


Abstract
The maintenance of telomere length is critical to longevity and survival. Specifically, the failure to properly replicate, resect, and/or form appropriate telomeric structures drives telomere shortening and, in turn, genomic instability. The endonuclease CtIP is a DNA repair protein that is well-known to promote genome stability through the resection of endogenous DNA double-stranded breaks. Here, we describe a novel role for CtIP. We show that in the absence of CtIP, human telomeres shorten rapidly to non-viable lengths. This telomere dysfunction results in an accumulation of fusions, breaks, and frank telomere loss. Additionally, CtIP suppresses the generation of circular, extrachromosomal telomeric DNA. These latter structures appear to arise from arrested DNA replication forks that accumulate in the absence of CtIP. Hence, CtIP is required for faithful replication through telomeres via its roles at stalled replication tracts. Our findings demonstrate a new role for CtIP as a protector of human telomere integrity.
INTRODUCTION
Telomeres, the specialized nucleoprotein complexes found at the ends of eukaryotic chromosomes, consist of a repetitive DNA sequence bound by a complex of proteins that protectively masks the chromosome end from being recognized as a DNA double-stranded break (DSB) (1). The telomere repeat is a 2–15 kb-long, double-stranded hexameric (TTAGGG:AATCCC) tract that terminates with a 3′ G-rich, single-stranded overhang (the G-overhang). Importantly, the G-overhang is folded back and imbedded into the repetitive double-stranded tract to form a 3-stranded structure termed a t-loop (2). Complexed to this unusual structure is a set of six telomere binding proteins (collectively called the Shelterin complex) that are required for the formation and maintenance of, the t-loop and telomere, respectively (1). Thus, the binding of Shelterin to the telomere and the subsequent formation of the t-loop structure generates a protective cap on the ends of mammalian chromosomes that makes them invisible to the cellular DSB repair machinery. Unfortunately, mammalian telomeres are subject to constant shortening due to the end replication problem and endogenous DNA damage (3). When telomeres become too short or too damaged, they fail to carry out their protective function, which greatly reduces cellular survival or facilitates oncogenic transformations (4,5).
One source of telomeric damage is DNA replication. Telomeric sequences are especially difficult to replicate due to their repetitive nature and their heterochromatic regions that are capable of forming unique secondary structures including G-quadruplexes (6). These obstacles can lead to stalled replication forks (7). Stalled forks, in turn, can collapse into DSBs, which are then resolved via non-homologous end joining (NHEJ) or homologous recombination (HR) (8). NHEJ puts the telomere at risk for undesirable telomere:telomere fusion events (5,9), while HR is viewed as an ‘error-free’ pathway. Thus, HR is generally the pathway of choice for recovery of a collapsed replication tract, including those within a telomere (10).
A key HR protein is the C-terminal binding protein (CtBP) interacting protein (CtIP) (11,12). CtIP generally functions in association with the meiotic recombination defective 11:radiation sensitive 50:Nijmegen breakage syndrome 1 protein (Mre11:RAD50:NBS1; MRN) complex and acts as a short-range resection endonuclease, generating short, single-stranded DNA overhangs, which can be further extended by exonuclease 1 (EXO1) or DNA replication helicase 2 (DNA2) and Bloom syndrome (BLM) (13–15). The initial resection of a DSB by CtIP is key in stimulating these downstream nucleases as well as in recruiting other DNA damage response proteins (16). Thus, DNA2 and EXO1 depend on the activity and presence of CtIP to perform proper resection at DNA breaks (17,18). These activities of CtIP may extend from DSBs to telomeres as EXO1 and DNA2 have also been implicated in the generation of the G-overhang at telomeric ends (19,20). Similarly, MRN functions at dysfunctional telomeres to promote 3′-overhang generation and suppress NHEJ at telomeric ends (21). In summary, while a direct role of CtIP at telomeres is lacking, some of CtIP’s interacting partners at least appear to be crucial for the maintenance of mammalian telomeres.
While CtIP’s best-characterized role is in the resection of DNA DSBs, it also has other important cellular functions. CtIP plays a role in the DNA replication of difficult regions, specifically those containing complex secondary structures (22,23). Most recently, CtIP was also shown to have a MRN-independent role in protecting stalled forks from DNA2-dependent degradation (24). Thus, depending upon the cellular context—either at a frank DSB or at a stalled replication fork—CtIP can either facilitate or inhibit DNA2-mediated resection, respectively. On an organismal level, CtIP plays a role(s) in tumorigenesis, although what that role is, is not entirely clear. Thus, in one study CtIP haploinsufficiency was linked to significant tumorigenesis in mice but this was not confirmed in a similar subsequent study (25,26). However, genetic mutations of human CtIP have been linked to breast and ovarian cancer (27) supporting a role in tumor suppression. Therefore, whether CtIP has tumor suppressor activity and whether this is directly related to its DNA resection activity on DNA DSBs and/or telomeres is currently unclear. Importantly, however, CtIP is clearly essential as its absence in mice is embryonically lethal (25,28).
All of this information, taken together, provides strong circumstantial evidence that CtIP may be (directly or indirectly) functioning at mammalian telomeres through resection and/or replication. In this study, we sought to address this issue using a CtIP inducibly-null human cell line. We demonstrate that severe telomere dysfunction arises in cells devoid of CtIP. Specifically, in the absence of CtIP human telomeres and their G-overhangs rapidly shorten. At least part of this telomere loss is due to an accumulation of t-circles—extrachromosomal, circular telomeric DNA arising from loss of the t-loop. Additionally, we document increased telomere replication fork stalling events upon the depletion of CtIP, which results in a decrease of fully-replicated telomeres. Our work therefore demonstrates that CtIP is an essential telomere maintenance protein in human cells — likely playing roles in both the replication and resection of telomeric DNA.
MATERIALS AND METHODS
Telomere restriction fragment (TRF) analyses
Genomic DNA was purified, digested with RsaI and HinfI restriction enzymes and separated via gel electrophoresis overnight at 35 V. Following electrophoresis, the gels were de-purinated, denatured, and neutralized. DNA was transferred to a Hybond XL membrane overnight by capillary transfer. After transfer, the membrane was irradiated with ultraviolet light to crosslink the DNA to the membrane and then the membrane was hybridized with a 32P-end-labeled oligonucleotide probe (CCCTAA)4 overnight. Membranes were exposed to PhosphoImager screens and scanned using a Typhoon FLA 9500 imager. Images were analyzed with FIJI software.
G-overhang assays
Genomic DNA was purified, and the negative control samples were pre-digested with bacterial Exo1. Subsequently, all of the samples were digested with RsaI and HinfI restriction enzymes. The samples were then subjected to gel electrophoresis for 2 h at 35 V. Following electrophoresis, the gels were dried with a Bio-Rad Model 583 gel dryer for 3 h. Dried gels were hybridized with a 32P-end-labeled oligonucleotide probe (CCCTAA)4 overnight. Following hybridization and washing, the gels were exposed to a PhosphoImager screen and scanned using a Typhoon FLA 9500 imager. After native imaging, the gels were denatured, neutralized, re-probed, and imaged in the denatured condition. Images were analyzed and quantified with FIJI software.
Two-dimensional (2D) gel electrophoresis
2D gel electrophoresis was performed as described (29).
C-circle assay
Genomic DNA was isolated and restriction enzyme digested as described above for the TRF analysis. C-circles were then amplified via rolling circle amplification with or without phi29 DNA polymerase as described (30). Samples were dot blotted onto a Hybond XL membrane with a Bio-Dot and hybridized with a 32P-end-labeled oligonucleotide probe (CCCTAA)4 overnight. Membranes were exposed to PhosphoImager screens and scanned using a Typhoon FLA 9500 Imager. Images were analyzed and quantified with the FIJI software.
