


Abstract
The CRISPR system provides adaptive immunity against mobile genetic elements (MGE) in prokaryotes. In type III CRISPR systems, an effector complex programmed by CRISPR RNA detects invading RNA, triggering a multi-layered defence that includes target RNA cleavage, licencing of an HD DNA nuclease domain and synthesis of cyclic oligoadenylate (cOA) molecules. cOA activates the Csx1/Csm6 family of effectors, which degrade RNA non-specifically to enhance immunity. Type III systems are found in diverse archaea and bacteria, including the human pathogen Mycobacterium tuberculosis. Here, we report a comprehensive analysis of the in vitro and in vivo activities of the type III-A M. tuberculosis CRISPR system. We demonstrate that immunity against MGE may be achieved predominantly via a cyclic hexa-adenylate (cA6) signalling pathway and the ribonuclease Csm6, rather than through DNA cleavage by the HD domain. Furthermore, we show for the first time that a type III CRISPR system can be reprogrammed by replacing the effector protein, which may be relevant for maintenance of immunity in response to pressure from viral anti-CRISPRs. These observations demonstrate that M. tuberculosis has a fully-functioning CRISPR interference system that generates a range of cyclic and linear oligonucleotides of known and unknown functions, potentiating fundamental and applied studies.
INTRODUCTION
The CRISPR system provides adaptive immunity against mobile genetic elements (MGE) in prokaryotes (reviewed in (1,2)). CRISPR systems are classified into two major classes and six major types: class 1 (types I, III and IV) have multi-subunit effector complexes based around a Cas7 backbone subunit, whilst class 2 (types II, V and VI) are single-subunit enzymes (3). The latter, encompassing Cas9, Cas12 and Cas13, have shown great utility in genome engineering applications due to their relative simplicity. However, class 1 systems are more widespread in prokaryotes. Type III effector systems are arguably the most complex, powerful and versatile of the characterized CRISPR interference complexes. Assembled around a Cas7 backbone that binds CRISPR RNA (crRNA) (4), they act as surveillance complexes in the cell, detecting target RNA molecules from MGE via base-pairing to the crRNA. Target RNA binding licenses the HD-nuclease domain of the Cas10 subunit to degrade single-stranded DNA, providing a mechanism for co-transcriptional target DNA degradation (5–10). In addition, the Cyclase/Palm domain of Cas10 is activated for synthesis of cyclic oligoadenylate (cOA) (11–13). cOA, which is assembled from 3–6 AMP monomers to generate species from cA3 to cA6, is a potent second messenger that in turn binds to downstream effector complexes via a CRISPR Associated Rossman Fold (CARF) domain (14). Diverse proteins with CARF domains have been identified computationally, but only one family has been studied extensively: the Csx1/Csm6 family that utilizes a C-terminal HEPN (Higher Eukaryotes and Prokaryotes, Nucleotide binding) domain to cleave RNA (15,16). These enzymes are important for CRISPR-based immunity in vivo (17–19).Thus, type III CRISPR systems are capable of directing a multi-faceted antiviral defence on detection of foreign RNA in the cell (Figure 1A).
Figure 1.

The CRISPR system of Mycobacterium tuberculosis. (A) The CRISPR locus of M. tuberculosis includes genes encoding Cas6 (crRNA processing), Csm1–5 (type III-A interference complex), Csm6 (ancillary ribonuclease), Cas1 and Cas2 (Adaptation). Cas6 cleaves the CRISPR RNA at the base of a short hairpin to generate mature crRNA that is bound by the Csm complex. On target RNA binding, the Csm complex is expected to display three enzymatic activities: target RNA cleavage by Cas7 (Csm3) (1), DNA cleavage by the HD domain (2) and cOA production by the cyclase domain (3). (B) Purified, recombinant CRISPR-associated proteins of M. tuberculosis. M: PageRuler Unstained (Thermo Scientific); 1: Csm1–5 interference complex; 2: Csm1–5, Csm1 D630A, D631A (Cy variant); 3: Csm1–5, Csm3 D35A (C3 variant); 4: Csm6; 5: Cas6.
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB), which imposes a huge burden on human health and on developing economies, with 10 million new infections and 1.6 million deaths in 2017 (WHO Global Tuberculosis Report, 2018). The majority of Mtb strains encode a complete CRISPR system (20), and indeed the type III-A (or Csm) CRISPR system was originally labelled as the ‘Mtube’ subtype based on the genes present in Mtb strain H37Rv (21). CRISPR loci vary between Mtb strains and have been used for many years as a strain typing (spoligotyping) tool (22), suggesting uptake of DNA derived from mycobacteriophage or other MGE by the CRISPR system occurs. The type III-A system encoded by Mtb H37Rv comprises Csm1–6, with Cas6 for crRNA processing and the adaptation proteins Cas1 and Cas2 next to the CRISPR array (Figure 1A). In Mycobacterium bovis (BCG), the organization is identical with the exception of a truncating mutation in the csm6 gene (20). Looking across the entirety of the prokaryotic domains of life, Mtb is a rare example of an isolated type III effector and associated adaptation system.
Little is known about the activity of the Mtb CRISPR system, although recent work suggests it can provide modest immunity against MGE (23). Here, we report the reconstitution and characterization of the Mtb system both in vitro and in vivo, in Escherichia coli. We demonstrate that the Mtb Csm complex provides immunity via a cA6-activated Csm6 ribonuclease, and that the system can be reprogrammed to provide cA4-modulated immunity by swapping the cOA-activated effector protein.
MATERIALS AND METHODS
Cloning
Enzymes were purchased from Thermo Scientific, BioLine, Promega or New England Biolabs and used according to manufacturer's instructions. Oligonucleotides (Supplementary Table S2) and synthetic genes (Supplementary Table S3) were obtained from Integrated DNA Technologies (Coralville, IA, USA). Synthetic genes were codon-optimized for E. coli and restriction sites for cloning incorporated. All constructs were verified by sequencing (GATC Biotech, Eurofins Genomics, DE) unless stated otherwise. Protein concentrations were determined by UV quantitation (NanoDrop 2000, Thermo Scientific) using calculated extinction coefficients (ExPASy, ProtParam software) unless stated otherwise.
CRISPR genes
All genes were obtained as synthetic genes. To obtain N-terminal His6-fusion proteins, Mtb cas10 (csm1), cas11 (csm2), cas7 (csm3), cas5 (csm4), csm5 and csm6 were digested with NcoI and XhoI and ligated into the corresponding sites of the expression vector pEHisTEV (24). The Mtb cas6 gene was cloned into the NdeI and XhoI sites of the same vector to obtain the C-terminal His6-fusion. We cloned a truncated version of cas6, starting at Met53 relative to NCBI annotation, because the N-terminal region showed no homology to non-mycobacterial Cas6 proteins, and a transcription start site was identified within the predicted cas6 gene (25). Thioalkalivibrio sulfidiphilus csx1 was cloned into the NcoI and BamHI sites of pEV5HisTEV to obtain a N-terminal His8-fusion (13).
CRISPR arrays
A CRISPR array containing three Mtb repeat sequences flanking two identical spacer sequences targeting the pUC19 MCS was assembled by BamHI digest of the double-stranded mtbCRISPR array oligonucleotide (Supplementary Table S2) followed by ligation into pCDFDuet™-1 (Novagen, Merck Millipore). Mtb cas6 was cloned into MCS-2 to give pCRISPR.
A CRISPR array consisting of four identical spacers targeting the tetracycline resistance gene, flanked by five Mtb repeat sequences, was assembled from phosphorylated oligonucleotides mtbCA_Rep and mtbCA_TetR-Sp (Supplementary Table S2). The array was ligated into MCS-1 of pCDFDuet™-1 containing cas6 in MCS-2 to give pCRISPR-Target.
