


Abstract
Genetic polymers that could plausibly govern life in the universe might inhabit a broad swath of chemical space. A subset of these genetic systems can exchange information with RNA and DNA and could therefore form the basis for model protocells in the laboratory. N3′→P5′ phosphoramidate (NP) DNA is defined by a conservative linkage substitution and has shown promise as a protocellular genetic material, but much remains unknown about its functionality and fidelity due to limited enzymatic tools. Conveniently, we find widespread NP-DNA-dependent DNA polymerase activity among reverse transcriptases, an observation consistent with structural studies of the RNA-like conformation of NP-DNA duplexes. Here, we analyze the consequences of this unnatural template linkage on the kinetics and fidelity of DNA polymerization activity catalyzed by wild-type and variant reverse transcriptases. Template-associated deficits in kinetics and fidelity suggest that even highly conservative template modifications give rise to error-prone DNA polymerase activity. Enzymatic copying of NP-DNA sequences is nevertheless an important step toward the future study and engineering of this synthetic genetic polymer.
INTRODUCTION
Chemical exploration has revealed a vast space of RNA and DNA alternatives that exhibit structural and functional homology to native nucleic acids (1). Many of these alternative genetic polymers can base pair with RNA, which could conceivably point to diverse prebiotic paths to ribose-based life (2) or for the emergence of life elsewhere in the universe. These alternatives could also form the basis for engineering laboratory-grown protocells that exhibit rapid genome copying reactions in the absence of evolved polymerases (2–6).
Alternative backbone chemistry yields a spectrum of geometries and thermodynamic stabilities, as well as other biochemical properties that have been exploited technologically (7). However, only a few of these alternative genetic polymers have been shown to be functionally active in catalysis, a foundational requirement for a Darwinian cell (1,8). Among the options, nucleotide phosphoramidates form relatively well-studied oligonucleotides with backbone linkages not found in nature, and they have shown promise as potential genetic materials for developing a model protocell in the laboratory. These oligonucleotides are characterized by the substitution of nitrogen-phosphorus bonds (NP) in place of the phosphodiester linkage found in native nucleic acids. Chemically activated amino sugar nucleotide derivatives can polymerize rapidly in aqueous copying reactions, yielding phosphoramidate-linked complementary strands on RNA, DNA or phosphoramidate-linked templates (3–5,9–11). Synthetic phosphoramidate DNA oligonucleotides incorporating the linkage regioisomer N3′→P5′ (NP-DNA, Figure 1A) and their sulfurized thiophosphoramidate derivatives have also been of technological interest in the development of RNA therapeutics (12,13) and have been shown to inhibit the reverse transcriptase activity of telomerase (14–16). NP-DNA is hydrolytically stable under basic conditions and is resistant to a range of nucleolytic activities, including those that require a 2′-OH and those that cleave the 3′-O-P bond (12,17). However, enzymatic tools to analyze and manipulate NP-DNA are not yet available. Besides telomerase, the activity of the broader class of reverse transcriptases (RTs) or other polymerases on NP-DNA remains unknown, to our knowledge. Identifying polymerases that operate on NP-DNA would permit analysis of product fidelity arising from non-enzymatic copying reactions with this highly reactive genetic system, and it could also yield the tools necessary to generate functional aptamers and catalysts incorporating this backbone chemistry.
Figure 1.

Reverse transcriptase activity on synthetic NP-DNA oligonucleotide templates. (A) Chemical structure of N3′→P5′ phosphoramidate-linked DNA (NP-DNA, left) versus native RNA (right). (B) Crystal structures of NP-DNA (left) and RNA (right) duplex helices, adapted from PDB structures 363D (19) and 1RNA (22). (C) Cartoon of primer extension assays for reverse transcriptase activity on RNA (black) and NP-DNA (magenta) templates. A FAM-labeled DNA primer (green, 5′-FAM-AGTGAGTAACGC, 2 μM) was annealed with one of three complementary strands (3 μM), consisting of an RNA primer binding site (black) 3′-r(UCACUCAUUGCG) followed by a template region of six ribothymidine residues (rT6), six NP-DNA 3′-aminothymidine residues (nT6) or three repeats of the NP-linked dinucleotide n(TG)3. (D) Primer extension with 50 nM wild-type MMLV or AMV RT enzyme at 37°C with either 0.5 mM of each dNTP or with 0.5 mM dATP alone, as indicated. For each reaction, samples were taken at 3, 20, 30 and 60 min (left-to-right lanes) and analyzed by 20% TBE-urea PAGE. (E) Primer extension reactions with 50 nM of the MMLV-derived RNase H activity-deficient RT mutant ProtoScript II (PS2) or the AMV RT-derived RNase H activity-deficient mutant ThermoScript (TS) with conditions as in panel (D), except samples were taken at 2, 10, 30 and 60 min (left-to-right lanes). Each enzyme was incubated in its supplied buffer formulation, which contained 3 mM Mg2+ for MMLV and PS2 (M buffer) or 8 mM Mg2+ for AMV and TS (A buffer).
Structural studies both in crystals and in solution suggest that duplex NP-DNA adopts an A-form helical conformation similar to that of RNA (Figure 1B), in which the 3′-amino-2′,3′-dideoxyribose exhibits a sugar pucker conformation similar to 3′-endo despite the absence of a 2′-substituent (18–20). Circular dichroism of DNA:NP-DNA duplexes also shows an A-form-like UV hyperchromicity near 270 nm, similar to that observed for RNA:DNA duplexes but with some apparent sequence-dependence (21). Given these observations of NP-DNA’s structural homology to RNA (22), we asked whether synthetic NP-DNA oligonucleotides might be templates for RNA-dependent DNA polymerase activity. Here we report that RT enzymes are indeed active on NP-DNA templates, and we characterize the consequences of this conservative linkage substitution on RT-catalyzed DNA polymerization activity.
