


Abstract
ATP-dependent chromatin remodeling factors of SWI/SNF2 family including ISWI, SNF2, CHD1 and INO80 subfamilies share a conserved but functionally non-interchangeable ATPase domain. Here we report cryo-electron microscopy (cryo-EM) structures of the nucleosome bound to an ISWI fragment with deletion of the AutoN and HSS regions in nucleotide-free conditions and the free nucleosome at ∼ 4 Å resolution. In the bound conformation, the ATPase domain interacts with the super helical location 2 (SHL 2) of the nucleosomal DNA, with the N-terminal tail of H4 and with the α1 helix of H3. Density for other regions of ISWI is not observed, presumably due to disorder. Comparison with the structure of the free nucleosome reveals that although the histone core remains largely unchanged, remodeler binding causes perturbations in the nucleosomal DNA resulting in a bulge near the SHL2 site. Overall, the structure of the nucleotide-free ISWI-nucleosome complex is similar to the corresponding regions of the recently reported ADP bound ISWI-nucleosome structures, which are significantly different from that observed for the ADP-BeFx bound structure. Our findings are relevant to the initial step of ISWI binding to the nucleosome and provide additional insights into the nucleosome remodeling process driven by ISWI.
INTRODUCTION
Genomic DNA of eukaryotic organisms is wrapped around the histone octamer core forming compact nucleosome units that in turn are tightly packed into higher-order chromatin structures to allow the genetic material to efficiently fit within the cell nucleus. A highly condensed form of genomic DNA is important for accurate segregation of the genetic material during cell division, however, the compaction also limits access to the genetic information by regulatory proteins that are critical for accomplishing fundamental cellular processes including transcription, repair, replication, and recombination. For example, in the context of chromatin, eukaryotic gene expression is highly regulated and requires various protein factors that ensure proper temporal control. Transcription is initiated by DNA-binding factors that recognize specific sequences in promoters and recruit multi-subunit coactivator complexes (1). These coactivators contain enzymes that can post-translationally modify histones and chromatin remodelers that alter the position of nucleosomes or evict histones from the DNA.
Chromatin remodelers are a family of ATP-dependent enzymes that utilize energy generated from ATP hydrolysis to facilitate access to genomic DNA (2). These remodeling factors are divided into four subfamilies (SWI/SNF: SWItching defective/Sucrose Non-Fermenting, CHD: Chromodomain Helicase DNA binding, INO80: INOsitol requiring 80, and ISWI: Imitation SWItch), all of which share a catalytic ATPase domain in addition to one or more accessory domains needed to perform subfamily specific functions (2,3). Malfunctions in these remodeling factors are associated with aging and cancer and this class of proteins has therefore generated great interest as therapeutic targets (3–6). Recent structural studies have provided molecular insights on how ISWI, SNF2, CHD1 and INO80/SWR1 interact with nucleosome (7–17) but a complete understanding of ISWI functional mechanism is still lacking (18,19). In the present study, we determined a cryo-EM structure of the complex formed between nucleosome and the ATPase domain of the Chaetomium thermophilum ISWI. We carried out detailed comparisons with the structure of the free nucleosome (also determined in the present study), previously available structures of the free ISWI (14), and the recently reported nucleotide-bound ISWI-nucleosome complexes (20) to provide a more comprehensive understanding of the functional mechanism of ISWI class of remodeling enzymes.
MATERIALS AND METHODS
Construction of expression plasmids
The codon-optimized DNA fragment encoding the ATPase fragment ISWI77–134-GSSG-ISWI167–722 of the Chaetomium thermophilum ISWI (hereafter referred to as CtISWI77-Δ-722) was synthesized (GENEWIZ, South Plainfield, NJ) and cloned into pProEX-Htb expression vector using restriction enzyme MfeI and BamHI, which had an N-terminal histidine tag and TEV protease cleavage site (M-AHHHHHHGHHH-ENLYFQG-SSS-ISWI77–134-GSSG-ISWI167–722). The flexible ‘GSSG’ linker is used to replace the L3 loop and α4 helix of the AutoN region to increase the binding affinity of the remodeler for the nucleosome (14).
Expression and purification of the CtISWI77-Δ-722 ATPase fragment
Escherichia coli BL21-CodonPlus(DE3)-RIPL (Agilent Technologies, Wilmington, DE, USA) was used for expression of the recombinant CtISWI77-Δ-722 ATPase fragment. Escherichia coli cells harboring the CtISWI77-Δ-722 ATPase fragment expression plasmid were grown at 37°C in 1× LB Broth (IPM Scientific) with 100 μg ml−1 Ampicillin until OD600 reached around 1.0. At this point, recombinant protein expression was induced by adding 0.3 mM IPTG (isopropyl β-d-1-thiogalactopyranoside, final concentration) for 18–20 h at 14°C. The cells were harvested at 4,000 RPM for 10 min, then washed twice with 40 ml wash buffer containing 20 mM Tris–HCl pH 8.0, 500 mM NaCl and 2 mM β-ME. Cells were resuspended in 40 ml binding buffer containing 20 mM Tris–HCl pH 8.0, 500 mM NaCl, 20 mM imidazole, 5 mM β-ME, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 U ml−1 Benzonase Nuclease (Sigma-Aldrich, Saint Louis, MO, USA) and 10 μg ml−1 Ribonuclease A (Sigma-Aldrich, Saint Louis, MO), followed by sonication on ice for a total of 10 min with a pulse of 5 s on, and 10 s off, and centrifugation at a speed of 35,000 RPM for 2 h and at 4°C. The protein was first purified using Ni-NTA agarose (QIAGEN, Valencia, CA, USA) following the protocol provided by the manufacturer and further purified by one round of size-exclusion chromatography (SEC) in a buffer containing 20 mM Tris–HCl pH 7.4, 200 mM NaCl, and 5 mM β-ME.
Nucleosome preparation
Drosophila melanogaster histones H2A, H2B, H3 and H4, and 147-mer Widom ‘601’ positioning sequence DNA were used to reconstitute nucleosome core particle (hereafter referred to as NCP 147) in vitro as described previously (21).
