

Abstract
Mitochondria are considered highly plastic organelles. This plasticity enables the mitochondria to undergo morphological and functional changes in response to cellular demands. Stem cells also need to remain functionally plastic (i.e. to have the ability to “decide” whether to remain quiescent or to undergo activation upon signaling cues to support tissue function and homeostasis). Mitochondrial plasticity is thought to enable this reshaping of stem cell functions, integrating signaling cues with stem cell outcomes. Indeed, recent evidence highlights the crucial role of maintaining mitochondrial plasticity for stem cell biology. For example, tricarboxylic acid (TCA) cycle metabolites generated and metabolized in the mitochondria serve as cofactors for epigenetic enzymes, thereby coupling mitochondrial metabolism and transcriptional regulation. Another layer of mitochondrial plasticity has emerged, pointing toward mitochondrial dynamics in regulating stem cell fate decisions. Imposing imbalanced mitochondrial dynamics by manipulating the expression levels of the key molecular regulators of this process influences cellular outcomes by changing the nuclear transcriptional program. Moreover, reactive oxygen species have also been shown to play an important role in regulating transcriptional profiles in stem cells. In this review, we focus on recent findings demonstrating that mitochondria are essential regulators of stem cell activation and fate decisions. We also discuss the suggested mechanisms and alternative routes for mitochondria-to-nucleus communications.
Keywords: mitochondria, metabolism, stem cells, epigenetics, nucleus, molecular dynamics, mitochondrial metabolism, mitochondrial DNA (mtDNA), histone modification, ROS signaling, self-renewal, cell signaling, metabolic cross-talk
Introduction
Stem cells have the unique ability to both self-renew and generate differentiated functional cell types in vivo through asymmetric cell division (1). Recent evidence has emerged to point out the importance of the stem cell niche in vivo for maintaining stemness, imposing a continuous requirement of stem cells to adapt to their environment (2, 3). The mitochondria are multifaceted organelles, mainly implicated in the regulation of energy and fuel homeostasis (4). Acting as central metabolic hubs, the mitochondria rapidly adapt to different environmental cues and metabolic alterations to meet the biogenetic demands of the cell, also termed mitochondrial plasticity (4). An important feature of maintaining mitochondrial plasticity are the ongoing fusion and fission events reshaping mitochondrial morphology termed mitochondrial dynamics (5). Owing to their highly dynamic nature and plasticity, the mitochondria constitute an essential mediator of environmental cues with fate decisions (6). As mitochondrial regulation of stem cell function is becoming increasingly recognized, mitochondrial metabolism in particular was shown to have a pivotal role in dictating whether a stem cell will proliferate, differentiate, or remain quiescent (7).
Increasing amounts of evidence support the notion of a cross-talk between mitochondrial metabolism and the epigenome (6–8). Accordingly, the abundance and availability of TCA3 cycle metabolites, that also function as epigenetic enzyme cofactors, reshape DNA and histones to establish an epigenetic landscape to initiate nuclear transcriptional reprogramming (8–10). Other evidence supports a secondary messenger in the form of reactive oxygen species (ROS) signaling to initiate transcriptional reprogramming (11, 12). An established notion of stem cell metabolism is the importance of maintaining a high glycolytic flux (i.e. the cytosolic conversion of glucose to pyruvate/lactate) as a critical determinant of stemness (13–15). Relying on aerobic glycolysis for ATP generation and biosynthetic demands, stem cells generally exhibit a fragmented mitochondrial network with underdeveloped cristae, although maintaining a functionally active electron transport chain (ETC) (16). On the other hand, terminally differentiated cells shift their reliance of bioenergetic demands to the mitochondria by utilizing oxidative phosphorylation (OXPHOS), the process of energy generation fueled by respiration and the ETC, characterized by a hyperfused mitochondrial network important for OXPHOS activity (17). Interestingly, these metabolic shifts are accompanied by profound changes in mitochondria morphology, and indeed mitochondrial dynamics and metabolism were shown to reciprocally influence each other during cellular processes (5, 18). Recent evidence indicates that the metabolic profile of stem cells is actually dependent on and can be manipulated by the molecular regulators of mitochondrial fusion and fission, leading to changes in stem cell fate (19). Different stem cell states, however, show distinct mitochondrial and metabolic profiles, pointing toward the complexity of the relationship between mitochondrial dynamics, metabolism, and consequent cell fate (19).
An important notion of the in vitro stem cells research is that these cells are self-renewed in culture in the presence of cytokines and small molecules acting directly on transcription factors and epigenetic enzymes. In vivo stem cells reside in specialized niches and are exposed to specific metabolic alterations. These factors are ruled out and are absent in vitro. Therefore, the physiological relevance of the in vitro models should be carefully interpreted.
In this review, we discuss the evolving mitochondrial mechanisms for self-renewal and stem cell differentiation, mainly focusing on mitochondrial dynamics, cellular metabolic programming, and epigenetic remodeling, placing the mitochondria at the center of stem cell fate decisions.
Stem cells and mitochondrial dynamics
Stem cells maintain tissue homeostasis by dividing asymmetrically, generating differentiated cell types, while self-renewing to maintain the stem cell pool (1). An interesting feature of stem cells is their distinct differentiation potential, as stem cells are compartmentalized according to their potency, defined by the variety of cell lineages they can differentiate into (20). The most potent cells are the primitive stem cells generated after zygote formation, termed pluripotent embryonic stem cells, which have the capacity to generate the entire embryo. Potency declines during development, and the fetal/adult stem cells comprising the hematopoietic and neuronal systems are multipotent (20). This section will describe the recent evidence supporting the importance of mitochondrial dynamics in pluripotent and multipotent stem cells, for maintaining mitochondrial plasticity and normal stem cell function.
Pluripotent stem cells (PSC) have the unique ability to differentiate into all three embryonic germ layers (21). In vitro naive pluripotency refers to embryonic stem cells (ESC) derived from the inner cell mass of preimplantation mouse blastocysts, whereas in vitro primed pluripotency is represented by epiblast stem cells (EpiSC) commonly derived from the late epiblast layer of the postimplantation embryo. These two cell types differ in their distinctive morphologies, cytokine dependence, gene expression, epigenetic regulation, and metabolic profiles (22, 23). Interestingly, another feature shown to be distinct between the naive and primed mouse pluripotent states is the mitochondrial network. Naive ESC exhibit fragmented, perinuclear mitochondrial morphology, characterized by underdeveloped cristae (24) (Fig. 1). Primed EpiSC mitochondria appear elongated, with relatively more developed cristae, critical determinants of OXPHOS and respiration (24). Surprisingly, measuring cell respiration using oxygen consumption rate as a readout showed that in both mouse and human models, primed PSC respire significantly less than naive ESC (24, 25), suggesting a role for mitochondria in these cells that is distinct from energy production and cell respiration.
Figure 1.

Mitochondria morphology and the molecular regulators of mitochondria dynamics in the regulation of transitioning between stem cells states and immune cells. The molecular regulators underlying fusion and fission events are depicted below the schematic representation of round/fragmented and elongated/fused mitochondria, respectively. Transitioning between the different stem cell states and immune cells was reported to be dependent upon the levels of these molecular regulators, according to the established mitochondria morphology characteristics with each state.
An indication for the importance of maintaining a fragmented mitochondrial network in ESC was reported during reprogramming of somatic cells to naive pluripotency, as an early wave of mitochondrial fission/fragmentation was recorded upon the expression of reprogramming factors (26). Importantly, inhibition of dynamin-related protein 1 (DRP1), a major pro-mitochondrial fission regulator, resulted in impaired generation of induced pluripotent stem cell (iPSC) colonies. Of note, in mouse embryonic fibroblasts, increased fatty acid (FA) synthesis as well as the exogenous addition of FA were reported to promote mitochondrial fission. Accordingly, lipogenesis was proven important for the maintenance of ESC and for the generation of iPSC, presumably by promoting mitochondrial fragmentation (27), thereby further supporting the notion that naive pluripotency actively requires the maintenance and establishment of a fragmented mitochondrial network.
