

Abstract
Purpose:
Manifestations of infection and the degree of influenza virus vary. We hypothesized that the nose/throat microbiota modifies the duration of influenza symptoms and viral shedding. Exploring these relationships may help identify additional methods for reducing influenza severity and transmission.
Methods:
Using a household transmission study in Nicaragua, we identified secondary cases of influenza virus infection, defined as contacts with detectable virus or a >4-fold change in hemagglutinin inhibition antibody titer. We characterized the nose/throat microbiota of secondary cases prior to infection and explored whether the duration of symptoms and shedding differed by bacterial community characteristics.
Results:
Among 124 secondary cases of influenza, higher bacterial community diversity prior to infection was associated with longer shedding duration (Shannon acceleration factor (AF): 1.61, 95% confidence interval (CI): 1.24, 2.10) and earlier time to infection (Shannon AF: 0.72, 95% CI: 0.53, 0.97; Chao1 AF: 0.992, 95% CI: 0.986, 0.998). Neisseria and multiple other oligotypes were significantly associated with symptom and shedding durations and time to infection.
Conclusions:
The nose/throat microbiota prior to influenza virus infection was associated with influenza symptoms and shedding durations. Further studies are needed to determine if the nose/throat microbiota is a viable target for reducing influenza symptoms and transmission.
Keywords: influenza, microbiota, signs and symptoms, virus shedding
Introduction
Clinical presentation of influenza virus infection can range from mild to severe [1]. Among the estimated 90 million new cases of influenza that occurred in young children in 2008, 20 million had acute lower respiratory infections, 1 million had severe acute respiratory lower infections, and 28,000–111,500 cases resulted in death [2]. Infectiousness, estimated by viral shedding, is not highly correlated with symptoms: viral shedding can be detected in asymptomatic cases but is often undetected in symptomatic cases [3–6]. A meta-analysis of challenge studies estimated that young adults shed for an average duration of 5 days after inoculation [3]. However, longer durations of shedding have been observed in more symptomatic cases [7] and in young children [4,8].
This heterogeneity in influenza illness and infectiousness has been largely attributed to the host immune response, which impacts pathogenicity and viral replication [9]. As the role of microbiota in stimulating host immunity has become evident [10–13], there have been increasing numbers of studies demonstrating associations between the microbiome and risk and severity of infectious diseases [14–16]. However, to our knowledge, no epidemiologic study has examined whether the microbiota is associated with symptoms or viral shedding during influenza virus infection. Identifying these links would lay groundwork for developing symbiotic approaches to reduce influenza severity and transmission. This study fills this gap using data from a household transmission study in Nicaragua.
Materials and methods
Study population
This analysis uses data and samples collected by the Nicaraguan Household Transmission Study conducted in Managua, Nicaragua, between 2012–2014. Household index cases of influenza virus infection were identified at a primary healthcare center using the following criteria: 1) a positive QuickVue Influenza A + B rapid diagnostic test, 2) symptom onset of febrile acute respiratory illness (fever or feverishness with a rhinorrhea, sore throat, and/or cough) within the past 48 hours, 3) residing in a household with at least one other member (household contact), and 4) no household contacts with influenza symptoms in the two weeks prior to symptom onset in the index case.
Index cases and household contacts were invited to participate and monitored through up to 5 home visits, conducted at 2–3 day intervals. Nasal and oropharyngeal swabs were collected and combined at each visit. Blood samples were collected at enrollment and 30–45 days later. A secondary case was defined as a household contact with a positive real-time reverse transcription polymerase chain reaction (RT-PCR) result or a ≥4-fold change in hemagglutination inhibition (HAI) antibody titers specific to the subtype/type identified in the index case.
A written informed consent or proxy consent was obtained for all participants. Verbal assent was obtained from children ≥5 years. The study was approved by the Institutional Review Boards at the University of Michigan and the Nicaraguan Ministry of Health.
Laboratory assays
Influenza type/subtype-specific RT-PCR was conducted on all samples using validated Centers for Disease Control and Prevention protocols [17]. Influenza type/subtype-specific HAI titers were measured using validated World Health Organization protocols [18].
