animals used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Massachusetts General Hospital. They were housed at the animal facility of MGH Center for Comparative Medicine. Two strains of mice carrying deletions of the Meg3 gene were created. Meg3Δ(1-4) mice contain a deletion of exons 1 to 4 of the Meg3 gene (Fig. S1)(Supplementary Data). To obtain this strain, a mouse strain carrying floxed exons 1-4 of the Meg3 gene (Meg3+/fl(1-4)) was created using services from inGenious Targeting Laboratories (iTL, Stony Brook, NY). To generate heterozygous mice carrying a deletion on the paternal allele (Meg3+/Δ(1-4)), male Meg3+/fl(1-4) mice were mated with female CRE transgenic mice (BALB/c-Tg(CMV-cre)1Cgn/J, Jackson Lab, Bar Harbor, ME). Meg3Δ(2-4) mice carry a deletion of exons 2 through 4 of the Meg3 gene (Fig. S2). To obtain this strain, a mouse strain carrying floxed exons 2-4 of the Meg3 gene (Meg3+/fl(2-4)) was created using services from Taconic (TaconicArtemis, Cologne, Germany). To generate heterozygous mice carrying a deletion on the paternal allele (Meg3+/Δ(2-4)), male Meg3+/fl(2-4) mice were mated with the female aforementioned CRE transgenic mice.
Gene expression:
Expression of maternally and paternally expressed genes were detected by SYBR quantitative RT-PCR. Total RNAs were isolated from 11.5 dpc embryos and placentas using Qiagen RNeasy Mini Kit with on column DNase treatment (Qiagen, Germantown, MD). For lncRNAs and mRNAs, reverse transcriptions were performed using the ProtoScript® First Strand cDNA Synthesis Kit from New England Biolabs (Ipswich, MA). To detect Rtl1 RNA, the primer used in reverse transcription was 5’GATACTCAAACTTTTCTGA3’. The primer for Gapdh, 5’TTGAGTGAGTTGTCATATT3’, was included as the internal control. The primers used in qPCR are listed in Table S1 and S2. To detect snoRNAs and miRNAs, reverse transcriptions were performed with the qScript mircoRNA DNA synthesis kit from Quantabio (Beverly, MA). The forward primers for small RNAs are listed in Table S2. The reverse primer was the PerfeCTa® Universal PCR Primer obtained from Quantabio. Quantitative PCRs were performed using PowerUp SYBR master mix from ThermoFisher Scientific (Grand Island, NY) per the manufacturer’s instruction. Fold changes of gene expression in knockout mice over the WT mice were calculated by comparative Ct method described by Schmittgen et al (17)(33) and detailed in the technical note by Haimes et al at Dharmacon of GE Healthcare (http://dharmacon.horizondiscovery.com/uploadedFiles/Resources/delta-cq-solaris-technote.pdf). Gapdh was used as the internal reference gene for lncRNAs and mRNAs. Sno202 was used as the reference for snoRNAs and miRNAs.
Bialleic expression:
Chromosome-specific gene expression was determined by SNP analysis. In Dlk1 cDNA (NM_010052), nucleotide 1207 is a G in C57BL/6 and an A in DBA/2, respectively. In Rtl1 cDNA (NM_184109), nt2061 is a T in C57BL/6 and a C in NOD/ShLit. DBA/2J and NOD/ShLitJ mice were obtained from the Jackson Laboratory. Bialleic expression of Dlk1 in embryos was done as previously described (14). Total RNA was isolated from 11.5 dpc embryos and placentas. Reverse transcriptions were done with the first strand cDNA synthesis kit from New England Biolabs using oligo dT as the RT primer for Dlk1 and Rtl1-RT (5’TCATCTCACTTTCCTTAAT3’) for Rtl1. The cDNA fragments containing SNPs for Dlk1 and Rtl1 were amplified by PCR. After gel purification, the fragments were sequenced to identify the SNPs. PCR primers for Dlk1 are 5’TCCTGAAGGTGTCCATGAAAGAGC3’ (forward) and 5’AAGCATAGCGTTCACTCGATTCCAC3’ (reverse); for Rtl1 5’TCTGATACCCACCTTAGGCTGGC3’ (forward) and 5’GACGCCGCTTTGATCACTGTCTC3’ (reverse).
DNA methylation analysis:
Methlyation status of the IG-DMR was determined by bisulfite sequencing (Bisulfite Sequencing Services, Active Motif, Carlsbad, CA). A region of 500 nt long containing 35 CpG sites, located approximately 12.4 Kb upstream of the Meg3 gene, was selected for the analysis. Genomic DNAs were isolated from 11.5 dpc embryos, including 3 Meg3Δ(1-4)/+, 3 Meg3+/(1-4) and 3 Meg3+/+, and their corresponding placentas. After bisulfite treatment, the converted genomic DNAs were amplified by PCR, processed into standard, barcoded Illumina sequencing libraries and sequenced in NextSeq 500 system (Illumia, San Diego, CA). Reads were analyzed using the bismark alignment program (v 0.7.7) (http://www.bioinformatics.babraham.ac.uk/projects/bismark/). The mouse chr12 (mm10 assembly) was used as the reference sequence. Between 4.7 and 7.05 million reads were analyzed per sample.
