

We demonstrate that β-N-acetyl-d-glucosaminyl-(1,4)-d-glucosamine (GlcNAc-GlcN) enhances the chitin-metabolizing ability of V. parahaemolyticus. Members of the genus Vibrio are chitin-degrading bacteria, and some species of this genus are associated with diseases affecting fish and animals, including humans (F. L. Thompson, T. Iida, and J. Swings, Microbiol Mol Biol Rev 68:403–431, 2004; M. Y. Ina-Salwany, N. Al-Saari, A. Mohamad, F.-A. Mursidi, et al., J Aquat Anim Health 31:3–22, 2019). Studies on Vibrio are considered important, as they may facilitate the development of solutions related to health, food, and aquaculture problems attributed to this genus. This report enhances the current understanding of chitin degradation by Vibrio bacteria.
KEYWORDS: induced gene expression, Vibrio parahaemolyticus, chitin heterodisaccharide, chitin metabolism, chitin oligosaccharide deacetylase
ABSTRACT
Vibrio parahaemolyticus RIMD2210633 secretes both chitinase and chitin oligosaccharide deacetylase and produces β-N-acetyl-d-glucosaminyl-(1,4)-d-glucosamine (GlcNAc-GlcN) from chitin. Previously, we reported that GlcNAc-GlcN induces chitinase production by several strains of Vibrio harboring chitin oligosaccharide deacetylase genes (T. Hirano, K. Kadokura, T. Ikegami, Y. Shigeta, et al., Glycobiology 19:1046–1053, 2009). The metabolism of chitin by Vibrio was speculated on the basis of the findings of previous studies, and the role of chitin oligosaccharide produced from chitin has been well studied. However, the role of GlcNAc-GlcN in the Vibrio chitin degradation system, with the exception of the above-mentioned function as an inducer of chitinase production, remains unclear. N,N′-Diacetylchitobiose, a homodisaccharide produced from chitin, is known to induce the expression of genes encoding several proteins involved in chitin metabolism in Vibrio strains (K. L. Meibom, X. B. Li, A. Nielsen, C. Wu, et al., Proc Natl Acad Sci U S A 101:2524–2529, 2004). We therefore hypothesized that GlcNAc-GlcN also affects the expression of enzymes involved in chitin metabolism in the same manner. In this study, we examined the induction of protein expression by several sugars released from chitin using peptide mass fingerprinting and confirmed the expression of genes encoding enzymes involved in chitin metabolism using real-time quantitative PCR analysis. We then confirmed that GlcNAc-GlcN induces the expression of genes encoding many soluble enzymes involved in chitin degradation in Vibrio parahaemolyticus. Here, we demonstrate that GlcNAc-GlcN enhances the chitin-metabolizing ability of V. parahaemolyticus.
IMPORTANCE We demonstrate that β-N-acetyl-d-glucosaminyl-(1,4)-d-glucosamine (GlcNAc-GlcN) enhances the chitin-metabolizing ability of V. parahaemolyticus. Members of the genus Vibrio are chitin-degrading bacteria, and some species of this genus are associated with diseases affecting fish and animals, including humans (F. L. Thompson, T. Iida, and J. Swings, Microbiol Mol Biol Rev 68:403–431, 2004; M. Y. Ina-Salwany, N. Al-Saari, A. Mohamad, F.-A. Mursidi, et al., J Aquat Anim Health 31:3–22, 2019). Studies on Vibrio are considered important, as they may facilitate the development of solutions related to health, food, and aquaculture problems attributed to this genus. This report enhances the current understanding of chitin degradation by Vibrio bacteria.
INTRODUCTION
Marine environments contain a tremendous amount of chitin, a water-insoluble β-(1,4)-linked polymer of N-acetyl-d-glucosamine (GlcNAc) that is a component of the shells of crabs and shrimp. Bacteria of the genus Vibrio are among the most abundant microorganisms in the marine biosphere, and they decompose chitin to its oligosaccharides, particularly N,N′-diacetylchitobiose [(GlcNAc)2], by secreting the enzyme chitinase (EC 3.2.1.14, 3.2.1.200, or 3.2.1.201) and taking up the oligosaccharide metabolites into the cells via several transporter systems (1).
Many chitinolytic enzymes and transporters involved in catabolizing chitin and chitin oligosaccharides in Vibrio have been described (1–4). The pathway of chitin metabolism in Vibrio was proposed on the basis of genomic and other experimental data (3). A number of recent studies have examined chitin disaccharide deacetylase (EC 3.5.1.105), also known as chitin oligosaccharide deacetylase (COD), which hydrolyzes the N-acetyl group at the reducing-end GlcNAc residue of (GlcNAc)2 to generate the heterodisaccharide β-N-acetyl-d-glucosaminyl-(1,4)-d-glucosamine (GlcNAc-GlcN). Although this enzyme has been identified in four species of Vibrio (5–8), not all members of this genus have a gene encoding COD. In addition, we recently identified CODs in two Shewanella species (9, 10). We reported that the product generated by COD from (GlcNAc)2, namely, GlcNAc-GlcN, strongly induces chitinase production in COD-producing Vibrio (11) and functions as a chemoattractant for these bacteria (12). We considered these phenomena very interesting from the perspective of demonstrating the possibility that GlcNAc-GlcN functions as a specific signaling molecule for COD-producing strains. Other researchers described the functions of chitin homo-oligosaccharides with respect to marine bacteria and showed that homo-oligomers of GlcNAc induce the expression of genes encoding a number of enzymes and other proteins involved in chitin degradation in Vibrio cholerae and that these mechanisms are part of global gene expression systems (13, 14). Although V. cholerae was also shown to produce COD (6), the effect of GlcNAc-GlcN had yet to be characterized; whether GlcNAc-GlcN functions the same as (GlcNAc)2 could not be determined. We therefore investigated in greater detail the role of GlcNAc-GlcN in chitin degradation by Vibrio in terms of the induction of protein production and gene expression.
Over 10 years ago, Hunt et al. (3) suggested a pathway in which chitin is degraded to fructose-6-phosphate by V. cholerae, based on the considerable available evidence regarding chitinolytic enzymes and proteins involved in chitin catabolism. Based on these data, we hypothesized that chitin is degraded as shown in Fig. 1, in which the identifiers shown correspond to sequences in the genome of Vibrio parahaemolyticus strain RIMD2210633. The proposed degradation mechanism is based on previous enzymologic research involving bacterial CODs, which are extracellular enzymes (11, 15) that primarily produce GlcNAc-GlcN from homodisaccharides from chitin (5–10, 16). The enzyme in Fig. 1 that deacetylates the acetamide bond of the nonreducing end of GlcNAc residues in (GlcNAc)2 or GlcNAc-GlcN in the periplasm remains unknown. We also hypothesize that GlcNAc-GlcN, rather than (GlcN)2, is the primary metabolite in the cytosol. However, at present, it seems most proper to show the pathway as indicated in Fig. 1.
FIG 1.

Proposed pathway of chitin degradation by Vibrio parahaemolyticus. The pathway is based on that proposed by Hunt et al. (3), with some modifications, particularly regarding COD (5–11, 15, 16), as described in the text. The numbers under the protein names and the corresponding numbers in parentheses are KEGG and UniProt accession numbers, respectively.
