

Abstract
CD8+ T cells recognize and eliminate tumors in an antigen-specific manner. Despite progress in characterizing the antitumor T cell repertoire and function, identifying their target antigens remains a challenge. Here, we describe the use of chimeric receptors called Signaling and Antigen-presenting Bifunctional Receptors (SABRs) in a novel cell-based platform for T Cell Receptor (TCR) antigen discovery. SABRs present an extracellular peptide-MHC complex and induce intracellular signaling via a TCR-like signal upon binding with a cognate TCR. We devised a strategy for antigen discovery using SABR libraries to screen thousands of antigenic epitopes. We validated this platform by identifying the targets recognized by public TCRs of known specificities. Moreover, we extended this approach for personalized neoantigen discovery. The antigen discovery platform reported here will provide a scalable and versatile way to develop novel targets for immunotherapy.
Introduction:
The ability of CD8+ T cells to recognize and kill cancer cells is well-established1 and exploited by immunotherapies such as vaccines2, checkpoint blockade3,4, and adoptive T cell therapies5. A CD8+ T cell encodes a unique surface T Cell Receptor (TCR) that recognizes 8-12 residue long peptide epitopes presented on class I Major Histocompatibility Complex (MHC) molecules, also known as Human Leukocyte Antigens (HLA) in humans6. When a TCR complex binds cognate peptide-MHC (pMHC), the associated CD247 (CD3ζ) chains dimerize to initiate downstream signaling. Multiple signaling cascades are activated, leading to rapid gene expression driven by the transcription factors NF-κB, AP-1, and NFAT7. In CD8+ T cells, TCR signaling induces expression of early activation markers (CD69 and LAMP1), release of cytotoxic granules, and secretion of cytokines, ultimately killing the target cell7. The interaction of cognate TCR and pMHC complexes generates a high degree of specificity towards a target antigen. T cells can recognize epitopes presented by tumor cells and infiltrate the tumor microenvironment. Antitumor T cells respond to two kinds of tumor-derived epitopes: 1) Public or private epitopes originating from non-mutated, tissue specific antigens or cancer-testis antigens; and 2) Private neoantigens originating from non-synonymous mutations8. Both endogenous antigens and neoantigens can be used to provide targets of immunotherapies.
Considerable progress has been made towards understanding the T cell repertoire9 and function10 using high throughput genomics, transcriptomics, and proteomics. However, one of the bottlenecks in the field of tumor immunology is the identification of the antigen recognized by a particular antitumor CD8+ T cell. Several techniques have been developed to identify cognate antigens for T cells. The most common approach uses pMHC multimers to identify antigen-specific T cells by flow cytometry11,12. Antigen discovery using pMHC multimers requires ab initio knowledge of the antigenic landscape, is not scalable beyond 103 antigens, but can identify multiple antigenic specificities simultaneously. This approach has been used to discover public tumor antigens13 as well as private neoantigens12,14. A recently reported approach uses yeast display of epitope libraries for antigen discovery15. However, this approach is technically challenging because of the requirement of soluble TCR, does not represent the physiological TCR-pMHC interaction, but is antigen-agnostic and scalable to 106-108 epitopes16. These limitations underscore the need for new techniques for T cell antigen discovery.
Here, we sought to develop novel antigen discovery techniques to address the unmet need. In two parallel studies (Li et al, accompanying manuscript), we present cell-based platforms for T cell antigen discovery. We describe chimeric receptors called Signaling and Antigen-presenting Bifunctional Receptors (SABRs) that allow identification of a successful TCR-pMHC interaction. We report a strategy for TCR antigen discovery using SABR libraries and demonstrate its use for known public TCRs and a private neoantigens-specific TCR. Through these studies, we describe a flexible and scalable method for T cell antigen discovery.
Results:
Signaling and Antigen-presenting Bifunctional Receptors (SABRs)
T cell activation upon recognizing a target antigen induces detectable gene expression. However, as MHC molecules lack signaling domains, detection of recognized Antigen Presenting Cells (APCs) is challenging. To address this, we constructed chimeric receptors called Signaling and Antigen-presenting Bifunctional Receptors (SABRs). The extracellular domain of a SABR is a covalently linked peptide-β2microglobulin-MHC trimer17, fused to an intracellular CD3ζ signaling domain with a CD28 co-stimulatory domain. We constructed two variations of SABRs, SABR-F and SABR-E, which contained the entire MHC molecule or only the extracellular part of the MHC molecule respectively (Supplementary Fig 1a). We hypothesized that upon interaction with a TCR, SABRs presenting its cognate antigen will induce an intracellular signal (Fig 1a). To detect the signal induced by SABRs, we used NFAT-GFP-Jurkat cells, which express GFP upon receiving a signal via CD3ζ. We transduced NFAT-GFP-Jurkat cells with SABRs presenting the EAAGIGILTV epitope (from the MART1/MLANA protein) on HLA-A*0201 (hereafter known as A2-MART1-SABR) or the KRWIILGLNK epitope (KK10, from the HIV-1 gag protein) from HIV-1 on HLA-B*2705 (hereafter known as B27-KK10-SABR). We co-incubated transduced NFAT-GFP-Jurkat cells with Jurkat cells expressing TCRs and measured GFP expression by flow cytometry after 8 hours. Specifically, we used F5 (recognizes A2-MART118), EC27 (recognizes B27-KK1019), SL9 (recognizes A2-SLYNTVATL20) TCRs or untransduced Jurkat cells. GFP expression was detected only in co-culture assays with the cognate TCR-SABR-F pairs (Fig 1b and Supplementary Fig 1b). The SABR-F construct showed higher signal than SABR-E, and therefore, was used for further experiments (Supplementary Fig 1c). SABR signaling was titratable and sensitive enough to detect at least as low as 10 F5+ Jurkat cells mixed with 10,000 untransduced Jurkat cells (Fig 1c). SABR signaling was rapid, as GFP signal was detectable within 3 hours of co-incubation, and reached saturation within 6-8 hours (Fig 1d and Supplementary Fig 2). Both the sensitivity and kinetics were conserved for four different TCRs, EC27, FC4.3, FC5.5, and CP7.9, all of which recognize B27-KK1019. To test if SABR signaling can be induced upon recognition of low-affinity TCR-pMHC interactions, we used M1, an A2-MART1-specific TCR. The affinity of M1 TCR was too low to be measured by Surface Plasmon Resonance (SPR) as described previously21. F5 and M1 TCRs were indistinguishable in their ability to induce GFP signaling in NFAT-GFP-Jurkat cells expressing A2-MART1-SABR (Supplementary Fig 3). Taken together, these results show that SABRs can induce signaling upon successful and specific TCR-pMHC interaction, allowing identification of recognized APCs.
