

Abstract
Glycyl radical enzymes (GREs) utilize a glycyl radical cofactor to carry out a diverse array of chemically challenging enzymatic reactions in anaerobic bacteria. Although the glycyl radical is a powerful catalyst, it is also oxygen sensitive such that oxygen exposure causes cleavage of the GRE at the site of the radical. This oxygen sensitivity presents a challenge to facultative anaerobes dwelling in areas prone to oxygen exposure. Once GREs are irreversibly oxygen-damaged, cells either need to make new GREs or somehow repair the damaged one. One particular GRE, pyruvate formate lyase (PFL), can be repaired through the binding of a 14.3 kDa protein, termed YfiD, which is constitutively expressed in E. coli. Herein, we have solved a solution structure of this ‘spare part’ protein using nuclear magnetic resonance (NMR) spectroscopy. These data, coupled with data from circular dichroism, indicate that YfiD has an inherently flexible N-terminal region (residues 1–60) that is followed by a C-terminal region (residues 72–127) that has high similarity to the glycyl radical domain of PFL. Reconstitution of PFL activity requires that YfiD bind within the core of the PFL barrel fold; however, modeling suggests that oxygen-damaged, cleaved, PFL cannot fully accommodate YfiD. We further report that a PFL variant that mimics the oxygen-damaged enzyme is highly susceptible to proteolysis, yielding additionally truncated forms of PFL. One such PFL variant of ~77 kDa makes an ideal scaffold for the accommodation of YfiD. A molecular model for the rescue of PFL activity by YfiD is presented.
Keywords: radical chemistry, glycyl radical enzyme, nuclear magnetic resonance, cofactor repair, circular dichroism
Graphical abstract
Introduction
Radical-based chemistry allows enzymes to catalyze an impressive array of molecular transformations such as those that involve cleavage and formation of C-C bonds (Fig. 1A) [1–5]. This chemistry comes at a price, however, as a radical species must be generated on the enzyme, a process that usually involves a metallocofactor [6]. The radical species must also be stored between turnovers, controlled during turnover, and protected from or repaired after oxygen damage. Some radical-generating cofactors have native protective mechanisms; adenosylcobalamin (AdoCbl) typically generates its highly reactive 5’-deoxyadenosyl (5’-dAdo) radical through Co-C bond homolysis only when substrate is present, and then reforms the Co-C bond when each turnover is complete, limiting both unwanted chemistry and interception of the radical species [3]. Although this strategy is relatively successful, AdoCbl is an expensive cofactor, requiring upwards of thirty enzymes for its biosynthesis [7,8]. In contrast, glycyl radical enzymes (GREs) have an inexpensive cofactor, a glycyl radical species (i.e.a glycine that is missing a hydrogen atom), which transiently forms the catalytically essential thiyl radical species during turnover (Fig. 1A). A glycyl radical species does require a metallocofactor-containing activase for its formation [9,10], but a single activase can post-translationally modify many of its associated GREs, and the modification need only happen once, as the glycyl radical is reformed from the thiyl radical on every round of turnover (Fig. 1A). Additionally, the activases are S-adenosylmethionine (AdoMet) radical enzymes [9], which have been referred to as ‘poor man AdoCbl enzymes’ since they use inexpensive cofactors, [4Fe–4S] and AdoMet, for radical generation [2,11–13]. GREs are thus an efficient option for radical-based enzymology, but unlike AdoCbl-dependent enzymes, they are very sensitive to inactivation by molecular oxygen. In these enzymes, oxygen-induced cleavage occurs at the glycyl radical site, splitting the enzyme into two pieces. For strictly anoxic-living microbes, GREs are an excellent option, powerful catalysts that are inexpensive for the cell to produce; however, for microbes that live under microaerobic conditions, GREs can be an acceptable option if damage repair is a possibility. Here we investigate a novel repair mechanism for the central microbial metabolic GRE, pyruvate formate lyase (PFL).
Figure 1:


PFL reaction, activation, and rescue by YfiD. A) PFL catalyzes conversion of pyruvate and CoA to acetyl-CoA and formate. The glycyl radical (G734) of PFL is generated through a direct hydrogen atom abstraction by PFL activating enzyme, or PFL-AE, a member of the AdoMet radical enzyme superfamily. Catalysis occurs through a putative catalytic thiyl radical. B) PFL-AE (orange) installs a glycyl radical on PFL in a reaction that is thought to require a conformational change of a region of PFL known as the glycyl radical domain (GRD) (maroon). Upon oxygen exposure, the C-terminus of PFL is cleaved at the glycyl radical site (star). YfiD (blue), which is also capable of being activated by PFL-AE, is able to bind to PFL and restore PFL activity.