MATERIALS AND METHODS
Materials
Reverse transcriptases from AMV and MMLV (M-MuLV), as well as the variant ProtoScript II (PS2), were obtained from New England Biolabs. ThermoScript (TS) RT was obtained from ThermoFisher. HIV-1 reverse transcriptase was purchased from Worthington Biochemical. Phosphoramidite synthesis reagents were purchased from Glen Research. Ribothymidine phosphoramidite and other canonical DNA and RNA phosphoramidites were purchased from ChemGenes. 5′-O-(2-cyanoethoxy(diisopropylamino)-phosphino)-3′-N-(4-methoxytriphenylmethyl)amino-2′,3′-dideoxythymidine was purchased from ChemGenes. 3′-Amino-2′,3′-dideoxyguanosine and 3′-amino-2′,3′-dideoxyadenosine were purchased from ThermoFisher. 3′-Amino-2′,3′-dideoxycytidine was purchased from Carbosynth. Solvents were purchased from Sigma Aldrich or ThermoFisher, were of anhydrous grade, and were used without further purification. Silica gel chromatography was performed using Redisep Rf Normal Phase silica cartridges on a CombiFlash system (Teledyne ISCO).
Primer extension reactions
Fluorescently labeled DNA primers were extended on RNA, NP-DNA or chimeric template strands using commercially available reverse transcriptase enzymes. For all reactions, fluorescein-labeled primer and template oligonucleotides were first denatured in RNase-free water containing 50 μM sodium EDTA by heating to 75°C briefly, and then cooled to room temperature. The primer/template was then equilibrated with reaction buffer and RT enzymes at the reaction temperature, as indicated, and primer extension was initiated by the addition of a dNTP mixture or a single nucleoside triphosphate substrate alone. Extension reactions were performed in one of two possible buffer formulations, indicated as AMV (A) or MMLV (M) RT buffers and diluted to 1× from the manufacturer’s concentrated stocks. Buffer A contained 50 mM Tris-acetate, 75 mM potassium acetate, 8 mM magnesium acetate, 10 mM DTT at pH 8.3. Buffer M contained 50 mM Tris-HCl, 75 mM KCl, 3 mM MgCl2 and 10 mM DTT at pH 8.3. Reaction samples were taken at the indicated times and quenched by diluting 25-fold into 90% formamide containing 10 mM sodium EDTA. Samples were denatured at 90°C before separation on 20% polyacrylamide TBE-urea gels. Primer fluorescence was visualized with a laser scanning imager (Typhoon, GE Amersham).
Oligonucleotide synthesis
NP-DNA oligonucleotides (Supplementary Table S3) were synthesized essentially as described in Nelson et al. (23,24) with minor modifications. Briefly, 1 μmol scale automated synthesis was performed on an Expedite 8909 oligonucleotide synthesizer in the ‘reverse’ 5′→3′ direction. Reverse protected 5′-phosphoramidites were dissolved at 67–100 mM in anhydrous MeCN and maintained under Ar. The deblocking reagent was 3% trichloroacetic acid in CH2Cl2. The cap A reagent was 1:9 (v/v) isobutyric anhydride in anhydrous THF, and cap B was anhydrous 1:1:8 (v/v/v) 1-methylimidazole:Py:THF. As reported, a low water oxidizer mix, 0.1 M iodine in 2:20:78 (v/v/v) H2O:Py:THF, is critical to the yield of NP-DNA oligonucleotides, likely due to the reversibility of the coupling reaction. The activator reagent was 0.25 M 5-benzylthio-1H-tetrazole (BTT) in anhydrous MeCN. Reverse synthesis proceeded from a CPG support displaying a preattached 3′-blocked nucleoside or from a universal support using the cycling conditions reported in Nelson et al. adapted for the Expedite synthesizer. The crude product was released from the support in concentrated (30%) ammonium hydroxide over 30 min at room temperature, and nucleobase deprotection was carried out by incubating the collected eluate at 55°C for 12–16 h in a sealed screw-top tube. Volatiles in the deprotection reaction were partially removed in vacuo, and then the crude product was flash frozen and dried fully by lyophilization. For chimeric sequences containing RNA positions, the 2′-O-silyl protection was removed by resuspending the lyophilized residue in 0.1 ml anhydrous DMSO, followed by 60 μl anhydrous TEA and 75 μl TEA:3HF. The vial was sealed and placed at 55°C for 3–4 h, quenched by addition of buffer (Glen Research RNA quenching reagent) and desalted by ion pairing using a C18 cartridge. The desalted oligonucleotides were further purified by PAGE and characterized by LC-HRMS. In some cases, an additional round of purification by reverse phase C18 ion-pairing HPLC was performed following PAGE, using a gradient of 20 mM triethylammonium bicarbonate (TEAB), pH 7, in water versus MeCN.
Circular dichroism spectroscopy
Oligonucleotide samples were prepared at 10 μM in 50 mM Tris-HCl, pH 8.3, 75 mM KCl in a quartz 1-mm cuvette at 25°C. Aliquots of 0.1 M MgCl2 in 50 mM Tris-HCl, pH 8.3, 75 mM KCl were added, and reported final concentrations were adjusted for the volume addition during titration. CD spectra between 320 and 200 nm were taken on an AVIV model 202 CD Spectrometer with a bandwidth of 1 nm, averaging time 0.1 s, settling time 0.33 s and were averaged from two scans. A baseline subtraction from buffer alone was performed, and the resulting spectra were smoothed by application of a quadratic Savitsky-Golay filter with a bandwidth of 5 nm.
Fluorescence spectroscopy
Intrinsic tryptophan fluorescence was measured in a quartz cuvette using a Cary Eclipse Fluorescence Spectrophotometer. Samples were excited at 285 nm with slit width 5 nm, and emission was monitored at 340 nm with slit width 20 nm. Enzyme stocks were diluted to 20 nM (MMLV wild-type and PS2) or 10 nM (TS) in a buffer composed of 50 mM Tris-Cl, pH 8, 75 mM KCl, 8 mM MgCl2 and 1 mM DTT. Template RNA or NP-DNA (14 nt) was added to a concentration of 500 nM, and the mixture was equilibrated at 23°C. Aliquots of a 9 nt DNA primer, prediluted into the same buffer, were then added sequentially to the cuvette in ∼1 min intervals, and emission intensity was measured at 10 s intervals for stability following each addition. Titrations were performed in duplicate, except where indicated. The raw intensities were corrected for dilution and the internal filter effect, and the resulting normalized fluorescence intensities were fit to a quadratic one-step ligand binding model, essentially as in Divita et al. (25).