Fig 1. Signaling and antigen-presenting bifunctional receptors.
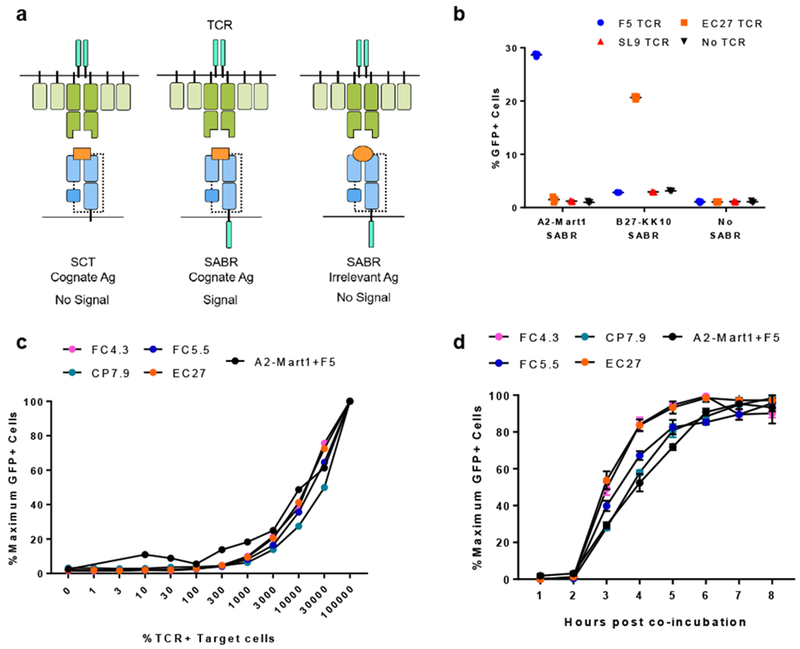
a. Schematics demonstrating SABRs and TCR-pMHC specific signaling. SCT – Single Chain Trimer, Ag - Antigen. Dotted lines indicate Gly-Ser linkers. b. GFP expression by SABR transduced NFAT-GFP-Jurkat cells upon co-culture with TCR-transduced Jurkat cells. Line and error bars indicate mean±sd, n=3 biologically independent cell culture replicates. c. Titration of SABR signal by measuring frequency of GFP+ cells in co-culture assays at 8 hours after co-culture. For FC4.3, FC5.5, CP7.9, and EC27 TCRs, NFAT-GFP-Jurkat cells transduced with B27-KK10-SABR were used. Dots indicate values from n=1 experiment. d. Timecourse of GFP expression by SABR transduced NFAT-GFP-Jurkat cells co-cultured with TCR-transduced Jurkat cells. For FC4.3, FC5.5, CP7.9, and EC27 TCRs, NFAT-GFP-Jurkat cells transduced with B27-KK10-SABR were used. Line and error bars indicate mean±sd, n=3 biologically independent cell culture replicates, n=4 for A2-Mart1.
SABRs allow different modes of antigen presentation
The endogenous MHC complexes present epitopes from newly translated proteins or endocytosed proteins via cross-presentation. To test if SABRs can also utilize these pathways for antigen presentation, we constructed an ‘empty’ version of SABRs that linked β2-microglobulin with HLA-A2 or B27, but did not genetically encode for an epitope (Supplementary Fig 4a). We incubated NFAT-GFP-Jurkat cells expressing the empty SABRs with soluble MART1 or KK10 peptides, and co-cultured them with Jurkat cells expressing F5 or EC27 TCRs. Both A2 and B27 empty SABRs induced a signal only in presence of the soluble peptide corresponding to the TCRs. Moreover, the signal induced by correct peptide-TCR combinations was comparable to the signal induced by the corresponding SABRs presenting covalently linked epitopes (Fig 2a). Next, we constructed pentameric tandem minigenes22 (TMGs) to express the KK10 epitope along with four irrelevant CMV-derived epitopes (Supplementary Fig 4a). We co-transduced NFAT-GFP-Jurkat cells with the empty B27-SABR and the KK10 TMG. Following co-incubation with EC27- or F5-expressing Jurkat cells, the empty SABRs were able to present the endogenously expressed epitopes and induce specific signaling (Fig 2b). However, the overall signal was lower than the corresponding empty SABRs pulsed with soluble peptide. These results show that SABRs can present non-covalently linked epitopes generated through endogenous antigen processing and presentation pathways.
Fig 2. Different modes of antigen presentation by SABRs.
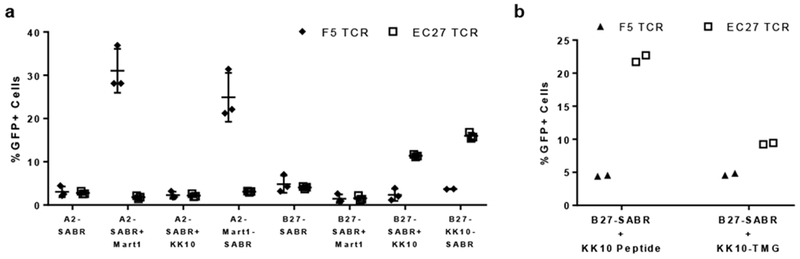
a. GFP expression by NFAT-GFP-Jurkats transduced with empty SABRs pulsed with soluble MART1 or KK10 peptides and co-cultured with Jurkat cells transduced with F5 or EC27 TCRs. Line and error bars indicate mean±sd, n=3 biologically independent cell culture replicates. b. GFP expression by NFAT-GFP-Jurkats co-transduced with empty B27-SABRs KK10-TMG or transduced with empty B27-SABR and pulsed with KK10 peptide, and co-cultured with Jurkat cells transduced with F5 or EC27 TCRs. Dots indicate individual values from n=2 biologically independent cell culture replicates.