Chitin is hydrolyzed by chitinases (KEGG accession numbers VP0619, VP2338, VPA0055, and VPA1177) to GlcNAc oligosaccharides with a small degree of polymerization outside of cells, and then (GlcNAc)2 is deacetylated to GlcNAc-GlcN by COD in V. parahaemolyticus. In this process, GlcN is also produced incidentally, as some residues of chitin are naturally deacetylated. While homo-oligosaccharides ranging in degree of polymerization from trimers to hexamers are taken up primarily via chitoporin (VP0769), disaccharides and monosaccharides may enter the cells through as yet unknown porins (2, 4). In the periplasm, longer homo-oligosaccharides are then hydrolyzed to disaccharides and/or trisaccharides by chitodextrinase (VPA0832, EC 3.2.1.202) and then hydrolyzed again to the monosaccharide GlcNAc by β-N-acetylhexosaminidases (VP2486 and VP0755, EC 3.2.1.52). The pathway by which these saccharides are converted to other sugars in the periplasm and then transported to and metabolized in the cytosol is thought to be the same pathway proposed by Hunt et al. (3) for V. cholerae, although some of the enzymes involved in cytosolic metabolism have not been identified. These metabolites are finally converted into fructose-6-phosphate, which then enters the pentose-phosphate pathway.
We hypothesize that GlcNAc-GlcN is a much stronger inducer of the expression of proteins involved in chitin metabolism in V. parahaemolyticus RIMD2210633 than (GlcNAc)2. We also hypothesize that the associated gene expression mechanism in V. parahaemolyticus RIMD2210633 is similar to that in V. cholerae reported previously (13, 14). To test this hypothesis and clarify the role of GlcNAc-GlcN in the chitin degradation process, we examined the effect on protein expression of several sugars released from chitin. We also evaluated the potential enhancement of gene expression by GlcNAc-GlcN and (GlcNAc)2. Our data demonstrate that the expression of genes encoding various soluble enzymes involved in chitin metabolism (Fig. 1) is induced by disaccharides produced outside of Vibrio cells. The results of this investigation suggest that the chitin degradation mechanism in V. parahaemolyticus involves GlcNAc-GlcN.
Some Vibrio species are pathogenic to marine organisms, such as fish and coral, in addition to animals, including humans (17, 18). Indeed, V. parahaemolyticus is a well-known human pathogen (17). Investigations of the ecology of Vibrio species, particularly as they relate to controlling Vibrio populations, will be helpful in addressing Vibrio-associated problems in health, food, and aquaculture.
RESULTS
Detection of chitin-metabolizing enzymes in strain RIMD2210633 that are upregulated by chitin-derived sugars.
Proteins detected in all cellular compartments of V. parahaemolyticus RIMD2210633 cultivated with chitin-derived sugars [GlcNAc, GlcN, (GlcNAc)2, (GlcN)2, or GlcNAc-GlcN] for 6 h were analyzed using a peptide mass fingerprinting (PMF) approach. We examined proteins consistently exhibiting sequence coverage of ≥20% in three independent experiments (see Table S1 in the supplemental material). We then focused on proteins involved in chitin metabolism. Some proteins were detected in several compartments; for example, some intracellular proteins were also identified in the culture supernatant. We attributed this result to leakage from cells during cultivation. Based on analyses of peak area data from the PMF experiments, we found that the production of several proteins was enhanced when the cells were cultivated with disaccharides. Moreover, when bacteria were cultivated with GlcNAc-GlcN, the production of numerous proteins was upregulated compared with their expression in cells cultivated with (GlcNAc)2 (Fig. 2). Two of four chitinases (VP0619 and VPA0055) were detected in the culture supernatant at high coverage. These chitinases appear to be constitutive enzymes, as they were also highly expressed by cells cultured without sugar. After cultivation with (GlcN)2 or GlcNAc-GlcN, both chitinases (VP0619 and VPA0055) were expressed at much higher levels than they were under the other conditions examined. Based on these data, we concluded that the expression of chitinases VP0619 and VPA0055 is induced by disaccharides in which at least one residue is GlcN.
FIG 2.
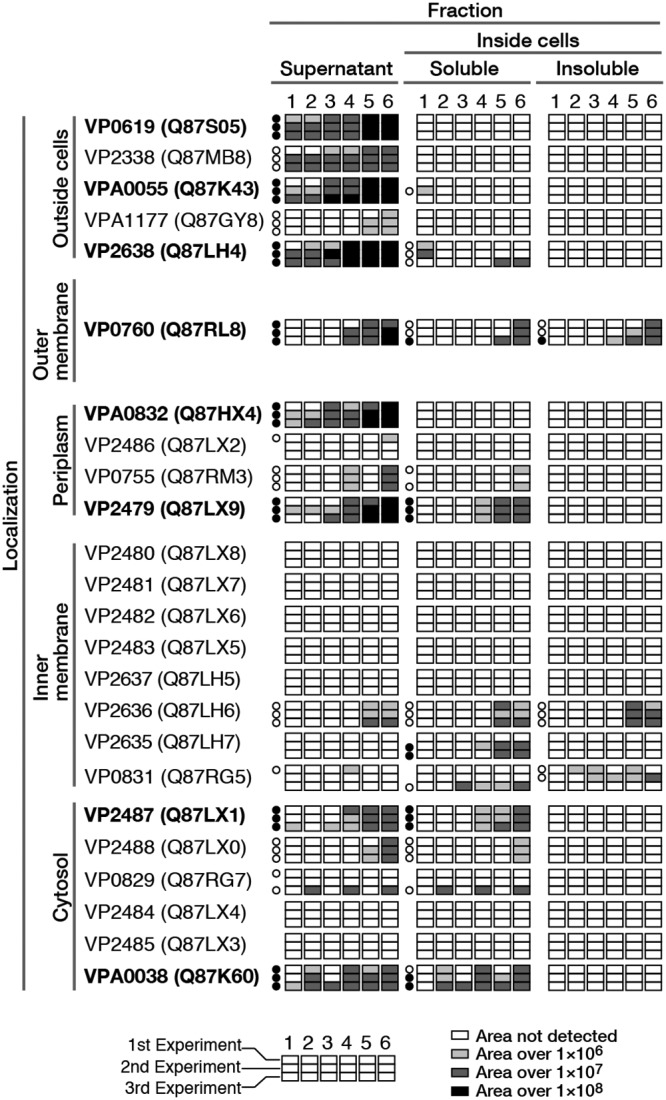
Expression levels of the proteins shown in Fig. 1, as determined by PMF analysis. The numbers at the top indicate the sugars used as inducers of protein expression: 1, control (no sugar); 2, GlcNAc; 3, GlcN; 4, (GlcNAc)2; 5, (GlcN)2; and 6, GlcNAc-GlcN. Each box represents the results of one experiment, and peak areas are indicated by light and dark shading. Circles indicate the coverage, as follows: black, ≥20%; white, <20%; and blank, not detected. Proteins detected at high coverage (≥20%) in three experiments are also shown in bold. Each coverage level is described in Table S1 in the supplemental material.