Mtb Csm complex for in vitro studies
Cas10 (csm1), csm2 and csm5 were cloned into pACYCDuet-1 (5′-NcoI, 3′-XhoI; Novagen, Merck Millipore), csm4 was cloned into pEHisTEV (5′-NcoI, 3′-XhoI) and csm3 was inserted into pEHisTEV (5′-NdeI, 3′-XhoI) by restriction enzyme cloning. These constructs were used as templates for assembly of M. tuberculosis CRISPR genes, including ribosome binding sites, into MultiColi™ (Geneva Biotech, Genève, CH) acceptor and donor vectors by sequence- and ligation-independent cloning (SLIC) as per manufacturer's instructions. Briefly, His6-csm4, csm2 and cas10 were introduced into the acceptor pACE; csm3, csm5 were cloned into pDK; Mca csm2 was inserted into pDC using restriction enzyme cloning (5′-NdeI, 3′-XhoI). The acceptor and donor plasmids were then recombined using Cre recombinase to give pCsm1–5. To obtain variant Csm complexes, His8-V5-csm4 (from pEHisV5TEV-Csm4), Mca csm2 (from pACYCDuet-Mca_Csm2) and cas10 (csm1) were introduced into pACE by SLIC; and csm3, csm5 were inserted into the donor plasmid pDK. Primer-directed mutagenesis was carried out on the assembled acceptor and donor plasmids, before Cre recombination to give pCsm1–5_C3 (Csm3 D35A) and pCsm1–5_Cy (Csm1 D630A/D631A). The identity of all donor and acceptor plasmids was verified by sequencing, and the final, recombined construct was analysed by restriction digest.
Mtb Csm complex for in vivo studies
The same templates as for construction of pCsm1–5_C3 were used with the addition of csm6 (pACYCDuet-1). The pACE assembly was as for pCsm1–5_C3. Csm3, csm5 and csm6 were introduced into pDK; for the ΔCsm6 plasmid, the donor plasmid en route to pCsm1–5 was used. Mutagenesis was carried out as before. All constructs were verified by sequencing. Suitable acceptor and donor plasmid combinations were then assembled by Cre recombination to give pCsm1–6, pCsm1–5_ΔCsm6, pCsm1–6_C3, pCsm1–6_Cy and pCsm1–6_HD (Supplementary Table S1). The control plasmid pCsm_Control was obtained by Cre recombination of pACE and pDK without inserts. The identity of the final, recombined constructs was confirmed by restriction digest analysis.
Construction of target plasmids
An arabinose-inducible pRSFDuet™-1 (Novagen, Merck Millipore) derivative with tetracycline resistance was constructed as follows. The araC and pBAD region was polymerase chain reaction (PCR)-amplified from pBADHisTEV (Huanting Liu, University of St Andrews, UK) using primer pair ara-AvrII-R/F and cloned into the XbaI and NdeI sites of pRSFDuet™-1 to give pRSFara. The tetracycline resistance gene was PCR-amplified from the MultiColi™ vector pACE2 using the TetR primer pair; the pRSFara-backbone lacking its resistance gene was PCR-amplified using primer pair RSFara. The two PCR products were digested with SphI and BamHI and joined through ligation to give pRAT. The pUC19 MCS / lacZα gene was introduced into pRAT by megaprimer PCR (primer pair lacZ-pRAT) to give pRAT-Target (Supplementary Table S1).
Protein production and purification
Mtb Cas6
C-terminally His6-tagged Cas6 was expressed in BL21 (DE3) grown in LB containing 50 μg ml-1 kanamycin and 50 μg ml−1 chloramphenicol. An overnight culture was diluted 100-fold into fresh medium and grown at 37°C, 180 rpm to an OD600 of 0.4. The temperature was lowered to 16°C, and incubation continued until the OD600 reached 0.8. Production was induced with 50 μM IPTG, and the cultures were continued for a further 20 h. Cells were harvested by centrifugation. The pellet was resuspended in 50 mM Tris–HCl, 250 mM NaCl, 10 mM imidazole, 10% glycerol, pH 7.5, sonicated and the cell debris removed by centrifugation. The cleared lysate was loaded onto a pre-equilibrated HisTrap Crude FF (GE Healthcare) column, washed with 50 mM Tris–HCl, 250 mM NaCl, 30 mM imidazole, 10% glycerol, pH 7.5 and eluted in a gradient to 50 mM Tris, 250 mM NaCl, 500 mM imidazole, 10% glycerol, pH 7.5. Cas6-containing fractions were pooled and further purified by gel filtration (HiLoad 16/60 Superdex pg 200, GE Healthcare) in 50 mM Tris–HCl, 250 mM NaCl, 10% glycerol, pH 7.5. Cas6 and Cas6 H47N were concentrated as required using an Amicon Ultra centrifugal filter (MWCO 10 kDa, Merck-Millipore).
Mtb Csm complex
Escherichia coli C43(DE3) cells were co-transformed with the MultiColi™ construct pCsm1–5 (or pCsm1–5_C3, pCsm1–5_Cy) and pCRISPR. Overnight cultures were diluted 100-fold into LB containing 100 μg ml−1 ampicillin and 50 μg ml−1 spectinomycin, incubated at 37°C, 180 rpm until the OD600 reached 0.8. After induction with 100 μM IPTG, incubation was continued at 25°C for 5 h. Cells were harvested by centrifugation and pellets stored at −20°C. Cells were resuspended in lysis buffer (50 mM Tris–HCl, 500 mM NaCl, 10 mM imidazole, 10% glycerol, pH 7.5) and lysed by sonication. The cleared lysate was loaded onto HisPur Ni-NTA resin (Thermo Scientific), washed with lysis buffer and eluted with lysis buffer containing 250 mM imidazole. Csm complex-containing fractions were diluted 2-fold with lysis buffer to prevent aggregation, pooled, then dialysed at 4°C overnight in the presence of 0.3 mg ml−1 TEV protease against lysis buffer. The protein solution was passed through His-Pur Ni-NTA resin a second time, and the flow-through was concentrated using an Amicon Ultracentrifugal filter (30 kDa MWCO, Merck-Millipore) and further purified by gel filtration (HiPrep 16/60 Sephacryl pg 300 HR, GE Healthcare) using 20 mM Tris–HCl, 250 mM NaCl, 10% glycerol, pH 7.5 as mobile phase. When necessary, the Csm complex was further purified on MonoS 4.6/100 PE (GE Healthcare) in 20 mM MES, 50 mM NaCl, pH 6 and eluted using a salt gradient to 1 M NaCl. Single-use aliquots were flash-frozen and stored at −80°C.
Mtb Csm6 and T. sulfidiphilus Csx1
Mtb Csm6 and TsuCsx1 were expressed in C43 cells. Cells were grown for 4 h at 25°C before harvest by centrifugation at 4000 rpm at 4°C for 15 min (JLA 8.1 rotor). Cells were lysed in buffer containing 50 mM Tris–HCl pH 8.0, 0.5 M NaCl, 10 mM imidazole and 10% glycerol supplemented with 1 mg/ml chicken egg lysozyme (Sigma-Aldrich) and one ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor tablet (Roche) by sonicating six times for 1 min with 1 min rest intervals on ice. Cell lysate was then ultracentrifugated at 40000 rpm (70 Ti rotor) at 4°C for 45 min and loaded onto a 5 ml HisTrap FF Crude column equilibrated with wash buffer containing 50 mM Tris-HCl pH 8.0, 0.5 M NaCl, 30 mM imidazole and 10% glycerol. After washing unbound protein with 20 CV wash buffer, recombinant Csm6 or TsuCsx1 was eluted with a linear gradient of wash buffer supplemented with 0.5 M imidazole across 16 CV, holding at 10% for 2 CV, 20% for 4 CV and 50% for 3 CV. Csm6 could not be concentrated and was therefore further purified using MonoQ (GE Healthcare) anion exchange chromatography, diluting protein from nickel affinity fractions directly in buffer containing 20 mM Tris-HCl pH 7.5 and 50 mM NaCl and eluting protein across a linear gradient of buffer containing 20 mM Tris-HCl pH 7.5 and 1M NaCl. For TsuCsx1, nickel affinity fractions containing the protein were concentrated using a 10 kDa MWCO ultracentrifugal concentrator, and further purified by size exclusion chromatography (S200 26/60; GE Healthcare) in buffer containing 20 mM Tris-HCl pH 8.0, 0.5 M NaCl and 1 mM DTT. All proteins were aliquoted, flash frozen with liquid nitrogen, and stored at −80°C.
