

Abstract
Permeability (Pm) across biological membranes is of fundamental importance and a key factor in drug absorption, distribution and development. Although the majority of drugs will be charged at some point during oral delivery, our understanding of membrane permeation by charged species is limited. The canonical model assumes that only neutral molecules partition into and passively permeate across membranes, but there is mounting evidence that these processes are also facile for certain charged species. However, it is unknown whether such ionizable permeants dynamically neutralize at the membrane surface or permeate in their charged form. To probe protonation-coupled permeation in atomic detail, we herein apply continuous constant-pH molecular dynamics along with free energy sampling to study the permeation of a weak base propranolol (PPL), and evaluate the impact of including dynamic protonation on Pm. The simulations reveal that PPL dynamically neutralizes at the lipid-tail interface, which dramatically influences the permeation free energy landscape and explains why the conventional model overestimates the assigned intrinsic permeability. We demonstrate how fixed-charge-state simulations can account for this effect, and propose a revised model that better describes pH-coupled partitioning and permeation. Our results demonstrate how dynamic changes in protonation state may play a critical role in the permeation of ionizable molecules, including pharmaceuticals and drug-like molecules, thus requiring a revision of the standard picture.
Graphical Abstract
INTRODUCTION
Permeation across biomembranes, the barriers that enable the compartmentalization and localization essential to life,1 is generally a prerequisite for the effective delivery of pharmaceuticals to target sites. Thus, permeability (Pm) is an essential pharmacological property to assess in preclinical drug discovery.2 It is often measured in vitro with cell-based assays (Caco-23 and MDCK4) or the parallel artificial membrane permeability assay (PAMPA).5 More than 60% of the human pharmaceuticals contain at least one ionizable group6–7 and ionize to varying magnitudes in response to the microenvironment pH, depending on the ionization constant (pKa), which in turn affects their in vivo distribution and availability to affect biological reactions. Given the considerable pH gradient along the gastrointestinal tract,8 a significant proportion of these ionizable compounds are charged at one point or another during delivery to their target destination. Therefore, knowledge of the titration behavior of a pharmaceutical or drug-like molecule is essential to predict and optimize its pharmacokinetic profile.9–10
The pH-partition hypothesis states that only the neutral form of an ionizable molecule can diffuse across lipophilic membranes.11–12 In analogy to the Henderson-Hasselbalch equation, the pH-dependent apparent or effective permeability coefficient (for a monoprotic base) is expressed in the form:8
| (1) |
where is the aqueous pKa and is the Pm of the neutral form, or the “intrinsic” Pm. Yet, a growing list of studies has revealed violations of the pH-partition hypothesis. For example, it has been shown that the permanently charged quaternary ammonium compounds are able to permeate cell membranes.13–14 The permeation in the charged form has also been reported for ionizable compounds, albeit at a much slower rate than the neutral form,15–17 and the abundance of the charged species can make this a significant contribution to the total measured Pm.18 In the context of cells, molecules can permeate via multiple pathways (e.g., paracellular, transcellular carrier-mediated, and passive transport),19 making it difficult to evaluate unambiguously the contribution of the passive diffusion of charged species. Palm et al. compared Caco-2 permeation of two weak bases with approximately the same and found that the one with a larger molecular weight has a higher for the charged form,18‘20 suggesting that the transcellular passive diffusion is nonnegligible because it is believed that larger molecules are not expected to permeate substantially via the paracellular route.15 Violation of the pH-partition hypothesis was also observed in PAMPA and liposomal assays, where the paracellular and active transport are absent. Fischer et al. measured the PAMPA of 12 quaternary ammonium compounds and found that the permanently charged molecules can have high passive transmembrane permeability, potentially due to the charge delocalization.21 Regev et al. measured liposomal of doxorubicin, a primary amine with a of 9.7, and found the increase in at high pH is significantly smaller than what eq. 1 predicted,22 but the origin of the deviation is not clear. All of these violations suggest that the neutral permeant assumed in eq. 1 is oversimplified.
The pH-partition hypothesis of course neglects the possibility that an ionizable molecule might neutralize at the membrane surface before entry. This behavior would provide an obvious explanation for the violations of the pH-partition hypothesis described above. However, this explanation has been challenged by several results showing that amphiphilic molecules migrate into membranes in their charged form23–31 and accumulate in the lipid headgroup and glycerol regions.32–33 The ionizable sites of the bound amphiphiles position in the water-accessible34 headgroup region35–36 where they stabilize their ionized states by forming a surface ion pair with the zwitterionic lipid headgroups.8, 2 (Note here the interesting difference between bases, which associate lower in the membrane with the negatively charged phosphates, and acids, which are closer to the membrane-water interface associating with the positively charged trimethylammonium groups.8, 29) By contrast, the neutralized species were observed in a deeper location,23–24 suggesting that a titration event could indeed occur somewhere inside the membrane during permeation. Two questions naturally arise: Can ionized molecules really neutralize before permeating through membranes as these results suggest, and, if so, where exactly within the membrane does this neutralization occur? The solution to these questions requires insight into permeant-membrane interactions at the atomic level, which is not easily accessible in experiments.
Molecular dynamics (MD) simulations can, with care, predict Pm and offer atomistic details of permeation. Since the work by Marrink and Berendsen,34 MD simulations have become a well-established method to study permeation.37–49 On the basis of the pH-partition hypothesis, simulations are mostly performed with neutralized permeants from which can be computed. However, using a fixed ionization state, whether it is charged or neutral, faces several limitations. First, the choice of the neutral form overlooks the above-mentioned evidence supporting membrane partitioning of charged species. Second, it assumes an unperturbed pKa inside the membrane, and thus no dynamic titration during permeation, despite the physically expected quite significant pKa shift. Given the observed location-dependent ionization state for some molecules,23 it could be more realistic to have multiple simulations at fixed locations along the permeation route with varying ionization states, which can be assigned by comparing the aqueous phase pH and the pKa at the location of interest. The pKa inside membrane might then be predicted by Poisson-Boltzmann (PB) electrostatic calculations, for example, the PB solver APBSmem.50 However, this approach often gives large errors in pKa predictions for highly buried ionizable sites.51 Moreover, calculations based on static structures suffer from the loss of the coupling between protonation and conformation equilibria, which has been shown to be essential in titration-coupled processes.52–54 Alternatively, the pKa inside a membrane can be derived from permeation free energies for both ionization states.55 Yet, this approach is computationally expensive and fails to offer a dynamic picture of the protonation in response to varying the membrane environment. Consequently, better modeling of membrane permeation of ionizable molecules requires a tool enabling dynamic protonation in response to the changes in microenvironment.
The past decade has witnessed the development of constant-pH molecular dynamics (pHMD), a class of methods that determines the charge states of ionizable sites during the conformational dynamics. On the basis of how charge states are represented and sampled, pHMD methods are categorized into the discrete pHMD (DpHMD) and continuous pHMD (CpHMD). In DpHMD,56 a standard MD simulation is periodically interrupted to update charge states using Monte Carlo methods, which determines if the randomly generated new charge states should be accepted or rejected based on the Metropolis criteria.57 MD simulation will resume with the new charge states if accepted or old charge states if rejected. Different DpHMD formulations mostly vary in the solvation model for evaluating the energetics of titration56, 58–62. Unlike DpHMD, CpHMD allows charge states to fluctuate in MD simulations by adding titration as an extra degree of freedom to an extended Hamiltonian.63 In the CpHMD formalism64–65 based on the λ-dynamics approach for free energy calculations,66 each ionizable site is assigned a λ, which is bounded between 0 (protonated form) and 1 (deprotonated form), and propagates simultaneously with spatial coordinates. The pHMD methods have succeeded in modeling a variety of pH-coupled systems.67–68 Using DpHMD, the Machuqueiro lab characterized the pKa values of ionizable amino acid at the membrane surface69 and the pKa shift (ΔpKa) in the pHLIP peptides during membrane insertion.70 The Roux lab investigated the translocation of a pentapeptide AADAA across a POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) membrane and found that the pKa of the central aspartic acid was shifted to 7.3 at the center of the membrane.71 The Brooks lab first applied CpHMD with an implicit description of solvent and membrane to study the protonation-linked structural rearrangement in rhodopsin activation.72 Later, the Brooks lab investigated the protonation equilibria of titratable amino acids in transmembrane helices using explicit-solvent CpHMD73 and found that the neutral states of those titratable residues were significantly favored in the context of membranes.74 More recently, the Shen lab developed hybrid-solvent CpHMD,75 which combines the accuracy of explicit solvent and membrane for conformational sampling with the efficiency of the implicit-solvent model for calculating the solvation free energies or pKa values of ionizable sites. This method has been validated in various pH-coupled systems, such as assembly and phase transition of fatty acids76–77 as well as polysaccharides,78 inhibitor-enzyme binding,79–81 and protein folding.82 With membrane description enabled in the underlying implicit-solvent model,83 the hybrid-solvent CpHMD has also afforded mechanistic insight into the proton-driven activation of the Na+/H+ antiporter NhaA,83 the influenza A M2 proton channel,84 and the multidrug efflux pump AcrB.85
Inspired by these successes, we combined the membrane-enabled hybrid-solvent CpHMD technique75, 83 with the umbrella sampling (US) method86 to investigate the permeation of a cationic amphiphile propranolol (PPL, Figure 1A) from the β-blocker family through a POPC bilayer. β-Blockers are a class of drugs primarily used to treat cardiovascular diseases. As the prototype of β-blockers, propranolol is listed by the World Health Organization as one of the most effective and safe medicines needed in a health system.87 The POPC lipids were chosen because they were used to determine Pm in the liposomal fluorescence assay.88–89 The liposome Pm measures the rate of passive permeation and is thus directly comparable to the MD-based Pm.90 We sought to answer the following questions: (1) how does PPL adjust its ionization state in response to the changing environment during permeation; and (2) how does the dynamic protonation of PPL impact its permeability? We find that PPL enters the POPC bilayer in the charged form but neutralizes at the lipid-tail interface and then permeates in its neutral form. Including dynamic protonation in the MD free energy sampling simulations not only confirms the permeation pathway but also predicts Pm with very high accuracy (~1–3-fold the experimental measures). Accordingly, we propose a revised pH-partition model to better describe the ionization-linked permeation process. The major findings of the present work yield new insight into the membrane permeation process at the atomic level, and the overall methodology will be applicable to other important ionization-coupled permeation phenomena.