In contrast, the coverage of chitinases VP2338 and VPA1177 was lower than that of chitinases VP0619 and VPA0055. However, we observed that the differences in the expression of these proteins between sugars were similar. COD (VP2638) was detected at a high coverage in the supernatant, and its expression appeared to be induced by disaccharides. Chitoporin (VP0760), located in the outer membrane, was detected in the culture supernatant. We speculated that this protein also leaked from cells during cultivation or other experimental procedures. Chitoporin was also detected in the soluble and insoluble fractions, although the protein coverage was low. The expression of this porin was thought to be induced by disaccharides, particularly GlcNAc-GlcN. No periplasmic enzymes were detected in the soluble fraction, although two such enzymes were isolated in the culture supernatant. The expression of chitodextrinase (VPA0832, EC 3.2.1.202) and periplasmic peptide-binding protein (VP2479) was higher when cells were cultivated with disaccharides. Bacteria cultivated with (GlcN)2 or GlcNAc-GlcN also exhibited increased expression of these proteins compared with cells cultivated with (GlcNAc)2. Two other enzymes, β-N-acetylhexosaminidases (VP2486 and VP0755), were not detected in this experiment, even though they are thought to be constitutively expressed (12). The above-mentioned periplasmic peptide-binding protein (VP2479), through which V. cholerae was thought to bind (GlcNAc)2 (14), functions as a component of the ABC transporter for uptake of (GlcNAc)2. We therefore examined our PMF data regarding the other component proteins located on the inner membrane. We did not detect the two permeases (VP2480 and VP2481) or two ATP-binding proteins (VP2482 and VP2483) in our three independent experiments. We also examined the data for proteins associated with two phosphotransferase systems (PTSs) thought to be involved in transporting mono- or disaccharides derived from chitin (3). One PTS, which is generally annotated as cellobiose specific, consists of three components, IIB (VP2637), IIC (VP2636), and IIA (VP2635), and it is thought to be (GlcN)2 specific in V. cholerae (13). Component IIB (VP2637) was not detected in our PMF analysis, but we did detect component IIC (VP2636) at a low coverage in each experiment and IIA (VP2635) at a high coverage only twice in the three independent experiments. Another PTS thought to be involved in the uptake of the monosaccharide GlcNAc by V. furnissii (19), namely, N-acetylglucosamine-specific component IIABC (VP0831) (Fig. 1), was detected at a low coverage only.
As for proteins located in the cytosol, N,N′-diacetylchitobiose phosphorylase (VP2487, EC 2.4.1.280) and glucosamine-6-phosphate deaminase (VPA0038, EC 3.5.99.6) were detected at a high coverage in both the culture supernatant and the soluble fraction. N,N′-Diacetylchitobiose phosphorylase expression was induced by disaccharides, particularly (GlcN)2 and GlcNAc-GlcN. In contrast, the expression of glucosamine-6-phosphate deaminase, which was detected at a high coverage in almost all experiments, exhibited no significant difference between sugars. Other cytosol proteins, including phosphoglucomutase (VP2488), N-acetylglucosamine-6-phosphate deacetylase (VP0829, EC 3.5.1.25), endoglucanase-like protein (VP2484), and ATP-dependent glucosamine kinase (a putative N-acetylglucosamine kinase; VP2485, EC 2.7.1.8), either were not detected or were detected at a low coverage. These data suggest that the enzymes that function in the initial steps of chitin catabolism are induced to a higher degree by deacetylated chitin disaccharides, namely, (GlcN)2 and GlcNAc-GlcN. We consider that (GlcN)2 or GlcNAc-GlcN specifically induces the expression of various proteins that function during the initial steps of chitin degradation, namely, oligosaccharide production from chitin outside the cell, and they also induce the expression of several soluble intracellular proteins.
Analysis of the transcription of genes encoding proteins involved in chitin degradation in strain RIMD2210633 using RT-qPCR.
We focused on soluble proteins and chitoporin in this investigation using a real-time quantitative PCR (RT-qPCR) technique. We chose (GlcNAc)2 and GlcNAc-GlcN as inducers because these disaccharides are produced outside the cell by this strain. We initially selected the induction time (the cultivation time after adding each sugar as an inducer) based on experiments examining the expression of four chitinase genes. Protein expression was clearly detected after 6 h of induction (Fig. 2); however, gene expression was detected much earlier, after only 2 h of cultivation (Fig. 3A). We were able to detect the transcription of all chitinase genes using both induction times. However, the expression levels of the chitinases after 2 h of induction were higher than those after 6 h of induction, and GlcNAc-GlcN exhibited a much more significant effect with a 2-h induction time (Fig. 3B). These data indicate that these disaccharides induced chitinase gene expression basally (Fig. 3A) and that GlcNAc-GlcN is a much more effective inducer than (GlcNAc)2 (Fig. 3B). We therefore decided to investigate the expression of other genes using cells induced for 2 h.
FIG 3.
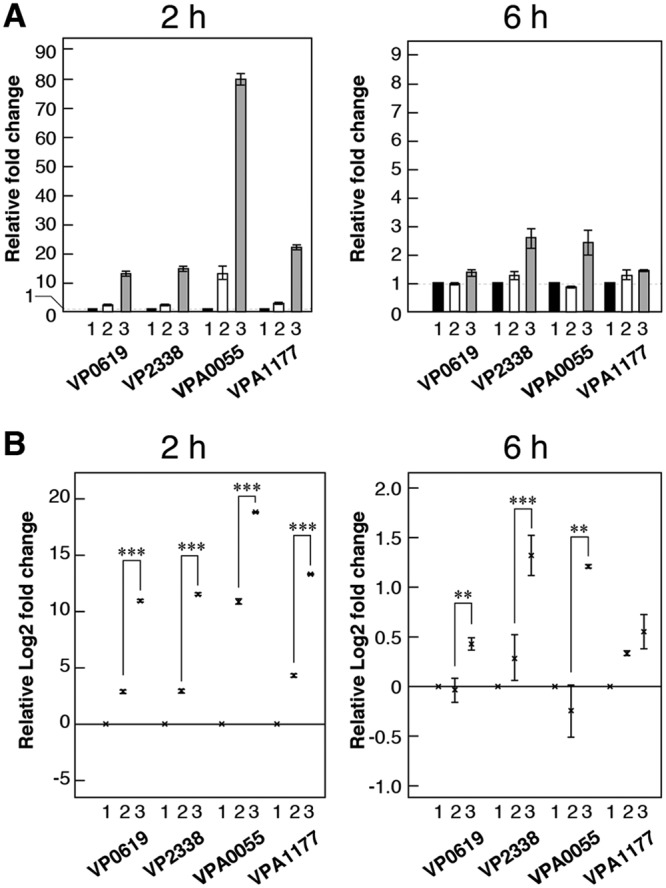
Comparison of chitinase gene expression levels using RT-qPCR. Gene expression levels were assessed after induction for 2 h or 6 h. (A) Gene expression levels are shown as the relative fold change. Bars indicate the mean ± SE (n = 3) fold change. (B) Gene expression levels shown as the relative log2 fold change. The × marks indicate the mean ± SE (n = 3) of the log2 fold change, and asterisks indicate P values, as follows: ***, P < 0.01; **, P < 0.05. The sugars used as inducers of gene expression were as follows: 1, control (no sugar); 2, (GlcNAc)2; and 3, GlcNAc-GlcN.
Most of the genes that we examined exhibited the same tendency; that is, disaccharides markedly induced their expression. The expression of genes encoding some proteins, such as chitoporin (VP0760), two β-N-acetylhexosaminidases (VP2486 and VP0755), periplasmic peptide-binding protein (VP2479), N,N′-diacetylchitobiose phosphorylase (VP2487), an endoglucanase-like protein (VP2484), and ATP-dependent glucosamine kinase (VP2485), was markedly induced by GlcNAc-GlcN, such that the mean fold change relative to the level of expression of the control was >100, and the relative fold change in the expression of COD (VP2638) and phosphoglucomutase (VP2488) was >50 (Fig. 4A). Moreover, the expression of all of the above-mentioned genes except that encoding VP0755 was induced by GlcNAc-GlcN to a much greater degree than by (GlcNAc)2 (Fig. 4B). Expression of the gene encoding chitodextrinase (VPA0832) was also induced much more significantly by GlcNAc-GlcN than by the other inducer (Fig. 4B), although the fold change in expression relative to that of the other genes was quite low (Fig. 4A). Expression of the genes encoding N-acetylglucosamine-6-phosphate deacetylase (VP0829) and glucosamine-6-phosphate deaminase (VPA0038) exhibited no significant difference under these conditions (Fig. 4B).
FIG 4.