Assays
Cyclic oligonucleotides
cA4 and cA6 were purchased from BIOLOG Life Science Institute (Bremen, DE).
Oligonucleotides
All RNA and DNA oligonucleotides as well as 6-FAM™-labelled RNA substrates were purchased from Integrated DNA Technologies (Leuven, BE). For nuclease assays, labelled oligonucleotides were gel purified as described previously (26). Duplexed DNA oligonucleotides were prepared by mixing equimolar amounts of ssDNA in 10 mM Tris-HCl, 50 mM NaCl, 0.5 mM EDTA, pH 8.0, heating to 95°C for 10 min, followed by slow cooling. Oligonucleotides were 5′-end-labelled with [γ-32P]-ATP (10 mCi ml−1, 3000 Ci mmol−1; Perkin Elmer) using polynucleotide kinase (Thermo Scientific). Oligonucleotides were quantified spectophotometrically using calculated extinction coefficients (OligoAnalyzer tool, IDT). RNA ladders were obtained by alkaline hydrolysis (Thermo Fisher Scientific, RNA Protocols).
Cas6 ribonuclease assay
The reaction mixture contained 3 μM Cas6 in 20 mM Tris–HCl, 100 mM potassium glutamate, 1 mM DTT, 5 mM EDTA, 0.1 U μl−1 SUPERase•In™ (Thermo Scientific), pH 7.5. Reactions were initiated by addition of 5′ 6-FAM™-labelled mtbRepeat_Cas6 RNA (Supplementary Table S2) to 50 nM final concentration. The samples were incubated for up to 1 h at 37 °C. The reaction was stopped by extraction with phenol-chloroform-isoamyl alcohol. RNA species were resolved on a 20% denaturing (7 M urea) acrylamide gel in 1X TBE buffer, and visualized by scanning (Typhoon FLA 7000, GE Healthcare).
Target RNA cleavage by the Csm complex
The ribonuclease activity of the Csm complex was assessed by adding 25 nM [5′-32P]-labelled target RNA (Supplementary Table S2) to 0.8 μM Csm, 5 mM MgCl2, 0.1 U μl−1 SUPERase•In™ (Thermo Scientific), 50 mM Tris–HCl, 50 mM NaCl, pH 8.0, and incubating at 30°C for up to 2 h. The reaction was stopped by phenol–chloroform–isoamyl alcohol extraction to remove proteins. P1.A26 RNA (Supplementary Table S2) was used as non-target RNA control. Products were separated by gel electrophoresis (20% denaturing polyacrylamide) and visualized by exposure to a BAS Storage Phosphor Screen (GE Healthcare).
DNase activity of the Csm complex
To test for DNase activity of the Csm complex, 25 nM 5′-32P-labelled DNA substrate was added to 0.8 μM Csm, 200 nM target RNA, 0.1 mM CoCl2, 0.1 U μl−1 SUPERase•In™ (Thermo Scientific), 50 mM Tris–HCl, 50 mM NaCl, pH 8.0. The solution was incubated at 30°C for 90 min, and the reaction stopped by phenol–chloroform extraction. Products were analysed as described above.
Cyclic oligoadenylate production and LC-MS analysis
Cyclic oligoadenylate production was triggered by adding 2 μM crude target RNA to 0.8 μM Csm, 1 mM adenosine triphosphate (ATP), 5 mM MgCl2, 0.1 U μl−1 SUPERase•In™ (Thermo Scientific), 50 mM Tris–HCl, 50 mM NaCl, pH 8.0, and incubating at 30°C for up to 2 h. The reaction was stoped by phenol–chloroform–isoamyl alcohol extraction. Reaction mixtures that were used for Csm6 or Tsu Csx1 activation assays were further extracted with chloroform–isoamyl alcohol to remove residual phenol. Liquid chromatography-high resolution mass spectrometry (LC-HRMS) analysis was performed on a Thermo Scientific Velos Pro instrument equipped with HESI source and Dionex UltiMate 3000 chromatography system. Samples were desalted on C18 cartridges (Harvard Apparatus). Compounds were separated on a Kinetex 2.6 μm EVO C18 column (2.1 × 5 mm, Phenomenex) using the following gradient of acetonitrile (B) against 20 mM ammonium bicarbonate (A): 0–2 min 2% B, 2–10 min 2–8% B, 10–11 min 8–95% B, 11–14 min 95% B, 14–15 min 95–2% B, 15–20 min 2% B at a flow rate of 350 μl min−1 and column temperature of 40°C. UV data were recorded at 254 nm. Mass data were acquired on the FT mass analyser in positive ion mode with scan range m/z 200–2000 at a resolution of 30 000.
Csm6 activation assay
Csm6 (125 nM dimer) was incubated with 25 nM 5′-radiolabelled P1.A26 RNA substrate together with 2 μM cold RNA in 50 mM MES, 100 mM potassium l-glutamate, pH 6.5. The reaction was started by addition of synthetic cA6 or 0.1 volumes protein-free reaction mixture from the cyclic oligoadenylate production above. The reaction was stopped after 45 min at 37°C by phenol–chloroform extraction. Products were analysed by 20% denaturing polyacrylamide gel electrophoresis.
TsuCsx1 activation assay
Csx1 (500 nM dimer) was incubated with 50 nM 5′-radiolabelled A1 RNA substrate and 1 μM cA4, 1 μM cA6 or 1 μl Csm-derived cOA (see above) in pH 7.5 buffer containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl and 1 mM DTT in a 10 μl reaction volume at 35°C. Reactions were stopped at 1, 5, 15 or 30 min by the addition of a reaction volume equivalent of phenol–chloroform and extracted products were analysed by denaturing gel electrophoresis as described above.
Plasmid immunity in vivo
pCsm1–6 and pCRISPR were co-transformed into E. coli C43(DE3). Plasmids were maintained by selection with 100 μg ml−1 ampicillin and 50 g ml−1 spectinomycin. Competent cells were prepared by diluting an overnight culture 50-fold into fresh, selective LB medium; the culture was incubated at 37°C, 220 rpm until the OD600 reached 0.8–1.0. Cells were collected by centrifugation and the pellet resuspended in an equal volume of 60 mM CaCl2, 25 mM MES, pH 5.8, 5 mM MgCl2, 5 mM MnCl2. Following incubation on ice for 1 h, cells were collected and resuspended in 0.1 volumes of the same buffer containing 10% glycerol. Aliquots (100 μl) were stored at −80°C. Target or control plasmid (100 ng pRAT-Target, 100 ng pRAT, respectively) were added to the competent cells, incubated on ice for 30 min and transformed by heat shock. LB medium (0.5 ml) was added and the transformation mixture incubated with shaking for 2.5 h before collecting the cells and resuspending in 100 μl LB. A total of 5 μl of a 10-fold serial dilution were applied in duplicate to LB agar plates supplemented with 100 μg ml−1 ampicillin and 50 g ml−1 spectinomycin when selecting for recipients only; transformants were selected on LB agar containing 100 μg ml−1 ampicillin, 50 g ml−1 spectinomycin, 25 μg ml−1 tetracycline, 0.2% (w/v) β-lactose and 0.2% (w/v) l-arabinose. Arabinose was omitted when transcriptional induction of target was not required. Plates were incubated at 37°C for 16–18 h. Colonies were counted manually and corrected for dilution and volume to obtain colony-forming units (cfu) ml−1, statistical analysis was performed with RStudio (RStudio Inc., Boston, USA, version 1.2.1335). The experiment was performed as two independent experiments with two biological replicates and at least two technical replicates.