SABRs initiate a bona fide TCR signal
SABRs use a CD3ζ-CD28 domain for intracellular signaling, similar to chimeric antigen receptors (CARs) and TCRs. Therefore, we asked whether the intracellular signaling ability of SABRs is comparable to TCRs. We first tested if SABRs induce early activation markers in NFAT-GFP-Jurkat cells. NFAT-GFP-Jurkat cells transduced with the B27-KK10-SABR expressed CD69 specifically upon co-culture with Jurkat cells transduced with CP4.3 TCR, implying that SABR signaling activates endogenous gene expression (Fig 3a). If SABRs induce a bona fide TCR signal, they should confer cytotoxic capabilities to primary T cells. We transduced activated primary T cells with A2-MART1-SABR and incubated them with CFSE-labeled target cells expressing F5 TCR. Transduced primary T cells lysed Jurkat cells or primary T cells expressing the F5 TCR specifically (Fig 3b and c). Next, we compared the antigen sensitivity of SABRs and TCRs. We transduced NFAT-GFP-Jurkat cells with either empty A2-SABR or F5 TCR, and used them as effectors. As targets, we used Jurkat cells transduced with A2-SABR or F5 TCR (Fig 3d). We co-cultured effectors and targets in presence of a range of concentrations of the MART1 peptide, and measured GFP expression. Antigen sensitivity was determined as the concentration of the peptide required for half-maximal signaling. The antigen sensitivity of SABRs was 30-fold lower as compared to TCRs (Fig 3e). We also interrogated the correlation between functional avidity of TCR-pMHC interactions and signaling induced by SABRs. We first measured functional avidity of EC27 TCR towards six variants of the KK10 peptide as described previously19. We measured the ability of the EC27 TCR to induce GFP expression upon co-culture with empty B27 SABRs pulsed with the variants of the KK10 peptide. We observed a correlation between functional avidity of TCR-pMHC interactions with SABR signaling. Importantly, approximately 180-fold reduction of functional avidity observed between R2T and R2Q variants was still able to induce detectable SABR signaling (Supplementary Fig 4b). Taken together, these results show that SABRs signal similar to TCRs, albeit with lower antigen sensitivity.
Fig 3. SABRs induce a bona fide TCR signal.
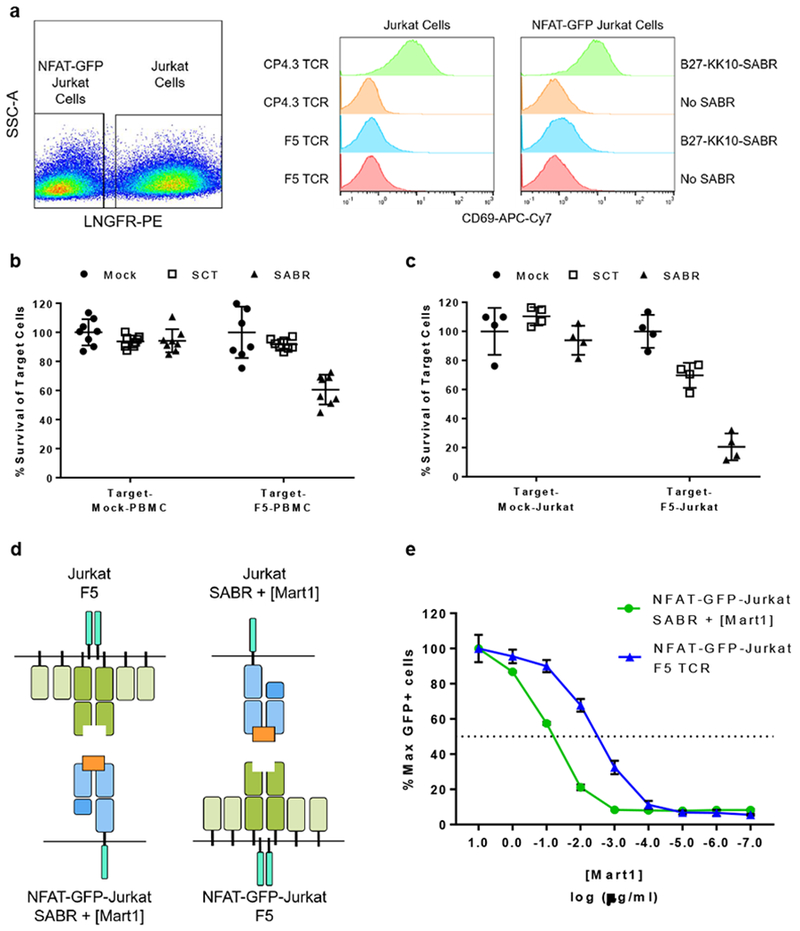
a. Induction of CD69 expression in co-culture assays with indicated TCR-SABR combinations is shown. The panel on the left is a representative plot showing discrimination of NFAT-GFP-Jurkat cells and Jurkat cells based on LNGFR expression. The panel on the right shows histogram for CD69 expression in the indicated populations. The shown data are representative of n=2 biologically independent cell culture replicates. b. Cytotoxicity induced by SABR-expressing primary T cells against Jurkat cells. Line and error bars indicate mean±sd for n=8 biologically independent cell culture replicates. c. Cytotoxicity induced by SABR-expressing primary T cells against autologous target cells. Line and error bars indicate mean±sd for n=4 biologically independent cell culture replicates. d. Schematic of the assay to measure antigen sensitivity of A2-SABR (left) and of F5-TCR (right). e. Antigen sensitivity of SABR and TCR signaling indicating GFP signal as a function of MART1 peptide. Dots and error bars indicate mean±sd for n=3 biologically independent cell culture replicates. The dotted horizontal line indicates half-maximal signal.
Proof-of-concept of using SABR libraries for antigen discovery
Next, we asked if SABR libraries presenting a large number of epitopes can be used to screen successful TCR-pMHC interactions. We designed a strategy to construct and use SABR libraries for T cell antigen discovery of ‘orphan’ TCRs with unknown antigens. First, a list of target epitopes was generated to encode for 12,055 peptides (A2-SABR-library) consisting of all known HLA-A*0201-restricted epitopes from the Immune Epitope Database23 (Supplementary Table 3). The protein sequences of the target epitopes were backtranslated to generate codon-optimized oligonucleotide sequences along with 15bp overhangs that overlap with the SABR vector. The entire list of oligonucleotides for the library was synthesized using pooled synthesis. The pooled library was then amplified and cloned using ligation-free cloning into the SABR vector plasmid. (Supplementary Fig 5 and Supplementary Fig 6a). The SABR libraries were packaged into lentiviral vectors and used to transduce NFAT-GFP-Jurkat cells. We first interrogated if the A2-SABR-library allows identification of the cognate antigen for F5 and SL9 TCRs. We transduced NFAT-GFP-Jurkat cells with the A2-SABR-library, and incubated them with Jurkat cells expressing the TCRs. After 10 hours of co-culture, we sorted GFP+CD69+ cells by FACS (Supplementary Fig 6b and Fig 4a) and extracted genomic DNA from them. The epitope portion of the SABRs was amplified and sequenced (Supplementary Fig 6c). The sequencing reads were aligned with the SABR vector backbone using Burrows-Wheeler alignment24. Aligned reads were translated to their protein sequences, and the number of reads corresponding to each epitope was counted and reported in a list. A minimum of three replicates of the co-incubation assay were performed. For each replicate, a numerical rank was given to each epitope based on descending order of the number of reads. The rank from three replicates for each assays was averaged and reported as ‘Average Rank’ (Supplementary Fig 6c). First, we plotted the average ranks of each of the epitopes from the SL9 sort against those from F5 sort (Fig 4b). The top six epitopes in the F5 sort were analogs of EAAGIGILTV, indicating successful identification of its antigen (Fig 4c). Six out of the top ten epitopes from the SL9 sort were analogs of SLYNTAVATL (Fig 4d). The average fold-enrichment of the top hits from the F5- and SL9-sorts over the Mock-sort was 296 and 70 respectively. The noise observed in the SL9 sort is possibly due to the higher number of analogs of the SLYNTVATL peptide. We compared the ranks of all the analogs of EAAGIGILTV and SLYNTVATL in the sorts. Six out of twenty-two EAAGIGILTV analogs were identified in the F5 sort (Supplementary Fig 7a), whereas nine out of sixty SLYNTVATL analogs were identified in the SL9 sort (Supplementary Fig 7b). The lack of identification of all the analogs is presumably due to reduced cross-reactivity of the F5 or SL9 TCRs towards them. Indeed, analogs SLYNTIATL (V6I) and SLFNTVATL (Y3F) are documented escape mutations in the SLYNTVATL epitope25,26. We validated the top six hits from the F5 sort by in vitro cytotoxicity assays. We observed that all six analogs of the Mart1 peptide were specifically recognized by the F5 TCR, leading to induction of cytotoxicity (Fig 4e). Nevertheless, these experiments showed that a SABR library approach could identify the cognate antigen of a TCR by screening thousands of epitopes.
