

Abstract
Anti-Müllerian hormone (AMH) is a member of the Transforming Growth Factor-β family of secreted signaling proteins. AMH is expressed in Sertoli cells of the fetal and adult testes and granulosa cells of the postnatal ovary. AMH is required for the regression of the Müllerian ducts in mammalian fetuses during male differentiation. AMH signals through its Type II receptor, AMHR2. AMHR2 is expressed in mesenchyme adjacent to the Müllerian ducts, and in Sertoli, Leydig, and granulosa cells. Although AMH and AMHR2 genes have been identified in numerous vertebrate species, spontaneous or engineered mutations or variants have been found or created in only a few mammals and teleost fishes. AMH or AMHR2 mutations in mammals lead to the development of Persistent Müllerian Duct Syndrome (PMDS), a recessive condition in which affected males are fully virilized but retain Müllerian duct-derived tissues, including a uterus and oviducts, and in human and dog, undescended testes. Amh mutant female mice had accelerated ovarian primordial follicle recruitment, suggesting a role for AMH in regulating germ cells, amh and amhr2 mutations have also been experimentally generated in various teleost fishes. Depending on the fish species, loss of AMH signaling results in infertility, germ cell tumors, or male-to-female sex reversal. Here we compare the spectrum of phenotypes caused by AMH and AMHR2 mutations in a variety of vertebrate species. There are both common and unique phenotypes between species, highlighting the range of biological processes regulated by AMH signaling.
Keywords: Müllerian duct regression, testicular descent, gametogenesis, fertility, sex determination
Introduction
Pioneering fetal transplant surgery experiments by Alfred Jost first established that a secreted protein from the fetal testis was required for the regression of the nascent female reproductive tract (Müllerian ducts) in males (Jost, 1953). Jost called this I’hormone inhibitrice that later became known as anti-Müllerian hormone (AMH) (Josso, 1973). In mammals, AMH, a Transforming Growth Factor-β (TGF-β) family member glycoprotein hormone, is expressed in Sertoli cells of fetal and adult testis and granulosa cells of postnatal ovary (Cate et al., 1986; Josso, 1973, 1974; Takahashi et al., 1986). AMH binds to Type I receptors shared with the bone morphogenetic protein (BMP) pathway (ACVR1 and BMPR1A) and its sole Type II receptor AMHR2 in the Müllerian duct mesenchyme, activating the AMH signaling pathway and initiating regression of the Müllerian ducts (reviewed in (Mullen and Behringer, 2014)). The absence of functional AMH or AMHR2 results in a rare recessive disorder known as Persistent Müllerian Duct Syndrome (PMDS) that is characterized by the presence of Müllerian duct-derived tissues including oviducts, uterus and vagina in a fully virilized male (Josso et al., 2005).
While the essential role of AMH in Müllerian duct regression led to its discovery, it is not the only function of AMH, nor what is postulated to be AMH’s evolutionarily ancient function. Analysis across vertebrate species, including mammals, birds and fishes, suggest that AMH initially evolved to regulate germ ceil proliferation (Adolfi et al., 2018). The phenotypes resulting from mutations in the AMH and AMHR2 genes across species highlight both shared roles for AMH-signaling and species-specific differences. Here we review AMH and AMHR2 mutant phenotypes in vertebrates, revealing various roles for AMH signaling in reproductive organ development.
Identification of AMH and AMHR2 mutations in humans with Persistent Müllerian Duct Syndrome
AMH and AMHR2 mutations have been identified in human males with PMDS (Josso et al., 2005). The first described mutation in the AMH gene in humans was found in three brothers of Moroccan descent. The brothers, aged 2 months, 5 years and 7 years, had been diagnosed with PMDS. No AMH was detected in their testicular tissue however AMH mRNA was present. DNA sequencing detected a homozygous mutation in the AMH gene that resulted in a premature stop codon in exon 5 (NM_000479.3: c.1144G>T, p. (Glu382*)). This mutation was expected to produce AMH protein lacking the bioactive C-terminus. Although AMH protein was not detected in the patients, a truncated AMH protein was secreted from cells transfected with a gene construct carrying the same mutation in vitro, suggesting rapid degradation of the mutant protein occurs in vivo (Knebelmann et al., 1991).
Since this first case, 64 additional unique mutations in the AMH gene have been found that result in PMDS. Together these include 38 missense mutations, 10 stop mutations, 1 non-stop mutation, 9 deletions, 2 insertions and 5 splicing mutations (Altincik et al., 2017; Picard et al., 2017). Mutations have occurred throughout the AMH gene with a slightly higher rate of mutation found in the biologically active C-terminal region. AMH mutations are homozygous in approximately 65% of patients and more commonly born of consanguineous parents (second cousins or closer). Nineteen mutations have occurred in 2 or more families. These recurrent mutations may be the result of founder effects in regional populations including Brazil, the Middle East and Northern Europe (Nishi et al., 2012; Picard et al., 2017).
AMHR2 mutations are suspected in PMDS patients that are positive for active AMH and/or have serum AMH levels within the normal range (Josso et al., 2005). The first described human AMHR2 gene mutation was found following a screen of 21 AMH-positive PMDS patients (Imbeaud et al., 1995). One patient had a homozygous AMHR2 gene mutation at the invariant GT dinucleotide of the splicing donor site 5’ of intron 2. His parents were both heterozygous for this mutation. His serum AMH levels were normal and bioactive AMH was found in his testicular biopsy. Reverse transcription PCR of his testicular biopsy showed two forms of AMHR2 mRNA: transcripts lacking exon 2 and transcripts generated by use of a cryptic splice site leading to an amino acid change (Gly78Asp) and the addition of 4 residues (EWQR) encoded at the end of exon 2.
To date, a total of 58 additional unique gene mutations in AMHR2 have been described. Together these include 36 missense, 11 stop, 8 deletions and 4 splicing defects (Picard et al., 2017; Ren et al., 2017). Mutations resulting in PMDS occur in all 11 exons of the AMHR2 gene and are found as both homozygous and compound heterozygous mutations. Ten AMHR2 gene mutations occur in multiple families (Picard et al., 2017). The most common, a 27-bp deletion in exon 10 (NM_020547.2:c.1332_1358del, p(Gly445_Glu453del) occurring in 30 patients, is found predominantly in Northern Europeans and presumed to be the result of a founder effect (Imbeaud et al., 1996).
Men with mutations in either the AMH or AMHR2 gene are fully virilized with male secondary sex characteristics. The presence of a uterus in these men is usually detected early in childhood because of cryptorchidism (undescended testes) and/or associated hernias (Josso et al., 1993a). In men with PMDS, the uterus is attached to the pelvis via the broad ligament and the testes are closely joined to the uterus. If the uterus is strongly attached to the pelvis, the testes will remain in an abdominal position. In cases where the uterus is loosely attached the testis descends into the scrotum along with the attached uterus (Miller et al., 2004). During testicular descent in unaffected males the gubernacular cord, tethered to the scrotum, thickens and shortens followed by constriction of the inguinal canal. In cases of human AMH or AMHR2 gene mutations, the gubernacular cord remains thin and elongated and the inguinal canal fails to constrict. This, in addition to the presence of the uterus, may also contribute to the predisposition of cryptorchidism in men with AMH or AMHR2 gene mutations (Hutson et al., 1994; Hutson and Lopez-Marambio, 2017).
