

Abstract
Breast cancer is the most common cancer in women worldwide. Despite recent developments in breast cancer detection and treatment, 1.38 million women each year are still affected. Breast cancer heterogeneity at the population and single-cell level, complexity and developing different metastases are setting several challenges to develop efficient breast cancer therapies. RNA interference (RNAi) represents an opportunity to silence gene expression and inhibit specific pathways in cancer cells. In order to reap the full advantages of RNAi-based therapy, different pathways that sustain cancer cells growth have been targeted using specific siRNAs. The present study investigated the ability of a set of cytotoxic siRNAs to inhibit growth of breast cancer cells. These siRNAs are targeting eukaryotic elongation factor 2 (EEF2), polo-like kinase 1 (PLK1), G protein-coupled receptor kinase 4 (GRK4) and sphingosine kinase interacting protein (SKIP5). To facilitate their targeted delivery, the human epidermal growth factor receptor-3 (HER3)-specific aptamer A30 was used. The in vitro results described in this work indicate that combining the highly specific HER3 aptamer with cytotoxic siRNAs targeting (EEF2, PLK1, GRK4 and SKIP5) can inhibit its activity and ultimately suppress proliferation of HER3 positive breast cancer cells.
Keywords: aptamer, RNA interference, breast cancer, aptamer-siRNA chimera, targeted cancer therapy, human epidermal growth factor receptor-3
Introduction
Breast cancer is the second most common malignancy worldwide accounting up to 29% of all new cancers in women and the leading cause of cancer-related mortality in women. In the last years, significant reduction in breast cancer mortality has been achieved in the developed world through implementing breast cancer screening methods, particularly mammography, and developing new therapeutic strategies. However, there are still 522,000 annual cases of death attributed to breast cancer (1).
Breast cancer is a complex and heterogeneous disease encompassing five distinct molecular subtypes. These include luminal A and B, human epidermal growth factor receptor 2 (HER2) positive, basal-like (lacking hormone receptor and HER2 expression and therefore also called ‘triple-negative’), and normal breast-like subtypes (2). In the last two decades, several molecular targeting therapies against estrogen receptor (ER), HER2 and vascular endothelial growth factor (VEGF) have been developed (3). However, the absence of these receptors in some breast cancer subtypes, inherent and acquired resistance to endocrine and/or cytostatic treatments as well as the breast cancer intra-tumor heterogeneity make it unlikely that targeting of a single pathway will be suitable for developing efficient and long-term breast cancer therapy (4).
RNA interference (RNAi) holds great promise for improving cancer management through inhibiting the pathways that promote or sustain the growth of cancer cells by degrading corresponding mRNAs in a sequence-specific manner (5). For the application of RNAi in cancer therapy, one major aim is to inhibit the expression of genes that are crucial for cell viability, allowing specific elimination of diseased cells (6) as well as to overcome multidrug resistance of cancer cells against chemotherapy and radiotherapy (7).
Due to the heterogeneity of breast cancer, we have selected a panel of siRNAs that can inhibit four vital pathways in cancer cells and enable the specific tailoring of therapies for particular, patient-specific needs. The siRNAs described have the ability to inhibit vital cell pathways by silencing the gene expression of eukaryotic elongation factor 2 (EEF2), Polo-like kinase 1 (PLK1), G-protein coupled receptor kinase 4 (GRK4), and sphingosine kinase interacting protein (SKIP5).
The activity of EEF2 is tightly regulated by a specific calmodulin-dependent protein kinase. ADP ribosylation of EEF2 by diphtheria toxin or Pseudomonas A toxin was shown to inhibit protein biosynthesis efficiently and thus induce apoptosis (8). These toxins are effective in various cell types and have been used for the development of targeted anti-cancer agents, so-called immunotoxins (8).
Several studies have reported elevated PLK-1 level in breast cancer cells (9), whereas it is solely overexpressed in healthy proliferative tissues (10). PLK1 is the best-characterized member of the human PLK family, which plays a pivotal role in mitotic entry, spindle assembly, and DNA damage response (11).
GRKs regulate the activity of G-protein coupled receptors (GPCRs) via phosphorylation of their intracellular domains after release and activation of their associated G-proteins (12). GRKs regulate cell signaling by phosphorylating heptahelical receptors, thereby promoting GPCR interaction with β-arrestins.
Sphingosine kinases (SKs) are conserved lipid kinases catalysing the formation of Sphingosine-1-phosphate (S1P) from the precursor sphingolipid, sphingosine. S1P is characterized as a signalling molecule with dual function. On one hand, it binds to five different S1P receptors that are coupled to a variety of G-proteins allowing to regulate diverse biological functions, on the other hand it appears to act as an intracellular second messenger but potential binding partner are still unknown (13). Inhibiting these pathways will reduce the cell viability and promote cell apoptosis.
Considering the high therapeutic potential of RNAi, the inefficient systemic delivery of siRNAs sets a real challenge in the development of RNAi-based cancer therapies in general (14). Incorporating siRNAs into different kind of nanocarriers has improved siRNA cell internalization and facilitate siRNA accumulation in tumor tissues passively due to the enhanced permeability and retention effect (EPR) (15).
Using aptamers to deliver siRNA to specific target cells is one of the promising approaches due to some advantages compared to nanocarriers and other ligands. For example, aptamers can be generated against any target including toxins and non-immunogenic targets through an in vitro chemical process. Furthermore, they are generally generated by chemical synthesis which reduces the structural variation from batch to batch and allows the chemical modification of aptamer molecules as well as unlimited shelf life (16).
In the present study, we have used the RNA aptamer A30 that binds to the extracellular domain of human epidermal growth factor receptor-3 (HER3) to deliver the described siRNAs into breast cancer cells. HER3 is a member of receptor tyrosine kinases (RTKs) family. These receptors are frequently overexpressed on the surface of several cancers including breast cancer cells, and are involved in several cell growth and differentiation pathways (17).
The RNA aptamer A30 binds to the extracellular domain of HER3 and does not compete with heregulin (HRG) binding or inhibit HRG-dependent HER2 phosphorylation (18). However, it does reduce cell proliferation by inhibiting HRG signaling. We set out to enhance the anti-cancer activity of the aptamer by using it to deliver cytotoxic siRNAs specifically to HER3-expressing breast cancer cells (19, 20).
We found that the combined siRNAs against EEF2, PLK-1, GRK4 and SKIP5 with aptamer A30, were taken up specifically by HER3-expressing breast cancer cells, induced target-specific gene silencing and ultimately suppress cell proliferation. The limited immunogenicity and minimal off-target effects achieved by the specific targeting of tumor cells suggest that these aptamer-siRNA chimeras could represent an option for cancer therapy.