Figure 1. PPL structure and permeation thermodynamics.

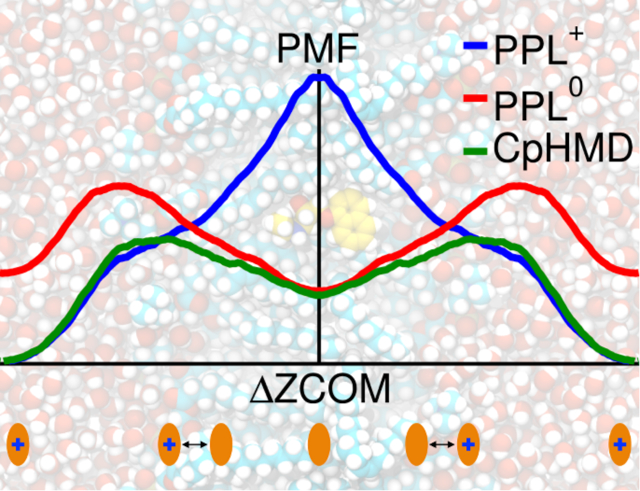
(A) Structure of PPL. The ionizable amine group is indicated by a blue circle. Experimental91 is shown below. (B) Potential of mean force (PMF) as a function of the distance from bilayer center ΔZCOM, which is defined as the projection of the center-of-mass (COM) distance between the heavy atoms of PPL and POPC on the bilayer normal (Z-axis). Charged (PPL+) and CpHMD (at pH 7) PMFs were set to zero in aqueous phase, while the neutral (PPL0) PMF was upshifted by the deprotonation free energy in aqueous phase at pH 7 (3.55 kcal/mol). Error bars indicate the standard deviations from block analysis and are displayed every 3 Å for clarity (see Figure S1 for details). A schematic POPC molecule is aligned below with its components colored and mapped along the membrane normal according to Figure 2B.
RESULTS AND DISCUSSION
Dynamic Protonation Delineates the Permeation Pathway.
We first investigated the thermodynamic impact of including dynamic titration on the PPL permeation. Figure 1B shows the potentials of mean force (PMFs), that is, free energy profiles, as a function of the distance from bilayer center ΔZCOM in both the charged (PPL+) and the neutral (PPL0) forms, as well as under the CpHMD environment. PPL+ experiences a large barrier of ~12 kcal/mol as it approaches the bilayer center, consistent with other computational work,55, 92–98 whereas PPL0 experiences a moderate barrier of ~3 kcal/mol around the POPC headgroups (ΔZCOM of 15–20 Å) where the charge and density of the system maximize.34, 55 The PPL+ and PPL0 PMFs experience a crossover at ΔZCOM of 12 Å. Consistent with previous computational studies,92, 95–98 this suggests a permeation path of the least effort (or lowest free energy), that is, entering the bilayer as PPL+ but permeating through it as PPL0. With CpHMD turned on, PPL was able to adjust its charge state in response to microenvironment. In the permeation direction, the CpHMD PMF coincides with PPL+ first until ΔZCOM of ~12 Å and then with PPL0 within statistical error. In other words, CpHMD reproduced the lowest free energy path of the combined fixed-ionization-state PMFs, a commonly adopted approach without rigorous validation.92, 95–98 Note that CpHMD delineated the path at one-half the computational cost.
PPL Neutralizes at the Hydrophobic Boundary of the POPC Bilayer.
Next, we examined the molecular details of the change of the PPL charge state during permeation. Although bound to biomembranes as a charged species,26–31 it has been shown that the PPL trans-liposome partition increases the internal pH, suggesting permeation in the neutral form and reprotonation inside liposome.99 To verify this, we plotted the free energy surface (FES) as a function of ΔZCOM and the titration coordinate of PPL (λPPL) in Figure 2A. λPPL describes the transition between the protonated (λ = 0, PPL+) and deprotonated states (λ = 1, PPL0). The distribution probabilities of various components of the system were computed on the basis of the scattering density profile model100 and are displayed in Figure 2B. Figures 2A and B indicate PPL+ exists from water down to the carbonyl and glycerol groups of the lipids (ΔZCOM ≈ 16.5 Å), but a neutral PPL0 exists inside the lipid hydrocarbon tails (ΔZCOM ≤ 10.5 Å), in good agreement with the experiments (around physiological pH amphiphiles accumulate in the lipid headgroup and glycerol region in their charged forms23–31 but become neutral once they migrate to deeper locations23–24) and the lowest free energy permeation path (Figure 1B). Notably, PPL titrates (λPPL changes from 0 to 1) in the ΔZCOM range of 10.5–16.5 Å, where the bilayer transitions from the carbonyl and glycerol groups (CG) to the hydrocarbon tails (HC). That is to say, PPL neutralizes at the hydrophobic boundary of the bilayer (14.6 Å, DC in Figure 2B) rather than at the bilayer surface. This finding, although contradicting the common notion that neutralization would occur at the water-membrane interface (20.7 Å, DB/2 in Figure 2B), is not especially surprising. Considering the zwitterionic nature of the lipid headgroups, the membrane-bound ionized amphiphiles likely have their hydrophilic groups pointing toward and salt-bridging with the lipid headgroups, while their hydrophobic moieties are buried in the vicinity of carbonyl and glycerol groups or hydrocarbon tail region to reduce their water contact (i.e., they likely follow the surface ion pair model8). PPL also binds to membranes in the same fashion, as can be inferred from experiments.32, 35–36 In our simulations, PPL partitioned into the POPC bilayer with its naphthalene moiety localized close to the carbonyl and glycerol region and amine group ion-paired with the phosphate groups (Figure S2). Such a partition pattern emerges approximately between the hydrophobic boundary (DC) and the bilayer surface (DB/2), which agrees with the experiments and lends support to the surface ion pair model. Furthermore, amphiphiles bound to the carbonyl and glycerol groups of a bilayer33 are likely to titrate there given the limited water accessibility in the region34 (Figures 2B–D). In fact, Boulanger et al. studied the pH-dependent interactions of two local anesthetics (bases) with eggPC vesicles using NMR.23–24 They found the ionized form at low pH preferred binding close to the membrane surface, while the neutral form at high pH strongly bound to a deeper site. This indicates an amphiphile can adjust its location inside the membrane (moving up and down) in response to varying ionization states induced by pH, leading to a “pH piston hypothesis”.29 In fact, the position-dependent ionization state revealed in this work agrees with Boulanger et al.’s findings as well as previous CpHMD results,69–71, 74 supporting the “pH piston hypothesis”.
Figure 2. Change of PPL protonation state during permeation.

(A) Free energy surface (FES) as a function of insertion depth (ΔZCOM) and the titration coordinate of PPL (λPPL) at pH 7. Contour lines are displayed every 1 kBT. The dashed vertical white lines mark where PPL titrates (λPPL changes from 0, i.e., PPL+, to 1, i.e., PPL0). The average standard deviation from block analysis is 0.3 kBT. (B) Volume probability distributions for the density profile model100 of components of the POPC bilayer. Componentization follows Kucerka et al.:101 water (H2O, brown), phosphate and CH2CH2N part of choline (PCN, red), 3xCH3 part of choline (CholCH3, green), carbonyl and glycerol groups (CG, black), methine group (CH, cyan), methylene groups (CH2, blue), methyl groups (CH3, magenta), and the total hydrocarbon tails (HC, orange). Data collected from the umbrella sampling run at pH 7 with PPL at ΔZCOM = 13.5 Å. The vertical purple and dark green dot-dashed lines mark the hydrophobic boundary (DC) and bilayer surface (DB/2),100 respectively. The background gray box highlights the titration region (same below). A schematic POPC molecule is mapped along the bilayer normal in the center with components colored accordingly. (C) The ΔpKa of PPL as a function of ΔZCOM. Black line was calculated from the PMFs in Figure 1B with error bars calculated from error propagation. Red circles represent the pKa values calculated from CpHMD with error bars calculated from three independent runs. (D) The hydration number of PPL, counted as the number of water oxygen atoms within 3.5 Å of the PPL titrating nitrogen (Figure 1A) as a function of ΔZCOM. Error bars indicate the standard deviations from block analysis and are shown every 3 Å for clarity.
From the permeation PMFs of PPL+ and PPL0 (Figure 1B), location-specific pKa can be computed using a thermodynamic cycle55 (Figure S3). As displayed in Figure 2C, the PPL pKa smoothly decreases from 9.5 in water to ~1 in the bilayer center. This is expected because it is energetically unfavorable to charge a base like PPL inside membranes. Remarkably, the pKa reaches 7 at ΔZCOM ≈ 12 Å, indicating this is where POPC-bound PPL deprotonates at pH 7, which agrees with Figure 2A. Using the Hill equation (eq. S3), we also calculated pKa values from the membrane-enabled hybrid-solvent CpHMD combined with the pH-based replica-exchange (pHREX) protocol,75, 83 which has been shown to predict pKa values with high accuracy and faster convergence.68 Consistent with the PMF pKa, the CpHMD pKa (Figure 2C) decreases as permeation proceeds and reaches 7 at ΔZCOM ≈ 14 Å. The PMF and CpHMD pKa values agree well at ΔZCOM ≥ 15 Å. Below 15 Å, the CpHMD pKa values are shifted to lower values until they plateau at ΔZCOM ≤ 10 Å. The second plateau absent in the PMF pKa maps to the hydrocarbon tail region of the bilayer (Figure 2B) and exhibits a pKa of 2.5–3. The decrease in pKa occurs in the ΔZCOM range of ~10–19.5 Å, consistent with the titration region revealed by Figure 2A. Moreover, the Hill coefficients calculated from the pH-REX CpHMD simulations are in the range of 0.7–0.8 inside the bilayer, an indication of anticooperativity in titration.102.