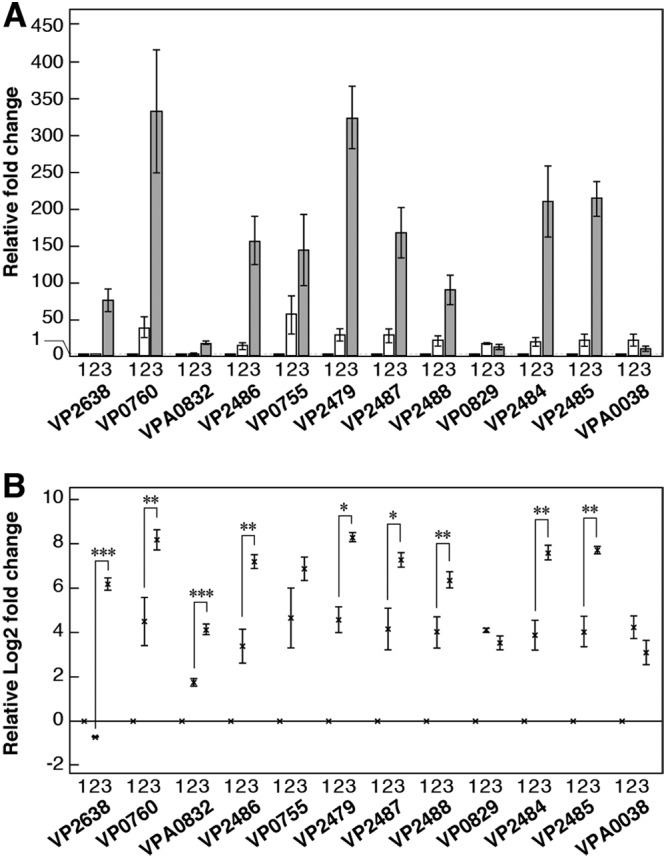
Comparison of gene expression levels using RT-qPCR. (A) Gene expression levels shown as relative fold change. Bars indicate the mean ± SE (n = 3). (B) Gene expression levels shown as relative log2 fold change. The × marks indicate the mean ± SE (n = 3), and asterisks indicate P values, as follows: ***, P < 0.01; **, P < 0.05; *, P < 0.1. The sugars used as inducers of gene expression were as follows: 1, control (no sugar); 2, (GlcNAc)2; and 3, GlcNAc-GlcN.
DISCUSSION
In our previous study, extracellular V. parahaemolyticus KN1699 chitinase activity was higher in the presence of GlcNAc-GlcN (11). In our current PMF analysis, the expression of two of four chitinases (VP0619 and VPA0055) of V. parahaemolyticus RIMD2210633 increased in the presence of disaccharides derived from chitin, and this tendency was greater in the presence of GlcNAc-GlcN and (GlcN)2 than in the presence of (GlcNAc)2 (Fig. 2). In contrast, the monosaccharides GlcNAc and GlcN did not upregulate chitinase expression in this study. The same tendency was observed with regard to COD (VP2638) (Fig. 2). Based on the results of our previous study involving V. parahaemolyticus KN1699, we had hypothesized that the most important factor in inducing chitinase and COD production was that the sugar was a disaccharide and that its reducing end was deacetylated (11). The results of PMF analyses in this study agree with that hypothesis. In addition to chitinases and COD, we identified other proteins for which their expression was upregulated in strain RIMD2210633 when the cells were cultivated with (GlcN)2 or GlcNAc-GlcN. The expression of some of these proteins appeared to increase in the presence of (GlcNAc)2, even though the expression level was comparatively low relative to that observed in the presence of (GlcN)2 and GlcNAc-GlcN. We suspect that (GlcNAc)2 added to the culture was deacetylated to GlcNAc-GlcN by COD, which was constitutively produced during the 6-h cultivation. The results of RT-qPCR analyses clearly indicated that GlcNAc-GlcN is a stronger inducer of chitinase and COD expression in COD-producing V. parahaemolyticus than (GlcNAc)2. Notably, GlcNAc-GlcN is expressly produced outside cells by a specific enzyme, COD. We speculate that COD genes are a dominant factor enabling marine bacteria to utilize chitin efficiently, as strains that produce COD can degrade chitin to oligosaccharide via chitinase faster than strains that do not produce COD. Moreover, GlcNAc-GlcN also induces expression of the gene encoding COD, and bacteria also produce GlcNAc-GlcN. This enhances the chitin-degrading efficiency of COD-producing bacteria.
The expression of all four chitinases increased in the presence of GlcNAc-GlcN, and this effect was more significant with cells induced for 2 h than with cells induced for 6 h (Fig. 3). The expression levels of other genes encoding chitin degradation enzymes also increased when the cells were cultivated with disaccharides. None of the chitin degradation-related genes that we examined exhibited significantly decreased expression in the presence of GlcNAc-GlcN or (GlcNAc)2. With the exception of the genes for N-acetylglucosamine-6-phosphate deacetylase (VP0829) and glucosamine-6-phosphate deaminase (VPA0038), gene expression levels were higher when cells were cultivated with GlcNAc-GlcN than when they were cultivated with (GlcNAc)2 (Fig. 4). However, the degree of difference in expression depended on the gene. We hypothesize that the mechanism underlying the increased expression of these proteins does not depend on a single operon, as these upregulated genes are carried on both of two chromosomes. Regarding the chitin degradation gene expression system in V. cholerae, Li and Roseman (14) reported that one of the mechanisms involves a two-component system in which the carbohydrate binding protein (CBP) of the ABC transporter (KEGG accession number VC0620) acts as a switch to activate the sensor kinase ChiS (VC0622), as follows. Once (GlcNAc)2 binds to CBP, which interacts with ChiS, CBP disassociates from ChiS, in turn activating global gene expression. Meibom et al. (13) also reported that the expression of many genes in V. cholerae increases in the presence of crab shell. Expression of genes in the cluster VC0620 to VC0611, which is located downstream of ChiS and which includes the components of ABC transporter and metabolic enzymes (see Fig. S1A in the supplemental material), is induced by GlcNAc oligomers, including (GlcNAc)2; however, this is not the case for mutants lacking the ChiS gene incubated in the presence of (GlcNAc)2 (13). These authors also reported that the genes of this cluster are expressed as an operon and speculated that the cluster functions as a regulon system, although the response regulator remains unidentified (13). This gene cluster including ChiS was also identified in the genome of V. parahaemolyticus RIMD2210633 (Fig. S1B). According to the results of our PMF analyses, only a few proteins encoded by this cluster were significantly induced by GlcNAc-GlcN (Fig. S1C). However, the expression of many genes of soluble enzymes, including those of the above-mentioned cluster, was clearly induced to a greater degree by GlcNAc-GlcN than by (GlcNAc)2, according to the results of RT-qPCR analyses (Fig. 3 and 4). Indeed, the enzymes encoded by this cluster in strain RIMD2210633 (VP2479, VP2484, VP2485, VP2486, VP2487, and VP2488) were investigated in this study. Although whether these enzymes (except for VP2479 and VP2487) were induced by GlcNAc-GlcN was not conclusive according to PMF analyses, the results of RT-qPCR analyses indicated that the expression of all of them was significantly upregulated by GlcNAc-GlcN rather than by (GlcNAc)2 (Fig. 2 and 4B). Based on these data, we hypothesize that these genes are regulated at the same time, and it is possible that they are part of a regulon similar to that reported by Meibom et al. (13) in V. cholerae; however, in this case, it is mainly under the control of GlcNAc-GlcN.