Materials and methods
siRNA preparation
Synthetic 21-nt RNAs were purchased from Qiagen GmbH (Hilden, Germany) in their deprotected, desalted and annealed form. The EEF2-specific siRNA sequences were: sequence siEEF2-1, 5′-AGGCCUAUCUGCCCGUCAAdTdT (sense) and 5′-UUGACGGGCAGAUAGGCCUdTdG (antisense); sequence siEEF2-2, 5′GCGCCAUCAUGGACAAGAAUUdTdT (sense) and 5′UUCUUGUCCAUGAUGGCGCGGdGdG (antisense). The PLK1, GRK-4 and SKIP5-specific siRNA sequences were provided by the Department of Molecular Biology, Max Planck Institute for Infection Biology (Berlin, Germany). siPLK1-1, 5′-CCAUAUGAAUUGUACAGAAdTdT (sense 1) and 5′-UUCUGUACAAUUCAUAUGGdTdG (antisense 1); siPLK1-2, 5′-GGAUCAAGAAGAAUGAAUAdTdT (sense 2) and 5′-UAUUCAUUCUUCUUGAUCCdGdG (antisense 2). siGRK-4-1, 5′-GGAUGUUACUCACCAAGAAdTdT (sense) and 5′-UUCUUGGUGAGUAACAUCCdTdG (antisense); siGRK4-2, 5′GGGUGUUUCAAAGACAUCAdTdT (sense) and 5′UGAUGUCUUUGAAACACCCdGdG (antisense). The siSKIP5 sequences, 5′-CGUCUGGCUGCUGAUGGAAdTdT (sense) and 5′-UUCCAUCAGCAGCCAGACGdTdT (antisense). Non-silencing controls were also purchased from Qiagen GmbH, with the following sequences: 5′-UUCUCCGAACGUGUCACGUdTdT (sense) and 5′-ACGUGACACGUUCGGAGAAdTdT (antisense). The siTOX siRNA (GE Healthcare Dharmacon, Inc., Lafayette, CO, USA) was used as a toxicity control.
Cell lines
The human HER3+ mammary adenocarcinoma cell line MCF-7 (ATCC HTB-22) (21–23) was cultured in GIBCO™ RPMI 1640 medium (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific, Inc.), 100 µg/ml penicillin and 100 µg/ml streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.). HER3– mammary carcinoma cell line MDA-MB-231 (HTB-26) (21–23) was maintained in DMEM tissue culture medium (Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS, 100 U/ml penicillin and 100 µg/ml streptomycin. The cells were maintained at 37°C in a humidified 5% CO2 atmosphere.
Transfections
All transfections were carried out using RNAifect (Qiagen GmbH) following the manufacturer's recommendations. Briefly, RNAifect (1.5 µl) was mixed with 50 µl RPMI medium containing the appropriate concentration of siRNA and incubated for 10 min at room temperature, then added dropwise in 96-well plates (Greiner Bio-One GmbH, Frickenhausen, Germany). Approximately 1–3×104 cells/well were seeded on top of the siRNA drop and grown overnight (final volume 100 µl). The cells were harvested after 12, 24 or 48 h incubation for gene silencing analysis. Cell viability was assayed after 24 or 48 h.
Gene silencing assay
Gene silencing was assessed by quantitative RT-PCR (qRT-PCR), with a Lightcycler Faststart DNA Master SYBR Green I (Roche Applied Science, Penzberg, Germany) and a Roche Lightcycler 1.0 system. The reaction volume was 20 µl. The primers for human GAPDH were: GAPDH forward, 5′-CTCACTGGCATGGCCTTCCGTG-3′; GAPDH reverse, 5′-GTACTCCAGGTGGTGGGACAACG-3′. The primers for EEF2 were: EEF2 forward, 5′-ATGGTGAACTTCACGGTAGAC-3′; EEF2 reverse, 5′-GACTTGATGGTGATGCAACGGACTTGATGGTGATGCAACG-3′. Primers for GRK-4, SKIP-5 and PLK-1 were the same as shown above. Briefly, 1–3×104 MCF-7 and MDA-MB-231 cells were seeded in 96-well plates (Greiner Bio-One GmbH) and incubated overnight at 37°C. Cells were transfected the following day with 200 nM non-silencing siRNA or varying amounts of siRNA (15–150 nM for siRNA sequences 1 and 2) using RNAifect Reagent (Qiagen GmbH) following the manufacturer's recommendations. Cells were lysed after 12 h. Total RNA was extracted using the RNeasy kit (Qiagen GmbH) and treated with DNase I (Qiagen GmbH). RNA was eluted with 30 µl of RNase-free water. Equal amounts of RNA were used for cDNA synthesis using random hexamer primers and Superscript III RNA polymerase (Invitrogen; Thermo Fisher Scientific, Inc.) following the manufacturer's recommendations. Appropriate amounts of cDNA (2 µl) were used for qRT-PCR. The relative amount of target gene mRNA was normalized to GAPDH mRNA by the ΔΔCq method (24). The specificity of amplified PCR products was verified through melting curve analysis.
Cell proliferation and viability assay
Cell proliferation and viability was assessed using the CellTiter 96® AQueous One solution cell proliferation assay (MTS; Promega GmbH, Mannheim, Germany). This assay contains a tetrazolium compound (MTS) and an electron coupling reagent, phenazine ethosulfate (PES). MTS can be bioreduced by cells into formazan, which is soluble in cell culture medium. The quantity of the formazan product as measured spectrophotometrically is directly proportional to the number of viable cells. The assay measures dehydrogenase enzyme activity found in metabolically active cells. MCF-7 or MDA-MB-231 cells were seeded in 96-well plates at a density of 1–3×104 cells per well in 100 µl RPMI 1640 medium, and incubated overnight at 37°C. The cells were washed 2 times with PBS then different concentration (10, 25, 50,75, 100, 250, 500, 1000 nM) of siRNAs were mixed with 1.5 µl RNAifect (Qiagen GmbH) and added at a final volume of 100 µl per well. After 48 h, the Aqueous One Solution containing MTS/PES was added and the optical density was measured at 490 nm using an ELx808 microplate reader (BioTek Instruments GmbH, Bad Friedrichshall, Germany) after 6 h. This experiment was performed in triplicate wells. The absorbance measured from control siRNA (siNON) treatment was normalized to 100% viability.
Predicting RNA secondary structure
The RNA structure program MFOLD (http://unafold.rna.albany.edu/?q=mfold/download-mfold) was used to predict the secondary structures of aptamer A30 and aptamer-siRNA transcripts. The negative control A30-siGFP was designed accordingly. The most stable structures with the lowest free energies for each construct were compared.