Results and Discussion
The mouse Meg3-DMR is approximately 3.5 kb long overlapping the promoter, exon 1 and intron 1 of the Meg3 gene. To clarify the role of the Meg3-DMR and Meg3 gene in imprinting regulation of the Dlk1-Dio3 locus, two mouse models were generated. Part of the Meg3 promoter as well as exons 1 through 4 of the Meg3 gene were deleted in Meg3Δ(1-4) mice (Fig. S1). Exons 2 through 4 of the gene were deleted in Meg3Δ(2-4) mice (Fig. S2). Mice carrying a paternal allele of Meg3Δ(1-4) (Meg3+/Δ(1-4)) or Meg3Δ(2-4) (Meg3+/Δ(2-4)) developed normally. All mice were bred by crossing female Meg3+/Δ(1-4) or Meg3+/Δ(2-4) with WT male B6 mice. Mice carrying a maternal allele of Meg3Δ(1-4) (Meg3Δ(1-4)/+) died embryonically starting at 12.5 day post coitus (dpc). All Meg3Δ(1-4)/+ embryos died by 13.5 dpc (Table S1). In contrast, Meg3Δ(2-4)/+ embryos developed to full term and were born alive. Therefore, we used 11.5 dpc embryos to assess the effects of deletions on imprinting of the Dlk1-Dio3 locus.
There are three Meg3 transcript variants (Fig. 1A). Variant 3 (NR_027652.1) was first reported by Schuster-Gossler et al (18). The transcript contains all 10 exons. Variant 1 (NR_003633.3) and 2 (NR_027651.2) are derived from multiple sequence entries in NCBI. They contain a long alternative exon 10 (10A). Using quantitative PCR with primers targeting specific variant transcripts, we quantified their expression in WT embryos (Fig. 1B). The ΔCt values of PCR #1 and 2, detecting all three variants, were similar to those of PCR #2 and 4, detecting variant 3 only, suggesting that the variant 3 is the predominant transcript of all variants. In agreement, the ΔCt values of PCR #5, 6 and 7, detecting variants 1 and 2, were significantly higher than ΔCt values from other 4 qPCRs. Their expression levels were less than 6% of total Meg3 expression (Fig. 1C). Compared with the variant 3 levels, the combined expression of variants 1 and 2 were also very low (less than 6%) (Fig. 1C). These data suggest that the variant 3 is the main functional transcript of the Meg3 gene. Therefore, variant 3 was used as the functional product of the Meg3 gene.
Figure 1.
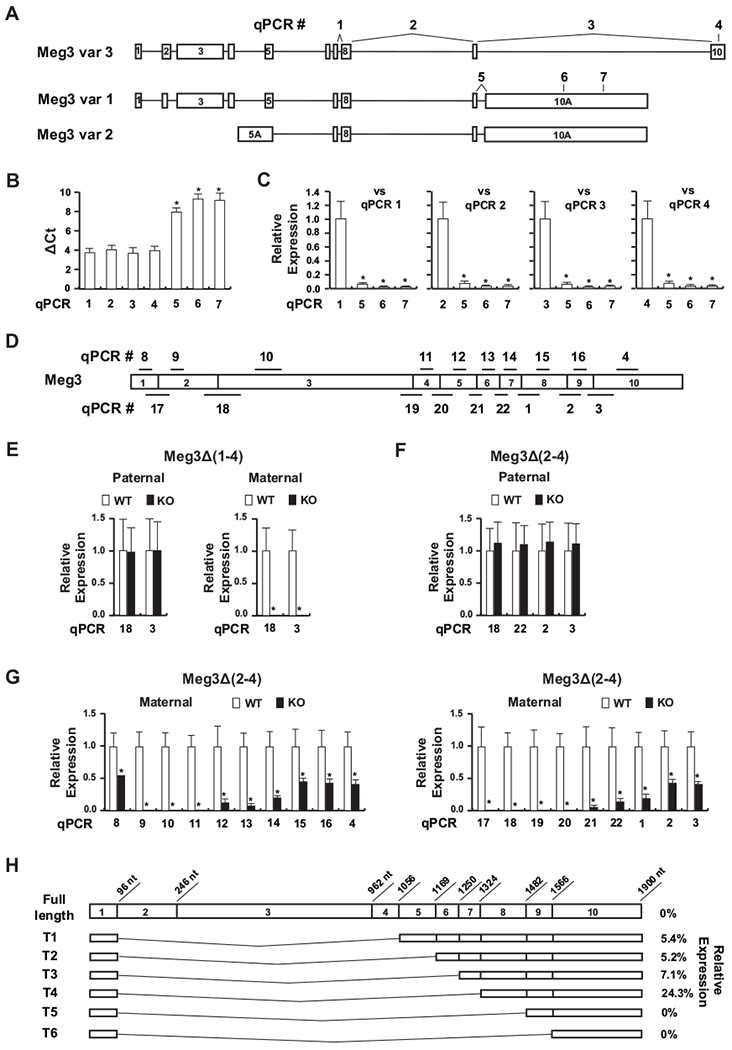
Meg3 expression in Meg3Δ(1-4) and Meg3Δ(2-4) mice. (A) There are three Meg3 transcript variants. Variant 3 contains 10 exons. Variants 1 and 2 contain a large alternative exon 10 (10A). Variant 2 has an alternative exon 5 (5A). Quantitative PCRs used to quantify their expression are indicated as numbers above each variant. (B) The SYBR based qRT-PCR was used to determine expression of the Meg3 variants in 11.5 dpc WT embryos (n=7). Gapdh was used as the internal reference gene. ΔCt values were presented as mean+/−SD. The ΔCt values from qPCR #5, 6 and 7 were compared with those from qPCR #1, 2, 3 and 4 using ANOVA multiple comparison tests. * p<0.05, considered to be statistically significant. (C) Relative expression levels of Meg3 variants 1 and 2 compared to all three variants (vs qPCR #1 and 2) or to variant 3 (vs qPCR #3 or 4). The values were calculated using 2-ΔΔCt method as described in the Material and Methods. Student t-test was used to compare values against the reference qPCR. * p<0.05, considered to be statistically significant. (D) Detection of exons in Meg3 transcript was done by qPCR. For example, exon 1 was detected by qPCR #8; exons 1–2 by qPCR #17. Other exons were similarly detected by their respective qPCRs as indicated. (E) Relative expression levels of Meg3 detected with qPCR #3 and 18 in 11.5 dpc Meg3Δ(1-4) embryos compared with their WT