RESULTS
Reverse transcription on synthetic NP-DNA templates
Reverse transcriptases catalyze both RNA-dependent and DNA-dependent DNA polymerization as key aspects of the retroviral life cycle (26). We began by screening several readily available polymerases for the ability to synthesize complementary DNA (cDNA) on NP-DNA oligonucleotides prepared with homopolymeric or heteropolymeric templating sequences. We used phosphoramidite chemistry to synthesize chimeric oligonucleotides containing NP-DNA template sequences (23,24,27) downstream of an RNA primer binding site (Figure 1C). A complementary fluorescently labeled DNA primer was extended on these templates in the presence of all four canonical deoxynucleotide triphosphates (dNTP) or dATP alone at 37°C, using buffer conditions recommended for each enzyme’s RNA-templated activity (Figure 1C–E and Supplementary Figure S1). NP-DNA oligonucleotides have traditionally been synthesized with 3′-amino-3′-deoxythymidine (nT) rather than the corresponding uridine derivative; for consistency with prior reports, we chose to do the same and to compare enzyme activities on synthetic NP-DNA templates versus RNA containing ribothymidine (rT) to control for the potential methylation effect on thermodynamic stability (28).
Wild-type Murine Moloney leukemia virus (MMLV or Mu-MLV) RT and a commercial RNase H activity-deficient variant ProtoScript II (PS2) exhibited qualitatively similar RT activity when extending a DNA primer on chimeric RNA/NP-DNA templates, which contain an RNA primer binding site followed by an NP-DNA template region (Figure 1D and E, upper panels). Both showed evidence of stalling at the +2 and +3 positions on an nT6 template at 37°C in buffer conditions typical for their native activities. Wild-type avian myeloblastoma virus (AMV) RT and a commercial RNase H− mutant ThermoScript (hereafter, TS) also showed activity on homopolymeric and repeat NP-DNA templates (Figure 1D and E, lower panels), but TS showed virtually no stalling on nT6 and substantially improved activity on the repeat n(TG)3 sequence, yielding 33% full-length product within 1 h. Moreover, in reactions containing dATP alone, mismatch read-through extension of all four RTs on n(TG)3 was significantly stalled at +1. In contrast, while the wild-type RT from HIV-1 exhibited activity on all templates, it also showed substantial untemplated 3′-tailing beyond the length of the template strand. HIV-1 RT also rapidly achieved and exceeded full-length extension on the n(TG)3 repeat template with dATP alone, indicating an extremely low fidelity RT activity (Supplementary Figure S1).
Reverse transcriptases also recognized primer/template complexes when the DNA primer is bound to an NP-DNA template, rather than RNA (Figure 2). Binding isotherms for titrated DNA primer were produced by monitoring tryptophan fluorescence of RT enzymes in the presence of excess template. For three tested RT enzymes, the macroscopic impact of the non-native NP-DNA template on the formation of ternary complexes was minimal, and the resulting binding curves showed remarkably similar affinities, with dissociation constants in the range 5–6 nM (Supplementary Table S1) in the presence of 8 mM Mg2+ with both RNA and NP-DNA templates for MMLV RT, PS2 (Figure 2A) and TS (Figure 2B) enzymes. These affinities estimated by tryptophan fluorescence were in the range of previously reported values for HIV-1 and AMV RTs on various templates (∼1–10 nM) (29–32). Interestingly, the relative magnitude of tryptophan fluorescence quenching was notably less pronounced upon complexation in the presence of excess NP-DNA template relative to excess RNA template. This could plausibly indicate a difference in the conformational state of bound complexes containing NP-DNA. However, since the titration was performed as primer addition to excess template, a condition chosen to minimize the concentration-dependence of primer/template duplex formation and the inner-filter effect as confounders, it remains possible that an interaction with single-stranded NP-DNA could explain the discrepancy. Nevertheless, since no apparent defect in macroscopic binding constants was associated with NP-DNA templates relative to native RNA, we conclude that the conditions for all RT reactions reported here occur in a regime that is saturated for primer/template binding.
Figure 2.

Recognition of NP-DNA/DNA heteroduplexes and templated primer extension activity of reverse transcriptases. (A and B) Equilibrium binding isotherms for DNA primer in the presence of excess template, given as normalized change in fluorescence intensity, 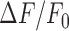 . Titrations of a 9 nt DNA primer (5′-GCGATCAGC) at 23°C into A buffer (8 mM Mg2+) containing 20 nM PS2 (A) or 10 nM TS (B) enzyme and excess template (0.5 μM) composed entirely of either RNA (5′-r[ACACGCUGAUCGCA], black) or NP-DNA (5′-n[ACACGCTGATCGCA], magenta) backbones, monitored by tryptophan fluorescence (λex = 285 nm, λem = 340 nm). For PS2, estimated dissociation constants, Kd, were 4.8 ± 0.3 nM (n = 2) or 6.1 ± 0.2 nM (n = 3) with RNA or NP-DNA templates, respectively (mean ± S.E.M.). For TS, Kd values were 6.0 ± 0.3 nM (n = 2) or 6.4 ± 2.0 nM (n = 2) for RNA or NP-DNA templates, respectively. Also see Supplementary Table S1 for these binding constant estimates compared to wild-type MMLV RT, not shown here. (C) A 5′ FAM-labeled (green star) DNA primer (gray sequence, 1 μM) was extended on the indicated fully NP-DNA template (magenta, 2 μM) at 42°C in A buffer (containing 8 mM Mg2+) using 250 nM TS enzyme. (D) Extension as in panel (C) at the indicated temperature with 250 nM PS2 enzyme with either A buffer or M buffer, containing 8 mM or 3 mM Mg2+, respectively. Reaction samples for panels (C) and (D) were quenched at 0, 15 min, 1 h, 2 h and 8 h (left-to-right lanes for each condition) and analyzed by 20% TBE-urea PAGE. (E) Reaction temperature optimization on mixed repeat template n(TG)3. Reaction conditions were as described in panel (C) for all reactions, but oligonucleotide concentrations were as in Figure 1 (2 μM primer, 3 μM template), and reaction temperatures were as indicated. Time points for each condition were taken and quenched at 10 min, 1 h, 2 h and 4 h (left to right for each indicated reaction temperature).