Microbiota characterization
Detailed methods used for microbiota characterization are discussed in Lee et al. [19]. Briefly, DNA was extracted from the first and last nasal/oropharyngeal sample collected from all index cases and household contacts. The V4 hypervariable region of the 16S rRNA gene was amplified and sequenced on an Illumina MiSeq System using a validated dual-indexing method [20]. Following alignment and quality filtering in mothur v1.38.1 [21] and oligotyping to assign reads to taxonomic units [22], Dirichlet multinomial mixture models [23] were used to assign all nasal/oropharyngeal samples to 5 bacterial community types (Supplemental Figure 1). Each community type represents a group of samples with similar taxa compositions. We determined the number of community types by estimating the Laplace approximation of the negative log models and identifying the point at which an increase in Dirichlet components resulted in minor reductions in model fit (Supplemental Figure 2). Considerations were placed on statistical power in downstream analyses. Taxonomy was assigned using the Human Oral Microbiome Database v14.51 [24] and blastn v2.2.23 [25].
Comparisons between community types were conducted using all available microbiota data from all index cases and household contacts (n=1,405 samples). β-diversity, representing within-group dissimilarity of samples, was estimated using Bray-Curtis dissimilarity and Jaccard distance. α-diversity, representing within-sample community diversity, was estimated using Shannon diversity index and Chao1 index. Shannon diversity accounts for both richness and evenness of taxa while Chao1 only accounts for richness.
Influenza shedding and symptom data
Household contacts with ≥1 positive RT-PCR result during follow-up were defined as secondary cases with viral shedding. Shedding duration was estimated as the time between the first positive RT-PCR result and a negative RT-PCR result.
Study participants completed a daily symptom diary documenting the presence of the following symptoms: fever or feverishness, rhinorrhea, sore throat, and cough. To reduce potential bias from symptoms unrelated to influenza virus infection, we defined an influenza-associated illness period for each participant using symptom onset and alleviation dates. Illness onset was defined as the earliest date of any symptom. However, symptoms were excluded if they were alleviated >1 day prior to onset of viral shedding. Illness alleviation was defined as the date on which all symptoms were alleviated. Any recurring symptoms were excluded if the symptom recurred ≥3 days after viral shedding cessation or if fever recurred ≥3 days after fever alleviation. The duration of each symptom was estimated within the defined illness period. Febrile acute respiratory illness was defined as the presence of fever plus rhinorrhea, sore throat, and/or cough and influenza-like illness was defined as fever plus sore throat and/or cough.
Statistical analysis
Accelerated failure time models using a generalized estimating equation approach were used to examine the relationship between bacterial community diversity and symptom duration, viral shedding duration, the serial interval, and time to shedding onset. Time to shedding onset was relative to symptom onset dates of index cases. Survival time was parameterized as a Weibull distribution in all models [26].
Models were repeated using community types. We further explored whether outcomes were associated with the relative abundance of the 15 oligotypes that contributed to >50% of the difference between community types. We ran single-oligotype models using log10-transformed relative abundance in consideration of the constant sum constraint [27] and the Benjamin-Hochberg method to correct for multiple testing.
We adjusted for age and sex in models estimating viral shedding and symptom. We adjusted for age, a smoker in the household, sex, and household crowding in models estimating time to shedding onset and estimating the serial interval. All models were adjusted for clustering by household. A summary of our models is available in Supplemental Table 1. All statistical analyses were conducted using R version 3.4.2 [28].
Availability of data and materials
Raw sequence reads have been deposited in a NCBI Sequence Read Archive repository (accession number PRJNA482032). Datasets generated and analyzed during the current study are available in an open-access Deep Blue Data repository (https://deepblue.lib.umich.edu/data/concern/generic_works/sb3979224?locale=en). Certain individual participant data have been excluded due to identifiability concerns.