Telomere dysfunction induced foci (TIF) assay
Cells were grown on chamber slides and fixed with 4% paraformaldehyde (PFA) for 10 min. Cells were then permeabilized with 0.5% NP-40. After fixing, cells were blocked with 0.2% fish gelatin and 0.5% bovine serum albumin in phosphate buffered saline (PBS). Slides were incubated with a γ-H2AX rabbit antibody, then incubated with a secondary anti-rabbit antibody AF488 in blocking solution. Antibodies were crosslinked and slides were denatured at 80°C. Following denaturation, the slides were incubated with a Cy3-conjugated C3TA2 PNA probe. Cells were counterstained with diamidino-phenylindole (DAPI) and imaged on a Zeiss TIRF microscope. The depth of Z-stacks was taken with the recommended optimization. Images were processed and analyzed with FIJI software.
Immunoblotting
Cells were collected, centrifuged, and re-suspended in radioimmunoprecipitation assay (RIPA) buffer plus protease inhibitors. The cells were then incubated in RIPA for 15 min at 4°C and centrifuged at 4°C for 15 min. Protein concentrations were determined using a Bradford protein assay. The samples were then resuspended in 4× lithium dodecyl sulfate sample buffer and denatured. Protein samples were electrophoresed on 4–20% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) gels and then transferred to nitrocellulose membranes in transfer buffer (20% methanol, 11% glycine, and 3.4% Tris) at 100 V. The membranes were blocked with 5% milk in Tris-buffered saline—0.1% Tween (TBST) at room temperature for 45 min. The membranes were then incubated overnight in 5% milk in TBST at 4°C with primary antibodies. Membranes were subsequently washed with TBST and exposed to horse radish peroxidase-conjugated secondary antibodies in 5% milk in TBST at room temperature for 1 hr. The membranes were developed and enhanced using an enhanced chemiluminescent kit.
The primary antibodies used were: CtIP (14-1; (31)), beta-actin (NB600-501SS, Novus), tubulin (3708-100, BioVision), Exo1 (ab95068, Abcam), DNA2 (PA5-23691, Invitrogen), phospho-Chk2 (2197S, Cell Signaling), phospho-p53 (9284, Cell Signaling) and GAPDH (sc-47724, Santa Cruz).
Telomere FISH
Prior to collection, cells were treated with colcemid at 10 mg/ml and incubated for 3 h. After collection, the cells were re-suspended in 75 mM KCl and incubated for 30 min. Cells were then treated with fixative (3:1 MeOH and glacial acetic acid) and dropped over a wet slide at a 45°angle. Slides were subsequently washed with fixative and dried overnight. After drying, the slides were rehydrated, fixed with 4% formaldehyde and then washed and dehydrated in an ethanol series at –20°C. Once dry, the slides were denatured at 80°C and hybridized with a Cy3-conjugated protein:nucleic acid (PNA) probe (C3TA2)3. The slides were then washed, counterstained with DAPI, and dehydrated. Slides were mounted using ProLong Gold. Images were acquired using a Zeiss TIRF microscope. The images were analyzed with FIJI software and scored in a double-blind manner.
Flow cytometry
Cells were re-suspended in PBS and fixed with 1% PFA and subsequently incubated in cold 70% ethanol. The cells were then washed and stained with propidium iodide. The cells were then processed with a BD LSR II cytometer and analyzed with FlowJo™ software.
Cell lines
Human RPE1 cells were obtained from the ATCC. These cells were grown in Dulbecco's modified Eagle's medium F12 (DMEM-F12) supplemented with l-glutamine, penicillin-streptomycin, and 10% fetal bovine serum. Cells were cultured at 37°C in a humidified atmosphere (5% CO2). HeLa LT cells were a gift from Dr. Titia de Lange (Rockefeller University). HeLa cells were cultured the same as RPE1 cells, with the exception of utilizing regular DMEM media. U-2 OS cells were obtained from the ATCC and cultured the same as RPE1 cells, with the exception of utilizing McCoy's media instead of DMEM-F12.
Generation of the CtIPf/− cell line and complementation
A single-guide RNA (sgRNA) targeting CtIP exon 2 was delivered along with Cas9 using a plasmid expression system (hSpCas9-2A-GFP/px459). A dsDNA donor template was designed containing two LoxP sites flanking CtIP exon 2 in the pAAV_MCS_Sept plasmid as described (32). Candidate clones were screened by PCR for LoxP site integration after electroporation with both the sgRNA/Cas9 and LoxP-containing donor template. CtIPf/+ clones were isolated after the first round of targeting and validated via Sanger sequencing. A second round of targeting was performed with the same sgRNA/Cas9 to induce a frameshift mutation on the remaining wild-type allele. Candidate clones were confirmed via PCR and, subsequently, by Sanger sequencing. The second round of targeting yielded five independent CtIPf/− clones. CreER was introduced into one of these using a plasmid expression system (CreER-PCDNA3.1-Zeocin) and clones stably expressing CreER were isolated and confirmed via western blot analyses. Treatment of these clones with 10 nM 4-OHT and subsequent western blot analysis for CtIP confirmed the successful integration and functionality of this system.
The CtIPf/− cell line was then complemented with a wild-type CtIP cDNA, which was cloned into a PiggyBac transposase vector containing a CAG (cytomegaloviral/beta-actin/beta-globin) promoter and a neomycin selection cassette. Individual clones were isolated and analyzed for CtIP expression via western blot analysis.
DNA combing
Dividing cells were labeled with 25 μM IdU for 1 h followed by a 2 h period without IdU, repeated four times. The cells were then embedded in 0.5% low-melt agarose plugs, proteinase K-treated, and DNA extracted. The plugs were subjected to restriction enzyme digestion with AluI to enrich for telomeric DNA. DNA was then combed onto vinyl silane-coated coverslips using a Genomic Vision system. Telomeres were identified with a AF488-conjugated PNA probe (C3TA2)3 by FISH. IdU was detected with a primary antibody (mouse anti-BrdU) and an AF647-conjugated secondary antibody (goat anti-mouse). Images were acquired with a Nikon TiE deconvolution microscope and analyzed with Fiji software in a blind manner.
RESULTS
Generation and characterization of a CtIP conditionally-null human cell line
We utilized a cyclization recombination/locus of crossover P1 (Cre/LoxP) recombination approach (33) along with clustered regularly interspaced short palindromic repeats/CRISPR-associated 9 (CRISPR/Cas9; (34)) to generate a CtIP inducibly-null allele in the telomerase-immortalized retinal pigment epithelial-1 (RPE1) cell line. RPE1 cells were chosen because they are an immortalized, but non-transformed, human epithelial cell line (35) that is diploid and has been successfully utilized for gene editing studies (36). We knocked in LoxP sites flanking exon 2 of the CtIP locus using a targeted single-guide RNA (sgRNA) and Cas9 nuclease coupled with a double-stranded DNA donor containing LoxP sites (Figure 1A and Supplemental Figure S1A). A second round of targeting with the sgRNA and Cas9 nuclease alone induced a frameshift mutation in the remaining wild type allele (Supplemental Figure S1B) generating a CtIPf/− cell line. An inducible Cre:estrogen receptor (CreER) cassette was subsequently stably integrated in the cell line to facilitate recombination between the flanking LoxP (floxed) sites upon tamoxifen (4-OHT) treatment, which eliminated the residual endogenous copy of CtIP (Figure 1A and Supplemental Figure S1C). The CtIPf/− cell line was subsequently also stably complemented with a wild type CtIP cDNA (CtIPf/-:CtIP). When this cell line was treated with 4-OHT the resulting cells (CtIP−/-:CtIP) were viable and were used to assess whether CtIP re-expression was sufficient to enable a functional rescue in all subsequent experiments. A western blot analysis was utilized to confirm that all of the experimental CtIP cell lines expressed CtIP protein at the expected levels (Figure 1B).
Figure 1.