The pKa inside a membrane can be inferred from the partition coefficient (logP)8, 103 (Figure S4). The pKa of PPL has been reported to downshift by 0.5–1 pH units with respect to based on the PC liposome logP values (Table S2). The same ΔpKa was reported by the cosolvent method using methanol/water mixture,104 whose dielectric constant is close to that of the lipid headgroups.8 The PPL-POPC interactions involved in the PPL membrane partition pattern35–36 discussed above become the most obvious in the ΔZCOM range of 14–16 Å (Figure S2). The predicted ΔpKa at this location is 1–2 pH units, which is ~1 pH units larger than the experimental values. This can be attributed to the absent ionic effect in our simulations, because the presence of salt enhances the partition of the charged species by attenuating their electrostatic repulsion on the bilayer surface,105 leading to a higher pKa. As indicated in Table S2, ΔpKa increases by ~0.5 pH units with the ionic strength reduced from 0.23 to 0.15 M. A further increase in ΔpKa is thus expected in the absence of salt. Note some membranes listed in Table S2 are anionic, which should increase the partitioning of PPL+ and result in a reduced Δ pKa as compared to the charge-neutral PC lipids. Octanol logP values are also frequently reported. Although octanol/water mixtures may resemble the bilayer surface better than the hydrophobic interior given their ability to form hydrogen bonds, the hydrocarbon chains create a solvent-inaccessible region with a dielectric constant similar to that of the bilayer interior.8 Thus, we reason that the octanol logP values could give some insights into the permeant pKa around the entry of the hydrocarbon tail region (ΔZCOM of 7–14 Å). The octanol ΔpKa has been measured to be ~3 pH units in the 0.15 M background ionic strength (Table S2) and may further increase by ~1.5 pH units for weak bases under marginal ionic strength.8 We then expect a ΔpKa of ~4.5 pH units, which falls within the range of the calculated ΔpKa values within the region. Collectively, the predicted ΔpKa values are in agreement with the experiments, underling the robustness of our calculations.
We now consider the discrepancy between the two sets of calculated ΔpKa values. As compared to the PMF approach, CpHMD predicted larger ΔpKa values in the ΔZCOM range of 10–14 Å (Figure 2C). Because of the limited solvent accessibility in the region (Figures 2B,D), there exists an equilibrium between the “open” and “closed” microstates, each coupled to a separate protonation equilibrium. Also, the calculated pKa values in Figure 2C are macroscopic values that incorporate the microscopic pKa values of both conformations.52–53 Because the effect of discrete water molecules around a buried site cannot be captured by the underlying generalized-Born (GB) model of the hybrid-solvent CpHMD, the error in the closed-state pKa may be nonnegligible.68 It has been shown that the closed-state pKa for a buried lysine could be underestimated by ~1.5 pH units, resulting in an overestimated ΔpKa by the same amount.52 Bearing this in mind, the CpHMD pKa values agree well with the PMF ΔpKa values in the lipid headgroup, carbonyl, and glycerol regions. The deviation becomes more significant in the hydrocarbon tail region that is devoid of water. On the CpHMD side, it is known that ΔpKa is overestimated for deeply buried titration sites due to the inaccuracy in the desolvation penalties by GB models.51, 106 This systematic error is less likely to improve with longer simulations. On the PMF side, the error in the PPL+ PMF maximizes around the bilayer center (Figure 1B), possibly due to the water defect (Figure 2D), and propagates uncertainty into the resulting ΔpKa values (Figure 2C). Although prolonged simulation may alleviate this error, within the present simulation length we have achieved qualitative agreement between the two sets of ΔpKa in this region. We note that CpHMD is much less computationally demanding as compared to the US method (pKa values converged within 3 ns per replica, Figure S5) and therefore a more suitable tool for predicting membrane pKa values.
To better understand the cause of the PPL deprotonation inside the POPC bilayer, we quantified its hydration by computing a hydration number, which was defined as the number of water oxygen atoms within 3.5 Å (first solvation shell) of the PPL titrating nitrogen (Figure 1A), as a function of ΔZCOM (Figure 2D). Similar to the change in charge state (Figure 2A), the resulting hydration profile from the CpHMD coincides with that for PPL+ until the entry of the bilayer, decreases within the titration region, and eventually matches that for PPL0 near the bilayer center. This suggests that the decrease in pKa is induced by the decreased hydration. To verify this, we investigated the correlation between λPPL and PPL hydration in the US window of ΔZCOM = 13.5 Å, the center of the titration region (Figure 2A) where the CpHMD pKa decreases to ~7 (Figure 2C). As plotted in Figure S6A, λPPL mostly samples values close to 1 (PPL0) when PPL is dehydrated (blue bars), but flips to values close to 0 (PPL+) when there are two or more water molecules within the first solvation shell (red bars). This anticorrelation (reduced solvation favors proton release) is also reflected in the Hill coefficients (0.7–0.8). Interestingly, if there is only one water molecule around, PPL0 and PPL+ are equally sampled (brown bars), an indication of titration event. A similar finding was reported by Bonhenry et al., who studied the permeation of a lysine analogue through a POPC bilayer by constructing the 2D-PMF as a function of ΔZCOM as well as hydration and found the charge state was tightly coupled to the hydration number.97 These results demonstrate that neutralization inside membrane correlates with desolvation.
A related question then arises: Where does the proton go after titrating off PPL+? In the absence of titratable lipid headgroups the obvious acceptor is water; but is that water also buried in the membrane, or can the proton diffuse into bulk? Note that the hydration number displayed in Figure 2D only quantifies the number of “solvating” water molecules in close proximity to the PPL titrating nitrogen and is insufficient to gauge the water distribution inside membrane or its connection to bulk water. Therefore, we counted the number of water molecules along the permeation pathway (Nwater), which was defined as the number of water oxygen atoms in 1-Å slices within a radius of 6.5 Å (the second solvation shell of the PPL titrating nitrogen) centered along the membrane normal. Figure S6C shows that water molecules are always present above the hydrophobic boundary (DC) and occasionally go below DC. By adding Nwater cumulatively, we find that on average there is always one water molecule slightly penetrating the POPC tails (4 Å below DC), and above DC the cumulative Nwater increases rapidly (Figure S6D). These results suggest that there are ample water molecules distributed along the permeation pathway to pass the proton from PPL+ to bulk water (Figure S6B). It is of course possible that titratable lipid headgroups could be an intermediate resting place for the proton. Such intermediates would influence the rate or proton transport out to bulk, but would not influence the pKa of PPL unless they were close enough to alter the relative stability of the charged/neutral drug, a perturbation that CpHMD would capture. No such headgroups were included in these simulations. Furthermore, on the basis of the assumption that the permeation profiles in both bilayer leaflets are symmetric, our results indicate there exists another titration region in the opposite leaflet (ΔZCOM from −16.5 to −10.5 Å) where PPL0 can reprotonate by the influx proton from the opposite bulk waters before leaving the POPC bilayer as PPL+, consistent with the experimental observation that PPL permeation increased the pH inside liposomes.99
PPL Permeates in a “Triple-Flip” Manner.
Next, we examined the molecular-scale conformational dynamics of PPL during permeation. To describe the orientation of PPL with respect to the bilayer, we defined an order parameter θ, the angle between the principal axis of the bulky rigid PPL naphthalene moiety and the bilayer normal (Figure 3A). The FES as a function of ΔZCOM and θ (Figure 3B) shows that the naphthalene moiety adopts an orientation parallel to the membrane surface (θ of ~90°) beyond the hydrocarbon tail region (ΔZCOM ≥ 14.5 Å). In the ΔZCOM range of 5.5–14.5 Å, the naphthalene moiety prefers to align with (θ of ~30). This observation is consistent with a result reported by Badaoui et al. using an equivalent orientational reaction coordinate.107 This region is where the cis double bonds in the POPC tails reside (CH in Figure 2B) and the contacts of naphthalene moiety with CH maximize (Figure S7). The same interaction was reported for amlodipine using NM R.108 No preference in the orientation is seen near the bilayer center (ΔZCOM ≤ 5.5 Å), as suggested by a broad minimum. Furthermore, the aliphatic chain can stay either above or below the naphthalene moiety when PPL is located near the bilayer center but favors the former conformation if PPL moves closer to water (Figure S8). Given the symmetry in bilayer, in the context of the entire bilayer, these results collectively suggest one flip of the naphthalene moiety around the lipid CH region in each bilayer leaflet and a flip of the aliphatic chain near the bilayer center (Figure S9), which is consistent with the “triple-flip” mechanism reported in our previous work.46 However, the accuracy of the permeation PMF as a function of ΔZCOM may be affected by hidden barriers involving permeant orientation,109 which might be solved by adding an orientational collective variable. However, because the free energy barrier related to PPL orientation is marginal (< 1 kBT, Figure 3B), using ΔZCOM alone should not cause significant error.
Figure 3. Change of PPL conformation during permeation.

(A) Variables characterizing PPL conformation. Angle θ is the angle between the principal axis of PPL naphthalene moiety and the bilayer normal . Angle ϕ is the angle between and the vector pointing from PPL alkoxy oxygen (O1) to titrating nitrogen (N1) is the distance between O1 and N1. (B) FES as a function of ΔZCOM and θ. Contour lines are displayed every 0.5 kBT. The vertical white dashed lines mark the region where the PPL naphthalene moiety is aligned with The average standard deviation from block analysis is 0.3 kBT. (C) FES as a function of ϕ and dO1−N1. Contour lines are displayed every 0.5 kBT. Representative structures of the extended, twisted, and collapsed states are shown. Data collected from all US windows at pH 7. The average standard deviation from block analysis is 0.1 kBT. (D) Occupancies of the E (orange), T (red), and C (blue) states as a function of ΔZCOM. See Figure S10 for state definitions.
The PPL conformation can be characterized by another two order parameters: ϕ and dO1−N1 (Figure 3A).ϕ is the angle between the PPL aliphatic chain and naphthalene moiety and quantifies the intrapermeant orientation. dO1−N1 is the distance between atoms O1 and N1 and describes the extension of the aliphatic chain. The FES as a function of ϕ and dO1−N1 reveals three local minima (Figure 3C). We refer to the three states as the extended (E), twisted (T), and collapsed (C) states. In the E state centered at ϕ = 65° and dO1−N1 = 5 Å, the dihedrals along the PPL aliphatic chain are in trans rotamers, resulting in an extended conformation. Separated from the E state by a barrier of 2 kcal/mol is the T state centered at ϕ = 95° and dO1−N1 = 4.3 Å. The T state has a “twisted” aliphatic chain as compared to the E state. The conformation with the aliphatic chain bending toward the naphthalene ring corresponds to the C state, which is separated from the T state by a 3 kcal/mol barrier and centered at ϕ = 110° and dO1−N1 = 2.8 Å. The occupancies of the three states display a ΔZCOM-dependent manner (Figure 3D). The T state (Figure 3D, red line) is dominant in water and about twice sampled as the C state (Figure 3D, blue line). After PPL enters the bilayer and deprotonates, the populations of the T and C states start to decrease and increase, respectively. Near the bilayer center, the C state is slightly favored over the T state. The E state (Figure 3D, orange line), although marginally sampled, becomes more populated inside the bilayer.