Results
Study population
A total of 144 index cases and 573 household contacts were enrolled in the Nicaraguan Household Transmission Study during 2012–2014. Following sequencing of the V4 region of the 16SrRNA of the first and last available nose/throat samples from all study participants, including both index cases and household contacts, and assignment to oligotypes, we used an unsupervised clustering technique to identify 5 bacterial community types (n=1,405 samples) (Supplemental Figures 1 & 2). Community types varied significantly in composition and structure as tested using PERMANOVA (beta diversity: Bray-Curtis dissimilarity, R2=0.207, p=0.001) and differed in alpha diversity ((Shannon: Wilcoxon rank-sum, p<0.001) (Figure 1); (Chao1: Wilcoxon rank-sum, p<0.001) (Supplemental Figure 3)). Most notably, community type 5 had the lowest alpha diversity. Clustering of samples into community types was largely explained by a few oligotypes, with 50% of the difference between the single community type and five-community type models attributed to 15 out of the total 230 oligotypes. The relative abundance of these oligotypes are depicted in Figure 2. The complete taxa composition is available in Supplemental Figure 4.
Figure 1.
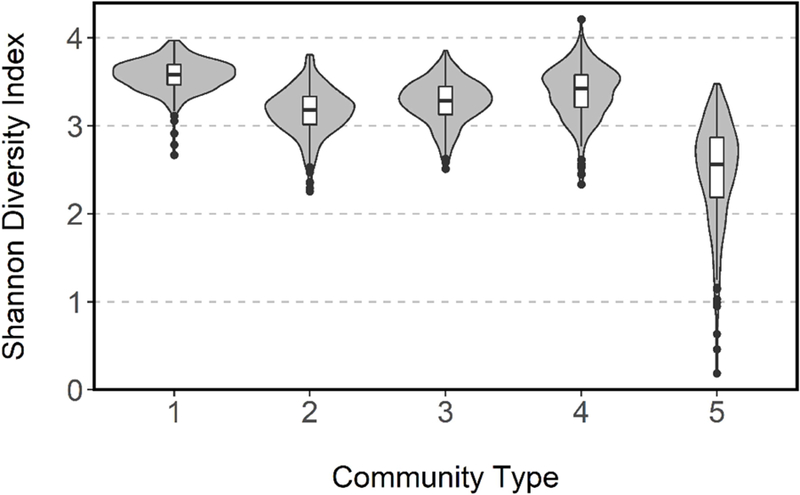
Shannon diversity of bacterial community types based on first and last nose/throat samples of 144 index cases and 573 household contacts from 144 households, Managua, Nicaragua, 2012–2014. Each violin plot contains a box plot with a kernel density estimation on either side depicting the distribution of data.
Figure 2.

Relative abundance of 15 oligotypes that contributed to 50% of difference between community types. Each square represents 0.05% relative abundance.
One hundred sixty secondary influenza virus infections were identified over a ≤13-day follow-up period using RT-PCR or a ≥4-fold increase in HAI titer specific to the influenza type/subtype of the household index case 30–45 days after enrollment. Thirty-two were positive only by RT-PCR, 53 were positive only by HAI titer, and 62 were positive by both methods. Thirty-six household contacts with a positive RT-PCR result at the first home visit were excluded as nose/throat samples were not available prior to infection. Analysis was conducted on the remaining 124 secondary cases: 71 were positive for influenza by RT-PCR (57%) and 92 (74%) were positive by HAI during follow-up. Half of all secondary cases were adults (48%) and most infections were symptomatic (61%). Thirty-six secondary cases experienced febrile acute respiratory illness (29%), including 34 with influenza-like illness (27%) (Table 1). Forty-one percent of households had more than 1 secondary case, suggesting clustering of secondary cases by household. Compared to persons with secondary infections without viral shedding (n=53), persons with secondary infections with viral shedding (n=71) were younger (mean: 16.7 years vs. 25.2 years, t-test, p=0.001), more likely to be symptomatic (75% vs. 43% with ≥1 symptom, χ2 test, p<0.001), and more likely to have febrile acute respiratory illness (42% vs. 6%, χ2 test, p<0.001).
Table 1.