Preparation of the CtISWI77-Δ-722-nucleosome complex
A complex of CtISWI77-Δ-722 with NCP 147 was prepared by adding 1 ml of 10 μM CtISWI77-Δ-722 protein, dropwise into 10 ml of 0.3 μM mononucleosome with a ratio of 3.3:1, in a buffer containing 10 mM Tris–HCl, pH 7.4, 80 mM NaCl and 2 mM β-ME, with gentle stirring for 30 min. The solution was concentrated to a final volume of 1 ml. The complex was purified by SEC with a Superdex 200 Increase 10/300 GL column (GE Healthcare) controlled by AKTA FPLC at a flow rate of 1 ml min−1 using a buffer containing 10 mM Tris–HCl, pH 7.4, 80 mM NaCl and 2 mM β-ME. Two fractions corresponding to 9.5–10 ml and 10–10.5 ml fractions were collected. They were concentrated to a final volume of 100 μl, with a final sample concentration of 9.5 μM and 9.6 μM, respectively.
Cryo-EM specimen preparation and image acquisition
Vitrified samples of CtISWI77-Δ-722-nucleosome complex were prepared by first cleaning Quantifoil R1.2/1.3, 200 mesh holey carbon grids (Quantifoil, Jena, Germany) using H2/O2 gas mixture for 10 s in a Solarus plasma cleaner (Gatan, Inc., Pleasanton, CA, USA) followed by addition of 2.8 μl of sample at a concentration of 2.0−2.5 μM. The sample was allowed to absorb for 5 s prior to blotting for 7 s followed by plunge freezing into liquid ethane cooled at liquid N2 temperature with a Leica EM GP (Leica Microsystems Inc., Buffalo Grove, IL, USA) operating at 20°C and 95% humidity. We did not utilize any crosslinking agents to stabilize the complex formation between the nucleosome and the ISWI remodeler fragment in our analysis to prevent any potential artifacts that could result from cross-linking.
Cryo-EM imaging was performed using a Titan Krios (FEI Company) electron microscope, operated at 300 kV, aligned for parallel illumination, and operated in zero-energy-loss mode with a slit width of 20 eV, with the specimen maintained at liquid nitrogen temperature. Images were recorded on K2 Summit camera equipped with the XP sensor (Gatan, Pleasanton), operated in super-resolution counting mode with a defocus range of –0.7 to –2.5 μm, corresponding to a pixel size of 0.42 Å at the specimen plane. Each exposure was recorded using automated data acquisition in Latitude software (Gatan Inc.) as a 38-frame movie, with dose rate and exposure time of ∼2.6 e Å−2 s−1 and 15.2 s, respectively.
Image processing
Movie frame alignment from the K2 Summit dataset were carried out as described previously (22) and the average micrographs were processed in the framework of RELION 2.1 (23). The integrated images were used for CTF estimation with CTFFIND3 as implemented in the RELION workflow. An initial set of ∼10k particles was selected manually to generate reference-free 2D class averages for semi-automated particle picking using RELION. A total of 570,521 particles were selected and extracted from the 1948 micrographs. The extracted particles were subjected to multiple rounds of 2D classifications and low-population or poorly-defined classes were discarded to remove junk or inconsistent particles. The 480,472 particles remaining after 2D classification were subjected to multiple rounds of 3D classifications with C1 symmetry to finally obtain two distinct classes corresponding to the CtISWI77-Δ-722-nucleosome complex and the free nucleosome. Three-dimensional classification was bootstrapped using previously determined low-pass filtered map of the free nucleosome as an initial model (24). The final selected 3D classes were refined within RELION workflow until convergence was reached. The refined maps of the CtISWI77-Δ-722-nucleosome complex and free nucleosome were sharpened using the Post-processing module within RELION workflow with automatically determined B-factor values of –144 and –141 Å2, respectively, to produce the final maps.
Model building and structure analysis
An initial model of the nucleosome core was generated with a rigid body fit of the histone octamer corresponding to the X-ray structure of the free nucleosome reconstituted with Drosophila histone proteins (PDB 4X23) (25), and 147-mer Widom 601 DNA sequence corresponding to the previously reported structure of the nucleosome complex (PDB 6BUZ) (24) into the cryo-EM density map of the free nucleosome using UCSF Chimera (26). This initial model was optimized by manual rebuilding in COOT (27), followed by real space refinement using PHENIX (28), and validation using Molprobity (29) and EMRinger (30).
For the CtISWI77-Δ-722-nucleosome structure, an initial model for the nucleosome core was built in a similar manner to that of the free nucleosome described above. In addition, additional residues at the N-terminal tail of histone H4 (A15-R23, Chain B) was modeled based on the structure of SNF2-nucleosome complex (PDB 5X0Y) (12). Since the density in the N-terminal tail of histone H4 is highly disordered these additional residues were pruned to Cβ atoms. We next generated a homology model for the nucleosome-bound CtISWI77-Δ-722 fragment based on the crystal structure of the free Myceliophthora thermophila ISWI (MtISWI81–723, PDB 5JXR) as the template (14) using SWISS-MODEL server (31). Finally, the nucleosome core and the individual domains of the CtISWI77-Δ-722 fragments were fitted into the CtISWI77-Δ-722-nucleosome density map to generate a combined molecular model that was optimized and validated similar to the free nucleosome structure (28–30). The final model for the bound CtISWI included residues F174–I436, and T450–Q640 that roughly corresponds to the ATPase domain of the remodeler. Several residues of the CtISWI77-Δ-722 fragment with poor or lack of side-chain density were pruned to Cβ. Other regions of the CtISWI construct including the majority of the AutoN domain (residues 80–187) and all of the NegC domain (residues 650–722) were not observed in the density map.
Fourier shell correlation (FSC) curves were generated based on the Post-processing output from RELION. Local resolution calculations were performed using RELION. Molecular figures were prepared using UCSF Chimera (26) and PyMol (32). Electrostatic calculations were performed using APBS (Adaptive Poisson-Boltzmann Solver) (33). Domain movement was analyzed using DynDom (34).
