


Abstract
Cellular response to oxidative stress is a crucial mechanism that promotes the survival of Pseudomonas aeruginosa during infection. However, the translational regulation of oxidative stress response remains largely unknown. Here, we reveal a tRNA modification-mediated translational response to H2O2 in P. aeruginosa. We demonstrated that the P. aeruginosa trmB gene encodes a tRNA guanine (46)-N7-methyltransferase that catalyzes the formation of m7G46 in the tRNA variable loop. Twenty-three tRNA substrates of TrmB with a guanosine residue at position 46 were identified, including 11 novel tRNA substrates. We showed that loss of trmB had a strong negative effect on the translation of Phe- and Asp-enriched mRNAs. The trmB-mediated m7G modification modulated the expression of the catalase genes katA and katB, which are enriched with Phe/Asp codons at the translational level. In response to H2O2 exposure, the level of m7G modification increased, consistent with the increased translation efficiency of Phe- and Asp-enriched mRNAs. Inactivation of trmB led to decreased KatA and KatB protein abundance and decreased catalase activity, resulting in H2O2-sensitive phenotype. Taken together, our observations reveal a novel role of m7G46 tRNA modification in oxidative stress response through translational regulation of Phe- and Asp-enriched genes, such as katA and katB.
INTRODUCTION
Posttranscriptional modification of tRNA is part of the tRNA maturation process during which nascent tRNAs are converted into functional, mature tRNA molecules. To date, >100 species-specific base modifications have been identified in mature tRNAs, which play a large variety of functions in different biological processes and cellular stress responses. Modifications in the anticodon region, especially at the wobble position (position 34) and position 37, commonly play a major role in ensuring efficient and accurate translation, whereas other modifications found along the tRNA body largely contribute to tRNA structure and stability (1–8).
7-Methylguanosine (m7G) tRNA modification is widely found in all three domains of life, and it occurs at position 46 in the tRNA variable loop, except in three rare cases: in the mitochondrial tRNA of squid (9) and starfish (10) and in chloroplast tRNA (11), wherein m7G modification takes place at the guanosine positions 34 and 36 in the anticodon loop. m7G modification is catalyzed by the tRNA guanine N7-methyltransferase, a member of the S-adenosyl methionine (SAM)-dependent RNA methyltransferase enzyme family (12–16). This modification is not essential for viability (12,16). However, it plays an important role in tRNA tertiary structure maintenance, tRNA thermal stability, and cell survival at high temperatures (15,17–19).
m7G tRNA modification is reported to strengthen tertiary interactions among the base triplet C13-G22-m7G46 in the elbow region of tRNAPhe, thereby maintaining the L-shaped structure of tRNA (17). In yeast, loss of m7G46 and 5-methylcytidine (m5C) modifications causes tRNAVal/AAC degradation via the rapid tRNA degradation (RTD) pathway, resulting in a temperature-sensitive phenotype (18,19). Furthermore, degradation of specific tRNAs as well as severe growth defects have been observed at high temperatures in an m7G-deficient Thermus thermophilus strain (15). At elevated temperatures, hypo-m7G46 modification in T. thermophilus additionally leads to depleted levels of 2′-O-methylguanosine (Gm18) and 1-methylguanosine (m1G37) modifications, resulting in specific degradation of tRNAPhe/GAA and tRNAIle/GAT (15). Thus, in combination with other modifications, m7G46 contributes to tRNA thermal stability and cellular survival at high temperatures.
In humans, m7G46 is involved in the pathogenicity of primordial dwarfism (PD). Significant reductions in m7G levels in tRNAPhe/GAA and tRNAVal/AAC are observed in PD patients carrying a missense mutation in the gene encoding Wdr4, a component of the human tRNA guanine N7-methyltransferase Wdr4/Mettl1 complex (20). It has recently been found that Wdr4/Mettl1-mediated m7G modification plays an essential role in self-renewal and differentiation of embryonic stem cells (ESCs) in mice (21). Mettl1 deficiency causes inefficient translation of the codons decoded by m7G-modified tRNAs, decreased mRNA levels of genes associated with cell differentiation and brain development, and impaired ESC colony formation (21).
To date, growing evidence demonstrates the functions of tRNA modifications in response to stresses, especially the modifications in the wobble positions (5,22,23). In our preliminary screening, we found a possible link between tRNA-modifying enzymes targeting non-wobble position and H2O2 resistance in Pseudomonas aeruginosa, a human pathogenic bacterium. To investigate the role of non-wobble tRNA modification in oxidative stress response, we characterize a P. aeruginosa tRNA-modifying enzyme, PaTrmB, which transfers methyl group to the guanosine residue, forming m7G at position 46 in the variable loop of tRNA. Moreover, we describe its novel role in the translational control of critical stress response proteins required for the H2O2 resistance of P. aeruginosa. Our data demonstrate the impact of m7G46 modification on the efficient translation of Phe- and Asp-enriched mRNAs. Translation of Phe- and Asp-enriched catalase genes, such as katA and katB, is positively controlled by TrmB in response to H2O2. Inactivation of trmB results in depleted catalase levels and in strong H2O2-sensitive phenotype, demonstrating the essential role of tRNA m7G modification in oxidative stress response.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and plasmids
The strains and plasmids used in this study are listed in Supplementary Table S1. P. aeruginosa strain UCBPP-PA14 (PA14) and Escherichia coli strains BW20767, DH5α and DE3 (BL21) were grown on lysogeny broth (LB) agar plate at 37°C and in LB medium at 37°C with shaking. When needed, antibiotics were added as appropriate at the following concentrations: for P. aeruginosa, carbenicillin (Cb) at 200 μg/ml and gentamicin (Gm) at 75 μg/ml; for E. coli, gentamicin (Gm) at 15 μg/ml, ampicillin (Ap) at 100 μg/ml, and chloramphenicol (Cm) at 15 μg/ml. All primers and synthesized oligonucleotides used in cloning are listed in Supplementary Table S2.
Construction of trmB mutant strain
PA14 trmB or PA14_05000 was disrupted through the insertional knockout technique by using the suicide plasmid pKNOCK-Gm (24). A 215 bp fragment located within trmB was amplified by PCR using Taq DNA polymerase and the primer pair BT4381/BT4382. The amplified fragment was cloned into pKNOCK-Gm plasmid digested with SmaI according to a standard restriction enzyme digestion and ligation protocol. A recombinant plasmid, pKN-trmB, was transferred into the recipient P. aeruginosa PA14 through biparental mating, with E. coli BW20767 as the donor strain. At the end of the experiment, the trmB mutant was verified by colony PCR and Southern blot analysis.
Construction of trmB complemented strain
The full length sequence of trmB was amplified with the primers BT4767, BT4768 and Pfu polymerase by PCR followed by cloning into the SmaI site of the broad-host-range vector pBBR1MCS-4 (25), producing a pBB-trmB-FL plasmid. A verified pBB-trmB-FL plasmid was electroporated into the trmB mutant strain, producing trmB complemented strain, which was selected by plating on LB agar plate that contains 200 μg/ml Cb and 75 μg/ml Gm.
Expression and purification of PaTrmB recombinant protein
The full length of PA14 trmB was PCR-amplified with Pfu DNA polymerase, with forward primer BT5330 containing NcoI site, and with reverse primer BT5331 containing XhoI site. A NcoI-XhoI fragment containing full-length trmB was subsequently cloned into the expression vector pETBLUE-2 (Novagen) at the NcoI–XhoI site. For protein production, the expression host E. coli DE3 (BL21) harboring a verified pET-trmB-FL plasmid was grown to the exponential phase and induced with 200 μM isopropyl-β-d-thiogalactopyranoside at 37°C for 2–4 h. 6XHis-tagged proteins were eluted with a gradient of 200–500 mM imidazole and analyzed by SDS-PAGE. The final yield of recombinant PaTrmB protein was measured by Bradford assay (26). The purity of the PaTrmB was estimated to be >99% on a Coomassie blue stained SDS-polyacrylamide gel (Supplementary Figure S1). Aliquots of the purified recombinant PaTrmB protein were stored at −20°C before use.