A little over half of patients with AMH or AMHR2 gene mutations have bilateral cryptorchidism where both testes fail to descend. Inguinal hernias are present when either one testis (hernia uteri inguinalis) or both testes (transverse testicular ectopia) along with the retained uterus are found in one inguinal sac (Fig. 1A). Approximately 20% of patients with AMH or AMHR2 mutations present with hernia uteri inguinalis. Transverse testicular ectopia occurs in about 25% of patients with AMH or AMHR2 mutations but has not been observed in cases of idiopathic PMDS and is considered indicative of AMH or AMHR2 gene mutations (Picard et al., 2017). The position of the testes is not specific for AMH or AMHR2 mutations and can vary between siblings and families sharing the same mutation (Abduljabbar et al., 2012; Knebelmann et al., 1991; Nalbantoglu et al., 2015).
Fig. 1. AMH signaling mutant phenotypes in human and mouse.
A, Human; B, C, mouse. A, Diagram showing variation in position of testes and uterus in human Persistent Müllerian Duct Syndrome. B, C. Images of male reproductive tract organs from control (B) and Amhr2-null (C) mice. Amh-null male mice have the same phenotype as Amhr2-null mice. Ep, epididymis; ft, fallopian tube; gb, gubernaculum; scr, scrotum; sv, seminal vesicle; t, testis; ut, uterus; vd, vas deferens. Diagram in A modified from Hutson, Thorup, & Beasley, 2016.
The majority of patients with AMH or AMHR2 gene mutations are infertile. However review of past literature has found that approximately 19% of patients have naturally fathered at least one child (Picard et al., 2017). In all cases, they had either transverse testicular ectopia or hernia uteri inguinalis. Infertility in PMDS patients can be overcome with testicular sperm extraction followed by intracytoplasmic sperm injection (Picard et al., 2017).
If cryptorchidism is not surgically corrected there is a higher risk of cancer development in the undescended testis. In a recent review, Picard et al. (2017) estimated that testicular cancer arose in 33% of adult PMDS patients, exceeding the risk associated with isolated cryptorchidism. Previous estimates have suggested risk of testicular cancer is similar in PMDS and isolated cryptorchidism (Bucci et al., 2002; Picard et al., 2017; Shamim, 2007). Although very rare, malignant transformation of the Müllerian-derived tissues has also been observed (Farikullah et al., 2012).
Thus far, female relatives carrying homozygous mutations of AMH or AMHR2 have no abnormal phenotypes and are fertile (Picard et al., 2017). However, heterozygous variants of the AMH and AMHR2 gene with reduced AMH signaling activity in vitro have been found in women with premature menopause (Alvaro Mercadal et al., 2015; Li et al., 2016). Thus, it is possible that women with homozygous mutations of AMH or AMHR2 may later undergo premature menopause but at present it is not known.
Generation of Amh and Amhr2 mutant mouse models
The development of gene targeting and embryonic stem (ES) cell technologies led to the generation of Amh and Amhr2 mutant mouse models (Arango et al., 2008; Arango et al., 1999; Behringer et al., 1994; Jamin et al., 2002; Mishina et al., 1996). These Amh and Amhr2 alleles are listed in Table 1. These mouse models recapitulate many aspects of human PMDS but there were differences and new insights into the biological roles of the AMH signaling pathway.
Table 1.
Amh and Amhr2 mutations in the mouse and dog.
| Mouse Amh | ||||
|---|---|---|---|---|
| Allele | Variant | Dominant/recessive | Phenotype | References |
| Exon 1, intron 1, exon 2 deletion with neo cassette (tm1Bhr) | induced | recessive | male PMDS, infertility, Leydig cell hyperplasia; female primordial follicle recruitment alterations | Behringer 1994; Durlinger 1999 |
| SF1-binding site mutation in promoter region with neo cassette in intron 1 (tm2Bhr) | induced | recessive | male PMDS | Arango 1999 |
| SF1-binding site mutation in promoter region (tm2. 1Bhr) | induced | recessive | wild-type, decreased Amh transcript levels | Arango 1999 |
| SOX9-binding site mutation in promoter region with neo cassette in intron 1 (tm3Bhr) | induced | recessive | male PMDS | Arango 1999 |
| SOX9-binding site mutation in promoter region (itm3. 1Bhr) | induced | recessive | male PMDS | Arango 1999 |
| SF1- & SOX9-binding site mutation in promoter region with neo cassette in intron 1 (tm4. 1Bhr) | induced | recessive | male PMDS | Arango 1999 |
| SF1- & SOX9-binding site mutation in promoter region (tm4.1Bhr) | induced | recessive | male PMDS | Arango 1999 |
| neo cassette in intron 1 (.tm5Bhr) | induced | recessive | male PMDS | Arango 1999 |
| loxP in intron 1 (tm5.1Bhr) | induced | N/A | wild-type | Arango 1999 |
| Mouse Arnhr2 | ||||
| Allele | Variant | Dominant/recessive | Phenotype | References |
| Exons 1-6 deletion with neo cassette (tm1Bhr) | induced | recessive | male PMDS | Mishina 1996 |
| Insertion of IRES-cre cassette with neo cassette into exon 5 with 50-bp deletion (tm3(cre)Bhr) | induced | recessive | male PMDS | Jamin 2002 |
| Insertion of IRES-lacZ cassette with neo cassette into exon 5 with 50-bp deletion (tm2Bhr) | induced | recessive | male PMDS | Arango 2008 |
| Dog AMHR2 | ||||
| Allele | Variant | Dominant/recessive | Phenotype | Reference |
| Exon 3, C241T | spontaneous | recessive | male PMDS; some males with undescended testes and infertility | Wu 2009 |
The first targeted mutation in the AMH pathway was achieved in the Amh locus (Behringer et al., 1994). This initial Amh knockout allele had a drug resistance gene expression cassette introduced into the Amh gene that simultaneously deleted ~0.6 kb, including a portion of exon 1, all of intron 1, and all of exon 2, encoding part of the N-terminal domain of AMH (Fig. 2, Table 1). This mutation resulted in a complete loss-of-function allele. Both male and female mice heterozygous for the Amh mutation were phenotypically normal and fertile. In addition, females homozygous for the mutation had morphologically normal uteri and ovaries and were fertile. Subsequent studies showed that Amh heterozygous and homozygous mutant females, despite morphologically normal ovaries, had alterations in primordial follicle recruitment in the ovary (Durlinger et al., 1999). 25-day old and 4-month old homozygous mutant females had more preantral and small antral follicles in their ovaries compared to wild-type females. At 13 months of age, the Amh homozygous mutant females had very few primordial follicles, i.e. an early depletion of primordial follicles compared to wild type.
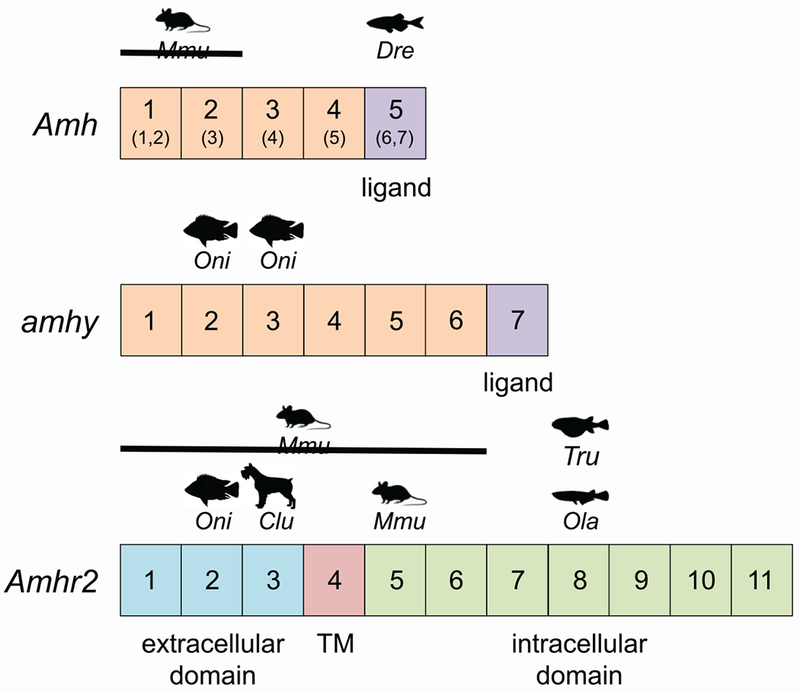
Fig. 2. Amh, amhy, and Amhr2 mutations in vertebrates.