RESULTS AND DISCUSSION
Structure of the CtISWI77-Δ-722-nucleosome complex
An important aspect of the ISWI functional mechanism involves specific recognition of the nucleosome substrate that couples with ATP binding and hydrolysis to facilitate the translocation step of the remodeling reaction. The ATPase domain of ISWI alone is sufficient to bind and translocate nucleosomes along the DNA and the binding per se is not regulated by the presence nucleotide (35,36), suggesting that the initial recognition of the nucleosome substrate is primarily driven by the remodeler. To determine structural aspects of the binding between nucleosome and the remodeling factor ISWI, we carried out cryo-EM analysis of the complex formed between the Chaetomium thermophilum ISWI encoding for residues 77–134 and residues 167–722 connected with a GSSG linker (hereafter referred to as CtISWI77-Δ-722) and the canonical nucleosome consisting of Drosophila melanogaster histones and the 147 bp widom ‘601’ DNA sequence. Residues K135-V166 that includes the L3 loop and α4 helix of the AutoN region of the ISWI remodeler were replaced by a GSSG linker from the present construct to improve its binding to the nucleosome (Figure 1) (14). The density maps of the 3D models revealed two distinct classes at overall resolutions of 4.07 Å (CtISWI77-Δ-722-nucleosome complex) and 3.95 Å (free nucleosome) (Figure 1A and B, Supplementary Figure S1 and Supplementary Table S1). In both classes, the nucleosome core is resolved at resolutions that are high enough to visualize side-chain densities for the histone proteins (Supplementary Figures S2 and S3). Although the bound remodeler in the CtISWI77-Δ-722-nucleosome complex is resolved at a lower resolution, secondary structural features and side-chain densities can be observed clearly for several segments of the remodeler (Supplementary Figure S4).
Figure 1.
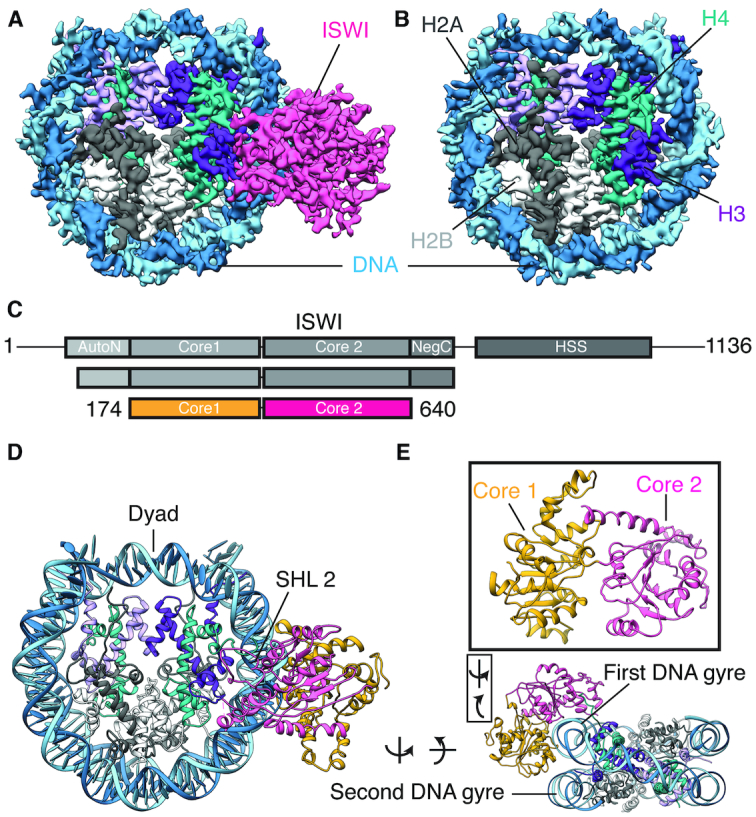
Structure of the ISWI-nucleosome complex. (A and B) Cryo-EM density maps of the CtISWI77-Δ-722-nucleosome complex (A) and the free nucleosome (B). The density corresponding to the bound CtISWI is shown in pink. H2A, H2B and H4 are colored in gray, white and teal respectively. H3 is shown in shades of purple to illustrate the ∼2-fold symmetry of histone core. The two strands of DNA are shown in cyan and blue. (C) Schematic representation of the functional domains of CtISWI (top panel), the CtISWI construct used for the present structural analysis (middle panel), and the regions of the CtISWI whose structure was modeled in the density map of CtISWI77-Δ-722-nucleosome (bottom panel). The two subdomains of CtISWI ATPase are labeled as core 1 (yellow) and core 2 (pink). (D and E) Different views of the structural model corresponding to the CtISWI77-Δ-722-nucleosome complex.
The density observed for the bound CtISWI77-Δ-722 is largely accounted by the core 1 (residues D187–V400) and core 2 (residues S407-Q640; G641-A651 of core 2 are disordered) of the ATPase domain of the remodeler (Figure 1C–E). Density corresponding to other regions of CtISWI77-Δ-722 are not observable, suggesting that they are disordered. The disordering of the remaining region of AutoN and NegC in the nucleosome-bound CtISWI77-Δ-722 is in contrast to the previously reported crystal structure of the M. thermophila ISWI (containing the catalytic core consisting of residues H81−S723, hereafter referred to as MtISWI81–723, PDB 5JXR) determined in the nucleosome-free form (14), suggesting that these domains become disordered upon nucleosome binding. It should be noted that in the present CtISWI77-Δ-722 construct, the L3 loop and the α4 helix were replaced by a flexible ‘GSSG’ linker. This is consistent with the nucleosome-bound conformation of CtISWI77-Δ-722 that is incompatible with the AutoN domain in the similar location as that in free MtISWI81–723 (Supplementary Figure S5A) (14), suggesting that large structural rearrangements of the AutoN domain occur in ISWI remodelers upon nucleosome binding. Overall, our structure allows a detailed structural examination of the molecular determinants responsible for the specific recognition of nucleosome substrate and the subsequent conformational changes that are induced upon remodeler binding.