(A) A CtIP conditionally-null cell line was generated in an RPE1 background using CRISPR/Cas9 technology (scissors). LoxP sites, represented as purple triangles, were knocked-in to flank exon 2 of CtIP. The second allele was knocked out by creating a frameshift mutation (lightning bolt). CreER was stably integrated and then used to conditionally recombine out the remaining CtIP allele creating a CtIP-null cell line. (B) Confirmation of a CtIP-null cell line and a complemented derivative by western analysis. The CtIP expression levels in the indicated experimental cell lines was assessed by western blot analysis. This analysis also validated the complementation of the CtIPf/- cell line with a CtIP cDNA. GAPDH was used as a loading control. (C) CtIP-deficient cells are proliferation defective. Growth analyses of RPE1 WT, CtIPf/−, CtIPf/- cells treated with 4-OHT for 2 days and CtIPf/-:CtIP over a period of 6 days. The data shown represent the average of three biological replicates. (D) CtIP-null cells show high levels of genomic instability. Representative images of intact nuclei, micronuclei, and fragmented nuclei from CtIPf/- cells treated with 4-OHT for 3 days. Nuclei were DAPI stained; scalebar = 30 μm. (E) Quantification of the number of micronuclei observed in CtIP-null cells. 150 cells similar to the ones shown in panel (D) were scored for the presence of micronuclei in each background.
CtIPf/− cells (i.e., functional heterozygotes) grew much slower than their wild type counterparts (Figure 1C). This growth defect was also observed in an independently generated CtIPf/– +CreER clone (Supplemental Figure S4A). Upon removal of the remaining endogenous allele of CtIP by tamoxifen treatment, the null cells ceased proliferating altogether within 1 to 2 cell cycles (Figure 1C). Again, this severe proliferation defect was confirmed in an independently generated CtIP−/– cell line (Supplemental Figure S4A). These data are consistent with previous findings that murine CtIP displays haploinsufficiency and is required for cellular survival (25). Importantly, however, we also noted that CtIP-null cells quickly enlarged relative to CtIP-proficient cells (Supplementary Figure S2A). This cellular bloating was accompanied by the accumulation of micronuclei and chromosomes so fragmented that over 25% of CtIP-deficient cells did not possess an intact nucleus (Figure 1D and E). These phenotypes are the classical hallmarks of the severe genomic instability generally associated with the loss of key DNA repair factors (37,38). Moreover, CtIP can function as a mediator between cell cycle checkpoints and the regulation of DNA repair pathway choices (39,40). Consistent with these findings, we documented a G2 cell cycle arrest in asynchronous cell populations after the loss of CtIP (Supplemental Figure S2B and C). These findings demonstrated that CtIP is an essential human gene and that CtIP-null cells succumb to a genomic instability-driven cellular death.
Chromosomal aberrations and DNA damage arise in the absence of CtIP
To confirm the presence of specific DNA damage, the accumulation of 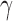 H2AX foci, a marker of DNA DSBs (41), was quantified for CtIP wild type, heterozygous, and null cells. Since the parental RPE1 cells are near normal and non-cancerous they exhibited very low levels of endogenous DNA damage and thus low levels of
H2AX foci, a marker of DNA DSBs (41), was quantified for CtIP wild type, heterozygous, and null cells. Since the parental RPE1 cells are near normal and non-cancerous they exhibited very low levels of endogenous DNA damage and thus low levels of 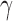 H2AX in non-challenged conditions (Figure 2A and B). However, the removal of CtIP resulted in an increase of
H2AX in non-challenged conditions (Figure 2A and B). However, the removal of CtIP resulted in an increase of 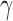 H2AX foci with CtIP-null cells having on average >8
H2AX foci with CtIP-null cells having on average >8 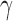 H2AX damage foci in otherwise unperturbed conditions (Figure 2A and B). To determine the effects of this DNA damage on chromosome stability, chromosome spreads were also analyzed for abnormalities. CtIPf/− cells treated with 4-OHT displayed a significant increase in breaks, which was accompanied by an increase in chromosome end-to-end fusions events and even the presence of radial structures (Figure 2C–E). These effects were due to the absence of CtIP since the restoration of CtIP expression completely suppressed them (Figure 2E). These data demonstrated that CtIP removal has a catastrophic effect on the chromosomes of an otherwise unperturbed human cell during normal proliferation.
H2AX damage foci in otherwise unperturbed conditions (Figure 2A and B). To determine the effects of this DNA damage on chromosome stability, chromosome spreads were also analyzed for abnormalities. CtIPf/− cells treated with 4-OHT displayed a significant increase in breaks, which was accompanied by an increase in chromosome end-to-end fusions events and even the presence of radial structures (Figure 2C–E). These effects were due to the absence of CtIP since the restoration of CtIP expression completely suppressed them (Figure 2E). These data demonstrated that CtIP removal has a catastrophic effect on the chromosomes of an otherwise unperturbed human cell during normal proliferation.
Figure 2.

(A) CtIP-null cells accumulate markers of DNA damage. Representative images of RPE1 WT, CtIPf/−, or CtIPf/− cells treated with 4-OHT for 2 days, and then stained for γ-H2AX. Scale bars, 30 μm. (B) Quantification of γ-H2AX foci per cell. Data from three independent experiments are shown, n = 150, paired t-test, P < .0001 = ****. (C) Gross chromosomal abnormalities are frequently observed in metaphase-arrested CtIP-null cells. Representative images of metaphase spreads from RPE1 WT, CtIPf/−, CtIPf/− treated with 4-OHT for 2 days, or CtIPf/-:CtIP. Scale bars, 45 μm. (D) Presence of radial chromosomes, fusions and chromosomal fragments in CtIP-null cells. The boxed representative abnormalities in CtIPf/− cells treated with 4-OHT from (C) are shown in expanded form. (E) Quantification of chromosomal breaks, fusions, and radials. Data from 3 independent experiments, n = 2200, one-way ANOVA statistical analysis and Tukey's test, P < .01 = **, P < .001 = ***.
Telomeres shorten and accumulate damage after loss of CtIP
The formation of chromosome end-to-end fusions is characteristic of telomere uncapping or loss (1). In particular, chromosomes can fuse end-to-end if they are not able to form a t-loop or if the telomere sequence is lost in an attempt to resolve what is perceived by the cell as essentially one half of a DSB (i.e. a one-ended DSB). Thus, the presence of elevated levels of chromosome end-to-end fusion events (Figure 2E) implied a telomere protection role for CtIP outside of its characterized genome maintenance functions.
To determine whether any of the DNA damage resulting from the loss of CtIP was localized at telomeres, the presence of telomere dysfunction-induced foci (TIFs) was assessed. Abolishing CtIP expression increased DNA damage at telomeres significantly (Figure 3A and B). Indeed, the majority of CtIPf/− cells treated with tamoxifen contained the colocalization of γH2AX with telomere signals (Figure 3A and B). Again, this phenotype could be completely complemented by the restoration of CtIP expression (Figure 3A and B).
Figure 3.

(A) IF analysis of telomere-associated damage in CtIP-null cells. TIFs in RPE1 WT, CtIPf/−, CtIPf/− treated with 4-OHT for 2 days, and CtIPf/-:CtIP cells were determined. Telomeres were labeled with a PNA probe and  -H2AX foci were visualized by IF staining. Scale bar in the bottom right panel is 10 μm, n = 300. (B) TIFs are highly elevated in CtIP-null cells. Quantification of TIFs in RPE1 WT, CtIPf/− untreated, CtIPf/- cells treated for 3 days with 4-OHT, and CtIPf/-:CtIP cells as shown in panel (A). The data are derived from three biological replicates; one-way ANOVA and Tukey's test, P < .001 = ***. (C) Telomere restriction fragment (TRF) analysis of RPE1 WT, CtIPf/−, and CtIPf/− cells treated with 4-OHT for 2 days. (D) Quantification of telomere length measurements from 3 independent TRF experiments such as shown in panel (C). Paired t-test, P < .05 = *, P < .0001 = ****. (E) Native G-overhang analysis using a 32P-radiolabeled (A2TC3)3 probe of DNA extracted from RPE1 WT, CtIPf/− untreated, and CtIPf/- cells 2 days after 4-OHT treatment. –Exo1 and +Exo1 refer to pre-treatment of the samples without or with, respectively, bacterial Exo1. (F) Quantification of G-overhang length measurements such as shown in panel (E) from 3 independent experiments. Paired t-test, P < .05 = *, P < .01 = **.