Diagram of Amh, amhy, and Amhr2 gene and protein domains. Exons, numbered boxes not to scale. Numbers in parentheses correspond to Dre exons. Exon encoding TGF-beta domain, purple; extracellular domain, blue; transmembrane domain (TM), red; intracellular region containing kinase domain, green. Species symbols are indicated over the exons containing spontaneous or targeted mutations or SNPs. Clu, Canis lupis/dog; Dre, Danio rerio/zebrafish; Mmu, Mus musculus/mouse; Ola, Oryzias latlpes/medaka; Oni, Oreochromis nitoticus/Patagonian pejerrey; Tru, Takifugu rubripes/tiger pufferfish. Lines associated with species symbols indicate multiple exons in a deletion.
Morphological abnormalities were found in males homozygous for the Amh mutation. These males had developed Müllerian duct derivatives, including a uterus, oviducts, and vaginal tissue, in addition to fully descended, normal sized testes and a properly differentiated Wolffian duct system (seminal vesicles, vasa deferentia, and epididymides) identical to the defects observed in Amhr2 mutants shown in Fig. 1B, C. In addition, the homozygous mutant males had fertility defects with only 13% of them able to sire litters. The mutant males were able to produce vaginal plugs in females. Spermatogenesis appeared normal because spermatozoa isolated from the epididymides were able to fertilize wild-type oocytes in vitro.
As predicted from human studies, the loss of Amh in male mice led to the formation of Müllerian duct derivatives, including the oviduct, uterus, and vaginal tissue. The normal descent of the testes in the mutant mice was in contrast to human PMDS that present with undescended testes or inguinal hernia (Josso et al., 1993b). This may be due in part to the bicornuate uterus of the mouse compared to the simplex uterus of human. The fertility defects observed in the Amh mutant male mice were thought to be caused by the mechanical repercussions of having a female reproductive tract superimposed onto a male reproductive tract.
A series of mutant Amh alleles were subsequently generated to investigate the requirement of cis-regulatory sequences located 5’ of the start of transcription (Table 1) (Arango et al., 1999; Shen et al., 1994). In vitro studies had indicated that the nuclear hormone orphan receptor, steroidogenic factor 1 (SF1, also known as NR5A1), bound a 20-bp motif within 100-bp upstream of the TATAA sequence that was essential for Amh transcription (Shen et al., 1994). Approximately 50-bp upstream of the SF1 binding site is a conserved high mobility group protein binding site that was shown to bind the SOX9 transcription factor to activate Amh transcription (De Santa Barbara et al., 1998). Point mutations were introduced into ES cells in the SF1, SOX9, or both binding sites to abolish binding. The SF1 binding site mutation was identical to the one used in the previous in vitro studies (Shen et al., 1994). These alleles were called R1 (SF1 binding site mutation), R2 (SOX9 binding site mutation), and R3 (SF1 and SOX9 binding site mutations) (Arango et al., 1999). In contrast to the in vitro studies, Amh R1/R1 males expressed Amh transcripts in fetal and post-natal testes but had 3-fold lower levels compared to wild type. Thus, the SF1 binding site mutation reduces but does not block Amh transcription and is considered a hypomorphic allele. Surprisingly, Müllerian duct regression occurred normally in Amh R1/R1 males. Amh R2/R2 and Amh R3/R3 mutant males had a complete block in Müllerian duct regression, essentially a phenocopy of the Amh-null male phenotype. These results suggest that SF1 contributes to Amh transcription and that SOX9 is essential for Amh transcription.
The initial gene targeting manipulations in ES cells to generate the Amh R1 allele required the inclusion of a neomycin drug resistance expression cassette that was placed in intron 1 in reverse orientation relative to the direction of Amh transcription (Arango et al., 1999). The allele called R1-neo was effectively null because Amh R1-neo/R1-neo males had a block in Müllerian duct regression. The Amh R1 hypomorphic allele was combined with the Amh R1-neo null allele to generate males with even more reduced levels of Amh transcripts compared to Amh R1/R1 males. Amh R1/R1-neo males developed some uterine tissue, indicating that Müllerian duct regression was incomplete. Thus, AMH levels must be severely reduced to observe partial Müllerian duct regression. Biologically, it appears that the levels of Amh transcription lead to more than sufficient AMH to regress the Müllerian system. Perhaps this is to ensure elimination of the Müllerian duct derivatives during male differentiation to prevent fertility defects.
The second targeted mutation in the AMH pathway was in the Amhr2 gene (Mishina et al., 1996). The mutation was designed to delete ~4.4 kb of the Amhr2 locus, including exons 1-6, replacing them with a drug resistance gene expression cassette (Fig. 2, Table 1). Amhr2 heterozygous mutant ES cells from two clones were injected into blastocysts to generate male chimeras that were bred with controls to produce progeny carrying the targeted allele. All mice that were heterozygous for the mutation were phenotypically normal and fertile. Homozygous mutant females were phenotypically normal and fertile. The homozygous mutant males all had normal sized, descended testes and normally differentiated Wolffian duct derivatives. They all developed Mullerian duct derivatives, including a uterus, oviducts, and partial vagina that were superimposed onto the male reproductive system (Fig. 1B, C). This phenotype was identical to that of Amh-null mice. Additionally, Amh mRNA was expressed in both Amhr2 heterozygous and homozygous mutant males at the stage for the onset of Mullerian duct regression. The homozygous mutant males were able to sire offspring but at less than a 50% rate, with decreased litter size relative to controls. Similar to Amh homozygous mutant males, Amhr2 homozygous mutant males initially had normal spermatogenesis. However, by two months of age they displayed focal atrophy of the germinal epithelium in seminiferous tubules, which led to significantly reduced spermatogenesis by 9 months of age (Mishina et al., 1996).
Two additional Amhr2 alleles have been generated by gene targeting in mouse ES cells (Fig. 2). Both are so-called “knock-in” alleles to express an introduced gene in a pattern similar to Amhr2 transcription. The bacterial lacZ gene was introduced into the Amhr2 locus to visualize Amhr2 expression using a simple histochemical stain and also to follow Müllerian duct regression (Arango et al., 2008; Arango et al., 1999). An intraribosomal entry site (IRES)-lacZ-pA cassette followed by a drug resistance expression cassette was introduced into exon 5 of the Amhr2 locus. The resulting mice expressed lacZ in an Amhr2 pattern, including Sertoli cells of the testes, preantral and antral ovarian follicles, Müllerian duct mesenchyme and postnatal uterine myometrium. Amhr2 lacZ/lacZ males exhibited PMDS exactly like Amhr2-null males, indicating that the lacZ insertion caused a loss-of-function mutation.
The bacterial Cre DNA recombinase gene was also introduced into the Amhr2 locus to genetically modify the target tissue for AMH action, the Amhr2-expressing Müllerian duct mesenchyme, Sertoli and granulosa cells (Jamin et al., 2002). Similar to the lacZ knock-in strategy, an IRES-Cre-pA cassette followed by a drug resistance expression cassette was introduced into exon 5 of the Amhr2 locus to generate the Amhr2 Cre allele. Crosses with mice carrying Cre reporter alleles demonstrated that Cre was expressed in Müllerian duct mesenchyme and fetal gonads. Amhr2 Cre/Cre males develop PMDS just like Amh- and Amhr2-null males (Mullen and Behringer, unpublished results). Thus, similar to the Amhr2-lacZ allele, Amhr2-Cre is likely a null allele.