. Titrations of a 9 nt DNA primer (5′-GCGATCAGC) at 23°C into A buffer (8 mM Mg2+) containing 20 nM PS2 (A) or 10 nM TS (B) enzyme and excess template (0.5 μM) composed entirely of either RNA (5′-r[ACACGCUGAUCGCA], black) or NP-DNA (5′-n[ACACGCTGATCGCA], magenta) backbones, monitored by tryptophan fluorescence (λex = 285 nm, λem = 340 nm). For PS2, estimated dissociation constants, Kd, were 4.8 ± 0.3 nM (n = 2) or 6.1 ± 0.2 nM (n = 3) with RNA or NP-DNA templates, respectively (mean ± S.E.M.). For TS, Kd values were 6.0 ± 0.3 nM (n = 2) or 6.4 ± 2.0 nM (n = 2) for RNA or NP-DNA templates, respectively. Also see Supplementary Table S1 for these binding constant estimates compared to wild-type MMLV RT, not shown here. (C) A 5′ FAM-labeled (green star) DNA primer (gray sequence, 1 μM) was extended on the indicated fully NP-DNA template (magenta, 2 μM) at 42°C in A buffer (containing 8 mM Mg2+) using 250 nM TS enzyme. (D) Extension as in panel (C) at the indicated temperature with 250 nM PS2 enzyme with either A buffer or M buffer, containing 8 mM or 3 mM Mg2+, respectively. Reaction samples for panels (C) and (D) were quenched at 0, 15 min, 1 h, 2 h and 8 h (left-to-right lanes for each condition) and analyzed by 20% TBE-urea PAGE. (E) Reaction temperature optimization on mixed repeat template n(TG)3. Reaction conditions were as described in panel (C) for all reactions, but oligonucleotide concentrations were as in Figure 1 (2 μM primer, 3 μM template), and reaction temperatures were as indicated. Time points for each condition were taken and quenched at 10 min, 1 h, 2 h and 4 h (left to right for each indicated reaction temperature).
On mixed sequence NP-DNA templates, RT enzymes can achieve full-length products on useful timescales using elevated enzyme concentrations. On a 23 nt NP-DNA template oligonucleotide, primer extension to or beyond full length (+11 or +12, respectively, from a 12 nt primer/template duplex) was reached in 53% yield with 250 nM TS in 1 h at 42°C and 8 mM MgCl2 (Figure 2C). However, virtually no activity could be observed with PS2 on this template in the canonical MMLV RT reaction buffer formulation (M buffer) that contains 3 mM Mg2+. Activity could be recovered by running the PS2 extension reaction in the canonical buffer for AMV RT derivatives (A buffer), which contains 8 mM Mg2+, with full-length product achievable in 47% yield of +11 or +12 products at 42°C in 1 h (Figure 2D). These reactions occurred below the expected melting temperature for the primer/template duplex, and no significant structural changes could be observed by circular dichroism (CD) in this range of Mg2+ concentrations for full-length DNA:NP-DNA duplexes (Supplementary Figure S2). Temperature optima on the fully NP-DNA template (Figure 2D) as well as the chimeric repeat n(TG)3 template (Figure 2E) were similar for both RNase H– RT variants in the presence of all four dNTPs. With 8 mM Mg2+ in the reaction buffer, PS2 showed maximal initial activity at 50°C on the chimeric n(TG)3 template, but showed little apparent reaction progress beyond early time points at this temperature, likely due to thermal inactivation of the enzyme. Similar behavior was observed on the fully mixed NP-DNA template. TS activity was comparable on both n(TG)3 and mixed sequence templates but was highest at 42°C.
We sought further insight into the mechanistic aspects of NP-DNA-templated RT activity by generating steady-state kinetic profiles as a function of dATP concentration on homopolymeric rT6 or nT6 templates (Table 1). At 37°C and in a reaction buffer containing 8 mM Mg2+, the substrate-dependent initial rates were reasonably well fit by a simple Michaelis–Menten model (Supplementary Figure S3). Apparent catalytic efficiencies, 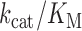 , for all three tested RTs were similar across enzymes for each template, but the catalytic rate associated with activity on an NP-DNA template was approximately one order-of-magnitude lower than on an RNA template. This drop in efficiency on nT6 versus rT6 was more pronounced for MMLV RT (∼27-fold) and its RNase H-deficient variant PS2 (∼51-fold), compared to the AMV RT variant TS (∼8-fold). Interestingly, the impact of phosphoramidate linkage substitution on steady-state poly-dA extension activity was not solely explicable by macroscopic KM at steady-state, implying that the linkage may have microscopic effects on the rate limiting step of catalysis.
, for all three tested RTs were similar across enzymes for each template, but the catalytic rate associated with activity on an NP-DNA template was approximately one order-of-magnitude lower than on an RNA template. This drop in efficiency on nT6 versus rT6 was more pronounced for MMLV RT (∼27-fold) and its RNase H-deficient variant PS2 (∼51-fold), compared to the AMV RT variant TS (∼8-fold). Interestingly, the impact of phosphoramidate linkage substitution on steady-state poly-dA extension activity was not solely explicable by macroscopic KM at steady-state, implying that the linkage may have microscopic effects on the rate limiting step of catalysis.
Table 1.