In vitro RNA transcription
The template DNAs for the aptamer-siRNA transcripts were synthesized by assembly-PCR using six overlapping primer sequences (Table I) as described by Rydzanicz et al (25). The aptamer (A30) and the aptamer-siRNA transcripts (Table II) were synthesized by in vitro transcription from a dsDNA template containing a T7 RNA polymerase promoter. Transcriptions were performed in a 100-µl reaction volume using T7 RNA transcription kit (Agilent Technologies GmbH, Waldbronn, Germany). Reactions were carried out overnight at 37°C and then treated with DNaseI for 20 min at 37°C. The RNA products were purified by denaturing (7 M urea) gel electrophoresis on an 8% polyacrylamide gel. RNA bands were excised, and RNA was eluted in 0.3 M sodium acetate for 1 h at 60°C and recovered by ethanol precipitation.
Table I.
DNA sequences of synthetic oligonucleotides for RNA transcripts.
| Primer name | Sequence (5′-3′) |
|---|---|
| A30 | |
| 5′ A30 | TAA TAC GAC TCA CTA TAG GGA ATT CCG CGT GTG CC |
| 3′ A30 | GAG GAT CCC GAA CGG ACC GCC |
| A30-1 | GGG AAT TCC GCG TGT GCC AGC GAA AGT TGC GTA TGG GTC ACA |
| A30-2 | ACG GAC CGC CCA GAT GAC ATG TGC CTG CGA TGT GAC CCA TAC GCA ACT T |
| A30-3 | CAT CTG GGC GGT CCG TTC GGG ATC CTC |
| A30-siEEF2 | |
| 5′ A30-EEF2 | 5′ A30 |
| 3′A30-EEF2 | CCG CGC CAT CAT GGA CAA GAA GAA GC |
| A30-EEF2-1 | GGG AAT TCC GCG TGT GCC AGC GAA AGT TGC GTA TGG GTC ACA |
| A30-EEF2-2 | ACG GAC CGC CCA GAT GAC ATG TGC CTG CGA TGT GAC CCA TAC GCA ACT T |
| A30-EEF2-3 | CAT CTG GGC GGT CCG TTC GGG ATC CTC GAA GCT AGC GCC ATC ATG GAC A |
| A30-EEF2-4 | ATG GAC AAG AAG AAG CTT CAA TTC TTG TCC ATG ATG GCG CTA G |
| A30-siGRK4 | |
| 5′ A30-GRK4-1 | 5′ A30 |
| 3′ A30-GRK4-1 | CAG GAT GTT ACT CAC CAA GAA GAA GC |
| A30-GRK4-1-1 | A30-EEF2-1 |
| A30-GRK4-1-2 | A30-EEF2-2 |
| A30-GRK4-1-3 | CAT CTG GGC GGT CCG TTC GGG ATC CTC GAA GCT AGG ATG TTA CTC ACC A |
| A30-GRK4-1-4 | CTC ACC AAG AAG AAG CTT CAA TTC TTG GTG AGT AAC ATC CTA G |
| 5′ A30-GRK4-2 | 5′ A30 |
| 3′ A30-GRK4-2 | CCG GGT GTT TCA AAG ACA TCA GAA GC |
| A30-GRK4-2-1 | A30-EEF2-1 |
| A30-GRK4-2-2 | A30-EEF2-2 |
| A30-GRK4-2-3 | CAT CTG GGC GGT CCG TCC GGG ATC CTC GAA GCT AGG GTG TTT CAA AGA C |
| A30-GRK4-2-4 | CAA AGA CAT CAG AGG CTT CAA TTC TTG GTG AGT ACC ATC CAT G |
| A30-siPLK1 | |
| 5′ A30-PLK1-1 | 5′ A30 |
| 3′ A30-PLK1-1 | CAC CAT ATG AAT TGT ACA GAA GAA GC |
| A30-PLK1-1-1 | A30-EEF2-1 |
| A30-PLK1-1-2 | A30-EEF2-2 |
| A30-PLK1-1-3 | CAT CTG GGC GGT CCG TTC GGG ATC CTC GAA GCT ACC ATA TGA ATT GTA C |
| A30-PLK1-1-4 | ATT GTA CAG AAG AAG CTT CAA TTC TGT ACA ATT CAT ATG GT G |
| 5′ A30-PLK1-2 | 5′ A30 |
| 3′ A30-PLK1-2 | CCG GAT CAA GAA GAA TGA ATA GAA GC |
| A30-PLK1-2-1 | A30-EEF2-1 |
| A30-PLK1-2-2 | A30-EEF2-2 |
| A30-PLK1-2-3 | CAT CTG GGC GGT CCG TTC GGG ATC CTC GAA GCT AGG ATC AAG AAG AAT G |
| A30-PLK1-2-4 | AAG AAT GAA TAG AAG CTT CAA TAT TCA TTC TTC TTG ATC CTA G |
| A30-siSKIP5 | |
| 5′ A30-SKIP5 | 5′ A30 |
| 3′ A30-SKIP5 | AAC GTC TGG CTG CTG ATG GAA GAA GC |
| A30-SKIP5-1 | A30-EEF2-1 |
| A30-SKIP5-2 | A30-EEF2-2 |
| A30-SKIP5-3 | CAT CTG GGC GGT CCG TTC GGG ATC CTC GAA GCT ACG TCT GGC TGC TGA T |
| A30-GFP | |
| 5’ A30-GFP | 5’ A30 |
| 3’ A30-GFP | CGG CAA GCT GAC CCT GAA GTT CCA AGC |
| A30-GFP-1 | A30-EEF2-1 |
| A30-GFP-2 | A30-EEF2-2 |
| A30-GFP-3 | CAT CTG GGC GGT CCG TTC GGG ATC CTC GGA AGC TTG CAA GCT GAC CCT G |
| A30-GFP-4 | CTG AAG TTC CAA GCT TCA TGA ACT TCA GGG TCA GCT TGC AAG C |
Table II.