-H2AX foci were visualized by IF staining. Scale bar in the bottom right panel is 10 μm, n = 300. (B) TIFs are highly elevated in CtIP-null cells. Quantification of TIFs in RPE1 WT, CtIPf/− untreated, CtIPf/- cells treated for 3 days with 4-OHT, and CtIPf/-:CtIP cells as shown in panel (A). The data are derived from three biological replicates; one-way ANOVA and Tukey's test, P < .001 = ***. (C) Telomere restriction fragment (TRF) analysis of RPE1 WT, CtIPf/−, and CtIPf/− cells treated with 4-OHT for 2 days. (D) Quantification of telomere length measurements from 3 independent TRF experiments such as shown in panel (C). Paired t-test, P < .05 = *, P < .0001 = ****. (E) Native G-overhang analysis using a 32P-radiolabeled (A2TC3)3 probe of DNA extracted from RPE1 WT, CtIPf/− untreated, and CtIPf/- cells 2 days after 4-OHT treatment. –Exo1 and +Exo1 refer to pre-treatment of the samples without or with, respectively, bacterial Exo1. (F) Quantification of G-overhang length measurements such as shown in panel (E) from 3 independent experiments. Paired t-test, P < .05 = *, P < .01 = **.
To directly assess CtIP’s contribution to total telomere length, a telomere restriction fragment (TRF) analysis was performed. CtIP heterozygous cells displayed a more heterogeneous telomere length profile compared to the parental RPE1 cells, but only possessed a slightly shorter mean telomere length. CtIPf/− cells treated with tamoxifen, however, exhibited extremely rapid telomere shortening, in which the overall size was reduced from ∼6 kb to under 2 kb within 2 days of CtIP removal (Figure 3C and D). This telomere shortening defect was confirmed in an independently generated CtIPf/- plus tamoxifen treated cell line (Supplemental Figure S4B). Indeed, the telomeres of CtIP-null cells were so short that they were no longer capable of forming TRF2 foci (Supplemental Figure S3). This unusually expedited telomere shortening—which is normally only observed with extreme telomere uncapping, breakage, or defects in telomere replication—in the absence of CtIP implied that telomere dysfunction was a contributing factor in CtIP-mediated death. Again, and importantly, these aberrant telomere phenotypes could be rescued by the restoration of CtIP expression in the CtIP-null cells (Supplemental Figure S5A).
Essential to the mechanism of telomere capping is the generation of a 3′ overhang at telomere ends to form the t-loop. In mammals and yeast, two nucleases, EXO1 and DNA2, contribute to the generation of the G-overhang (20,42). To determine the contribution, if any, of CtIP to this overhang, G-overhang lengths were assayed by a conventional non-denaturing in-gel hybridization analysis. Pre-treatment of the samples either without (−Exo1) or with (+Exo1) purified bacterial Exo1 (a 3′ > 5′ exonuclease) was used as a control to demonstrate that the ssDNA that hybridized in this assay corresponded to a G-overhang. G-overhangs were shortened significantly in the absence of CtIP, with CtIP heterozygous cells also displaying a diminished 3′ overhang relative to total telomere signal (Figure 3E and F). Because CtIP is known to regulate resection by EXO1 and DNA2 (17,18,24), expression of these nucleases was assessed in CtIP-null conditions. DNA2 levels were unaltered regardless of CtIP expression levels, however EXO1 displayed a marked decrease in expression after the loss of CtIP (Supplemental Figure S6A and B). Thus, the decrease in EXO1 may contribute to defective 3′ overhang generation in the CtIP-null background. In summary, the loss of CtIP in human somatic cells manifested itself in rapid and dramatic telomere dysfunction.
CtIP protects against telomere loss and fusion
Telomere loss and defective G-overhang generation is commonly associated with telomere fusion events, an increase in which we had already documented in the CtIP-null cells (Figure 2E). To investigate the status of individual telomeres, telomere fluorescent in situ hybridization (T-FISH) was performed. Gross aberrations were observed following the removal of CtIP compared to CtIP-proficient and heterozygous backgrounds (Figure 4A). Chromosome ends that were free of telomere signals was the most abundant abnormality (Figure 4B and C), which was consistent with the rapid shortening seen by the TRF analysis. Chromosome breaks containing a telomere signal as well as internalized telomeres, presumably arising from these breaks fusing to other chromosomes, were observed at a frequency of ∼5% (Figure 4C and E). Increased fusions events, both sister chromatid and chromosome-to-chromosome types, were also observed, a typical outcome of signal-free chromosomal ends (Figure 4D and E). As a marker for non-telomere-related genomic instability, centromere breaks were scored. While centromere breaks were elevated in CtIP-deficient cells (Figure 4C), this increase was not statistically significant. This telomere dysfunction phenotype was also confirmed in an independently generated CtIPf/– cell line (Supplemental Figure S4C). These data imply that CtIP’s roles in general genomic stability is distinct from, and perhaps not as important as, its role(s) at telomeres.
Figure 4.

(A) Representative images of Telomere-FISH (T-FISH) from RPE1 WT, CtIPf/−, CtIPf/− + 4-OHT 2 days and CtIPf/-:CtIP using a (T2AG3)3 PNA telomere probe (red), as well as a AT2CGT2G2A3CG3A PNA centromere probe (green). Scale bars are 20 μm. (B) Representative images of sister chromatid fusions, chromosome:chromosome fusions, and internalized telomere signals. (C) Quantification of the telomere abnormalities such as those shown in panel (B). n = 2000, one-way ANOVA statistical test and Tukey’s test, P < .01 = **. (D) Representative images of signal free ends, centromere breaks, and breaks containing a telomere signal. (E) Quantification of the telomere abnormalities such as those shown in panel (D). The data from three independent experiments is shown. n = 2,000, one-way ANOVA and Tukey’s test, P < .01 = **, P < .001 = ***. (F) Quantification of total telomere abnormalities in CtIPf/- cells after treatment with 4-OHT for 36 h and Mirin or ATRN-119 for 24 h. The telomere aberrations scored include: signal free ends, fusions, internalized telomeres, and breaks including telomere signals. The data shown is from three independent experiments, n = 1750; one-way ANOVA and Tukey's test were used with P < .05 = *. (G) Quantification of γ-H2AX foci in CtIPf/- cells after treatment with 4-OHT for 36 h and Mirin or ATRi for 24 h. n = 250; one-way ANOVA and Tukey’s test were used, P < .01 = **.
To determine the mechanism of CtIP-null telomere dysfunction, we assessed the impact of inhibiting the ATR and ATM pathways in the absence of CtIP. Functional inhibition of ATR decreased the telomere aberrations caused by the absence of CtIP (Figure 4F). In contrast, inhibition of Mre11, which presumably recruits ATM, had no effect on the number of telomere defects observed (Figure 4F). Total levels of DNA damage followed this trend, with ATR inhibition lowering the overall damage observed and Mre11 inhibition having no effect (Figure 4G). The Mirin utilized in these studies was functional for its inhibition of Mre11 as validated by a strong induction of phosphorylated Chk2 and p53 in WT RPE1 cells (Supplemental Figure S7D). Interestingly, inhibition of Mre11 did change the type of telomere abnormalities observed (Supplemental Figure S7A and B). When Mre11 was inhibited and CtIP expression ablated, telomeres were more likely to fuse with another telomere or chromosome end, resulting in a decrease of lone, signal-free ends. These data imply that in the absence of CtIP and Mre11, NHEJ-mediated telomere fusion increases, but that CtIP’s role in preventing telomere loss is distinct from its interaction with Mre11. Additionally, the reduction of telomere defects upon ATR inhibition indicates that CtIP’s primary telomere role may be related to DNA replication.