Extraction and purification of in vivo total tRNA
Cells were harvested and extracted using TRIzol® Reagent (Thermo Fisher Scientific). Large RNA species were precipitated in a cold solution of 35% absolute ethanol. Pellets of these large RNA species were separated from small RNA species by centrifugation at 12 000 × g for 5 min at 4°C. The aqueous phase containing the small RNA species was subsequently transferred into a new tube, precipitated using 2 volumes of absolute ethanol at −70°C for 2 h, and resuspended in 50 μl of RNase-free water. The total tRNA was isolated by HPLC as previously described (27).
In vitro preparation of tRNA
Thirty-eight tRNA types were synthesized in vitro using a MEGAshortscriptTM Kit (Ambion). Double-stranded DNA templates for the MEGAshortscriptTM Kit were prepared from synthesized fragments containing the T7 promoter located upstream of the candidate target tRNA (Supplementary Table S2). Each synthesized fragment was amplified by PCR using Phusion DNA polymerase, T7 forward primer, and a specific reverse primer listed in Supplementary Table S2. The reactions were assembled and incubated at 37°C for 4 h. The tRNA transcripts were purified from the reactions by HPLC (27).
Analysis of the modified ribonucleosides by LC-QQQ
Five micrograms of total tRNA was treated with Benzonase nuclease (Sigma), bacterial alkaline phosphatase (Invitrogen), and phosphodiesterase in the presence of deaminase inhibitors (0.5 μg/ml coformycin and 5 μg/ml tetrahydrouridine) and antioxidants (50 μM desferrioxamine and 50 μM butylatedhydroxytoluene) at 37°C overnight to generate ribonucleoside products. Proteins were removed from the digested product by filtration with 10K MWCO columns (Ambion). The ribonucleosides were fractionated by using a reversed-phase column (Thermo Hypersil Gold aQ column) with a gradient made of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile) prior to introduction into an electrospray ionization triple quadrupole mass spectrometer (Agilent 6470), which was operated in the positive ion mode as described previously (27,28). The modified ribonucleosides were identified by their HPLC retention times, CID fragmentation patterns, fragmentor voltages, and collision energies in comparison with available chemical synthetic standards. The level of each modified ribonucleoside was quantified from the MRM signal intensity and normalized by dividing by the summed signals of adenosine, guanosine, cytidine and uridine from the sample.
tRNA methyltransferase assays
In vitro methyltransferase assays (MTase assay) were performed using the MTase-Glo™ Bioluminescent Assay Kit (Promega) to assess methyltransferase activity and to identify the tRNA substrates of PaTrmB. The 20 μl MTase reaction mixtures consisting of 3 μg tRNA, 5 μM recombinant PaTrmB protein, 50 mM Tris–HCl (pH 8.0), 5 mM MgCl2, 50 mM KCl, 50 μM SAM and 1x MTase-Glo™ reagent. For detection, 1 volume of MTase-Glo™ detection solution was added. The light signal corresponding to the amount of S-adenosyl homocysteine (SAH) produced by the PaTrmB methyltransferase activity was subsequently measured using a microplate luminometer (Thermo Fisher Scientific). The reactions without PaTrmB were used as negative control.
Localization of the modified ribonucleosides by LC-QTOF
The 80 μl MTase reaction mixtures consisting of 10 μg T7 in vitro tRNA transcript, 5 μM recombinant PaTrmB protein, 50 mM Tris–HCl (pH 8.0), 5 mM MgCl2, 50 mM KCl and 50 μM SAM (Sigma) were assembled and incubated at 37°C for 1 h. The m7G-modified tRNA was subsequently purified from the MTase reaction mixtures by filtration using 10K MWCO columns (Ambion). Five micrograms of methylated tRNA was digested into fragments with RNase T1 or RNase U (Invitrogen) and then dephosphorylated with bacterial alkaline phosphatase (Invitrogen) at 37°C for 4 h in the presence of deaminase inhibitors and antioxidants. To purify the digested fragments from the solution, 100 μl of the digestion reaction was applied to Strata™ solid-phase extraction columns (Phenomenex). The purified fragments were then fractionated on TSK-gel Amide-80 column (2.0 mm ID × 150 mm, 3 μm particle size) with a gradient made of solvent A (8 mM ammonium acetate) and solvent B (100% acetonitrile) before sequencing using a quadrupole time-of-flight mass spectrometer (Agilent 6520) operated in negative ion mode as previously described (27).
Viability analysis to assess H2O2 sensitivity
Exponential-phase cultures were diluted in LB medium to a final OD600 of 0.1. H2O2 was added to the treated cultures, and cultures without H2O2 exposure served as the untreated control. After H2O2 addition, the cultures were incubated at room temperature without shaking for 25 min. Serial dilutions of the treated and untreated cultures were prepared, and 10 μl aliquots were dropped onto LB agar plates without antibiotic. The cells were grown on the plates at 37°C, and the sensitivity of the treated cultures to H2O2 was observed within 24 h.
Codon reporter assay
The effects of m7G46 modification on the translation of transcripts enriched with codons decoded by the tRNA substrates of PaTrmB were assayed in vivo using β-galactosidase reporter plasmids (29,30). The experimental reporters containing codon bias fragment were used to examine the translation of the codons of interest, whereas the control reporter without codon bias was used to examine the general translation and to normalize the translation background. In constructing the experimental and control reporters, DNA fragments with codon bias or a control fragment without codon bias (Supplementary Table S2) was cloned in-frame with the lacZ gene in the pPR9TT translational fusion plasmid (31) under the control of ampicillin resistance promoter (PApR). Recombinant plasmids (Supplementary Table S1) were introduced into the wild type and trmB mutant cells. The exponential-phase cultures of the transformants were treated with 10 mM H2O2 for 25 min. β-Galactosidase activity was measured using the Miller's method (32), and the activity of empty pPR9TT plasmid was subtracted from the β-galactosidase activity of the experimental and control reporters before calculating the translation efficiency. Translational efficiency was calculated as the ratio of the β-galactosidase activities (Miller unit) of the experimental reporters to the activity of control reporter without codon bias.
Total catalase activity assay
Cell pellets were harvested by centrifugation at 3400 × g for 10 min at 4°C and were washed three times with cold 50 mM phosphate buffer. Crude protein was extracted by sonication. Protein concentration was measured by Bradford assay. Specific catalase activity (U/mg protein) was measured by a spectrophotometric method (A420, light path = 1 cm) using 10 mM H2O2 in 50 mM phosphate buffer (pH 7.0) as substrate. U was defined as one unit of catalase decomposing 1.0 μmol of H2O2 per minute at pH 7.0 at 25°C.
Catalase gel activity assay
Thirty micrograms of total protein was separated on an 8% native polyacrylamide gel. Gel staining for catalase activity was performed using the di-amino benzidine (DAB) staining method (33). The gel was soaked in renaturation buffer [40 mM Tris–HCl (pH 8.0), 2 mM MgCl2 and 0.1% v/v Triton-X 100] at 4°C overnight and then washed twice with 50 mM phosphate buffer (pH 6.0) before soaking in 50 mM phosphate buffer (pH 6.0) containing 50 μg/μl horseradish peroxidase (Sigma, USA) for 45 min at room temperature. The gel was subsequently incubated in 50 mM phosphate buffer (pH 6.0) containing 10 mM H2O2 for 10 min, washed with distilled water, and then soaked in 50 mM potassium phosphate buffer (pH 6.0) containing 500 μg/μl DAB to develop a brown background. Colorless bands indicate the area where H2O2 is decomposed by catalase.