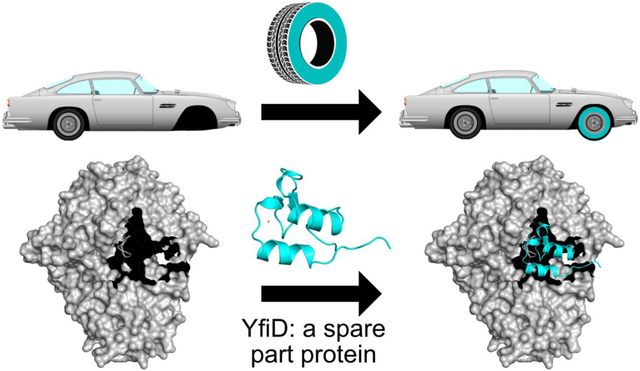
PFL cleaves the C-C bond of pyruvate to produce formate and acetyl-CoA, providing acetyl-CoA to metabolic pathways including the citric acid cycle during anaerobic glycolysis (Fig. 1A) [14,15]. Although the glycyl radical of PFL is oxygen sensitive, the enzyme is constitutively expressed, albeit with about tenfold higher PFL expression under anoxic conditions [9,16,17]. Under transient microaerobic environments, Escherichia coli upregulate expression of the yfid gene product YfiD [18,19], a small protein that appears to be able to rescue the enzymatic activity of oxygen-damaged PFL [20]. Key regulators of anaerobiosis and pH in E. coli, arcA, fnr and fur, appear to be responsible for YfiD upregulation [21–24]. YfiD has 127 amino acids, the last sixty of which share 77% sequence identity to the last sixty residues of E. coli PFL, the portion of PFL that contains both the glycyl radical (Gly734) and a region of the enzyme (residues 713–759), referred to as the glycyl radical domain (GRD) (Fig. 2) [25]. The GRD was previously shown by circular dichroism (CD) spectroscopy to undergo a conformational change when PFL binds to its activating enzyme (PFL-AE), presumably to expose the buried Gly734 for radical installation (Fig. 1B) [26]. It is also this region (734–759) of the activated protein that is cleaved upon oxygen exposure. The ability of a highly homologous small protein to restore activity to a much larger enzyme suggests that YfiD is the biological equivalent of a ‘spare part’ (Fig. 1B) [20].
Figure 2:

Structure of PFL (PDB ID: 2PFL) monomer and sequence alignment of the C-terminal region of PFL and YfiD. A) Crystal structure of PFL showing residues 633–711 in blue and residues 712–759 that comprise the glycyl radical domain (GRD) in red. The thiyl radical loop is in yellow, and the rest of the PFL monomer is in gray. B) Topographical representation of PFL structure. C) Sequence of YfiD and PFL residues 633–759, showing the secondary structural elements of PFL below the sequences.
The molecular mechanism by which YfiD is able to restore activity is unknown. Previous work established that PFL-AE can install a glycyl radical on YfiD [20], and we assume that activated YfiD must bind to cleaved PFL (cPFL) to restore activity, but exactly how this restoration takes place is unclear. The cleavage that occurs cuts the loop containing Gly734 in half. This glycyl radical loop is positioned next to the loop that contains the transient thiyl radical species in the core of PFL’s 10-stranded β-barrel (Fig. 2A, B). YfiD is much longer, at 127 residues, than the cleaved portion of PFL (734–759), and it is uncertain if more residues of cPFL will be displaced as a result of YfiD binding or if only a small fraction of YfiD will bind to cPFL. A sequence alignment of PFL GRD and YfiD shows that PFL Gly734 is directly aligned with YfiD Glyl02. The residues in this region of the alignment are completely conserved for a stretch of 22 amino acids. Furthermore, the sequence similarity between YfiD and PFL is restricted to the last sixty residues, raising the question of whether YfiD will adopt a PFL-like fold in the non-conserved segments (Fig. 2C). Although the exact number of residues involved in binding remains unknown, YfiD must be bound tightly enough to cPFL that the inserted glycyl radical is stable. To investigate these questions, we have taken a structural approach using nuclear magnetic resonance (NMR) spectroscopy and circular dichroism (CD) spectroscopy to determine the solution structure of the E. coli YfiD protein and explore its thermal stability. We have further created a truncated form of PFL that mimics the oxygen-cleaved enzyme (cPFL) and have investigated whether cellular components cause additional proteolysis of cPFL in the presence of YfiD. Our results taken together allow us to propose a model for how YfiD binds and restores activity to cPFL.
Results
YfiD is a partially structured/partially unstructured protein
Representative spectra of YfiD from 2D 1H-15N heteronuclear single quantum coherence (HSQC) experiments show good peak separation indicative of a folded protein (Fig. 3A). To solve the structure, standard triple-resonance experiments were used for backbone and sidechain assignments (see Materials and Methods), and distance restraints based on nuclear Overhauser effects (NOEs) were obtained from 1H-15N-NOSEY-HSQC and 1H-13C-NOSEY-HSQC experiments.
Figure 3:

Solution structure of YfiD. A) HSQC of YfiD. B) Ten lowest energy NMR structures of residues 68–127 of YfiD (PDB ID 60WR; backbone RMSD 0.07 Å). The N-terminal region of YfiD shows fewer crosspeaks and is unable to be assigned, thus indicating this region is disordered, as depicted by the dotted line. C) Sequence and secondary structure of YfiD (cyan) and residues 633–679 of PFL (colored as in Figure 2).
The calculations of the YfiD NMR structure converge well, as indicated by the ensemble of the ten lowest energy structures shown in Fig. 3B. However, the solved structure only contains the C-terminal half of YfiD (residues 68–127). Very few cross peaks could be identified for the N-terminal region of YfiD (residues 1–65), suggesting that this region is unstructured. In contrast to the N-terminal half, the C-terminal residues of YfiD have a high level of sequence similarity and share the same fold as the last sixty residues of PFL. YfiD has one β-strand (y-βl) and two α-helices (y-α1a and y-α1b), with a Gly radical loop situated between the two helices (Fig. 3B, C). Superposition of the structure of YfiD onto PFL reveals that the structure of YfiD y-βl strand overlaps with the PFL βl0 loop, and YfiD y-α1a and y-α1b helices overlap with PFL α10a and α10b helices (Fig. 3C).