During in vitro differentiation of naive ESC to cardiomyocytes, robust mitochondrial elongation was detected (28). This mitochondrial elongation was accompanied by an increase in the protein levels of the promitochondrial fusion regulators, mitofusin 2 (MFN2) and optic atrophy 1 (OPA1), mediating outer and inner mitochondrial membrane fusion, respectively (28). Ablation of MFN2 or OPA1 resulted in impaired differentiation of naive ESC into beating cardiomyocytes, which was restored upon their re-expression (28). Moreover, combined ablation of mitochondrial outer membrane pro-fusion proteins, mitofusin 1 (MFN1) and MFN2, restricted to the neonatal murine heart, resulted in embryonic lethality (28). In addition, conditional ablation of MFN1 and MFN2 in the adult heart led to mitochondrial fragmentation, followed by heart failure and lethality 7–8 weeks after knockout induction (29). Imbalanced processing of OPA1 by mitochondrial protease OMA1, thereby increasing the levels of short OPA1 (S-OPA1) over the unprocessed long OPA1 (L-OPA1), the latter required for inner membrane mitochondrial fusion, results in mitochondrial fragmentation and heart failure in mice (30). Altogether, these findings support the notion that mitochondrial elongation is essential for ESC differentiation to cardiomyocytes and for normal cardiac development and function.
Naive ESC acutely deleted of mitochondrial carrier homolog 2 (MTCH2), an outer mitochondrial membrane protein, exhibited a compromised ability to exit naive pluripotency upon the induction of brief in vitro priming to epiblast-like cells (31). Further exploring this phenomenon, we found that MTCH2 acts as a novel regulator of mitochondrial fusion. Accordingly, MTCH2−/− ESC possess a hyperfragmented mitochondrial morphology and fail to properly elongate mitochondria upon priming. Forcing mitochondrial elongation in WT and MTCH2−/− ESC by expressing MFN2 or by expressing a dominant negative form of DRP1 was sufficient to drive the exit from naive pluripotency, as evident by the down-regulation of naive markers (31).
Long-term hematopoietic stem cells (LT-HSC) were recorded to exhibit highly fragmented mitochondria, whereas slightly differentiated short-term HSC (ST-HSC) undergo an increase in mitochondrial length (32). Interestingly, MFN2 expression levels are up-regulated specifically during the transition from LT-HSC to ST-HSC, and preventing mitochondrial fusion in HSC by MFN2 ablation results in an increase in the levels of LT-HSC and loss of ST-HSC. These events further lead to deficiency of the lymphoid lineage, giving rise to the acquired immune system, but not the myeloid lineage giving rise to the innate immune system, as determined by divergent competitive transplantation assays.
Neuronal stem cells (NSC) were reported to exhibit an elongated mitochondria morphology in vivo (33). Furthermore, progression of NSC to a more committed fate was accompanied by the transition of the mitochondria to a fragmented state, which was once again elongated upon terminal differentiation of these progenitors to postmitotic neurons. In the case of somatic cells, T cells of the immune system, which require the intrinsic capacity to constantly shape and adapt to their environment, were also shown to reside in different cell states, depending on their mitochondrial dynamics properties. In this context, effector T (TE) cells were shown to exhibit a fragmented mitochondrial morphology, whereas memory T (TM) cells maintain a fused mitochondrial network (34). Importantly, ablation of mitochondrial fusion in TE cells impaired the generation of TM cells both in vitro and in vivo after viral infection, indicating that mitochondrial fusion is necessary for TM cell generation.
Altogether, the findings described above clearly show the importance of maintaining a plastic mitochondrial network to dictate and mediate cellular processes such as differentiation and the acquisition of specialized functions, highlighting the role of mitochondrial dynamics in mediating the different stem cell states (PSC, HSC, and NSC). Mitochondrial dynamics and metabolism are known to reciprocally affect each other (35); it is therefore likely that the direct changes in mitochondria dynamics described above trigger metabolic reprogramming, which in turn enables plasticity in cell fate decisions.
Metabolic control of stem cell fate
Metabolic plasticity is required for stem cells to match their energy demands, depending on available nutrients and energy sources (36). Early work has shown that during early embryonic development, the human embryo is exposed to different levels of metabolites (37), suggesting a necessity of stem cells for metabolic adaptation during these early developmental stages. However, beyond a role in energy support, recent evidence highlights stem cell “metabolic shifts” as an essential process in mediating stem cell fate decisions (36). A major challenge to this field is to establish a direct cross-talk between changes in the metabolic flux and stem cell fate.
Multiple lines of evidence support the paradigm of preferentially glycolytic self-renewing stem cells undergoing a metabolic switch to activate mitochondria and OXPHOS during differentiation and lineage priming, placing glycolysis as an established determinant of stemness, utilized by stem cells even in the presence of oxygen and functionally active mitochondria (38). An indication for the importance of glycolysis for the acquisition of stemness was recorded during reprogramming to iPSC, as somatic cells are required to undergo a metabolic shift from OXPHOS to glycolysis upon the introduction of reprogramming factors (39) (Fig. 2). Importantly, the described metabolic shift precedes the expression of pluripotency markers, and stimulation of glycolysis was reported to promote iPSC reprogramming efficiency (39). Interestingly, the NAD-dependent deacetylase Sirtuin-2 (Sirt2) was reported to facilitate the described metabolic shift during iPSC reprogramming (40). Sirt2 expression was down-regulated during reprogramming to iPSC, followed by an increase in the acetylation levels of specific glycolytic genes, rendering these enzymes more active, thereby enhancing glycolysis (40). Sirt2 overexpression enhanced ESC differentiation potential, whereas Sirt2 knockdown increased iPSC reprogramming efficiency (40).
Figure 2.
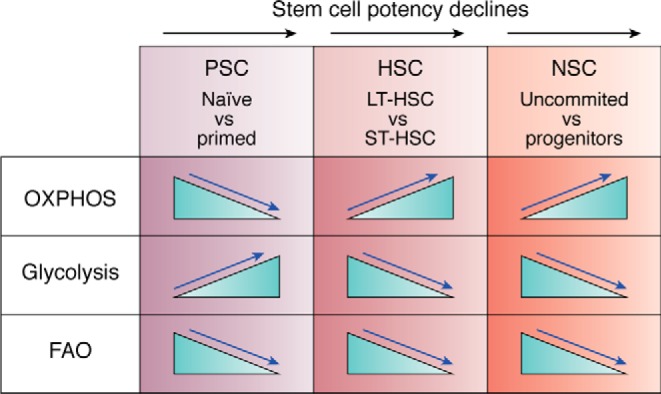
Metabolic requirements underlying the transition between the different stem cell states. The metabolic shifts underlying the transition between the different stem cell states are depicted above, as blue arrows point out the directionality of the metabolic shift, which was reported necessary for achieving the new stem cell state. The black arrow marks the decline in stem cell potency. Note that whereas the requirements for OXPHOS and glycolysis are diverse, the decrease in FAO levels is shared between the different stem cells.
Mechanisms for balancing between mitochondrial clearance and mitochondrial biogenesis were reported to facilitate the metabolic shifting between OXPHOS and glycolysis, thereby dictating stem cell outcomes. Accordingly, reprogramming efficiency of somatic cells to iPSC, which was accompanied by decreased mitochondrial mass and mitochondrial DNA (mtDNA) levels (41), was increased upon autophagy enhancement, followed by increased lactate levels, indicative of higher glycolytic flux. Importantly, blocking autophagy specifically during the early stages of reprogramming abolished iPSC generation (41). Limiting mitochondrial expansion was recorded to be important for preserving ESC fitness. Accordingly, acetyl-CoA (Ac-CoA) generation by threonine dehydrogenase (TDH), expressed only in undifferentiated ESC, was found to acetylate Kif1-binding protein (KBP), thereby degrading KBP and limiting mitochondrial biogenesis (42). As mitochondrial biogenesis is initiated upon ESC differentiation, stabilizing KBP in ESC results in an unscheduled early increase in mitochondrial biogenesis, followed by increased respiration and ROS levels, whereas perturbing this pathway, thereby limiting mitochondrial accumulation, impairs ESC differentiation capacity (42).
In the bone marrow, LT-HSC reside in hypoxic niches and are dependent on glycolysis to maintain a nonproliferative quiescent state (43). Interestingly, pyruvate dehydrogenase kinase isoforms, phosphorylating pyruvate dehydrogenase (PDH) and thereby suppressing pyruvate influx into the mitochondria, were recorded to be highly expressed in LT-HSC as compared with ST-HSC, in concordance with the higher glycolytic flux and lower OXPHOS in these cells (43). Accordingly, loss of autophagy, which led to the accumulation of mitochondria, was followed by an increase in OXPHOS and loss of HSC self-renewal and regenerative capacity.