Western blot analysis
Total protein was extracted from cells by sonication. Fifty micrograms of total protein was separated using 12.5% SDS-PAGE electrophoresis and then transferred onto polyvinylidene difluoride (PVDF) membranes (GE Healthcare). The membranes were blocked in TBST buffer [20 mM Tris–HCl (pH 7.5), 150 mM NaCl and 0.05% Tween 20] containing 5% skim milk on an orbital shaker for 1 h at room temperature followed by incubation in an antibody solution (HRP-conjugated antibody diluted in 3% skim milk and 1% BSA in TBST) for 1 h. The membranes were washed three times for 30 min with TBST buffer. The target protein was detected using a chemiluminescent (ECL) detection reagent kit (GE Healthcare) according to the manufacturer's instructions.
For western blot analysis of KatB, a his-tag approach was used given that anti-KatB antibody was unavailable in our laboratory. The full length katB gene was amplified with the BT5515/BT5755 primer pair by PCR and cloned in-frame with the C-terminus hexahistidine tag in the SmaI–XhoI site of the pETBlue-2 plasmid (Novagen), producing the recombinant plasmid pET-katB-6XHis. The katB-6XHis fragment was further amplified from the pET-katB-6XHis plasmid using the pETUP/pETDOWN primer pair and subcloned into the EcoRV site of the pBBR1MCS-4 vector for gene expression in PA14. The resulting plasmid, pBB-katB-6XHis, was verified by sequencing before introduction into the wild type and trmB mutant strains for Western blot analysis.
Semiquantitative real-time PCR
Total RNA was extracted according to a hot acid phenol extraction protocol (34). The residual DNA in the sample was eliminated by DNase treatment (Thermo Fisher Scientific). For cDNA synthesis, a reverse transcription reaction was performed using random hexa-primers and a RevertAidTM M-MuLV Reverse Transcriptase Kit (Thermo Fisher Scientific). Semiquantitative real-time PCR was performed using KAPA SYBR FAST reagent (KAPA Biosystems) and a StepOnePlusTM real-time PCR system (Thermo Fisher Scientific) according to the manufacturers’ instructions. The specific primers for trmB, oxyR, katA, katB, katE, ahpB, ahpC, ankB and recG are listed in Supplementary Table S2. 16S ribosomal RNA gene was used as internal control to normalize the gene expression.
Statistical analysis
Statistical analysis was performed using Graphpad Prism (Graphpad Software). t-test was used to determine the statistical significance. The levels of statistical significance were indicated in each experiment.
RESULTS
P. aeruginosa trmB gene encodes a tRNA guanine-N7-methyltransferase that catalyzes m7G formation in tRNA
PaTrmB (PA14_05000) displays high identity (60%) and similarity (73%) scores with the experimentally identified E. coli TrmB, which is a tRNA guanine-N7-methyltransferase (12). Analysis of the PaTrmB sequence revealed the presence of GXGXG, a SAM-binding site (35) at residue 71–75; GXGXG belongs to the Rossmann-fold SAM-dependent methyltransferase superfamily (Figure 1). To test whether PaTrmB functions as a tRNA guanine N7-methyltransferase, PaTrmB was purified for in vitro assays.
Figure 1.

PaTrmB shows high identity and similarity scores with Escherichia coli TrmB. A sequence alignment of PaTrmB and E. coli K12 TrmB was performed using ENDscript 2.0 (ENDscript 2.0; http://endscript.ibcp.fr/ESPript/ENDscript/). PaTrmB shows identity and similarity scores of 60% (126/209) and 73% (154/209), respectively, with the E. coli TrmB. Black box, identity; white box, similarity in a group.
The methyltransferase activity of trmB was assessed in vitro using an MTase-GloTM Methyltransferase Assay Kit (Promega). Recombinant PaTrmB protein was incubated with the in vivo tRNA extracted from the wild type, trmB mutant, or complemented strains in the presence of SAM as methyl donor. As shown in Figure 2A, the amount of the reaction product S-adenosyl homocysteine (SAH) represents the degree of methyltransferase activity. We found that the recombinant PaTrmB protein effectively functioned in vitro in the presence of the hypo-methylated tRNA substrate and of the methyl donor SAM. After 1 h of incubation at 37°C, the total tRNA extracted from the trmB mutant strain produced the highest amount of SAH (3785±474 nM), which is significantly higher (P ≤ 0.001) than that obtained when the tRNA from the wild type or complemented strains was used as substrate (219±6 and 2441±82 nM, respectively). Although we expected that the reaction containing the tRNA from the wild type and complemented strains would share similar SAH levels, the amount of SAH produced in the reaction containing the tRNA from the complemented strain was higher, indicating partial methylation when trmB is expressed from a multiple-copy plasmid (pBB-trmB-FL) after reintroduction into the trmB mutant strain.
Figure 2.
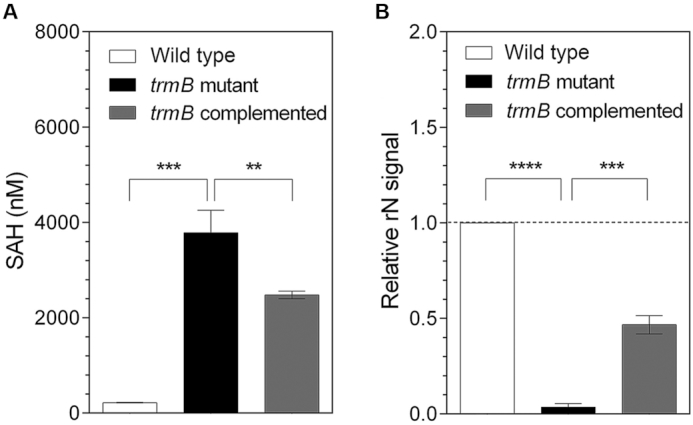
PaTrmB functions as tRNA methyltransferase in vitro and is responsible for m7G tRNA formation in vivo. (A) The recombinant PaTrmB protein effectively methylates the tRNA isolated from the trmB mutant strain. The amount of SAH produced by the PaTrmB methyltransferase activity was measured using an MTase-Glo™ Methyltransferase Assay Kit (Promega). The reactions without PaTrmB served as negative control. (B) Inactivation of trmB leads to specific m7G hypo-modification. The relative levels of m7G modification (as compared with that in the wild type strain) in the total tRNA extracted from the wild type, trmB mutant, or trmB complemented strains were assessed using LC-QQQ and were calculated based on the normalized signal intensity of its own sample. Data represent the mean ± SD for three biological replicates. Asterisks denote a significant difference by t-test (**P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
Next, to determine whether trmB is responsible for the m7G tRNA modification, the individual-modified ribonucleosides from the in vivo total tRNA of the wild type, trmB mutant, or complemented strains were identified and quantified using an LC-QQQ with 24 modified nucleoside standards; 20 modifications were identified in the P. aeruginosa total tRNA. Among the modifications, only the m7G modification was significantly reduced (P ≤ 0.001) in the total tRNA isolated from the trmB mutant strain compared with the modification in the tRNA isolated from the wild type and complemented strains (Figure 2B and Supplementary Table S3). In good agreement with the partial methylation observed in the in vitro MTase assay (Figure 2A), the abundance of m7G in the tRNA extracted from trmB complemented strain was ∼50% lower than the abundance of m7G extracted from the wild type strain (Figure 2B).