Fig 4. Proof-of-concept of using SABR libraries for TCR antigen discovery.
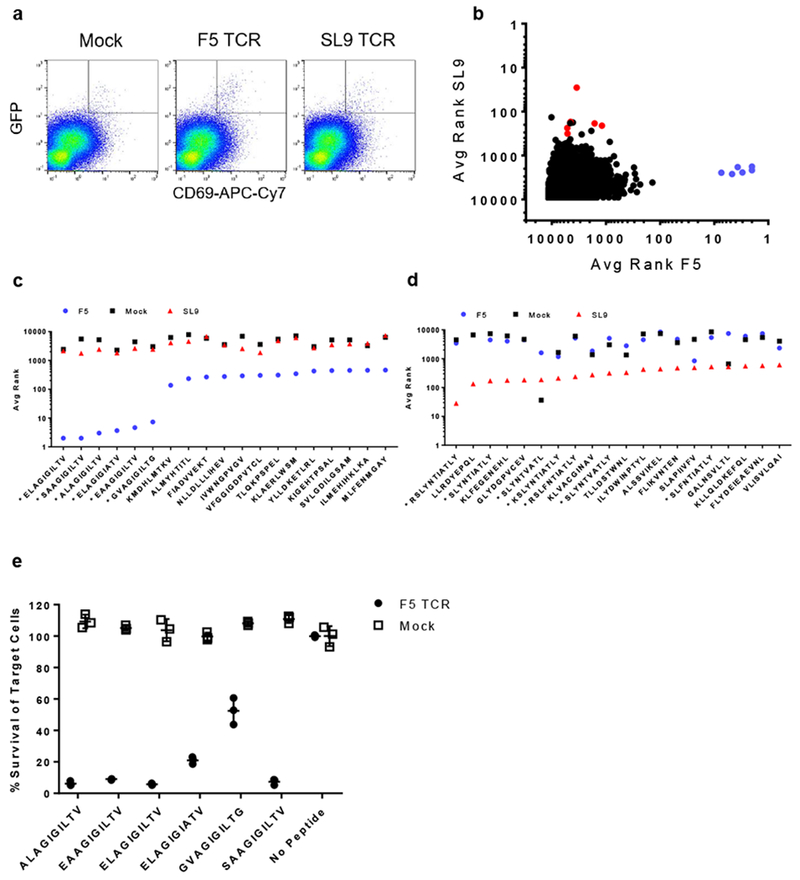
a. Sorting A2-SABR library cells based on GFP and CD69 expression in co-culture assays. Representative flow cytometry plots from one out of three biologically independent cell culture replicates are shown. The rectangle in the top right corner of each flow plot shows the gate used for the sort. Frequency of cells in the sort gate is indicated as percentage. b. Average ranks from F5 and SL9 sorts. Each dot represents the average rank for a unique epitope. Purple dots indicate EAAGIGILTV analogs and red dots indicate SLYNTVATL analogs. c. Average ranks for the top 20 hits from the F5 sort. Epitopes with asterisks indicate EAAGIGILTV analogs. d. Average ranks for the top 20 hits from the SL9 sort. Epitopes with asterisks indicate SLYNTVATL analogs. e. Validation of the top hits in the F5 sort by cytotoxicity assays performed on K562 cells expressing HLA-A2.1 and pulsed with the indicated peptides. Line and error bars indicate means±s.d. for n=3 biologically independent cell culture replicates.
Personalized neoantigen discovery using SABRs
To further demonstrate the versatility of SABR libraries, we used a personalized approach for neoantigen discovery. A recent study identified a neoantigen-specific, tumor-reactive TCR from a melanoma patient using DNA-barcoded tetramers. The identified TCR (neoTCR) recognizes a non-synonymous mutation in the USP7 protein (S.Peng, J.M.Zaretsky, A.H.C.Ng, W.Chour, M.T.Bethune, A.Hsu, E.Holman, X.Ding, K.Guo, J.Kim, A.M.Xu, J.E.Heath, W.Noh, J.Zhou, Y.Su, Y.Lu, J.Mclaughlin, D.Cheng, O.N.Witte, D.Baltimore, A.Ribas and J.R.Heath, unpublished data). We generated a SABR library presenting 3,251 predicted HLA-A*0201-restricted epitopes (NeoAg-SABR library) corresponding to 108 non-synonymous mutations found in the tumor (Supplementary Table 4). We used the neoTCR as a surrogate for a tumor-reactive orphan TCR and used the NeoAg-SABR library to identify its antigen. We co-cultured Jurkat cells transduced with neoTCR and NFAT-GFP-Jurkat cells expressing the NeoAg-SABR library, sorted GFP+CD69+ cells, then sequenced and ranked epitopes from the sorted cells (Fig 5a). For each epitope, we plotted the average ranks from neoTCR sort against mock-sort (Fig 5b). The top seven hits in the neoTCR sort were epitopes derived from USP7, demonstrating successful identification of the neoantigen using our approach (Fig 5c). The non-synonymous D798Y mutation in USP7 was predicted to generate thirty overlapping neoepitopes, out of which we identified seven as cognate epitopes of neoTCR (Supplementary Fig 8). To validate the seven detected epitopes, we constructed individual SABRs to present them. NFAT-GFP-Jurkat cells transduced with these SABRs induced GFP expression upon co-culture with Jurkat cells expressing neoTCR (Fig 5d). Unexpectedly, the SABR presenting the unmutated DLYHRVDVIF epitope also induced signaling upon recognition by neoTCR. Primary T cells transduced with neoTCR were able to specifically kill target cells pulsed with the peptides corresponding to seven detected neoepitopes, but not cells pulsed with the unmutated peptide (Fig 5e). We posit that by covalently linking the unmutated epitope, we may enforce its binding to MHC, unlike peptide pulsing. While this may lead to a false identification of the unmutated peptide as a ‘hit’, we expect subsequent validation by orthogonal assays to erase this concern. These results demonstrate validation as well as successful personalization of the antigen discovery approach presented here.