Dynamic Protonation Dramatically Affects the Membrane Permeability.
Finally, we examined the impact of including protonation dynamics on Pm prediction. The Pm values were calculated on the basis of the solubility-diffusion model from the permeation PMF and calculated local diffusivity D (eq. S4–S6).34, 47 We note that overall there is no significant difference in D values of PPL+ and PPL0 (Figure S11). Yet around the bilayer center PPL0 is twice as diffusive as PPL+ due to the increased population of the C state (Figure 3D). Because of the presence of a large energy barrier, Pm of PPL+ (−7.38, log scale, Table S3) is ~30,000 times smaller than that of PPL0 (−2.50), indicating protonation of PPL strongly disfavors permeation and likewise deprotonation greatly enhances it. This observation agrees with other computation studies92, 94 and the pH-dependent PAMPA Pm profile of PPL110 (Figure 4B). As compared to PPL0, CpHMD at pH 7 predicts a Pm ≈ 60% smaller (−2.92, magenta rhombus in Figure 4A), because PPL0 underestimates the permeation barrier of bilayer entry relative to its stability in bulk (Figure 1B). The Pm from liposomal fluorescence assay88 (Figure 4A, orange squares) offers a good experimental measure of Pm solely due to passive diffusion.90 The CpHMD Pm (−2.92) is 3.8-fold (or 2.8 times larger than) the Pm reported by Eyer et al. 88 (−3.50 log scale, Table S3), and 1.7-fold (or 0.74 times larger than) the Pm determined with a larger liposome (−3.16).89 The origin of the deviation with experiment is mainly 2-fold: (1) the experiments were conducted at pH 6 while the CpHMD was run at pH 7; and (2) the titration dynamics in CpHMD are accelerated due to the underlying biasing potential, which effectively removes the aqueous titration free energy barrier.64–65 In other words, CpHMD predicts Pm at the limit where the titration of a permeant becomes instantaneous relative to its permeation, which is the case when liposomes with sufficiently large radii (20 μm or larger) are used.111 Thus, the Pm determined with a largest liposome to date (−3.16 was measured with a liposome radius of 239 nm)89 is approaching this limit, and is expected to increase closer to our calculated value (−2.92) in even larger liposomes that better reflect most biological membranes.
Figure 4. pH-dependent permeability coefficient (Pm) of PPL.

(A) Comparison of the calculated “hybrid” (●) and experimental liposome Pm values (orange ■).88–89 The red solid line is the best fit of the “hybrid” Pm to eq. 2. The brown solid line was predicted from eq. 1. (B) Reassessment of the experimental PAMPA Pm values  .110 The solid red line is the best fit to eq. S10. The dashed lines refer to the corrected for the aqueous boundary layer (brown based on eq. 1 and red based on eq. 2, check the Supporting Information section 3.2 for details).
.110 The solid red line is the best fit to eq. S10. The dashed lines refer to the corrected for the aqueous boundary layer (brown based on eq. 1 and red based on eq. 2, check the Supporting Information section 3.2 for details).
Following the lowest free energy path along the PMF (Figure 1B), we calculated Pm from PPL+ and PPL0.92 This “hybrid” Pm at pH 7 (−2.92) agrees with the CpHMD prediction and the value at pH 6 (−3.37) is approximately consistent with the liposome Pm values (Table S3). Consequently, we were able to build the pH-dependent profile (Figure 4A, ●) that qualitatively agrees with experiments (Figure 4B,  ), i.e., increases at low pH values and plateaus at high pH values, resulting in a hyperbolic shape. Note that the PAMPA includes the aqueous boundary layer contribution (, check Supporting Information section 3.2 for details). , is absent in liposome method and simulations, making their Pm measures directly comparable. The calculated Pm values cannot fit to eq. 1 (Figure 4A, brown line), an indication of the violation of the pH partition hypothesis. However, by incorporating the Hill coefficient n and replacing with effective :
), i.e., increases at low pH values and plateaus at high pH values, resulting in a hyperbolic shape. Note that the PAMPA includes the aqueous boundary layer contribution (, check Supporting Information section 3.2 for details). , is absent in liposome method and simulations, making their Pm measures directly comparable. The calculated Pm values cannot fit to eq. 1 (Figure 4A, brown line), an indication of the violation of the pH partition hypothesis. However, by incorporating the Hill coefficient n and replacing with effective :
| (2) |
the best fit (n = 0.61 and ) was obtained (Figure 4A, red line). As revealed in this work, the assumption that only PPL0 contributes to underestimates the amount of “effective” PPL0 because PPL+ deprotonates at the hydrophobic boundary rather than at the bilayer surface (Figures 2A,B). Thus, a reduced pKa is expected with the “effective” PPL0 taken into account. An n < 1 indicates anticooperativity in the pH-dependent permeation. Note that n is not just a scaling factor. Instead, it reflects the sensitivity of Pm to the change in pH, which originates from how the microenvironment affects the titration of the membrane-associated permeant (check Supporting Information section 3.3 for details). Using the revised model (eq. S10) based on eq. 2, we reassessed the experimental PAMPA values. The best fit (Figure 4B, red solid line) gives a of 7.63 and an n of 0.99. The PAMPA agrees with the “hybrid” prediction, but an n close to 1 indicates little anticooperativity, which can be attributed to the high ionic strength in the experiment. The new is 80 times smaller than the value (0.06) overestimated by the original model (eq. S9) based on eq. 1 and is 4.5-fold (3.5 times larger than) the “hybrid” value (−2.50), potentially due to the different composition in the PAMPA membrane.28, 112 Note that we cannot predict from the liposome measures at single pH using eq. 2, but we feel the pH-dependent profile should also have a close to 7 as well as an n close to 1 under 0.2 M ionic strength, leading to an estimated around −2.
CONCLUDING DISCUSSION
We have employed CpHMD simulations combined with US free energy surface analysis to study the ionization-coupled permeation of the drug molecule PPL through a POPC lipid bilayer. Two fixed ionization states, PPL+ and PPL0, were also simulated for comparison. With dynamic protonation included, CpHMD largely reproduced the permeation path with the lowest free energy (Figure 1B) as delineated by jointly considering PPL+ and PPL0.92, 95–98 Contrary to what is commonly assumed, the permeation pathway indicates PPL ionizes inside membrane rather than at the surface, which was confirmed by the ΔZCOM-dependent pKa values (Figure 2C). The pKa values, either calculated from the permeation PMFs or predicted by pH-REX CpHMD, are in line with the experimental values inferred from logP values (Table S2). As the molecule partitions into the lipid bilayer, the ΔpKa monotonically increases due to the desolvation effect (Figures 2D and S6A), in agreement with previous fixed-ionization-state55, 93 and CpHMD69–71, 74 simulations. Notably, the pKa reaches 7 (the aqueous pH) in the neighborhood of the carbonyl and glycerol moieties of the lipids (Figures 2B,C). These results are consistent with the partitioning experiments showing that amphiphilic molecules migrate into membrane as ionized species but permeate in the neutral form,23–31, 99 and lend support to the surface ion pair model8 as well as the “pH piston hypothesis”, which states that ionizable molecules associate with either the zwitterionic headgroups or glycerols/lipid tails depending on their ionization states.29 These findings do not necessarily override the pH-partition hypothesis (PPL still permeates through the lipid tail region in its neutral form) but instead point to an essential complementary understanding (that dynamic neutralization increases the permeable molecules to include some proportion of those charged in solution).
Collectively, this work demonstrates the importance of considering dynamic protonation in permeation studies. With dynamic protonation, CpHMD was able to predict passive diffusion Pm within a factor of ~1–3 of the experimental measurements88–89 (Table S3). Following the lowest free energy permeation PMF from the fixed-ionization-state PMFs, which was validated by CpHMD simulations, we were able to delineate Pm as a function of pH. The resulting pH-dependent Pm values qualitatively agree with those determined by PAMPA,110 with some expected deviation due to the different PAMPA membrane composition.28, 112 Importantly, we found the pH-dependent Pm profile cannot be described by the conventional model (eq. 1). By assuming that dynamic ionization does not occur at the membrane surface, the conventional model underestimates the number of permeable molecules, which results in an overestimated based on the experimentally measured . To better describe membrane partitioning and permeation of ionizable molecules, we modified eq. 1, adding the Hill coefficient n to reflect the cooperativity/anticooperativity in the proton-coupled permeation and replacing by to reflect the molecule’s ionizability inside the membrane (eq. 2). This modified model helps explain violations of the pH-partition hypothesis. For example, Regev et al. found that the trans-liposome permeation of doxorubicin increased only 2-fold from pH 7.4 to 9.7, while eq. 1 predicted an increase of 68-fold.22 Such a deviation is generally treated as a clue of charged species permeation. However, eq. 2 estimates an increase of ~1.4 with of 7 and n of 1 (0.2 M ionic strength), suggesting that this “pseudo-violation” originates from the assumption that only neutral species in solution permeate.