The first 60 residues of YfiD dramatically affect YfiD’s thermal denaturation
To investigate the thermal stability of a protein that appears to be partially unstructured, we employed far-UV circular dichroism (CD) spectroscopy. We also created a truncated form of YfiD (truncYfiD) in which the majority of residues found to be unstructured by NMR (residues 1–60) were deleted. Thermal denaturation measurements were carried out on both YfiD and truncYfiD to determine the protein melting temperatures under two different buffering conditions. The denaturation curves for YfiD reveal that the protein does not undergo a denaturation typical of a well-folded protein, as illustrated by the non-sigmoidal shape to the denaturation curve (Fig. 4A, B). This unusual unfolding behavior is consistent with a protein that has a large unstructured region, where that unstructured region dominates the CD spectrum [27,28]. As a control, we tested full length YfiD expressed in the presence of PFL to determine if co-expression leads to a better ordered YfiD protein; it did not. An analogous CD spectrum was obtained (Fig. S1). However, when the unstructured part of YfiD is genetically removed, a classic sigmoidal shape for the denaturation curve is observed (Fig. 4C, D). In other words, truncYfiD (residues 61–127) behaves as well-ordered, folded protein. Again, this result is consistent with the NMR structure, in which residues 68–127 are structured.
Figure 4:

Circular dichroism spectroscopy thermal denaturation measurements for YfiD in the presence and absence of the first 60 residues. A) and B) The thermal denaturation curves for intact YfiD in two different buffers (MES, pH 6.5 and HEPES, pH 7.2). For the YfiD in HEPES buffer, a melting temperature cannot be determined as the algorithm does not converge. C) and D) The thermal dénaturation curves for a YfiD variant that is missing the first 60 residues of the protein (truncYfiD) in two buffers (MES, pH 6.5 and HEPES, pH 7.2). Tm values are listed in panels.
The last 62 residues of YfiD form an autonomous glycyl radical cofactor
Given that the oxygen-cleavage site on PFL is in the middle of the Gly radical loop, it is not surprising that YfiD duplicates the full Gly radical loop. A glycine residue at the N-terminus of a protein would likely not be able to form a stable glycyl radical species [29], even if it were a substrate for PFL-AE. Thus, it was expected that YfiD would contain an intact Gly radical loop, including the PFL C-terminal αl0b helix that is cleaved, but it was unclear how much more of the PFL structure would be duplicated in YfiD. Although the high sequence homology between YfiD and PFL starts at the beginning of PFL’s β10 strand, the residues preceding this strand could have formed similar secondary structure with a different sequence, but that does not appear to be the case. We do find, however, a full duplication of the PFL β10 strand and α10a helix, some thirty residues more than the part of PFL that is cleaved by oxygen. The inclusion of these ‘extra’ residues could serve multiple functions, including: providing binding affinity to PFL-AE; protecting the Gly radical during transfer from PFL-AE to cPFL; providing binding affinity to cPFL; and securing the Gly radical loop in the active site barrel for proper radical transfer to the Cys loop to initiate catalysis. In terms of binding to either PFL-AE for activation or PFL for restoration of enzyme activity, the use of a highly identical sequence in this structural unit of YfiD (a BLAST alignment of YfiD and PFL yields 77% identity for YfiD residues 64–127 aligned to PFL residues 696–759), allows YfiD to swap in for PFL residues with shape and charge complementary intact.
To further investigate how YfiD might interact with PFL-AE, we manually docked YfiD onto the X-ray structure of PFL-AE, emulating the proposed mode of binding of PFL to PFL-AE (Fig. 5) [25]. We observe that the two helices and the Gly radical loop of YfiD fit well to PFL-AE; however, YfiD’s y-β1 strand is not accommodated in this docking mode (Fig. 5).
Figure 5:

YfiD manually docked with PFL-AE. The structure of YfiD (teal) is superimposed onto the docked structure of PFL GRD (purple) bound to PFL-AE (orange) (PDB ID: 3CB8). Catalytic Gly are shown as spheres. AdoMet is shown in sticks. The 90° rotated view (right) shows that the y-β1 strand clashes with PFL-AE.
Proteolysis of PFL follows oxygen-induced cleavage
The discovery that YfiD is structurally homologous to sixty residues of the C-terminus of PFL, rather than just to the twenty-five residues of PFL (734–759) that are cleaved upon oxidative damage, indicates that more residues of PFL might need to be displaced from the active site for YfiD to bind in the location that places the Gly radical loop correctly for chemistry to occur. Given that one cleavage event often leads to protein unfolding, which can cause previously buried cleavage sites to be exposed, we wanted to investigate whether cPFL is further unfolded by examining its susceptibility to proteolysis by cellular proteases. We co-expressed His-tagged YfiD and a truncated version of PFL that mimics the oxygen-damaged enzyme (cPFL), and purified the resulting complex by immobilized metal affinity chromatography (IMAC). In addition to full length cPFL (81 kDa), we observe bands for two truncation products (Fig. 6), a more intense band that is consistent with a 77 kDa fragment, and a less intense band that is consistent with a 71 kDa fragment. Cleavage occurring between residues 690–695 (tlPFL) and residues 635–640 (t2PFL) would be expected to generate fragments of 77 kDa and 71 kDa, respectively. A common E coli proteolytic enzyme, chymotrypsin-like protease I, has sequence specificity consistent with cleavage in these regions. In fact, the ExPASy PeptideCutter prediction algorithm predicts two cleavage sites between residues 690 and 695 with > 73% probability of being cleaved by E. coli chymotrypsin-like protease I, and one cleavage site between residues 635 and 640 with 58% probability of being cleaved by this common E. coli protease (Fig. 6B). These results are consistent with cPFL undergoing further protein unfolding events following oxygen-induced cleavage at Gly734, which exposes previously buried proteolysis sites.
Figure 6:

cPFL is subject to proteolysis. A) SDS-PAGE of cPFL. Molecular weights of cPFL, 81 kDa, and two additional truncation products at 77 kDa (tlPFL) and 71 kDa (t2PFL) were extrapolated using a 10–200 kDa protein standard. These molecular weight bands correspond to the expected length of cPFL ending at residue 733, PFL truncated between residues 690–695 (tlPFL), and PFL truncated between 635–640 (t2PFL), respectively. B) Crystal structure of the PFL monomer showing residues 734–759 (end of C-terminus) in maroon, residues 695–733 in blue, the catalytic Cys loop in purple, and the rest of the monomer in gray. Upon exposure to oxygen, PFL is cleaved at Gly734. This cleavage exposes two truncation sites for E. coli chymotrypsin-like protease I activity. Positions of cleavage sites are indicated with stars.