Activation of the peroxisome proliferator–activated receptor δ (PPAR-δ) which was found essential for the maintenance and function of LT-HSC, was recorded to preserve LT-HSC potential by inducing mitochondrial autophagy (mitophagy) (44). Importantly, PPAR-δ agonist treatment in vivo was found to induce preferential symmetric self-renewal of HSC, hence generating two daughter cells that are stem cells, thereby expanding the HSC pool (44). Blocking mitophagy by inhibiting the molecular regulators of this process abolishes HSC reconstitution capacity, indicating that HSC stemness depends upon successful mitochondrial clearance (44). Finally, measuring mitophagy of the mouse brain in vivo revealed higher rates of autophagy specifically in the dentate gyrus, areas enriched for NSC (45).
These studies highlight the role of metabolic reprogramming as a driving force of stem cell fate outcomes, preceding the establishment of nuclear identity, also regulated by mitochondrial homeostasis mechanisms as well as post-translational modifications, ensuring high metabolic plasticity upon demand.
Mitochondrial OXPHOS activation was described during the differentiation of PSC and exit from quiescence of HSC. In PSC, expression levels of uncoupling protein 2 (UCP2), which regulates OXPHOS levels by shunting pyruvate out of the mitochondria, thereby facilitating glycolysis, were found in reverse correlation with mitochondrial respiration levels, decreasing upon PSC differentiation (46). Importantly, ectopic expression of UCP2 during PSC spontaneous differentiation to embryoid bodies was significantly impaired, arguing that perturbing the metabolic switch from glycolysis to OXPHOS blocks PSC differentiation (46). In HSC, loss of MTCH2 was shown to increase mitochondrial OXPHOS and trigger LT-HSC exit from quiescence and entry into cycle (47). Also, deletion of PTEN-like mitochondrial phosphatase 1 in HSC impaired respiration capacity while enhancing glycolysis by inducing UCP2 activity and blocking differentiation of HSC while expanding the HSC pool, leading to hematopoietic failure and death (48).
The mitochondrial pyruvate carrier, which resides in the inner mitochondrial membrane, is required for efficient mitochondrial pyruvate uptake, coupling glycolysis and the TCA cycle (49, 50). Interestingly, overexpression of mitochondrial pyruvate carrier in glycolytic cancer cells or in intestinal stem cells was shown to decrease proliferation and self-renewal capacity, respectively (51, 52). During compromised supply of pyruvate into the mitochondria, glutamine oxidation (glutaminolysis) engages to support TCA cycle intermediates, as glutamine-driven OXPHOS was shown to be a major source of ATP during decreased glucose oxidation (53). In addition, both embryonic and adult NSC were shown to be predominantly glycolytic as opposed to OXPHOS-activated committed progenitors and neurons. Moreover, glycolytic neuronal progenitor cells (NPC) initiate OXPHOS upon neuronal differentiation, and forcing glycolysis during differentiation leads to neuronal cell death. Finally, high energy–demanding postmitotic cells, which are terminally differentiated, rely on the mitochondria and OXPHOS for a continuous supply of ATP (54, 55).
All of the above findings highlight the role of metabolic reprogramming as a driving force of stem cell fate outcomes, preceding the establishment of nuclear identity, also regulated by mitochondrial homeostasis mechanisms and function as well as post-translational modifications of glycolytic enzymes, ensuring high metabolic plasticity upon demand.
These studies make an attractive claim of a unidirectional metabolic pattern of stem cells switching from glycolysis to OXPHOS upon differentiation signal. However, deepening our understanding of stem cell metabolism revealed that in the case of PSC, naive ESC were shown to be bioenergetically bivalent, utilizing both glycolysis and OXPHOS for energy production, whereas slightly differentiated primed EpiSC were shown to be exclusively glycolytic, failing to grow and generate PSC colonies in the presence of 2-DG, a competitive inhibitor of glycolysis (24). Additionally, both human and mouse primed PSC were shown to accumulate lipid droplets, as a consequence of impaired ability of fatty acid β-oxidation, which led to reduced respiration (56). The conversion from primed to naive PSC was reported to be mediated by STAT3 activation of mitochondrial respiration (57). The RNA-binding protein LIN28 was shown to facilitate the conversion from naive to primed pluripotency by specifically binding to mRNAs important for OXPHOS, reducing their protein abundance and thereby maintaining the low mitochondrial function associated with primed pluripotency (58). Importantly, loss of LIN28 resulted in delayed exit from naive pluripotency (58).
Of note, the small-molecule inhibitor of DRP1, mdivi1 (59), was recently demonstrated to inhibit also mitochondrial complex I, which does not regulate mitochondrial fission (60). In fact, somatic cell reprogramming to pluripotency in the presence of mdivi1 results in a completely diminished ability of iPSC generation, suggesting an important role of functionally active ETC during this process (26). Indeed, an OXPHOS burst was recorded during the early reprogramming of somatic cells to iPSC and proven essential for this process, as the inhibition of complex I by the small molecule inhibitor rotenone led to impaired iPSC generation (61).
In the hematopoietic system, loss of the mitochondrial respiratory complex III subunit, leading to impaired respiration and albeit mitochondrial dysfunction, resulted in the loss of HSC quiescence and impaired differentiation (62). This study showed that mitochondrial dysfunction of HSC in vivo results in increased BrdU incorporation and HSC exhaustion (62). PPAR-δ was found essential for the maintenance and function of LT-HSC, through the respiration-mediated fatty acid oxidation (FAO) pathway (63). The inhibition of FAO was reported to result in the symmetric commitment of HSC daughter cells (63) and is found in correlation with the activation of this pathway in naive ESC (56). Adult quiescent NSC show increased levels of FAO as compared with proliferating NSC as well as in mature neurons. The inhibition of FAO in a fully established and continuous quiescent state was sufficient to enhance robust proliferation in a dose-dependent manner (64). Interestingly, in PSC, HSC, and NSC, the levels of carnitine palmitoyltransferase 1 (CPT1), which is the rate-limiting enzyme for the FAO pathway, were reported to be expressed at higher levels as compared with their more differentiated progenies.
Taken together, we can conclude that switching from glycolysis to OXPHOS upon differentiation is not a linear process and that the different stem cell types exhibit different metabolic requirements in a cell context–dependent manner, similarly to the high plasticity observed with mitochondrial dynamics. Importantly, the fact that these metabolic alterations precede transcriptional and cell fate changes suggests that these metabolic shifts serve as checkpoints for the integrity and plasticity of cell functions.
Regulation of stem cells by mitochondrial ROS/oxidative metabolism
During the transport of electrons through the respiratory complexes (RC), oxygen-derived free radicals, termed ROS, are generated. These ROS can have detrimental consequences on proteins, lipids, and DNA molecules, specifically in stem cells that can acquire DNA mutations that will be passed on to their cell decedents (65) and therefore must be counterbalanced by the cell. One example of the importance of balancing ROS levels was shown for uncommitted NSC. In this model, upon NSC commitment to NPC, OXPHOS and ROS levels increase, essential for activating the developmental gene expression profile (33). Interestingly, the transcription factor nuclear factor erythroid 2–related factor 2 (NRF2), a known regulator of cellular resistance to oxidative damage, was proven essential for the ROS-mediated activation of the developmental program both in vitro and in vivo (33). These findings are consistent with the idea that ROS acts as a second messenger that activates redox-sensitive transcription factors, thereby mediating transcriptional reprogramming (66). Interestingly, upon NSC commitment to NPC, mitochondrial fragmentation was recorded. This appears to be counterintuitive to elevated OXPHOS levels, yet because high ROS levels are essential for neuronal differentiation, increased OXPHOS combined with mitochondrial fission and cristae relaxation (34) might be beneficial for this differentiation process by increasing ROS levels.