Sequences of RNA constructs.
| Aptamer | Sequence (5′-3′) |
|---|---|
| A30 | GGGAAUUCCGCGUGUGCCAGCGAAAGUUGCGUAUGGGUCACAUCGCA GGCACAUGUCAUCUGGGCGGUCCGUUCGGGAUCCUC |
| A30-siEEF2 | GGGAAUUCCGCGUGUGCCAGCGAAAGUUGCGUAUGGGUCACAUCGCA GGCACAUGUCAUCUGGGCGGUCCGUUCGGGAUCCUCGAAGCUAGCGC CAUCAUGGACAAGAAUUGAAGCUUCUUCUUGUCCAUGAUGGCGCGG |
| A30-siPLK1-1 | GGGAAUUCCGCGUGUGCCAGCGAAAGUUGCGUAUGGGUCACAUCGCA GGCACAUGUCAUCUGGGCGGUCCGUUCGGGAUCCUCGAAGCUAGGGU GUUUCAAAGACAUCAUUGAAGCUUCUGAUGUCUUUGAAACACCCGG |
| A30-siPLK1-2 | GGGAAUUCCGCGUGUGCCAGCGAAAGUUGCGUAUGGGUCACAUCGCA GGCACAUGUCAUCUGGGCGGUCCGUUCGGGAUCCUCGAAGCUAGGAU CAAGAAGAAUGAAUAUUGAAGCUUCUAUUCAUUCUUCUUGAUCCGG |
| A30-siSKIP5 | GGGAAUUCCGCGUGUGCCAGCGAAAGUUGCGUAUGGGUCACAUCGCA GGCACAUGUCAUCUGGGCGGUCCGUUCGGGAUCCUCGAAGCUACGUC UGGCUGCUGAUGGAAUUGAAGCUUCUUCCAUCAGCAGCCAGACGUU |
| A30-siGRK4-1 | GGGAAUUCCGCGUGUGCCAGCGAAAGUUGCGUAUGGGUCACAUCGCA GGCACAUGUCAUCUGGGCGGUCCGUUCGGGAUCCUCGAAGCUAGGAU GUUACUCACCAAGAAUUGAAGCUUCUUCUUGGUGAGUAACAUCCUG |
| A30-siGRK4-2 | GGGAAUUCCGCGUGUGCCAGCGAAAGUUGCGUAUGGGUCACAUCGCA GGCACAUGUCAUCUGGGCGGUCCGUUCGGGAUCCUCGAAGCUAGGGU GUUUCAAAGACAUCAUUGAAGCUUCUGAUGUCUUUGAAACACCCGG |
Fluorescence labeling of RNA
In order to fluorescently labeling the 5′-ends of generated RNAs, 25 mM guanosine-5′-O-monophosphothioate (GMPS) was added to the transcription reaction. After DNase I digestion the thiol group of GMPS was reduced using 100 mM DTT and incubated for 1 h. Then, 1.5 nmol of RNA transcript was incubated with 0.5 mg/ml 5′-iodoacetamidofluoresceine (IAF), 10 mM EDTA, 1 M urea, 100 mM Tris-HCl (pH 7.4) and 10% v/v dimethylformamide s(DMF). The reaction was incubated at 4°C in the dark overnight. Labeled transcripts were purified by ethanol precipitation, washed several times with 70% ethanol and then resuspended in RNase free water.
Cell surface binding studies of aptamer-siRNA transcripts
Specific binding of the aptamer transcripts to HER3+ cells was determined by flow cytometry. Briefly, IAF-labeled RNA transcripts were first heated to 80°C for 3 min and then incubated at 37°C for 10 min in binding buffer (20 mM HEPES, 150 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, pH 7.4). HER3+ MCF-7 cells and HER3– MDA-MB-231 cells were washed twice in ice-cold PBS and resuspended in binding buffer, then 2–5×105 cells were incubated at 4°C for 1 h with the IAF-labeled aptamer A30 at a concentration of 100 nM or aptamer-siRNA transcripts at a concentration of 10 and 100 nM, respectively. After an additional washing step, FACS profiles were acquired based on the analysis of at least 10,000 events in a FACSCalibur (Becton Dickinson, Heidelberg, Germany). Data analysis was performed with CellQuest Software (Becton Dickenson). HER3– control cell lines were treated as described above using RNA transcript concentrations of 100 nM.
Internalization assay
HER3+ MCF-7 and MDA-MB-231 control cells (2×105) were washed with 500 µl HEPES binding buffer and incubated with 100 nM FITC-labeled aptamer-siRNA in binding buffer supplemented with 16 mM glucose. The ‘non-internalization’ controls for each transcript were kept on ice. All other samples were incubated in the dark at 37°C for 30–120 min. Thereafter, cells were cooled in ice for 5 min. In order to remove aptamer-siRNA transcripts bound to HER3 on cell surface, 0.5 mg/ml proteinase K was added to the cells in HEPES binding buffer. For the ‘100% internalization’ value of each transcript, the proteinase K treatment was omitted so that the maximum number of internalized transcripts reflected the amount of bound fluorescence. After 15 min, cells were collected, washed twice, resuspended in 400 µl binding buffer and analyzed by flow cytometry. For each transcript, data were normalized to the untreated samples (100% internalization) and the no internalization controls (0% internalization).
Aptamer-siRNA transcripts-mediated cytotoxicity
Cell proliferation and viability assay was performed using MTS assay as described before. Briefly, Approximately, 2×104 MCF-7 or MDA-MB-231 cells were seeded in 96-well plates, and allowed to attach overnight at 37°C. The cells were washed 2 times with PBS then the cells were treated with increased concentrations (10, 25, 50, 75, 100, 250, 500, 1000 nM) of A30-siRNAs. After 48 h, the Aqueous One Solution containing MTS/PES was added and the optical density was measured at 490 nm using an ELx808 microplate reader after 6 h. The absorbance measured from control siRNA (siNON) treatment was normalized to 100% viability.
Interferon assay
In order to analyze the aptamer-siRNA immunogenicity, the secretion of interferon β into the supernatant of MCF-7 cells was examined. This was done after treating the cells with the aptamer-siRNA chimera using the human interferon β ELISA kit (PBL Biomedical Laboratories, New Jersey, United States). Briefly, 1×104 MCF-7 cells were seeded in 96-well plates and incubated overnight at 37°C. Thereafter, 1 µM of A30 or aptamer-siRNA was added to the cells and incubated for a further 48 h. Then 100 µl of the supernatant was transferred to a pre-coated ELISA plate and as a positive control different concentration (25, 50, 500, 1000 and 2000 pg/ml) of human IFNβ have been used and incubated for further 24 h at room temperature. Interferon β was detected using an antibody specific to human Interferon β following the manufacturer's recommendations.
Data analysis
GraphPad Prism software v5.00 for Windows (GraphPad Software, San Diego, California, USA) was used for statistical analyses and curve fitting. Data represent the average of triplicates ± SEM. The Student's t-test and and two-way repeated-measure ANOVA followed by Sidak's multiple comparisons test were used to assess the significance of independent experiments. P<0.05 was considered to indicate a statistically significant difference.