RESULTS
Production of the functional Mtb Csm complex
Although the Mtb type III-A effector complex was described very early in the classification of CRISPR systems (21), there have been no subsequent publications describing studies of the recombinant system. This is surprising given the importance of Mtb as a human pathogen and the relative lack of model type III CRISPR systems outwith the hyperthermophiles. We took the initial approach of expressing synthetic genes encoding Csm1–5 individually in E. coli using the expression vector pEhisV5Tev (27). Most of the subunits were expressed strongly, but the Csm2 (Cas11) subunit was completely insoluble under all induction conditions tested. We also expressed the Mtb Csm6 and Cas6 enzymes in soluble form (Figure 1B). Cas6 was observed to cleave a CRISPR RNA substrate to generate the expected 8 nt 5′-handle (Supplementary Figure S1). This activity was stimulated by the addition of a divalent cation, as observed previously (23). However, the ability of Ca2+ to support catalysis is suggestive of a role for the metal ion in RNA folding and stability rather than a direct role in the catalytic mechanism, and all other Cas6 enzymes studied are metal-independent enzymes (reviewed in (28)).
We next constructed a plasmid to express all five subunits of Mtb Csm simultaneously using the MultiColi™ system (Geneva Biotech), along with a compatible pCDFDuet-1 derivative expressing an Mtb mini-CRISPR and the Cas6 enzyme (pCRISPR, Supplementary Table S1) to allow for co-production of the crRNA-charged interference complex in E. coli. Using a polyhistidine tag on the Csm4 (Cas5) subunit, it was noted that all subunits except Csm2 could be identified. Coupled with the observation that Csm2 was insoluble in isolation, we hypothesized that heterologous expression of this subunit was a barrier to successful expression of the Csm complex. We therefore added a gene encoding the Csm2 homologue from Mycobacterium canettii (Mca) to the Csm expression vector (pCsm1–5, Supplementary Table S1). The two proteins are 71% identical at the amino acid level. Gratifyingly, the Csm complex could then be purified using affinity chromatography followed by gel filtration (Figure 1B). Mass spectrometry confirmed that all subunits were present. In all subsequent Csm expression plasmids, the Mtb csm2 gene was therefore replaced by Mca csm2. Protein expression plasmids were also constructed carrying site directed mutations targeted to the active sites of the Csm complex: a Cyclase (Cy) variant (GGDD to GGAA, pCsm1–5_Cy) and a Csm3 D35A variant (C3, pCsm1–5_C3, Supplementary Table S1), as described in the methods. The variant interference complexes were expressed and purified as described for the wild-type (WT).
Target RNA and DNA cleavage by Mtb Csm
The CRISPR array used in heterologous production of the Csm effector complex contained two copies of the same artificial spacer to minimize heterogeneity. We first evaluated the ability of Csm to cleave target RNA that was complementary to the crRNA. Incubation of Csm effector complex with 5′-radiolabelled target RNA (Figure 2 and Supplementary Figure S2) in the presence of Mg2+ resulted in five cleavage products (B1–B5) with the characteristic 6 nt spacing, consistent with other type III effectors (29–32). This suggests that 5 Cas7 subunits are present in the complex, although we have not determined the subunit stoichiometry definitively and it will likely vary with crRNA length. Non-complementary RNA was not cleaved by the effector complex (Supplementary Figure S2A); however, anti-tag RNA with full-length complementarity to the crRNA (including 5′-handle), that mimics antisense RNA derived from the CRISPR locus, produced the same cleavage pattern as observed for target RNA (Supplementary Figure S2B). A variant Csm complex with a mutation targeted to the cyclase domain (Cy) was indistinguishable from the WT protein for RNA cleavage. However, a variant (C3) with the Csm3 (Cas7) D35A mutation was not competent for target RNA cleavage, as expected (Figure 2) (30). We also investigated DNA cleavage by the Mtb Csm effector complex. We observed weak cleavage of ssDNA substrates by the WT complex; however, a variant with a mutation in the HD nuclease domain was still capable of degrading ssDNA, whilst the Csm3 D35A variant showed no DNase activity (Supplementary Figure S3). No detectable hydrolysis was observed for DNA bubbles or dsDNA and the observed DNase activity was not dependent on the presence of target RNA. Together, these data suggest there is no functional HD nuclease activity in the recombinant Mtb Csm complex in vitro. Given that the complex is recombinant and chimeric, with an orthologous Csm2 subunit, we cannot rule out the possibility that the HD nuclease domain plays a more prominent role in immunity in vivo in M. tuberculosis.
Figure 2.
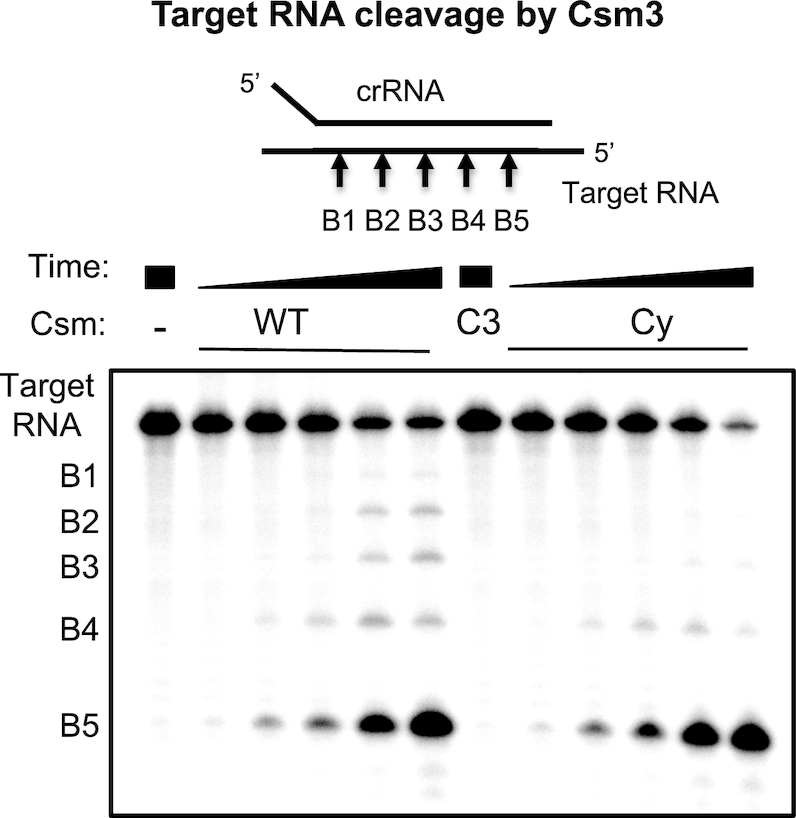
Target RNA cleavage by the Csm complex. WT, Csm3 D35A variant (C3) or cyclase variant (Cy) Csm were incubated with 5′-32P-target RNA under single-turnover conditions for 0.5, 2, 5, 30, 90 min and analysed by denaturing PAGE. Five RNA cleavage sites (B1–B5) were observed.
Cyclic oligoadenylate synthesis by the Csm complex
Most type III CRISPR systems are closely associated with a gene encoding a Csm6 or Csx1 enzyme, which is activated by cOA generated by the Cas10 cyclase domain on target RNA binding (11,12) and required for immunity (17,33,34). To test for cOA production, we incubated the Csm interference complex with cold target RNA and ATP and analysed the products by LC-MS (Figure 3A). Mtb Csm produced cA3, cA4, cA5, cA6 in decreasing order of abundance. The identity of cA4 and cA6 was further confirmed by comparison to synthetic standards (data not shown). We also detected substantial amounts of the corresponding linear OA triphosphates that are likely to be intermediates of cOA biosynthesis. When the same analysis was performed on metabolites produced by the Csm Csm3 D35A complex, a significant shift in cOA profile towards larger ring sizes was observed.
Figure 3.