Fig 5. Personalized neoantigen discovery using SABR libraries.
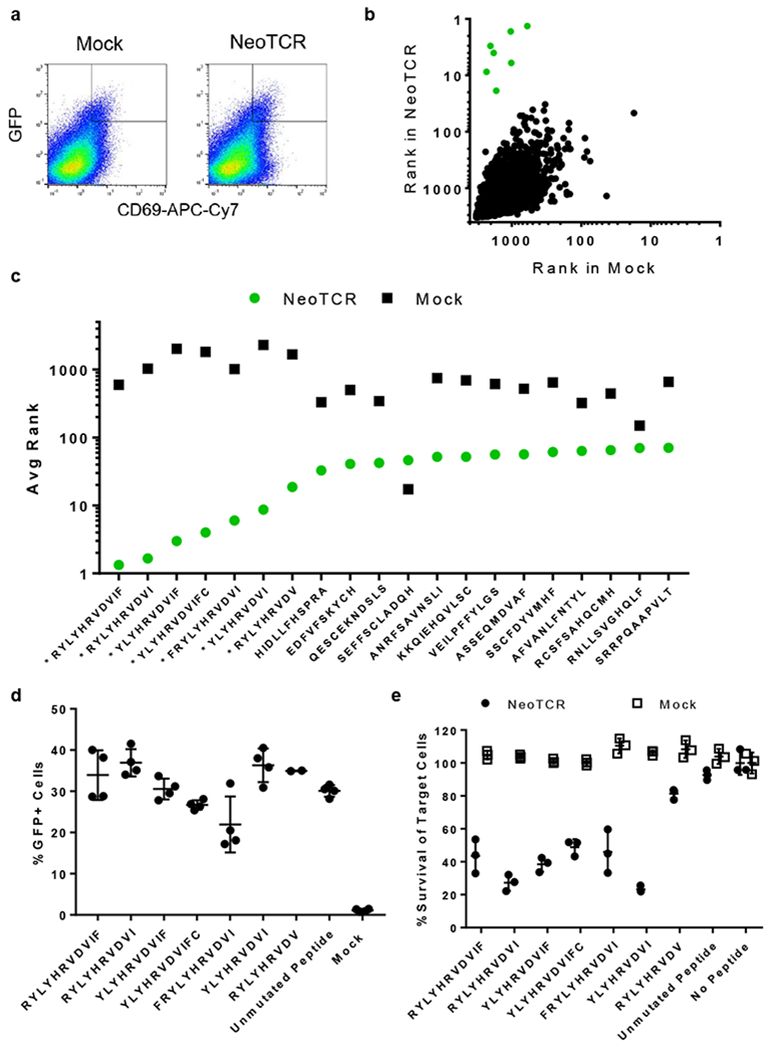
a. Sorting NeoAg-SABR library cells based on GFP and CD69 expression in co-culture assays. Representative flow cytometry plots from one out of three biologically independent cell culture replicates are shown. The rectangle in the top right corner of each flow plot shows the gate used for the sort. Frequency of cells in the sort gate is indicated as percentage. b. Average ranks from neoTCR and mock sorts. Each dot represents the average rank for a unique epitope. Green dots indicate USP7-derived epitopes. c. Average ranks for the top 20 hits from the neoTCR sort. Epitopes with asterisks indicate USP7-derived epitopes. d. Validation of top hits identified in the NeoAg-SABR screen by measuring GFP expression in co-culture assays. Line and error bars represent mean±sd, n=4 biologically independent cell culture replicates, n=2 for RYLYHRVDV. e. Validation of the top hits in the Neo TCR sort by cytotoxicity assays performed on K562 cells expressing HLA-A2.1 and pulsed with the indicated peptides. Line and error bars indicate means±s.d. for n=3 biologically independent cell culture replicates.
Discussion:
In this study, we report a novel, cell-based technique for TCR antigen discovery. We invented chimeric receptors called SABRs that combine antigen presentation and signaling to allow identification of APCs recognized by a given TCR. Using SABR libraries presenting thousands of epitopes, we showed unbiased identification of the targets of known melanoma- and HIV-specific TCRs. Moreover, we adapted this strategy for personalized neoantigen discovery. We envision employing the SABR library approach for antigen discovery of antitumor CD8+ T cells (Supplementary Fig 6). The flexibility of this approach allows customizations required for antigen discovery for public or private TCRs. A SABR library based on shared tumor gene expression among patients can identify the antigen of a shared, public TCR, whereas, a SABR library based on a single patient’s tumor mutanome can interrogate the specificity of a private TCR. While we have shown data for HLA-A*0201 and HLA-B*2705, SABRs can presumably be constructed for any class I HLA. Further testing and optimization may be required to expand the use of SABRs to all HLA-A, B, and C alleles from a patient. Furthermore, this approach can be used for antigen discovery for pathogen-reactive or self-reactive TCRs. Finally, we posit that SABRs based on class II MHC alleles will be able to signal similarly, and therefore be used for antigen discovery of TCRs from CD4+ T cells. This will greatly aid antigen discovery of autoimmune CD4+ T cells and regulatory T cells27,28. Beyond antigen discovery, SABRs can also be purposed for different applications. SABR libraries incorporating variants of a single epitope may be used to interrogate TCR crossreactivity and to identify altered peptide ligands or heteroclitic peptides29. The signaling ability of SABRs may also be used to impart functional attributes to primary T cells for therapeutic use. For instance, arming primary T cells with SABRs may allow them to eliminate autoreactive T cells via fratricide, or to suppress autoreactive T cells by inducing a tolerogenic signal30.