P. aeruginosa TrmB catalyzes m7G46 formation in the tRNA variable loop
To identify the tRNA substrates of PaTrmB, all 38 tRNA species in P. aeruginosa (http://gtrnadb2009.ucsc.edu) were in vitro synthesized using a MEGAshortscript™ Kit (Ambion). The in vitro synthetized tRNAs were subsequently in vitro methylated by recombinant PaTrmB. In P. aeruginosa, 26 tRNAs contain G at position 46 (G46) (Figure 3, black bar). We found that 23 of the 26 G46-containing tRNAs gave higher SAH signals compared with the G46-lacking tRNAs (Figure 3, white bar), namely, tRNAAla/GGC, tRNAAla/UGC, tRNAArg/ACG, tRNAArg/CCG, tRNAArg/CCU, tRNAArg/UCU, tRNAAsn/GUU, tRNAAsp/GUC, tRNAGly/GCC, tRNAHis/GUG, tRNAIle/GAU, tRNALys/UUU, tRNAMet/CAU, tRNAPhe/GAA, tRNAPro/CGG, tRNAPro/GGG, tRNAPro/UGG, tRNAThr/CGU, tRNAThr/GGU, tRNAThr/UGU, tRNATrp/CCA, tRNAVal/GAC and tRNAVal/UAC. However, there was little, if any, SAH signals when three other G46-containing tRNAs (tRNALeu/CAG, tRNALeu/GAG, and tRNASec(p)/UCA) were used as substrate, suggesting that these tRNAs might not be substrates of PaTrmB (Figure 3). As expected, all tRNAs lacking G46 yielded considerably low amount of SAH (Figure 3). Thus, we hypothesized that PaTrmB transfers a methyl group strictly to the G residue at position 46.
Figure 3.

Identification of PaTrmB tRNA substrates. In vitro transcribed tRNAs were used as substrates for the purified PaTrmB, with SAM as methyl donor. Methylation reaction was assessed using an MTase-Glo™ Methyltransferase Assay Kit (Promega) according to the manufacturer's instructions. The reaction without PaTrmB served as control. The tRNA transcripts containing G46 are shown as black bars, whereas those lacking G46 are shown as white bars. Data represent the mean ± SD for three biological replicates.
To confirm whether PaTrmB catalyzes m7G modification at position 46, LC-QTOF mass spectrometric analysis was performed. Six PaTrmB substrates, the synthetic T7 transcription products of tRNAAsn/GUU, tRNAAsp/GUC, tRNAGly/GCC, tRNAPhe/GAA, tRNAThr/GGU and tRNAVal/GAC, were individually incubated with recombinant PaTrmB, with SAM as methyl donor. Following in vitro methylation, each methylated tRNA was digested into fragments with RNase T1 or RNase U, dephosphorylated, and subjected to mass spectrometric analysis. Analysis of the methylated tRNA treated with RNase revealed that the m7G-modified base was located at position 46 in the tRNA variable loop (Figure 4). After RNase T1 digestion, the sequence of the tRNA fragment containing G46 is GUCG, covering the base positions 46–49 for tRNAAsp/GUC, and UGUCG, covering the base positions 45–49 for tRNAPhe/GAA. The extracted ion chromatogram of the RNase T1-digested fragments from tRNAAsp/GUC and tRNAPhe/GAA are shown in Figure 4A and D, respectively. Ion chromatograms of m7GUCG (m/z of 625.6008) from tRNAAsp/GUC and of Um7GUCG (m/z of 778.6142) from tRNAPhe/GAA were detected at 21.9 and 21.7 min, respectively. Mass spectra indicating a doubly charged negative ion (M – 2H)−2 of the m7GUCG and Um7GUCG fragments are shown in Figure 4B and E, respectively. The w1, w2, y1 and y3 ion products of m/z 625.6008 and the w1, w3 and a2-B2 ion products of m/z 778.6142 were consistent with the m7GUCG and Um7GUCG sequences, respectively, indicating the presence of m7G at position 46 (Figure 4C and F). Furthermore, m7G formation at position 46 by PaTrmB in tRNAAsn/GUU, tRNAGly/GCC, tRNAThr/GGU and tRNAVal/GAC was also confirmed (Supplementary Figures S2 and S3).
Figure 4.
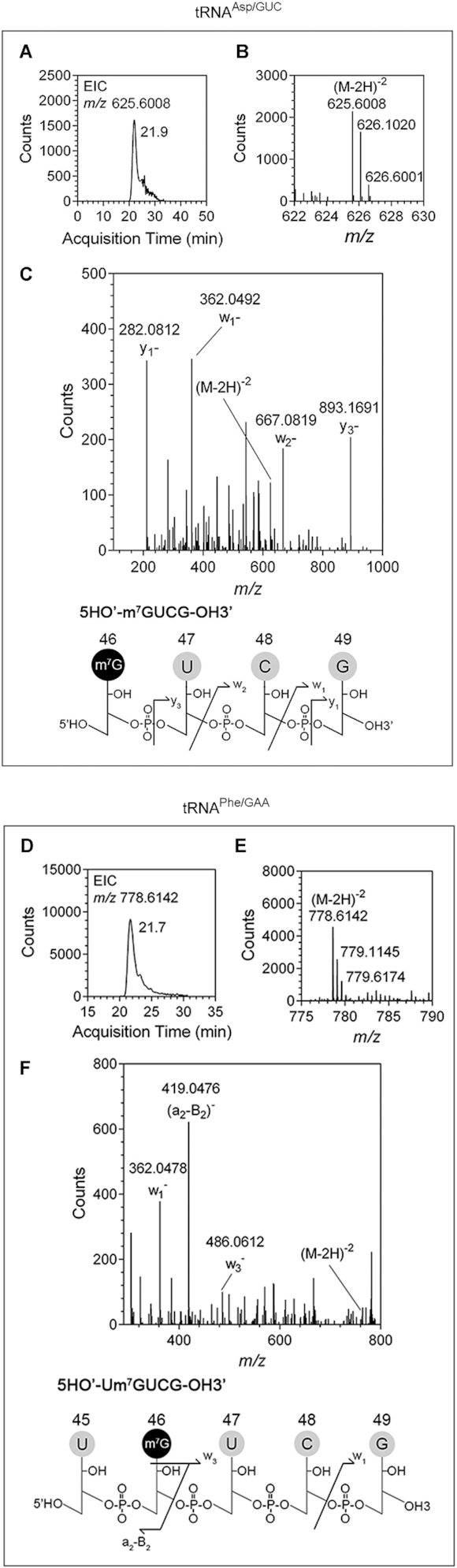
PaTrmB catalyzes m7G formation at position 46 in the variable loops of tRNAAsp/GUC and tRNAPhe/GAAin vitro. (A) Extracted ion chromatogram of the m7GUCG fragment with an m/z of 625.6008; this fragment is an RNase T1-digestion product of tRNAAsp/GUC. (B) The mass spectrum of the m7GUCG fragment shows a doubly charged negative ion with an m/z of 625.6008. (C) CID spectrum and CID fragmentation pattern of m/z 625.6008 with w1, w2, y1 and y3 ion products. (D) Extracted ion chromatogram of the Um7GUCG fragment with an m/z of 778.6142; this fragment is an RNase T1-digestion product of tRNAPhe/GAA. (E) The mass spectrum of the Um7GUCG fragment shows a doubly charged negative ion with an m/z of 778.6142. (F) CID spectrum and CID fragmentation pattern of m/z 778.6142 with w1, w3 and a2-B2 ion products.
Taken together, these results clearly demonstrated that P. aeruginosa trmB encodes a tRNA guanine (46)-N7-methyltransferase that catalyzes the transfer reaction of a methyl group from SAM to the N7 atom of guanine at position 46 in the tRNA variable loop.
Lack of m7G46 modification leads to decreased translation efficiency of transcripts enriched with codons decoded by tRNAPhe/GAA and tRNAAsp/GUC
To investigate the role of trmB in protein synthesis, we examined in vivo the impact of m7G46 tRNA modification on the translation of transcripts enriched with codons decoded by PaTrmB tRNA substrates, including the Ala-GCC, Arg-CGU, Asn-AAC, Asp-GAC, Asp-GAU, Ile-AUC, Phe-UUC, Phe-UUU and Val-GUC codons, by using a codon reporter assay. We cloned a control fragment without codon bias as well as experimental fragments containing 5X codon runs decoded by PaTrmB tRNA substrates and a negative control fragment carrying a repeat of the Gln-CAA codon into the pPR9TT vector under the control of the ampicillin resistance promoter and in frame with the lacZ gene (Figure 5A). The abilities of the wild type and trmB mutant strains to translate the engineered fragments were expressed as the translation efficiency, which was calculated by taking the ratio of the β-galactosidase activity of the experimental reporter to the activity of the control reporter without codon bias.