Note that our findings should be generalized with a degree of caution. Weak acids are also likely to bind the POPC bilayer as charged species, neutralize (protonate) before diffusing through lipid tails, and reionize (deprotonate) upon leaving lipid tails in the opposite leaflet. So the surface ion pair model8 and the “pH piston hypothesis”29 should still apply. However, slight deviations are anticipated, given the association of acids with the positively charged trimethylammonium groups at the POPC-water interface, as opposed to the negatively charged and more buried phosphates to which weak bases associate. In the permeation direction, a continual increase of acid pKa is expected, and the protonation occurs where the pKa upshifts to 7 (aqueous pH). This has been demonstrated for acidic amino acids using the fixed-ionization-state55 and DpHMD simulations.69–71 Given that only neutral species diffuse through lipid tails, acid plateaus at low pH values and then decreases at high pH values.8, 112–114 Yet an overestimated by the conventional model (eqs. 1 and S9) is also anticipated by ignoring the membrane-associated charged species. Ampholytes (with at least one acidic and one basic group) are interesting cases because they may adopt three or more charge states. However, how an ampholyte adjusts its charge state during passive permeation cannot be readily deduced from the current work and may be the focus of future work. In the case of a strong acid with very low or a base with very high , its pKa inside the membrane may not be adequately shifted for it to neutralize. For instance, the p ^ a of arginine reduces from 12.5 in water to ~7 at the center of bilayer,55, 115–118 indicating that arginine could remain ionized inside membrane. Also, the permeation may be facilitated via transient water pores,119 which can stabilize the ionized species and reduce the permeation barrier for a charged molecule.93, 118, 120–121 For weak acids or bases, however, our findings suggest that they are likely to permeate biomembranes as neutral species, which is consistent with other computational studies55, 69, 71, 74, 93, 116 and explains why human drugs typically have a in the range of 3.5−10.5.6 Also note that this study was conducted using a pure bilayer with a single drug. Introducing ionizable membrane “impurities” may charge the bilayer and affect drug permeation. It has been demonstrated that the presence of anionic lipids or fatty acids increases the partition (Table S2) and permeability (Table S4) of PPL in PC liposomes,26, 28, 30, 114, 122 which is not surprising given the strong electrostatic attraction between PPL+ and the negatively charged membranes. If multiple PPLs were present in close proximity, their ionizations are likely to be coupled in an anticooperative manner due to the great electrostatic repulsion between PPL+s inside low-dielectric membrane. Smaller pKa and the Hill coefficient are thus expected, which have been captured in the titration of the fatty acid76–77 and polysaccharide78 aggregates by CpHMD. However, how multiple drugs would affect the MD-based Pm prediction is not clear at this stage since the impact may be nonadditive.123
As compared to the fixed-ionization-state simulations, pHMD not only offers a more realistic scenario via real-time update of charge states during permeation, but also is computationally more efficient, especially in the presence of ampholytic and/or multiple permeants whose charge states inside membranes may vary in an unpredictable way. There are, however, a few limitations of using pHMD in the permeation studies, such as the absence of explicit proton transfer because pHMD models the relative deprotonation free energy or ΔpKa of an ionizable site relative to that in bulk. In reality, when deprotonation occurs inside membranes, an excess proton will be accepted by the surrounding water molecules and transported via Grotthuss shuttling124 to bulk water or to an ionizable headgroup. However, the only way in which this process would alter the drug’s permeation free energy profile is if the protonated waters or lipid headgroup were attractive, long-lived, and in close enough proximity to shift the drug’s local stability in a kinetically significant way (i.e., to interact strongly with and thereby slow the drug’s permeation). This is unlikely in the presented scenario given the weak interaction expected between the neutral drug and such protonated groups. On the basis of our previous work on carbon nanotubes, a good mimic of permeation pathway inside membranes, using the multiscale reactive molecular dynamics (MS-RMD) simulations, an excess proton (H3O+ delocalized over multiple waters) inside a hydrophobic environment is highly energetically unfavorable.125 Given ample waters connecting the titrating drug and bulk water (Figures S6B-D), the release of H3O+ to bulk should be fast. Thus, we estimated Pm on the basis of the assumption that the titration is fast relative to permeation. A more rigorous assessment can be done by adding explicit proton transfer using the MS-RMD technique developed in our group,126–130 which may be the focus of future work. Because the phosphate groups of POPC lipids can occasionally ion-pair with the titrating nitrogen atom of PPL+ (Figure S2), it will be interesting to see how these moieties are involved in the excess proton’s release to bulk water. Turning on titration for lipids131–132 and the drug-bound waters133 in pHMD will be an additional way to explore their local stability. For the present work, above pH 4 (as in Figure 4A), the POPC lipids remain zwitterionic,25, 134 limiting their influence to an intermediate during proton release to bulk water.
Another limitation of pHMD is that it ignores the role of altered pH in local microenvironments, such as at the membrane surface.135–140 Turning to the hybrid-solvent CpHMD method applied in this work, we note that it systematically overestimates the ΔpKa inside membranes, especially near the center of membrane (Figure 2C), due to the underestimated desolvation of the underlying GB model.51, 75 However, on the basis of the previous CpHMD studies,51–52, 54, 75 we believe the direction of the ΔpKa is robust. In addition, hybrid-solvent CpHMD neglects any explicit interactions with ions.75 Although the current work is free of ions and not influenced by them, hybrid-solvent CpHMD may be less suitable to study the ion pair permeation.141 Other potential limitations, as in all MD-based permeation studies, are the need to include bilayer asymmetry, membrane undulations, and polarizability. The simulations in this study on a symmetric bilayer focused sampling on one-half of the system (one leaflet) and generated the permeation profile for the exiting leaflet by mirroring those in the entering leaflet (eq. S4). Given the symmetric lipid distribution and converged sampling (Figure S16), the permeation profiles should be symmetric.142 The reaction coordinate in this study (ΔZCOM) was defined on the basis of all lipid molecules, which might lead to size-dependent permeation PMFs due to the artifact from membrane undulations.109 However, given the system size chosen in this work (26 lipids per leaflet), undulation should be insignificant, and the resulting PMFs should be relatively size insensitive.42 Molecules experience significant change in dielectric environment during permeation,8 so the description by polarizable force fields may be more physically realistic. Yet using nonpolarizable force fields should not impair the major findings of this work, that is, dynamic protonation should be considered when investigating facile membrane permeation of ionizable molecules, which is also supported by a study including the polarization effects.96
In summary, we have applied CpHMD, where protonation and conformation equilibria are dynamically coupled, to study the permeation of an ionizable drug PPL through a POPC lipid bilayer. We find that PPL migrates into the bilayer in the charged form and deprotonates at the hydrophobic boundary, and that by involving dynamic protonation the Pm was predicted with high accuracy. What is commonly assumed in the permeation studies is that an ionizable molecule must neutralize in solution before membrane partitioning. As such, the amount of the “effective” permeant is underestimated, leading to an overestimated intrinsic Pm. We have also presented a revised model to better describe the pH-dependent partitioning and permeation. While the generality of our findings awaits further testing on a larger data set, these findings demonstrate how adding dynamic protonation can advance our understanding of the permeation of ionizable pharmaceuticals and drug-like molecules and hence improve the accuracy of MD-based Pm prediction. Last, this work demonstrates the utility of CpHMD in permeation studies.
METHODS
Unless otherwise stated, simulations were conducted and analyzed using the CHARMM package143 (version c42b2). Lipids and waters were represented by CHARMM36144 and CHARMM modified TIP3P model,145 respectively. PPL was parametrized by CHARMM General Force Field146 (CGenFF, version 3.0.1) using ParamChem147–148 (version 1.0.0). The CpHMD parameters for PPL were derived using the protocol described by Lee et al.64 A PPL0 molecule was inserted in an explicit POPC bilayer with a 15-Å water layer added to the top and bottom. No explicit ions were added. The system was equilibrated in three stages, on the basis of the protocol previously established.85 In stage 1, after energy minimization, a 500 ps restrained simulation was performed to relax the system, following the CHARMM-GUI protocol.149–150 In stage 2, the POPC bilayer was equilibrated for ~100 ns using the GROMACS package151 (version 5.1.4). In stage 3, 21 replicated systems were generated with PPL restrained at various locations along the membrane normal (Z-axis) and equilibrated for 26 ns with titration enabled on PPL at pH 7 using the membrane-enabled hybrid-solvent CpHMD method75, 83 implemented in CHARMM. Following the equilibration, production runs were performed at 310 K and 1 atm using the US method.86 The equilibrated 21 configurations were further branched into 60 systems with PPL positioned from 0 to 29.5 Å with an interval of 0.5 Å. The reaction coordinate (ΔZCOM) was chosen as the Z-component of the COM distance between the PPL and POPC heavy atoms. US simulations were performed for PPL+, PPL0, and CpHMD at pH 7. All US windows were run for 70.5 ns. Detailed simulation protocols and additional analyses are given in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Cheng-Chieh Tsai and Fang-Yu Lin of the University of Maryland School of Pharmacy for their advice on compound force field parametrization. The personnel were supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH Grant R01 GM053148). The computational resources were provided by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant number ACI-1053575, and the University of Chicago Research Computing Center (RCC).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b04387. Details of the computational protocols and additional analyses.