To date, many reports regarding chitin metabolic enzymes in members of the genus Vibrio and their expression systems have been published. However, a number of issues remain unclear, such as the reaction through which (GlcN)2 is produced from (GlcNAc)2 or GlcNAc-GlcN in the periplasm (Fig. 1). It is possible that some unknown deacetylase acts on the nonreducing end GlcNAc residue of the disaccharide within the cells. On the other hand, there is perhaps no (GlcN)2 in the periplasm if such an enzyme does not exist, and GlcNAc-GlcN appears to be transported into the cytosol via the PTS (VP2635 to VP2637). According to Meibom et al. (13), the expression of genes encoding this PTS (VC1281 to VC1286) and COD (VC1280), which is located adjacent to PTS and in the opposite transcription direction, in V. cholerae (Fig. S2A) increased in the presence of (GlcN)2, and the authors speculated that this gene cluster (VC1281 to VC1286) was an operon controlled by (GlcN)2 (13). This gene cluster is also encoded in the genome of V. parahaemolyticus RIMD2210633 (Fig. S2B). Our PMF analyses suggested that some of the genes of this cluster are upregulated by (GlcN)2 or GlcNAc-GlcN (Fig. S2C). We therefore hypothesize that GlcNAc-GlcN instead of (GlcN)2 also controls the expression of these genes, as (GlcN)2 is not present in the periplasm. However, we did not assess the expression of these genes in this study because we focused on soluble metabolic enzymes in order to characterize the role of GlcNAc-GlcN in chitin metabolism. In a future study, we should examine the expression of membrane protein genes and downstream genes of this PTS (VP2635 to VP2637), especially the endoglucanase-like protein (VP2484), which in V. cholerae does not hydrolyze (GlcNAc)2 or (GlcN)2 (20). It is speculated that the disaccharide, carried via PTS and phosphorylated, is then hydrolyzed by another enzyme, such as 6-phospho-β-glucosidase (VP2634, EC 3.2.1.86) (Fig. S2D), which is a component of the PTS cluster that was detected twice at a low coverage in our PMF analyses (Fig. S2C).
According to our RT-qPCR results, there was no difference in the gene expression levels of N-acetylglucosamine-6-phosphate deacetylase (VP0829) and glucosamine-6-phosphate deaminase (VPA0038) between the inducers (GlcNAc)2 and GlcNAc-GlcN (Fig. 4B). The genes encoding these enzymes are under the control of the same system, known as the nag regulon. Some studies have examined this gene cluster (21, 22), and there are reports of chitin-derived sugars inducing the expression of these genes (13, 23). nagE, nagA, nagC, and nagB correspond to the N-acetylglucosamine-specific IIABC component (VP0831), N-acetylglucosamine-6-phosphate deacetylase (VP0829), the N-acetylglucosamine repressor (VP0828), and glucosamine-6-phosphate deaminase (VPA0038), respectively. As expression of these genes is upregulated to a greater degree by GlcNAc or the GlcNAc oligomer in V. cholerae than by other sugars (13), it is appropriate that these genes, which are involved in the uptake of GlcNAc, exhibited an expression pattern in the presence of GlcNAc-GlcN different from that of the other genes that we examined in this research (Fig. S3). We hypothesize that there are several gene expression systems involved in chitin metabolism in Vibrio species and that these systems are probably controlled by different sugars. The findings of a previous study considering the possibility of a relationship between the PTS and chemotaxis are very interesting (24). In addition, the expression of genes encoding some chemotaxis-related proteins, chitin adhesion proteins, and GlcNAc-binding protein (GbpA), which are involved in Vibrio colonization of biotic environmental surfaces (25) and which exhibit lytic polysaccharide monooxygenase activity (26), was shown to be induced by GlcNAc in V. cholerae (13). Besides producing chitin degradation enzymes, adhesion to the chitin surface and chemotaxis are important for the survival of Vibrio in marine environments (1). We are particularly interested in chitin metabolism by Vibrio species because this plays a critical role in the ecological dominance of this genus. Such knowledge will be helpful in addressing Vibrio-associated problems in health, food, and aquaculture.
In this study, we focused on the role of GlcNAc-GlcN in mediating the expression of soluble chitin metabolic enzymes and found that this heterodisaccharide strongly induces the expression of several enzymes. We also found, however, that GlcNAc-GlcN does not induce the expression of all related enzymes. These data, along with those from previous reports by many researchers, strongly suggest that chitin degradation by Vibrio species is controlled by several regulon systems. In the pathway shown in Fig. 1, there are two routes by which metabolites can enter the pentose-phosphate pathway from chitin: via the N-acetylglucosamine-specific IIABC component of the PTS (VP0831) or via the peptide ABC transporter (VP2479 to VP2483) and cellobiose-specific PTS (VP2635 to VP2637). The soluble enzymes of the latter pathway are activated by GlcNAc-GlcN, which appears to be under the control of the ChiS regulon system (Fig. S3). It is still unclear why GlcNAc-GlcN is a better inducer of the expression of some enzyme genes than (GlcNAc)2. It may depend on the ease of interaction with CBP of the disaccharide or of saccharide transportation via each transporter, and we think that these issues should be elucidated.
We found that the heterodisaccharide GlcNAc-GlcN is an important factor for enhancing the chitin-metabolizing ability of V. parahaemolyticus in the marine environment. However, some issues remain to be clarified. For example, the effect of saccharides from chitin, including GlcNAc-GlcN, on the expression of proteins associated with sugar transport systems is also worthy of investigation. Such studies would enhance our understanding of chitin degradation by marine bacteria.
MATERIALS AND METHODS
Bacterial strains.
Vibrio parahaemolyticus RIMD2210633 was obtained from the Research Institute for Microbial Diseases, Osaka University (Japan).
Preparation of saccharides.
GlcNAc and GlcN were obtained from Tokyo Chemical Industry, (Tokyo, Japan) and Fujifilm Wako Pure Chemical (Osaka, Japan), respectively. (GlcNAc)2 was purified from a mixture of GlcNAc and GlcNAc oligomers (chitin oligosaccharides containing N-acetylglucosamine; Tokyo Chemical Industry) using a modification of previously reported methods (27), as follows. GlcNAc oligomers dissolved in H2O were loaded onto a charcoal (particle size, 63 to 300 mm; Fujifilm Wako Pure Chemical) column. After elution of unbound oligomers with H2O, the (GlcNAc)2 that adhered to charcoal was eluted with 7% (vol/vol) ethanol in water. Fractions containing only (GlcNAc)2 were collected, and the solvent was evaporated to obtain the solid. The solid was dissolved in a small amount of H2O, and (GlcNAc)2 was recrystallized by adding 2-propanol. We also loaded hydrolyzed products from the above-mentioned GlcNAc and GlcNAc oligomer produced by Denazyme CBB-P1 (Nagase chemteX, Osaka, Japan), which produces (GlcNAc)2 from the GlcNAc oligomer, onto a charcoal column and eluted the column using the same purification steps described above. The mass spectra of the obtained powder revealed peaks at m/z 425, 447, and 871, corresponding to [M + H+], [M + Na+], and [2M + Na+], respectively, using an LCMS-2020 liquid chromatography (LC)-mass spectrometry (MS) system (Shimadzu, Kyoto, Japan) under dual ion source (DUIC) ionization, confirming that (GlcNAc)2 was obtained. We also qualitatively assessed (GlcNAc)2 using high-performance liquid chromatography (HPLC) analysis, which revealed a single peak with the same retention time as the GlcNAc dimer (Fig. S4). GlcNAc-GlcN was produced from (GlcNAc)2 using recombinant COD of V. parahaemolyticus KN1699 and purified using the same method described in a previous paper (11). (GlcN)2 was obtained from the hydrolysate of GlcN oligomers (chitosan oligosaccharides; Tokyo Chemical Industry) treated with chitosanase L (HBI Enzymes, Hyogo, Japan) in 0.5 M sodium acetate buffer (pH 5.2). (GlcN)2 dissolved in buffer was purified using two types of ion-exchange column chromatography columns: a column with Dowex 50W-X8 column with 200- to 400-mesh resin (Fujifilm, Wako Pure Chemical) and a column with IRA96SB resin (Organo, Tokyo, Japan). After the (GlcN)2-containing eluate was concentrated by evaporation using a rotary evaporator, purified (GlcN)2 was collected by acetone precipitation and then dissolved in a small amount of H2O. The details of (GlcN)2 preparation are described in Method S1 in the supplemental material. The mass spectra of the solution obtained revealed peaks at m/z 341, 363, and 703, corresponding to [M + H+], [M + Na+], and [2M + Na+], respectively, using an LCMS-2020 LC-MS system (Shimadzu) under DUIC ionization conditions, indicating that pure (GlcN)2 was obtained. We also qualitatively assessed (GlcN)2 by HPLC analysis and observed a single peak with the same retention time as the GlcN dimer (Fig. S5). We calculated the (GlcN)2 concentration of this solution by HPLC analysis using the peak area value of (GlcN)2·2HCl (chitobiose; Seikagaku Kogyo, Tokyo, Japan) in H2O as a standard solution.