Characteristics of 124 secondary influenza cases from 70 households, Managua, Nicaragua, 2012–2014, by bacterial community type. Secondary cases were defined as household contacts of index cases with a positive RT-PCR result for influenza or ≥4-fold change in HAI titer during follow-up.
| Characteristics | All (n=124a) |
Community Type 1 (n=35) |
Community Type 2 (n=31) |
Community Type 3 (n=30) |
Community Type 4 (n=14) |
Community Type 5 (n=7) |
|---|---|---|---|---|---|---|
| No. (%) | No. (%) | No. (%) | No. (%) | No. (%) | No. (%) | |
| Influenza type/subtype (RT-PCR) | ||||||
| H1N1 | 12 (10) | 2 (6) | 3 (10) | 2 (7) | 4 (29) | 0 (0) |
| H3N2 | 37 (30) | 12 (34) | 9 (29) | 9 (30) | 0 (0) | 5 (71) |
| B | 21 (17) | 6 (17) | 7 (23) | 4 (13) | 1 (7) | 1 (14) |
| Co-infection | 1 (1) | 0 (0) | 1 (3) | 0 (0) | 0 (0) | 0 (0) |
| None | 53 (43) | 15 (43) | 11 (35) | 15 (50) | 9 (64) | 1 (14) |
| Influenza type/subtype (HAI) | ||||||
| H1N1 | 18 (15) | 4 (11) | 5 (16) | 4 (13) | 4 (29) | 0 (0) |
| H3N2 | 48 (39) | 13 (43) | 8 (26) | 15 (50) | 6 (43) | 1 (0) |
| B | 26 (21) | 6 (17) | 8 (26) | 7 (23) | 3 (21) | 1 (14) |
| Co-infection | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| None | 23 (19) | 7 (20) | 7 (23) | 4 (13) | 1 (7) | 2 (29) |
| Missing | 9 (7) | 3 (9) | 3 (10) | 0 (0) | 0 (0) | 3 (43) |
| Age (years) | ||||||
| 0–5 | 19 (15) | 5 (14) | 6 (19) | 1 (3) | 0 (0) | 5 (71) |
| 6–17 | 45 (36) | 16 (46) | 10 (32) | 13 (43) | 4 (29) | 1 (14) |
| ≥18 | 60 (48) | 14 (40) | 15 (48) | 16 (53) | 10 (71) | 1 (14) |
| Female | 80 (65) | 20 (57) | 23 (74) | 19 (63) | 10 (71) | 4 (57) |
| Influenza vaccinationb | 6 (5) | 1 (3) | 3 (10) | 2 (7) | 0 (0) | 0 (0) |
| Smoker in household | 59 (54) | 16 (52) | 14 (52) | 17 (61) | 6 (55) | 4 (57) |
| Oseltamivir use | 10 (8) | 2 (6) | 4 (13) | 2 (7) | 0 (0) | 1 (14) |
| Symptoms | ||||||
| Fever/feverishness | 44 (35) | 11 (31) | 12 (39) | 8 (27) | 7 (50) | 3 (43) |
| Rhinorrhea | 53 (43) | 14 (40) | 16 (52) | 10 (33) | 7 (50) | 3 (43) |
| Sore throat | 35 (28) | 8 (23) | 12 (39) | 6 (20) | 5 (36) | 2 (29) |
| Cough | 60 (48) | 14 (40) | 18 (58) | 14 (47) | 9 (64) | 3 (43) |
| FARIc | 36 (29) | 7 (20) | 11 (35) | 6 (20) | 6 (43) | 3 (43) |
| ILId | 34 (27) | 7 (20) | 11 (35) | 6 (20) | 6 (43) | 2 (29) |
Includes secondary cases with undefined community types
Prior to enrollment and in same year as index case
Fever/feverishness with rhinorrhea, sore throat or cough
Fever/feverishness with sore throat or cough
Bacterial community diversity prior to infection and symptom and shedding durations
We explored whether α-diversity prior to influenza virus infection was associated with symptom and shedding durations. We found no statistically significant associations between α-diversity and symptom durations (Supplemental Table 2). Shannon diversity was positively associated with shedding duration (AF: 1.61; 95% CI: 1.24, 2.10) (Figure 3; Supplementary Table 3). The mean predicted durations at the 25th and 75th quartiles of Shannon diversity (distribution among secondary cases) were 3.1 and 3.6 days, respectively.
Figure 3.