REFERENCES
- (1).van Meer G; Voelker DR; Feigenson GW Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol 2008, 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev 1997, 23, 3–26. [DOI] [PubMed] [Google Scholar]
- (3).Hilgers AR; Conradi RA; Burton PS Caco-2 Cell Monolayers as a Model for Drug Transport Across the Intestinal Mucosa. Pharm. Res 1990, 07, 902–910. [DOI] [PubMed] [Google Scholar]
- (4).Irvine JD; Takahashi L; Lockhart K; Cheong J; Tolan JW; Selick HE; Grove JR MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci 1999, 88, 28–33. [DOI] [PubMed] [Google Scholar]
- (5).Kansy M; Senner F; Gubernator K Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem 1998, 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- (6).Manallack DT The pKa Distribution of Drugs: Application to Drug Discovery. Perspect. Med. Chem 2007, 1, 25–38. [PMC free article] [PubMed] [Google Scholar]
- (7).Manallack DT; Prankerd RJ; Nassta GC; Ursu O; Oprea TI; Chalmers DK A chemogenomic analysis of ionization constants--implications for drug discovery. ChemMedChem 2013, 8, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Avdeef A Physicochemical profiling (solubility, permeability and charge state). Curr. Top. Med. Chem 2001, 1, 277–351. [DOI] [PubMed] [Google Scholar]
- (9).Manallack DT; Prankerd RJ; Yuriev E; Oprea TI; Chalmers DK The significance of acid/base properties in drug discovery. Chem. Soc. Rev 2013, 42, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Charifson PS; Walters WP Acidic and basic drugs in medicinal chemistry: a perspective. J. Med. Chem 2014, 57, 9701–9717. [DOI] [PubMed] [Google Scholar]
- (11).Shore PA; Brodie BB; Hogben CA The gastric secretion of drugs: a pH partition hypothesis. J. Pharmacol. Exp. Ther 1957, 119, 361–369. [PubMed] [Google Scholar]
- (12).Schanker LS; Tocco DJ; Brodie BB; Hogben CA Absorption of drugs from the rat small intestine. J. Pharmacol. Exp. Ther 1958, 123, 81–88. [PubMed] [Google Scholar]
- (13).Langguth P; Kubis A; Krumbiegel G; Lang W; Merkle HP; Wachter W; Spahn-Langguth H; Weyhenmeyer R Intestinal absorption of the quaternary trospium chloride: permeability-lowering factors and bioavailabilities for oral dosage forms. Eur. J. Pharm. Biopharm 1997, 43, 265–272. [Google Scholar]
- (14).Chen Y; Pant AC; Simon SM P-Glycoprotein Does Not Reduce Substrate Concentration from the Extracellular Leaflet of the Plasma Membrane in Living Cells. Cancer Res. 2001, 61, 7763–7769. [PubMed] [Google Scholar]
- (15).Adson A; Raub TJ; Burton PS; Barsuhn CL; Hilgers AR; Audus KL; Ho NF Quantitative approaches to delineate paracellular diffusion in cultured epithelial cell monolayers. J. Pharm. Sci 1994, 83, 1529–1536. [DOI] [PubMed] [Google Scholar]
- (16).Adson A; Burton PS; Raub TJ; Barsuhn CL; Audus KL; Ho NFH Passive Diffusion of Weak Organic Electrolytes across Caco-2 Cell Monolayers: Uncoupling the Contributions of Hydrodynamic, Transcellular, and Paracellular Barriers. J. Pharm. Sci 1995, 84, 1197–1204. [DOI] [PubMed] [Google Scholar]
- (17).Pade V; Stavchansky S Estimation of the Relative Contribution of the Transcellular and Paracellular Pathway to the Transport of Passively Absorbed Drugs in the Caco-2 Cell Culture Model. Pharm. Res 1997, 14, 1210–1215. [DOI] [PubMed] [Google Scholar]
- (18).Palm K; Luthman K; Ros J; Grasjo J; Artursson P Effect of molecular charge on intestinal epithelial drug transport: pH-dependent transport of cationic drugs. J. Pharmacol. Exp. Ther 1999, 291, 435–443. [PubMed] [Google Scholar]
- (19).Artursson P; Palm K; Luthman K Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Delivery Rev 2001, 46, 27–43. [DOI] [PubMed] [Google Scholar]
- (20).Nagahara N; Tavelin S; Artursson P Contribution of the paracellular route to the pH-dependent epithelial permeability to cationic drugs. J. Pharm. Sci 2004, 93, 2972–2984. [DOI] [PubMed] [Google Scholar]
- (21).Fischer H; Kansy M; Avdeef A; Senner F Permeation of permanently positive charged molecules through artificial membranes—influence of physico-chemical properties. Eur. J. Pharm. Sci 2007, 31, 32–42. [DOI] [PubMed] [Google Scholar]
- (22).Regev R; Eytan GD Flip-Flop of Doxorubicin across Erythrocyte and Lipid Membranes. Biochem. Pharmacol 1997, 54, 1151–1158. [DOI] [PubMed] [Google Scholar]
- (23).Boulanger Y; Schreier S; Leitch LC; Smith ICP Multiple binding sites for local anesthetics in membranes: characterization of the sites and their equilibria by deuterium NMR of specifically deuterated procaine and tetracaine. Can. J. Biochem 1980, 58, 986–995. [DOI] [PubMed] [Google Scholar]
- (24).Boulanger Y; Schreier S; Smith ICP Molecular details of anesthetic-lipid interaction as seen by deuterium and phosphorus-31 nuclear magnetic resonance. Biochemistry 1981, 20, 6824–6830. [DOI] [PubMed] [Google Scholar]
- (25).Pauletti GM; Wunderli-Allenspach H Partition coefficients in vitro: artificial membranes as a standardized distribution model. Eur. J. Pharm. Sci 1994, 1, 273–282. [Google Scholar]
- (26).Krämer SD; Wunderli-Allenspach H The pH-Dependence in the Partitioning Behaviour of (RS)-[3H]Propranolol Between MDCK Cell Lipid Vesicles and Buffer. Pharm. Res 1996, 13, 1851–1855. [DOI] [PubMed] [Google Scholar]
- (27).Ottiger C; Wunderli-Allenspach H Partition behaviour of acids and bases in a phosphatidylcholine liposome-buffer equilibrium dialysis system. Eur. J. Pharm. Sci 1997, 5, 223–231. [Google Scholar]
- (28).Krämer SD; Jakits-Deiser C; Wunderli-Allenspach H Free Fatty Acids Cause pH-Dependent Changes in Drug-Lipid Membrane Interactions Around Physiological pH. Pharm. Res 1997, 14, 827–832. [DOI] [PubMed] [Google Scholar]
- (29).Avdeef A; Box KJ; Comer JEA; Hibbert C; Tam KY pH-Metric logP 10. Determination of Liposomal Membrane-Water Partition Coefficients of lonizable Drugs. Pharm. Res 1998, 15, 209–215. [DOI] [PubMed] [Google Scholar]
- (30).Krämer SD; Braun A; Jakits-Deiser C; Wunderli-Allenspach H Towards the predictability of drug-lipid membrane interactions: the pH-dependent affinity of propanolol to phosphatidylinositol containing liposomes. Pharm. Res 1998, 15, 739–744. [DOI] [PubMed] [Google Scholar]
- (31).Ottiger C; Wunderli-Allenspach H Immobilized Artificial Membrane (lAM)-HPLC for Partition Studies of Neutral and Ionized Acids and Bases in Comparison with the Liposomal Partition System. Pharm. Res 1999, 16, 643–650. [DOI] [PubMed] [Google Scholar]
- (32).Först G; Cwiklik L; Jurkiewicz P; Schubert R; Hof M Interactions of beta-blockers with model lipid membranes: molecular view of the interaction of acebutolol, oxprenolol, and propranolol with phosphatidylcholine vesicles by time-dependent fluorescence shift and molecular dynamics simulations. Eur. J. Pharm. Biopharm 2014, 87, 559–569. [DOI] [PubMed] [Google Scholar]
- (33).Paloncýová M; DeVane R; Murch B; Berka K; Otyepka M Amphiphilic drug-like molecules accumulate in a membrane below the head group region. J. Phys. Chem. B 2014, 118, 1030–1039. [DOI] [PubMed] [Google Scholar]
- (34).Marrink S-J; Berendsen HJC Simulation of water transport through a lipid membrane. J. Phys. Chem 1994, 98, 4155–4168. [Google Scholar]
- (35).Herbette L; Katz AM; Sturtevant JM Comparisons of the interaction of propranolol and timolol with model and biological membrane systems. Mol. Pharmacol 1983, 24, 259–269. [PubMed] [Google Scholar]
- (36).Herbette L; Trumbore M; Chester D; Katz A Possible molecular basis for the pharmacokinetics and pharmacodynamics of three membrane-active drugs: Propranolol, nimodipine and amiodarone. J. Mol. Cell. Cardiol 1988, 20, 373–378. [DOI] [PubMed] [Google Scholar]
- (37).Xiang TX; Anderson BD Liposomal drug transport: a molecular perspective from molecular dynamics simulations in lipid bilayers. Adv. Drug Delivery Rev 2006, 58, 1357–1378. [DOI] [PubMed] [Google Scholar]
- (38).Awoonor-Williams E; Rowley CN Molecular simulation of nonfacilitated membrane permeation. Biochim. Biophys. Acta, Biomembr 2016, 1858, 1672–1687. [DOI] [PubMed] [Google Scholar]
- (39).Shinoda W Permeability across lipid membranes. Biochim. Biophys. Acta, Biomembr 2016, 1858, 2254–2265. [DOI] [PubMed] [Google Scholar]
- (40).Lopes D; Jakobtorweihen S; Nunes C; Sarmento B; Reis S Shedding light on the puzzle of drug-membrane interactions: Experimental techniques and molecular dynamics simulations. Prog. Lipid Res 2017, 65, 24–44. [DOI] [PubMed] [Google Scholar]
- (41).Venable RM; Krämer A; Pastor RW Molecular Dynamics Simulations of Membrane Permeability. Chem. Rev 2019, 119, 5954–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Comer J; Schulten K; Chipot C Calculation of Lipid-Bilayer Permeabilities Using an Average Force. J. Chem. Theory Comput 2014, 10, 554–564. [DOI] [PubMed] [Google Scholar]
- (43).Comer J; Schulten K; Chipot C Diffusive Models of Membrane Permeation with Explicit Orientational Freedom. J. Chem. Theory Comput 2014, 10, 2710–2718. [DOI] [PubMed] [Google Scholar]
- (44).Chipot C; Comer J Subdiffusion in Membrane Permeation of Small Molecules. Sci. Rep 2016, 6, 35913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lee CT; Comer J; Herndon C; Leung N; Pavlova A; Swift RV; Tung C; Rowley CN; Amaro RE; Chipot C; Wang Y; Gumbart JC Simulation-Based Approaches for Determining Membrane Permeability of Small Compounds. J. Chem. Inf. Model 2016, 56, 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Sun R; Dama JF; Tan JS; Rose JP; Voth GA Transition-Tempered Metadynamics Is a Promising Tool for Studying the Permeation of Drug-like Molecules through Membranes. J. Chem. Theory. Comput 2016, 12, 5157–5169. [DOI] [PubMed] [Google Scholar]
- (47).Sun R; Han Y; Swanson JMJ; Tan JS; Rose JP; Voth GA Molecular transport through membranes: Accurate permeability coefficients from multidimensional potentials of mean force and local diffusion constants. J. Chem. Phys 2018, 149, 072310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Tse CH; Comer J; Wang Y; Chipot C Link between Membrane Composition and Permeability to Drugs. J. Chem. Theory Comput 2018, 14, 2895–2909. [DOI] [PubMed] [Google Scholar]