Detection of enzymes involved in chitin metabolism that are induced by chitin-derived sugars in strain RIMD2210633.
The following experiment was conducted three times independently (1st, 2nd, and 3rd experiments). Cells of V. parahaemolyticus RIMD2210633 were cultivated in M9-lactate liquid medium, which consisted of 10 ml of salt solution (49 mM Na2HPO4·H2O, 22 mM KH2PO4, 19 mM NH4Cl, 8.6 mM NaCl), 100 μl of 50% (wt/vol) sodium lactate solution, 10 μl of 1 M MgSO4, and 1 μl of 1 M CaCl2, at 30°C with shaking (160 rpm) for 24 h. The cells were collected by centrifugation at 3,200 × g for 10 min and resuspended in a fresh salt solution consisting of the same components as M9-lactate liquid medium but without sodium lactate, MgSO4, and CaCl2 in order to adjust the optical density at 600 nm (OD600) to approximately 0.6. Then, we mixed 5 ml of cell suspension, 4 ml of fresh salt solution, and 1 ml of 10 mM sugar dissolved in the same solution. After adding 100 μl of 50% (wt/vol) sodium lactate solution, 10 μl of 1 M MgSO4, and 1 μl of 1 M CaCl2, the mixture (10 ml) was incubated at 30°C for 6 h with shaking (160 rpm). Salt solution was used instead of sugar solution as a control. The OD600 values before and after incubation of each culture were very similar. The OD600 was approximately 0.3 in the three independent experiments when the cultivation was started, and after cultivation for 6 h, the values were between 0.7 and 1.0.
Seven milliliters of supernatant was collected from each culture solution by centrifugation at 6,000 × g for 10 min at 4°C, and then the supernatant was centrifuged again to remove the remaining cells. Approximately 4 ml of the collected supernatant was used. After adding 20 μl of protease inhibiter solution (cOmplete, Mini, EDTA free [Roche Molecular Systems, Inc., CA], dissolved in 250 μl of H2O), the volume of each supernatant to use in the next experiment was determined. We used a volume of 3,000 to 4,000 μl so as to equalize the quantity of cells that was calculated on the basis of the wet volume of cells from a 4-ml culture. Each culture supernatant was concentrated to <174 μl using an Amicon Ultra-15 centrifugal filter device (Merck KGaA, Darmstadt, Germany), 24 μl of protease inhibiter solution (cOmplete, Mini, EDTA free, in 300 μl H2O) was added, and then the volume was made up to 200 μl with H2O for use as the supernatant protein solution.
Using an Ez-Apply 2D kit (ATTO, Tokyo, Japan), soluble and insoluble proteins were extracted from the cells. Before extraction, we adjusted the cell number based on the wet weight of cells collected from a full culture under each condition. The proper quantity of wash buffer from the kit was added (100 mg of cells/ml), and we used the same volume of cell suspension for each culture condition in the same experiment. Finally, after operation according to the kit manual, solutions of soluble and insoluble proteins were obtained from the following wet weights of cells in the three experiments: 1st experiment, 50 mg; 2nd experiment, 40 mg; and 3rd experiment, 40 mg. The concentrations of these protein solutions were measured and calculated using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, MA), and the volume of protein solution for LC-tandem MS (LC-MS/MS) analysis was determined. Next, the protein solutions from the same compartment were added at the same volume. Each solution was heated at 95°C for 5 min with the same volume of 2× sample buffer, which consisted of 4% SDS, 10% glycerol, 125 mM Tris-HCl buffer (pH 6.8), 0.2 M dithiothreitol, and 0.02% bromophenol blue, and then an appropriate volume equivalent to the same weight of cells for each cultivation condition was loaded onto a polyacrylamide gel (E-R 520L e-PAGEL 5 to 20% gel; ATTO). After electrophoresis, using an SDS-PAGE system until the proteins moved to 5 mm from the bottom of the well on the gel, the gel was stained using AE-1360 EzStain silver (ATTO), and stained protein bands were excised. The proteins in the gel fragments were alkylated using iodoacetamide and ammonium hydrogen carbonate. After digestion by treatment with trypsin, the protein fragments were extracted from the gel fragments. These procedures are described in detail in Method S2. LC-MS/MS analysis was performed using an Advance LC (Bruker-Michrom, CA) equipped with a Zaplous alpha Pep C18 column (0.2 [inside diameter] by 50 mm; AMR, Tokyo, Japan), a PAL HTS-xt autosampler (CTC Analytic AG, Zwingen, Switzerland), and a Q Exactive mass spectrometer (Thermo Fisher Scientific). For each analysis, we loaded 1 μl of sample onto the column, and the peptides were separated at a flow rate of 1,500 nl/min at 35°C for 30 min using a linear gradient composed of solution A (0.1% formic acid in water) and solution B (acetonitrile) in the following steps. The gradient was started at 5% solution B and ended at 65% solution B over 20 min, was increased up to 95% solution B in 1 min and maintained for 3 min, and was decreased to 5% solution B in 1 min and then maintained for 5 min. The Q Exactive mass spectrometer parameters were set as follows: polarity, positive; resolution for full-scan MS, 70,000; mass scan range, 350 to 1,800 m/z; and resolution for MS/MS, 17,500. The data files obtained from the above-mentioned operations were analyzed using Proteome Discoverer (version 2.0) software (Thermo Fisher Scientific) and searched against all protein sequence data for V. parahaemolyticus RIMD2210633 (NCBI taxonomy accession number 223926) deposited in the UniProt database (http://www.uniprot.org/) as of 17 July 2015. For the database searches, we set the parameters as follows: enzyme name, trypsin; precursor mass tolerance, 10 ppm; fragment mass tolerance, 0.02 Da; dynamic modification, oxidation/+15.995 Da (M); and static modification, carbamidomethyl/+57.021 Da (C). Other parameters were set at the default.
RT-qPCR analysis of transcription of genes encoding proteins involved in chitin degradation in strain RIMD2210633.