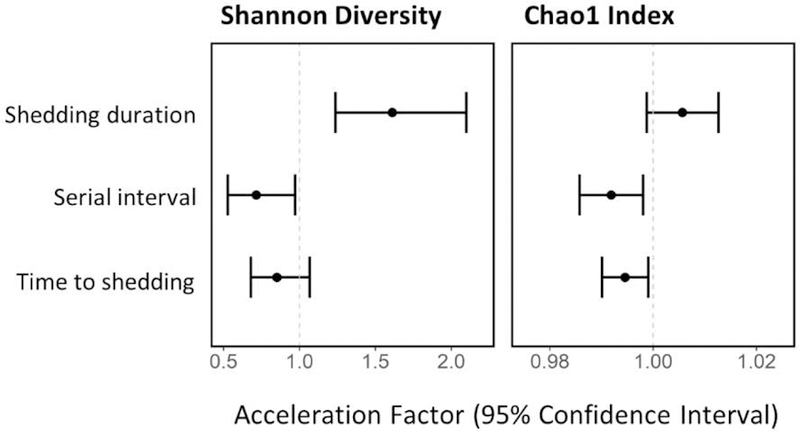
Accelerated failure time models examining relationship between α-diversity and shedding duration, serial interval and time to shedding onset among 124 secondary cases from 70 households, Managua, Nicaragua, 2012–2014. Models are not specific to influenza type/subtype.
Bacterial community diversity prior to infection and time to infection
We examined whether α-diversity was associated with time to infection using two different proxy measures, serial interval (defined as the time between onset of symptoms between an index case and a secondary case) and time to shedding onset, after adjusting for age, sex, a smoker in the household, household crowding, and clustering by household. The serial interval was negatively associated with Shannon diversity (AF: 0.72; 95% CI: 0.53, 0.97) and Chao1 (AF: 0.992; 95% CI: 0.986, 0.998). The mean serial interval was 3.7 and 3.2 days at the 25th and 75th quartiles of Shannon diversity, respectively, and 3.8 and 3.0 days at the 25th and 75th quartiles of Chao1, respectively. Chao1 was associated with earlier time to shedding onset (AF: 0.995; 95% CI: 0.990, 0.999). Mean serial interval was 5.8 and 5.3 days at the 25th and 75th quartiles of Chao1 index, respectively.
Exploring community types
We repeated our models using community types as our primary exposure variable. Due to small sample sizes of certain community types, we also assessed whether our models were robust. We generated a bootstrapped dataset with 100 iterations of randomly reassigned community types and ran the viral shedding duration model for each iteration. The most uncommon community type was statistically significant in 26% of iterations suggesting an inflated type I error rate. All model results are included in the appendix for exploratory purposes and should be interpreted as such (Supplemental Figure 5).
The role of individual taxa
To explore the role of individual taxa, we examined whether the relative abundance of 15 oligotypes were associated with the duration of symptoms and viral shedding. We specifically focused on the oligotypes that contributed most to the difference between community types (>50% of the difference) and used the Benjamin-Hochberg method to correct for multiple testing.
Duration of fever was negatively associated with Veillonella parvula / rogosae / atypica / denticariosi / dispar (AF: 0.66; 95% CI: 0.50, 0.86) (Supplemental Table 4). Duration of runny nose was positively associated with Neisseria (AF: 1.41; 95% CI: 1.25, 1.60) and Prevotella melaninogenica / scopos / sp. / histicola / veroralis (AF: 2.01; 95% CI: 1.46, 2.75). Duration of sore throat was positively associated with Prevotella sp. / veroralis / fusca / histicola / scopos / melaninogenica, Megasphaera micronuciformis, and Prevotella salivae.
Shedding duration was positively associated with the abundance of Fusobacterium (AF: 1.14; 95% CI: 7%, 22%), Neisseria (AF: 1.16; 95% CI: 1.06, 1.27), and Haemophilus (AF: 1.13; 95% CI: 1.04, 1.23). Shedding duration was negatively associated with the abundance of Streptococcus vestibularis / salivarius / gordonii / sp (AF: 0.61; 95% CI: 0.49, 0.77) and Streptococcus australis / parasanguinis II / parasanguinis I/ sp. / oligofermentans / cristatus / sinensis / sanguinis / gordonii / lactarius / peroris / oralis (AF: 0.59; 95% CI: 0.39, 0.91). Fusobacterium (AF: 0.89; 95% CI: 0.83, 0.95) and Neisseria (AF: 0.87; 95% CI: 0.79, 0.95) were associated with a shorter serial interval.