cOA production and Csm6 activation. (A) Extracted ion chromatograms of oligoadenylates produced by WT and Csm3 D35A variant (C3) Csm complex. Identical y-scaling throughout. The identity of the cyclic OAs (cAn) and respective 5′-triphosphates (P3-An) is indicated by the number of AMP subunits. Production of cOAs, cA5 and cA6 in particular, was significantly increased for the Csm3 D35A (C3) Csm complex. (B) Csm6 ribonuclease activation by cyclic oligoadenylates. Mtb Csm6 (125 nM) was incubated with 200 nM 5′-[32P]-substrate RNA at 37°C for 30 min in the presence of synthetic oligoadenylates (cA4, cA6) or a 10-fold dilution of Csm-derived cOAs. Csm WT (800 nM), cyclase variant (Cy, 800 nM) or Csm3 D35A (C3, 200 nM) was incubated with 1 mM ATP and 200 nM target (T), non-target (NT) or anti-tag (AT) RNA for 0.5, 2 (WT, C3), or 90 min (all others) at 30°C. Only cA6 activated Csm6. For WT and C3 Csm, sufficient cA6 was produced within 0.5 min to switch on Csm6 ribonuclease activity; whereas no detectable amount of cA6 was produced under all other conditions even after prolonged reaction time.
Activation of M. tuberculosis Csm6 requires cA6
The cOA-dependent ribonucleases studied to date are activated by either cA6 or cA4 (11–13). To determine which cOA species is required for activation of Mtb Csm6, we incubated the recombinant protein with synthetic cA4 and cA6, along with a radioactively labelled substrate RNA (Figure 3B). cA4 at 50 nM did not activate the ribonuclease activity, but in contrast cA6 showed strong activation at 5 nM and supported complete degradation of substrate RNA at 50 nM. This confirms cA6 as the relevant activator of Mtb Csm6. To investigate the activation of Csm6 by the Csm complex, we set up an assay where RNA binding and cOA synthesis by Csm are coupled to activation of Csm6 (13). In the presence of WT Csm and target RNA, strong activation of Csm6 was observed, consistent with the generation of cA6 by the Csm complex. Non-target (NT) control DNA did not activate Csm6, and neither did an RNA matching the 5′-handle of the crRNA (anti-tag, AT). The variant Csm with a mutation that inactivates the cyclase domain (Cy), as expected, did not activate Csm6, whereas the variant deficient in target RNA cleavage (C3) was, as expected, proficient in Csm6 activation, confirming previous findings for a variety of type III systems (11,13).
In vivo activity of the Csm complex
We next set out to test if the Csm complex was active in vivo. A recent study of plasmid transformation in M. tuberculosis yielded 3- to 10-fold less transformants when the introduced plasmid carried one of the spacer sequences of the native CRISPR array, suggesting that the Mtb CRISPR system is functional in its native host (23). We tested the capability of Csm to confer immunity against incoming plasmids in the heterologous host E. coli. The genes for the Csm interference complex were assembled together with csm6 (pCsm1–6, Supplementary Table S1). pCsm1–6 was co-transformed with pCRISPR, harbouring the CRISPR array with cas6 and two identical spacers targeting the pUC19 multiple cloning site (MCS) portion of the lacZα gene. We constructed plasmid pRAT, which combines the origin of replication of pRSF-Duet-1 (Novagen), compatible with both Csm/CRISPR constructs, with tetracycline resistance and an arabinose-inducible MCS, and introduced pUC19 lacZα, containing the target sequence, into pRAT to give pRAT-Target. Plasmid maps are shown in Supplementary Figure S4.
An E. coli C43 strain harbouring the complete Mtb Csm interference module, was transformed with 100 ng target plasmid pRAT-Target or with pRAT as negative control. Serial dilutions of the transformation mixture were applied to plates with three different conditions (Figure 4A): selecting for pCsm1–6 and pCRISPR only, additionally selecting for the incoming plasmid without induction of target expression, and finally selecting for the incoming plasmid with induction of target expression. The first condition provided the number of recipient cells and served to establish that differences in transformation efficiency were not due to different cell concentrations. The other two conditions were designed to establish if target transcription was essential for plasmid immunity. There was no significant difference in number of recipients. The number of transformants on uninduced plates also showed no significance difference between the control and target plasmid. When target expression was induced, however, ∼100 times fewer transformants were observed for the target plasmid relative to control or uninduced conditions (Figure 4A). This demonstrates that the Csm-mediated immunity is functional in the heterologous host, and that immunity is dependent on target transcription as observed in other type III systems (35).
Figure 4.

Plasmid immunity assays of Mtb Csm in heterologous host Escherichia coli. (A) C43 harbouring pCsm1–6 and pCRISPR are transformed with pRAT (control plasmid) or with pRAT-target (plasmid with pUC19 MCS target). Target transcription was arabinose-inducible. In the absence of arabinose, the transformation efficiency was independent of the presence or absence of target; whereas when target transcription was induced, a 2-log reduction of transformation efficiency was observed for the target plasmid compared to the control plasmid. *** P-value (Welch two sample test, two tailed) < 1e-05; significance threshold at < 0.05. (B) C43 harbouring pCsm1–6 or indicated mutants and pCRISPR_TetR (target tetracycline resistance gene, constitutively expressed) were transformed with pRAT. Vector control (Ctrl): backbone vectors without inserts; WT Csm, Csm1–5 only (ΔCsm6, C6), Csm3 D35A (C3), Csm1 (Cas10) D630A/D631A (cyclase mutant, Cy), Cas10 H18A/D19A (HD mutant, HD).
Thus far, the observed plasmid immunity was less efficient by one to three orders of magnitude compared to those in other type III systems (5,19,33,35,36). To test whether targeting a gene essential for plasmid maintenance would provide a higher degree of immunity than when targeting a non-essential one as before, we constructed a new CRISPR array with spacers targeting the tetracyline resistance gene of pRAT. The new array together with Cas6 was cloned into pCDFDuet-1 to give pCRISPR_TetR and co-transformed with the complete Mtb Csm module (pCsm1–6) into E. coli C43 as before. Constructs where csm6 had been omitted (pCsm1–5_ ΔCsm6) or that carried inactivating mutations in the Csm3 subunit (pCsm1–6_ C3), the cyclase (pCsm1–6_ Cy) or the HD domain (pCsm1–6_ HD) were also constructed and introduced into E. coli together with pCRISPR_TetR. As a control, empty backbone (pCsm_Control) was co-transformed with pCDF-Duet-1. Cells were transformed with pRAT, and a 10-fold serial dilution was plated selecting for recipients (selection for pCsm and pCRISPR_TetR only) and for transformants (additional selection for incoming plasmid and induction of CRISPR-Cas genes).
We observed three orders of magnitude fewer transformants for the full WT Csm module (including Csm6) compared to the empty vector control (Figure 4B). As expected, the Csm3 active site variant (C3) was as active as WT Csm; however, so was the Csm HD variant. No plasmid immunity was observed for the Csm cyclase domain variant (Cy) or when Csm6 was not present. This is strong indication that cOA induced Csm6 ribonuclease activity is the driving force behind plasmid immunity for this system. It also demonstrates that target RNA cleavage is not required for immunity, nor is DNase activity, at least not in a heterologous host such as E. coli.
Reprogramming the Mtb Csm system as a cA4-mediated defence
The observation that the Mtb Csm complex generates a range of cOA species from cA3-cA6 is consistent with previous findings for type III systems (11–13). Thus, the cOA species relevant for downstream immunity should be defined by the CARF-family effector protein(s) present in the organism. For Mtb, the native Csm6 enzyme is activated by cA6. We sought to determine whether we could reprogramme the Mtb CRISPR system as a cA4-mediated defence, by replacing Csm6 with an alternative effector protein. A candidate Csx1 protein from the gammaproteobacterium Thioalkalivibrio sulfidiphilus (37) was selected and expressed in E. coli from a synthetic gene. The activity of the purified recombinant protein (TsuCsx1) was tested against a labelled RNA substrate in the absence and presence of cOA species (Figure 5A). TsuCsx1 was strongly activated by synthetic cA4 but not by cA6, demonstrating that this enzyme is cA4 specific. Strong activation was also observed when the enzyme was incubated with cOA generated by the Mtb Csm complex, consistent with the observation that cA4 is a significant component of this mixture (Figure 3A).
Figure 5.