Results
Silencing potency of selected siRNAs
RNAi-based genome-wide screens have provided a powerful tool for the identification of cytotoxic siRNAs. We have recognized siRNAs that induced the knock-down of PLK1-1 (83.6 ± 8.6%), PLK1-2 (91.5 ± 1.3%), and SNW1-5 (86.9 ± 1.2%) in HeLa cells (data not shown). A further target, GRK4, was discovered by phenotypic analysis of different cell lines (data not shown). Short interfering RNA sequences targeting EEF2, PLK1, GRK4 and SNW1 mRNA were designed by Qiagen GmbH, using their HP OnGuard siRNA Design algorithm. Their silencing activity in MCF-7 and MDA-MB-231 human mammary adenocarcinoma cells was evaluated at two concentrations (15 and 150 nM). The corresponding mRNA levels were monitored by quantitative real time RT-PCR (qPCR) 12 h post-transfection, to ensure that any knock-down effects were siRNA-specific and not artifacts caused by interferon-induced apoptosis. At 150 nM, all the tested siRNAs significantly reduced the expression of their target mRNAs with the exception of the non-silencing control (siNON). Among them, the siPLK1-1 and siSNW1-5 constructs were the most potent, resulting in up to 80% knock-down of target mRNA levels. Transfection of siEEF2 and siGRK4-1 reduced the target mRNA levels up to 75% (Fig. 1A).
Figure 1.

Gene silencing activity of cell-death promoting siRNAs. (A) RT-qPCR analysis of MCF-7 and MDA-MB-231cells transfected with siRNA. Both cell lines were transfected with 15 and 150 nM siRNA against different target genes (EEF2, PLK1, GRK4 and SNW1 mRNA). A siNON was used as the negative control and reference for normalisation. After 12 h, total RNA was isolated and cDNA synthesized using random hexamer primers (Invitrogen; Thermo Fisher Scientific, Inc.). The mRNA levels were normalized to GAPDH mRNA. Error bars represent SEM (n=3). (B) Toxic activity of cell-death promoting siRNAs. The cytotoxicity of siEEF2, siPLK1-1, siPLK1-2, siSKIP5, siGRK4-1 and siGRK4-2, was evaluated against MCF-7 and MDA-MB-231 cells using different concentrations (10, 25, 50,75, 100, 250, 500, 1000) nM of each siRNA. A siNON was used as the negative control and the siTOX as the positive control at the same concentrations. EEF2, eukaryotic elongation factor 2; PLK1, polo-like kinase 1; GRK4, G protein-coupled receptor kinase 4; siNON, non-silencing siRNA; siTOX, toxic siRNA
siRNA toxic activity
The potent ability of six selected siRNAs to eliminate breast cancer cells was determined by analyzing the cell proliferation and viability of treated cells. To determine the cytotoxicity of selected siRNAs to MCF-7 and MDA-MB-231 cells, cell proliferation and viability assay was performed 48 h after transfection. The viability of treated cells was reduced significantly, in a concentration-dependent manner. The IC50 values ranged from 79 nM for SKIP-5 to 158 nM for siEEF2. Exposing MCF-7 and MDA-MB-231 cells to negative control siRNA had no effect on cell viability, while universally cytotoxic siTOX siRNA (Dharmacon, Chicago, USA) showed comparable toxic effect on treated cells (Table III and Fig. 1B).
Table III.
IC50 values of cytotoxic siRNAs.
| siRNA | IC50 |
|---|---|
| siSKIP-5 | 79 nM |
| siPLK1-1 | 129 nM |
| siGRK4-1 | 130 nM |
| siPLK1-2 | 131 nM |
| siGRK4-2 | 140 nM |
| siEEF2 | 158 nM |
| siTOX | 126 nM |
| siNON | −− |
Design of aptamer-siRNA transcripts
The predicted secondary structure of the A30 aptamer using the MFOLD web server showed that the full-length aptamer folded into four main stem loop structures (Fig. 2). No significant alteration of predicted secondary structure of the A30 aptamer has been observed after adding siRNA sequence to the 3′ end of the aptamer as a short hairpin RNA (Fig. 3). The putative stabilization loop (Fig. 3B) has been shown to have an important impact for functional folding of both the (Fig. 3A) aptamer domain and (Fig. 3C) shRNA stem loop. A non-silencing control construct was generated with a siRNA sequence against GFP (siGFP).
Figure 2.

Representative RNA secondary structure prediction of the A30 aptamer using the MFOLD server. (A) The potential aptamer binding domain and (B) the putative minor stabilization stem loop are depicted by frames.
Figure 3.

Representative RNA secondary structure prediction of the A30-siEEF2 construct. (A) represent the potential aptamer binding domain, (B) represent the putative minor stabilization stem loop and (C) the major shRNA stem loop.
Binding and cellular uptake of aptamer-siRNA transcripts
The binding properties of A30 aptamer to HER3 positive MCF-7 cells and MDA-MB-231 negative cells were analyzed using flow cytometry. We observed specific binding of IAF-labeled aptamer-siRNA transcripts to the HER3+ MCF-7 cells, as shown exemplarily for A30-siEEF2 transcripts (Fig. 4). The binding activity of IAF-labeled aptamer-siRNA transcripts was increased in a dose-dependent manner. This was confirmed by using two different RNA concentrations of 10 and 100 nM. Furthermore, IAF-labeled RNA transcripts were solely bound to HER3 receptor as no fluorescence signal has been detected using the antigen-negative MDA-MB-231 cells.
Figure 4.

Flow cytometric binding analysis of HER3-targeting RNA transcripts. (A) HER3+ MCF-7 cells and (B) HER3− MDA-MB-231 cells (filled red curve) were incubated with 10 nM (blue curve) and 100 nM (green curve) IAF-labeled A30-siEEF2 transcript.
To determine whether aptamer-siRNA transcripts were taken up by HER3+ MCF-7 cells, the rate of aptamer-siRNA internalization was studied. Here, proteinase K was used to remove the HER3 extracellular domain and attached transcripts, allowing to detect only internalized IAF-labeled RNA transcripts. The results of this experiment revealed that the amount of internal A30-siRNA increased steadily until 60% was internalized (Fig. 5). This indicates that the aptamer-siRNA transcripts were internalized via a receptor-mediated endocytosis process in HER3+ cancer cells.
Figure 5.
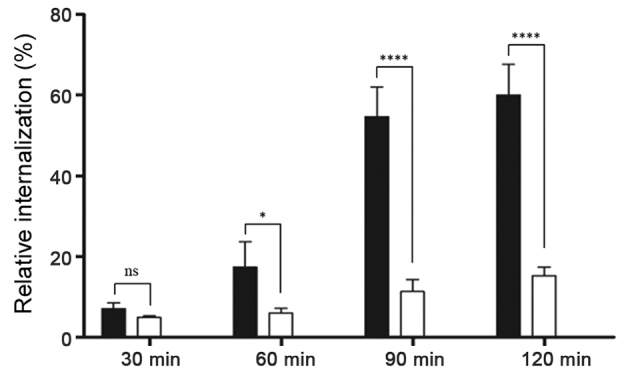
Representative analysis of HER3-receptor internalization using flow cytometry. HER3+ MCF-7 (black columns) and HER3− MDA-MB-231 cells (white columns) were incubated with 100 nM IAF-labeled A30-siEEF2 for 30, 60, 90 and 120 min at 37°C. Additionally cells were treated with proteinase K to remove bound RNA allowing only internalized RNA to be detected by flow cytometry. The diagram shows the percentage of internalized RNA against time. Students t-test indicated that P<0.05 at 60, 90 and 120 min, i.e., the differences were statistically significant. *P<0.05 and ****P<0.0001.