littermates. (F) Relative expression levels of Meg3 in 11.5 dpc paternal Meg3Δ(2-4) embryos compared with their WT littermates. (G) Relative expression levels of Meg3 in 11.5 dpc maternal Meg3Δ(2-4) embryos compared with their WT littermates. The levels of Meg3 transcripts detected by qPCR in KO embryos were normalized against the levels in their respective WT littermates, which were designated as 1. A minimum of 6 embryos for each genotype from at least 2 litters were used for gene expression analysis. Student t-test was used to compare values between KO mice and their WT littermates. * p<0.05, considered to be statistically significant. (H) Expression levels of predicted truncated Meg3 transcripts in Meg3Δ(2-4)/+ embryos. The percentage of each transcript in KO embryos compared to their WT littermates were deduced from data presented in (G).
To determine Meg3 expression in Meg3 KO embryos, we performed multiple qPCRs to quantify Meg3 transcripts with primers targeting specific exons as indicated in Fig. 1D. In Meg3Δ(1-4)/+ mice, no Meg3 transcripts were detected with PCR #3 and 18 (Fig. 1E). In Meg3Δ(2-4)/+ mice, no full-length Meg3 transcript was detected. Instead, multiple short truncated transcripts were detected in embryos, likely due to alternative RNA splicing. Their expression was very low compared to the level at which the full length Meg3 was expressed in the WT littermates (Fig. 1G). The highly possible transcripts were predicted (Fig. 1H). Among them, the most prominent transcript contained exons 1, 8, 9 and 10, which retains approximately 40% of the full-length Meg3 RNA (Fig. 1H). Previously, we have shown that deletion of less than a third of MEG3 RNA in the 5’-, 3’-end or middle of the molecule functionally inactivates MEG3 in p53 activation (19). Therefore, the data suggest that the function of the Meg3 gene in Meg3Δ(2-4)/+ mice is most likely diminished. Because the function of Meg3 RNA has not been fully understood, however, our data could not rule out the possibility that the low-level expression of truncated Meg3 transcripts is enough to maintain imprinting of the Dlk1-Dio3 locus.
The mouse Dlk1-Dio3 domain contains nearly 60 known maternally expressed genes (MEGs) and 3 known paternally expressed genes (PEGs). We determined expression of 13 MEGs across the maternal locus in embryos and their corresponding placentas (Fig. 2A). In Meg3Δ(1-4)/+ embryos and placentas, expression of all MEGs was completely abolished compared to their WT littermates (Fig. 2B). Conversely, expression levels of Dlk1, Rtl1 and Dio3 were increased by 1.94, 3.24 and 2.36 fold, respectively, in Meg3Δ(1-4)/+ embryos (Fig. 2B). Increases in Dlk1 and Rtl1 expression were also observed in Meg3Δ(1-4)/+ placentas (Fig. 2B). However, the level of Dio3 expression was not changed in Meg3Δ(1-4)/+ placentas compared to WT placentas (Fig. 2B). Taking advantage of SNPs in the Dlk1 and Rtl1 genes among mouse strains, we found that both paternal and maternal alleles of the genes were expressed in Meg3Δ(1-4)/+ embryos and placentas (Fig. 3A). Therefore, the transcriptional activation of their respective maternal alleles plays a role in increase in expression of Dlk1 and Rtl1. In addition, the maternally expressed miR-127 and miR-136 are anti-sense to Rtl1, which have been suggested to down regulate Rtl1 transcript (20). Therefore, lack of their expression is also likely to play a role in the increase in Rtl1 RNA levels in Meg3Δ(1-4)/+ embryos and placenta (Fig 2B). These data demonstrated that deletion of the exons 1 through 4 of the Meg3 gene leads to LOI in the Dlk1-Dio3 locus.