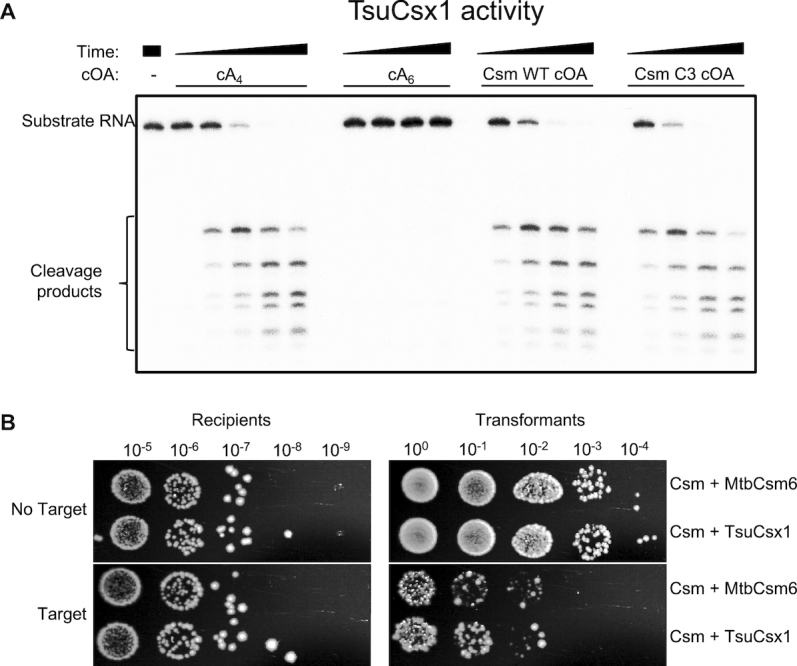
Re-programming the Mtb Csm system for cA4-responsive immunity. (A) In vitro activity of TsuCsx1. The reaction contained 0.5 μM TsuCsx1 dimer, 100 nM 5′-32P-labelled RNA A1, 20 mM Tris, 150 mM NaCl, 1 mM DTT, pH 7.5 and was conducted at 35°C for 1, 5, 15, 30 min. Activators were added as indicated; Csm-derived cOAs were from a 2 h reaction, otherwise as described for Figure 3. TsuCsx1 was activated by cA4 but not cA6, and Csm-derived cOAs are able to induce Csx1 ribonuclease activity in vitro. (B) Plasmid immunity assay using pUC19 lacZα-targeting Csm effector complex in Escherichia coli C43 (pCsm1–5_Csm6/tsuCsx1 and pCRISPR, Supplementary Figure S4). The ribonuclease was either the cognate Csm6 or TsuCsx1. Cells were transformed with pRAT (control plasmid) or pRAT-Target (target plasmid) and 10-fold serial dilutions were plated on selective plates containing arabinose for induction of target transcription. TsuCsx1 confers the same level of plasmid immunity as the cognate Mtb Csm6.
We proceeded to add the gene encoding TsuCsx1 to the Mtb csm1–5 genes (pCsm1–5_Csx1). As before, E. coli C43 harbouring pCsm1–5_tsuCsx1 and pCRISPR, were transformed with the target plasmid (pRAT-Target) and pRAT as the control lacking the target sequence. The 10-fold serial dilutions were applied to selective, inducing plates to determine the number of transformants; the cell count for recipients was carried out as before. We observed similar levels of plasmid immunity for the cognate Mtb Csm6 and the heterologous TsuCsx1 systems (Figure 5B), demonstrating that the downstream effector proteins determine which cyclic oligoadenylate species is active for immunity, and that the Csm interference complex is not specific for a given ribonuclease or vice versa.
DISCUSSION
The type III-A (Csm) CRISPR system of M. tuberculosis provides immunity via cOA signalling
M. tuberculosis harbours the archetypal type III-A (‘Mtube’) CRISPR system, and the variability of CRISPR loci in mycobacteria (spoligotyping) has been utilized to classify isolates for over 20 years. Despite this, little has been reported on the characterization of the Mtb CRISPR system, particularly in vitro. Initial work on reconstituting the Mtb Csm complex in E. coli encountered problems due to poor solubility of the Csm2 (Cas11) subunit. We surmounted this hurdle by swapping the problematic Mtb csm2 gene with that from the closely related species Mycobacterium cannetti, allowing expression of Mtb Csm complex that is functional both in vitro and in vivo. Such an approach may find utility in other circumstances when studying multi-protein complexes.
Having succeeded in constructing an active Mtb Csm complex, we could dissect the contributions of the various enzymatic activities of this complex to CRISPR-based immunity. We could detect no strong evidence for target RNA dependent DNA cleavage by the HD nuclease domain of the Mtb Csm complex (Supplementary Figure S3). Furthermore, targeted mutation of the HD nuclease active site did not affect the ability of the Mtb CRISPR system to provide immunity against plasmid transformation in E. coli, and immunity was lost when the Cyclase active site was inactivated and the HD nuclease unaffected (Figure 4). Target RNA dependent DNA cleavage by the HD nuclease domain of Cas10 has been demonstrated in vitro for the type III-A complexes of Staphylococcus epidermidis (38), Streptococcus thermophilus (7) and Thermus thermophilus (39), and type III-B complexes of Thermatoga maritima (6), Pyrococcus furiosus (5) and Sulfolobus islandicus (9). On the other hand, not all type III CRISPR effectors support robust DNase activity in vitro (32), and not all type III systems have an intact HD nuclease domain (3). HD nuclease activity is often not required for plasmid immunity in vivo, although the cyclase activity of Cas10 and the presence of a cOA effector nuclease clearly is (9,17–19). Recent analysis of plasmid clearance by the type III-A system of S. epidermidis revealed that highly transcribed plasmid targets were cleared efficiently by DNA targeting, whilst low levels of transcription resulted in a requirement for RNA cleavage by the Csm6 nuclease. This suggests that these two aspects of type III defence systems can work in tandem, and assume differing importance for immunity, depending on the biology of the invading genetic element (36). Although we see no target-activated role for the HD nuclease domain of Mtb Csm, either in vitro or in E. coli, we cannot rule out a role in vivo in the homologous host. It is possible that the recombinant system generated here has resulted in deactivation of the HD nuclease, for example, or that a missing component is crucial for full function in M. tuberculosis.
Cyclic oligoadenylate synthesis has been studied for a variety of type III CRISPR systems. One emerging paradigm is that a range of cOA molecules, from cA3 to cA6, are synthesized—at least for recombinant systems in vitro. We observed a predominance of the smaller species for WT Mtb Csm (Figure 3A), consistent with observations for the type III-A complex from S. thermophilus (11). Notably, when target RNA cleavage was abolished by introduction of a D35A mutation in the Csm3 subunit, the levels and distribution of cOA species changed dramatically. A significant increase in the amounts of the larger cA6 and cA5 species was observed, with cA6 coming to predominate. This reflects the finding that target RNA cleavage by the Csm3 subunit is the event that deactivates the cyclase domain (13,40). However, even with target RNA cleavage abolished, a range of cOA species are synthesized. This leads to the question: is this a feature or a bug? Or in other words, has the ability to generate a range of cOA species been selected for by evolution, or just not selected against?
Ancillary defence enzymes are replaceable parts for Type III CRISPR systems
A number of Csx1/Csm6 family proteins have now been studied structurally and/or biochemically (Table 1). As already noted, proteins in this family have an N-terminal CARF domain and a C-terminal HEPN domain. They tend to be named as ‘Csm6’ proteins if their genes are closely associated with those encoding a type III-A/Csm CRISPR effector complex, and Csx1 proteins if they are not, but it is not clear that this division relates to any intrinsic property or sequence of family members. The enzymes from the Archaeal, Deinococcus-Thermus and Proteobacterial phlya that have been studied to date all utilize cA4 as an activator (Table 1), with examples of cA6-dependent enzymes only characterized so far in the Firmicutes and Actinobacteria. As cA4 and cA6 have quite significantly different structures and sizes, this is likely to be reflected in the structures of the corresponding CARF domains that bind these ligands. To date however, there is no structural information available for any cA6-binding CARF domain—this is a priority for future studies.
Table 1.