Aptamer-siRNA-mediated cellular cytotoxicity
The ability of A30-siEEF2, A30-siPLK1, A30-siSKIP5 and A30-siGRK4 to inhibit HER3+ cell proliferation was analyzed using a MTS-based colorimetric cell proliferation assay with the HER3+ cell line MCF-7 and Her3- MDA-MB-231 cells as a negative control. The cell viability of HER3+ MCF-7 cells was found to decrease significantly in a concentration dependent manner after incubation with the different A30-siRNA constructs. The IC50 values were 89 nM for A30-siSKIP-5, 125 nM for A30-siPLK1-1, 199 nM for A30-siEEF2 and 501 nM for A30-siGRK4-1, respectively (Fig. 6A and Table IV). MDA-MB-231 cells remained unaffected even when treated with 1000 nM of the aptamer-siRNA transcripts (Fig. 6B). Furthermore, neither A30-siGFP nor A30 aptamer alone exerted any cytotoxicity to both tumor cell lines.
Figure 6.

Toxic activity of cell-death promoting aptamer-siRNA transcripts. The cytotoxicity of A30-siEEF2, A30-siPLK1-1, A30-siPLK1-2, A30-siSKIP5, A30-siGRK4-1 and A30-siGRK4-2 was evaluated by cell viability assays using (A) HER3+ MCF-7 cells and (B) HER3− MDA-MB-231 cells. As a negative control siRNA against GFP (siGFP) and untreated cells were used.
Table IV.
IC50 values of aptamer-siRNA transcripts.
| siRNA | IC50 |
|---|---|
| A30-siSKIP-5 | 89 nM |
| 30-siPLK1-1 | 125 nM |
| A30-siGRK4-1 | 501 nM |
| A30-siPLK1-2 | 316 nM |
| A30-siEEF2 | 199 nM |
| siGFP | −− |
Interferon assay
A major safety concern regarding the use of siRNA-based therapy is the induction of nonspecific inflammatory responses and subsequent cellular cytotoxicity. Therefore, we determined the interferon β level after treating MCF-7 cells with all aptamer-siRNA transcripts for 24 h. The interferon β levels in treated cells were lower than the background levels observed in untreated cells, indicating that under our experimental conditions the aptamer-siRNA transcripts did not trigger a type I interferon response (Fig. 7).
Figure 7.

Induction of Interferon β production assay using sandwich interferon β ELISA kit. (A) Positive control: MCF-7 cells treated with different concentrations of poly(I:C) RNA; (B) MCF-7 cells were incubated for 48 h with 1 µM aptamer-siRNA transcripts. Students t-test indicated that no significant differences were observed in Interferon β production after treating the cells with aptamer-siRNA transcripts at P<0.05.
Discussion
Since the first description of the RNAi process by Fire et al (26), significant progress has been made in understanding the technical details and in selecting siRNA against a wide range of target genes. The use of siRNA has several therapeutic advantages compared to conventional drugs. RNAi is an endogenous natural process generally used by cells to selectively down-regulate gene expression or as defense mechanism against viral infection (27,28). As cancer diseases are associated with the up-regulation of specific genes, their specific knock down in cancer cells with the help of siRNA emerged as a highly promising therapeutic option. Furthermore, siRNA-based therapies have the ability to overcome multidrug resistance of cancer cells against chemotherapy, photodynamic therapy and radiotherapy. Remarkable progress has been made to select siRNA against target genes (27).
Due to breast cancer heterogeneity at the population and single-cell level, its complexity and its metastasizing potential (29), targeting different pathways that could inhibit growth and invasion of cancer cells is essential for developing effective therapies. Here we have tested several siRNAs targeting not only one but a set of different pathways to allow tailored development of siRNA-based therapeutic agents to ideally hit the vast majority of breast cancer cells and subtypes. As promising siRNAs candidates against four gene products overexpressed and/or sustaining cancer cell growth have been investigated. The corresponding toxic siRNAs are directed against PLK-1 (30–32) and EEF-2 (33), as well as GRK4 and SKIP-5 (34). All tested siRNAs induced toxic effect in breast cancer cells, however siSKIP-5 was two times more toxic than other tested siRNAs. Recent reports indicated that sphingosine kinase is overexpressed in breast cancer cells and serve as an oncogene in tumorigenesis by enhancing tumor cell growth and invasion and reducing cell apoptosis (35).
As described in previous studies, siSKIP-5 was able to reduce the sphingosine kinase expression level and induce cell death in HER3+ breast cancer cells (36). Although, this could reflect the high toxic activity of siSKIP-5 comparing to other siRNAs, several reasons could also play an essential role in knock down experiments.
The inefficient systemic delivery of siRNAs, due to their hydrophilicity, negative charge and sensitivity to nucleases, poses challenges in the development of RNAi-based cancer therapies in general (14). To solve the aforementioned problems, siRNAs have been armed with several classes of specific cell receptor ligands to direct them to the target cells. These ligands include folic acid, aptamers, monoclonal antibodies and their fragments (37–39).
Over the past years, several studies have reported that arming of siRNAs with aptamers could significantly improve therapeutic efficacy and pharmacokinetic properties of siRNA (31).
In the present study, we chose the HER3-specific aptamer A30 for the cell type-specific delivery of novel cytotoxic siRNAs. HER3 is a member of the EGFR family, and is overexpressed in diverse human cancers; it is associated with poor prognosis in breast, lung and ovarian cancer (40,41). Several publications emphasize the therapeutic potential of HER3 targeting, particularly for the treatment of drug-resistant tumors (42). Here, several RNA transcripts have been developed by fusing the HER3-specific aptamer A30 to different cytotoxic siRNAs. The resulting transcripts maintained high binding activity for cell-surface HER3. In comparison to aptamer A30 alone, aptamer-siRNA conjugates showed an increased binding to target cells. This improved binding activity might be due to potential alterations in their RNA secondary structure that increased duplex stability and nuclease resistance and thus prevents degradation by RNases. However, the binding specificity of A30 and A30-siRNA was confirmed using MDA-MB-231 cells which express very low levels of HER3. The binding specificity need to be further determined after knocking down HER3 gene in MCF-7 cells.
A30 aptamer-siRNA constructs were rapidly taken up specifically into HER3+ MCF-7 cells even in the absence of heregulin (HRG), which is normally required to trigger receptor mediated endocytosis of HER3 (43). Furthermore, the inhibition of HRG signaling in vivo by aptamer A30 could have synergistic antitumor effects thus increasing its therapeutic potential (18).