AMH pathway mutations in dogs
Numerous AMH and AMHR2 recessive mutations have been reported in humans with PMDS (Picard et al., 2017). Amh and Amhr2 knockouts generated by gene targeting in ES ceils in mice have also resulted in PMDS (Behringer et al., 1990; Mishina et al., 1996). The only other mammalian species in which an AMH pathway gene mutation has been identified is the dog (Wu et al., 2009).
There are numerous reports of PMDS in dogs (Meyers-Wallen, 2012). However, a causative mutation has only been identified in AMHR2 (MISRII) in the miniature schnauzer breed (Fig. 3A) (Wu et al., 2009). A non-sense mutation was identified in exon 3 of AMHR2 (C241T) (Fig. 2, Table 1). This is predicted to result in a truncated protein, lacking part of the extracellular region and the transmembrane and serine/threonine kinase domain and therefore no ability to transduce AMH signals. Males homozygous for C241T have uterine horns attached to and located parallel to the vasa deferentia (Fig. 3B, C). 50% of the males C241T homozygotes had unilateral or bilateral cryptorchidism. Mutant males with at least one scrotal testis were fertile (Fig. 3D). The female C241T homozygotes were described as normal (Wu et al., 2009). Two female C241T homozygous produced progeny (Vicki Meyers-Wallen, personal communication). The frequency of this mutant allele was found to be 16% in a cohort of 216 miniature schnauzers (Smit et al., 2018).
Fig. 3. PMDS in the miniature schnauzer.

A. miniature schnauzer. B. reproductive tract from 60-day old male homozygous for AMHR2 missense mutation. C. H&E-stained histological section of vas deferens and connected uterine horn. D. H&E-stained histological section of seminiferous tubule from testis of PMDS male, showing productive spermatogenesis. Image in A from Wikipedia. Images in B-D modified from Wu et al., 2009.
A male German shepherd with PMDS was screened for AMH or AMHR2 mutations (De Lorenzi et al., 2018). Single base pair variants were identified in AMHR2 exons 6 and 7. However, neither variant resulted in a predicted amino acid change. No mutations were found in exons 1-4 of AMH. However, technical difficulties precluded obtaining sequence for 21 bp at the 3’ end of exon 4 and all of exon 5. Thus, it is formally possible that the causative mutation resides in these regions. A male Belgian Malinois with PMDS was identified (Smit et al., 2018). However, no coding or splicing mutations were found in AMH or AMHR2. Likewise, two male Basset Hounds were found to have PMDS but sequencing of their AMHR2 exons did not identify coding mutations (Pop et al., 2017).
The regulatory sequences that direct Amhr2 transcription in the Müllerian duct mesenchyme have not been identified. Thus, it is possible that mutations in the Amh promoter region or the predicted Amhr2 Müllerian duct mesenchyme-specific enhancer could lead to PMDS. More studies are required to identify the Müllerian duct mesenchyme transcriptional enhancer. The miniature schnauzer AMHR2 model and other PMDS dog models provide useful complementary information relative to human PMDS patients and mouse knockout models. Comparisons of these diverse mammalian species may reveal how AMH signaling to conserved target tissues (Müllerian duct mesenchyme, Seroli cells, Leydig cells, granulosa cells) results in a spectrum of reproductive phenotypes influenced by species-specific anatomy.
AMH ligand and receptor gene mutations in teleost fishes
Teleost fish genomes possess amh and amhr2 orthoiogous genes (Pfennig et al., 2015). At first, this seems counterintuitive because teleosts do not possess Müllerian ducts that give rise to reproductive tract organs (Suzuki and Shibata, 2004). In mammals, in addition to its role in Müllerian duct regression, AMH has been shown to regulate germ cell development. As mentioned above, Amh regulates primordial follicle recruitment in the mouse ovary (Durlinger et al., 1999). Amh and amhr2, have been shown to be expressed in at least 20 different teleost fish species (Pfennig et al., 2015). amh transcripts are detected in Sertoli cells of the testis and granulosa cells of the ovary. In some species, amh expression has also been detected in extragonadal tissues, including pituitary gland, brain and heart (Halm et al., 2007). amhr2 expression has been reported in four teleost species: tiger pufferfish (Takifugu rubripes), black porgy (Acanthopagrus schlegeli), medaka (Oryzias latipes) and Nile tilapia (Oreochromis niloticus) (Pfennig et al., 2015). Interestingly, amhr2 expression in Nile tilapia is found in the brain and gonads; amhr2 expression in medaka and fugu is gonad-specific. Thus, amh and amhr2 expression in somatic gonadal cells is conserved between fish and mammals. Does the AMH signaling pathway regulate germ cell development in teleosts? Does it regulate other processes?
The first non-mammalian vertebrate in which the function of an AMH signaling pathway gene was studied was the Japanese rice fish, medaka (Oryzias latipes). An N-ethyl-N-nitrosourea (ENU) recessive mutagenesis screen for gonadal defects identified a mutation in the autosomal amhr2 gene (Morinaga et al., 2007). Chromosomal mapping and sequencing identified an A to G mutation in exon 9 of amhr2. This point mutation results in an amino acid substitution from Tyr to Cys in the receptor kinase domain (Fig. 2, Table 2). Phenotypically, XX female homozygous mutants had enlarged abdomens due to excessive proliferation of germ cells and therefore, enlarged ovaries (Fig. 4A–C). One-half of XY homozygous mutants showed sex reversal, and the remaining half displayed enlarged abdomens due to enlarged testis with overproliferation of germ cells. Analysis of adult fish showed that both male and female mutants where infertile and had a shorter lifespan compared to wild-type controls. These findings suggest that amhr2 regulates germ cell proliferation and male development in medaka.
Table 2.
amh, amhy, and amhr2 mutations in teleost fishes.
| Fish amh | |||||
|---|---|---|---|---|---|
| Species | Allele | Variant | Dominant/recessive | Phenotype | Reference |
| zebrafish (Danio rerio) | Exon 6, 5-bp deletion | induced | recessive | enlarged gonads, germ ceil proliferation; female-biased sex ratio | Lin 2017 |
| zebrafish (Danio rerio) | Exon 6, 17-bp insertion | induced | recessive | enlarged gonads, germ cell proliferation; female-biased sex ratio | Lin 2017 |
| Fish amhy | |||||
| Species | Allele | Variant | Dominant/recessive | Phenotype | Reference |
| Patagonian pejerrey (Odonthestes hatchteri) | Intron 3, 557-bp insertion | spontaneous | Y-linked | Male to female sex reversal (morpholino knockdown) | Hattori 2012 |
| Nile tilapia (Oreochromis niloticus) | Exons 2 and 3, indels (amhΔ-y) | induced | Y-linked | wild-type | Li 2015 |
| Nile tilapia (Oreochromis niloticus) | Exons 2 and 3, indels (amhy) | induced | Y-linked | Male to female sex reversal | Li 2015 |
| Fish amhr2 | |||||
| Species | Allele | Variant | Dominant/recessive | Phenotype | Reference |
| Medaka (Oryzias latipes) | Exon 9, Y390C | induced | recessive | enlarged gonads, germ cell proliferation; male to female sex reversal | Morinaga 2007 |
| Tiger pufferfish (‘Takifugu rubripes) | Exon 9, H384D | variant | male heterozygotes; female homozygotes | N/A | Kamiya 2012 |
| Nile tilapia (Oreochromis niloticus) | Exon 2, indels | induced | recessive | male to female sex reversal | Li 2015 |
| Nile tilapia (Oreochromis niloticus) | Exon 3, indels | induced | recessive | male to female sex reversal | Li 2015 |
Fig. 4. Germ cell tumors and sex reversal in AMH signaling mutant teleost fish.