Comparison of characterized Csx1/Csm6 family proteins
| Species | Phylum | Gene | CRISPR subtype | PDB | cOA activator | Reference |
|---|---|---|---|---|---|---|
| S. solfataricus | Crenarchaeota | Sso1389 | III-D | 2I71 | cA4 | (13) |
| S. islandicus | Crenarchaeota | SiRe_0884 | III-B | cA4 | (49) | |
| P. furiosus | Euryarchaeota | PF1127 | III-B | 4EOG | ?* | (50) |
| M. thermautotrophicus | Euryarchaeota | MTH1076 | III-A | cA4 | (32) | |
| T. thermophilus | Deinococcus-Thermus | TTHB152 | III-A | 5FSH | cA4 | (12) |
| T. thermophilus | Deinococcus-Thermus | TTHB144 | III-A | cA4 | (48) | |
| T. sulfidiphilus | Proteobacteria | Tgr7_1803 | III-B | cA4 | This work | |
| S. thermophilus | Firmicutes | StCsm6 | III-A | cA6 | (11) | |
| E. italicus | Firmicutes | EiCsm6 | III-A | cA6 | (23) | |
| M. tuberculosis | Actinobacteria | Rv2818c | III-A | cA6 | This work |
*Activator has not been determined for PF1127, but predicted to be cA4.
Based on the data collated in Table 1, it is already apparent that the cOA activator utilized by any particular type III system does not correlate with the CRISPR subtype. Although only type III-A systems are known to utilize cA6, there are well-studied examples (such as T. thermophilus and Methanothermobacter thermautotrophicus) of type III-A systems with ancillary ribonucleases activated by cA4. All type III systems studied to date have the capability of making a range of cyclic oligoadenylates from cA3-cA6 (11–13,40). As we have shown here, the immunity provided by a type III system can be readily converted from a cA6 to a cA4-based mechanism, simply by replacing the ancillary enzyme. This suggests there is no direct cross-talk between the type III complex and the ribonuclease, other than via the cOA activator. Given the extent of gene loss and gain in CRISPR loci, the capture of a new CARF-family effector protein might be sufficient to potentiate a switch from one cOA signalling molecule to another. It is equally possible that distinct effector proteins, activated by cA4 and cA6, could operate in parallel in a single host species. This could provide enhanced levels of immunity by supporting multiple ancillary defence proteins, many of which remain uncharacterized so far. Furthermore, the ability to switch between reliance on cA4 or cA6 could be an effective mechanism to maintain immunity in response to (putative) viral anti-CRISPRs which disrupt the cOA signalling pathway.
Cyclic and linear oligoadenylate signalling—more to discover?
Another pressing question for type III CRISPR systems concerns the role (if any) for cA3 molecules, which are often the most abundant species synthesized, at least in vitro (11,12). Given the lack of 2-fold symmetry for cA3, recognition of this molecule may be accomplished by a sensing domain quite distinct from the dimeric CARF domain found in many CRISPR-associated proteins (13). Recent discoveries of novel cyclic trinucleotides synthesiszed by bacterial cGAS-like enzymes and detected by vertebrate innate immune surveillance proteins (41,42) make this question pertinent. This is particularly true for organisms such as M. tuberculosis, which has an intracellular lifestyle in vertebrate hosts with which it participates in a two-way communication via cyclic nucleotides (43,44). Furthermore, the significant quantities of nanoRNAs (eg A3-triphosphate) synthesized by the Csm complex should not be discounted as dead-end side products. NanoRNAs have been shown capable of priming transcription initiation in bacteria (45). Recently, the CnpB (Rv2837c) protein, which can degrade nanoRNAs, controls the transcription of the csm genes and CRISPR RNA in M. tuberculosis (46). Deletion of the cnpB gene leads to massive transcriptional upregulation of the csm genes, which is consistent with a model whereby nanoRNAs generated by the Csm cyclase domain play a role in sculpting the transcriptional response to phage infection in cells with a type III CRISPR system by activating defence gene expression. Clearly, there are still many avenues to explore.
Finally, there is the requirement to re-set the cell to a ground-state when infection has been cleared, which probably requires degradation of the cOA generated during infection. Specialized ‘ring nucleases’ that degrade cA4 have been identified in archaea (47), and recent work has shown that Csm6 enzymes can also slowly degrade cA4 in their CARF domain, acting as self-limiting ribonucleases (48). There is no published evidence yet for cA6 degradation enzymes, but the most obvious candidates are the CARF domains of Csm6 family proteins. Alternatively, given the larger size and increased flexibility of cA6 compared to cA4, the former may be degraded effectively by promiscuous ribonucleases in the cell.
Supplementary Material
ACKNOWLEDGEMENTS
Thanks to Christophe Rouillon, Marialena Varympopioti and Chloe Jones for helpful discussions.
Notes
Present address: Tess Hoogeboom, ProQR Therapeutics N.V., Zernikedreef 9, 2333 CK Leiden, Netherlands.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Royal Society Challenge Grant [REF: CH160014 to M.F.W.]; Biotechnology and Biological Sciences Research Council [REF: BB/S000313/1 to M.F.W.]. Funding for open access charge: Institutional Block Grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wright A.V., Nunez J.K., Doudna J.A.. Biology and applications of CRISPR systems: harnessing nature's toolbox for genome engineering. Cell. 2016; 164:29–44. [DOI] [PubMed] [Google Scholar]
- 2. Koonin E.V., Makarova K.S.. Mobile genetic elements and evolution of CRISPR-cas systems: all the way there and back. Genome Biol. Evol. 2017; 9:2812–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J., Charpentier E., Haft D.H. et al.. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rouillon C., Zhou M., Zhang J., Politis A., Beilsten-Edmands V., Cannone G., Graham S., Robinson C.V., Spagnolo L., White M.F.. Structure of the CRISPR interference complex Csm reveals key similarities with Cascade. Mol. Cell. 2013; 52:124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elmore J.R., Sheppard N.F., Ramia N., Deighan T., Li H., Terns R.M., Terns M.P.. Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR-Cas system. Genes Dev. 2016; 30:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Estrella M.A., Kuo F.T., Bailey S.. RNA-activated DNA cleavage by the Type III-B CRISPR-Cas effector complex. Genes Dev. 2016; 30:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kazlauskiene M., Tamulaitis G., Kostiuk G., Venclovas C., Siksnys V.. Spatiotemporal control of type III-A CRISPR-Cas immunity: coupling DNA degradation with the target RNA recognition. Mol. Cell. 2016; 62:295–306. [DOI] [PubMed] [Google Scholar]
- 8. Jung T.Y., An Y., Park K.H., Lee M.H., Oh B.H., Woo E.. Crystal structure of the Csm1 subunit of the Csm complex and its single-stranded DNA-specific nuclease activity. Structure. 2015; 23:782–790. [DOI] [PubMed] [Google Scholar]
- 9. Han W., Li Y., Deng L., Feng M., Peng W., Hallstrom S., Zhang J., Peng N., Liang Y.X., White M.F. et al.. A type III-B CRISPR-Cas effector complex mediating massive target DNA destruction. Nucleic Acids Res. 2017; 45:1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson K., Learn B.A., Estrella M.A., Bailey S.. Target sequence requirements of a type III-B CRISPR-Cas immune system. J. Biol. Chem. 2019; 294:10290–10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kazlauskiene M., Kostiuk G., Venclovas C., Tamulaitis G., Siksnys V.. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science. 2017; 357:605–609. [DOI] [PubMed] [Google Scholar]
- 12. Niewoehner O., Garcia-Doval C., Rostol J.T., Berk C., Schwede F., Bigler L., Hall J., Marraffini L.A., Jinek M.. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature. 2017; 548:543–548. [DOI] [PubMed] [Google Scholar]