Figure 5.

TrmB affects the translation of codons decoded by tRNAPhe/GAA and tRNAAsp/GUC. (A) Design of the reporter system. Each synthesized fragment was cloned into the pPR9TT-PApR vector in frame with lacZ under the control of the PApR constitutive promoter. Relative translation efficiencies of (B) 5XGln-CAA reporter (negative control), (C) 5XPhe-UUC reporter, (D) 5XPhe-UUU reporter, (E) 5XAsp-GAC reporter and (F) 5XAsp-GAU reporter in the wild type and trmB mutant strains after H2O2 exposure. Data represent the mean ± SD for four biological replicates. Asterisks denote a significant difference by t-test (**P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). Q, glutamine; F phenylalanine; D, aspartic acid.
Comparison of translation efficiency between the wild type and trmB mutant strains showed that loss of trmB specifically affected the translation of Phe and Asp codons decoded by the m7G-modified tRNAPhe/GAA and the m7G-modified tRNAAsp/GUC, respectively, but it exerted no effect on the translation of Gln-CAA (the negative control codon) (Figure 5B). Despite the fact that the Ala-GCC, Arg-CGU, Asn-AAC, Asp-GAC, Asp-GAU, Ile-AUC, Phe-UUC, Phe-UUU and Val-GUC codons are all recognized by the PaTrmB tRNA substrates, we did not observe any differences in translation efficiency between the wild type and trmB mutant strains for the 5XAla-GCC, 5XArg-CGU, 5XAsn-AAC, 5XIle-AUC and 5XVal-GUC reporters (Supplementary Figure S4). By contrast, the translation efficiency of the 5XPhe-UUC, 5XPhe-UUU, 5XAsp-GAC and 5XAsp-GAU reporters significantly decreased (P ≤ 0.01) in the trmB mutant strain under both normal and H2O2 stress conditions (Figure 5C–F). Furthermore, we found that the translation efficiency of these four reporters significantly increased (P ≤ 0.01) in the wild type strain after treatment with 10 mM H2O2 (Figure 5C–F), suggesting a relationship between trmB-mediated Phe and Asp codon translation and oxidative stress response.
trmB plays an important role in the H2O2 stress response in P. aeruginosa
We examined the survival of the wild type, trmB mutant, and complemented strains after H2O2 exposure. Exponential-phase cultures were exposed to 1, 5, 10 or 20 mM H2O2 for 25 min at 37°C. Untreated cultures were used as control (which showed 100% survival). The trmB mutant strain, which lacks tRNA m7G46 modification, displayed a hypersensitive phenotype to H2O2. The survival levels of trmB mutant cells exposed to 10 and 20 mM H2O2 dropped by more than 50% and 90%, respectively, relative to that of the wild type strain. Importantly, the % survival of the trmB complemented strain was recovered during H2O2 stress, indicating that the H2O2-sensitive phenotype is trmB-dependent (Figure 6A). To investigate the pattern of m7G tRNA modification after H2O2 exposure in the wild type PA14, the abundance of m7G modification was monitored by using LC-MS/MS. We found a slight but significant (P ≤ 0.0001) increase in the level of m7G tRNA modification in the total tRNA after H2O2 exposure. m7G modification in total tRNA was induced by approximately 12% and 23% after exposure to 1 and 10 mM H2O2, respectively (Figure 6B). We further measured the mRNA level of trmB and found a 2.6-fold increase when the wild type cells were exposed to H2O2 (Figure 6C), suggesting that the increased m7G level is due to the upregulation of trmB. Taken together, these results demonstrated an important role of trmB in the H2O2 stress response in P. aeruginosa.
Figure 6.

trmB plays an important role in H2O2 stress response. (A) Survival of the wild type, trmB mutant, and complemented strains after H2O2 treatment. Cultures without H2O2 treatment served as the untreated control (100% survival). (B) Induction percentages of m7G tRNA modification after exposure to 1 or 10 mM H2O2 in wild type cells compared with those in untreated cells. (C) Relative expression of trmB in wild type cells after H2O2 exposure. Data represent the mean ± SD for three biological replicates for (A) and (C) and the mean ± SD for six biological replicates for (B). Asterisks denote a significant difference by t-test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
KatA and KatB protein levels are reduced in the trmB mutant strain
To obtain insight into the mechanism as to how TrmB mediates the H2O2 stress response and to identify the TrmB-regulated genes, we first assessed the impact of loss of trmB on the activity of the catalase enzymes given that catalase is a major H2O2-detoxifying enzyme (36,37). The results showed that inactivation of trmB led to reduced catalase activity. As shown in Figure 7A, the total catalase activity of the trmB mutant strain was significantly lower (P ≤ 0.01) than that of the wild type and complemented strains. In contrast to that of the wild type strain, whose total catalase activity increased by two-fold in response to H2O2 treatment, the total catalase activity of the trmB mutant strain did not change under any of the conditions regardless of whether H2O2 was added or not. Complementation of trmB mutation fully recovered the ability of the trmB-deficient mutant to increase its total catalase activity back to the H2O2-induced level of the wild type strain; these results demonstrated that the change in the total catalase activity in response to H2O2 exposure is trmB-dependent. In P. aeruginosa, three catalases are found, namely, KatA, KatB and KatE; KatA and KatB play important roles during logarithmic growth (37,38). We further investigated the activity of KatA, the major catalase during the logarithmic growth, using catalase gel staining assay. Consistent with the total catalase activity, KatA activity decreased in the trmB mutant strain compared with that in the wild type strain under both normal and H2O2 treatment conditions (Figure 7B). However, the activity of KatB in PA14 was dramatically low and could not be detected in the catalase gel even after H2O2 treatment or even when the amount of the total protein loaded was increased to 100 μg (data not shown).
Figure 7.

trmB exerts effects on catalases. (A) Total catalase activities of the wild type, trmB mutant, and complemented strains upon exposure to H2O2. (B) Comparison of KatA activities of the wild type and trmB mutant strains. KatA activity was examined by a catalase gel activity assay. The colorless band indicates the area where H2O2 was decomposed; M indicates the protein molecular weight markers. (C) Western blot analysis of OxyR, KatA and KatB proteins. OxyR and KatA levels were assessed using anti-OxyR and anti-KatA antibodies, respectively. The amount of KatB protein overexpressed from the pBB-katB-6XHis plasmid was assessed using anti-6X-His antibody. The amount of total protein loaded in each gel was shown as loading control. (D) Relative expression levels of katA, katB, katB-His, and oxyR in the wild type and trmB mutant strains upon H2O2 exposure. Data represent the mean ± SD for three biological replicates for (A) and for five biological replicates for (D). Asterisks denote a significant difference by t-test (* P ≤ 0.05, ** P ≤ 0.01).
To test whether the low catalase activity is due to reduced catalase protein levels, we performed a western blot analysis to detect the protein levels of the catalases. As shown in Figure 7C, a clear reduction in KatA (seven-fold decrease at 0 mM H2O2 and three-fold decrease at 5 mM H2O2) and KatB (2-fold decrease at 0 mM H2O2 and 3-fold decrease at 5 mM H2O2) protein levels was observed in the trmB mutant strain compared with that in the wild type strain under both normal and oxidative stress conditions. Semiquantitative real-time PCR analysis revealed that the transcript levels of katA, katB and katB-His were trmB-independent throughout the H2O2 treatment (Figure 7D). These results suggested that trmB plays a role in mediating the H2O2 stress response in P. aeruginosa at the translational level by positively controlling the levels of KatA and KatB.