Figure 2.
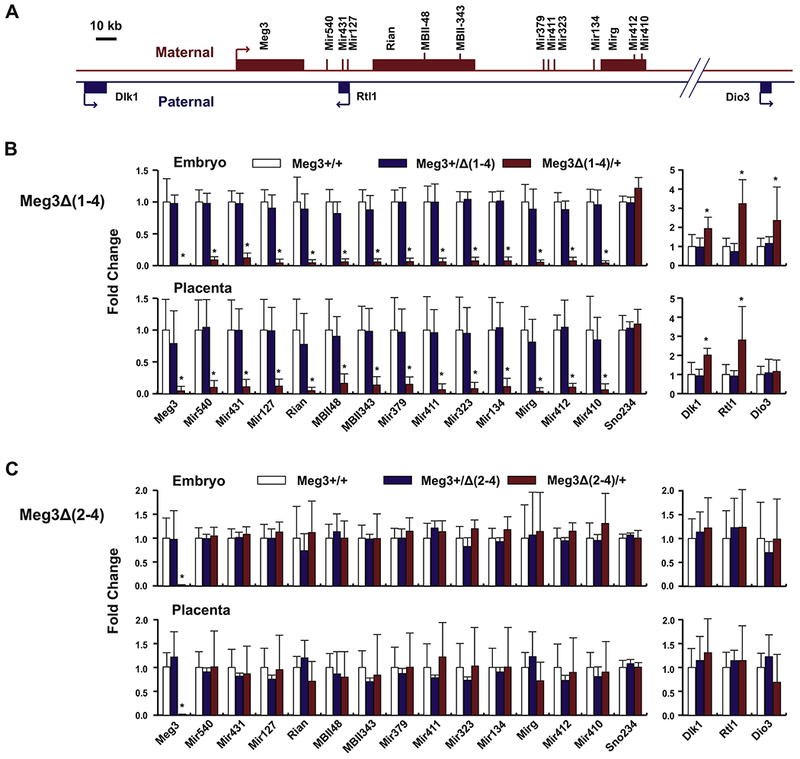
Maternal deletion of Meg3Δ(1-4), not Meg3Δ(2-4), silences MEGs and increases PEG expression. (A) Schematic representation of locations for selected 14 MEGs and 3 PEGs in the Dlk1-Dio3 locus. (B) Relative expression levels of the selected genes in 11.5 dpc Meg3Δ(1-4) embryos (upper panel) or placentas (lower panel) compared with their WT littermates. (C) Relative expression levels of the selected genes in 11.5 dpc Meg3Δ(2-4) embryos (upper panel) or placentas (lower panel). Gene expression levels were determined by qRT-PCR. Sno202 was used as the internal reference gene for miRNAs and snoRNAs. Gapdh was the reference gene for lncRNAs and mRNAs. Sno234 is also included as an internal control gene. The expression levels for each gene in KO embryos and placentas were normalized against the levels in their respective WT littermates, which were designated as 1. For each genotype, a minimum of 7 embryos and their corresponding placentas from at least 2 litters were used for gene expression analysis. Student t-test was used to compare values between KO mice and their WT littermates. * p<0.05, considered to be statistically significant.
Figure 3.

Bialleic expression of Dlk1 and Rtl1. Bialleic expression of these two genes were observed in Meg3Δ(1-4) mice (A), not in Meg3Δ(2-4) mice (B). Chromosome-specific gene expression was determined by SNP. To detect SNP in Dlk1, Meg3Δ(1-4) or Meg3Δ(2-4) mice in C57BL/6 background (B6Δ(1-4) or B6Δ(2-4)) were crossed with WT DBA/2J (D2+/+) mice. To detect SNP in Rtl1, B6Δ(1-4) or B6Δ(2-4) mice were crossed with WT NOD/ShLit (NOD+/+) mice. Total RNAs were extracted from embryos and placentas of 11.5 dpc. After reverse transcription, cDNA fragments containing SNPs for Dlk1 and Rtl1 were amplified by PCR, purified and sequenced. For each genotype, a minimum of 4 embryos and their corresponding placentas from at least 2 litters were used for SNP analysis. A purple arrow indicates detected bialleic expression.