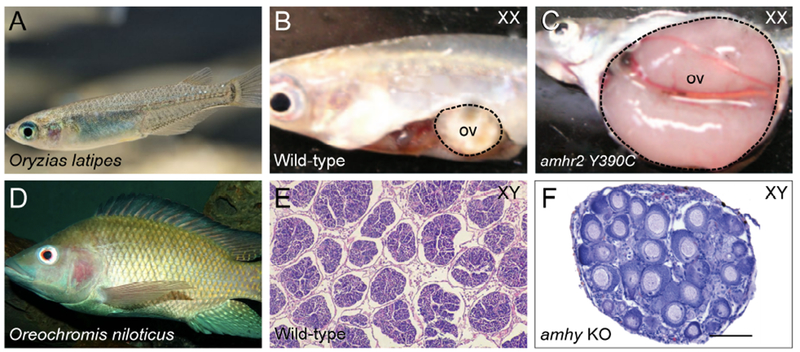
A-C, Japanese rice fish, also known as medaka (Oryzias latipes). B, Wild-type female dissection, showing ovary (ov, dotted line). C, Amhr2 Y390C/Y390C female, showing hypertrophic ovary (dotted line). D-F, Nile tilapia (Oreochromis niloticus). E, Histological section of testis from wild-type XY. F, Histological section of gonad, showing typical ovarian histology in amhy knockout XY. Image in A modified from www.seriouslyfish.com. Images B and C modified from Morinaga et al., 2007, copyright 2007 National Academy of Sciences. Image in D from Wikipedia. Image in E from El-Sayed et al., 2014. Image in F modified from Li et al., 2015.
The tiger pufferfish (Takifugu rubripes) uses an XX-XY sex determination system (Kikuchi et al., 2007). Genetic mapping for the sex-determining locus identified a single nucleotide polymorphism (SNP) in the amhr2 gene (Kamiya et al., 2012). The SNP (C to G) in exon 9 produces an amino acid substitution at position 384 from His to Asp and is located in the receptor kinase domain (Fig. 2, Table 2). Biochemical studies suggest that the amhr2H384 allele has reduced activity compared to amhr2D384. All males were heterozygous (amhr2H384/D384) and all females were homozygous for amhr2H384. This amhr2 SNP is also present in Takifugu pardalis and Takifugu poecilonotus and shows a perfect correlation with sex phenotypes. These results suggest that AMH signaling is required for male development in three species of Takifugu pufferfish. Interestingly, the T. rubripes amhr2 SNP is not found in the green spotted freshwater pufferfish, Tetraodon nigroviridis (Kamiya et al., 2012).
The Patagonian pejerrey (Odonthestes hatcheri) has an XX-XY system of sex determination (Hattori et al., 2010). A male-specific duplication of the amh gene was identified in O. hatched, designated amhy (Y chromosome-specific amh) (Hattori et al., 2012). amhy encodes the mature AMH ligand, is expressed earlier than autosomal amh, and was found to have a 557-bp insertion in intron 3. The authors performed an amhy morpholino knockdown and found that XY embryos resulted in male-to-female sex reversal and upregulation of two genes important for ovarian development: foxl2 and cyp19a1a. Sex-reversed XY fish had ovaries with oocytes that were identical to wild-type XX female fish.
The Nile tilapia (Oreochromis niloticus) also uses an XX-XY sex determination system (Eshel et al., 2011; Lee et al., 2003). The amh gene resides on the X chromosome. Interestingly, O. niloticus has two male-specific duplications of amh designated amhy and amhΔ-y (Li et al., 2015). The coding region of amhy was found to be identical to amh except for a SNP (C to T) in exon 2 that causes an amino acid change from Ser to Leu in the N-terminal domain, amhy has also lost a large (5,608-bp) portion of the promoter region compared to the amh locus. In contrast, amhΔ-y has a 5-bp insertion in exon 6 that produces a frameshift that leads to a premature stop codon. In addition, amhΔ-y also has a 233-bp deletion in exon 7 and an insertion and numerous deletions 5’ of the start codon. The authors used CRISPR/Cas9 technologies to knockout amh, amhy and amhΔ-y genes in XY fish. Guide RNAs were targeted individually to exons 2 and 3 of all three genes (Fig. 2, Table 2). All amhy mutants showed male-to-female sex reversal (Fig. 4D–F), whereas amhΔ-y had no sex-specific mutant phenotype. No results were reported on amh single mutants. In addition, overexpression of amhy but not amh in XX individuals led to female-to-male sex reversal. Furthermore, CRISPR/Cas9-induced mutations in exons 2 and 3 of amhr2 led to 100% male-to-female sex reversal (Fig. 2, Table 2). These results suggest that AMH signaling is required for male sex determination in Nile tilapia.
In zebrafish (Danio rerio), amh was mutated within the AB line genetic background, using CRISPR/Cas9 technology (Lin et al., 2017). Guide RNAs were designed to target exon 6 that would disrupt the expression of the mature AMH ligand (Fig. 2, Table 2). Two alleles were chosen for analysis: a 5-bp deletion and a 17-bp insertion; both produced premature stop codons. Homozygotes for the amh mutant alleles displayed a female-biased (−70%) sex ratio compared to wild-type siblings that were 40% female. Both male and female homozygous mutants showed an enlarged abdomen secondary to hypertrophic gonads. Histological analysis of ovaries and testes revealed increased germ cell proliferation with abnormal differentiation. These findings demonstrate that amh regulates germ cell proliferation and sex ratios in zebrafish. Interestingly, zebrafish does not have an amhr2 gene (Adolfi et al., 2018), suggesting that AMH signals through a different Type 2 receptor in this teleost.
Summary and perspectives
AMH was discovered because of its role in Müllerian duct regression in mammals (Cate et al., 1986; Jost, 1953; Knebelmann et al., 1991; Picard et al., 1986). In humans, PMDS is predominantly caused by recessive mutations in either AMH or AMHR2 (Picard et al., 2017). Similar genetic observations are found in the mouse and dog (Behringer et al., 1994; Mishina et al., 1996; Wu et al., 2009). Perhaps this is not surprising because human, dog and mouse are eutherian mammals. Mouse knockout studies also indicated a role for AMH signaling in regulating germ cell development, specifically in primordial follicle recruitment (Durlinger et al., 1999). However, there are differences among these mammals that may be related to species-specific anatomy and physiology, including testicular descent and male fertility. PMDS has been observed in other mammals (Meyers-Wallen, 2012; Panasiewicz et al., 2015). However, Amh or Amhr2 mutations have not yet been found in those species.
amh and amhr2 genes have also been identified in non-mammalian species, including teleost fishes, i.e. animals that do not form a Müllerian duct. A primary role for the AMH pathway in germ cell proliferation has emerged through studies of amh and amhr2 mutants in teleost species. In teleost fishes, the AMH pathway also regulates gonadal sex determination. Thus, it appears that the biological function of AMH in germ cell development precedes the evolution of Müllerian ducts. Interestingly, many sex-determining genes in mammals, reptiles, birds, and frogs, encode transcription factors (Capel, 2017). The above studies demonstrate that the AMH signaling pathway is a non-transcription factor sex-determining system at least in some species of teleost fishes (Hattori et al., 2012; Kamiya et al., 2012; Li et al., 2015). There is a wide variety of teleost and other fish species in which amh and amhr2 are expressed (Pfennig et al., 2015). Further mutant studies in other fishes may provide insights into the evolutionary origins of these genes and their roles distinct from Müllerian duct regression.