Interactions with the nucleosomal DNA
In the CtISWI77-Δ-722-nucleosome complex, the ATPase domain of the remodeler binds at the super-helical location 2 (SHL 2) site of the nucleosomal DNA (Figure 1D and E), a feature consistent with the other members of this family except the INO80 that binds at the SHL 6–SHL 7 position (7–15). The core 1 of the CtISWI ATPase domain intercalates between the two gyres of nucleosomal DNA while the core 2 domain traverses through the first DNA gyre and reaches towards the lateral face of the nucleosome core (Figure 2A and B). Inspection of the CtISWI77-Δ-722-nucleosome structure reveals that several positively charged residues including K243, K269, R272, H273, R297, H301, K303, K304, H316, R317 and K319 from core 1 and R460, K461, H541, R544, R566, K622, R626 and K630 from core 2 of the ATPase domain face the negatively charged backbone phosphates of nucleosomal DNA that are largely contributed by SHL 2 region on the first DNA gyre, and the SHL –5 region on the second DNA gyre (Figure 2B and C).
Figure 2.
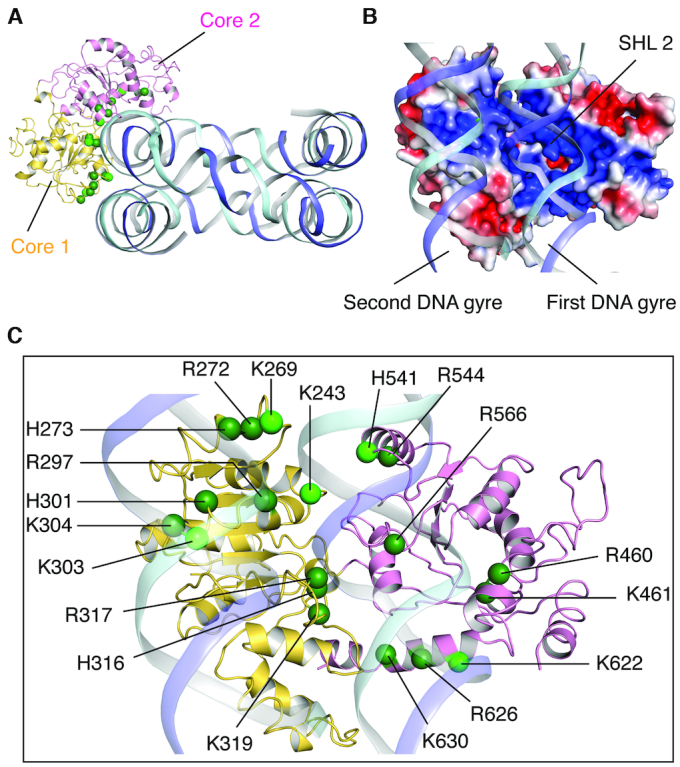
Interaction between ISWI and nucleosomal DNA. (A) Overview of the CtISWI77-Δ-722-binding to the nucleosomal DNA. Histone core is removed for clarity. See more details in panel (C). (B) Close-up and ∼90° rotated view of the panel (A). CtISWI77-Δ-722 is shown in surface representation colored using electrostatic potential (blue – positive, red – negative). (C) Close-up and ∼90° rotated view of the panel (A). Positively charged residues at the interface of the nucleosomal DNA are shown as spheres. Light green represents residues that are highly conserved within the ISWI subfamily.
To further examine the conservation of the electrostatic interactions between the nucleosomal DNA and the ISWI remodeler, we carried out sequence-based comparison using remodelers from all the four subfamilies. Among the residues that were identified to interact with the nucleosomal DNA, R272, K319, R460–K461, R544, R566, R626, K630 were found to be highly conserved. Residues H273, R297, K304 and H316–R317 also show high conservation though substitutions were observed at a few of these locations (Supplementary Figure S6). Intriguingly, residues K243, K269, K303, H541 and K622 (shown in light green in Figure 2C) are conserved within the ISWI subfamily but corresponding substitutions at these locations also show high conservation within the respective subfamily (Supplementary Figure S6). Altogether, the structure of the CtISWI77-Δ-722-nucleosome in conjunction with sequence analysis revealed important interactions that are conserved among all the remodelers, further reinforcing the idea of overall mechanistic similarity with family specific substitutions to fine-tune the function of ATP-dependent chromatin remodelers (2,3).
Interaction with histones
We next examined the CtISWI77-Δ-722-nucleosome complex for interactions formed between the remodeler and the histone octamer. The ATPase domain of CtISWI forms a few, but critical interactions with histones H4 and H3, and no interactions with histones H2A or H2B (Figure 3). The N-terminal tail of H4 contains a patch of basic residues (16–KRHRK–20 of H4) that protrudes out of the nucleosome core and binds to the acidic patch on the remodeler formed by residues E470 and E473, D519 (also part of motif IV), E522 and E523 (Figure 3A and B, and Supplementary Figure S7). All of the negatively charged residues forming the acidic patch are highly conserved (Asp or Glu) in all the remodeler subfamilies beside the exception of G728 in ScChd1 (in place of E522 of CtISWI). Noticeably, this substitution of E to G is largely preserved within the Chd1 subfamily, highlighting another subfamily-specific change in the otherwise conserved feature among all the remodelers of this family. Furthermore, in the nucleosome-free MtISWI81–723 structure, the same acidic patch is occupied by the L3 loop of AutoN domain that contains a patch of basic residues (149–RRRR–152 of MtISWI) similar to the N-terminal tail of histone H4 (Figure 3C) (14,37). This suggests a direct competition between the L3 loop of AutoN domain in ISWI and the N-terminal tail of H4 to bind to the common site on the ATPase domain of the remodeler (Supplementary Figure S5B). This is consistent with the role of N-terminal tail of H4 in the activation of the ATPase domain by likely destabilizing or preventing the AutoN binding to the acidic patch in the nucleosomal environment (38).
Figure 3.