Reactive oxidants are counterbalanced by antioxidant defense systems. An important antioxidant for maintaining the cellular redox state is GSH, a reducing agent that protects the cell from oxidative damage (67). Upon exposure to increased oxidative stress, GSH converts to the oxidized GSH form (GSSG), a reaction catalyzed by GSH peroxidase, and thus monitoring the ratio of GSH/GSSG serves as an indicator of the cell redox state (68). Interestingly, a differential redox state was observed upon spontaneous differentiation of mouse naive ESC, showing a significant decrease in the ratio of GSH/GSSG, indicative of an increase in oxidative stress and in concordance with mitochondrial OXPHOS activation (69). Importantly, ESC were characterized by abundant metabolites with highly unsaturated structures, which are highly active under oxidative conditions. Indeed, inhibiting oxidative pathways in ESC delayed the loss of pluripotency upon neuronal differentiation, as shown by the improper levels of two established core pluripotency markers, OCT4 and Nanog (69). Furthermore, the addition of exogenous fatty acids, thereby inducing OXPHOS and ROS through the mitochondrial β-oxidation pathway, promoted ESC cardiac and neuronal differentiation (69). Taken together, the unique metabolic profile of ESC, characterized by highly reactive unsaturated metabolites, was proposed to confer “chemical plasticity” important in mediating differentiation upon activation of oxidative pathways.
Interestingly, withdrawal of glutamine, a precursor of GSH synthesis, led to a significant decrease in the GSH/GSSG ratio in ESC and to an increase of ROS levels (70). As a consequence, oxidized OCT4 levels were significantly increased, leading to a decrease in OCT4 protein levels and to a decrease in its cysteine-dependent DNA-binding activity (70). This suggests that glutamine regulates OCT4 levels and activity by imposing an oxidized OCT4 redox state. In fact, during the naive-to-primed pluripotency transition, which is not accompanied by increased OXPHOS and ROS (24, 56), OCT4 levels remain high, whereas the levels of other core pluripotency factors are strongly down-regulated (22).
Mitochondrial DNA control of stem cells
In embryonic stem cells and during early development
Maintaining balanced ROS levels for preserving mtDNA integrity and ETC function was shown to be a critical feature of cell fate (67). The mtDNA, packaged into histone-free nucleoids, carries 13 protein-coding RC genes, along with the mitoribosome and tRNA noncoding genes important for RC expression (71). Distribution of mtDNA molecules is regulated by mitochondrial dynamics, specifically by mitochondrial fission or division, segregating mtDNA to daughter mitochondria (72). The mtDNA levels of ESC were recorded to transiently increase during ESC spontaneous differentiation, in parallel with the up-regulation of mtDNA synthesis and mitochondrial transcription factor A (TFAM) enzyme (73). Knocking out TFAM in mice results in mtDNA depletion and abolished OXPHOS, leading to postgastrulation embryonic lethality (74). Interestingly, mtDNA possess a higher mutation rate than nuclear DNA, mainly because of the absence of protective histone molecules and due to the close proximity of the mitochondrial genome to the inner mitochondrial membrane, where ROS are routinely generated (75). Nonetheless, due to mitochondrial fusion activity and content mix exchange, functional complementation of pathogenic mtDNA molecules, which are mtDNA molecules having large-scale deletions or point mutations resulting in mitochondrial respiration abnormalities, renders cells highly tolerant to mtDNA mutations (75). The expression of an error-prone mitochondrial polymerase, PolgAD257A, which causes cells to accumulate mutations in an accelerated rate, together with an MFN1-deficient background, results in synthetic embryonic lethality (76). PolgAD257A alone was not sufficient to induce lethality, pointing out to the importance of mitochondrial fusion in the exchange of pathogenic mtDNA molecules and thereby maintaining a nonpathological embryonic state (76). In addition, this study demonstrates that mtDNA integrity, essential for ETC activity and function, regulates embryonic development.
In adult stem cells
Differential distribution of aged mitochondria was also recorded during asymmetric cell division of mammary epithelial stem-like cells (77). Old mitochondria, labeled using photoactivatable GFP, were shown to preferentially reside perinuclearly in mother cells. Strikingly, during asymmetric epithelial stem-like cell division, and not symmetrical cell division of normal epithelial cells, daughter cells designated for differentiated progeny were distributed mainly with old mitochondria, whereas daughter stem cells asymmetrically apportioned young mitochondria, in a mitochondrial fission-dependent manner (77). This supports the notion of the importance of maintaining a “young mitochondria” pool in stem cells to avoid accumulation of mtDNA mutations, leading to the perturbation of normal ETC activity, regulated by mitochondrial dynamics. Indeed, iPSC generated from somatic cells of patients containing exclusively WT mtDNA displayed normal metabolic functions compared with iPSC with exclusively mutant mtDNA (78). Moreover, mtDNA somatic mutations accumulated in an age-related manner (79). Accordingly, iPSC derived from young human adults exhibit fewer mtDNA mutations as opposed to elderly adults, showing that accumulated mutations negatively impact mitochondrial metabolism (79).
Mitochondria-to-nucleus communication in stem cells
Metabolic regulation of DNA and histone methylation
Epigenetic mechanisms ensure the highly dynamic chromatin state enabling cellular plasticity. As cellular metabolism provides the essential metabolites for the direct regulation of DNA and histone modifications, as well as cofactors and allosteric inhibitors of epigenetic enzymes, the study of metabolism-driven chromatin regulation, mainly in PSC, has emerged in recent years. Pioneering work in this field pointed out the importance of metabolic regulation for the survival of mouse ESC in vitro (80). Strikingly, the use of culture media individually deprived of each of the 20 amino acids led to the discovery that ESC are critically dependent on one essential amino acid, threonine (80). In addition, TDH, which converts threonine to glycine, and Ac-CoA were shown to be expressed specifically during the undifferentiated state of ESC and in explanted blastocyst embryos (80).
Follow-up work investigated the importance of threonine metabolism in ESC, revealing a mechanistic link between cellular metabolism and the epigenetic state during pluripotency (81). Interestingly, threonine catabolism was proven essential for generating S-adenosylmethionine (SAM) from glycine via the one-carbon metabolism pathway, a universal substrate for all protein methylation reactions in the cell, and threonine deprivation led to reduced H3K4me3 levels important for the euchromatin state (81) (Fig. 3). Surprisingly, rescuing ESC growth as well as H3K4me3 levels following threonine deprivation was dependent upon the exogenous addition of both glycine and pyruvate-derived Ac-CoA, which feed into the TCA cycle and do not directly participate in SAM generation. H3 acetylation levels were not impaired in the absence of threonine, suggesting other pathways for Ac-CoA generation, as opposed to a more prominent dependence on the threonine-SAM pathway for histone methylation (81). Finally, TDH knockdown in ESC results in more robust differentiation upon spontaneous differentiation culture conditions (81).
Figure 3.

Metabolic control of DNA and histone methylation. Metabolites and amino acids implicated in the generation of SAM and in the regulation of histone/DNA demethylase activity in stem cells, generated or metabolized within the mitochondria, are depicted above. Note that enriched pathways depicted in blue will support, whereas those depicted in red will repress, histone/DNA demethylase activity. The activity of histone/DNA methyltransferase depends upon the abundance and availability of nuclear SAM levels. SAH, S-adenosylhomocysteine; FH, fumarate hydratase; SDHA, succinate dehydrogenase A; NNMT, nicotinamide N-methyltransferase; 1-MNA, 1-methylnicotinamide.
Deprivation of methionine, which participates in the generation of SAM downstream to threonine metabolism, was also found essential for the maintenance and differentiation of human PSC, decreasing SAM levels and thereby negatively regulating methylation marks, followed by a decrease in Nanog levels (82). Recent work has shown that nicotinamide N-methyltransferase is expressed specifically in naive human ESC, consuming SAM and thereby rendering this metabolite unavailable for histone methylation. Nicotinamide N-methyltransferase specifically represses H3K27me3 and hypoxia-inducible factor α (HIF-1α) levels, the latter important for the naive-to-primed transition of PSC by elevating the glycolytic flux (56).
Ground-state naive ESC grown in culture media containing the two kinase inhibitors (i.e. 2i; inhibitors for the GSK3 and MEK pathways, which were shown to stabilize the core naive pluripotency network) as opposed to cells grown without 2i (slightly committed naive ESC), survive without the addition of exogenous glutamine to the culture medium, pointing toward 2i-dependent metabolic regulation (83). Indeed, metabolic flux analysis showed that more committed ESC decrease mitochondrial TCA cycle glucose utilization while increasing glutaminolysis, rendering these cells more glutamine-dependent to support TCA cycle intermediates. Ground-state ESC also showed higher contribution of labeled glucose to the generation of glutamine, which indicates higher generation with the lower consumption of glutamine in these cells. As a consequence, α-ketoglutarate (αKG) levels increased, whereas succinate levels decreased, indicative of reduced levels of αKG feeding into the TCA cycle and suggesting a higher αKG/succinate ratio in ground-state ESC. As αKG is an important cofactor for the activity of demethylases, growing ESC in 2i conditions without glutamine decreases αKG levels, resulting in increased repressive histone methylation marks associated with ESC differentiation. Importantly, the addition of exogenous αKG increases the efficiency of somatic cell reprogramming to pluripotency, concluding that αKG is essential for maintaining pluripotency and self-renewal of ESC (83).