Transcription of katA and katB is positively regulated by OxyR, an H2O2-sensing transcriptional regulator (39,40). To confirm that trmB directly regulates the translation levels of KatA and KatB and that it has no influence on oxyR expression, we examined the effects of trmB inactivation on oxyR mRNA and OxyR protein levels. Expectedly, the oxyR mRNA and OxyR protein levels did not change in the trmB mutant strain compared with those in the wild type strain with and without H2O2 exposure (Figure 7C and D). Most likely, the expression levels of katA and katB were directly modulated by trmB at the translational level.
DISCUSSION
In this study, we reveal an important role of PaTrmB-catalyzed m7G46 tRNA modification in the oxidative stress response of P. aeruginosa; such a role is most likely facilitated through positive regulation of the translation efficiency of catalase genes via a biased codon usage mechanism. PaTrmB catalyzes the formation of m7G tRNA modification at position 46 in the tRNA variable loop (Figures 2 and 4). Twenty-three tRNAs were identified as PaTrmB substrates, including eleven novel substrates, namely, tRNAAla/GGC, tRNAArg/ACG, tRNAArg/CCG, tRNAArg/CCU, tRNAAsp/GUC, tRNAHis/GUG, tRNAIle/GAU, tRNAPro/GGG, tRNAThr/CGU, tRNAThr/GGU and tRNAVal/GAC (12,13,15,16,21,41,42). Analysis of all P. aeruginosa tRNA sequences revealed an RGGUC (G represents G46) motif at the variable loop site in PaTrmB substrates. The sequence of P. aeruginosa m7G modification motif is slightly different from that of the RAGGU motif in m7G-modified tRNAs isolated from mouse ESCs (21). However, RGGUC motif was not detected in G46-containing tRNAs that do not serve as substrates or are poor substrates for PaTrmB; these tRNAs include tRNALeu/CAG, tRNALeu/GAG and tRNASec(p)/UCA. Furthermore, analysis of tRNA secondary structure revealed that these three tRNAs contain a long variable loop (11–19 ribonucleotide long) belonging to class II tRNA (Supplementary Figure S5). Thus, PaTrmB may not simply recognize the G46 residue as its substrate. Both the sequence and the structure of the variable loop in the tRNA substrate may be essential for the methyl transfer reaction.
The impact of tRNA modifications on the efficiency of translation is mostly studied in anticodon loop (5,22,23). Here, we report that trmB-mediated m7G modification at position 46 in the tRNA variable loop is specifically necessary for the optimal translation of Phe- and Asp-enriched mRNAs (Figure 5). To our knowledge, only a few reports have shown the impact of tRNA modifications outside the anticodon region on the efficiency of protein synthesis. In E. coli, disruption of the gene responsible for the formation of Ψ55 further reduces the translation efficiency of Arg-CGA-enriched mRNA (30). Ribosome footprinting analysis in mouse ESCs has revealed an essential role of m7G modification in the efficient recognition of codons decoded by m7G-modified tRNAs during the elongation step (21). However, the manner by which m7G46 specifically affects the translation of the codons decoded by tRNAPhe/GAA and tRNAAsp/GUC remains unknown.
Here, we demonstrate the essential role of trmB in oxidative stress response of P. aeruginosa and reveal that trmB-mediated m7G modification directly regulates katA and katB at the translational level (Figures 6 and 7). KatA and KatB are the major H2O2-detoxifying enzymes in P. aeruginosa (37,38). The expression of katA and katB in P. aeruginosa are positively regulated by OxyR, a H2O2-sensing transcriptional regulator (39,40). In addition to the induction of katA and katB mRNA levels by H2O2 via OxyR regulation, our data suggest an additional regulation of katA and katB expression via the enhancement of translation efficiency. This finding is supported by the codon reporter assay results (Figure 5). The translation efficiency of Phe- and Asp-enriched mRNAs increased upon exposure to H2O2, consistent with the H2O2-induced increased in m7G modification (Figures 5 and 6B, C). Interestingly, a comparison of the codon usage of katA, katB and oxyR revealed that the Phe-UUC, Asp-GAC and Asp-GAU codons were enriched in katA and katB compared with the full genome (Supplementary Figure S6). However, these codons are not enriched in oxyR, consistent with the observation that trmB had no effect on oxyR expression at either the transcriptional or translational level (Figure 7). Moreover, examination of the distribution of Phe and Asp codons showed that the Phe and Asp codons were clustered in katA (161FKWDFF, 179DFSD and 417DDD) and katB (182DDDSRRFDFF and 297FDFDPLD) but were separately distributed in oxyR. These observations strongly supported that translational regulation by TrmB depends on the usage and distribution of the Phe and Asp codons in the target genes of TrmB.
The percentage of cell survival (Figure 6A) and total activity of catalase (Figure 7A) of the trmB mutant strain were fully complemented when compared with those of the wild type strain, whereas the level of total m7G modification in trmB complemented strain was partially recovered (Figure 2). It has been established that the overexpression of plasmid DNA can alter cellular metabolism (43). Therefore, we hypothesized that the lack of full complementation may arise from trmB overexpression, which is driven by the lacZ promoter in the medium to high copy number pBBR1MCS-4 plasmid. In addition, the level of certain modifications varies between individual tRNAs in P. aeruginosa (44). The m7G modification level in tRNATrp/CCA was higher than that in tRNAArg/UCG, suggesting variation in the methyl acceptance activity of each tRNA type (44). Despite the lower total m7G level in the trmB complemented strain compared with that in the wild type (Figure 2B), the level of m7G in tRNAs that contributed to the translational control by TrmB, e.g. tRNAPhe/GAA and tRNAAsp/GUC, in the trmB complemented strain may be high enough to fully restore catalase activity and survival under H2O2 exposure.
In addition to trmB, few other tRNA-modifying genes play important roles in cell responses to oxidative stress. Our previous work on P. aeruginosa has described two examples of such genes, namely, trmJ and ttcA. trmJ gene encodes 2′-O-methyl transferase, which catalyzes tRNA modifications at position 32, such as 2′-O-methylcytidine (Cm32), 2′-O-methyluridine (Um32), and 2′-O-methyladenosine (Am32) (27). ttcA gene encodes putative 2-thiocytidine synthetase, which catalyzes the formation of 2-thiocytidine (s2C) (45). Mutation of either trmJ or ttcA alters the expression of H2O2-defense genes and confers H2O2 sensitivity in P. aeruginosa (27,45). Moreover, H2O2-induced reprogramming of tRNA modification has been found in yeast trm4. Increase in Trm4-mediated m5C34 modification after exposure to H2O2 selectively enhances the translation of rpl22a gene enriched with the Leu-UUG codon. The rpl22a gene, which encodes the ribosomal protein Rpl22a, is involved in H2O2 resistance, allowing yeast cells to grow following H2O2 treatment (29). In addition, defects in E. coli trmM (46), yeast trm7 (47) and Drosophila dnmt2 (48), which are responsible for the formation of tRNA 6-methyladenosine (m6A37), 2′-O-methylcytidine (Cm32 and Cm34), and 5-methylcytidine (m5C38), respectively, also result in H2O2 sensitivity.
We observed an ∼2.6-fold increase in the transcription level of trmB following H2O2 treatment, and this finding might explain the slight increase in m7G level and the increased translation efficiency of Phe- and Asp-enriched transcripts following the same treatment (Figures 5 and 6B, C). To date, regulation of the expression of trmB and most tRNA-modifying genes has not yet been studied. Our previous study on ttcA gene, which encodes a putative 2-thiocytidine tRNA synthetase, has demonstrated that ttcA transcription is directly modulated by OxyR, a H2O2-sensing transcriptional regulator, via the derepression mechanism in response to H2O2 exposure (45). However, no putative OxyR-binding site was detected in the upstream region of trmB. The regulation of trmB expression needs further investigation.