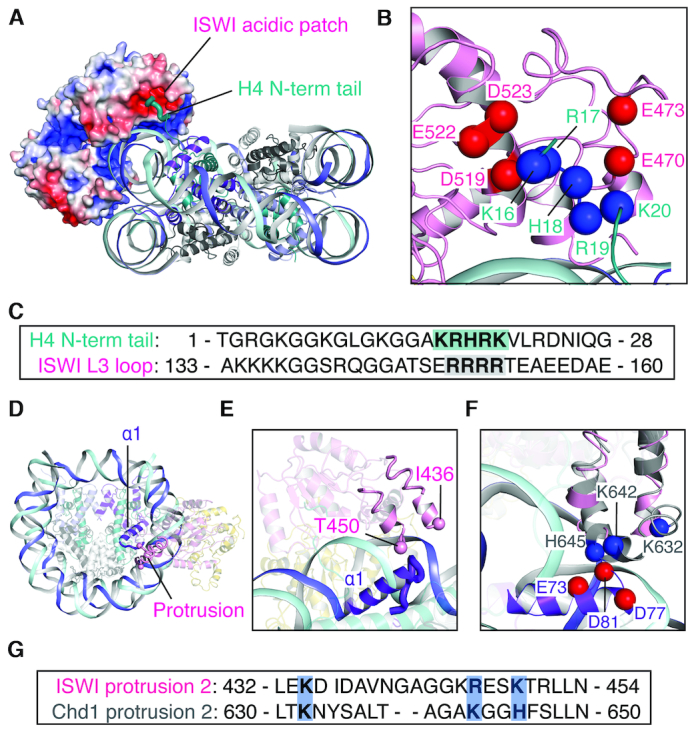
Interaction between ISWI and histones H4 and H3. (A and B) Overall (A) and close-up (B) views of the interactions formed between the acidic patch of CtISWI and the positively charged segment at the N-terminal tail of the histone H4. (C) Sequence alignment between the N-terminal tail of H4 and the L3 loop of ISWI. The conserved stretch of basic residues is highlighted. (D and E) Overall (D) and close-up (E) views of the proximity between the protrusion region of ISWI and the α1 helix of histone H3 CtISWI77-Δ-722-nucleosome complex. The sphere depicts the position of the two residues that connect the missing loop in the protrusion region. (F) Details of interactions formed between the protrusion region and the α1 helix of H3. The superimposed ScChd1 of the nucleosome-bound form (PDB 6FTX) is shown in gray. (G) Sequence alignment of the protrusion region from CtISWI and ScChd1 to indicate conservation of the positively charged residues involved in the interaction with the α1 helix of histone H3.
In addition to interaction with histone H4, a segment of CtISWI consisting of residues S421–P466 is located in close proximity to histone H3 (Figure 3D–F). This segment includes two antiparallel helices connected by a loop region and corresponds to a subfamily-specific protrusion observed in the ATP-dependent chromatin remodelers (Supplementary Figure S6) (3). In the CtISWI77-Δ-722-nucleosome complex, the two antiparallel helices are ordered but the loop region is disordered (Figure 3E). However, a comparative structural analysis suggests that it may occupy a position similar to that observed for the homologous Saccharomyces cerevisiae Chd1 (ScChd1, residues S619-P662) in the nucleosome-bound form (PDB 6FTX) (Figure 3F) (8). In the ScChd1-nucleosome structure, residues K632, K642, and H645 of the remodeler are in close proximity to residues E73, D77 and D81 of histone H3 (Figure 3F). In CtISWI, the equivalent positions correspond to residues K434, R446, and K449, indicating a similar set of interactions could be formed between the ISWI remodeler and the histone H3 (Figure 3F and G). Deletion of K632–K636 loop in the ScChd1 obliterates the nucleosome sliding function of the remodeler (8). Conservation of these interactions in ISWI suggest a similar functional role. Thus, our analysis shows that the interactions between the ATPase domain of ISWI and the histone core are likely to play important roles in selecting the nucleosome rather than naked DNA as the substrate by the remodeler.
Conformational transition upon nucleosome binding
We next compared structures of the nucleosome-bound CtISWI77-Δ-722 with the nucleosome-free MtISWI81–723 (14), both of which share ∼90% sequence identity, to examine overall conformational rearrangements that occur in the remodeler upon nucleosome binding. The structural superpositions, based on residues from either core 1 or core 2 domains, revealed an overall rigid-body motion between the two cores of the ATPase domain upon nucleosome binding (Figure 4A). When core 1 is fixed, core 2 rotates by an angle of ∼137° with E405−K406 as the approximate anchor/bending residues. These two residues are conserved among the subfamilies with an exception of HsINO80 in which K406 of CtISWI is substituted by N753 (Supplementary Figure S6) (34). Furthermore, in the nucleosome-free form of MtISWI81–723 (14), a segment including the helix α5 (residues R187−N203) of core 1 is stacked against core 2 helices α20 (residues T566−G572, motif V) and α21 (residues P590-R599, arginine fingers or motif VI) (14). In the CtISWI77-Δ-722-nucleosome complex, the helices α20 and α21 moved away from the helix α5 subsequently bringing motif I (residues A209−Q219, P-loop) in core 1 closer to motif V (residues L562−N573) and motif VI (residues M596−I602, arginine fingers) of core 2 (Figure 4B). The nucleosome-bound conformation of CtISWI77-Δ-722 is compatible with the nucleotide binding (Figure 4C), suggesting that the CtISWI77-Δ-722-nucleosome complex is primed to carry out the remodeling reaction.
Figure 4.
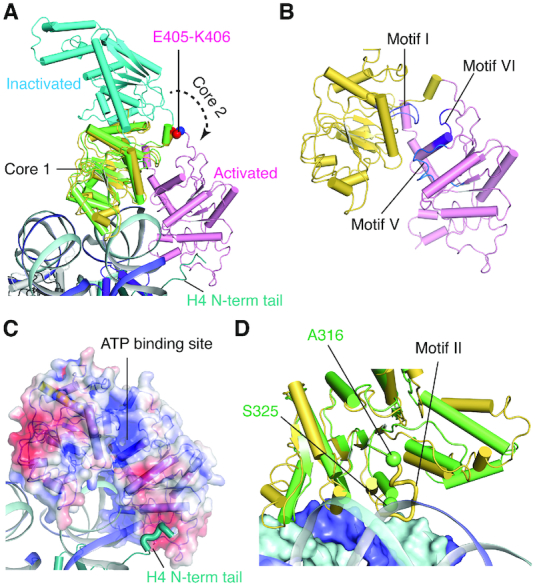
Conformational transition of the ISWI ATPase domain upon nucleosome binding. (A) Superimposition of the nucleosome-free form of MtISWI81–723 (PDB 5JXR, core 1 – green, core 2 – cyan) on the nucleosome-bound form of CtISWI77-Δ-722 (present study, core 1 – yellow, core 2 – pink). The superimposition was performed based on core 1 coordinates. For clarity, only the ATPase domain of the nucleosome free form of MtISWI81–723 (PDB 5JXR) is shown. The residues E405-K406 of CtISWI are shown as spheres. (B) Structure of the nucleosome-bound CtISWI77-Δ-722 highlighting positions of the helicase motifs I (light blue), V (blue), and VI (purple). The view is the same as in the panel (A). (C) Surface representation of the nucleosome-bound CtISWI77-Δ-722 colored by electrostatic potential. Same view as in the panel (A). (D) A rotated view as in panel (A) to depict the positioning of a loop containing the helical motif II between the two DNA gyre. First and second DNA gyres are shown as ribbon and surface. Core 2 of the ATPase domain is removed for clarity. The two spheres depict residues on each side of the disordered loop in the MtISWI81–723 structure (PDB 5JXR).