Our approach presents several advantages over the current TCR antigen discovery techniques. The scale offered by SABRs, currently up to 106 epitopes, is considerably higher than a multimer or a functional screen approach31,32. SABR libraries do not rely on synthesis of peptides or MHC molecules, and are therefore far easier to construct, and unlike yeast display systems, do not require production of soluble TCRs15, which is technically challenging and non-robust. Moreover, antigen discovery via yeast display requires several rounds of selection, outgrowth, and sequencing, whereas SABR libraries allow antigen discovery in a single, short-duration assay. Finally, SABR libraries do not require any specialized reagents, and can be employed by any standard immunology laboratory with access to facilities for molecular cloning, cell culture, FACS, and high-throughput sequencing.
There are also some technical limitations of using SABR libraries. Unlike yeast display systems that can present up to 106-108 epitopes15, SABRs are limited to 106 epitopes without requiring specialized cell culture facilities. Due to this limitation, the optimal use of SABRs will still require ab initio knowledge of antigenic epitopes. In their current form, SABRs allow antigen discovery for a single TCR in one assay, unlike multimers. In the future, this throughput may be increased by performing the screen on primary T cell samples directly, by multiplexing sequencing samples, or by multiplexing SABR libraries. However, if primary T cells were to be used for screening, cytotoxicity induced by the primary T cells towards SABR-expressing cells needs to be mitigated for optimization of this approach. We are currently developing this technique for multiplexed neoantigen discovery at a higher scale. At the current scale, the SABR library screens described here did not yield false positive hits for F5 and NeoAg TCRs, but did yield false positive hits identified in the SL9 sort. This indicates that further optimization may be required to reduce false discovery rate and to improve signal:noise ratio in the case of atypical situations such as the SL9 TCR. Using more sensitive and specific techniques, such as single cell sorting and sequencing, the signal:noise ratio is expected to improve.
Collectively, we demonstrate that SABR libraries are versatile and powerful tools for antigen discovery. The simplicity and scalability of SABRs will greatly aid the development of novel antigen-guided immunotherapies.
Online Methods:
Detailed step-by-step protocol
A detailed step-by-step protocol for SABR library generation and screening can be found in Supplementary Protocol. The same protocol can be found at Protocol Exchange (DOI: 10.1038/protex.2018.126).
Reagents and oligonucleotide primers
The specific reagents used in the methods are detailed in Supplementary Table 1. The oligonucleotide primers used for cloning and sequencing are listed in Supplementary Table 2. The lists of epitopes in the A2-SABR and the NeoAg-SABR libraries are in Supplementary Tables 3 and 4.
Cell lines and peptides
Jurkat cells and K562 cells (ATCC) were cultured in R10 (RPMI1640 (Corning) supplemented with 10% fetal bovine serum (Corning) and Penicillin/Streptomycin (Corning)). NFAT-GFP-Jurkat cells were a kind gift from Arthur Weiss and Yvonne Chen, and were cultured in R10 supplemented with 2 mg/ml G-418 (Corning). GXR-B27+ cells were a gift from Bruce D. Walker and were cultured in R10. Primary T cells were obtained from the CFAR virology core at University of California, Los Angeles, activated in R10 supplemented with Immunocult CD3/28 (StemCell Technologies) and 40 U/ml IL-2 (Miltenyi Biotec). HEK-293T cells (ATCC) were cultured in D10 (DMEM (Corning) supplemented with 10% fetal bovine serum (Corning) and Penicillin/Streptomycin (Corning)). All indicated peptides were synthesized by Pierce Thermo Fisher.
Construction of SABRs
Single molecules encoding for β2-microglobulin and HLA were synthesized as gBlocks (IDT) and amplified using primers SS-Fwd and CD28-Overlap-HLA-Rev. CD3ζ/CD28 signaling domains were cloned from the J3 CAR (A gift from Pin Wang) using primers CD28Intracell-Fwd and XhoI-CD3z-Rev. The two parts of SABRs were assembled via PCR or via InFusion HD cloning kit (Takara). A synthetic 2kb fragment of non-specific stuffer DNA (IDT) flanked by BsmBI sites was cloned in place of the epitope. To clone a given epitope into a SABR vector, the vector was first linearized by BsmBI digestion (NEB) and gel purified using Nucleospin Gel and PCR kit (Takara). A single stranded oligonucleotide containing overlaps with the vector and the epitope was synthesized (IDT). The oligonucleotides were amplified using KOD polymerase (Milipore) and Oligo-Insert-Fwd and Oligo-Insert-Rev. Amplified oligonucleotide was cloned into the linearized SABR vector using InFusion HD cloning kit (Takara). All cloning reactions were transformed into Stellar competent cells (Takara), grown on LB+Agar plates containing 100 μg/ml Carbenicillin (Life Technologies), and individual colonies were inoculated in liquid culture. Plasmid minipreps were performed using Zyppy Miniprep kit (Zymo). Plasmids were verified by Sanger sequencing (Laragen).
Cloning TCRs
Sequences for the F518, SL920, and neoTCR (S.Peng, J.M.Zaretsky, A.H.C.Ng, W.Chour, M.T.Bethune, A.Hsu, E.Holman, X.Ding, K.Guo, J.Kim, A.M.Xu, J.E.Heath, W.Noh, J.Zhou, Y.Su, Y.Lu, J.Mclaughlin, D.Cheng, O.N.Witte, D.Baltimore, A.Ribas and J.R.Heath, unpublished data) were synthesized as gBlocks (IDT) and cloned in the pCCLc-MND-X backbone (a kind gift from Donald B. Kohn) along with a truncated form of LNGFR gene as described previously19. EC27, FC5.5, FC4.3, and CP7.9 TCRs were cloned as described previously19.
Generation and cloning of SABR libraries
To generate lists of epitopes to be clone into SABR vectors, two approaches were taken. In the universal A2-SABR library, all HLA-A*0201-restricted epitopes from Immune Epitope Database23 (IEDB) were downloaded. In the neoantigen SABR library, HLA-A*0201-restricted neoepitopes generated from the tumor mutanome data reported previously (S.Peng, J.M.Zaretsky, A.H.C.Ng, W.Chour, M.T.Bethune, A.Hsu, E.Holman, X.Ding, K.Guo, J.Kim, A.M.Xu, J.E.Heath, W.Noh, J.Zhou, Y.Su, Y.Lu, J.Mclaughlin, D.Cheng, O.N.Witte, D.Baltimore, A.Ribas and J.R.Heath, unpublished data) were used. Protein sequences were back-translated to nucleotide sequences using the most abundant codon for each amino acid based on the GenScript Codon Usage Frequency Table Tool (GenScript). Oligonucleotides encoding for the epitopes and containing overlaps with the SABR vector were synthesized in pooled single stranded oligonucleotide libraries (Twist Biosciences). Oligonucleotide libraries were amplified and cloned into the SABR vector as described previously. To ensure sufficient coverage, bacterial cells transformed with the cloning reaction were inoculated directly into 500 ml liquid cultures overnight. The plasmid DNA containing the libraries was prepared using Nucleobond Xtra Maxi Plus EF (Takara).