Overall, we present herein the regulatory function of TrmB in the translational control of biased codon usage transcript in response to oxidative stress. We propose a possible model to explain the TrmB-mediated translational control of oxidative stress response. The amounts of m7G-modified tRNAs, such as m7G modified-tRNAPhe/GAA and m7G modified-tRNAAsp/GUC, are increased by H2O2 treatment due to trmB upregulation, which subsequently enhances the translation efficiency of the transcripts enriched with the relevant Phe and Asp codons. As a consequence, the levels of KatA and KatB, in which Phe and Asp codons are enriched and which are critical for H2O2 resistance, are increased, enabling P. aeruginosa cells to survive during oxidative stress (Figure 8). Taken together, our work describes a novel function of m7G tRNA modification in translational regulation of the oxidative stress response of P. aeruginosa, and it highlights the importance of tRNA modification outside the anticodon region in the translational control.
Figure 8.
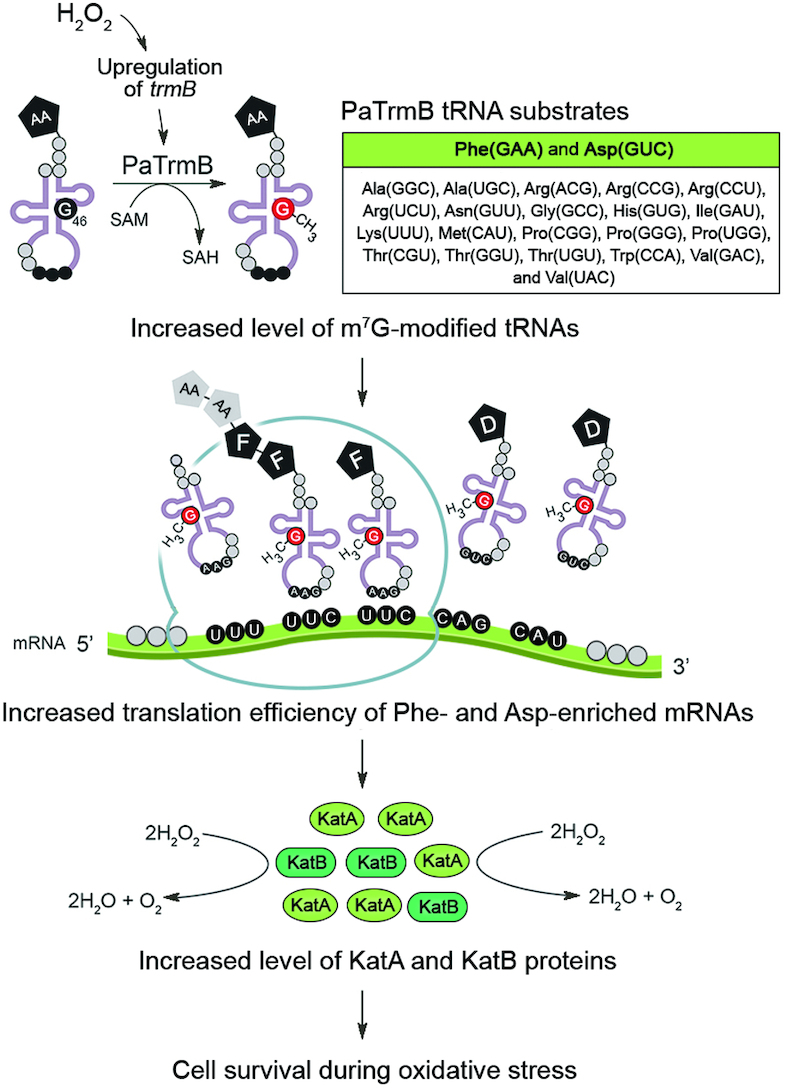
Proposed model for the TrmB-mediated translational control of the oxidative stress response of P. aeruginosa. The increased level of m7G-modified tRNA produced by PaTrmB following H2O2 exposure subsequently enhances the translation of genes encoding the H2O2-degrading enzymes, namely, KatA and KatB, in which Phe and Asp codons are enriched, allowing cell survival during oxidative stress. AA, amino acid; F, phenylalanine; D, aspartic acid.
Supplementary Material
ACKNOWLEDGEMENTS
The authors very much appreciate Professor Peter C. Dedon from Massachusetts Institute of Technology, USA for providing the modified ribonucleoside standards and Dr Chayasith Uttamapinant from Vidyasirimedhi Institute of Science and Technology, Thailand for critical reading of the manuscript.
Notes
Present address: Thanyaporn Srimahaeak, Faculty of Science, University of Copenhagen, Denmark.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Chulabhorn Research Institute [to M.F., S.M., P.V.]; Chulabhorn Graduate Institute [to N.T., ABS-01 to M.F.]. Funding for open access charge: Chulabhorn Graduate Institute; Chulabhorn Research Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1. Yacoubi B.E., Bailly M., Crécy-Lagard V.d. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012; 46:69–95. [DOI] [PubMed] [Google Scholar]
- 2. Motorin Y., Helm M.. RNA nucleotide methylation. Wiley Interdiscip. Rev. 2011; 2:611–631. [DOI] [PubMed] [Google Scholar]
- 3. Hori H. Methylated nucleosides in tRNA and tRNA methyltransferases. Front. Genet. 2014; 5:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duechler M., Leszczynska G., Sochacka E., Nawrot B.. Nucleoside modifications in the regulation of gene expression: focus on tRNA. Cell Mol. Life Sci. 2016; 73:3075–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gustilo E.M., Vendeix F.A.P., Agris P.F.. tRNA’s modifications bring order to gene expression. Curr. Opin. Microbiol. 2008; 11:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Agris P.F. Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008; 9:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang H.-Y., Hopper A.K.. Multiple layers of stress-induced regulation in tRNA biology. Life. 2016; 6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Motorin Y., Helm M.. tRNA stabilization by modified nucleotides. Biochem. 2010; 49:4934–4944. [DOI] [PubMed] [Google Scholar]
- 9. Tomita K., Ueda T., Watanabe K.. 7-Methylguanosine at the anticodon wobble position of squid mitochondrial tRNASerGCU: molecular basis for assignment of AGA/AGG codons as serine in invertebrate mitochondria1. BBA-Gene Struct. Expr. 1998; 1399:78–82. [DOI] [PubMed] [Google Scholar]
- 10. Matsuyama S., Ueda T., Crain P.F., McCloskey J.A., Watanabe K.. A novel wobble rule found in starfish mitochondria presence of 7-methylguanosine at the anticodon wobble position expands decoding capability of tRNA. J. Biol. Chem. 1998; 273:3363–3368. [DOI] [PubMed] [Google Scholar]
- 11. Jakab G.b., Kis M.l., Palfi Z., Solymosy F.. Nucleotide sequence of chloroplast tRNA (Leu)/UA m7G/from Chlamydomonas reinhardtii. Nucleic Acids Res. 1990; 18:7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Bie L.G., Roovers M., Oudjama Y., Wattiez R., Tricot C., Stalon V., Droogmans L., Bujnicki J.M.. The yggH gene of Escherichia coli encodes a tRNA (m7G46) methyltransferase. J. Bacteriol. 2003; 185:3238–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zegers I., Gigot D., Van Vliet F., Tricot C., Aymerich S., Bujnicki J.M., Kosinski J., Droogmans L.. Crystal structure of Bacillus subtilis TrmB, the tRNA (m7G46) methyltransferase. Nucleic Acids Res. 2006; 34:1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Okamoto H., Watanabe K., Ikeuchi Y., Suzuki T., Endo Y., Hori H.. Substrate tRNA recognition mechanism of tRNA (m7G46) methyltransferase from Aquifex aeolicus. J. Biol. Chem. 2004; 279:49151–49159. [DOI] [PubMed] [Google Scholar]
- 15. Tomikawa C., Yokogawa T., Kanai T., Hori H.. N7-Methylguanine at position 46 (m7G46) in tRNA from Thermus thermophilus is required for cell viability at high temperatures through a tRNA modification network. Nucleic Acids Res. 2010; 38:942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alexandrov A., Martzen M.R., Phizicky E.M.. Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA. 2002; 8:1253–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Robertus J., Ladner J.E., Finch J., Rhodes D., Brown R., Clark B., Klug A.. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 1974; 250:546. [DOI] [PubMed] [Google Scholar]
- 18. Alexandrov A., Chernyakov I., Gu W., Hiley S.L., Hughes T.R., Grayhack E.J., Phizicky E.M.. Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell. 2006; 21:87–96. [DOI] [PubMed] [Google Scholar]