The structural comparison further provided insights into a potential mechanism of conformational transition from the nucleosome free state to nucleosome-bound state of ATP-dependent chromatin remodelers. As depicted in Figure 4A, core 1 can directly bind to the nucleosomal DNA in the nucleosome-free state without requiring any major conformational changes. Upon this initial binding, core 2 needs to undergo a dramatic conformational change to attain the nucleosome bound state. Although it is not obvious what may drive this conformational transition, the new set of interactions formed between core 1 and nucleosomal DNA likely helps shift the equilibrium towards the nucleosome-bound state (Figure 4D). We propose that AutoN and NegC in the free enzyme may transiently unfold to allow the ATPase domain of ISWI to interact with the nucleosome. Consistent with this hypothesis, the N-terminal tail of H4 can bind to the acidic patch of the remodeler further stabilizing the nucleosome-bound state (Figures 4A and C). Intriguingly, we also identified a segment (residues A315-S324 of CtISWI) which is disordered in the corresponding nucleosome-free form of MtISWI81–723 (14). However, in the CtISWI77-Δ-722-nucleosome complex, this segment became ordered and formed contacts with the first and second gyre of the nucleosomal DNA (Figure 4D). Noticeably, this segment is part of the motif II (Supplementary Figure S6) and H316, R317 and K319 of this region can form stabilizing interactions with nucleosomal DNA (Figure 2C), suggesting that the H316-K319 region plays an important role in the remodeling reaction.
Implications of ATPase binding on DNA remodeling
In the cryo-EM image processing of the CtISWI77-Δ-722-nucleosome samples, we were able to computationally separate the free nucleosome from the CtISWI77-Δ-722-nucleosome complex (Figures 1A and B, and Supplementary Figure S1), allowing a direct comparison between the two forms derived from the same biochemical preparation. Most regions of the nucleosome structures in the two forms are superimposable but significant differences are observable in the region in contact with the CtISWI77-Δ-722 including the H4 tail and the nucleosomal DNA near the SHL2 (Figure 5). The resolution of the density maps is good enough to allow us to observe the deviations, in particular, around the SHL 2 region where the DNA bulges out of the regular path in the CtISWI77-Δ-722-nucleosome complex (Figure 5C–E), consistent with an earlier observation on the Snf2 remodeler (12). Importantly, since we did not include any nucleotides (or analogs) in the preparation, our structure reveals that these structural changes are driven solely by remodeler binding to the nucleosome.
Figure 5.
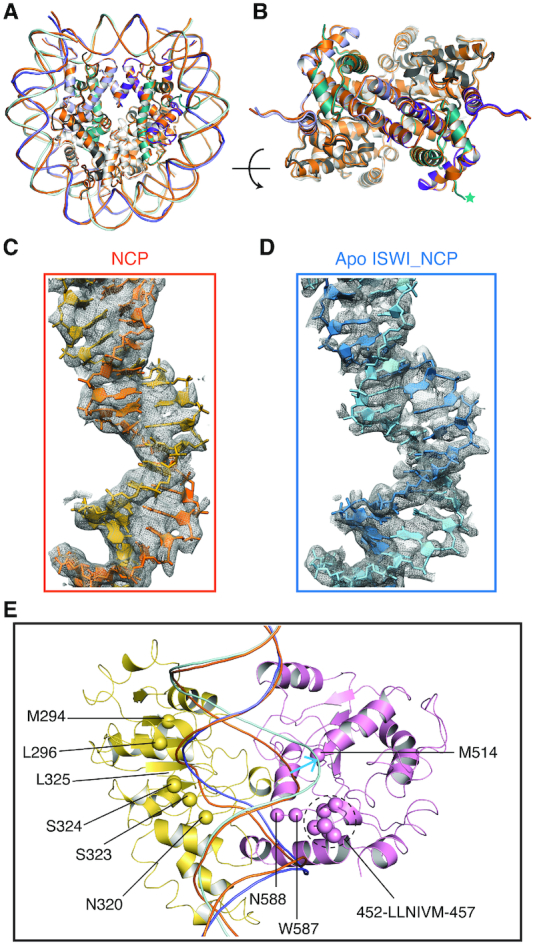
Implications of ISWI ATPase binding on DNA remodeling. (A) Structural superimpositions between the free nucleosome (orange) and the CtISWI77-Δ-722-nucleosome complex. The bound remodeler in the apo CtISWI77-Δ-722-nucleosome complex is omitted for clarity. (B) Rotated view to that of panel (A) to highlight the superimposition in the histone core region. Nucleosomal DNA is omitted for clarity. The asterisk indicates the position of the N-terminal tail of histone H4. (C and D) Quality of the density and fitting of the nucleosomal DNA near the SHL2 site of the free nucleosome (C) or in the apo CtISWI77-Δ-722-nucleosome complex (D). (E) Close up view to highlight the deviations in the path of nucleosomal DNA when compared between the free nucleosome (orange) and the CtISWI77-Δ-722-nucleosome complex. Additionally, hydrophobic/polar residues that are in close proximity of nucleosomal DNA near the SHL 2 region in the apo CtISWI77-Δ-722-nucleosome complex are also shown as spheres. The DNA of the free nucleosome was superimposed on the CtISWI77-Δ-722-nucleosome structure and the blue arrow mark highlights the location of DNA bulge near the SHL2 site.