Contrary to the findings in Meg3Δ(1-4)/+ mice, expression levels of all MEGs downstream of the Meg3 gene in Meg3Δ(2-4)/+ embryos and placentas were comparable to their respective levels in WT littermates (Fig. 2C). There were no changes in expression of PEGs in Meg3Δ(2-4)/+ embryos and placentas (Fig. 2C). Furthermore, bialleic expression analysis indicated that the maternal alleles of Dlk1 and Rtl1 genes were not activated in Meg3Δ(2-4)/+ embryos and placentas (Fig. 3B). These data demonstrate that deletion of exons 2 through 4 does not result in LOI in the Dlk1-Dio3 locus. Taken together, therefore, the LOI is caused by the deletion of the Meg3-DMR not the Meg3 gene.
We previously generated Meg3-5kb mice, in which exons 1-5 of the Meg3 gene were deleted (14). A maternal deletion of Meg3-5kb results in LOI in the Dlk1-Dio3 locus, which is similar to the finding in Meg3Δ(1-4)/+. In mat-Meg3-5Kb mice, the IG-DMR is completely methylated (14). We examined the methylation status of the IG-DMR core region in Meg3Δ(1-4) mice (Fig. 4). The methylation status of IG-DMR in Meg3Δ(1-4)/+ embryos was very similar to those found in WT and paternal KO embryos, which were approximately 50% (Fig. 4B). There were also no obvious differences in methylation patterns among their respective placentas (Fig. 4C). These data indicate that deletion of the Meg3-DMR does not affect the methylation patterns of the IG-DMR. This raises the possibility that the mechanisms of LOI in these two mouse models are different. Because IG-DMR deletion results in LOI in the Dlk1-Dio3 locus (12), it is very likely that the reason for LOI in mat-Meg3-5kb is methylation of the IG-DMR. This is further supported by the fact that mat-Meg3-5kb mice display identical phenotypes to ΔIG-DMR/+ mice, including skeletal muscle defects and death right after birth (12, 14). The Meg3-5kb mice were generated using a conventional recombination technology in which the targeted region was replaced by a neomycin resistant gene (Neor) cassette. The transcription of Neor is toward the IG-DMR (14). It has been found that transcription-through is a common mechanism of DNA methylation in imprinted loci, such as the Gnas locus (21, 22) and the Peg3 locus (23). Therefore, it is most likely that IG-DMR methylation in the mat-Meg3-5kb mice is caused by transcription from the Neor cassette. Taken together, these data indicate that the underlying mechanisms for LOI in the Dlk1-Dio3 locus between mat-Meg3-5kb mice and Meg3Δ(1-4)/+ mice are different: inactivation of IG-DMR in the former and inactivation of Meg3-DMR in the latter.
Figure 4.
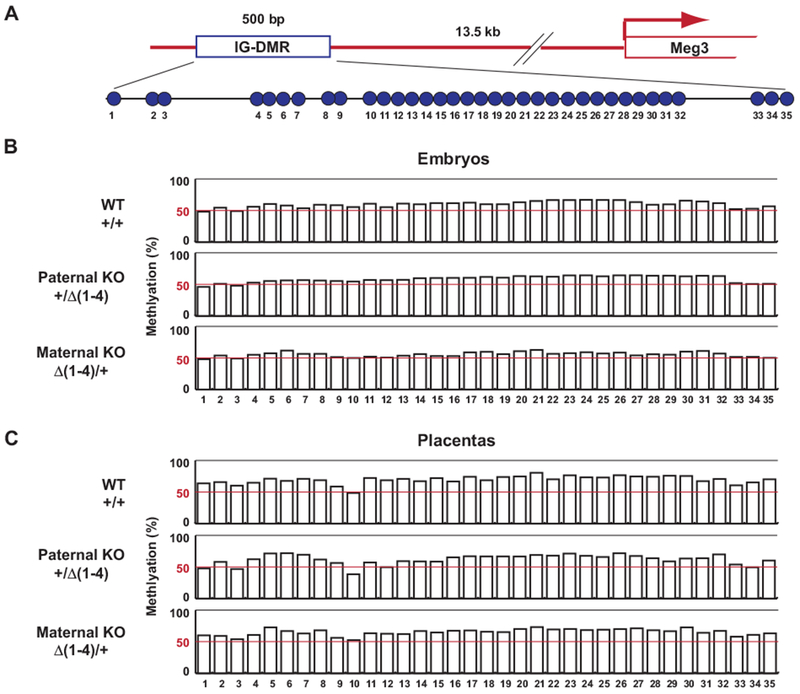
Deletion of Meg3-DMR does not affect methylation status of IG-DMR. (A) Schematic representation of CpG locations (open circle) in the core region of IG-DMR. (B) Methylation status in embryos of WT, paternal KO (+/Δ(1-4)) and maternal KO (Δ(1-4)/+). (C) Methylation status in placentas of WT, paternal KO (+/Δ(1-4)) and maternal KO (Δ(1-4)/+). The values were derived from 3 WT, 3 Meg3Δ(1-4)/+ and 3 Meg3+/Δ(1-4) embryos and their corresponding placentas.