Steady-state kinetic parameters from initial rates of dATP polymerization on homopolymeric templates rT6 or nT6 as in Figure 1C
| Template | Enzyme |
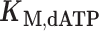 (μM) (μM) |
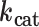 (min−1) (min−1) |
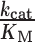 (μM−1 min−1) (μM−1 min−1) |
|---|---|---|---|---|
| MMLV | 20.9 ± 0.3 | 241 ± 31 | 12 | |
| rT6 | PS2 | 21 ± 2 | 336 ± 9 | 16 |
| TS | 19 ± 6 | 78 ± 7 | 4.2 | |
| MMLV | 110 ± 22 | 48 ± 3 | 0.43 | |
| nT6 | PS2 | 160 ± 40 | 49 ± 5 | 0.31 |
| TS | 128 ± 16 | 68 ± 3 | 0.53 |
Kinetic measurements were performed at 37°C in the A buffer formulation (8 mM Mg2+) containing 25 nM enzyme, 1 μM primer, 2 μM template and initiated by addition of varying concentrations of dATP. Parameter estimates are given as mean ± s.e.m. for n = 2 titrations (see Supplementary Figure S3).
The appearance of stalling products during extension on mixed sequences and the kinetic defect associated with NP-DNA polymerase activity could indicate important effects on the fidelity of extended products. If stalling arises from an elevated rate of mismatch incorporation during primer extension, then any resumption of extension beyond a mismatch will result in the incorporation of mutations into full-length products. Viral reverse transcriptases generally show high in vitro mutation rates even on their native templates, in the range of 10−4–10−5 (29,33,34), owing in part to the absence of proofreading domains that are characteristic of high fidelity replicative DNA-dependent DNA polymerases (35). We therefore sought to characterize the fidelity of NP-DNA-templated RT activity by high-throughput sequencing of full-length products (Figure 3). We generated synthetic templates containing two flanking DNA primer binding sites surrounding either an NP-DNA region or a continuous DNA region of interest, and we analyzed the mutations observed in the complementary DNA strand generated by RT primer extension (Supplementary Figures S4 and S5). Relative to typical conditions, the primer extension reactions performed for this assay used elevated concentrations of Mg2+ (8 mM), enzyme (250 nM) and dNTPs (1 mM) at 42°C, chosen based on preliminary experiments to yield full-length products on NP-DNA templates. In Figure 3, mutation matrices show each expected base on the primer-extended strand (rows) and the rate of observed mutations to every other possible base (columns). The mutation rates on a DNA template were on same order of magnitude as those reported for wild-type AMV RT activity on RNA and DNA templates (∼10−4) (30,34), but somewhat higher than those reported on DNA templates in a recent single-molecule high-throughput analysis of RT activity (5–6 × 10−5) (36). In contrast, the mean substitution error rates observed on NP-DNA templating regions were on the order of 3–4 × 10−3 for both RNase H– RT variants. Relative to a DNA template, a substantial increase in mutation rate was observed for both RT homologs: 14-fold for PS2 (Figure 3C) and 7-fold for TS (Figure 3F), respectively. Misincorporation errors on NP-DNA templates were elevated for all possible mutations in DNA products generated by both enzymes, but unique patterns of mutation emerged for the two enzymes. For instance, while both enzymes show large increases in A→C and T→G mutation rates, the effect was opposite in magnitude between the two enzymes: PS2 showed a larger effect on T→G, whereas TS showed a larger effect on A→C. The wobble pairs T:G and G:T, resulting in the respective G→A and C→T transitions, appear to be significant drivers of mutation for both enzymes on NP-DNA, but the largest relative increases in mutation rate on NP-DNA versus DNA templates were transversions for both enzymes. Finally, we repeated the analysis using a 1:1 mol/mol blend of the two enzymes (Figure 3G–I). The resulting mutation matrix was more similar to TS than PS2, but intermediate rates of G→A and C→T mutations on the product strand were evident.
Figure 3.

Mutation matrices for RT primer extension products on synthetic templates. Expected (rows) versus observed (columns) base calls for the extended RT product strand for DNA polymerization activity of either PS2 (A–C), TS (D–F) or a 1:1 blend of both enzymes (G–I) on either NP-DNA (A, D and G) or DNA (B, E and H) synthetic templates. Each entry shows the rates (×10−4) of each observed substitution mutation relative to the reference sequence. The right-most column in each table is the summation (Σ) of all mutation rates for each expected base, and the overall mean of these values is given below each table. In this analysis, only base substitution calls were considered. See Supplementary Figure S4 for methodological details. (C, F and I) Fold-change in mutation rate for NP-DNA versus DNA templates for PS2, TS, or a 1:1 blend, respectively. Color in each cell is mapped between the maximum (red) to minimum (green) normalized within each table. Extension reactions contained 250 nM PS2 (A–C), 250 nM TS (D–F) enzyme or a blend of 125 nM PS2 and 125 nM TS (G–I). In all cases, the reaction was carried out at 42°C with 1 mM each dNTP in buffer A, containing 8 mM Mg2+.