The genomic DNA used for constructing a standard curve for RT-qPCR analysis was extracted and purified from V. parahaemolyticus RIMD2210633 cells using a Nexttec 1-step DNA isolation kit for bacteria (Nexttec GmbH, Leverkusen, Germany). The cells were cultivated in 10 ml of high-nutrition liquid medium containing 0.5% (wt/vol) Bacto peptone (BD Biosciences, CA), 0.1% (wt/vol) Bacto yeast extract (BD Biosciences), 0.5 M NaCl, 62.5 mM MgCl2, 62.5 mM MgSO4, and 50 mM KCl for 16 h at 30°C with shaking. The concentration of the DNA in the solution was prepared in order to be appropriate for constructing standard curves. Cell cultivation and RNA extraction from the cells were carried out three times independently, and RT-qPCR analysis was conducted using each RNA sample (1st, 2nd, and 3rd experiments). Cells of V. parahaemolyticus RIMD2210633 were cultivated in 10 ml of M9-lactate liquid medium at 30°C with shaking (160 rpm) for 16 h. Then, 500 μl of this preculture was added to 50 ml of fresh medium and cultivated at 30°C with shaking (160 rpm) for 40 h, and at that time, the OD600 of the culture was >1.5. The cells were collected by centrifugation at 3,200 × g for 10 min and resuspended in fresh salt solution in order to adjust the OD600 to approximately 0.6. Then, we mixed 5 ml of cell suspension, 4 ml of fresh salt solution, and 1 ml of 10 mM sugar dissolved in the same solution. After adding 100 μl of 50% (wt/vol) sodium lactate solution, 10 μl of 1 M MgSO4, and 1 μl of 1 M CaCl2, the mixture (10 ml; OD600, approximately 0.3) was incubated at 30°C for 6 h or 2 h with shaking (160 rpm). We used salt solution instead of sugar solution as a control. In this experiment, the absorbance at 600 nm was considered indicative of the number of cells. We calculated the values using the following formula: absorbance (OD600) × volume of cell suspension (in milliliters). We took an adequate volume of the cell suspension and added fresh medium in order to adjust the value. Each cell suspension in a 1.5-ml tube was centrifuged at 10,000 × g for 1 min in order to collect the cells. The supernatant was removed; the cells were suspended in 100 μl of Tris-EDTA buffer solution (Sigma-Aldrich, MO), to which 10 μl of 10 mg/ml lysozyme (Sigma-Aldrich) solution was added; and the components were mixed using a vortex mixer. Then, the solution was incubated at 37°C for 10 min. Approximately 60 μl of RNA solution was collected from the cells using a NucleoSpin RNA kit (Macherey-Nagel GmbH & Co. KG, Düren, Germany). However, this solution was still contaminated with genomic DNA. We therefore repeated the DNA digestion and purification steps according to the NucleoSpin RNA kit manual. We then collected the RNA solution, which contained only a minimal amount of DNA that had little or no influence on RT-qPCR. No DNA contamination was judged to be present from a comparison of the results of RT-qPCR analyses with and without cDNA synthesis against the RNA polymerase sigma factor RpoD gene (VP0404), which was selected as a reference to normalize gene expression levels. We also confirmed the quality and concentration of total RNA using an Experion RNA StdSens analysis kit (Bio-Rad, CA) and an Experion automated electrophoresis system (Bio-Rad) before the synthesis of cDNA, which was prepared by reverse transcription-PCR. We used approximately 1 μg of total RNA for each sample per 20 μl of reaction mixture for cDNA synthesis, which was carried out using an iScript Advanced cDNA synthesis kit for RT-qPCR (Bio-Rad). The primers for RT-qPCR were designed, and it was confirmed that each target gene was amplified by the corresponding primer set shown in Table S2. The expression level of each gene was quantified relative to the levels on a standard curve. RT-qPCR was conducted as follows. We prepared 20 μl of the RT-qPCR mixture using SsoAdvanced Universal SYBR green supermix (Bio-Rad) containing 1 μl of 10 μM forward (Fw) and reverse (Rv) PCR primers and the required volume of the cDNA reaction mixture. For preparing the standard curve, genomic DNA was used instead of the above-mentioned cDNA solution. Amplification and detection were carried out in 96-well plates (Multiplate 96-well PCR plates; low profile, unskirted, white; Bio-Rad) sealed with film (Microseal B adhesive sealing films; Bio-Rad) using a CFX96 PCR detection system (Bio-Rad) in association with CFX Manager (version 2.0) software (Bio-Rad). The PCR cycle was programmed for 98°C for 2 min and 46 cycles of 98°C for 10 s and an appropriate annealing temperature for 30 s. After every annealing step, the plate was scanned to collect the data. After the amplification steps, the plate was heated at 95°C for 10 s, and then a melting curve was generated by heating from 65 to 95°C at 0.5°C per 5-s increment. The annealing temperature for each primer set is shown in Table S2. RT-qPCR was performed in duplicate or triplicate, and two values were adopted for the average calculation for each experiment. The standard curve was prepared using genomic DNA as a template in the same 96-well plate. The reactions for standard curves were performed in duplicate or triplicate, and two values were adopted for the calculation of the average. The concentration of the templates for RT-qPCR was set in order that the cycle number of each threshold was between 21 and 36. The expression level of each target gene was determined using the following steps. The starting quantity (SQ) of the target genes and the RpoD gene was calculated from each standard curve using CFX Manager software, and the means of the SQ for duplicate data (mean SQ) were used for the calculation of expression levels. Next, the mean SQ for the target gene was divided by the mean the SQ for the RpoD gene. The expression level of the control (the no-sugar condition) was treated as a standard value of 1.0, and then we derived the relative fold change in expression when sugars were added in each experiment. Data are expressed as the mean ± standard error (SE) based on the data from the three experiments. We also calculated the log2 fold change in expression and expressed the data as the mean ± SE based on the data from the three experiments. The significance of the differences in target gene expression in the presence of (GlcNAc)2 or GlcNAc-GlcN as an inducer was determined by the paired Student's t test (two-tailed) using the log2 fold change values. Statistical analyses were performed using Excel software (Microsoft Office 365; Microsoft, WA).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (grant number 16K18751) from 2016 to 2018 and was also funded by our university through the Life Science Research Center of College of Bioresource Sciences, Nihon University (2015), and by a grant from the College of Bioresource Sciences, Nihon University (2014).
We thank Tomoyo Kiuchi, Saki Ebashi, Maiko Abe, Mai Ito, and Mika Kashiwagi of the General Research Institute of our college for support in operating the LC-MS/MS and DNA sequencing systems. We also appreciate the contributions of students Saya Kanzawa and Mio Nakamura for assistance with the production and purification of disaccharides.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00270-19.