- 13. Rouillon C., Athukoralage J.S., Graham S., Grüschow S., White M.F.. Control of cyclic oligoadenylate synthesis in a type III CRISPR system. Elife. 2018; 7:e36734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Makarova K.S., Anantharaman V., Grishin N.V., Koonin E.V., Aravind L.. CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front. Genet. 2014; 5:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Niewoehner O., Jinek M.. Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein Csm6. RNA. 2016; 22:318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sheppard N.F., Glover C.V. 3rd, Terns R.M., Terns M.P.. The CRISPR-associated Csx1 protein of Pyrococcus furiosus is an adenosine-specific endoribonuclease. RNA. 2016; 22:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang W., Samai P., Marraffini L.A.. Degradation of phage transcripts by CRISPR-associated RNases enables type III CRISPR-Cas immunity. Cell. 2016; 164:710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng L., Garrett R.A., Shah S.A., Peng X., She Q.. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol. Microbiol. 2013; 87:1088–1099. [DOI] [PubMed] [Google Scholar]
- 19. Foster K., Kalter J., Woodside W., Terns R.M., Terns M.P.. The ribonuclease activity of Csm6 is required for anti-plasmid immunity by Type III-A CRISPR-Cas systems. RNA Biol. 2019; 16:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Freidlin P.J., Nissan I., Luria A., Goldblatt D., Schaffer L., Kaidar-Shwartz H., Chemtob D., Dveyrin Z., Head S.R., Rorman E.. Structure and variation of CRISPR and CRISPR-flanking regions in deleted-direct repeat region Mycobacterium tuberculosis complex strains. BMC Genomics. 2017; 18:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haft D.H., Selengut J., Mongodin E.F., Nelson K.E.. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005; 1:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brudey K., Driscoll J.R., Rigouts L., Prodinger W.M., Gori A., Al-Hajoj S.A., Allix C., Aristimuno L., Arora J., Baumanis V. et al.. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006; 6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wei W., Zhang S., Fleming J., Chen Y., Li Z., Fan S., Liu Y., Wang W., Wang T., Liu Y. et al.. Mycobacterium tuberculosis type III-A CRISPR/Cas system crRNA and its maturation have atypical features. FASEB J. 2019; 33:1496–1509. [DOI] [PubMed] [Google Scholar]
- 24. Liu H., Naismith J.H.. A simple and efficient expression and purification system using two newly constructed vectors. Protein Expr. Purif. 2009; 63:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cortes T., Schubert O.T., Rose G., Arnvig K.B., Comas I., Aebersold R., Young D.B.. Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep. 2013; 5:1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rollie C., Schneider S., Brinkmann A.S., Bolt E.L., White M.F.. Intrinsic sequence specificity of the Cas1 integrase directs new spacer acquisition. Elife. 2015; 4:e08716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rouillon C., Athukoralage J.S., Graham S., Grüschow S., White M.F.. Investigation of the cyclic oligoadenylate signalling pathway of type III CRISPR systems. Methods Enzymol. 2019; 616:191–218. [DOI] [PubMed] [Google Scholar]
- 28. Hochstrasser M.L., Doudna J.A.. Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem. Sci. 2015; 40:58–66. [DOI] [PubMed] [Google Scholar]
- 29. Staals R.H., Zhu Y., Taylor D.W., Kornfeld J.E., Sharma K., Barendregt A., Koehorst J.J., Vlot M., Neupane N., Varossieau K. et al.. RNA targeting by the type III-A CRISPR-Cas Csm complex of Thermus thermophilus. Mol. Cell. 2014; 56:518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tamulaitis G., Kazlauskiene M., Manakova E., Venclovas C., Nwokeoji A.O., Dickman M.J., Horvath P., Siksnys V.. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol. Cell. 2014; 56:506–517. [DOI] [PubMed] [Google Scholar]
- 31. Hale C.R., Cocozaki A., Li H., Terns R.M., Terns M.P.. Target RNA capture and cleavage by the Cmr type III-B CRISPR-Cas effector complex. Genes Dev. 2014; 28:2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang J., Graham S., Tello A., Liu H., White M.F.. Multiple nucleic acid cleavage modes in divergent type III CRISPR systems. Nucleic Acids Res. 2016; 44:1789–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hatoum-Aslan A., Maniv I., Samai P., Marraffini L.A.. Genetic characterization of antiplasmid immunity through a Type III-A CRISPR-Cas system. J. Bacteriol. 2014; 196:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Foster K., Kalter J., Woodside W., Terns R.M., Terns M.P.. The ribonuclease activity of Csm6 is required for anti-plasmid immunity by Type III-A CRISPR-Cas systems. RNA Biol. 2019; 16:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ichikawa H.T., Cooper J.C., Lo L., Potter J., Terns R.M., Terns M.P.. Programmable type III-A CRISPR-Cas DNA targeting modules. PLoS One. 2017; 12:e0176221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rostol J.T., Marraffini L.A.. Non-specific degradation of transcripts promotes plasmid clearance during type III-A CRISPR-Cas immunity. Nat. Microbiol. 2019; 4:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sorokin D.Y., Muntyan M.S., Panteleeva A.N., Muyzer G.. Thioalkalivibrio sulfidiphilus sp. nov., a haloalkaliphilic, sulfur-oxidizing gammaproteobacterium from alkaline habitats. Int. J. Syst. Evol. Microbiol. 2012; 62:1884–1889. [DOI] [PubMed] [Google Scholar]
- 38. Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A.. Co-transcriptional DNA and RNA cleavage during type III CRISPR-Cas immunity. Cell. 2015; 161:1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu T.Y., Iavarone A.T., Doudna J.A.. RNA and DNA targeting by a reconstituted Thermus thermophilus Type III-A CRISPR-Cas system. PLoS One. 2017; 12:e0170552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nasef M., Muffly M.C., Beckman A.B., Rowe S.J., Walker F.C., Hatoum-Aslan A., Dunkle J.A.. Regulation of cyclic oligoadenylate synthesis by the S. epidermidis Cas10-Csm complex. RNA. 2019; 25:948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Whiteley A.T., Eaglesham J.B., de Oliveira Mann C.C., Morehouse B.R., Lowey B., Nieminen E.A., Danilchanka O., King D.S., Lee A.S.Y., Mekalanos J.J. et al.. Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature. 2019; 567:194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McFarland A.P., Luo S., Ahmed-Qadri F., Zuck M., Thayer E.F., Goo Y.A., Hybiske K., Tong L., Woodward J.J.. Sensing of bacterial cyclic dinucleotides by the oxidoreductase RECON promotes NF-kappaB activation and shapes a proinflammatory antibacterial state. Immunity. 2017; 46:433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dey R.J., Dey B., Zheng Y., Cheung L.S., Zhou J., Sayre D., Kumar P., Guo H., Lamichhane G., Sintim H.O. et al.. Inhibition of innate immune cytosolic surveillance by an M. tuberculosis phosphodiesterase. Nat. Chem. Biol. 2017; 13:210–217. [DOI] [PubMed] [Google Scholar]
- 44. Johnson R.M., McDonough K.A.. Cyclic nucleotide signaling in Mycobacterium tuberculosis: an expanding repertoire. Pathog. Dis. 2018; 76:doi:10.1093/femspd/fty048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goldman S.R., Sharp J.S., Vvedenskaya I.O., Livny J., Dove S.L., Nickels B.E.. NanoRNAs prime transcription initiation in vivo. Mol. Cell. 2011; 42:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Y., Yang J., Bai G.C.. Regulation of the CRISPR-associated genes by Rv2837c (CnpB) via an orn-like activity in tuberculosis complex mycobacteria. J. Bacteriol. 2018; 200:doi:10.1128/JB.00743-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Athukoralage J.S., Rouillon C., Graham S., Grüschow S., White M.F.. Ring nucleases deactivate Type III CRISPR ribonucleases by degrading cyclic oligoadenylate. Nature. 2018; 562:277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Athukoralage J.S., Graham S., Grüschow S., Rouillon C., White M.F.. A type III CRISPR ancillary ribonuclease degrades its cyclic oligoadenylate activator. J. Mol. Biol. 2019; 431:2894–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Han W., Stella S., Zhang Y., Guo T., Sulek K., Peng-Lundgren L., Montoya G., She Q.. A Type III-B Cmr effector complex catalyzes the synthesis of cyclic oligoadenylate second messengers by cooperative substrate binding. Nucleic Acids Res. 2018; 46:10319–10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim Y.K., Kim Y.G., Oh B.H.. Crystal structure and nucleic acid-binding activity of the CRISPR-associated protein Csx1 of Pyrococcus furiosus. Proteins. 2013; 81:261–270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.