CtIP protects against extrachromosomal, circular telomere accumulation
The robust telomere loss observed in CtIP-deficient cells implied a mechanism that was both fast-acting and substantial. T-circle generation, a process in which DNA repair factors aberrantly act on a t-loop and liberate large portions of the telomere in the form of extrachromosomal, circular pieces of DNA, was a possible mechanism (29,43,44) for the massive telomere attrition we observed. To experimentally determine whether CtIP-deficient cells contained t-circles, genomic DNA from these and control cells was subjected to neutral/neutral 2D gel electrophoresis. Before electrophoresis, the genomic DNA was digested to completion with the RsaI and HinfI restriction enzymes. Importantly, because t-circular DNA consists solely of repetitions of TTAGGG, it should be refractory to this digestion. The restriction enzyme digests were then subjected to 2D gel electrophoresis. Open circular (i.e., t-circle) DNA should migrate slower in the second dimension and migrate as a retarded arc in the gel. No such arc was observed in the RPE1 parental or CtIPf/− cell lines (Figure 5A). In contrast a t-circle arc was present in the CtIP null cells (48 h after they had been treated with 4-OHT) as well as in the positive control cell line, U-2 OS, an alternative lengthening of telomeres (ALT) human sarcoma cell line that is known to contain t-circular DNA (43,45).
Figure 5.

(A) 2D gel analysis of telomeric DNA extracted from U-2 OS (positive control), RPE1 WT, CtIPf/-, and CtIPf/- treated with 4-OHT for 2 days. (B) Western blot analysis to confirm the siCtIP efficiency in U-2 OS cells. Actin was used as a loading control. (C) Dot blot analysis of Phi29-amplified (+Phi) or non-amplified (–Phi) c-circles from U-2 OS WT, U-2 OS +scrambled siRNA, and U-2 OS +siCtIP cells. Amplification without DNA (–DNA) served as a negative control. (D) Quantification of c-circles in U-2 OS WT, U-2 OS +scrambled siRNA, and U-2 OS +siCtIP such as those shown in panel (C) from three independent experiments. Paired t-test, P < .001 = ***.
C-circle formation is another hallmark of ALT and is a second example of extrachromosomal, circular telomeric DNA (46). While the mechanism and purpose of c-circle generation is still unclear, several theories have been proposed. One idea is that c-circles are generated in ALT cells by impaired telomere replication and recovery (47,48). Thus, stalled replication forks resulting in reversal and/or collapse within the telomere contribute to this phenotype. The presence of c-circles was not detected in CtIPf/− + 4-OHT or RPE1 WT backgrounds (Supplemental Figure S5B). This was not surprising, as c-circles are considered atypical in hTERT-immortalized human cells, particularly those with short telomeres. Nonetheless, to pursue the possibility that CtIP might have a role in c-circle formation in ALT-immortalized cells, we addressed this question experimentally in an ALT-positive background. Correspondingly, the level of CtIP was knocked down using RNA interference (RNAi) in U-2 OS cells (Figure 5B), and this resulted in a two-fold increase in the formation of c-circles (Figure 5C and D). These findings demonstrated that CtIP plays important telomere maintenance roles in both telomerase- and ALT-positive backgrounds: in both genetic backgrounds CtIP is required to suppress extrachromosomal telomeric DNA accumulation.
CtIP depletion drives telomere replication fork stalling
Replication through a telomere is innately difficult due to the telomere's G-rich, repetitive nature and as a consequence this renders them prone to replication fork stalling (6). If replication does not resume, a stalled replication fork can be processed to collapse or reverse (49). Compromised DNA replication forks are frequently resolved via HR-mediated repair with a pivotal first step being nucleolytic processing (50). To experimentally assess DNA replication fork status at CtIP-deficient telomeres, telomere DNA combing was performed (51). Unfortunately, the RPE1 CtIPf/− cell line, when treated with 4-OHT, develops such short telomeres (∼2 kb) that they are simply not suitable for telomere combing due to the insufficient signal that a short, 2 kb linear fragment will produce. Instead, we utilized a clonal HeLa long telomere (LT) cell line. HeLa LT cells have very long telomeres (Supplemental Figure S8A), which makes them an ideal tool for telomere fiber studies. HeLa LT cells depleted of CtIP for 48 h (Figure 6B) were analyzed either for stalled, partially replicated, and fully replicated tracts at the telomere (Figure 6A and C). Depletion of CtIP in HeLa LT resulted in a two-fold increase in DNA replication forks completely stalled in the sub-telomere and also in a two-fold increase in partially replicated telomeres (Figure 6C). CtIP knockdown did not have a significant effect on the number of telomeres that were unreplicated, indicating that replication origins were firing normally in all conditions (Figure 6D). To confirm that HeLa LT cells, like RPE1 cells, also display gross telomere defects when CtIP is removed, we knocked down CtIP in this background and measured telomere length (Supplemental Figure S9D). Telomere shortening was observed after CtIP depletion and presumably by the same mechanism as in RPE1, as evidenced by the concomitant formation of extrachromosomal, circular telomeric DNA (Supplemental Figure S9C and E). Finally, to confirm that the observed telomere replication defects equated to individual telomere dysfunction, fragile telomeres were measured (Supplemental Figure S9A and B). CtIP knockdown in HeLa LT increased telomere fragility dramatically, as was also observed in the RPE1 background (Supplemental Figure S7C). Altogether, this data demonstrates that CtIP is a key player in telomere replication and it strongly implies that replication fork stalling eventually leads to replication fork collapse, which, in turn, ultimately leads to t-circle formation.
Figure 6.

(A) Representative images of arrested, partially replicated, and completely replicated telomeres in HeLa LT cells depleted of CtIP for 2 days. Replication tracts (IdU) are displayed in blue and the telomere (Telo) tracts are displayed in purple. The scale bar is 20 kb. (B) Western blot analysis confirms the siCtIP knockdown efficacy in HeLa LT cells. Cells were left untreated or treated with siRNAs (scrambled or siCtIP) and then harvested 2 days post-transfection for immunoblot analysis. (C) Quantification of telomere replication forks that were arrested, partially replicated, or completely replicated as shown in panel (A). n = 225 telomere fibers from 3 biological replicates; one-way ANOVA and Tukey's test, P < .05 = *, P < .01 = **. (D) Quantification of telomeres that were not replicated during the interval of the analysis for panel (A) in HeLa LT, HeLa LT +scrambled siRNA, and HeLa LT +siCtIP siRNA treated cells.
DISCUSSION
CtIP is an essential gene in the mouse (25,28) and it has been inferred to be essential in humans as well (24,52). To date, virtually all of the work carried out with human somatic cells has involved technology to knockdown CtIP or stably reduce its expression. Unfortunately, low levels of CtIP expression are sufficient to maintain viability and suppress most of the dysfunctions related to the absence of CtIP (24) making unequivocal analyses difficult. To overcome this biological hurdle, we generated a novel tool (which should be of significant utility for the field) to study CtIP: an inducibly-null human somatic cell culture model. This experimental approach permitted the total ablation of CtIP expression and it enabled us to observe the immediate, acute cellular responses to the absence of CtIP. Specifically, using CtIP conditionally-null cells we were able to demonstrate unequivocally that CtIP is an essential gene in human epithelial cells and—much more surprisingly—that the absence of CtIP triggers a rapid and massive telomere loss.
CtIP is essential for human telomere maintenance
Our study documents the first bona fide role for CtIP in human telomere maintenance. In the absence of CtIP, rapid telomere catastrophe arises in the form of shortened, fused, and damaged telomeres. Interestingly, this reduction is also accompanied by a shortened 3′ G-overhang. This observation is consistent with CtIP’s known role in DNA DSB repair, where it is required for short-range resection that, in turn, stimulates longer range resection (53). Human G-overhangs are known to be generated by a combination of C-strand fill-in on the lagging strand and C-strand resection of a blunt telomere on the leading strand (54). Thus, our data imply that either CtIP is directly involved in G-overhang formation and/or that it may be stimulating other nucleases such as EXO1 and DNA2 (19,20) or Apollo (55) that have been implicated in the generation of yeast and murine telomeric G-overhangs. We favor the latter model since EXO1-null human cells also show evidence of shortened G-overhangs (Stroik et al., unpublished data). Regardless of whether the mechanism is direct or indirect, the loss of CtIP reduced the 3′ G-overhang length.