A comparison of AMH and AMHR2 mutant phenotypes in mammals and teleost fishes highlights the various biological processes regulated by AMH signaling, including Müllerian duct regression, primordial follicle recruitment, germ cell proliferation, and gonadal sex determination. CRISPR-CAS9 gene editing technologies have opened up new opportunities to create targeted mutations in species that previously were not amenable for this type of mutagenesis (Bier et al., 2018; DeLay et al., 2018; Farboud, 2017; Moravec and Pelegri, 2019). It will be very interesting to explore the spectrum of phenotypes for AMH and AMHR2 mutations in other vertebrate species. Amniotes, including birds, reptiles, and mammals form Müllerian ducts that differentiate into organs of the female reproductive tract (Roly et al., 2018). However, AMH and AMHR2 mutations have only been observed in eutherian mammals among amniote species. It seems reasonable to expect that Müllerian duct regression in males would be blocked in such mutants. However, would there be a germ cell phenotype? Would gonadal sex determination be altered as in teleost fishes? Amh and Amhr2 genes are also found in amphibians (Jansson et al., 2016; Piprek et al., 2013). Müllerian ducts have been reported in Xenopus laevis and Xenopus tropicalis (Jansson et al., 2016). What Müllerian duct, germ cell, and gonadal sex determination phenotypes might occur in an amphibian if AMH signaling was ablated by mutation (Tandon et al., 2017)? It will be very interesting to determine the spectrum of reproductive phenotypes associated with Amh and Amhr2 mutations that block AMH signaling across vertebrate species.
Acknowledgements
We thank Jace Aloway and Diana Machado for helpful comments on the manuscript. This research was supported by National Institutes of Health (NIH) grant HD030284 and the Ben F. Love Endowment to R.R.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in it’s final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abduljabbar M, Taheini K, Picard JY, Cate RL, & Josso N (2012). Mutations of the AMH type II receptor in two extended families with persistent Mullerian duct syndrome: lack of phenotype/genotype correlation. Horm Res Paediatr, 77(5), 291–297. doi: 10.1159/000338343 [DOI] [PubMed] [Google Scholar]
- Adolfi MC, Nakajima RT, Nobrega RH, & Schartl M (2018). Intersex, Hermaphroditism, and Gonadal Plasticity in Vertebrates: Evolution of the Mullerian Duct and Amh/Amhr2 Signaling. Annu Rev Anim Biosci. doi: 10.1146/annurev-animal-020518-114955 [DOI] [PubMed] [Google Scholar]
- Altincik A, Karaca F, & Onay H (2017). Persistent Mullerian duct syndrome: A novel mutation in the Alphanti-Mullerian Etaormone gene. Hormones (Athens), 16(2), 205–208. doi: 10.14310/horm.2002.1735 [DOI] [PubMed] [Google Scholar]
- Alvaro Mercadal B, Imbert R, Demeestere I, Gervy C, De Leener A, Englert Y, … Delbaere A (2015). AMH mutations with reduced in vitro bioactivity are related to premature ovarian insufficiency. Hum Reprod, 30(5), 1196–1202. doi: 10.1093/humrep/dev042 [DOI] [PubMed] [Google Scholar]
- Arango NA, Kobayashi A, Wang Y, Jamin SP, Lee HH, Orvis GD, & Behringer RR (2008). A mesenchymal perspective of Mullerian duct differentiation and regression in Amhr2-lacZ mice. Mol Reprod Dev, 75(7), 1154–1162. doi: 10.1002/mrd.20858 [DOI] [PubMed] [Google Scholar]
- Arango NA, Lovell-Badge R, & Behringer RR (1999). Targeted mutagenesis of the endogenous mouse Mis gene promoter: in vivo definition of genetic pathways of vertebrate sexual development. Cell, 99(4), 409–419. doi: 10.1016/S0092-8674(00)81527-5 [DOI] [PubMed] [Google Scholar]
- Behringer RR (1994). The in vivo roles of mullerian-inhibiting substance. Curr Top Dev Biol, 29, 171–187. doi: 10.1016/S0070-2153(08)60550-5 [DOI] [PubMed] [Google Scholar]
- Behringer RR, Cate RL, Froelick GJ, Palmiter RD, & Brinster RL (1990). Abnormal sexual development in transgenic mice chronically expressing mullerian inhibiting substance. Nature, 345(6271), 167–170. doi: 10.1038/345167a0 [DOI] [PubMed] [Google Scholar]
- Behringer RR, Finegold MJ, & Cate RL (1994). Mullerian-inhibiting substance function during mammalian sexual development. Cell, 79(3), 415–425. doi: 10.1016/0092-8674(94)90251-8 [DOI] [PubMed] [Google Scholar]
- Bier E, Harrison MM, O’Connor-Giles KM, & Wildonger J (2018). Advances in Engineering the Fly Genome with the CRISPR-Cas System. Genetics, 208(1), 1–18. doi: 10.1534/genetics.117.1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci S, Liguori G, Buttazzi L, Bussani R, &Trombetta C (2002). Bilateral testicular carcinoma in patient with the persistent mullerian duct syndrome. J Urol, 167(4), 1790. doi: 10.1016/S0022-5347(05)65207-X [DOI] [PubMed] [Google Scholar]
- Capel B (2017). Vertebrate sex determination: evolutionary plasticity of a fundamental switch. Nat Rev Genet, 18( 11), 675–689. doi: 10.1038/nrg.2017.60 [DOI] [PubMed] [Google Scholar]
- Cate RL, Mattaliano RJ, Hession C, Tizard R, Farber NM, Cheung A, … Donahoe PK (1986). Isolation of the Bovine and Human Genes for Mullerian Inhibiting Substance and Expression of the Human-Gene in Animal-Cells. Cell, 45(5), 685–698. doi: 10.1016/0092-8674(86)90783-X [DOI] [PubMed] [Google Scholar]
- De Lorenzi L, Arrighi S, Groppetti D, Bonacina S, & Parma P (2018). Persistent Mullerian Duct Syndrome in a German Shepherd Dog. Sex Dev. doi: 10.1159/000492037 [DOI] [PubMed] [Google Scholar]
- De Santa Barbara P, Bonneaud N, Boizet B, Desclozeaux M, Moniot B, Sudbeck P, … Berta P (1998). Direct interaction of SRY-related protein SOX9 and steroidogenic factor 1 regulates transcription of the human anti-Mullerian hormone gene. Mol Cell Biol, 18(11), 6653–6665. doi: 10.1128/MCB.18.11.6653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLay BD, Corkins ME, Hanania HL, Salanga M, Deng JM, Sudou N, … Miller RK (2018). Tissue-Specific Gene Inactivation in Xenopus laevis: Knockout of Ihx1 in the Kidney with CRISPR/Cas9. Genetics, 208(2), 673–686. doi: 10.1534/genetics.117.300468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durlinger AL, Kramer P, Karels B, de Jong FH, Uilenbroek JT, Grootegoed JA, & Themmen AP (1999). Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology, 140( 12), 5789–5796. doi: 10.1210/endo.140.12.7204 [DOI] [PubMed] [Google Scholar]
- El-Sayed Ali T, Abdel-Aziz SH, El-Sayed AF, & Zeid S (2014). Structural and functional effects of early exposure to 4-nonylphenol on gonadal development of Nile tilapia (Oreochromis niloticus): b-histological alterations in testes. Fish Physiol Biochem, 40(5), 1495–1507. doi: 10.1007/s10695-014-9944-5 [DOI] [PubMed] [Google Scholar]