Sensitivity analysis
To investigate whether the criteria used to define illness periods affected our results, we reran our α-diversity models with three sets of modified criteria: 1) illness period does not exclude symptoms if fever recurs ≥3 days after fever alleviation; 2) illness period only considers influenza-like symptoms; and, 3) all symptoms during follow-up contribute to illness period. Most model estimates remained the same or had minor differences that did not affect our overall conclusions (Supplemental Table 5). The exception was in our serial interval models using criteria set 2 and 3, in which associations were no longer statistically significant. The direction of association did not change for Shannon diversity models, but the strength of the association was attenuated. For Chao1 models, the association shifted towards the null.
Discussion
We explored whether the nose/throat microbiota prior to influenza virus infection influenced the duration of symptoms, viral shedding, and time to infection among secondary influenza cases identified by RT-PCR or a ≥4-fold increase in HAI titers. Community diversity prior to influenza virus infection was associated with viral shedding duration. Secondary cases with less diverse bacterial communities had a longer period of viral shedding and signs of infection were observed earlier.
Several oligotypes were associated with symptoms and shedding. Among them a Neisseria oligotype was of particular interest as it was associated with multiple outcomes including earlier signs of infection (by serial interval and viral shedding), longer durations of symptoms and longer viral shedding. There is little information regarding biological interactions between Neisseria and influenza. However, there is some evidence that the outer membrane vesicles of Neisseria can act as a mucosal adjuvant. Findings from intranasal influenza vaccine study demonstrated substantial increases in both IgG and IgA antibodies when inactivated Neisseria meningitidis was added as an adjuvant to the vaccine [29].
Our findings are consistent with results of murine experiments demonstrating a relationship between the gut microbiome and influenza symptoms and viral shedding. Mice treated with antibiotics prior to inoculation with influenza virus expressed enhanced disease severity and increased risk of death [13]. Among mice with microbiomes disrupted by antibiotics, macrophages expressed defective responses to type I and type II IFNs [13] and exhibited defective T-cell and B-cell responses linked to reduced priming of inflammasome-dependent cytokines [11]. These impairments resulted in higher viral replication [11,13]. However, these studies did not characterize the microbiota using an untargeted 16S rRNA taxonomic screen, making it difficult to connect our epidemiologic findings with specific biological mechanisms.
Replication and exploration of the mechanisms underlying our results are needed to evaluate whether our observations are causal. A particular limitation of our study is we were unable to determine if the characteristics of nose/throat microbiota are risk factors or risk markers, that is, whether host factors leading to differences in clinical outcomes of influenza also lead to the observed nose/throat microbiota. Larger studies that oversample young children and assess immune response and animal models are needed to clarify the relationship. Moreover, a more comprehensive assessment of the microbiota (metagenomics and metabolomics) would enable evaluation of other microbiota characteristics beyond α-diversity and selected taxa.
Our study has several strengths. As a case-ascertained study, we were able to characterize the nose/throat microbiota of secondary cases a few days prior to infection. By using both RT-PCR and HAI, we were able to improve our detection of secondary cases. Lastly, daily symptom diaries and regular RT-PCR testing over follow-up allowed us to estimate time-to-event outcomes related to influenza symptoms and viral shedding. However, there are also several potential limitations. Any criteria used to define an influenza-associated illness period is subject to misclassification. However, sensitivity analysis indicates our criteria did not meaningfully affect our results. Lastly, our study does not consider the infectiousness of index cases in time to infection estimates and we were inadequately powered to examine influenza subtype-specific relationships.