Opposing theories have been proposed for how 3′ G-overhang length can or can’t contribute to telomere shortening, genomic stability and cellular senescence (56,57). Inconsistent with a role for the shortened G-overhang being primarily involved in the formation of CtIP-null associated telomere aberrations, however, was the observation that CtIP-deficient cells displayed a rapid telomere attrition, implying that telomeres were not being lost via slow erosion, as would have been expected if shortened overhangs occasionally impaired telomere capping. Rather, the absence of CtIP seemed predominately to trigger t-circle accumulation (Figure 5), which, is associated with rapid telomere shortening (43). We propose that these extrachromosomal t-circles are arising due to impaired replication fork progression. Telomeric and sub-telomeric DNA are notoriously difficult to replicate due to the elevated presence of G-quadraplexes and heterochromatin, which result in the stalling of the replication fork machinery (Figure 7). A number of HR proteins have been identified that are responsible for fork restart and replication machinery escort during DNA replication (50,58,59). Our data suggest that CtIP is one of these HR factors and that it safeguards replication by rescuing replication forks that have stalled at the telomere by facilitating their re-start (59). Consistent with this interpretation, stalled forks accumulated in the absence of CtIP (Figure 6). Importantly, however, while we have demonstrated that CtIP is an important player for telomeric replication repair, our data also suggests—consistent with other studies (50,58,59)—that CtIP not the sole regulator of this repair. Thus, ATR inhibition partially rescued the gross telomere abnormalities observed in the absence of CtIP (Figure 4). This data implies that there are ATR-regulated backup pathways that can be activated in the absence of CtIP. These backup pathways, which are likely mutagenic, are presumably normally suppressed by ATR and only become activated when canonical CtIP-dependent rescue cannot occur. These backup pathways notwithstanding, our data is consistent with the idea that CtIP is critical to facilitate rescue of stalled replication forks.
Figure 7.
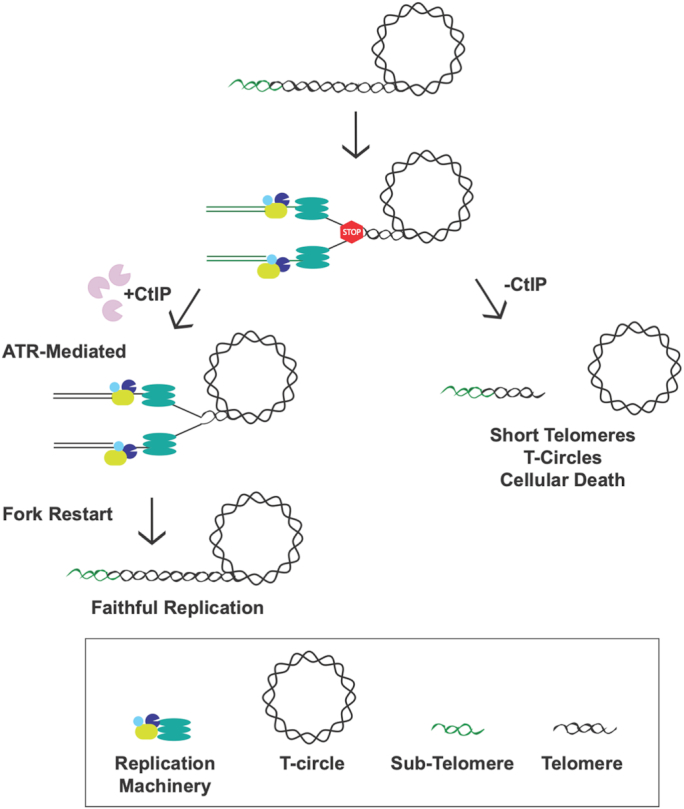
The replication fork machinery frequently stalls (STOP sign) when it encounters the telomeric sequence due to the heterochromatic nature, secondary structures, and telomere-bound proteins present. CtIP safeguards replication by facilitating either repair, restart, and/or rescue of these stalled forks leading to faithful, complete replication through the telomere. In the absence of CtIP, stalled forks persist in the telomere region eventually leading to fork collapse. Collapsed forks commonly lead to DSBs that may result in the loss of the t-loop, which, in turn, generates radically shortened telomeres and t-circles in just a few cell cycles.
Even in the presence of CtIP, however, replication re-start does not always properly occur. Upon prolonged stalling, some of these stalled forks are likely converted into breaks resulting in t-circles (Figure 7). The precise mechanism of t-circle formation is unknown, but it has repeatedly been proposed that it is likely due to the aberrant activity of HR factors on the t-looped DNA (29,43,48,60). Alternatively, and consistent with our findings, is that impaired telomere replication can also result in the accumulation of circular, telomeric DNA (61), probably through a looping-out mechanism of the replication fork whose resolution generates the t-circle and which predominately involves NHEJ factors (62). In addition, it has been proposed that a stalled and reversed fork can be bound by the canonical NHEJ protein Ku (63), and that CtIP may be needed to cleave Ku off the ends of the reversed fork in order to initiate re-start (64). Whether the copious formation of t-circles in CtIP-null cells requires either HR factors (mediators of classic t-circle formation) or NHEJ factors (mediators of replication-dependent t-circle formation) can ultimately be addressed by epistasis experiments. Finally, a recent study (24) suggested that CtIP participates primarily in the protection of a reversed replication fork through its ability to impede nuclease DNA2 degradation of that fork. Our model (Figure 7) is consistent with this possibility and indeed we postulate that in the absence of CtIP, stalled replication forks can become so aberrantly processed that t-circle formation can result (Figure 7). However, if CtIP solely had a fork protection activity, it is difficult to explain the increase in the number of stalled forks that we observe in human telomeres in the absence of CtIP (Figure 6). Thus, our data are most consistent with a role of CtIP in telomeric fork re-start, but they do not exclude an overlapping role in fork protection.
Previous work has indicated that the yeast homolog of CtIP, SAE2, together with the helicase, SGS1, also functions in telomere maintenance. In S. cerevisiae, SGS1 and SAE2 double mutants display significant telomere shortening upon extended subculturing (65). This study also documented a role for these two repair proteins in telomere replication, specifically in preventing excess ssDNA from accumulating presumably when replication goes awry. A second group additionally identified a role for SAE2 in both successful telomere elongation as well as 3′ overhang generation at telomeric termini (66). This telomere end processing by SAE2 was important for the downstream processing by other nucleases such as DNA2 and EXO1. In addition, since CtIP/SAE2 processing represents an early, commitment step in HR-mediated repair, it follows that removal of SAE2 would convey DNA damage sensitivity, which it does. Interestingly, however, the sensitivity of a SAE2 mutant to ionizing radiation can be rescued by removal of the NHEJ factor, Ku70 (13). In contrast, in our experiments, Ku70 depletion did not rescue the viability of CtIP-null cells (data not shown). Therefore, while some of CtIP’s functions in telomere maintenance, replication and repair are conserved across species, in humans at least some of CtIP’s functions are distinct from that in yeast. This is additionally evidenced by the essential nature of CtIP in mammals; a phenotype not observed with SAE2 mutants in yeast.
CtIP and telomere maintenance in ALT cells
ALT-positive, CtIP-depleted cells also accumulate another form of extrachromosomal, circular telomeric DNA: c-circles. C-circles are considered to be a specific hallmark of ALT cancers (67), and CtIP depletion clearly increased c-circle abundance in the U-2 OS ALT cell line (Figure 5). Unfortunately, the mechanism of c-circle production is still unclear and it has been proposed that these circles may be accumulating through either a replication or recombination dependent- (or perhaps both) mechanism(s) (46,48). Importantly, our data would imply that whatever the mechanism of c-circle production is, it is regulated by CtIP. Moreover, these data demonstrated that CtIP prevents free, circular telomeric DNA from accumulating in both ALT- (U-2 OS; c-circles) and hTERT- (RPE1; t-circles) positive backgrounds, implying that CtIP’s contribution to telomere maintenance is universal in immortalized cells. Perhaps most important mechanistically was the observation that c-circles were not formed in RPE1 cells, regardless of whether they were proficient or deficient in CtIP. This implies that while the mechanism for c-circle generation is distinct from t-circle generation, CtIP nonetheless contributes to both. Whether the absence of CtIP promotes the formation of telomeric circular DNA in normal (i.e. non-telomerase-positive and non-ALT cells) human cells is unknown, but will be of therapeutic importance to determine.