Lin et al reported that methylation of the Meg3-DMR was significantly increased in ΔIG-DMR/+ mice (12)(Fig. S3). A similar phenomenon was observed in a Kagami-Ogata patient carrying a microdeletion overlapping the IG-DMR (6). Conversely, the methylation status of the IG-DMR is unaffected by a Meg3-DMR deletion as shown in Meg3Δ(1-4)/+ mice (Fig. 4) and in Kagami-Ogata patients with microdeletions containing the 5’ part of the MEG3 gene (6, 24). Considering that inactivation of either IG-DMR or Meg3-DMR results in LOI of the Dlk1-Dio3 locus, these data suggest that imprinting regulation by IG-DMR is mediated by the Meg3-DMR. Luo et al identified an element 10 kb upstream of the Meg3 gene involved in regulating expression of MEGs (25). This element, known as the Meg3-proximal enhancer, contains binding sites for transcription factor ZFP281 (25, 26). ZFP281 recruits AFF3, a component of RNA polymerase II elongation complex, to the enhancer on the maternal chromosome where the nearby IG-DMR is not methylated (25, 26). This recruitment of AFF3 promotes transcription activation from the Meg3 promoter (25). On the paternal chromosome, AFF3 is sequestered to the methylated IG-DMR via ZFP57 (26), which binds specifically to its methylated motifs (27). These data further indicate that the IG-DMR regulates imprinting of the Dlk1-Dio3 locus through Meg3-DMR.
Data from previous studies suggest that all maternally expressed genes in the Dlk1-Dio3 domain are transcribed as one large transcript (15, 25, 28). Considering that all MEGs are downstream of the Meg3 promoter, it is very likely that the Meg3 promoter controls MEG expression. This is consistent with our finding that all MEG expression is silenced in Meg3Δ(1-4)/+ mice. The Meg3-DMR overlaps with the Meg3 promoter. Because DNA methylation is a common mechanism in gene silencing, it is logical to hypothesize that the main function of the Meg3-DMR is to modulate Meg3 promoter activity to control the parental origin-dependent expression of downstream MEGs. It turns on the Meg3 promoter on the maternal chromosome; while it turns the promoter off on the paternal chromosome. Our results indicate that deletion of the Meg3-DMR inactivates the Meg3 promoter which in turn silences expression of all MEGs (18), implying that the MEG products may play a role in suppression of PEG expression in the Dlk1-Dio3 locus. Sanli et al observed that Meg3 transcripts colocalized with the Dlk1 gene on the maternal chromosome and the maternal Dlk1 promoter was activated in Δpromoter−/− and Δintron1−/− cells. They suggested that Meg3 RNAs play a role in regulation of Dlk1 expression (15). However, all ES clones in the study contained deletions in either the Meg3 promoter or Meg3 intron 1, which overlap with the Meg3-DMR. Therefore, their data did not distinguish whether the effect was caused by lack of Meg3 expression or inactivation of Meg3-DMR. In contrast, data from our Meg3Δ(2-4) mice demonstrated that the reduced Meg3 expression did not affect Dlk1 expression (Fig. 2), which is consistent with data from the Meg3-10kb mice by Takahashi et al (16). The Meg3 expression was completely abolished in mice carrying a maternal Meg3-10kb deletion. However, the Dlk1 expression did not increase at all in those KO mice compared with their WT littermates (16). Furthermore, no changes in Meg3-DMR methylation patterns were observed in Meg3Δ(2-4)/+ embryos compared with those in Meg3+/Δ(2-4) and WT embryos (Fig. S4). Taken together, these data strongly indicate that Meg3 RNAs are not essential in imprinting regulation.
Anti-Rtl1 encodes seven miRNAs, such as miR-127 and miR-136, which are antisense to Rtl 1 (20, 29). These miRNAs were shown to reduce Rtl1 RNA transcripts in vivo by RISC-mediated cleavage of RNAs (29). However, all Rtl1 RNAs were transcribed from the paternal allele in mice with the miR-127 deletion (30). These data suggest that these miRNAs are dispensable in Rtl1 imprinting. Approximately 80% of miRNAs in the Dlk1-Dio3 locus were clustered between miR-379 and miR-410, which including miRNAs encoded by Mirg (31). Labialle et al reported that a maternal deletion of the miR-379/miR-410 cluster did not affect expression levels of Dlk1 and Dio3 (32). In agreement, Gao et al found that a smaller deletion between miR-379 and miR-544 did not affect the RNA levels of Dlk1, although the Dlk1 protein levels were increased by 1.6-fold in mice with a maternal deletion (33). These findings indicate that the vast majority of miRNAs in the Dlk1-Dio3 locus are not involved in inhibition of PEG transcription.