A single proteolysis event following oxygen-induced cleavage of PFL creates a PFL variant optimal for YfiD docking
As described above, the YfiD structure duplicates all of the Gly radical domain (red in Fig. 7A), as well as β10 strand (blue in Fig. 7A) and α10a helix (red in Fig. 7A), but does not duplicate β9 strand and α8b helix (blue in Fig. 7A, YfiD teal in Fig. 7B). The active site in cPFL would still be relatively buried (Fig. 7C) with no room for YfiD to bind. A single proteolysis event at the β9-α9 loop (Fig. 7A) would result in loss of β10 strand and α10a helix as well as the remaining residues of the GRD. A space filling illustration of such a PFL (tlPFL) shows an open active site that is ready for YfiD binding (Fig. 7D). Modeling YfiD into tlPFL shows an impressive fit with very little need for structural rearrangement (Fig. 7D–E). When YfiD is positioned as shown in 7E, the Gly radical loop is located suitably for radical transfer to the thiyl radical loop, and y-β1 strand is situated such that it serves as a strand to complete the 10-stranded barrel of PFL (Fig. 7B).
Figure 7:

Docking of YfiD to PFL variants. A) Topology diagram for PFL showing proteolytic cut sites identified in Fig. 6 (purple arrows). B) Topology diagram for YfiD (teal) bound to tlPFL. Second cut site is also shown (purple arrow). C) Space filling depiction of cPFL. D) Space filling depiction of tlPFL. E) YfiD manually docked into the pocket of tlPFL. On the right panel, YfiD is depicted in ribbons. PFL models generated from PDB ID: 2PFL.
Discussion
Since the discovery of YfiD by Knappe and coworkers in 2001 [20], the idea that enzymes have ‘spare parts’ has been a source of fascination. Here we present the NMR structure of YfiD and consider the molecular basis by which this ‘spare part protein’ may function in the repair of the major microbial metabolic enzyme PFL. First and foremost, radical chemistry requires precise positioning of cofactors, residues, and substrates. Thus, the restoration of catalysis in PFL requires that an activated YfiD be precisely positioned in the active site of PFL such that the glycyl radical species is stable, allowing for reversible radical transfer to Cys418 and multiple turnovers. Prior to this work, it was not clear how much of PFL would be duplicated in the YfiD structure and whether the binding of the YfiD structural unit to cPFL would require conformational rearrangements. Although a structure of YfiD:PFL has not yet been obtained despite considerable effort, we do not believe that the observed proteolysis of cPFL is a coincidence. Due to the fact that the major truncated form of cPFL (t1PFL) is a species that contains an opening that is perfectly sized for YfiD (Fig. 7D.E), we now propose that oxygen-induced cleavage to form cPFL is followed by proteolysis to form t1PFL, and that t1PFL is the enzyme variant to which YfiD binds (Fig. 8, steps 1–4).
Figure 8:

A revised model for YfiD rescue. When exposed to oxygen, activated PFL is cleaved at its glycyl radical site, G734 (cPFL). The cleaved region of PFL (G734 through the C-terminus) is depicted in red. In order for YfiD to fit into the pocket, additional residues that include residues of βlO strand of PFL (purple) must be lost, forming a tIPFL variant. From here, we propose that the y-βl strand of YfiD completes the 10-stranded barrel of PFL and the disordered N-terminus of YfiD helps secure this spare part protein to tIPFL. After YfiD radical domain (GRD) flips out, allowing PFL-AE to activate YfiD. Once activated, the GRD of YfiD enters the open PFL active site, resulting in a reactivated PFL:YfiD complex. This complex should remain stable for multiple rounds of turnover with YfiD acting as a noncovalent subunit.
Of course, YfiD binding cannot reconstitute PFL activity if YfiD does not contain a glycyl radical species. Thus, YfiD requires post-translational modification by PFL-AE to install a radical at position Glyl02 [20] before YfiD assumes its catalytically competent position inside the PFL barrel structure. Although it has been shown that YfiD is a substrate for PFL-AE [20], little is known about the nature of the complex that is formed. In general, our understanding of glycyl radical formation is limited [1]. Currently, the only structural data we have on how a glycyl radical domain is activated by an activase is from the crystal structure of a 7-mer peptide substrate bound to PFL-AE [25]. We do not know how much of the PFL glycyl radical domain binds to PFL-AE to afford activation nor do we know the exact binding mode of PFL to PFL-AE.
Previously, we presented a model for the binding of residues 712–759 of PFL to PFL-AE that was based on shape and charge complementarity [25]. If we now superimpose the NMR structure of YfiD onto this PFL:PFL-AE docked structure, we observe that the two helices and the Gly radical loop of YfiD fit well, but the y-βl strand does not (Fig. 5). It is possible that the docked PFL:PFL-AE structure is not an accurate model for how YfiD binds to PFL-AE; another possibility is that YfiD undergoes a conformational rearrangement of the y-β1 strand before binding to PFL-AE, potentially assisted by the binding of YfiD to t1PFL in a pre-activation step (Fig. 8, steps 3–4).