Interestingly, we also identified several hydrophobic segments including M294, L296, N320, 323–SSL–325, 452–LLNIVM–457, M514, W587–N588 that are in close proximity of the nucleosomal DNA (Figure 5E). The deformation or destabilization of the regular base pairing in the nucleosomal DNA is a likely prerequisite to carry out the remodeling reaction which suggests a possibility that the hydrophobic residues from the remodeler could make stabilizing interactions with the exposed/deformed nucleotide bases to facilitate the remodeling reaction. Consistent with this possibility, these residues show a high level of conservation and are suitably placed around the SHL 2 site (Figure 5E and Supplementary Figure S6).
CONCLUSIONS
The ATPase domain of ISWI is sufficient to bind and translocate nucleosomes along the DNA while the other domains play an important role in regulation (35). To further understand the structural mechanism of nucleosome recognition by the ISWI remodeler, we determined the cryo-EM structures of the apo form of the CtISWI77-Δ-722 bound to the nucleosome and the free nucleosome. We carried out detailed comparisons with the previously available crystal structure of the nucleosome-free state of MtISWI (14) and the free nucleosome. Our analysis reveals important interactions formed between the ISWI remodeler and the nucleosome and illustrates the role of structural epitopes from histones H4 and H3 in the activation and regulation of the ATPase domain. We propose that the disordering of AutoN and NegC regions of the ISWI remodeler may occur transiently and releases the ATPase domain from its constraints to undergo a large conformational transition that allows it to bind to the nucleosome. The binding alone is sufficient to cause deformation in the nucleosomal DNA near the SHL 2 site to prime the translocation step.
Our structure of the apo ISWI-nucleosome complex captures the initial step of ISWI binding to the nucleosome along with the recently reported nucleotide-bound structures of the ScISWI-nucleosome complexes (20) provides insights into the processes of nucleosome remodeling by ISWI. Comparison of these structures reveal the major differences in the conformation of the nucleosomal DNA at the SHL 2 region (Figure 6). Intriguingly, the DNA bulge observed at the SHL 2 site upon binding of the apo CtISWI77-Δ-722 is similar to that of the ADP bound conformation of the ScISWI-nucleosome complex (PDB 6IRO) while the ADP-BeFx bound ScISWI-nucleosome complex (PDB 6IRM) shows a conformation that is close to that of the free nucleosome. Our result is consistent with the recently proposed mechanism for the DNA translocation for ISWI and SNF2 remodelers (20,39), revealing a remarkable mechanistic similarity found between the ATP-dependent chromatin remodelers, which is likely due to the high conservation and autonomous nature of the ATPase domain shared by these remodelers (2,3,35). However, the systematic amino acid substitutions in the ATPase domain of these remodelers, which is likely important for fine-tuning of the subfamily specific functions, may have caused sufficient divergence to render these enzymatic machineries non-interchangeable between subfamilies (40,41).
Figure 6.

Comparison of the nucleosomal DNA near the SHL2 site of the free nucleosome, and the apo and nucleotide-bound ISWI-nucleosome complexes. Close up view of structural superimposition between the (A) free nucleosome (shown in shades of orange) and the apo CtISWI77-Δ-722-nucleosome complex (shades of blue), (B) free nucleosome and the ADP-BeFx bound ScISWI-nucleosome complex (shades of green, PDB 6IRM) (left panel) and the apo CtISWI77-Δ-722-nucleosome complex and ADP-BeFx bound ScISWI-nucleosome complex (right panel), (C) free nucleosome and the ADP bound ScISWI-nucleosome complex (shades of gray, PDB 6IRO) (left panel) and the apo CtISWI77-Δ-722-nucleosome complex and the ADP bound ScISWI-nucleosome complex (right panel), to highlight the changes in the conformation of the nucleosomal DNA at the SHL2 site during different states of remodeling activity by ISWI. The blue arrow mark highlights the location of DNA bulge around the SHL2 site.
DATA AVAILABILITY
The cryo-EM maps and structural coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and Protein Data Bank (PDB) under accession codes EMD-20506 and 6PWE (nucleosome) and EMD-20507 and 6PWF (CtISWI77-Δ-722-nucleosome complex).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Tara Fox for assistance with cryo-EM data collection. This work utilized the computational resources of the NIH High-Performance Computing (HPC) Biowulf cluster (http://hpc.nih.gov).