Conclusions
In conclusion, our study identified associations between the duration of influenza symptoms, viral shedding and the nose/throat bacterial microbiota prior to influenza infection. By extension, the microbiota may influence influenza transmission, which likely is dependent on both the duration and level of viral shedding and the presence of symptoms. Current methods for reducing influenza transmission and disease severity involve reducing exposure to the virus, vaccination, and antiviral treatment. However, complementary strategies should be explored to reduce the 3–5 million cases of severe illness [30] and 400,000 deaths [31] estimated to occur each year. The microbiome may provide opportunities for reducing this burden. Randomized controlled studies have shown drastic reductions in respiratory tract infections among newborns given synbiotics, which are estimated to cost around $1 per person for 1 week of treatment [14,15]. Future studies should investigate causal pathways between the microbiome and respiratory infections and evaluate the impact of synbiotics in different populations.
Supplementary Material
Acknowledgments
This work was supported by NIH NIAID (grant no. R21 AI119463). Data collection was supported by NIH (grant no. U01 AI088654]), the Fogarty International Center [grant no. K02 TW009483] and National Institutes of Health Department of Health and Human Services under the contract number HHSN272201400031C. Additional funding came from the Tinker Foundation, University of Michigan Center for Latin America and Caribbean Studies, University of Michigan International Institute, and University of Michigan Rackham Graduate School. We are grateful to the participants of the Nicaraguan Household Transmission Study and our collaborators at the Nicaraguan Ministry of Health and Sustainable Sciences Institute. We thank the University of Michigan Microbial Systems Laboratories for 16S rRNA sequencing. We also thank Dr. Marie Griffin for reviewing an earlier draft of this manuscript.
List of abbreviations and acronyms
- AF
acceleration factor
- CI
confidence interval
- CT
community type
- FARI
febrile acute respiratory illness
- HAI
hemagglutination inhibition
- ILI
influenza-like illness
- PERMANOVA
permutational multivariate analysis of variance
- RT-PCR
real-time reverse transcription polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Widdowson M-A, Monto AS. Epidemiology of influenza. In: Webster RG, Monto AS, Braciale TJ, Lamb RA, editors. Textb. Influenza 2nd ed., Wiley; 2013. [Google Scholar]
- [2].Nair H, Brooks WA, Katz M, Roca A, Berkley JA, Madhi SA, et al. Global burden of respiratory infections due to seasonal influenza in young children: a systematic review and meta-analysis. The Lancet 2011;378:1917–30. doi: 10.1016/S0140-6736(11)61051-9. [DOI] [PubMed] [Google Scholar]
- [3].Carrat F, Vergu E, Ferguson NM, Lemaitre M, Cauchemez S, Leach S, et al. Time lines of infection and disease in human influenza: a review of volunteer challenge studies. Am J Epidemiol 2008;167:775–85. doi: 10.1093/aje/kwm375. [DOI] [PubMed] [Google Scholar]
- [4].Ng S, Lopez R, Kuan G, Gresh L, Balmaseda A, Harris E, et al. The Timeline of Influenza Virus Shedding in Children and Adults in a Household Transmission Study of Influenza in Managua, Nicaragua. Pediatr Infect Dis J 2016;35:583–6. doi: 10.1097/INF.0000000000001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cowling BJ, Chan KH, Fang VJ, Lau LLH, So HC, Fung ROP, et al. Comparative epidemiology of pandemic and seasonal influenza A in households. N Engl J Med 2010;362:2175–84. doi: 10.1056/NEJMoa0911530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ferguson NM, Cummings DAT, Cauchemez S, Fraser C, Riley S, Meeyai A, et al. Strategies for containing an emerging influenza pandemic in Southeast Asia. Nature 2005;437:209. doi: 10.1038/nature04017. [DOI] [PubMed] [Google Scholar]
- [7].Ip DKM, Lau LLH, Leung NHL, Fang VJ, Chan K-H, Chu DKW, et al. Viral Shedding and Transmission Potential of Asymptomatic and Paucisymptomatic Influenza Virus Infections in the Community. Clin Infect Dis Off Publ Infect Dis Soc Am 2017;64:736–42. doi: 10.1093/cid/ciw841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Loeb M, Singh PK, Fox J, Russell ML, Pabbaraju K, Zarra D, et al. Longitudinal study of influenza molecular viral shedding in Hutterite communities. J Infect Dis 2012;206:1078–84. doi: 10.1093/infdis/jis450. [DOI] [PubMed] [Google Scholar]