Although biochemical data have not yet been obtained to demonstrate that YfiD, t1PFL, and PFL-AE form a ternary complex prior to activation, the formation of such a complex would explain how two potential pitfalls could be avoided. First, there must be a mechanism that protects the glycyl radical species on YfiD from being quenched before YfiD is positioned into the protective PFL active site. Formation of a ternary complex could provide this protection. Second, it would not be in the best interest of the cell to generate a glycyl radical species on YfiD if there were no damaged PFL to repair. Requiring that complex formation with a damaged PFL be the first step in the YfiD activation process ensures that YfiD is not activated when it isn’t needed. In this sense, YfiD’s y-β1 can be thought of as a safety lever, preventing radical formation by PFL-AE until a damaged PFL ‘pulls’ the lever down (Fig. 5). With these ideas in mind, we have revised our proposed mechanism of PFL rescue by YfiD (Fig. 8, Electronic Supplementary Material Video 1). In particular, we now show both the unstructured region and the y-β1 strand of YfiD bound to t1PFL with the unstructured region affording general binding affinity and the y-βl strand replacing the lost β10, completing the 10-standed barrel fold. Collectively, these parts of YfiD play a functional role in ‘sensing’ PFL damage and in tethering YfiD to t1PFL prior to YfiD activation by PFL-AE (Fig. 8, steps 3–5). This hypothesized tethering would serve to stabilize both the YfiD:PFL-AE and the YfiD:PFL interactions, ensuring that YfiD is activated only when needed, and protect the newly formed glycyl radical species from exposure to solvent as it transitions from its binding site on PFL-AE to its binding location inside of the PFL barrel (Fig. 8, steps 5–6).
The structural and biochemical analysis described herein is performed on YfiD from E. coli; however, it is reasonable to conclude that other microbes may use a similar repair mechanism involving spare part proteins for their corresponding oxygen-damaged PFL enzymes. Beyond the crucial role of PFL enzymes in microbial metabolism, enzymes within the larger glycyl radical enzyme (GRE) superfamily function in both primary and secondary metabolism, catalyzing a wide range of reactions. Other GREs include benzylsuccinate synthase (BSS), which catalyzes the radical addition of toluene to fumarate [30–32], glycerol/propanediol dehydratase (GDH), which catalyzes glycerol reduction to 3-hydroxypropanaldehyde [33], and class III ribonucleotide reductase (RNR), which catalyzes the formation of deoxyribonucleotides from ribonucleotides [5,34]. Though the reactions performed by these enzymes vary greatly, all use a common glycyl radical cofactor and are prone to the same type of oxidative damage. It is therefore tempting to wonder if similar spare part proteins have evolved to repair any of these other numerous enzymes.
Materials and Methods
Plasmid Construction
We received YfiD in a p-CAL-n-EK vector as a gift from the Broderick lab. To prepare the truncated YfiD (truncYfiD) construct used in the circular dichroism experiments, the Q5® Site-Directed Mutagenesis Kit (New England Biolabs) protocol was used to delete the first 180 base pairs from the YfiD gene (corresponding to a deletion of 60 amino acids at the N-terminus), in the p-CAL-n-EK vector. The following primers were used for this deletion: forward primer, 5’- GTG AAA CCA GAA GTT CGC −3’, and reverse primer, 5’- CAT ATG TAT ATC TCC TTC TTA AAG −3’. After the deletion was performed, a hexahistidine-TEV cleavage site was inserted at the 5’ end of truncYfiD, again using the NEB Q5® Site-Directed Mutagenesis Kit. All primers were designed using NEBaseChanger™. All mutagenesis experiments were confirmed through Sanger sequencing by GENEWIZ, Inc.
To prepare the cPFL:YfiD co-expression plasmid used in the SDS-PAGE experiment, the YfiD gene was inserted into the second MCS of pCOLA-Duet using Ndel and Xhol cut sites, and a hexahistadine-TEVcleavage site was added at the 5’ end of this gene (Genscript). Next, a synthesized 733-PFL (mimicking cleaved PFL, or cPFL) insert (Genscript) was mutated to add a Pcil cut site at the 5’ end and a Sall cut site at the 3’ end and inserted into the first MCS of pCOLA-duet at the Ncol and Sall cut sites (Ncol and Pcil have compatible sticky ends).
Protein Expression and Purification
YfiD, truncYfiD, and cPFL:YfiD plasmids were all transformed into T7 Express cells (New England Biolabs) for expression. The following expression protocol was used for all three constructs: Samples were prepared in growth media supplemented with 100 μg/mL ampicillin (Affymetrix). Unlabeled YfiD, truncYfiD, and cPFL:YfiD were expressed in Luria-Bertini broth. Starter cultures were inoculated with single colonies and incubated at 37 °C overnight at 220 RPM. 10 mL of starter culture was used to inoculate 1 L of media, which was incubated at 37 °C at 220 RPM until the optical density at 600 nm was 0.6. Expression was induced by addition of 1 mM IPTG (RPI Corp.), followed by an additional 3 hours of incubation at 37 °C at 220 RPM. Cells were spun down, and cell paste was frozen and stored at −80 °C prior to purification.
Uniformly 15N labeled YfiD (U-15N-YfiD) and uniformly 13C15N labeled YfiD (U-13C15N-YfiD) for NMR experiments were expressed in M9 minimal media supplemented with 10 mM magnesium sulfate (EMD-Millipore) and 0.03 g/L thiamine hydrochloride (Sigma-Aldrich). I g/L 15N-ammonium chloride (Cambridge Isotopes Laboratories) and 25 mL of 40% glucose were added to provide 15N and C for U-15N-YfiD. 1 g/L 15N-ammonium chloride (Cambridge Isotopes Laboratories) and 3 g/L13C-D-glucose (Cambridge Isotopes Laboratories) were added to provide 15N and 13C for U-13C15N-YfiD. Note, the deuterium from the 13C-D-glucose exchanges rapidly in the non-deuterated media. Aside from growth media, the expression protocol was the same as described above for unlabeled YfiD.