Lentiviral vector production and transduction
Lentiviral vectors to express SABRs or TCRs were packaged using previously described procedures19. Briefly, 5×106 HEK-293T cells were plated on poly-L-Lysine coated plates for 24 hours, followed by transfection of a mixture of the lentiviral shuttle plasmid, pMDG-VSVG, and pCMV-RD8.9 (a kind gift from Donald B. Kohn) using TransIT-293 (Mirus Bio) and OPTI-MEM (Life Technologies). Viruses were filtered through 0.45 micron syringe filters (Milipore) and stored at −80°C until further use. To transduce Jurkat, NFAT-GFP-Jurkat, or Primary T cells, 2-5×105 cells were plated in culture medium and mixed with an equal volume of thawed virus in 12-well plates for three days. For NFAT-GFP-Jurkat cells, G-418 was added to the transduction mixture 48 hours later.
Retroviral vector production and transduction
RD114-pseudotyped retroviruses encoding for the B27-KK10-specific TCRs were produced in HEK-293T cells by transient transfection of three plasmids – an MSCV-based shuttle plasmid (a gift from R.A. Morgan), pHIT60, and pRD114. HEK-293T cells were plated at 5×106 cells per 10 cm plate (Corning / BD Falcon) in D10. The cells were transfected using TransIT-293 (Mirus Bio) using the manufacturer’s protocol with 7.5 μg of shuttle plasmid, 7.5 μg of pHIT60, and 5 μg of pRD114. The cell free supernatant was harvested 72 hours later and filtered through a 0.45 micron filter (EMD Millipore), aliquoted, and stored at −80 °C until further use. To transduce primary human T cells, PBMCs were first activated for 7 days as described previously. The cells were mixed with retroviral vector at 1×106 cells per well in 12-well non tissue culture treated plates coated with 10 μg/ml Retronectin (Takara Bio) and spinfected at 1111g for 1.5 hours at room temperature. The cells were incubated for 72 hours post-transduction and harvested for functional assays.
Co-culture assays
For co-culture assays to test SABR signaling, 1-5×104 transduced NFAT-GFP-Jurkat cells were incubated with equal number of transduced Jurkat cells on 96 well flat or round bottom plates for 8-10 hours. The cells were then acquired on MACSQuant (Miltenyi) or stained with anti-CD69-APC-Cy7 (Biolegend) and then acquired on MACSQuant (Miltenyi). For SABR library assays, 1.5×106 SABR library cells were incubated with 1.5×106Jurkat cells in each well of a 6 well plate. At 8-10 hours after co-culture, cells were harvested, stained with anti-CD69-APC-Cy7 (Biolegend), and sorted on a BD FACS SORP (Becton-Dickinson). Cytotoxicity assays were performed using target cells labeled with CFSE (Biolegend) as described previously19. For empty SABR assays, transduced NFAT-GFP-Jurkat cells were incubated with 100 μg/ml of soluble peptide for 2 hours at 37 °C. Equal numbers of transduced Jurkat cells were then added to the cells, followed by 8-10 hours of co-culture. Gating strategies for these assays is shown in supplementary Fig 9. Functional avidity assays were performed using KK10 variant peptides as described previously19.
High throughput sequencing and analysis
Genomic DNA was extracted from sorted cells immediately following sorting using PureLink genomic DNA extraction kit (Life Technologies). The SABR vectors were amplified using KOD polymerase (Milipore) and 10:1:10 mixture of primers TruSeq-Univ-SCTfixed-F, TruSeq-Read2-SCTfixed-R, and Truseq-Adapter-Index respectively. For each sample, 5-10 reactions using 1-30 ng of genomic DNA were performed for 30-35 cycles. The reactions were pooled and purified using Nucleospin Gel and PCR purification kit (Takara). The purified PCR product was analyzed on Bioanalyzer (Agilent) and subjected to sequencing on HiSeq 2500 (Illumina). Unaligned reads generated by the sequencer were stored in FASTQ files. The reads were first aligned to the SABR vector using Burrows-Wheeler Alignment with a mismatch penalty of 124. For each aligned read, the epitope was translated and counted. All epitopes were ranked according to the number of reads, and an average rank was calculated for each read. The average rank was then used for further analysis.
Statistical analysis
Flow cytometry plots were analyzed on FlowJo (Treestar). Statistical analyses and graphical representations were generated by Microsoft Excel (Microsoft) and GraphPad Prism (Graphpad).
Data availability
The data that support the findings of this study are available from the corresponding author upon request. The raw data can be found in source data file. The list of epitopes in the SABR libraries can be found in supplementary tables 3 and 4. The plasmids for HLA-A*0201-SABR backbone (pCCLc-MND-A0201-SABR-Backbone, ID 119050), HLA-B*2705-SABR backbone (pCCLc-MND-B2705-SABR-Backbone, ID 119051), A2-Mart1-SABR (pCCLc-MND-A0201-Mart1-SABR, ID 119052), and B27-KK10-SABR (pCCLc-MND-B2705-KK10-SABR, ID 119053) are available through Addgene Inc. The sequencing data are deposited in Sequence Read Archive (SRR8207921: amplicon sequencing of A2-SABR-library co-incubated with F5 TCR, SRR8207922: amplicon sequencing of A2-SABR-library co-incubated with SL9 TCR, SRR8207923: amplicon sequencing of A2-SABR-library co-incubated with No TCR, SRR8207924: amplicon sequencing of A2-NeoAg-library co-incubated with Neo TCR, SRR8207925: amplicon sequencing of A2-NeoAg-library co-incubated with No TCR). The code used to analyze sequences is deposited at GitHub (https://github.com/Baltimore-Lab/nat-methods-SABR-trogo). The data availability statement can be found in Life Sciences Reporting Summary.
Supplementary Material
Acknowledgements:
We thank Igor Antoshechkin at the Millard and Muriel Jacobs Genetics and Genomics Laboratory for Illumina sequencing and Alexander Spalla at the Analytical Cytometry Core at the City of Hope for help with FACS. This work was funded by California Institute for Regenerative Medicine DISC2-09123 award, Caltech Rothenberg Innovation Initiative award, and National Cancer Institute 1U54 CA199090-01 grant.