- (49).Aydin F; Sun R; Swanson JMJ Mycolactone Toxin Membrane Permeation: Atomistic versus Coarse-Grained MARTINI Simulations. Biophys. J 2019, 117, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Callenberg KM; Choudhary OP; de Forest GL; Gohara DW; Baker NA; Grabe M APBSmem: a graphical interface for electrostatic calculations at the membrane. PLoS One 2010, J, e12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wallace JA; Wang Y; Shi C; Pastoor KJ; Nguyen BL; Xia K; Shen JK Toward accurate prediction of pKa values for internal protein residues: the importance of conformational relaxation and desolvation energy. Proteins: Struct., Funct., Genet 2011, 79, 3364–3373. [DOI] [PubMed] [Google Scholar]
- (52).Shi C; Wallace JA; Shen JK Thermodynamic coupling of protonation and conformational equilibria in proteins: theory and simulation. Biophys. J 2012, 102, 1590–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Goh GB; Laricheva EN; Brooks CL III Uncovering pH-dependent transient states of proteins with buried ionizable residues. J. Am. Chem. Soc 2014, 136, 8496–8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Huang Y; Yue Z; Tsai CC; Henderson JA; Shen J Predicting Catalytic Proton Donors and Nucleophiles in Enzymes: How Adding Dynamics Helps Elucidate the Structure-Function Relationships. J. Phys. Chem. Lett 2018, 9, 1179–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).MacCallum JL; Bennett WF; Tieleman DP Distribution of amino acids in a lipid bilayer from computer simulations. Biophys. J 2008, 94, 3393–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Baptista AM; Teixeira VH; Soares CM Constant-pH molecular dynamics using stochastic titration. J. Chem. Phys 2002, 117, 4184–4200. [Google Scholar]
- (57).Metropolis N; Rosenbluth AW; Rosenbluth MN; Teller AH; Teller E Equation of State Calculations by Fast Computing Machines. J. Chem. Phys 1953, 21, 1087–1092. [Google Scholar]
- (58).Mongan J; Case DA; McCammon JA Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem 2004, 2J, 2038–2048. [DOI] [PubMed] [Google Scholar]
- (59).Machuqueiro M; Baptista AM Constant-pH molecular dynamics with ionic strength effects: protonationconformation coupling in decalysine. J. Phys. Chem. B 2006, 110, 2927–2933. [DOI] [PubMed] [Google Scholar]
- (60).Machuqueiro M; Baptista AM Acidic range titration of HEWL using a constant-pH molecular dynamics method. Proteins: Struct., Funct., Genet 2008, 72, 289–98. [DOI] [PubMed] [Google Scholar]
- (61).Swails JM; York DM; Roitberg AE Constant pH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation. J. Chem. Theory Comput 2014, 10, 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Chen Y; Roux B Constant-pH Hybrid Nonequilibrium Molecular Dynamics-Monte Carlo Simulation Method. J. Chem. Theory Comput 2015, 11, 3919–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mertz JE; Pettitt BM Molecular Dynamics At a Constant pH. Int. J. Supercomput. Appl. High Perform. Comput 1994, 8, 47–53. [Google Scholar]
- (64).Lee MS; Salsbury FR Jr.; Brooks CL III Constant-pH molecular dynamics using continuous titration coordinates. Proteins: Struct., Funct., Genet 2004, J6, 738–752. [DOI] [PubMed] [Google Scholar]
- (65).Khandogin J; Brooks CL III Constant pH molecular dynamics with proton tautomerism. Biophys. J 2005, 89, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Kong X; Brooks CL III X-dynamics: A new approach to free energy calculations. J. Chem. Phys 1996, 10J, 2414–2423. [Google Scholar]
- (67).Mongan J; Case DA Biomolecular simulations at constant pH. Curr. Opin. Struct. Biol 2005, 1J, 157–163. [DOI] [PubMed] [Google Scholar]
- (68).Chen W; Morrow BH; Shi C; Shen JK Recent development and application of constant pH molecular dynamics. Mol. Simul 2014, 40, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Teixeira VH; Vila-Vifosa D; Reis PB; Machuqueiro M pKa Values of Titrable Amino Acids at the Water/Membrane Interface. J. Chem. Theory Comput 2016, 12, 930–934. [DOI] [PubMed] [Google Scholar]
- (70).Vila-Viçosa D; Silva TFD; Slaybaugh G; Reshetnyak YK; Andreev OA; Machuqueiro M Membrane-Induced pKa Shifts in wt-pHLIP and Its L16H Variant. J. Chem. Theory Comput 2018, 14, 3289–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Radak BK; Chipot C; Suh D; Jo S; Jiang W; Phillips JC; Schulten K; Roux B Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. J. Chem. Theory Comput 2017, 13, 5933–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Laricheva EN; Arora K; Knight JL; Brooks CL III Deconstructing activation events in rhodopsin. J. Am. Chem. Soc 2013, 13J, 10906–10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Goh GB; Hulbert BS; Zhou H; Brooks CL III Constant pH molecular dynamics of proteins in explicit solvent with proton tautomerism. Proteins: Struct., Funct., Genet 2014, 82, 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Panahi A; Brooks CL III Membrane environment modulates the pKa values of transmembrane helices. J. Phys. Chem. B 2015, 119, 4601–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Wallace JA; Shen JK Continuous Constant pH Molecular Dynamics in Explicit Solvent with pH-Based Replica Exchange. J. Chem. Theory Comput 2011, 7, 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Morrow BH; Koenig PH; Shen JK Atomistic simulations of pH-dependent self-assembly of micelle and bilayer from fatty acids. J. Chem. Phys 2012, 137, 194902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Morrow BH; Koenig PH; Shen JK Self-assembly and bilayer-micelle transition of fatty acids studied by replica-exchange constant pH molecular dynamics. Langmuir 2013, 29, 14823–14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Morrow BH; Payne GF; Shen J pH-Responsive Self-Assembly of Polysaccharide through a Rugged Energy Landscape. J. Am. Chem. Soc 2015, 137, 13024–13030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Ellis CR; Shen J pH-Dependent Population Shift Regulates BACE1 Activity and Inhibition. J. Am. Chem. Soc 2015, 137, 9543–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Ellis CR; Tsai CC; Hou X; Shen J Constant pH Molecular Dynamics Reveals pH-Modulated Binding of Two Small-Molecule BACE1 Inhibitors. J. Phys. Chem. Lett 2016, 7, 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Harris RC; Tsai CC; Ellis CR; Shen J Proton-Coupled Conformational Allostery Modulates the Inhibitor Selectivity for p-Secretase. J. Phys. Chem. Lett 2017, 8, 4832–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Yue Z; Shen J pH-Dependent cooperativity and existence of a dry molten globule in the folding of a miniprotein BBL. Phys. Chem. Chem. Phys 2018, 20, 3523–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Huang Y; Chen W; Dotson DL; Beckstein O; Shen J Mechanism of pH-dependent activation of the sodium-proton antiporter NhaA. Nat. Commun 2016, 7, 12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Chen W; Huang Y; Shen J Conformational Activation of a Transmembrane Proton Channel from Constant pH Molecular Dynamics. J. Phys. Chem. Lett 2016, 7, 3961–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Yue Z; Chen W; Zgurskaya HI; Shen J Constant pH Molecular Dynamics Reveals How Proton Release Drives the Conformational Transition of a Transmembrane Efflux Pump. J. Chem. Theory Comput 2017, 13, 6405–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Torrie GM; Valleau JP Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys 1977, 23, 187–199. [Google Scholar]
- (87).WHO. Model List of Essential Medicines, 20th ed.; The World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- (88).Eyer K; Paech F; Schuler F; Kuhn P; Kissner R; Belli S; Dittrich PS; Kramer SD A liposomal fluorescence assay to study permeation kinetics of drug-like weak bases across the lipid bilayer. J. Controlled Release 2014, 173, 102–109. [DOI] [PubMed] [Google Scholar]
- (89).Hermann KF; Neuhaus CS; Micallef V; Wagner B; Hatibovic M; Aschmann HE; Paech F; Alvarez-Sanchez R; Kramer SD; Belli S Kinetics of lipid bilayer permeation of a series of ionisable drugs and their correlation with human transporter-independent intestinal permeability. Eur. J. Pharm. Sci 2017, 104, 150–161. [DOI] [PubMed] [Google Scholar]
- (90).Dickson CJ; Hornak V; Pearlstein RA; Duca JS Structure-Kinetic Relationships of Passive Membrane Permeation from Multiscale Modeling. J. Am. Chem. Soc 2017, 139, 442–452. [DOI] [PubMed] [Google Scholar]
- (91).Avdeef A pH-Metric log P. II: Refinement of Partition Coefficients and Ionization Constants of Multiprotic Substances. J. Pharm. Sci 1993, 82, 183–190. [DOI] [PubMed] [Google Scholar]
- (92).Ulander J; Haymet ADJ Permeation Across Hydrated DPPC Lipid Bilayers: Simulation of the Titrable Amphiphilic Drug Valproic Acid. Biophys. J 2003, 85, 3475–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Johansson AC; Lindahl E Titratable amino acid solvation in lipid membranes as a function of protonation state. J. Phys. Chem. B 2009, 113, 245–253. [DOI] [PubMed] [Google Scholar]
- (94).Boggara MB; Krishnamoorti R Partitioning of nonsteroidal antiinflammatory drugs in lipid membranes: a molecular dynamics simulation study. Biophys. J 2010, 98, 586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Bonhenry D; Tarek M; Dehez F Effects of Phospholipid Composition on the Transfer of a Small Cationic Peptide Across a Model Biological Membrane. J. Chem. Theory Comput 2013, 9, 5675–5684. [DOI] [PubMed] [Google Scholar]
- (96).Zhu Q; Lu Y; He X; Liu T; Chen H; Wang F; Zheng D; Dong H; Ma J Entropy and Polarity Control the Partition and Transportation of Drug-like Molecules in Biological Membrane. Sci. Rep 2017, 7, 17749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Bonhenry D; Dehez F; Tarek M Effects of hydration on the protonation state of a lysine analog crossing a phospholipid bilayer — insights from molecular dynamics and free-energy calculations. Phys. Chem. Chem. Phys 2018, 20, 9101–9107. [DOI] [PubMed] [Google Scholar]
- (98).Škulj S; Vazdar M Calculation of apparent pKa values of saturated fatty acids with different lengths in DOPC phospholipid bilayers. Phys. Chem. Chem. Phys 2019, 21, 10052–10060. [DOI] [PubMed] [Google Scholar]