Notes
Present address: Jingjun Hong, Hefei National Laboratory for Physical Sciences at Microscale and School of Life Sciences, University of Science and Technology of China, Hefei, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural programs of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda; Canada Excellence Research Chair Award (to S.S.). Funding for open access charge: National Institutes of Health/National Cancer Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1. Weake V.M., Workman J.L.. Inducible gene expression: diverse regulatory mechanisms. Nat. Rev. Genet. 2010; 11:426–437. [DOI] [PubMed] [Google Scholar]
- 2. Hopfner K.P., Gerhold C.B., Lakomek K., Wollmann P.. Swi2/Snf2 remodelers: hybrid views on hybrid molecular machines. Curr. Opin. Struct. Biol. 2012; 22:225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartholomew B. Regulating the chromatin landscape: structural and mechanistic perspectives. Annu. Rev. Biochem. 2014; 83:671–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilson B.G., Roberts C.W.. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer. 2011; 11:481–492. [DOI] [PubMed] [Google Scholar]
- 5. Aydin O.Z., Vermeulen W., Lans H.. ISWI chromatin remodeling complexes in the DNA damage response. Cell Cycle. 2014; 13:3016–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mayes K., Qiu Z., Alhazmi A., Landry J.W.. ATP-dependent chromatin remodeling complexes as novel targets for cancer therapy. Adv. Cancer. Res. 2014; 121:183–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Willhoft O., Ghoneim M., Lin C.L., Chua E.Y.D., Wilkinson M., Chaban Y., Ayala R., McCormack E.A., Ocloo L., Rueda D.S. et al.. Structure and dynamics of the yeast SWR1-nucleosome complex. Science. 2018; 362:doi:10.1126/science.aat7716. [DOI] [PubMed] [Google Scholar]
- 8. Sundaramoorthy R., Hughes A.L., El-Mkami H., Norman D.G., Ferreira H., Owen-Hughes T.. Structure of the chromatin remodelling enzyme Chd1 bound to a ubiquitinylated nucleosome. Elife. 2018; 7:doi:10.7554/eLife.35720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eustermann S., Schall K., Kostrewa D., Lakomek K., Strauss M., Moldt M., Hopfner K.P.. Structural basis for ATP-dependent chromatin remodelling by the INO80 complex. Nature. 2018; 556:386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ayala R., Willhoft O., Aramayo R.J., Wilkinson M., McCormack E.A., Ocloo L., Wigley D.B., Zhang X.. Structure and regulation of the human INO80-nucleosome complex. Nature. 2018; 556:391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sundaramoorthy R., Hughes A.L., Singh V., Wiechens N., Ryan D.P., El-Mkami H., Petoukhov M., Svergun D.I., Treutlein B., Quack S. et al.. Structural reorganization of the chromatin remodeling enzyme Chd1 upon engagement with nucleosomes. Elife. 2017; 6:doi:10.7554/eLife.22510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu X., Li M., Xia X., Li X., Chen Z.. Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature. 2017; 544:440–445. [DOI] [PubMed] [Google Scholar]
- 13. Farnung L., Vos S.M., Wigge C., Cramer P.. Nucleosome-Chd1 structure and implications for chromatin remodelling. Nature. 2017; 550:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yan L., Wang L., Tian Y., Xia X., Chen Z.. Structure and regulation of the chromatin remodeller ISWI. Nature. 2016; 540:466–469. [DOI] [PubMed] [Google Scholar]
- 15. Xia X., Liu X., Li T., Fang X., Chen Z.. Structure of chromatin remodeler Swi2/Snf2 in the resting state. Nat. Struct. Mol. Biol. 2016; 23:722–729. [DOI] [PubMed] [Google Scholar]
- 16. Nguyen V.Q., Ranjan A., Stengel F., Wei D., Aebersold R., Wu C., Leschziner A.E.. Molecular architecture of the ATP-dependent chromatin-remodeling complex SWR1. Cell. 2013; 154:1220–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tokuda J.M., Ren R., Levendosky R.F., Tay R.J., Yan M., Pollack L., Bowman G.D.. The ATPase motor of the Chd1 chromatin remodeler stimulates DNA unwrapping from the nucleosome. Nucleic Acids Res. 2018; 46:4978–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bowman G.D. Mechanisms of ATP-dependent nucleosome sliding. Curr. Opin. Struct. Biol. 2010; 20:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sabantsev A., Levendosky R.F., Zhuang X., Bowman G.D., Deindl S.. Direct observation of coordinated DNA movements on the nucleosome during chromatin remodelling. Nat. Commun. 2019; 10:1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan L., Wu H., Li X., Gao N., Chen Z.. Structures of the ISWI-nucleosome complex reveal a conserved mechanism of chromatin remodeling. Nat. Struct. Mol. Biol. 2019; 26:258–266. [DOI] [PubMed] [Google Scholar]
- 21. Kato H., van Ingen H., Zhou B.R., Feng H., Bustin M., Kay L.E., Bai Y.. Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bartesaghi A., Matthies D., Banerjee S., Merk A., Subramaniam S.. Structure of beta-galactosidase at 3.2-A resolution obtained by cryo-electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:11709–11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scheres S.H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012; 180:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chittori S., Hong J., Saunders H., Feng H., Ghirlando R., Kelly A.E., Bai Y., Subramaniam S.. Structural mechanisms of centromeric nucleosome recognition by the kinetochore protein CENP-N. Science. 2018; 359:339–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kato H., Jiang J., Zhou B.R., Rozendaal M., Feng H., Ghirlando R., Xiao T.S., Straight A.F., Bai Y.. A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science. 2013; 340:1110–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 27. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barad B.A., Echols N., Wang R.Y., Cheng Y., DiMaio F., Adams P.D., Fraser J.S.. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods. 2015; 12:943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L. et al.. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018; 46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeLano Scientific The PyMOL Molecular Graphics System, Version 1.8. 2002; Schrödinger, LLC. [Google Scholar]
- 33. Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A.. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hayward S., Berendsen H.J.. Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins. 1998; 30:144–154. [PubMed] [Google Scholar]
- 35. Mueller-Planitz F., Klinker H., Ludwigsen J., Becker P.B.. The ATPase domain of ISWI is an autonomous nucleosome remodeling machine. Nat. Struct. Mol. Biol. 2013; 20:82–89. [DOI] [PubMed] [Google Scholar]
- 36. Al-Ani G., Briggs K., Malik S.S., Conner M., Azuma Y., Fischer C.J.. Quantitative determination of binding of ISWI to nucleosomes and DNA shows allosteric regulation of DNA binding by nucleotides. Biochemistry. 2014; 53:4334–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hwang W.L., Deindl S., Harada B.T., Zhuang X.. Histone H4 tail mediates allosteric regulation of nucleosome remodelling by linker DNA. Nature. 2014; 512:213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clapier C.R., Cairns B.R.. Regulation of ISWI involves inhibitory modules antagonized by nucleosomal epitopes. Nature. 2012; 492:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li M., Xia X., Tian Y., Jia Q., Liu X., Lu Y., Li M., Li X., Chen Z.. Mechanism of DNA translocation underlying chromatin remodelling by Snf2. Nature. 2019; 567:409–413. [DOI] [PubMed] [Google Scholar]
- 40. Dechassa M.L., Hota S.K., Sen P., Chatterjee N., Prasad P., Bartholomew B.. Disparity in the DNA translocase domains of SWI/SNF and ISW2. Nucleic Acids Res. 2012; 40:4412–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fan H.Y., Trotter K.W., Archer T.K., Kingston R.E.. Swapping function of two chromatin remodeling complexes. Mol. Cell. 2005; 17:805–815. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM maps and structural coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and Protein Data Bank (PDB) under accession codes EMD-20506 and 6PWE (nucleosome) and EMD-20507 and 6PWF (CtISWI77-Δ-722-nucleosome complex).