Interestingly, a different study showed that whereas a higher αKG/succinate ratio in primed PSC excreted global histone and DNA demethylation, αKG alone accelerated the differentiation of primed PSC (84). Thus, similar cellular epigenetic mechanisms can lead to different outcomes, indicating a differential role for αKG in mediating cell fate, depending on cellular context. In HSC, the metabolite 2-hydroxyglutarate, which is an antagonist of αKG and thus competitively inhibits the activity of αKG-dependent demethylases, was shown to increase in HSC upon respiration deficiency, leading to increased DNA and histone methylation and exit from quiescence (62). Of note, fumarate and succinate were also shown to competitively inhibit the activity of αKG-dependent demethylases (85). Specific mutations of the enzymes utilizing TCA cycle metabolites fumarate and succinate as substrates led to the accumulation of these metabolites in cancer cells, consequently altering global DNA and histone methylation and inducing cancer progression (85).
Metabolic regulation of histone acetylation
In the case of histone acetylation, the nuclear levels of the central metabolite Ac-CoA, in concordance with the enzymatic activity of histone acetyl transferase and histone deacetylase, determine the global histone acetylation levels (Fig. 4). The regulation of glycolysis-derived Ac-CoA was recorded to mediate early PSC differentiation by modulating histone acetylation levels (86). Accordingly, upon spontaneous differentiation of PSC, histone acetylation levels decrease, together with decreased glycolytic flux and reduced levels of Ac-CoA originating from labeled glucose (86). During self-renewal conditions, inhibiting glycolysis upstream of Ac-CoA generation decreased histone acetylation levels and slightly decreased stemness markers (86). Importantly, blocking histone deacetylation during differentiation by the addition of exogenous acetate, a precursor for Ac-CoA generation, impaired PSC exit from pluripotency (86). Surprisingly, these findings show that the importance of maintaining high glycolytic flux during PSC self-renewal is attributable to the coupling of glycolysis to the TCA cycle and the generation of pyruvate-derived Ac-CoA rather than the traditional uncoupling Warburg effect (84).
Figure 4.

Metabolic control of histone acetylation levels. Metabolites and lipids implicated in the generation of Ac-CoA within the mitochondria and the nucleus of stem cells are depicted above. Note that enriched pathways depicted in blue will support the generation of cytosolic and nuclear pools of Ac-CoA and subsequent histone acetylation levels, whereas those depicted in red will repress the generation of cytosolic Ac-CoA and histone acetylation levels. Pathways shown in pink were reported to support bioenergetics of stem cells in response to the continuous citrate efflux. Blocking pyruvate entry into the mitochondria, allowing for nuclear-pyruvate accumulation together with active nuclear PDC, will support the generation of nuclear Ac-CoA pools. MPC, mitochondrial pyruvate carrier.
Because Ac-CoA cannot freely diffuse from the mitochondria, cytosolic as well as nuclear pools of Ac-CoA are dependent upon mitochondria-derived citrate efflux, as the enzyme ATP citrate lyase (ACLY) degrades citrate to generate extramitochondrial Ac-CoA (and oxaloacetate) (87). ACLY nuclear localization was reported to have a critical role in the regulation of global histone acetylation levels (88). Silencing of ACLY led to decreased levels of histone acetylation in cancer cells and impaired lipid accumulation in differentiating adipocytes (88). Silencing ACLY also led to reduced glucose consumption and to decreased expression of glycolytic enzymes, rescued by the addition of exogenous acetate, a precursor for histone acetylation in the absence of Ac-CoA (88). These studies indicate that ACLY links cellular metabolism, mainly glucose-derived Ac-CoA, with histone acetylation and cell outcomes.
Human PSC were recorded to inefficiently generate citrate downstream metabolites originating from labeled glucose, whereas glutamine oxidation efficiently generates these metabolites and can be further enhanced upon glucose withdrawal to support respiration and ATP generation (89). In support of these findings, the addition of exogenous αKG, but not pyruvate, rescued PSC self-renewal and survival (89). Most importantly, human iPSC were recorded to express low levels of the enzymes utilizing citrate and isocitrate as TCA cycle–feeding substrates, followed by high expression levels of ACLY, as compared with their differentiated cardiomyocytes (89). These results further emphasize the importance of glucose-derived citrate to the generation of Ac-CoA in PSC rather than supporting bioenergetics for cell survival (89).
Pyruvate dehydrogenase complex (), which is a three-subunit enzymatic complex converting pyruvate to Ac-CoA, was also reported to localize in the nucleus of different cell types, including primary somatic and cancer cell lines (90). Importantly, nuclear PDC was proven functionally active and important for histone acetylation, as isolated nuclei treated with labeled pyruvate generated Ac-CoA in a dose-dependent manner, decreasing histone acetylation levels upon PDC silencing (90). Nuclear PDC fraction was shown to dynamically translocate from the mitochondria rather than being a newly translated product from the endoplasmic reticulum (90). In addition, mitochondria-to-nucleus translocation of PDC was detected during G1-S phase progression, characterized by increased histone acetylation as a hallmark, without changing total PDC levels (90). Finally, PDH together with a subset of mitochondrial TCA cycle enzymes were shown to be localized and active within the nucleus of early stage embryos (91). In the absence of exogenous pyruvate, explanted zygotes failed to progress to the two-cell stage and to initiate zygote genome activation (91). Isolated nuclei of two-cell embryos treated with labeled pyruvate generated labeled CO2, indicative of active nuclear PDH (91). In fact, nuclear localization of TCA cycle enzymes depended upon pyruvate supplementation, whereas eliminating pyruvate excreted diminished specific methylation and acetylation histone marks (91). Taken together, these studies point out that cellular metabolism has a direct effect on the epigenome, thereby generating an epigenome-mediated mitochondria-to-nucleus cross-talk, influencing the chromatin state and cell fate decisions. However, the main challenge remains to identify and establish direct links between changes in mitochondrial dynamics and function and cell fate decisions.
Future directions: Alternative mitochondria-to-nucleus communication routes
The major and most-studied route for communication between the mitochondria and the nucleus is the one in which TCA cycle metabolites act as cofactors for epigenetic enzymes. Are there other routes? Another possible route of communication is mtDNA. Recently, it has been demonstrated that mtDNA released into the cytosol, following mitochondrial DNA stress, activates the innate immune response (92). Perhaps mtDNA release into the cytosol can also find its way into the nucleus (and interact with nuclear DNA or certain nuclear factors) or to an interaction with nuclear surface receptors that have the capabilities to bind/sense mtDNA. We also discussed the role of mitochondrial dynamics in regulating mitochondrial metabolism, thereby indirectly regulating nuclear epigenetics. However, there is another possibility that we find extremely attractive: the mitochondria go through fantastic morphological changes during fusion and fission events, and perhaps the nucleus has the means to “sense” these changes. Perhaps the nucleus possesses receptors that sense the mitochondrial morphology changes (long/elongated mitochondria that can physically “wrap” the nucleus in large areas as opposed to small/fragmented mitochondria that have limited areas of interaction with the nucleus). As mentioned above, several mitochondrial enzymes (e.g. PDH) have been shown to also localize to the nucleus and thus to generate metabolites (Ac-CoA) close to the sites in which they have a regulatory effect (histone acetylation) (91). Is it possible that single mitochondria enter the nucleus and thereby carry the entire mitochondrial enzymatic machinery into the nucleus? If this is indeed the case, then the generation and location of many mitochondrial metabolites could be controlled by the nucleus. This would avoid the need to reconstruct the mitochondrial enzymatic network from scratch in the nucleus.
The authors declare that they have no conflicts of interest with the contents of this article.