Gene silencing by transcription has been demonstrated in several imprinted loci (34). For example, In the Igf2r locus, the paternal allele of Igf2r is silenced by the continuous transcription of long non-coding RNA Airn (35, 36). The Airn promoter is inside a DMR located in the intron 2 of the Igf2r gene. The transcription of Airn over the Igf2r promoter blocks the transcription initiation from the promoter (35). Similarly, the Nesp gene is transcriptionally silenced in the Gnas locus by the transcription of its anti-sense Nespas gene (37). In the Dlk1-Dio3 locus, the paternally expressed genes Rtl1 and Dio3 are downstream of the Meg3. Therefore, a likewise mechanism may be used by the Meg3-DMR to regulate the imprinting, which is to maintain an active transcription from the Meg3 promoter, whereby to silence Rtl1 and Dio3. This could be tested by inserting a polyadenylation (polyA) cassette before or after the Rtl1 gene, which stops transcriptions from the Meg3-DMR before or after the Rtl1 gene. This technique has been used to determine transcriptions of Airn or Nespas in imprinting regulation (36, 38, 39). Although the resultant RNA transcripts will contain full-length Meg3 RNAs, we expect that the maternal allele of the Rtl1 gene will be silenced in models with polyA inserted after the gene, not in models with poly A inserted before the gene. A caveat of this hypothesis does not explain how the transcription from the Meg3-DMR silences the Dlk1 expression. Because not all MEGs within the locus have been thoroughly investigated for their roles in regulating PEG expression, we speculate that Dlk1 may be silenced by one or more of the MEG products, similar to the inhibition of Slc22a3 by Airn lncRNA in the Igf2r locus (40).
Supplementary Material
Highlights.
Deletion of Meg3 exons 1-4, overlapping the Meg3-DMR, results in loss of imprinting.
Deletion of Meg3 exons 2-4, downstream of the Meg3-DMR, does not affect imprinting.
Meg3-DMR, not the Meg3 gene, regulates imprinting of the Dlk1-Dio3 locus.
Acknowledgments
Funding:
This work was supported in part by the National Institutes of Health (R01 CA193520 to A.K.) and the Jarislowsky Foundation (A.K.). None of the funding sources was involved in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- 1.Barlow DP, Bartolomei MS. Genomic imprinting in mammals. Cold Spring Harb Perspect Biol. 2014. February 1;6(2):a018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008. June;24(6):306–16. [DOI] [PubMed] [Google Scholar]
- 3.Takada S, Tevendale M, Baker J, Georgiades P, Campbell E, Freeman T, et al. Delta-like and gtl2 are reciprocally expressed, differentially methylated linked imprinted genes on mouse chromosome 12. Curr Biol. 2000. September 21;10(18):1135–8. [DOI] [PubMed] [Google Scholar]
- 4.Miyoshi N, Wagatsuma H, Wakana S, Shiroishi T, Nomura M, Aisaka K, et al. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells. 2000. March;5(3):211–20. [DOI] [PubMed] [Google Scholar]
- 5.Ogata T, Kagami M. Kagami-Ogata syndrome: a clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. Journal of human genetics. 2016. February;61(2):87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kagami M, O’Sullivan MJ, Green AJ, Watabe Y, Arisaka O, Masawa N, et al. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet. 2010. June 17;6(6):e1000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ioannides Y, Lokulo-Sodipe K, Mackay DJ, Davies JH, Temple IK. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet. 2014. August;51(8):495–501. [DOI] [PubMed] [Google Scholar]
- 8.Georgiades P, Watkins M, Surani MA, Ferguson-Smith AC. Parental origin-specific developmental defects in mice with uniparental disomy for chromosome 12. Development. 2000. November;127(21):4719–28. [DOI] [PubMed] [Google Scholar]
- 9.Kumamoto S, Takahashi N, Nomura K, Fujiwara M, Kijioka M, Uno Y, et al. Overexpression of microRNAs from the Gtl2-Rian locus contributes to postnatal death in mice. Hum Mol Genet. 2017. October 1;26(19):3653–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, et al. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010. May 13;465(7295):175–81. Epub 2010/04/27. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011. July 1;3(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, et al. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet. 2003. September;35(1):97–102. [DOI] [PubMed] [Google Scholar]
- 13.Lin SP, Coan P, da Rocha ST, Seitz H, Cavaille J, Teng PW, et al. Differential regulation of imprinting in the murine embryo and placenta by the Dlk1-Dio3 imprinting control region. Development. 2007. January;134(2):417–26. Epub 2006/12/15. eng. [DOI] [PubMed] [Google Scholar]
- 14.Zhou Y, Cheunsuchon P, Nakayama Y, Lawlor MW, Zhong Y, Rice KA, et al. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development. 2010. August;137(16):2643–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanli I, Lalevee S, Cammisa M, Perrin A, Rage F, Lleres D, et al. Meg3 Non-coding RNA Expression Controls Imprinting by Preventing Transcriptional Upregulation in cis. Cell reports. 2018. April 10;23(2):337–48. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi N, Okamoto A, Kobayashi R, Shirai M, Obata Y, Ogawa H, et al. Deletion of Gtl2, imprinted non-coding RNA, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum Mol Genet. 2009. May 15;18(10):1879–88. Epub 2009/03/07. eng. [DOI] [PubMed] [Google Scholar]