The A30-siRNAs specifically reduced the mRNA levels of four different target genes to the same extent as the transfected siRNAs indicating that the siRNAs also retained their activities as part of a fusion transcript. In HER3+ MCF-7 cells, the knock-down of each target mRNA was sufficient to reduce the cell viability whereas HER– MDA-MB-231 cells were not affected. The IC50 values for the overall cytotoxicity varied from 89 nM for SKIP-5 to 501 nM for GRK4-1, which was comparable to previous reports with other cytotoxic siRNAs, and a previously reported aptamer-siRNA targeting PSMA (31,33). In the cell viability assay only MCF-7 cells were affected, and the toxicity was dose-dependent. To confirm that these effects reflected specific RNAi and not simply the binding of A30 to the cell surface, we also analyzed A30 without a siRNA fusion and A30 fused to a non-silencing siRNA. Cytotoxic effects were observed only in cells incubated with RNA transcripts that containing cytotoxic siRNA.
Furthermore, simultaneous targeting of HER family members might represent a promising therapeutic option to overcome the heterogeneity of breast cancer. Nucleic acid aptamer-siRNA constructs targeting HER2 and HER3 have recently demonstrated their promising therapeutic properties in vivo against different breast cancer cell lines. Therefore, combining siRNAs targeting different gene transcripts with dual targeting aptamers could allow the development of novel therapeutic options especially for breast cancer patients (44).
Cell type-specific siRNA delivery is particularly important for cytotoxic siRNAs because nonspecific uptake could also kill healthy cells. HER3 is therefore a promising target for cell-specific drug delivery because it is predominantly expressed on breast, ovarian and lung cancer cells. Although more work is clearly required to further confirm these results in vivo, these initial experiments suggest that combining the highly specific HER3 aptamer with cytotoxic siRNAs allows the development of highly efficient agents for selective elimination of HER3-expressing malignant cells.
Acknowledgements
The authors would like to thank Dr. Richard M. Twyman (Twyman Research Management Ltd.) for critically reading the manuscript.
Glossary
Abbreviations
- DMF
dimethylformamide
- EEF2
eukaryotic elongation factor 2
- EGFR
epidermal growth factor receptor
- EPR
enhanced permeability and retention effect
- ER
estrogen receptor
- IAF
iodoacetamidofluoresceine
- GPCRs
G-protein coupled receptors
- GRK4
G protein-coupled receptor kinase 4
- HER2
human epidermal growth factor receptor-2
- HER3
human epidermal growth factor receptor-3
- HRG
heregulin
- PES
phenazine ethosulfate
- PLK1
polo-like kinase 1
- RNAi
RNA interference
- RTKs
receptor tyrosine kinases
- S1P
sphingosine-1-phosphate
- siGFP
siRNA against GFP
- siNON
control siRNA
- siRNA
small interfering RNA
- siTox
toxic siRNA
- SKIP5
sphingosine kinase interacting protein
- SKs
sphingosine kinases
- VEGF
vascular endothelial growth factor
Funding
The present study was partly supported by the Florindon Foundation, the German Research foundation, DFG (Graduiertenkolleg ‘Biointerface’ 1035), and by funding under the Sixth Research Framework Programme of the European Union, Project RIGHT (grant no. LSHB-CT-2004-005276).
Availability of data and materials
All data generated or analyzed during the present study are included in this published article and its supplementary information files.
Authors' contributions
SB, RF and MKT conceived the study. IN performed the experiments with help from UW. NM and TFM identified the cytotoxic siRNAs. SB, MKT, AFH, SG and IM discussed, analyzed and interpreted the results. AFH and MKT wrote the manuscript with essential input from all authors.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Conflicts of interest
The authors declare that they have no competing interest.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Subik K, Lee JF, Baxter L, Strzepek T, Costello D, Crowley P, Xing L, Hung MC, Bonfiglio T, Hicks DG, et al. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer (Auckl) 2010;4:35–41. [PMC free article] [PubMed] [Google Scholar]
- 3.Toss A, Cristofanilli M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res. 2015;17:60. doi: 10.1186/s13058-015-0560-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlotter CM, Vogt U, Allgayer H, Brandt B. Molecular targeted therapies for breast cancer treatment. Breast Cancer Res. 2008;10:211. doi: 10.1186/bcr2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo W, Chen W, Yu W, Huang W, Deng W. Small interfering RNA-based molecular therapy of cancers. Chin J Cancer. 2013;32:488–493. doi: 10.5732/cjc.012.10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim DH, Rossi JJ. Strategies for silencing human disease using RNA interference. Nat Rev Genet. 2007;8:173–184. doi: 10.1038/nrg2006. [DOI] [PubMed] [Google Scholar]
- 7.Karagiannis TC, El-Osta A. RNA interference and potential therapeutic applications of short interfering RNAs. Cancer Gene Ther. 2005;12:787–795. doi: 10.1038/sj.cgt.7700857. [DOI] [PubMed] [Google Scholar]
- 8.Hristodorov D, Mladenov R, von Felbert V, Huhn M, Fischer R, Barth S, Thepen T. Targeting CD64 mediates elimination of M1 but not M2 macrophages in vitro and in cutaneous inflammation in mice and patient biopsies. MAbs. 2015;7:853–862. doi: 10.1080/19420862.2015.1066950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maire V, Némati F, Richardson M, Vincent-Salomon A, Tesson B, Rigaill G, Gravier E, Marty-Prouvost B, De Koning L, Lang G, et al. Polo-like kinase 1: A potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res. 2013;73:813–823. doi: 10.1158/0008-5472.CAN-12-2633. [DOI] [PubMed] [Google Scholar]
- 10.King SI, Purdie CA, Bray SE, Quinlan PR, Jordan LB, Thompson AM, Meek DW. Immunohistochemical detection of Polo-like kinase-1 (PLK1) in primary breast cancer is associated with TP53 mutation and poor clinical outcom. Breast Cancer Res. 2012;14:R40. doi: 10.1186/bcr3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10:825–841. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