REFERENCES
- 1.Keyhani NO, Roseman S. 1999. Physiological aspects of chitin catabolism in marine bacteria. Biochim Biophys Acta 1473:108–122. doi: 10.1016/s0304-4165(99)00172-5. [DOI] [PubMed] [Google Scholar]
- 2.Keyhani NO, Li X, Roseman S. 2000. Chitin catabolism in the marine bacterium Vibrio furnissii. Identification and molecular cloning of a chitoporin. J Biol Chem 275:33068–33076. doi: 10.1074/jbc.M001041200. [DOI] [PubMed] [Google Scholar]
- 3.Hunt DE, Gevers D, Vahora NM, Polz MF. 2008. Conservation of chitin utilization pathway in the Vibrionaceae. Appl Environ Microbiol 74:44–51. doi: 10.1128/AEM.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suginta W, Chumjan W, Mahendran KR, Janning P, Schulte A, Winterhalter M. 2013. Molecular uptake of chitooligosaccharides through chitoporin from the marine bacterium Vibrio harveyi. PLoS One 8:e55126. doi: 10.1371/journal.pone.0055126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohishi K, Yamagishi M, Ohta T, Motosugi M, Izumida H, Sano H, Adachi K, Miwa T. 1997. Purification and properties of two deacetylases produced by Vibrio alginolyticus H-8. Biosci Biotechnol Biochem 61:1113–1117. doi: 10.1271/bbb.61.1113. [DOI] [Google Scholar]
- 6.Li X, Wang L, Wang X, Roseman S. 2007. The chitin catabolic cascade in the marine bacterium Vibrio cholerae: characterization of a unique chitin oligosaccharide deacetylase. Glycobiology 17:1377–1387. doi: 10.1093/glycob/cwm096. [DOI] [PubMed] [Google Scholar]
- 7.Kadokura K, Sakamoto Y, Saito K, Ikegami T, Hirano T, Hakamata W, Oku T, Nishio T. 2007. Production of a recombinant chitin oligosaccharide deacetylase from Vibrio parahaemolyticus in the culture medium of Escherichia coli cells. Biotechnol Lett 29:1209–1215. doi: 10.1007/s10529-007-9386-6. [DOI] [PubMed] [Google Scholar]
- 8.Hirano T, Maebara Y, Uehara R, Sakaki Y, Shiraishi H, Ichimura S, Hakamata W, Nishio T. 2013. Chitin oligosaccharide deacetylase from Vibrio harveyi ATCC BAA-1116: gene cloning, overexpression, purification, and characterization. Chitin Chitosan Res 19:321–324. [Google Scholar]
- 9.Hirano T, Uehara R, Shiraishi H, Hakamata W, Nishio T. 2015. Chitin oligosaccharide deacetylase from Shewanella woodyi ATCC51908. J Appl Glycosci 62:153–157. doi: 10.5458/jag.jag.JAG-2015_014. [DOI] [Google Scholar]
- 10.Hirano T, Shiraishi H, Ikejima M, Uehara R, Hakamata W, Nishio T. 2017. Chitin oligosaccharide deacetylase from Shewanella baltica ATCC BAA-1091. Biosci Biotechnol Biochem 81:547–550. doi: 10.1080/09168451.2016.1254529. [DOI] [PubMed] [Google Scholar]
- 11.Hirano T, Kadokura K, Ikegami T, Shigeta Y, Kumaki Y, Hakamata W, Oku T, Nishio T. 2009. Heterodisaccharide 4-O-(N-acetyl-β-d-glucosaminyl)-d-glucosamine is a specific inducer of chitinolytic enzyme production in Vibrios harboring chitin oligosaccharide deacetylase genes. Glycobiology 19:1046–1053. doi: 10.1093/glycob/cwp088. [DOI] [PubMed] [Google Scholar]
- 12.Hirano T, Aoki M, Kadokura K, Kumaki Y, Hakamata W, Oku T, Nishio T. 2011. Heterodisaccharide 4-O-(N-acetyl-β-d-glucosaminyl)-d-glucosamine is an effective chemotactic attractant for Vibrio bacteria that produce chitin oligosaccharide deacetylase. Lett Appl Microbiol 53:161–166. doi: 10.1111/j.1472-765X.2011.03083.x. [DOI] [PubMed] [Google Scholar]
- 13.Meibom KL, Li XB, Nielsen A, Wu C, Roseman S, Schoolnik GK. 2004. The Vibrio cholerae chitin utilization program. Proc Natl Acad Sci U S A 101:2524–2529. doi: 10.1073/pnas.0308707101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Roseman S. 2004. The chitinolytic cascade in vibrios is regulated by chitin oligosaccharides and a two-component chitin catabolic sensor/kinase. Proc Natl Acad Sci U S A 101:627–631. doi: 10.1073/pnas.0307645100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kadokura K, Rokutani A, Yamamoto M, Ikegami T, Sugita H, Itoi S, Hakamata W, Oku T, Nishio T. 2007. Purification and characterization of Vibrio parahaemolyticus extracellular chitinase and chitin oligosaccharide deacetylase involved in the production of heterodisaccharide from chitin. Appl Microbiol Biotechnol 75:357–365. doi: 10.1007/s00253-006-0831-6. [DOI] [PubMed] [Google Scholar]
- 16.Sakamoto Y, Kuno E, Tomiyama A, Hirano T, Kumaki Y, Tanaka A, Kanda H, Furuya K, Hakamata W, Oku T, Nishio T. 2009. A chitin oligosaccharide deacetylase from Vibrio sp. SN184: gene cloning, overexpression, purification and characterization. J Appl Glycosci 56:273–276. doi: 10.5458/jag.56.273. [DOI] [Google Scholar]
- 17.Thompson FL, Iida T, Swings J. 2004. Biodiversity of vibrios. Microbiol Mol Biol Rev 68:403–431. doi: 10.1128/MMBR.68.3.403-431.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ina-Salwany MY, Al-Saari N, Mohamad A, Mursidi F-A, Mohd-Aris A, Amal MNA, Kasai H, Mino S, Sawabe T, Zamri-Saad M. 2019. Vibriosis in fish: a review on disease development and prevention. J Aquat Anim Health 31:3–22. doi: 10.1002/aah.10045. [DOI] [PubMed] [Google Scholar]
- 19.Bouma CL, Roseman S. 1996. Sugar transport by the marine chitinolytic bacterium Vibrio furnissii. Molecular cloning and analysis of the glucose and N-acetylglucosamine permeases. J Biol Chem 271:33457–33467. doi: 10.1074/jbc.271.52.33457. [DOI] [PubMed] [Google Scholar]
- 20.Park JK, Wang L, Patel HV, Roseman S. 2002. Molecular cloning and characterization of unique β-glucosidase from Vibrio cholerae. J Biol Chem 277:29555–29560. doi: 10.1074/jbc.M202978200. [DOI] [PubMed] [Google Scholar]
- 21.Yamano N, Oura N, Wang J, Fujishima S. 1997. Cloning and sequencing of the genes for N-acetylglucosamine use that construct divergent operons (nagE-nagAC) from Vibrio cholerae non-O1. Biosci Biotechnol Biochem 61:1349–1353. doi: 10.1271/bbb.61.1349. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh S, Rao KH, Sengupta M, Bhattacharya SK, Datta A. 2011. Two gene clusters co-ordinate for a functional N-acetylglucosamine catabolic pathway in Vibrio cholerae. Mol Microbiol 80:1549–1560. doi: 10.1111/j.1365-2958.2011.07664.x. [DOI] [PubMed] [Google Scholar]
- 23.Thompson FL, Neto AA, Santos EO, Izutsu K, Iida T. 2011. Effect of N-acetyl-d-glucosamine on gene expression in Vibrio parahaemolyticus. Microbes Environ 26:61–66. doi: 10.1264/jsme2.ME10152. [DOI] [PubMed] [Google Scholar]
- 24.Yu C, Bassler BL, Roseman S. 1993. Chemotaxis of the marine bacterium Vibrio furnissii to sugars. A potential mechanism for initiating the chitin catabolic cascade. J Biol Chem 268:9405–9409. [PubMed] [Google Scholar]
- 25.Stauder M, Huq A, Pezzati E, Grim CJ, Ramoino P, Pane L, Colwell RR, Pruzzo C, Vezzulli L. 2012. Role of GbpA protein an important virulence-related colonization factor, for Vibrio cholerae’s survival in the aquatic environment. Environ Microbiol Rep 4:439–445. doi: 10.1111/j.1758-2229.2012.00356.x. [DOI] [PubMed] [Google Scholar]
- 26.Loose JSM, Forsberg Z, Fraaije MW, Eijsink VGH, Vaaje-Kolstad G. 2014. A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide. FEBS Lett 588:3435–3440. doi: 10.1016/j.febslet.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 27.Minamoto T, Takahashi N, Kitahara S, Shinozaki Y, Hirano T, Hakamata W, Nishio T. 2015. Saccharification of β-chitin from squid pen by a fermentation method using recombinant chitinase-secreting Escherichia coli. Appl Biochem Biotechnol 175:3788–3799. doi: 10.1007/s12010-015-1547-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.