- 19. Chernyakov I., Whipple J.M., Kotelawala L., Grayhack E.J., Phizicky E.M.. Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5′–3′ exonucleases Rat1 and Xrn1. Genes Dev. 2008; 22:1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shaheen R., Abdel-Salam G.M., Guy M.P., Alomar R., Abdel-Hamid M.S., Afifi H.H., Ismail S.I., Emam B.A., Phizicky E.M., Alkuraya F.S.. Mutation in WDR4 impairs tRNA m7G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol. 2015; 16:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin S., Liu Q., Lelyveld V.S., Choe J., Szostak J.W., Gregory R.I.. Mettl1/Wdr4-Mediated m7G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol. Cell. 2018; 71:244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ranjan N., Rodnina M.V.. tRNA wobble modifications and protein homeostasis. Translation. 2016; 4:e1143076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Endres L., Dedon P.C., Begley T.J.. Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol. 2015; 12:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alexeyev M.F. The pKNOCK series of broad-host-range mobilizable suicide vectors for gene knockout and targeted DNA insertion into the chromosome of gram-negative bacteria. BioTechniques. 1999; 26:824–826. [DOI] [PubMed] [Google Scholar]
- 25. Kovach M.E., Elzer P.H., Hill D.S., Robertson G.T., Farris M.A., Roop R.M., Peterson K.M.. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene. 1995; 166:175–176. [DOI] [PubMed] [Google Scholar]
- 26. Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976; 72:248–254. [DOI] [PubMed] [Google Scholar]
- 27. Jaroensuk J., Atichartpongkul S., Chionh Y.H., Wong Y.H., Liew C.W., McBee M.E., Thongdee N., Prestwich E.G., DeMott M.S., Mongkolsuk S.. Methylation at position 32 of tRNA catalyzed by TrmJ alters oxidative stress response in Pseudomonas aeruginosa. Nucleic Acids Res. 2016; 44:10834–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Su D., Chan C.T., Gu C., Lim K.S., Chionh Y.H., McBee M.E., Russell B.S., Babu I.R., Begley T.J., Dedon P.C.. Quantitative analysis of ribonucleoside modifications in tRNA by HPLC-coupled mass spectrometry. Nat. Protoc. 2014; 9:828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chan C.T.Y., Pang Y.L.J., Deng W., Babu I.R., Dyavaiah M., Begley T.J., Dedon P.C.. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2012; 3:937–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Urbonavičius J., Durand J.M., Björk G.R.. Three modifications in the D and T arms of tRNA influence translation in Escherichia coli and expression of virulence genes in Shigella flexneri. J. Bacteriol. 2002; 184:5348–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Santos P.M., Di Bartolo I., Blatny J.M., Zennaro E., Valla S.. New broad-host-range promoter probe vectors based on the plasmid RK2 replicon. FEMS Microbiol. Lett. 2001; 195:91–96. [DOI] [PubMed] [Google Scholar]
- 32. Miller H. 1972; Cold Spring Harbor: CSH Laboratory Press. [Google Scholar]
- 33. Wayne L.G., Diaz G.A.. A double staining method for differentiating between two classes of mycobacterial catalase in polyacrylamide electrophoresis gels. Anal. Biochem. 1986; 157:89–92. [DOI] [PubMed] [Google Scholar]
- 34. Chomczynski P., Sacchi N.. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987; 162:156–159. [DOI] [PubMed] [Google Scholar]
- 35. Kozbial P.Z., Mushegian A.R.. Natural history of S-adenosylmethionine-binding proteins. BMC Struct. Biol. 2005; 5:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hassett D., Charniga L., Bean K., Ohman D., Cohen M.. Response of Pseudomonas aeruginosa to pyocyanin: mechanisms of resistance, antioxidant defenses, and demonstration of a manganese-cofactored superoxide dismutase. Infect. Immun. 1992; 60:328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee J.-S., Heo Y.-J., Lee J.K., Cho Y.-H.. KatA, the major catalase, is critical for osmoprotection and virulence in Pseudomonas aeruginosa PA14. Infect. Immun. 2005; 73:4399–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brown S.M., Howell M.L., Vasil M.L., Anderson A.J., Hassett D.J.. Cloning and characterization of the katB gene of Pseudomonas aeruginosa encoding a hydrogen peroxide-inducible catalase: purification of KatB, cellular localization, and demonstration that it is essential for optimal resistance to hydrogen peroxide. J. Bacteriol. 1995; 177:6536–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ochsner U.A., Vasil M.L., Alsabbagh E., Parvatiyar K., Hassett D.J.. Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J. Bacteriol. 2000; 182:4533–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heo Y.-J., Chung I.-Y., Cho W.-J., Lee B.-Y., Kim J.-H., Choi K.-H., Lee J.-W., Hassett D.J., Cho Y.-H.. The major catalase gene (katA) of Pseudomonas aeruginosa PA14 is under both positive and negative control of the global transactivator OxyR in response to hydrogen peroxide. J. Bacteriol. 2010; 192:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alexandrov A., Grayhack E.J., Phizicky E.M.. tRNA m7G methyltransferase Trm8p/Trm82p: evidence linking activity to a growth phenotype and implicating Trm82p in maintaining levels of active Trm8p. RNA. 2005; 11:821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cartlidge R.A., Knebel A., Peggie M., Alexandrov A., Phizicky E.M., Cohen P.. The tRNA methylase METTL1 is phosphorylated and inactivated by PKB and RSK in vitro and in cells. EMBO J. 2005; 24:1696–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Silva F., Queiroz J.A., Domingues F.C.. Evaluating metabolic stress and plasmid stability in plasmid DNA production by Escherichia coli. Biotechnol. Adv. 2012; 30:691–708. [DOI] [PubMed] [Google Scholar]
- 44. Grobe S., Doberenz S., Ferreira K., Krueger J., Brönstrup M., Kaever V., Häußler S.. Identification and quantification of (t)RNA modifications in Pseudomonas aeruginosa by liquid chromatography–tandem mass spectrometry. ChemBioChem. 2019; 20:1430–1437. [DOI] [PubMed] [Google Scholar]
- 45. Romsang A., Duang-nkern J., Khemsom K., Wongsaroj L., Saninjuk K., Fuangthong M., Vattanaviboon P., Mongkolsuk S.. Pseudomonas aeruginosa ttcA encoding tRNA-thiolating protein requires an iron-sulfur cluster to participate in hydrogen peroxide-mediated stress protection and pathogenicity. Sci. Rep. 2018; 8:11882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Golovina A.Y., Sergiev P.V., Golovin A.V., Serebryakova M.V., Demina I., Govorun V.M., Dontsova O.A.. The yfiC gene of E. coli encodes an adenine-N6 methyltransferase that specifically modifies A37 of tRNA1Val (cmo5UAC). RNA. 2009; 15:1134–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chan C.T.Y., Dyavaiah M., DeMott M.S., Taghizadeh K., Dedon P.C., Begley T.J.. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010; 6:e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schaefer M., Pollex T., Hanna K., Tuorto F., Meusburger M., Helm M., Lyko F.. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010; 24:1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.