To purify U-15N-YfiD and U-13C15N-YfiD, cell paste was thawed and lysed at 4 °C while nutating for 1 hour with 10 mL lysis buffer for each 5 g of cell paste (lysis buffer: 20mM HEPES (Fisher Bioreagents) pH 7.2, 10 mM magnesium chloride (EMD-Millipore), 5% W/V glycerol (VWR Analytical), 1% V/V Triton X-100 (Sigma-Aldrich), 1 mini cOmplete ULTRA, mini, EDTA-free tablet (Roche) and luL Benzonase Nuclease (EMD-Millipore)). The resulting suspension was sonicated with 1/4” tip at 60% power for 4×2 minute cycles of 2 sec on and 2 sec off. Lysate was clarified at 18,000 RPM, and the protein was eluted from a DEAE column (GE Healthcare) in a linear gradient from 0 mM to 500 mM sodium chloride (Macron Fine Chemicals), 20mM HEPES, pH 7.2. Fractions containing YfiD were buffer exchanged into 1 M ammonium sulfate (Fisher Bioreagents), 20 mM HEPES, pH 7.2 and eluted from a high sub-phenyl sepharose HIC column (GE Healthcare) in a linear gradient from 1 M to 0 M ammonium sulfate, 20 mM HEPES, pH 7.2. YfiD elutes in nearly all fractions, but only those fractions at the end of the gradient and in the final wash were pooled and used, as they are the only fractions free of contaminating proteins. Protein was concentrated in a 10 kDa Amicon spin concentrator (EMD-Millipore).
To purify truncYfiD and unlabeled YfiD, cell paste was thawed and lysed following the same protocol as described for U-15N-YfiD and U-13C15N-YfiD. Protein was eluted from a complete His-Tag purification column (Roche) in a linear gradient from 0 mM to 500 mM imidazole, 20 mM HEPES, pH 7.2. Fractions containing truncYfiD were buffer exchanged into imidazole-free 20 mM HEPES, pH 7.2. truncYfiD was treated with hexahistidine-tagged TEV protease at a 1:100 (protease to truncYfiD) concentration ratio and incubated overnight at 4 °C. TEV protease was separated from untagged truncYfiD by collecting the flow-through and low imidazole fractions from a cOmplete His-Tag purification column (Roche). Protein was concentrated in a 10 kDa Amicon spin concentrator (EMD-Millipore).
cPFL:YfiD cell paste was thawed and lysed following the same protocol as described for U-15N-YfiD and U-13C15 N-YfiD. Lysate was clarified at 18,000 RPM. The cPFL:YfiD complex, with a hexahistidine-tag on the N-terminus of YfiD, was purified from a His SpinTrap column (GE Healthcare Life Sciences) by increasing the concentration of imidazole from 0 to 500 mM imidazole, with 20 mM NaCI, 20 mM HEPES, pH 7.2. Fractions of cPFL:YfiD eluted at 100 mM imidazole were pooled.
Protein concentration was determined by absorbance at 280 nm using the molar extinction coefficients of 11460 M−1cm−1 for YfiD, 5960 M−1cm−1 for tYfiD, and 49565 M−1cm1 for cPFL:YfiD.
SDS-PAGE
An SDS-PAGE gel was run in order to distinguish the different cleavage products of cPFL. SDS-PAGE was analyzed using a 7.5% Mini-PROTEAN® TGX™ precast gel (Biorad). Unstained Protein Standard, Broad Range (10–200 kDa, New England Biolabs) was used as the molecular weight standard. Although cPFL was purified with YfiD, YfiD had to be run off the gel in order to observe sufficient band separation of the cPFL cleavage products.
Circular Dichroism
Similar far-UV CD spectra were recorded on an Aviv Model 202 CD Spectrometer for experiments found in Fig. 4 and a JASCO Model J-1500 Circular Dichroism Spectrometer for experiments shown in Fig. S1. Both spectrometers are equipped with a jaacketed cell holder connected to a circulating water bath, and 0.1-cm path length cuvettes were used (Starna). Protein samples were prepared of 80 μM YfiD or 80 μM truncYfiD in 10 mM HEPES, pH 7.2 and 10 mM MES, pH 6.5 (Sigma-Aldrich). The thermal denaturation experiments recorded the CD signal at 220 nm at 5 °C intervals with a 30 sec equilibration time at each temperature range, over the temperature range 20–90 °C. Thermal denaturation curves were obtained by plotting the CD signal at 220 nm as a function of temperature and the melting temperature was determined by fitting the curves to the following two-state model equation:
where y is the measured ellipticity, R = 8.3145 J K−1 mol−1, yf and yu are the intercepts of the pre- and post-transition baselines, mf and mu are the slopes of the pre- and post-transition baselines, T is the temperature (K), ΔHm is the enthalpy at the unfolding transition and Tm is the melting temperature (K). The equation is fit in R Statistical Software [35] using nonlinear least squares to fit all parameters.
NMR Spectroscopy
NMR spectra of 0.7 mM U-15N-YfiD (18 mM HEPES pH 7.2, 2.7 mM (NH4)2S04 10% V/V D20) were collected on CMR 600 E, a 600 MHz NMR spectrometer at Massachusetts Institute of Technology’s Francis Bitter Magnet Lab. NMR spectra of 1.3 mM U-13C15N-YfiD (18 mM HEPES pH 7.2, 2.7 mM (NH4)2S04 10% V/V D20) were collected on Varian Inova 800 MHz spectrometer, equipped with a cryogenic triple-resonance probe, at University of Connecticut Health Center NMR facility. Chemical shifts were referenced to water. All NMR data was processed using NMRPipe [36] and analyzed using NMRFAM-SPARKY [37]. Standard triple-resonance experiments were used for backbone and sidechain assignments, including 2D 1H-15N HSQC and HSQC, and 3D HNCO, HNCA, HNCACB, CBCACONH, CCHTOCSY, HBHACONH, and 15N resolved 1H-1H TOCSY. Distance restraints based on nuclear Overhauser effect (NOE) were obtained from 1H-15N-NOESY-HSQC and 1H-13C-NOESY-HSQC. HSQC spectra were obtained between each experiment to assess quality of the protein. Peak picking and NOE assignments were performed using NMRFAM-SPARKY [37], CCPN [38], and CYANA [39]. Structure calculations were performed using NOE distances and dihedral angle constraints, and refined with molecular dynamics. The backbone RMSD from the medoid lowest energy structure is 0.07 A for the well-defined core residues of the protein, indicating convergence of the NMR structure solution. NMR assignments and details have been deposited to the Biological Magnetic Resonance Bank (ID 30611) and the 10 lowest energy conformers have been deposited to the PDB (ID 60WR).