- TCA
- tricarboxylic acid
- ROS
- reactive oxygen species
- ETC
- electron transport chain
- OXPHOS
- oxidative phosphorylation
- FA
- fatty acid
- FAO
- fatty acid oxidation
- PSC
- pluripotent stem cell(s)
- ESC
- embryonic stem cell(s)
- EpiSC
- epiblast stem cell(s)
- iPSC
- induced pluripotent stem cell(s)
- HSC
- hematopoietic stem cell(s)
- LT-HSC and ST-HSC
- long- and short-term HSC, respectively
- NSC
- neuronal stem cell(s)
- TE and TM cells
- effector T and memory T cells, respectively
- mtDNA
- mitochondrial DNA
- TDH
- threonine dehydrogenase
- KBP
- Kif1-binding protein
- PDH
- pyruvate dehydrogenase
- PPAR
- peroxisome proliferator–activated receptor δ
- NPC
- neuronal progenitor cell(s)
- RC
- respiratory complex(es)
- TFAM
- mitochondrial transcription factor A
- SAM
- S-adenosylmethionine
- H3K4me3 and H3K27me3
- histone H3 Lys-4 and -27 trimethylation, respectively
- αKG
- α-ketoglutarate
- ACLY
- ATP citrate lyase
- PDC
- pyruvate dehydrogenase complex.
References
- 1. Knoblich J. A. (2008) Mechanisms of asymmetric stem cell division. Cell 132, 583–597 10.1016/j.cell.2008.02.007 [DOI] [PubMed] [Google Scholar]
- 2. Morrison S. J., and Spradling A. C. (2008) Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 132, 598–611 10.1016/j.cell.2008.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maryanovich M., Zahalka A. H., Pierce H., Pinho S., Nakahara F., Asada N., Wei Q., Wang X., Ciero P., Xu J., Leftin A., and Frenette P. S. (2018) Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 24, 782–791 10.1038/s41591-018-0030-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ahola S., Langer T., and MacVicar T. (2019) Mitochondrial proteolysis and metabolic control. Cold Spring Harb. Perspect. Biol. 11, a033936 10.1101/cshperspect.a033936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mishra P., and Chan D. C. (2016) Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 212, 379–387 10.1083/jcb.201511036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lisowski P., Kannan P., Mlody B., and Prigione A. (2018) Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 19, e45432 10.15252/embr.201745432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shyh-Chang N., Daley G. Q., and Cantley L. C. (2013) Stem cell metabolism in tissue development and aging. Development 140, 2535–2547 10.1242/dev.091777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schvartzman J. M., Thompson C. B., and Finley L. W. S. (2018) Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 217, 2247–2259 10.1083/jcb.201803061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chandel N. S., Jasper H., Ho T. T., and Passegué E. (2016) Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol. 18, 823–832 10.1038/ncb3385 [DOI] [PubMed] [Google Scholar]
- 10. Mathieu J., and Ruohola-Baker H. (2016) Metabolic RemodeLIN of pluripotency. Cell Stem Cell 19, 3–4 10.1016/j.stem.2016.06.016 [DOI] [PubMed] [Google Scholar]
- 11. Perales-Clemente E., Folmes C. D. L., and Terzic A. (2014) Metabolic regulation of redox status in stem cells. Antioxid. Redox Signal. 21, 1648–1659 10.1089/ars.2014.6000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tonelli F. M. P., Santos A. K., Gomes D. A., Da Silva S. L., Gomes K. N., Ladeira L. O., and Resende R. R., (2012) Stem cells and calcium signaling. Adv. Exp. Med. Biol. 740, 891–916 10.1007/978-94-007-2888-2_40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ito K., and Suda T. (2014) Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 15, 243–256 10.1038/nrm3772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J., Nuebel E., Daley G. Q., Koehler C. M., and Teitell M. A. (2012) Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 11, 589–595 10.1016/j.stem.2012.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Folmes C. D. L., and Terzic A. (2016) Energy metabolism in the acquisition and maintenance of stemness. Semin. Cell Dev. Biol. 52, 68–75 10.1016/j.semcdb.2016.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shyh-Chang N., and Ng H. H. (2017) The metabolic programming of stem cells. Genes Dev. 31, 336–346 10.1101/gad.293167.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Riester M., Xu Q., Moreira A., Zheng J., Michor F., and Downey R. J. (2018) The Warburg effect: persistence of stem-cell metabolism in cancers as a failure of differentiation. Ann. Oncol. 29, 264–270 10.1093/annonc/mdx645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sebastián D., and Zorzano A. (2018) Mitochondrial dynamics and metabolic homeostasis. Curr. Opin. Physiol. 3, 34–40 10.1016/j.cophys.2018.02.006 [DOI] [Google Scholar]
- 19. Chen H., and Chan D. C. (2017) Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 26, 39–48 10.1016/j.cmet.2017.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singh V. K., Saini A., Kalsan M., Kumar N., and Chandra R. (2016) Describing the stem cell potency: the various methods of functional assessment and in silico diagnostics. Front. Cell Dev. Biol. 4, 134 10.3389/fcell.2016.00134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hanna J. H., Saha K., and Jaenisch R. (2010) Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell 143, 508–525 10.1016/j.cell.2010.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hayashi K., Ohta H., Kurimoto K., Aramaki S., and Saitou M. (2011) Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146, 519–532 10.1016/j.cell.2011.06.052 [DOI] [PubMed] [Google Scholar]
- 23. Weinberger L., Ayyash M., Novershtern N., and Hanna J. H. (2016) Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 17, 155–169 10.1038/nrm.2015.28 [DOI] [PubMed] [Google Scholar]
- 24. Zhou W., Choi M., Margineantu D., Margaretha L., Hesson J., Cavanaugh C., Blau C. A., Horwitz M. S., Hockenbery D., Ware C., and Ruohola-Baker H. (2012) HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 31, 2103–2116 10.1038/emboj.2012.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takashima Y., Guo G., Loos R., Nichols J., Ficz G., Krueger F., Oxley D., Santos F., Clarke J., Mansfield W., Reik W., Bertone P., and Smith A. (2015) (2014) Erratum: Resetting transcription factor control circuitry toward ground-state pluripotency in human (Cell 158 (1254–1269)). Cell 162, 452–453 10.1016/j.cell.2015.06.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prieto J., León M., Ponsoda X., Sendra R., Bort R., Ferrer-Lorente R., Raya A., López-García C., and Torres J. (2016) Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 7, 11124 10.1038/ncomms11124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang L., Zhang T., Wang L., Cai Y., Zhong X., He X., Hu L., Tian S., Wu M., Hui L., Zhang H., and Gao P. (2017) Fatty acid synthesis is critical for stem cell pluripotency via promoting mitochondrial fission. EMBO J. 36, 1330–1347 10.15252/embj.201695417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kasahara A., Cipolat S., Chen Y., Dorn G. W. 2nd, and Scorrano L. (2013) Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and notch signaling. Science 342, 734–737 10.1126/science.1241359 [DOI] [PubMed] [Google Scholar]
- 29. Chen Y., Liu Y., and Dorn G. W. 2nd (2011) Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109, 1327–1331 10.1161/CIRCRESAHA.111.258723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wai T., García-Prieto J., Baker M. J., Merkwirth C., Benit P., Rustin P., Rupérez F. J., Barbas C., Ibañez B., and Langer T. (2015) Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350, aad0116 10.1126/science.aad0116 [DOI] [PubMed] [Google Scholar]
- 31. Bahat A., Goldman A., Zaltsman Y., Khan D. H., Halperin C., Amzallag E., Krupalnik V., Mullokandov M., Silberman A., Erez A., Schimmer A. D., Hanna J. H., and Gross A. (2018) MTCH2-mediated mitochondrial fusion drives exit from naive pluripotency in ESC. Nat. Commun. 9, 5132 10.1038/s41467-018-07519-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luchsinger L. L., de Almeida M. J., Corrigan D. J., Mumau M., and Snoeck H. W. (2016) Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 529, 528–531 10.1038/nature16500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khacho M., Clark A., Svoboda D. S., Azzi J., MacLaurin J. G., Meghaizel C., Sesaki H., Lagace D. C., Germain M., Harper M. E., Park D. S., and Slack R. S. (2016) Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell 19, 232–247 10.1016/j.stem.2016.04.015 [DOI] [PubMed] [Google Scholar]