- 12.Keever LB, Jones JE, Andresen BT. G protein-coupled receptor kinase 4gamma interacts with inactive Galpha(s) and Galpha13. Biochem Biophys Res Commun. 2008;367:649–655. doi: 10.1016/j.bbrc.2007.12.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strub GM, Maceyka M, Hait NC, Milstien S, Spiegel S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv Exp Med Biol. 2010;688:141–155. doi: 10.1007/978-1-4419-6741-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dominska M, Dykxhoorn DM. Breaking down the barriers: siRNA delivery and endosome escape. J Cell Sci. 2010;123:1183–1189. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- 15.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci USA. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lakhin AV, Tarantul VZ, Gening LV. Aptamers: Problems, solutions and prospects. Acta Naturae. 2013;5:34–43. doi: 10.32607/20758251-2013-5-4-34-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 18.Chen CH, Chernis GA, Hoang VQ, Landgraf R. Inhibition of heregulin signaling by an aptamer that preferentially binds to the oligomeric form of human epidermal growth factor receptor-3. Proc Natl Acad Sci USA. 2003;100:9226–9231. doi: 10.1073/pnas.1332660100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carraway KL, III, Sliwkowski MX, Akita R, Platko JV, Guy PM, Nuijens A, Diamonti AJ, Vandlen RL, Cantley LC, Cerione RA. The erbB3 gene product is a receptor for heregulin. J Biol Chem. 1994;269:14303–14306. [PubMed] [Google Scholar]
- 20.Sliwkowski MX, Schaefer G, Akita RW, Lofgren JA, Fitzpatrick VD, Nuijens A, Fendly BM, Cerione RA, Vandlen RL, Carraway KL., III Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J Biol Chem. 1994;269:14661–14665. [PubMed] [Google Scholar]
- 21.Kim S, Han J, Shin I, Kil WH, Lee JE, Nam SJ. A functional comparison between the HER2(high)/HER3 and the HER2(low)/HER3 dimers on heregulin-β1-induced MMP-1 and MMP-9 expression in breast cancer cells. Exp Mol Med. 2012;44:473–482. doi: 10.3858/emm.2012.44.8.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salameh A, Fan X, Choi BK, Zhang S, Zhang N, An Z. HER3 and LINC00052 interplay promotes tumor growth in breast cancer. Oncotarget. 2017;8:6526–6539. doi: 10.18632/oncotarget.14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suga J, Izumiyama K, Tanaka N, Saji S. Estradiol promotes rapid degradation of HER3 in ER-positive breast cancer cell line MCF-7. Biochem Biophys Rep. 2018;16:103–109. doi: 10.1016/j.bbrep.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Rydzanicz R, Zhao XS, Johnson PE. Assembly PCR oligo maker: A tool for designing oligodeoxynucleotides for constructing long DNA molecules for RNA production. https://doi.org/10.1093/nar/gki380. Nucleic Acids Res. 2005;33(web server issue):W521–525. doi: 10.1093/nar/gki380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 27.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: Advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2919-c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hussain AF, Wullner U, Neef I, Tur MK, Barth S. Topics in Anti-Cancer-Research. Bentham Science Publishers; Sharjah, U.A.E.: 2013. Targeted delivery of short interfering RNAs - Strategies for in vivo application; pp. 228–253. [Google Scholar]
- 29.Martelotto LG, Ng CK, Piscuoglio S, Weigelt B, Reis-Filho JS. Breast cancer intra-tumor heterogeneity. Breast Cancer Res. 2014;16:210. doi: 10.1186/bcr3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bu Y, Yang Z, Li Q, Song F. Silencing of polo-like kinase (Plk) 1 via siRNA causes inhibition of growth and induction of apoptosis in human esophageal cancer cells. Oncology. 2008;74:198–206. doi: 10.1159/000151367. [DOI] [PubMed] [Google Scholar]
- 31.Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara JO, II, Giangrande PH. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27:839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McNamara JO, II, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, Sullenger BA, Giangrande PH. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 33.Wullner U, Neef I, Eller A, Kleines M, Tur MK, Barth S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr Cancer Drug Targets. 2008;8:554–565. doi: 10.2174/156800908786241078. [DOI] [PubMed] [Google Scholar]
- 34.Machuy N, Thiede B, Rajalingam K, Dimmler C, Thieck O, Meyer TF, Rudel T. A global approach combining proteome analysis and phenotypic screening with RNA interference yields novel apoptosis regulators. Mol Cell Proteomics. 2005;4:44–55. doi: 10.1074/mcp.M400089-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Wang Y, Wan Z, Liu S, Cao Y, Zeng Z. Sphingosine kinase 1 and cancer: A systematic review and meta-analysis. PLoS One. 2014;9:e90362. doi: 10.1371/journal.pone.0090362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Y, Dong B, Huang J, Kong W, Xue W, Zhu Y, Zhang J, Huang Y. Sphingosine kinase 1 is overexpressed and promotes adrenocortical carcinoma progression. Oncotarget. 2016;7:3233–3244. doi: 10.18632/oncotarget.6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hussain AF, Tur MK, Barth S. An aptamer-siRNA chimera silences the eukaryotic elongation factor 2 gene and induces apoptosis in cancers expressing αvβ3 integrin. Nucleic Acid Ther. 2013;23:203–212. doi: 10.1089/nat.2012.0408. [DOI] [PubMed] [Google Scholar]
- 38.Li TS, Yawata T, Honke K. Efficient siRNA delivery and tumor accumulation mediated by ionically cross-linked folic acid-poly(ethylene glycol)-chitosan oligosaccharide lactate nanoparticles: For the potential targeted ovarian cancer gene therapy. Eur J Pharm Sci. 2014;52:48–61. doi: 10.1016/j.ejps.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 39.Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 40.Reschke M, Mihic-Probst D, van der Horst EH, Knyazev P, Wild PJ, Hutterer M, Meyer S, Dummer R, Moch H, Ullrich A. HER3 is a determinant for poor prognosis in melanoma. Clin Cancer Res. 2008;14:5188–5197. doi: 10.1158/1078-0432.CCR-08-0186. [DOI] [PubMed] [Google Scholar]
- 41.Servidei T, Riccardi A, Mozzetti S, Ferlini C, Riccardi R. Chemoresistant tumor cell lines display altered epidermal growth factor receptor and HER3 signaling and enhanced sensitivity to gefitinib. Int J Cancer. 2008;123:2939–2949. doi: 10.1002/ijc.23902. [DOI] [PubMed] [Google Scholar]
- 42.Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM. A central role for HER3 in HER2-amplified breast cancer: Implications for targeted therapy. Cancer Res. 2008;68:5878–5887. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
- 43.Jeschke M, Wels W, Dengler W, Imber R, Stöcklin E, Groner B. Targeted inhibition of tumor-cell growth by recombinant heregulin-toxin fusion proteins. Int J Cancer. 1995;60:730–739. doi: 10.1002/ijc.2910600527. [DOI] [PubMed] [Google Scholar]
- 44.Yu X, Ghamande S, Liu H, Xue L, Zhao S, Tan W, Zhao L, Tang SC, Wu D, Korkaya H, et al. Targeting EGFR/HER2/HER3 with a three-in-one aptamer-siRNA chimera confers superior activity against HER2+ breast cancer. Mol Ther Nucleic. 2018;10:317–330. doi: 10.1016/j.omtn.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during the present study are included in this published article and its supplementary information files.