Supplementary Material
Acknowledgements:
This work was supported in part by the National Institutes of Health (NIH) GM069857 (C.L.D.), R35 GM126982 (C.L.D.), F32 GM129882 (M.C.A.), and F32 GM099257 (S.E.J.B.), Analog Devices (C.M.S.), MIT-IBM Watson Lab (C.M.S.), MIT J-Clinic (C.M.S.), and the National Science Foundation (NSF) Graduate Research Fellowship under Grant No. 1122374 (L.R.F.B.). C.L.D is a Howard Hughes Medical Institute (HHMI) Investigator. S.Y. and L.R.F.B. were part of the HHMI EXROP program and the Bio-MIT Summer Research Program (MSRP) program. A.C. was also part of the MSRP program. L.R.F.B. is a recipient of a Dow Fellowship at MIT and a Gilliam Fellowship from HHMI. The Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (NSF-0070319) is gratefully acknowledged. We would like to thank University of Connecticut Health Center NMR facility and Massachusetts Institute of Technology’s Francis Bitter Magnet Lab for instrument use. We acknowledge Miranda Lynch for helping with R scripting, and Laurel Kinman for helping with editing.
Abbreviations
- 5’-dAdo
5’-deoxyadenosyl
- AdoCbl
adenosylcobalamin
- AdoMet
S-adenosylmethionine
- BSS
benzylsuccinate synthase
- CD
circular dichroism
- cPFL
cleaved pyruvate formate lyase
- GDH
glycerol/propanediol dehydratase
- GrcA
autonomous glycyl radical cofactor
- GRD
glycyl radical domain
- GRE
glycyl radical enzyme
- HSQC
heteronuclear single quantum coherence
- IMAC
immobilized metal affinity chromatography
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- PFL
pyruvate formate lyase
- PFL-AE
pyruvate formate lyase activating enzyme
- RNR
ribonucleotide reductase
- t1PFL
PFL truncation product 1 at 77 kDa
- t2PFL
PFL truncation product 2 at 71 kDa
- truncYfiD
truncated YfiD with 60 N-terminal residues removed
References
- 1.Backman LRF, Funk MA, Dawson CD, Drennan CL. (2017) New tricks for the glycyl radical enzyme family. Crit Rev Biochem Mol Biol 52, 674–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Broderick JB, Duffus BR, Duschene KS, Shepard EM. (2014) Radical S-adenosylmethionine enzymes. Chem Rev 114, 4229–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee R (2003) Radical carbon skeleton rearrangements: catalysis by coenzyme B12-dependent mutases. Chem Rev 103, 2083–2094 [DOI] [PubMed] [Google Scholar]
- 4.Hausinger RP. (2004) Fell/alpha-ketoglutarate-dependent hydroxylases and related enzymes.Crit Rev Biochem Mol Biol 39, 21–68 [DOI] [PubMed] [Google Scholar]
- 5.Stubbe J, van Der Donk WA. (1998) Protein Radicals in Enzyme Catalysis. Chem Rev 98,705–762 [DOI] [PubMed] [Google Scholar]
- 6.Frey PA. (1990) Importance of Organic Radicals in Enzymatic Cleavage of Unactivated C-H Bonds. Chem Rev 90,1343–1357 [Google Scholar]
- 7.Warren MJ, Raux E, Schubert HL, Escalante-Semerena JC. (2002) The biosynthesis of adenosylcobalamin (vitamin B12). Nat Prod Rep 19, 390–412 [DOI] [PubMed] [Google Scholar]
- 8.Scott AI, Roessner CA. (2002) Biosynthesis of cobalamin (vitamin B(12)). Biochem Soc Trans 30, 613–620 [DOI] [PubMed] [Google Scholar]
- 9.Conradt H, Hohmann-Berger M, Hohmann HP, Blaschkowski HP, Knappe J (1984) Pyruvate formate-lyase (inactive form) and pyruvate formate-lyase activating enzyme of Escherichia coli: isolation and structural properties. Arch Biochem Biophys 228,133–142 [DOI] [PubMed] [Google Scholar]
- 10.Henshaw TF, Cheek J, Broderick JB. (2000) The [4Fe-4S]1+cluster of pyruvate formate-lyase activating enzyme generates the glycyl radical on pyruvate formate-lyase: EPR-detected single turnover. J Am Chem Soc 122, 8331–8332 [Google Scholar]
- 11.Frey PA. (1993) Lysine 2,3-aminomutase: is adenosylmethionine a poor man’s adenosylcobalamin? FASEBJ 7, 662–670 [DOI] [PubMed] [Google Scholar]
- 12.Bridwell-Rabb J, Grell TAJ, Drennan CL. (2018) A Rich Man, Poor Man Story of S-Adenosylmethionine and Cobalamin Revisited. Annu Rev Biochem 87, 555–584 [DOI] [PubMed] [Google Scholar]
- 13.Frey PA, Ballinger MD, Reed GH. (1998) S-adenosylmethionine: a ‘poor man’s coenzyme B12’ in the reaction of lysine 2,3-aminomutase. Biochem Soc Trans 26, 304–310 [DOI] [PubMed] [Google Scholar]
- 14.Knappe J, Blaschkowski HP, Grobner P, Schmittm. (1974) Pyruvate formate-lyase of Escherichia coli: the acetyl-enzyme intermediate. Eur J Biochem 50, 253–263 [DOI] [PubMed] [Google Scholar]
- 15.Knappe J, Wagner AF. (1995) Glycyl free radical in pyruvate formate-lyase: synthesis, structure characteristics, and involvement in catalysis. Methods Enzymol 258, 343–362 [DOI] [PubMed] [Google Scholar]
- 16.Knappe J, Sawers G (1990) A radical-chemical route to acetyl-CoA: the anaerobically induced pyruvate formate lyase system of Escherichia coli. FEMS Microbiol Rev 6, 383–398 [DOI] [PubMed] [Google Scholar]