To validate the fidelity seen in full-length products, we sought to measure the kinetics of PS2 catalyzed incorporation of either correct (dCTP) or incorrect (TTP) substrates on an nG template position (C→T), which corresponded to the highest mutation rate observed in the sequencing dataset. To do so under conditions similar to those used in sequencing studies, we used an elevated enzyme concentration and measured initial reaction velocities as a function of substrate concentration. However, the analysis was complicated by the observation that the transient phase rate constant, 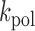 , for correct single-addition of dCTP on nG at high enzyme concentration, 250 nM, appeared substantially lower, 1.2 ± 0.4 min−1 (Supplementary Figure S6), than that measured at steady-state for dATP on nT using 10-fold lower enzyme concentration (Table 1). Indeed, the steady-state
, for correct single-addition of dCTP on nG at high enzyme concentration, 250 nM, appeared substantially lower, 1.2 ± 0.4 min−1 (Supplementary Figure S6), than that measured at steady-state for dATP on nT using 10-fold lower enzyme concentration (Table 1). Indeed, the steady-state 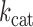 for single dCTP addition on nG was 11 ± 0.9 min−1 at 25 nM enzyme, ∼10-fold higher rate constant at 10-fold lower enzyme concentration (Supplementary Figure S6). This suggests that the elevated enzyme concentration used to achieve full-length extension may be partially inactivating for PS2. Nevertheless, if we estimate the misincorporation frequency,
for single dCTP addition on nG was 11 ± 0.9 min−1 at 25 nM enzyme, ∼10-fold higher rate constant at 10-fold lower enzyme concentration (Supplementary Figure S6). This suggests that the elevated enzyme concentration used to achieve full-length extension may be partially inactivating for PS2. Nevertheless, if we estimate the misincorporation frequency,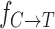 , using kinetic parameters obtained at the elevated enzyme concentration used for sequencing with the expression
, using kinetic parameters obtained at the elevated enzyme concentration used for sequencing with the expression 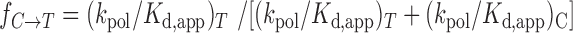 (37,38), where (
(37,38), where (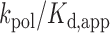 ) is the catalytic efficiency for addition of either T or dC obtained under noncompetitive transient conditions, we obtain a misincorporation rate of 2.4 × 10−3 for the C→T transition (Supplementary Table S2). This value compares favorably to the C→T mutation rate 3.5 × 10−3 obtained by sequencing full-length products of PS2 activity (Figure 3A).
) is the catalytic efficiency for addition of either T or dC obtained under noncompetitive transient conditions, we obtain a misincorporation rate of 2.4 × 10−3 for the C→T transition (Supplementary Table S2). This value compares favorably to the C→T mutation rate 3.5 × 10−3 obtained by sequencing full-length products of PS2 activity (Figure 3A).
DISCUSSION
The observation that reverse transcriptases exhibit template-directed polymerase activity on NP-DNA indicates the high degree of structural and functional homology retained by this conservative linkage substitution relative to RNA. By comparison, phosphodiester-linked nucleic acid derivatives with unnatural sugars (e.g. TNA, HNA, FANA, CeNA and LNA) have generally required more extensive polymerase variant screening to achieve significant RT activity (39,40), although substantial activity was recently reported on TNA templates with the wild-type polymerase from Bacillus stearothermophilus (Bst) (41). With NP-DNA templates, we observed that the macroscopic binding affinity of reverse transcriptases for primer/template complexes was largely unaffected by the linkage substitution. RTs nevertheless showed clear defects in kinetics and fidelity, implying exquisite sensitivity of the polymerase reaction cycle to even highly conservative chemical substitutions in template nucleic acids. The effect of magnesium concentration on NP-DNA-templated activity, the altered tryptophan fluorescence upon ternary complex formation, the presence of stalled extension products and reduced fidelity, and the large effect on steady-state kinetics generally point to a constellation of effects on the polymerase reaction cycle. These likely include effects of the bound template on the enzyme’s affinity for substrate and magnesium ion (42,43), on the magnesium-dependent stabilization of the active complex (44), and/or on the kinetics of conformational changes in the reaction cycle (45).
Although we observed significant decreases in fidelity on NP-DNA templates, it is important to note that these values set only an upper bound on the mutation rate observable in full-length RT products generated on this unnatural template linkage, since we could not differentiate the contribution of synthetic template oligonucleotide impurities on the assay (see Experimental Procedures in Supplementary Data). Of technical relevance, we indeed observed isolated site-specific errors during sequencing analysis, which appeared in the same position and to a similar extent across the two enzymes used for reverse transcription on synthetic NP-DNA templates (Supplementary Figure S5). We suspect that this effect is due to errors in oligonucleotide synthesis by phosphoramidite chemistry. These apparent mutation hot-spots remained observable even after exhaustive template purifications—a likely result of the low-yielding existing methodologies for NP-DNA oligonucleotide synthesis. The technical consequences of impurities in synthetic DNA oligonucleotide preparations have been reviewed elsewhere, particularly in the context of gene synthesis (46). Nevertheless, distinctions in mutation patterns between the two enzymes indicate that the mutation matrices reported here are likely to arise at least in part from altered enzyme fidelity on NP-DNA templates, not solely template-derived error. Furthermore, for the most prevalent mutation in the dataset, we validated using kinetic methods that the fidelity observed by sequencing full-length products is similar to the value estimated by denaturing gel analysis of primer extension products.
The ability to synthesize complementary DNA from NP-DNA templates is a key step toward the in vitro selection of functional aptamers and ribozymes composed of NP-DNA, as well as for efforts to characterize the products of non-enzymatic copying reactions that produce NP-DNA oligonucleotides. The predominant scaffolds for engineering backbone-modified polymerases have been thermophilic DNA-dependent DNA polymerases, such as the DNA polymerase I homologs from Thermus aquaticus (Taq) or Thermococcus gorgonarius (Tgo) (47), enzymes that are amenable to thermal cycling. On the other hand, the intrinsic ability of retroviral polymerases to catalyze both DNA- and RNA-templated primer extension is well known (48–50), implying that these enzymes could be appropriate starting points for laboratory evolution of enzymes that operate on alternative genetic polymers exhibiting predominantly A-form geometry.
Supplementary Material
Notes
Present address: Enver Cagri Izgu, Department of Chemistry and Chemical Biology, Rutgers University, Piscataway, New Jersey, 08854, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Howard Hughes Medical Institute (to J.W.S.); Simons Foundation [290363 to J.W.S.]; NSF [CHE-1607034 to J.W.S.]; Fonds de Recherche du Quebec−Nature et Technologies (FRQNT) (fellowship to D.K.O.); Canadian Institutes of Health Research (CIHR) (fellowship to D.K.O.). Funding for open access charge: HHMI.
Conflict of interest statement. None declared.