Footnotes
Competing Financial Interests Statements:
A.V.J., M.T.L., M.T.B., and D.B. are co-inventors on a patent application concerning the described technology. D.B. is a consultant of PACT and head of their scientific advising board. J.R.H. and A.R. are directors and consultants of PACT; M.T.B. and S.P are employees of PACT; J.M.Z. is a consultant of PACT; and each of the foregoing individuals have equity interests in PACT. The other authors declare that no competing interests exist.
References:
- 1.Shankaran V et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410, 1107–1111, doi: 10.1038/35074122 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Lollini PL, Cavallo F, Nanni P & Forni G Vaccines for tumour prevention. Nature reviews. Cancer 6, 204–216, doi: 10.1038/nrc1815 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Leach DR, Krummel MF & Allison JP Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996). [DOI] [PubMed] [Google Scholar]
- 4.Dong H et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature medicine 8, 793–800, doi: 10.1038/nm730 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Yee C et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proceedings of the National Academy of Sciences of the United States of America 99, 16168–16173, doi: 10.1073/pnas.242600099 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis MM & Bjorkman PJ T-cell antigen receptor genes and T-cell recognition. Nature 334, 395–402, doi: 10.1038/334395a0 (1988). [DOI] [PubMed] [Google Scholar]
- 7.Weiss A & Littman DR Signal transduction by lymphocyte antigen receptors. Cell 76, 263–274 (1994). [DOI] [PubMed] [Google Scholar]
- 8.Bethune MT & Joglekar AV Personalized T cell-mediated cancer immunotherapy: progress and challenges. Curr Opin Biotechnol 48, 142–152, doi: 10.1016/j.copbio.2017.03.024 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Woodsworth DJ, Castellarin M & Holt RA Sequence analysis of T-cell repertoires in health and disease. Genome Med 5, 98, doi: 10.1186/gm502 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchholz VR, Schumacher TN & Busch DH T Cell Fate at the Single-Cell Level. Annual review of immunology 34, 65–92, doi: 10.1146/annurev-immunol-032414-112014 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Klenerman P, Cerundolo V & Dunbar PR Tracking T cells with tetramers: new tales from new tools. Nature reviews. Immunology 2, 263–272, doi: 10.1038/nri777 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Castle JC et al. Exploiting the mutanome for tumor vaccination. Cancer research 72, 1081–1091, doi: 10.1158/0008-5472.CAN-11-3722 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Boon T & van der Bruggen P Human tumor antigens recognized by T lymphocytes. The Journal of experimental medicine 183, 725–729 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsushita H et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404, doi: 10.1038/nature10755 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gee MH et al. Antigen Identification for Orphan T Cell Receptors Expressed on Tumor-Infiltrating Lymphocytes. Cell 172, 549–563 e516, doi: 10.1016/j.cell.2017.11.043 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birnbaum ME et al. Deconstructing the Peptide-MHC Specificity of T Cell Recognition. Cell 157, 1073–1087, doi: 10.1016/j.cell.2014.03.047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu YY, Netuschil N, Lybarger L, Connolly JM & Hansen TH Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells. Journal of immunology 168, 3145–3149 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Morgan RA et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314, 126–129 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joglekar AV et al. T cell receptors for the HIV KK10 epitope from patients with differential immunologic control are functionally indistinguishable. Proceedings of the National Academy of Sciences of the United States of America 115, 1877–1882, doi: 10.1073/pnas.1718659115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett MS, Joseph A, Ng HL, Goldstein H & Yang OO Fine-tuning of T-cell receptor avidity to increase HIV epitope variant recognition by cytotoxic T lymphocytes. Aids 24, 2619–2628, doi: 10.1097/QAD.0b013e32833f7b22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bethune MT, Comin-Anduix B, Hwang Fu YH, Ribas A & Baltimore D Preparation of peptide-MHC and T-cell receptor dextramers by biotinylated dextran doping. BioTechniques 62, 123–130, doi: 10.2144/000114525 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Sahin U et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226, doi: 10.1038/nature23003 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Vita R et al. The immune epitope database (IEDB) 3.0. Nucleic acids research 43, D405–412, doi: 10.1093/nar/gku938 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, doi: 10.1093/bioinformatics/btp324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yokomaku Y et al. Impaired processing and presentation of cytotoxic-T-lymphocyte (CTL) epitopes are major escape mechanisms from CTL immune pressure in human immunodeficiency virus type 1 infection. Journal of virology 78, 1324–1332 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorrell L et al. Distinct recognition of non-clade B human immunodeficiency virus type 1 epitopes by cytotoxic T lymphocytes generated from donors infected in Africa. Journal of virology 73, 1708–1714 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peakman M et al. T cell clones generated from patients with type 1 diabetes using interleukin-2 proliferate to human islet antigens. Autoimmunity 17, 31–39 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Tang Q & Bluestone JA The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nature immunology 9, 239–244, doi: 10.1038/ni1572 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia KC et al. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science 279, 1166–1172 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Suwandi JS, Nikolic T & Roep BO Translating Mechanism of Regulatory Action of Tolerogenic Dendritic Cells to Monitoring Endpoints in Clinical Trials. Frontiers in immunology 8, 1598, doi: 10.3389/fimmu.2017.01598 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bentzen AK & Hadrup SR Evolution of MHC-based technologies used for detection of antigen-responsive T cells. Cancer immunology, immunotherapy : CII 66, 657–666, doi: 10.1007/s00262-017-1971-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran E et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645, doi: 10.1126/science.1251102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request. The raw data can be found in source data file. The list of epitopes in the SABR libraries can be found in supplementary tables 3 and 4. The plasmids for HLA-A*0201-SABR backbone (pCCLc-MND-A0201-SABR-Backbone, ID 119050), HLA-B*2705-SABR backbone (pCCLc-MND-B2705-SABR-Backbone, ID 119051), A2-Mart1-SABR (pCCLc-MND-A0201-Mart1-SABR, ID 119052), and B27-KK10-SABR (pCCLc-MND-B2705-KK10-SABR, ID 119053) are available through Addgene Inc. The sequencing data are deposited in Sequence Read Archive (SRR8207921: amplicon sequencing of A2-SABR-library co-incubated with F5 TCR, SRR8207922: amplicon sequencing of A2-SABR-library co-incubated with SL9 TCR, SRR8207923: amplicon sequencing of A2-SABR-library co-incubated with No TCR, SRR8207924: amplicon sequencing of A2-NeoAg-library co-incubated with Neo TCR, SRR8207925: amplicon sequencing of A2-NeoAg-library co-incubated with No TCR). The code used to analyze sequences is deposited at GitHub (https://github.com/Baltimore-Lab/nat-methods-SABR-trogo). The data availability statement can be found in Life Sciences Reporting Summary.