- Eshel O, Shirak A, Weller JI, Slossman T, Hulata G, Cnaani A, & Ron M (2011). Fine-mapping of a locus on linkage group 23 for sex determination in Nile tilapia (Oreochromis niloticus). Anim Genet, 42(2), 222–224. doi: 10.1111/j.1365-2052.2010.02128.x [DOI] [PubMed] [Google Scholar]
- Farboud B (2017). Targeted genome editing in Caenorhabditis elegans using CRISPR/Cas9. Wiley Interdiscip Rev Dev Biol, 6(6). doi: 10.1002/wdev.287 [DOI] [PubMed] [Google Scholar]
- Farikullah J, Ehtisham S, Nappo S, Patel L, & Hennayake S (2012). Persistent Mullerian duct syndrome: lessons learned from managing a series of eight patients over a 10-year period and review of literature regarding malignant risk from the Mullerian remnants. BJU Int, 110(11 Pt C), E1084–1089. doi: 10.1111/j.1464-410X.2012.11184.x [DOI] [PubMed] [Google Scholar]
- Halm S, Rocha A, Miura T, Prat F, & Zanuy S (2007). Anti-Mullerian hormone (AMH/AMH) in the European sea bass: its gene structure, regulatory elements, and the expression of alternatively-spliced isoforms. Gene, 388(1–2), 148–158. doi: 10.1016/j.gene.2006.10.018 [DOI] [PubMed] [Google Scholar]
- Hattori RS, Murai Y, Oura M, Masuda S, Majhi SK, Sakamoto T, … Strussmann CA (2012). A Y-linked anti-Mullerian hormone duplication takes over a critical role in sex determination. Proc Natl Acad Sci USA, 109(8), 2955–2959. doi: 10.1073/pnas.1018392109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori RS, Oura M, Sakamoto T, Yokota M, Watanabe S, & Strussmann CA (2010). Establishment of a strain inheriting a sex-linked SNP marker in Patagonian pejerrey (Odontesthes hatcheri), a species with both genotypic and temperature-dependent sex determination. Anim Genet, 41(1), 81–84. doi: 10.1111/j.1365-2052.2009.01948.x [DOI] [PubMed] [Google Scholar]
- Hutson JM, Davidson PM, Reece LA, Baker ML, & Zhou BY (1994). Failure of Gubernacular Development in the Persistent Mullerian Duct Syndrome Allows Herniation of the Testes. Pediatric Surgery International, 9(8), 544–546. doi: 10.1007/BF00179676 [DOI] [Google Scholar]
- Hutson JM, & Lopez-Marambio FA (2017). The possible role of AMH in shortening the gubernacular cord in testicular descent: A reappraisal of the evidence. J Pediatr Surg, 52(10), 1656–1660. doi: 10.1016/j.jpedsurg.2017.05.021 [DOI] [PubMed] [Google Scholar]
- Hutson JM, Thorup JM, & Beasley SW (2016). Transabdominal Migration of the Testis Descent of the Testis (pp. 17–28). Cham: Springer International Publishing. [Google Scholar]
- Imbeaud S, Belville C, Messika-Zeitoun L, Rey R, di Clemente N, Josso N, & Picard JY (1996). A 27 base-pair deletion of the anti-mullerian type II receptor gene is the most common cause of the persistent mullerian duct syndrome. Hum Mol Genet, 5(9), 1269–1277. doi: 10.1093/hmg/5.9.1269 [DOI] [PubMed] [Google Scholar]
- Imbeaud S, Faure E, Lamarre I, Mattei MG, di Clemente N, Tizard R, … Picard JY (1995). Insensitivity to anti-mullerian hormone due to a mutation in the human anti-mullerian hormone receptor. Nat Genet, 11(4), 382–388. doi: 10.1038/ng1295-382 [DOI] [PubMed] [Google Scholar]
- Jamin SP, Arango NA, Mishina Y, Hanks MC, & Behringer RR (2002). Requirement of Bmpr1a for Mullerian duct regression during male sexual development. Nat Genet, 32(3), 408–410. doi: 10.1038/ng1003 [DOI] [PubMed] [Google Scholar]
- Jansson E, Mattsson A, Goldstone J, & Berg C (2016). Sex-dependent expression of anti-Mullerian hormone (amh) and amh receptor 2 during sex organ differentiation and characterization of the Mullerian duct development in Xenopus tropicalis. Gen Comp Endocrinol, 229, 132–144. doi: 10.1016/j.ygcen.2016.03.018 [DOI] [PubMed] [Google Scholar]
- Josso N (1973). In vitro synthesis of mullerian-inhibiting hormone by seminiferous tubules isolated from the calf fetal testis. Endocrinology, 93(4), 829–834. doi: 10.1210/endo-93-4-829 [DOI] [PubMed] [Google Scholar]
- Josso N (1974). Mullerian-inhibiting activity of human fetal testicular tissue deprived of germ cells by in vitro irradiation. Pediatr Res, 8(8), 755–758. doi: 10.1203/00006450-197408000-00004 [DOI] [PubMed] [Google Scholar]
- Josso N, Belville C, di Clemente N, & Picard JY (2005). AMH and AMH receptor defects in persistent Mullerian duct syndrome. Hum Reprod Update, 77(4), 351–356. doi: 10.1093/humupd/dmi014 [DOI] [PubMed] [Google Scholar]
- Josso N, Cate RL, Picard JY, Vigier B, di Clemente N, Wilson C, … et al. (1993). Anti-mullerian hormone: the Jost factor. Recent Progress in Hormone Research, 48, 1–59. doi: 10.1016/B978-0-12-571148-7.50005-1 [DOI] [PubMed] [Google Scholar]
- Josso N, Picard JY, Imbeaud S, Carre-Eusebe D, Zeller J, & Adamsbaum C (1993). The persistent mullerian duct syndrome: a rare cause of cryptorchidism. Eur J Pediatr, 152 Suppl2, S76–78. doi: 10.1007/BF02125444 [DOI] [PubMed] [Google Scholar]
- Jost A (1953). Problems of Fetal Endocrinology - the Gonadal and Hypophyseal Hormones. Recent Progress in Hormone Research, 8, 379–418. [DOI] [PubMed] [Google Scholar]
- Kamiya T, Kai W, Tasumi S, Oka A, Matsunaga T, Mizuno N, … Kikuchi K (2012). A trans-species missense SNP in Amhr2 is associated with sex determination in the tiger pufferfish, Takifugu rubripes (fugu). PLoS Genet, 8(7), e1002798. doi: 10.1371/journal.pgen.1002798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Kai W, Hosokawa A, Mizuno N, Suetake H, Asahina K, & Suzuki Y (2007). The sex-determining locus in the tiger pufferfish, Takifugu rubripes. Genetics, 175(4), 2039–2042. doi: 10.1534/genetics.106.069278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knebelmann B, Boussin L, Guerrier D, Legeai L, Kahn A, Josso N, & Picard JY (1991). Anti-Mullerian hormone Bruxelles: a nonsense mutation associated with the persistent Mullerian duct syndrome. Proc Natl Acad Sci USA, 88(9), 3767–3771. doi: 10.1073/pnas.88.9.3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BY, Penman DJ, & Kocher TD (2003). Identification of a sex-determining region in Nile tilapia (Oreochromis niloticus) using bulked segregant analysis. Anim Genet, 34(5), 379–383. doi: 10.1046/j.1365-2052.2003.01035.x [DOI] [PubMed] [Google Scholar]
- Li L, Zhou X, Wang X, Wang J, Zhang W, Wang B, … Kee K (2016). A dominant negative mutation at the ATP binding domain of AMHR2 is associated with a defective anti-Mullerian hormone signaling pathway. Mol Hum Reprod, 22(9), 669–678. doi: 10.1093/molehr/gaw040 [DOI] [PubMed] [Google Scholar]