CtIP and Fanconi anemia-dependent repair
An outcome of prolonged fork stalling is that if the fork is not rescued in a timely manner it may result in a DSB, which can be repaired by error-prone NHEJ (5). Besides resulting in t-circle formation, this repair pathway can also generate chromosome:chromosome fusions, frank telomere loss, and genomic instability. Very interestingly, we observed a highly significant increase in the formation of radial chromosomes in CtIP-null cells (Figure 2). Radial formation is exceedingly rare and normally only associated with defects in the Fanconi Anemia (FA) pathway (68). Indeed, the presence of radial chromosomes alone is normally enough to confirm a diagnosis of FA (69). These radials are thought to be primarily generated by DNA interstrand crosslinks, and their repair often involves the transient formation of a DNA DSB to resolve the structure (70,71). Importantly, we (59) and others (22) have demonstrated that CtIP interacts with FA complementation group D2 (FANCD2), a key FA repair protein and that this interaction is important for replication restart and for faithful resolution of DNA interstrand crosslinks, respectively. Thus, the presence of radial chromosomes in CtIP-null cells strongly supports these earlier studies and it also argues that in CtIP-null cells the FA (and HR) pathway is likely defective or impaired. Thus, CtIP removal has catastrophic effects on a cell's ability to cope with these unique replication roadblocks and this—in addition to the telomere-specific defects—may also contribute to the general genetic instability (Figure 2) of CtIP-null cells.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the flow cytometry facility and university imaging center at the University of Minnesota for providing access to the Typhoon FLA, Zeiss TIRF, BD LSR II and Nikon TiE instrumentation. We also thank Dr. Vincent Leong and Taylor Takasugi (Yale University) for help and advice with the CtIPf/+ cell line generation.
Author contributions: S.S. and E.A.H. conceived and designed the overall project. S.S. and K.K. performed metaphase analyses, western blots, T-FISH, and immunofluoresence assays. K.K. performed micronuclei and c-circle assays. S.S. performed 2D gels, TRFs, G-overhang, flow cytometry, and DNA combing experiments. E.A.H., S.S. and K.K. prepared and edited the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute on Aging-funded pre-doctoral fellowship [T32 AG029796 to S.S.]; National Institutes of General Medicine [GM088351 to E.A.H.]; National Cancer Institute [CA154461 to E.A.H.]. Funding for open access charge: National Institute on Aging-funded pre-doctoral fellowship [T32 AG029796 to S.S.]; National Institutes of General Medicine [GM088351 to E.A.H.]; National Cancer Institute [CA154461 to E.A.H.].
Conflict of interest statement. E.A.H. sits on the Scientific Advisory Boards of Horizon Discovery, Ltd and Intellia Therapeutics.
REFERENCES
- 1. de Lange T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018; 52:223–247. [DOI] [PubMed] [Google Scholar]
- 2. Griffith J.D., Comeau L., Rosenfield S., Stansel R.M., Bianchi A., Moss H., de Lange T.. Mammalian telomeres end in a large duplex loop. Cell. 1999; 97:503–514. [DOI] [PubMed] [Google Scholar]
- 3. Harley C.B., Futcher A.B., Greider C.W.. Telomeres shorten during ageing of human fibroblasts. Nature. 1990; 345:458–460. [DOI] [PubMed] [Google Scholar]
- 4. Bertuch A.A. The molecular genetics of the telomere biology disorders. RNA Biol. 2016; 13:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baird D.M., Hendrickson E.A.. Telomeres and chromosomal translocations: there's a ligase at the end of the translocation. Adv. Exp. Med. Biol. 2018; 1044:89–112. [DOI] [PubMed] [Google Scholar]
- 6. Martinez P., Blasco M.A.. Replicating through telomeres: a means to an end. Trends Biochem. Sci. 2015; 40:504–515. [DOI] [PubMed] [Google Scholar]
- 7. Liao H., Ji F., Helleday T., Ying S.. Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018; 19:e46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muraki K., Murnane J.P.. The DNA damage response at dysfunctional telomeres, and at interstitial and subtelomeric DNA double-strand breaks. Genes Genet. Syst. 2018; 92:135–152. [DOI] [PubMed] [Google Scholar]
- 9. Liddiard K., Ruis B., Takasugi T., Harvey A., Ashelford K.E., Hendrickson E.A., Baird D.M.. Sister chromatid telomere fusions, but not NHEJ-mediated inter-chromosomal telomere fusions, occur independently of DNA ligases 3 and 4. Genome Res. 2016; 26:588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tubbs A., Nussenzweig A.. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017; 168:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Andres S.N., Williams R.S.. CtIP/Ctp1/Sae2, molecular form fit for function. DNA Repair (Amst.). 2017; 56:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biehs R., Steinlage M., Barton O., Juhasz S., Kunzel J., Spies J., Shibata A., Jeggo P.A., Lobrich M.. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell. 2017; 65:671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mimitou E.P., Symington L.S.. DNA end resection: many nucleases make light work. DNA Repair (Amst.). 2009; 8:983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Makharashvili N., Tubbs A.T., Yang S.H., Wang H., Barton O., Zhou Y., Deshpande R.A., Lee J.H., Lobrich M., Sleckman B.P. et al.. Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol. Cell. 2014; 54:1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anand R., Ranjha L., Cannavo E., Cejka P.. Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Mol. Cell. 2016; 64:940–950. [DOI] [PubMed] [Google Scholar]
- 16. Gnugge R., Symington L.S.. Keeping it real: MRX-Sae2 clipping of natural substrates. Genes Dev. 2017; 31:2311–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Daley J.M., Jimenez-Sainz J., Wang W., Miller A.S., Xue X., Nguyen K.A., Jensen R.B., Sung P.. Enhancement of BLM-DNA2-mediated long-range DNA end resection by CtIP. Cell Rep. 2017; 21:324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eid W., Steger M., El-Shemerly M., Ferretti L.P., Pena-Diaz J., Konig C., Valtorta E., Sartori A.A., Ferrari S.. DNA end resection by CtIP and exonuclease 1 prevents genomic instability. EMBO Rep. 2010; 11:962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Longhese M.P., Bonetti D., Manfrini N., Clerici M.. Mechanisms and regulation of DNA end resection. EMBO J. 2010; 29:2864–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nanbu T., Nguyen L.C., Habib A.G., Hirata N., Ukimori S., Tanaka D., Masuda K., Takahashi K., Yukawa M., Tsuchiya E. et al.. Fission yeast Exo1 and Rqh1-Dna2 redundantly contribute to resection of uncapped telomeres. PLoS One. 2015; 10:e0140456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dimitrova N., de Lange T.. Cell cycle-dependent role of MRN at dysfunctional telomeres: ATM signaling-dependent induction of nonhomologous end joining (NHEJ) in G1 and resection-mediated inhibition of NHEJ in G2. Mol. Cell Biol. 2009; 29:5552–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murina O., von Aesch C., Karakus U., Ferretti L.P., Bolck H.A., Hanggi K., Sartori A.A.. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep. 2014; 7:1030–1038. [DOI] [PubMed] [Google Scholar]
- 23. Wang H., Li Y., Truong L.N., Shi L.Z., Hwang P.Y., He J., Do J., Cho M.J., Li H., Negrete A. et al.. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell. 2014; 54:1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Przetocka S., Porro A., Bolck H.A., Walker C., Lezaja A., Trenner A., von Aesch C., Himmels S.F., D’Andrea A.D., Ceccaldi R. et al.. CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell. 2018; 72:568–582. [DOI] [PubMed] [Google Scholar]
- 25. Chen P.L., Liu F., Cai S., Lin X., Li A., Chen Y., Gu B., Lee E.Y., Lee W.H.. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol. Cell Biol. 2005; 25:3535–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reczek C.R., Shakya R., Miteva Y., Szabolcs M., Ludwig T., Baer R.. The DNA resection protein CtIP promotes mammary tumorigenesis. Oncotarget. 2016; 7:32172–32183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rebbeck T.R., Mitra N., Domchek S.M., Wan F., Friebel T.M., Tran T.V., Singer C.F., Tea M.K.M., Blum J.L., Tung N. et al.. Modification of BRCA1-associated breast and ovarian cancer risk by BRCA1-interacting genes. Cancer Res. 2011; 71:5792–5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Polato F., Callen E., Wong N., Faryabi R., Bunting S., Chen H.T., Kozak M., Kruhlak M.J., Reczek C.R., Lee W.H. et al.. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med. 2014; 211:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y., Ghosh G., Hendrickson E.A.. Ku86 represses lethal telomere deletion events in human somatic cells. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:12430–12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henson J.D., Lau L.M., Koch S., Martin La Rotta N., Dagg R.A., Reddel R.R.. The C-circle assay for alternative-lengthening-of-telomeres activity. Methods. 2017; 114:74–84. [DOI] [PubMed] [Google Scholar]