- (99).Åkerman KE; Jårvisalo JO Effect of propranolol and related drugs on transmembraneous pH differences in liposomes. Acta Pharmacol. Toxicol 1977, 40, 497–504. [DOI] [PubMed] [Google Scholar]
- (100).Kučerka N; Nagle JF; Sachs JN; Feller SE; Pencer J; Jackson A; Katsaras J Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophys. J 2008, 95, 2356–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Kučerka N; Gallová J; Uhríková D; Balgavý P; Bulacu M; Marrink SJ; Katsaras J Areas of monounsaturated diacylphosphatidylcholines. Biophys. J 2009, 97, 1926–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Onufriev A; Case DA; Ullmann GM A novel view of pH titration in biomolecules. Biochemistry 2001, 40, 3413–3419. [DOI] [PubMed] [Google Scholar]
- (103).Miyazaki J; Hideg K; Marsh D Interfacial ionization and partitioning of membrane-bound local anaesthetics. Biochim. Biophys. Acta, Biomembr 1992, 1103, 62–68. [DOI] [PubMed] [Google Scholar]
- (104).Avdeef A; Comer JEA; Thomson SJ pH-Metric log P. 3. Glass electrode calibration in methanol-water, applied to pKa determination of water-insoluble substances. Anal. Chem 1993, 65, 42–49. [Google Scholar]
- (105).Austin RP; Barton P; Davis AM; Manners CN; Stansfield MC The effect of ionic strength on liposome-buffer and 1-octanol-buffer distribution coefficients. J. Pharm. Sci 1998, 87, 599–607. [DOI] [PubMed] [Google Scholar]
- (106).Khandogin J; Brooks CL III Toward the accurate first-principles prediction of ionization equilibria in proteins. Biochemistry 2006, 45, 9363–9373. [DOI] [PubMed] [Google Scholar]
- (107).Badaoui M; Kells A; Molteni C; Dickson CJ; Hornak V; Rosta E Calculating Kinetic Rates and Membrane Permeability from Biased Simulations. J. Phys. Chem. B 2018, 11571–11578. [DOI] [PubMed] [Google Scholar]
- (108).Bäeuerle HD; Seelig J Interaction of charged and uncharged calcium channel antagonists with phospholipid membranes. Binding equilibrium, binding enthalpy, and membrane location. Biochemistry 1991, 30, 7203–7211. [DOI] [PubMed] [Google Scholar]
- (109).Neale C; Pomes R Sampling errors in free energy simulations of small molecules in lipid bilayers. Biochim. Biophys. Acta, Biomembr 2016, 1858, 2539–2548. [DOI] [PubMed] [Google Scholar]
- (110).Avdeef A; Artursson P; Neuhoff S; Lazorova L; Grasjo J; Tavelin S Caco-2 permeability of weakly basic drugs predicted with the double-sink PAMPA pKa(flux) method. Eur. J. Pharm. Sci 2005, 24, 333–349. [DOI] [PubMed] [Google Scholar]
- (111).Sezer D; Oruf T Protonation Kinetics Compromise Liposomal Fluorescence Assay of Membrane Permeation. J. Phys. Chem. B 2017, 121, 5218–5227. [DOI] [PubMed] [Google Scholar]
- (112).Avdeef A; Tsinman O PAMPA--a drug absorption in vitro model 13. Chemical selectivity due to membrane hydrogen bonding: in combo comparisons of HDM-, DOPC-, and DS-PAMPA models. Eur. J. Pharm. Sci 2006, 28, 43–50. [DOI] [PubMed] [Google Scholar]
- (113).Avdeef A; Nielsen PE; Tsinman O PAMPA--a drug absorption in vitro model 11. Matching the in vivo unstirred water layer thickness by individual-well stirring in microtitre plates. Eur. J. Pharm. Sci 2004, 22, 365–374. [DOI] [PubMed] [Google Scholar]
- (114).Avdeef A Absorption and Drug Development: Solubility, Permeability, and Charge State, 2nd ed.; John Wiley & Sons: Hoboken, NJ, 2012. [Google Scholar]
- (115).Li L; Vorobyov I; MacKerell AD Jr.; Allen TW Is arginine charged in a membrane? Biophys. J 2008, 94, L11–L13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Li L; Vorobyov I; Allen TW Potential of mean force and pKa profile calculation for a lipid membrane-exposed arginine side chain. J. Phys. Chem. B 2008, 112, 9574–9587. [DOI] [PubMed] [Google Scholar]
- (117).Yoo J; Cui Q Does arginine remain protonated in the lipid membrane? Insights from microscopic pKa calculations. Biophys. J 2008, 94, L61–L63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Li L; Vorobyov I; Allen TW The different interactions of lysine and arginine side chains with lipid membranes. J. Phys. Chem. B 2013, 117, 11906–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Volkov AG; Paula S; Deamer DW Two mechanisms of permeation of small neutral molecules and hydrated ions across phospholipid bilayers. Bioelectrochem. Bioenerg 1997, 42, 153–160. [Google Scholar]
- (120).Dorairaj S; Allen TW On the thermodynamic stability of a charged arginine side chain in a transmembrane helix. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 4943–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Wang Y; Hu D; Wei D Transmembrane Permeation Mechanism of Charged Methyl Guanidine. J. Chem. Theory Comput 2014, 10, 1717–1726. [DOI] [PubMed] [Google Scholar]
- (122).Surewicz WK; Leyko W Interaction of propranolol with model phospholipid membranes Monolayer, spin label and fluorescence spectroscopy studies. Biochim. Biophys. Acta, Biomembr 1981, 643, 387–397. [DOI] [PubMed] [Google Scholar]
- (123).MacCallum JL; Bennett WF; Tieleman DP Transfer of arginine into lipid bilayers is nonadditive. Biophys. J 2011, 101, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Agmon N The Grotthuss mechanism. Chem. Phys. Lett 1995, 244, 456–462. [Google Scholar]
- (125).Peng Y; Swanson JM; Kang SG; Zhou R; Voth GA Hydrated Excess Protons Can Create Their Own Water Wires. J. Phys. Chem. B 2015, 119, 9212–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Swanson JM; Maupin CM; Chen H; Petersen MK; Xu J; Wu Y; Voth GA Proton solvation and transport in aqueous and biomolecular systems: insights from computer simulations. J. Phys. Chem. B 2007, 111, 4300–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Knight C; Lindberg GE; Voth GA Multiscale reactive molecular dynamics. J. Chem. Phys 2012, 137, 22A525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Nelson JG; Peng Y; Silverstein DW; Swanson JM Multiscale Reactive Molecular Dynamics for Absolute pKa Predictions and Amino Acid Deprotonation. J. Chem. Theory Comput 2014, 10, 2729–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Lee S; Liang R; Voth GA; Swanson JM Computationally Efficient Multiscale Reactive Molecular Dynamics to Describe Amino Acid Deprotonation in Proteins. J. Chem. Theory Comput 2016, 12, 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Chen C; Arntsen C; Voth GA Development of reactive force fields using ab initio molecular dynamics simulation minimally biased to experimental data. J. Chem. Phys 2017, 147, 161719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Santos HA; Vila-Vifosa D; Teixeira VH; Baptista AM; Machuqueiro M Constant-pH MD Simulations of DMPA/DMPC Lipid Bilayers. J. Chem. Theory Comput 2015, 11, 5973–5979. [DOI] [PubMed] [Google Scholar]
- (132).Vila-Viçosa D; Teixeira VH; Baptista AM; Machuqueiro M Constant-pH MD Simulations of an Oleic Acid Bilayer. J. Chem. Theory Comput 2015, 11, 2367–2376. [DOI] [PubMed] [Google Scholar]
- (133).Chen W; Wallace JA; Yue Z; Shen JK Introducing titratable water to all-atom molecular dynamics at constant pH. Biophys. J 2013, 105, L15–L17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Moncelli MR; Becucci L; Guidelli R The intrinsic pKa values for phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine in monolayers deposited on mercury electrodes. Biophys. J 1994, 66, 1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Amdursky N; Lin Y; Aho N; Groenhof G Exploring fast proton transfer events associated with lateral proton diffusion on the surface of membranes. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Brändén M; Sandén T; Brzezinski P; Widengren J Localized proton microcircuits at the biological membrane-water interface. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 19766–197770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Gennis RB Proton Dynamics at the Membrane Surface. Biophys. J 2016, 110, 1909–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Smondyrev AM; Voth GA Molecular dynamics simulation of proton transport near the surface of a phospholipid membrane. Biophys. J 2002, 82, 1460–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Xu L; Öjemyr LN; Bergstrand J; Brzezinski P; Widengren J Protonation Dynamics on Lipid Nanodiscs: Influence of the Membrane Surface Area and External Buffers. Biophys. J 2016, 110, 1993–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Yamashita T; Peng Y; Knight C; Voth GA Computationally Efficient Multiconfigurational Reactive Molecular Dynamics. J. Chem. Theory Comput 2012, 8, 4863–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Neubert R Ion Pair Transport Across Membranes. Pharm. Res 1989, 06, 743–747. [DOI] [PubMed] [Google Scholar]
- (142).Allen TW; Andersen OS; Roux B Ion permeation through a narrow channel: using gramicidin to ascertain all-atom molecular dynamics potential of mean force methodology and biomolecular force fields. Biophys. J 2006, 90, 3447–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Brooks BR; Brooks CL III; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M CHARMM: the biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Durell SR; Brooks BR; Ben-Naim A Solvent-Induced Forces between Two Hydrophilic Groups. J. Phys. Chem 1994, 98, 2198–2202. [Google Scholar]
- (146).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD Jr. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem 2009, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Vanommeslaeghe K; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 2012, 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Vanommeslaeghe K; Raman EP; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 2012, 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Jo S; Kim T; Im W Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One 2007, 2, e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Wu EL; Cheng X; Jo S; Rui H; Song KC; Dávila-Contreras EM; Qi Y; Lee J; Monje-Galvan V; Venable RM; Klauda JB; Im W CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem 2014, 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Abraham MJ; Murtola T; Schulz R; Pall S; Smith JC; Hess B; Lindahl E GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.