- 34. Buck M. D., O'Sullivan D., Klein Geltink R. I., Curtis J. D., Chang C. H., Sanin D. E., Qiu J., Kretz O., Braas D., van der Windt G. J. J. W., Chen Q., Huang S. C., O'Neill C. M., Edelson B. T., Pearce E. J., et al. (2016) Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 10.1016/j.cell.2016.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liesa M., and Shirihai O. S. (2013) Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491–506 10.1016/j.cmet.2013.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Folmes C. D. L., Dzeja P. P., Nelson T. J., and Terzic A. (2012) Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 11, 596–606 10.1016/j.stem.2012.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gardner D. K., Lane M., Calderon I., and Leeton J. (1996) Environment of the preimplantation human embryo in vivo: metabolite analysis of oviduct and uterine fluids and metabolism of cumulus cells. Fertil. Steril. 65, 349–353 10.1016/S0015-0282(16)58097-2 [DOI] [PubMed] [Google Scholar]
- 38. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Folmes C. D. L., Nelson T. J., Martinez-Fernandez A., Arrell D. K., Lindor J. Z., Dzeja P. P., Ikeda Y., Perez-Terzic C., and Terzic A. (2011) Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 14, 264–271 10.1016/j.cmet.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cha Y., Han M. J., Cha H. J., Zoldan J., Burkart A., Jung J. H., Jang Y., Kim C. H., Jeong H. C., Kim B. G., Langer R., Kahn C. R., Guarente L., and Kim K. S. (2017) Metabolic control of primed human pluripotent stem cell fate and function by the miR-200c-SIRT2 axis. Nat. Cell Biol. 19, 445–456 10.1038/ncb3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ma T., Li J., Xu Y., Yu C., Xu T., Wang H., Liu K., Cao N., Nie B. M., Zhu S. Y., Xu S., Li K., Wei W. G., Wu Y., Guan K. L., and Ding S. (2015) Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat. Cell Biol. 17, 1379–1387 10.1038/ncb3256 [DOI] [PubMed] [Google Scholar]
- 42. Donato V., Bonora M., Simoneschi D., Sartini D., Kudo Y., Saraf A., Florens L., Washburn M. P., Stadtfeld M., Pinton P., and Pagano M. (2017) The TDH–GCN5L1–Fbxo15–KBP axis limits mitochondrial biogenesis in mouse embryonic stem cells. Nat. Cell Biol. 19, 341–351 10.1038/ncb3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takubo K., Nagamatsu G., Kobayashi C. I., Nakamura-Ishizu A., Kobayashi H., Ikeda E., Goda N., Rahimi Y., Johnson R. S., Soga T., Hirao A., Suematsu M., and Suda T. (2013) Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61 10.1016/j.stem.2012.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ito K., Turcotte R., Cui J., Zimmerman S. E., Pinho S., Mizoguchi T., Arai F., Runnels J. M., Alt C., Teruya-Feldstein J., Mar J. C., Singh R., Suda T., Lin C. P., Frenette P. S., and Ito K. (2016) Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 354, 1156–1160 10.1126/science.aaf5530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sun N., Yun J., Liu J., Malide D., Liu C., Rovira I. I., Holmström K. M., Fergusson M. M., Yoo Y. H., Combs C. A., and Finkel T. (2015) Measuring in vivo mitophagy. Mol. Cell 60, 685–696 10.1016/j.molcel.2015.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang J., Khvorostov I., Hong J. S., Oktay Y., Vergnes L., Nuebel E., Wahjudi P. N., Setoguchi K., Wang G., Do A., Jung H. J., McCaffery J. M., Kurland I. J., Reue K., Lee W. N. P., et al. (2011) UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 30, 4860–4873 10.1038/emboj.2011.401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maryanovich M., Zaltsman Y., Ruggiero A., Goldman A., Shachnai L., Zaidman S. L., Porat Z., Golan K., Lapidot T., and Gross A. (2015) An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat. Commun. 6, 7901 10.1038/ncomms8901 [DOI] [PubMed] [Google Scholar]
- 48. Yu W. M., Liu X., Shen J., Jovanovic O., Pohl E. E., Gerson S. L., Finkel T., Broxmeyer H. E., and Qu C. K. (2013) Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 12, 62–74 10.1016/j.stem.2012.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bricker D. K., Taylor E. B., Schell J. C., Orsak T., Boutron A., Chen Y. C., Cox J. E., Cardon C. M., Van Vranken J. G., Dephoure N., Redin C., Boudina S., Gygi S. P., Brivet M., Thummel C. S., and Rutter J. (2012) A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100 10.1126/science.1218099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Herzig S., Raemy E., Montessuit S., Veuthey J. L., Zamboni N., Westermann B., Kunji E. R. S., and Martinou J. C. (2012) Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96 10.1126/science.1218530 [DOI] [PubMed] [Google Scholar]
- 51. Schell J. C., Olson K. A., Jiang L., Hawkins A. J., Van Vranken J. G., Xie J., Egnatchik R. A., Earl E. G., DeBerardinis R. J., and Rutter J. (2014) A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol. Cell 56, 400–413 10.1016/j.molcel.2014.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schell J. C., Wisidagama D. R., Bensard C., Zhao H., Wei P., Tanner J., Flores A., Mohlman J., Sorensen L. K., Earl C. S., Olson K. A., Miao R., Waller T. C., Delker D., Kanth P., et al. (2017) Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 19, 1027–1036 10.1038/ncb3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fan J., Kamphorst J. J., Mathew R., Chung M. K., White E., Shlomi T., and Rabinowitz J. D. (2013) Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 9, 712 10.1038/msb.2013.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zheng X., Boyer L., Jin M., Mertens J., Kim Y., Ma L., Ma L., Hamm M., Gage F. H., and Hunter T. (2016) Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 5, e13374 10.7554/eLife.13374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Agostini M., Romeo F., Inoue S., Niklison-Chirou M. V., Elia A. J., Dinsdale D., Morone N., Knight R. A., Mak T. W., and Melino G. (2016) Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 23, 1502–1514 10.1038/cdd.2016.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sperber H., Mathieu J., Wang Y., Ferreccio A., Hesson J., Xu Z., Fischer K. A., Devi A., Detraux D., Gu H., Battle S. L., Showalter M., Valensisi C., Bielas J. H., Ericson N. G., Margaretha L., Robitaille A. M., Margineantu D., Fiehn O., Hockenbery D., Blau C. A., Raftery D., Margolin A. A., Hawkins R. D., Moon R. T., Ware C. B., and Ruohola-Baker H. (2015) The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 17, 1523–1535 10.1038/ncb3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carbognin E., Betto R. M., Soriano M. E., Smith A. G., and Martello G. (2016) Stat3 promotes mitochondrial transcription and oxidative respiration during maintenance and induction of naive pluripotency. EMBO J. 35, 618–634 10.15252/embj.201592629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang J., Ratanasirintrawoot S., Chandrasekaran S., Wu Z., Ficarro S. B., Yu C., Ross C. A., Cacchiarelli D., Xia Q., Seligson M., Shinoda G., Xie W., Cahan P., Wang L., Ng S. C., et al. (2016) LIN28 regulates stem cell metabolism and conversion to primed pluripotency. Cell Stem Cell 19, 66–80 10.1016/j.stem.2016.05.009 [DOI] [PubMed] [Google Scholar]