- Li M, Sun Y, Zhao J, Shi H, Zeng S, Ye K, … Wang D (2015). A Tandem Duplicate of Anti-Mullerian Hormone with a Missense SNP on the Y Chromosome Is Essential for Male Sex Determination in Nile Tilapia, Oreochromis niloticus. PLoS Genet, 77(11), e1005678. doi: 10.1371/journal.pgen.1005678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Mei J, Li Z, Zhang X, Zhou L, & Gui JF (2017). Distinct and Cooperative Roles of amh and drmrt1 in Self-Renewal and Differentiation of Male Germ Cells inZebrafish. Genetics, 207(3), 1007–1022. doi: 10.1534/genetics.117.300274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers-Wallen VN (2012). Gonadal and sex differentiation abnormalities of dogs and cats. Sex Dev, 6(1–3), 46–60. doi: 10.1159/000332740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A, Hong MK, & Hutson JM (2004). The broad ligament: a review of its anatomy and development in different species and hormonal environments. Clin Anat, 17(3), 244–251. doi: 10.1002/ca.10173 [DOI] [PubMed] [Google Scholar]
- Mishina Y, Rey R, Finegold MJ, Matzuk MM, Josso N, Cate RL, & Behringer RR (1996). Genetic analysis of the Mullerian-inhibiting substance signal transduction pathway in mammalian sexual differentiation. Genes Dev, 10(20), 2577–2587. doi: 10.1101/gad.10.20.2577 [DOI] [PubMed] [Google Scholar]
- Moravec CE, & Pelegri FJ (2019). An Accessible Protocol for the Generation of CRISPR-Cas9 Knockouts Using INDELs in Zebrafish. Methods Mol Biol, 1920, 377–392. doi: 10.1007/978-1-4939-9009-2_23 [DOI] [PubMed] [Google Scholar]
- Morinaga C, Saito D, Nakamura S, Sasaki T, Asakawa S, Shimizu N, … Kondoh H (2007). The hotei mutation of medaka in the anti-Mullerian hormone receptor causes the dysregulation of germ cell and sexual development. Proc Natl Acad Sci U S A, 104(23), 9691–9696. doi: 10.1073/pnas.0611379104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RD, & Behringer RR (2014). Molecular genetics of Mullerian duct formation, regression and differentiation. Sex Dev, 8(5), 281–296. doi: 10.1159/000364935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbantoglu O, Demir K, Korkmaz HA, Buyukinan M, Yildiz M, Tunc S, & Ozkan B (2015). A novel mutation of AMH in three siblings with persistent Mullerian duct syndrome. J Pediatr Endocrinol Metab, 28(11–12), 1379–1382. doi: 10.1515/jpem-2014-0501 [DOI] [PubMed] [Google Scholar]
- Nishi MY, Domenice S, Maciel-Guerra AT, Zaba Neto A, Silva MA, Costa EM, … Mendonca BB (2012). Analysis of anti-Mullerian hormone (AMH) and its receptor (AMHR2) genes in patients with persistent Mullerian duct syndrome. Arq Bras Endocrinol Metabol, 56(8), 473–478. doi: 10.1590/S0004-27302012000800002 [DOI] [PubMed] [Google Scholar]
- Panasiewicz G, Zamojska A, Bieniek M, Gizejewski Z, & Szafranska B (2015). Persistent Mullerian duct syndrome (PMDS) in the Polish free-ranged bull populations of the European bison (Bison bonasus L.). Anim Reprod Sci, 152, 123–136. doi: 10.1016/j.anireprosci.2014.11.012 [DOI] [PubMed] [Google Scholar]
- Pfennig F, Standke A, & Gutzeit HO (2015). The role of Amh signaling in teleost fish--Multiple functions not restricted to the gonads. Gen Comp Endocrinol, 223, 87–107. doi: 10.1016/j.ygcen.2015.09.025 [DOI] [PubMed] [Google Scholar]
- Picard JY, Benarous R, Guerrier D, Josso N, & Kahn A (1986). Cloning and expression of cDNA for anti-mullerian hormone. Proc Natl Acad Sci USA, 83(15), 5464–5468. doi: 10.1073/pnas.83.15.5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard JY, Cate RL, Racine C, & Josso N (2017). The Persistent Mullerian Duct Syndrome: An Update Based Upon a Personal Experience of 157 Cases. Sex Dev, 11(3), 109–125. doi: 10.1159/000475516 [DOI] [PubMed] [Google Scholar]
- Piprek RP, Pecio A, Laskowska-Kaszub K, Kubiak JZ, & Szymura JM (2013). Sexual dimorphism of AMH, DMRT1 and RSP01 localization in the developing gonads of six anuran species. Int J Dev Biol, 57(11–12), 891–895. doi: 10.1387/ijdb.130192rp [DOI] [PubMed] [Google Scholar]
- Pop AR, Henegariu 0, Micu R, Sonea A, Irimie A, Henegariu A, & Groza IS (2017). Hormone receptor type 2 antimullerian gene role in dogs with Persistent Mullerian Ducts Syndrome. Romanian Biotechnological Letters, 22(6), 13029–13034. doi: 10.25083/rbl [DOI] [Google Scholar]
- Ren X, Wu D, & Gong C (2017). Persistent Mullerian duct syndrome: A case report and review. Exp Ther Med, 14(6), 5779–5784. doi: 10.3892/etm.2017.5281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roly ZY, Backhouse B, Cutting A, Tan TY, Sinclair AH, Ayers KL, … Smith CA (2018). The cell biology and molecular genetics of Mullerian duct development. Wiley Interdiscip Rev Dev Biol, 7(3), e310. doi: 10.1002/wdev.310 [DOI] [PubMed] [Google Scholar]
- Shamim M (2007). Persistent Mullerian duct syndrome with transverse testicular ectopia presenting in an irreducible recurrent inguinal hernia. J Pak Med Assoc, 57(8), 421–423. [PubMed] [Google Scholar]
- Shen WH, Moore CC, Ikeda Y, Parker KL, & Ingraham HA (1994). Nuclear receptor steroidogenic factor 1 regulates the mullerian inhibiting substance gene: a link to the sex determination cascade. Cell, 77(5), 651–661. doi: 10.1016/0092-8674(94)90050-7 [DOI] [PubMed] [Google Scholar]
- Smit MM, Ekenstedt KJ, Minor KM, Lim CK, Leegwater P, & Furrow E (2018). Prevalence of the AMHR2 mutation in Miniature Schnauzers and genetic investigation of a Belgian Malinois with persistent Mullerian duct syndrome. Reprod Domest Anim, 53(2), 371–376. doi: 10.1111/rda.13116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, & Shibata N (2004). Developmental process of genital ducts in the medaka, Oryzias latipes. Zoolog Sci, 21(4), 397–406. doi: 10.2108/zsj.21.397 [DOI] [PubMed] [Google Scholar]
- Takahashi M, Hayashi M, Manganaro TF, & Donahoe PK (1986). The ontogeny of mullerian inhibiting substance in granulosa cells of the bovine ovarian follicle. Biol Reprod, 35(2), 447–453. doi: 10.1095/biolreprod35.2.447 [DOI] [PubMed] [Google Scholar]
- Tandon P, Conlon F, Furlow JD, & Horb ME (2017). Expanding the genetic toolkit in Xenopus: Approaches and opportunities for human disease modeling. Dev Biol, 426(2), 325–335. doi: 10.1016/j.ydbio.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wan S, Pujar S, Haskins ME, Schlafer DH, Lee MM, & Meyers-Wallen VN (2009). A single base pair mutation encoding a premature stop codon in the MIS type II receptor is responsible for canine persistent Mullerian duct syndrome. J Androl, 30(1), 46–56. doi: 10.2164/jandrol.108.005736 [DOI] [PMC free article] [PubMed] [Google Scholar]