REFERENCES
- 1. Eschenmoser A. Chemical etiology of nucleic acid structure. Science. 1999; 284:2118–2124. [DOI] [PubMed] [Google Scholar]
- 2. Orgel L.E. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004; 39:99–123. [DOI] [PubMed] [Google Scholar]
- 3. Lohrmann R., Orgel L.E.. Template-directed synthesis of high molecular weight polynucleotide analogues. Nature. 1976; 261:342–344. [DOI] [PubMed] [Google Scholar]
- 4. Zielinski W.S., Orgel L.E.. Oligomerization of activated derivatives of 3′-amino-3′-deoxyguanosine on poly(C) and poly(dC) templates. Nucleic Acids Res. 1985; 13:2469–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mansy S.S., Schrum J.P., Krishnamurthy M., Tobé S., Treco D.A., Szostak J.W.. Template-directed synthesis of a genetic polymer in a model protocell. Nature. 2008; 454:122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schrum J.P., Zhu T.F., Szostak J.W.. The origins of cellular life. Cold Spring Harbor Perspect. Biol. 2010; 2:a002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bell N.M., Micklefield J.. Chemical modification of oligonucleotides for therapeutic, bioanalytical and other applications. ChemBioChem. 2009; 10:2691–2703. [DOI] [PubMed] [Google Scholar]
- 8. Joyce G.F., Orgel L.E.. Prospects for understanding the origin of the RNA world. Cold Spring Harbor Monograph Series. 1999; 37:49–78. [Google Scholar]
- 9. Zhang S., Zhang N., Blain J.C., Szostak J.W.. Synthesis of N3′-P5′-linked phosphoramidate DNA by nonenzymatic Template-Directed primer extension. J. Am. Chem. Soc. 2013; 135:924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang S., Blain J.C., Zielinska D., Gryaznov S.M., Szostak J.W.. Fast and accurate nonenzymatic copying of an RNA-like synthetic genetic polymer. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:17732–17737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cagri Izgu E., Soo Oh S., Szostak J.W.. Synthesis of activated 3′-amino-3′-deoxy-2-thio-thymidine, a superior substrate for the nonenzymatic copying of nucleic acid templates. Chem. Commun. 2016; 52:3684–3686. [DOI] [PubMed] [Google Scholar]
- 12. Gryaznov S. Oligonucleotide N3′→P5′ phosphoramidates as antisense agents. Nucleic Acids Res. 1996; 24:1508–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gryaznov S.M. Oligonucleotide N3′ → P5′ phosphoramidates as potential therapeutic agents. Biochim. Biophys. Acta. 1999; 1489:131–140. [DOI] [PubMed] [Google Scholar]
- 14. Gryaznov S., Pongracz K., Matray T., Schultz R., Pruzan R., Aimi J., Chin A., Harley C., Shea-Herbert B., Shay J. et al.. Telomerase inhibitors–oligonucleotide phosphoramidates as potential therapeutic agents. Nucleosides Nucleotides Nucleic Acids. 2001; 20:401–410. [DOI] [PubMed] [Google Scholar]
- 15. Shea-Herbert B., Pongracz K., Shay J.W., Gryaznov S.M.. Oligonucleotide N3′ → P5′ phosphoramidates as efficient telomerase inhibitors. Oncogene. 2002; 21:638–642. [DOI] [PubMed] [Google Scholar]
- 16. Herbert B.-S., Gellert G.C., Hochreiter A., Pongracz K., Wright W.E., Zielinska D., Chin A.C., Harley C.B., Shay J.W., Gryaznov S.M.. Lipid modification of GRN163, an N3′ → P5′ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene. 2005; 24:5262–5268. [DOI] [PubMed] [Google Scholar]
- 17. Hanna R.L., Gryaznov S.M., Doudna J.A.. A phosphoramidate substrate analog is a competitive inhibitor of the Tetrahymena group I ribozyme. Chem. Biol. 2000; 7:845–854. [DOI] [PubMed] [Google Scholar]
- 18. Ding D., Gryaznov S.M., Lloyd D.H., Chandrasekaran S., Yao S., Ratmeyer L., Pan Y., Wilson W.D.. An oligodeoxyribonucleotide N3′ → P5′ phosphoramidate duplex forms an A-type helix in solution. Nucleic Acids Res. 1996; 24:354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tereshko V., Gryaznov S., Egli M.. Consequences of replacing the DNA 3′-Oxygen by an Amino Group: High-Resolution crystal structure of a fully modified N3′ → P5′ Phosphoramidate DNA dodecamer duplex. J. Am. Chem. Soc. 1998; 120:269–283. [Google Scholar]
- 20. Ding D., Gryaznov S.M., Wilson W.D.. NMR solution structure of the N3′ → P5′ Phosphoramidate Duplex d(CGCGAATTCGCG) 2 by the iterative relaxation matrix approach. Biochemistry. 1998; 37:12082–12093. [DOI] [PubMed] [Google Scholar]
- 21. Rigl C.T., Lloyd D.H., Tsou D.S., Gryaznov S.M., Wilson W.D.. Structural RNA Mimetics: N3′ → P5′ Phosphoramidate DNA analogs of HIV-1 RRE and TAR RNA Form A-Type helices that bind specifically to Rev and Tat-Related peptides. Biochemistry. 1997; 36:650–659. [DOI] [PubMed] [Google Scholar]
- 22. Dock-Bregeon A.C., Chevrier B., Podjarny A., Johnson J., de Bear J.S., Gough G.R., Gilham P.T., Moras D.. Crystallographic structure of an RNA helix: [U(UA)6A]2. J. Mol. Biol. 1989; 209:459–474. [DOI] [PubMed] [Google Scholar]
- 23. Nelson J.S., Fearon K.L., Nguyen M.Q., McCurdy S.N., Frediani J.E., Foy M.F., Hirschbein B.L.. N3′ → P5′ oligodeoxyribonucleotide phosphoramidates: a new method of synthesis based on a phosphoramidite amine-exchange reaction. J. Org. Chem. 1997; 62:7278–7287. [DOI] [PubMed] [Google Scholar]
- 24. Fearon K.L., Nelson J.S.. Synthesis and purification of oligonucleotide N3′→P5′ Phosphoramidates and their phosphodiester and phosphorothioate chimeras. Curr. Protoc. Nucleic Acid Chem. 2000; 3:4.7.1–4.7.40. [DOI] [PubMed] [Google Scholar]