- 59. Cassidy-Stone A., Chipuk J. E., Ingerman E., Song C., Yoo C., Kuwana T., Kurth M. J., Shaw J. T., Hinshaw J. E., Green D. R., and Nunnari J. (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204 10.1016/j.devcel.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bordt E. A., Clerc P., Roelofs B. A., Saladino A. J., Tretter L., Adam-Vizi V., Cherok E., Khalil A., Yadava N., Ge S. X., Francis T. C., Kennedy N. W., Picton L. K., Kumar T., Uppuluri S., et al. (2017) The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev. Cell 40, 583–594.e6 10.1016/j.devcel.2017.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kida Y. S., Kawamura T., Wei Z., Sogo T., Jacinto S., Shigeno A., Kushige H., Yoshihara E., Liddle C., Ecker J. R., Yu R. T., Atkins A. R., Downes M., and Evans R. M. (2015) ERRs mediate a metabolic switch required for somatic cell reprogramming to pluripotency. Cell Stem Cell 16, 547–555 10.1016/j.stem.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ansó E., Weinberg S. E., Diebold L. P., Thompson B. J., Malinge S., Schumacker P. T., Liu X., Zhang Y., Shao Z., Steadman M., Marsh K. M., Xu J., Crispino J. D., and Chandel N. S. (2017) The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 19, 614–625 10.1038/ncb3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ito K., Carracedo A., Weiss D., Arai F., Ala U., Avigan D. E., Schafer Z. T., Evans R. M., Suda T., Lee C. H., and Pandolfi P. P. (2012) A PML-PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 18, 1350–1358 10.1038/nm.2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Knobloch M., Pilz G. A., Ghesquière B., Kovacs W. J., Wegleiter T., Moore D. L., Hruzova M., Zamboni N., Carmeliet P., and Jessberger S. (2017) A fatty acid oxidation-dependent metabolic shift regulates adult neural stem cell activity. Cell Rep. 20, 2144–2155 10.1016/j.celrep.2017.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bigarella C. L., Liang R., and Ghaffari S. (2014) Stem cells and the impact of ROS signaling. Development 141, 4206–4218 10.1242/dev.107086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Forman H. J., Maiorino M., and Ursini F. (2010) Signaling functions of reactive oxygen species. Biochemistry 49, 835–842 10.1021/bi9020378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schieber M., and Chandel N. S. (2014) ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 10.1016/j.cub.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Armstrong J. S., Steinauer K. K., Hornung B., Irish J. M., Lecane P., Birrell G. W., Peehl D. M., and Knox S. J. (2002) Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 9, 252–263 10.1038/sj.cdd.4400959 [DOI] [PubMed] [Google Scholar]
- 69. Yanes O., Clark J., Wong D. M., Patti G. J., Sánchez-Ruiz A., Benton H. P., Trauger S. A., Desponts C., Ding S., and Siuzdak G. (2010) Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 6, 411–417 10.1038/nchembio.364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Marsboom G., Zhang G. F., Pohl-Avila N., Zhang Y., Yuan Y., Kang H., Hao B., Brunengraber H., Malik A. B., and Rehman J. (2016) Glutamine metabolism regulates the pluripotency transcription factor OCT4. Cell Rep. 16, 323–332 10.1016/j.celrep.2016.05.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Friedman J. R., and Nunnari J. (2014) Mitochondrial form and function. Nature 505, 335–343 10.1038/nature12985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lewis S. C., Uchiyama L. F., and Nunnari J. (2016) ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353, aaf5549 10.1126/science.aaf5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Facucho-Oliveira J. M., Alderson J., Spikings E. C., Egginton S., and St. John J. C. (2007) Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J. Cell Sci. 120, 4025–4034 10.1242/jcs.016972 [DOI] [PubMed] [Google Scholar]
- 74. Larsson N. G., Wang J., Wilhelmsson H., Oldfors A., Rustin P., Lewandoski M., Barsh G. S., and Clayton D. A. (1998) Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 18, 231–236 10.1038/ng0398-231 [DOI] [PubMed] [Google Scholar]
- 75. Nakada K., Sato A., and Hayashi J. (2009) Mitochondrial functional complementation in mitochondrial DNA-based diseases. Int. J. Biochem. Cell Biol. 41, 1907–1913 10.1016/j.biocel.2009.05.010 [DOI] [PubMed] [Google Scholar]
- 76. Chen H., Vermulst M., Wang Y. E., Chomyn A., Prolla T. A., McCaffery J. M., and Chan D. C. (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289 10.1016/j.cell.2010.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Katajisto P., Döhla J., Chaffer C. L., Pentinmikko N., Marjanovic N., Iqbal S., Zoncu R., Chen W., Weinberg R. A., and Sabatini D. M. (2015) Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348, 340–343 10.1126/science.1260384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ma H., Folmes C. D. L., Wu J., Morey R., Mora-Castilla S., Ocampo A., Ma L., Poulton J., Wang X., Ahmed R., Kang E., Lee Y., Hayama T., Li Y., Van Dyken C., et al. (2015) Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 524, 234–238 10.1038/nature14546 [DOI] [PubMed] [Google Scholar]
- 79. Kang E., Wang X., Tippner-Hedges R., Ma H., Folmes C. D. L., Gutierrez N. M., Lee Y., Van Dyken C., Ahmed R., Li Y., Koski A., Hayama T., Luo S., Harding C. O., Amato P., et al. (2016) Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human iPSCs. Cell Stem Cell 18, 625–636 10.1016/j.stem.2016.02.005 [DOI] [PubMed] [Google Scholar]
- 80. Wang J., Alexander P., Wu L., Hammer R., Cleaver O., and McKnight S. L. (2009) Dependence of mouse embryonic stem cells on threonine catabolism. Science 325, 435–439 10.1126/science.1173288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shyh-Chang N., Locasale J. W., Lyssiotis C. A., Zheng Y., Teo R. Y., Ratanasirintrawoot S., Zhang J., Onder T., Unternaehrer J. J., Zhu H., Asara J. M., Daley G. Q., and Cantley L. C. (2013) Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339, 222–226 10.1126/science.1226603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shiraki N., Shiraki Y., Tsuyama T., Obata F., Miura M., Nagae G., Aburatani H., Kume K., Endo F., and Kume S. (2014) Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 19, 780–794 10.1016/j.cmet.2014.03.017 [DOI] [PubMed] [Google Scholar]
- 83. Carey B. W., Finley L. W. S., Cross J. R., Allis C. D., and Thompson C. B. (2015) Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416 10.1038/nature13981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. TeSlaa T., Chaikovsky A. C., Lipchina I., Escobar S. L., Hochedlinger K., Huang J., Graeber T. G., Braas D., and Teitell M. A. (2016) α-Ketoglutarate accelerates the initial differentiation of primed human pluripotent stem cells. Cell Metab. 24, 485–493 10.1016/j.cmet.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sciacovelli M., Gonçalves E., Johnson T. I., Zecchini V. R., da Costa A. S. H., Gaude E., Drubbel A. V., Theobald S. J., Abbo S. R., Tran M. G. B., Rajeeve V., Cardaci S., Foster S., Yun H., Cutillas P., et al. (2016) Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547 10.1038/nature19353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Moussaieff A., Rouleau M., Kitsberg D., Cohen M., Levy G., Barasch D., Nemirovski A., Shen-Orr S., Laevsky I., Amit M., Bomze D., Elena-Herrmann B., Scherf T., Nissim-Rafinia M., Kempa S., et al. (2015) Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 21, 392–402 10.1016/j.cmet.2015.02.002 [DOI] [PubMed] [Google Scholar]
- 87. Sivanand S., Viney I., and Wellen K. E. (2018) Spatiotemporal control of acetyl-CoA metabolism in chromatin regulation. Trends Biochem. Sci. 43, 61–74 10.1016/j.tibs.2017.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wellen K. E., Hatzivassiliou G., Sachdeva U. M., Bui T. V., Cross J. R., and Thompson C. B. (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 10.1126/science.1164097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tohyama S., Fujita J., Hishiki T., Matsuura T., Hattori F., Ohno R., Kanazawa H., Seki T., Nakajima K., Kishino Y., Okada M., Hirano A., Kuroda T., Yasuda S., Sato Y., et al. (2016) Glutamine oxidation is indispensable for survival of human pluripotent stem cells. Cell Metab. 23, 663–674 10.1016/j.cmet.2016.03.001 [DOI] [PubMed] [Google Scholar]
- 90. Sutendra G., Kinnaird A., Dromparis P., Paulin R., Stenson T. H., Haromy A., Hashimoto K., Zhang N., Flaim E., and Michelakis E. D. (2014) A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 158, 84–97 10.1016/j.cell.2014.04.046 [DOI] [PubMed] [Google Scholar]
- 91. Nagaraj R., Sharpley M. S., Chi F., Braas D., Zhou Y., Kim R., Clark A. T., and Banerjee U. (2017) Nuclear localization of mitochondrial TCA cycle enzymes as a critical step in mammalian zygotic genome activation. Cell 168, 210–223.e11 10.1016/j.cell.2016.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. West A. P., Khoury-Hanold W., Staron M., Tal M. C., Pineda C. M., Lang S. M., Bestwick M., Duguay B. A., Raimundo N., MacDuff D. A., Kaech S. M., Smiley J. R., Means R. E., Iwasaki A., and Shadel G. S. (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 10.1038/nature14156 [DOI] [PMC free article] [PubMed] [Google Scholar]