- [9].Hayden FG, de Jong MD. Human influenza: Pathogenesis, clinical features, and management. In: Webster RG, Monto AS, Braciale TJ, Lamb RA, editors. Textb. Influenza 2nd ed., Wiley; 2013. [Google Scholar]
- [10].Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009;139:485–98. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A 2011;108:5354. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shaw MH, Kamada N, Kim Y-G, Núñez G. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med 2012;209:251–8. doi: 10.1084/jem.20111703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012;37:158–70. doi: 10.1016/j.immuni.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Panigrahi P, Parida S, Nanda NC, Satpathy R, Pradhan L, Chandel DS, et al. A randomized synbiotic trial to prevent sepsis among infants in rural India. Nature 2017;advance online publication. doi: 10.1038/nature23480. [DOI] [PubMed]
- [15].Luoto R, Ruuskanen O, Waris M, Kalliomäki M, Salminen S, Isolauri E. Prebiotic and probiotic supplementation prevents rhinovirus infections in preterm infants: A randomized, placebo-controlled trial. J Allergy Clin Immunol 2014;133:405–13. doi: 10.1016/j.jaci.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schuijt TJ, Lankelma JM, Scicluna BP, de Sousae Melo F, Roelofs JJTH, de Boer JD, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut 2016;65:575–83. doi: 10.1136/gutjnl-2015-309728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team, Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, et al. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 2009;360:2605–15. doi: 10.1056/NEJMoa0903810. [DOI] [PubMed] [Google Scholar]
- [18].World Health Organization. WHO Manual on Animal Influenza Diagnosis and Surveillance Geneva, Switzerland: World Health Organization; 2002. [Google Scholar]
- [19].Lee KH, Gordon A, Shedden K, Kuan G, Ng S, Balmaseda A, et al. The respiratory microbiome and susceptibility to influenza virus infection. PloS One 2019;14:e0207898. doi: 10.1371/journal.pone.0207898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl Environ Microbiol 2013;79:5112–20. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009;75:7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Eren AM, Morrison HG, Lescault PJ, Reveillaud J, Vineis JH, Sogin ML. Minimum entropy decomposition: unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. ISME J 2015;9:968–79. doi: 10.1038/ismej.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Holmes I, Harris K, Quince C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PloS One 2012;7:e30126. doi: 10.1371/journal.pone.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen T, Yu W-H, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database J Biol Databases Curation 2010;2010:baq013. doi: 10.1093/database/baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- [26].Cowling BJ, Fang VJ, Riley S, Peiris JSM, Leung GM. Estimation of the serial interval of influenza. Epidemiol Camb Mass 2009;20:344–7. doi: 10.1097/EDE.0b013e31819d1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Aitchison J The Statistical Analysis of Compositional Data. J R Stat Soc Ser B Methodol 1982;44:139–77. [Google Scholar]
- [28].R Core Team. R: A language and environment for statistical computing Vienna, Austria: R Foundation for Statistical Computing; 2017. [Google Scholar]
- [29].Berstad AKH, Andersen SR, Dalseg R, Drømtorp S, Holst J, Namork E, et al. Inactivated meningococci and pertussis bacteria are immunogenic and act as mucosal adjuvants for a nasal inactivated influenza virus vaccine. Vaccine 2000;18:1910–9. doi: 10.1016/S0264-410X(99)00442-9. [DOI] [PubMed] [Google Scholar]
- [30].World Health Organization. Influenza. Immun Vaccines Biol Influenza n.d http://www.who.int/immunization/topics/influenza/en/ (accessed February 17, 2017).
- [31].Iuliano AD, Roguski KM, Chang HH, Muscatello DJ, Palekar R, Tempia S, et al. Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. The Lancet 2017;0. doi: 10.1016/S0140-6736(17)33293-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence reads have been deposited in a NCBI Sequence Read Archive repository (accession number PRJNA482032). Datasets generated and analyzed during the current study are available in an open-access Deep Blue Data repository (https://deepblue.lib.umich.edu/data/concern/generic_works/sb3979224?locale=en). Certain individual participant data have been excluded due to identifiability concerns.