- 25. Divita G., Mueller B., Immendoerfer U., Gautel M., Rittinger K., Restle T., Goody R.S.. Kinetics of interaction of HIV reverse transcriptase with primer/template. Biochemistry. 1993; 32:7966–7971. [DOI] [PubMed] [Google Scholar]
- 26. Goff S.P. Retroviral reverse transcriptase: synthesis, structure, and function. J. Acquir. Immune Defic. Syndr. 1990; 3:817–831. [PubMed] [Google Scholar]
- 27. McCurdy S.N., Nelson J.S., Hirschbein B.L., Fearon K.L.. An improved method for the synthesis of N3′ → P5′ phosphoramidate oligonucleotides. Tetrahedron Lett. 1997; 38:207–210. [Google Scholar]
- 28. Wang S., Kool E.T.. Origins of the large differences in stability of DNA and RNA Helixes: C-5 Methyl and 2′-Hydroxyl effects. Biochemistry. 1995; 34:4125–4132. [DOI] [PubMed] [Google Scholar]
- 29. Kati W.M., Johnson K.A., Jerva L.F., Anderson K.S.. Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 1992; 267:25988–25997. [PubMed] [Google Scholar]
- 30. Yu H., Goodman M.F.. Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 1992; 267:10888–10896. [PubMed] [Google Scholar]
- 31. Creighton S., Huang M.M., Cai H., Arnheim N., Goodman M.F.. Base mispair extension kinetics. Binding of avian myeloblastosis reverse transcriptase to matched and mismatched base pair termini. J. Biol. Chem. 1992; 267:2633–2639. [PubMed] [Google Scholar]
- 32. Lanchy J.M., Ehresmann C., Le Grice S.F., Ehresmann B., Marquet R.. Binding and kinetic properties of HIV-1 reverse transcriptase markedly differ during initiation and elongation of reverse transcription. EMBO J. 1996; 15:7178–7187. [PMC free article] [PubMed] [Google Scholar]
- 33. Roberts J.D., Preston B.D., Johnston L.A., Soni A., Loeb L.A., Kunkel T.A.. Fidelity of two retroviral reverse transcriptases during DNA-dependent DNA synthesis in vitro. Mol. Cell Biol. 1989; 9:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Preston B.D., Poiesz B.J., Loeb L.A.. Fidelity of HIV-1 reverse transcriptase. Science. 1988; 242:1168–1171. [DOI] [PubMed] [Google Scholar]
- 35. Ellefson J.W., Gollihar J., Shroff R., Shivram H., Iyer V.R., Ellington A.D.. Synthetic evolutionary origin of a proofreading reverse transcriptase. Science. 2016; 352:1590–1593. [DOI] [PubMed] [Google Scholar]
- 36. Potapov V., Fu X., Dai N., Corrêa I.R., Tanner N.A., Ong J.L.. Base modifications affecting RNA polymerase and reverse transcriptase fidelity. Nucleic Acids Res. 2018; 46:5753–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fersht A. Enzyme Structure and Mechanism. 1985; NY: W H Freeman & Co. [Google Scholar]
- 38. Bertram J.G., Oertell K., Petruska J., Goodman M.F.. DNA polymerase Fidelity: Comparing direct competition of right and wrong dNTP substrates with steady state and Pre-Steady state kinetics. Biochemistry. 2010; 49:20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pinheiro V.B., Taylor A.I., Cozens C., Abramov M., Renders M., Zhang S., Chaput J.C., Wengel J., Peak-Chew S.-Y., McLaughlin S.H. et al.. Synthetic genetic polymers capable of heredity and evolution. Science. 2012; 336:341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pinheiro V.B., Holliger P.. The XNA world: progress towards replication and evolution of synthetic genetic polymers. Curr. Opin. Chem. Biol. 2012; 16:245–252. [DOI] [PubMed] [Google Scholar]
- 41. Dunn M.R., Chaput J.C.. Reverse transcription of threose nucleic acid by a naturally occurring DNA polymerase. ChemBioChem. 2016; 17:1804–1808. [DOI] [PubMed] [Google Scholar]
- 42. Beechem J.M., Otto M.R., Bloom L.B., Eritja R., Reha-Krantz L.J., Goodman M.F.. Exonuclease−polymerase active site partitioning of Primer−Template DNA strands and equilibrium Mg 2+ binding properties of bacteriophage T4 DNA polymerase. Biochemistry. 1998; 37:10144–10155. [DOI] [PubMed] [Google Scholar]
- 43. Raper A.T., Reed A.J., Suo Z.. Kinetic mechanism of DNA polymerases: contributions of conformational dynamics and a third divalent metal ion. Chem. Rev. 2018; 118:6000–6025. [DOI] [PubMed] [Google Scholar]
- 44. DeStefano J.J., Bambara R.A., Fay P.J.. Parameters that influence the binding of human immunodeficiency virus reverse transcriptase to nucleic acid structures. Biochemistry. 1993; 32:6908–6915. [DOI] [PubMed] [Google Scholar]
- 45. Johnson K.A. The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta. 2010; 1804:1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kosuri S., Church G.M.. Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods. 2014; 11:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Houlihan G., Arangundy-Franklin S., Holliger P.. Exploring the chemistry of genetic information storage and propagation through polymerase engineering. Acc. Chem. Res. 2017; 50:1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huber H.E., McCoy J.M., Seehra J.S., Richardson C.C.. Human immunodeficiency virus 1 reverse transcriptase. Template binding, processivity, strand displacement synthesis, and template switching. J. Biol. Chem. 1989; 264:4669–4678. [PubMed] [Google Scholar]
- 49. Loeb L.A., Tartof K.D., Travaglini E.C.. Copying natural RNAs with E. coli DNA polymerase I. Nat. New Biol. 1973; 242:66–69. [DOI] [PubMed] [Google Scholar]
- 50. Karkas J.D. Reverse transcription by Escherichia coli DNA polymerase I. Proc. Natl Acad. Sci. U.S.A. 1973; 70:3834–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.