- 17.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–8. [DOI] [PubMed] [Google Scholar]
- 18.Schuster-Gossler K, Bilinski P, Sado T, Ferguson-Smith A, Gossler A. The mouse Gtl2 gene is differentially expressed during embryonic development, encodes multiple alternatively spliced transcripts, and may ΔCt as an RNA. Dev Dyn. 1998. June;212(2):214–28. [DOI] [PubMed] [Google Scholar]
- 19.Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R, et al. Activation of p53 by MEG3 noncoding RNA. J Biol Chem. 2007. August 24;282(34):24731–42. [DOI] [PubMed] [Google Scholar]
- 20.Seitz H, Youngson N, Lin SP, Dalbert S, Paulsen M, Bachellerie JP, et al. Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet. 2003. July;34(3):261–2. [DOI] [PubMed] [Google Scholar]
- 21.Chotalia M, Smallwood SA, Ruf N, Dawson C, Lucifero D, Frontera M, et al. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 2009. January 1;23(1):105–17. Epub 2009/01/13. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mehta S, Williamson CM, Ball S, Tibbit C, Beechey C, Fray M, et al. Transcription driven somatic DNA methylation within the imprinted Gnas cluster. PLoS One. 2015;10(2):e0117378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bretz CL, Kim J. Transcription-driven DNA methylation setting on the mouse Peg3 locus. Epigenetics. 2017;12(11):945–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beygo J, Elbracht M, de Groot K, Begemann M, Kanber D, Platzer K, et al. Novel deletions affecting the MEG3-DMR provide further evidence for a hierarchical regulation of imprinting in 14q32. Eur J Hum Genet. 2015. February;23(2):180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo Z, Lin C, Woodfin AR, Bartom ET, Gao X, Smith ER, et al. Regulation of the imprinted Dlk1-Dio3 locus by allele-specific enhancer activity. Genes Dev. 2016. January 1;30(1):92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Shen Y, Dai Q, Yang Q, Zhang Y, Wang X, et al. A permissive chromatin state regulated by ZFP281-AFF3 in controlling the imprinted Meg3 polycistron. Nucleic Acids Res. 2017. February 17;45(3):1177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011. November 4;44(3):361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tierling S, Dalbert S, Schoppenhorst S, Tsai CE, Oliger S, Ferguson-Smith AC, et al. High-resolution map and imprinting analysis of the Gtl2-Dnchc1 domain on mouse chromosome 12. Genomics. 2006. February;87(2):225–35. [DOI] [PubMed] [Google Scholar]
- 29.Davis E, Caiment F, Tordoir X, Cavaille J, Ferguson-Smith A, Cockett N, et al. RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Peg11 locus. Curr Biol. 2005. April 26;15(8):743–9. [DOI] [PubMed] [Google Scholar]
- 30.Ito M, Sferruzzi-Perri AN, Edwards CA, Adalsteinsson BT, Allen SE, Loo TH, et al. A trans-homologue interaction between reciprocally imprinted miR-127 and Rtl 1 regulates placenta development. Development. 2015. July 15;142(14):2425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seitz H, Royo H, Bortolin ML, Lin SP, Ferguson-Smith AC, Cavaille J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004. September;14(9):1741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Labialle S, Marty V, Bortolin-Cavaille ML, Hoareau-Osman M, Pradere JP, Valet P, et al. The miR-379/miR-410 cluster at the imprinted Dlk1-Dio3 domain controls neonatal metabolic adaptation. EMBO J. 2014. October 1;33(19):2216–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao YQ, Chen X, Wang P, Lu L, Zhao W, Chen C, et al. Regulation of DLK1 by the maternally expressed miR-379/miR-544 cluster may underlie callipyge polar overdominance inheritance. Proc Natl Acad Sci U S A. 2015. November 3;112(44):13627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanduri C Long noncoding RNAs: Lessons from genomic imprinting. Biochim Biophys Acta. 2016. January;1859(1):102–11. [DOI] [PubMed] [Google Scholar]
- 35.Santoro F, Mayer D, Klement RM, Warczok KE, Stukalov A, Barlow DP, et al. Imprinted Igf2r silencing depends on continuous Airn lncRNA expression and is not restricted to a developmental window. Development. 2013. March;140(6):1184–95. [DOI] [PubMed] [Google Scholar]
- 36.Latos PA, Pauler FM, Koerner MV, Senergin HB, Hudson QJ, Stocsits RR, et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science. 2012. December 14;338(6113):1469–72. [DOI] [PubMed] [Google Scholar]
- 37.Williamson CM, Ball ST, Dawson C, Mehta S, Beechey CV, Fray M, et al. Uncoupling antisense-mediated silencing and DNA methylation in the imprinted Gnas cluster. PLoS Genet. 2011. March;7(3):e1001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tibbit CJ, Williamson CM, Mehta S, Ball ST, Chotalia M, Nottingham WT, et al. Antisense Activity across the Nesp Promoter is Required for Nespas-Mediated Silencing in the Imprinted Gnas Cluster. Non-coding RNA. 2015. November 30;1(3):246–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santoro F, Pauler FM. Silencing by the imprinted Airn macro lncRNA: transcription is the answer. Cell Cycle. 2013. March 1;12(5):711–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008. December 12;322(5908):1717–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.