- 31. Yu X., Baer R.. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J. Biol. Chem. 2000; 275:18541–18549. [DOI] [PubMed] [Google Scholar]
- 32. Kohli M., Rago C., Lengauer C., Kinzler K.W., Vogelstein B.. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004; 32:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sauer B., Henderson N.. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:5166–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sternberg S.H., Doudna J.A.. Expanding the biologist's toolkit with CRISPR-Cas9. Mol. Cell. 2015; 58:568–574. [DOI] [PubMed] [Google Scholar]
- 35. Bodnar A.G., Ouellette M., Frolkis M., Holt S.E., Chiu C.P., Morin G.B., Harley C.B., Shay J.W., Lichtsteiner S., Wright W.E.. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998; 279:349–352. [DOI] [PubMed] [Google Scholar]
- 36. Katoh Y., Michisaka S., Nozaki S., Funabashi T., Hirano T., Takei R., Nakayama K.. Practical method for targeted disruption of cilia-related genes by using CRISPR/Cas9-mediated, homology-independent knock-in system. Mol. Biol. Cell. 2017; 28:898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith L.E., Nagar S., Kim G.J., Morgan W.F.. Radiation-induced genomic instability: radiation quality and dose response. Health Phys. 2003; 85:23–29. [DOI] [PubMed] [Google Scholar]
- 38. Vral A., Fenech M., Thierens H.. The micronucleus assay as a biological dosimeter of in vivo ionising radiation exposure. Mutagenesis. 2011; 26:11–17. [DOI] [PubMed] [Google Scholar]
- 39. Yun M.H., Hiom K.. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009; 459:460–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. You Z., Bailis J.M.. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010; 20:402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sharma A., Singh K., Almasan A.. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol. Biol. 2012; 920:613–626. [DOI] [PubMed] [Google Scholar]
- 42. Wu P., Takai H., de Lange T.. Telomeric 3′ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell. 2012; 150:39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang R.C., Smogorzewska A., de Lange T.. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 2004; 119:355–368. [DOI] [PubMed] [Google Scholar]
- 44. Li B., Jog S.P., Reddy S., Comai L.. WRN controls formation of extrachromosomal telomeric circles and is required for TRF2DeltaB-mediated telomere shortening. Mol. Cell Biol. 2008; 28:1892–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cesare A.J., Griffith J.D.. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol. Cell Biol. 2004; 24:9948–9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Plantinga M.J., Pascarelli K.M., Merkel A.S., Lazar A.J., von Mehren M., Lev D., Broccoli D.. Telomerase suppresses formation of ALT-associated single-stranded telomeric C-circles. Mol. Cancer Res. 2013; 11:557–567. [DOI] [PubMed] [Google Scholar]
- 47. Poole L.A., Zhao R., Glick G.G., Lovejoy C.A., Eischen C.M., Cortez D.. SMARCAL1 maintains telomere integrity during DNA replication. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:14864–14869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rivera T., Haggblom C., Cosconati S., Karlseder J.. A balance between elongation and trimming regulates telomere stability in stem cells. Nat. Struct. Mol. Biol. 2017; 24:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Petermann E., Helleday T.. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010; 11:683–687. [DOI] [PubMed] [Google Scholar]
- 50. Costes A., Lambert S.A.. Homologous recombination as a replication fork escort: fork-protection and recovery. Biomolecules. 2012; 3:39–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herrick J., Bensimon A.. Introduction to molecular combing: genomics, DNA replication, and cancer. Methods Mol. Biol. 2009; 521:71–101. [DOI] [PubMed] [Google Scholar]
- 52. Qvist P., Huertas P., Jimeno S., Nyegaard M., Hassan M.J., Jackson S.P., Borglum A.D.. CtIP mutations cause seckel and jawad syndromes. PLos Genet. 2011; 7:e1002310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Symington L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016; 51:195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dai X., Huang C., Bhusari A., Sampathi S., Schubert K., Chai W.. Molecular steps of G-overhang generation at human telomeres and its function in chromosome end protection. EMBO J. 2010; 29:2788–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu P., van Overbeek M., Rooney S., de Lange T.. Apollo contributes to G overhang maintenance and protects leading-end telomeres. Mol. Cell. 2010; 39:606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chai W., Shay J.W., Wright W.E.. Human telomeres maintain their overhang length at senescence. Mol. Cell Biol. 2005; 25:2158–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rahman R., Forsyth N.R., Cui W.. Telomeric 3′-overhang length is associated with the size of telomeres. Exp. Gerontol. 2008; 43:258–265. [DOI] [PubMed] [Google Scholar]
- 58. Michel B., Boubakri H., Baharoglu Z., LeMasson M., Lestini R.. Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst.). 2007; 6:967–980. [DOI] [PubMed] [Google Scholar]
- 59. Yeo J.E., Lee E.H., Hendrickson E.A., Sobeck A.. CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum. Mol. Genet. 2014; 23:3695–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Compton S.A., Choi J.H., Cesare A.J., Ozgur S., Griffith J.D.. Xrcc3 and Nbs1 are required for the production of extrachromosomal telomeric circles in human alternative lengthening of telomere cells. Cancer Res. 2007; 67:1513–1519. [DOI] [PubMed] [Google Scholar]
- 61. Margalef P., Kotsantis P., Borel V., Bellelli R., Panier S., Boulton S.J.. Stabilization of reversed replication forks by telomerase drives telomere catastrophe. Cell. 2018; 172:439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang T., Zhang Z., Li F., Hu Q., Liu H., Tang M., Ma W., Huang J., Songyang Z., Rong Y. et al.. Looping-out mechanism for resolution of replicative stress at telomeres. EMBO Rep. 2017; 18:1412–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Teixeira-Silva A., Ait Saada A., Hardy J., Iraqui I., Nocente M.C., Freon K., Lambert S.A.E.. The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat. Commun. 2017; 8:1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ait Saada A., Lambert S.A.E., Carr A.M.. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair (Amst.). 2018; 71:135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hardy J., Churikov D., Geli V., Simon M.N.. Sgs1 and Sae2 promote telomere replication by limiting accumulation of ssDNA. Nat. Commun. 2014; 5:5004. [DOI] [PubMed] [Google Scholar]
- 66. Bonetti D., Martina M., Clerici M., Lucchini G., Longhese M.P.. Multiple pathways regulate 3′ overhang generation at S. cerevisiae telomeres. Mol. Cell. 2009; 35:70–81. [DOI] [PubMed] [Google Scholar]
- 67. Henson J.D., Cao Y., Huschtscha L.I., Chang A.C., Au A.Y., Pickett H.A., Reddel R.R.. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009; 27:1181–1185. [DOI] [PubMed] [Google Scholar]
- 68. Sasaki M.S., Tonomura A.. A high susceptibility of Fanconi's anemia to chromosome breakage by DNA cross-linking agents. Cancer Res. 1973; 33:1829–1836. [PubMed] [Google Scholar]
- 69. Kee Y., D’Andrea A.D.. Molecular pathogenesis and clinical management of Fanconi anemia. J. Clin. Invest. 2012; 122:3799–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kottemann M.C., Smogorzewska A.. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013; 493:356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McCabe K.M., Olson S.B., Moses R.E.. DNA interstrand crosslink repair in mammalian cells. J. Cell Physiol. 2009; 220:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.