- 17.Sawers G, Suppmann B (1992) Anaerobic induction of pyruvate formate-lyase gene expression is mediated by the ArcA and FNR proteins. J Bacteriol 174, 3474–3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Partridge JD, Sanguinetti G, Dibden DP, Roberts RE, Poole RK, Green J (2007) Transition of Escherichia coli from aerobic to micro-aerobic conditions involves fast and slow reacting regulatory components. J Biol Chem 282,11230–11237 [DOI] [PubMed] [Google Scholar]
- 19.Han MJ, Yoon SS, Lee SY. (2001) Proteome analysis of metabolically engineered Escherichia coli producing Poly(3-hydroxybutyrate). J Bacteriol 183, 301–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wagner AF, Schultz S, Bomke J, Pils T, Lehmann WD, Knappe J (2001) YfiD of Escherichia coli and Y06I of bacteriophage T4 as autonomous glycyl radical cofactors reconstituting the catalytic center of oxygen-fragmented pyruvate formate-lyase. Biochem Biophys Res Commun 285,456–462 [DOI] [PubMed] [Google Scholar]
- 21.Marshall FA, Messenger SL, Wyborn NR, Guest JR, Wing H, Busby SJ, Green J (2001) A novel promoter architecture for microaerobic activation by the anaerobic transcription factor FNR. Mol Microbiol 39, 747–753 [DOI] [PubMed] [Google Scholar]
- 22.Wyborn NR, Messenger SL, Henderson RA, Sawers G, Roberts RE, Attwood MM, Green J(2002) Expression of the Escherichia coli yfiD gene responds to intracellular pH and reduces the accumulation of acidic metabolic end products. Microbiology 148,1015–1026 [DOI] [PubMed] [Google Scholar]
- 23.Kumar R, Shimizu K (2011) Transcriptional regulation of main metabolic pathways of cyoA, cydB, fnr, and fur gene knockout Escherichia coli in C-limited and N-limited aerobic continuous cultures. Microb Cell Fact 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green J, Baldwin ML. (1997) HlyX, the FNR homologue of Actinobacillus pleuropneumoniae, is a [4Fe-4S]-containing oxygen-responsive transcription regulator that anaerobically activates FNR-dependent class I promoters via an enhanced AR1 contact. Mol Microbiol 24, 593–605 [DOI] [PubMed] [Google Scholar]
- 25.Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL. (2008) Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc Natl Acad Sci U S A 105, 16137–16141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng Y, Veneziano SE, Gillispie GD, Broderick JB. (2010) Pyruvate formate-lyase, evidence for an open conformation favored in the presence of its activating enzyme. J Biol Chem 285, 27224–27231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z (2001) Intrinsically disordered protein. J Mol Graph Model 19, 26–59 [DOI] [PubMed] [Google Scholar]
- 28.Kelly SM, Jess TJ, Price NC. (2005) How to study proteins by circular dichroism. Biochim Biophys Acta 1751,119–139 [DOI] [PubMed] [Google Scholar]
- 29.Easton CJ, Hay MP. (1986) Preferential reactivity of glycine residues in free radical reactions of amino acid derivatives. J Chem Soc Chem Commun 1, 55–57 [Google Scholar]
- 30.Funk MA, Judd ET, Marsh EN, Elliott SJ, Drennan CL. (2014) Structures of benzylsuccinate synthase elucidate roles of accessory subunits in glycyl radical enzyme activation and activity. Proc Natl Acad Sci U S A 111, 10161–10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Patterson DP, Fox CC, Lin B, Coschigano PW, Marsh EN. (2009) Subunit structure of benzylsuccinate synthase. Biochemistry 48,1284–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coschigano PW, Bishop BJ. (2004) Role of benzylsuccinate in the induction of the tutE tutFDGH gene complex of T. aromatica strain T1. FEMS Microbiol Lett 231, 261–266 [DOI] [PubMed] [Google Scholar]
- 33.O’Brien JR, Raynaud C, Croux C, Girbal L, Soucaille P, Lanzilotta WN. (2004) Insight into the mechanism of the B12-independent glycerol dehydratase from Clostridium butyricum: preliminary biochemical and structural characterization. Biochemistry 43,4635–4645 [DOI] [PubMed] [Google Scholar]
- 34.Sun X, Ollagnier S, Schmidt PP, Atta M, Mulliez E, Lepape L, Eliasson R, Graslund A, Fontecave M, Reichard P, Sjoberg BM. (1996) The free radical of the anaerobic ribonucleotide reductase from Escherichia coli is at glycine 681. J Biol Chem 271, 6827–6831 [PubMed] [Google Scholar]
- 35.Team, RC. (2017) R: A language and environment for statistical computing.
- 36.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomal NMR 6, 227293. [DOI] [PubMed] [Google Scholar]
- 37.Lee W, Tonelli M, Markley JL. (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31,1325–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, lonides J, Laue ED. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–6 [DOI] [PubMed] [Google Scholar]
- 39.Guntert P (2009) Automated structure determination from NMR spectra. Eur biophys J 38, 129–143 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.