

Abstract
Performing selective transformations on complex substrates remains a challenge in synthetic chemistry. These difficulties often arise due to cross-reactivity, particularly in the presence of similar functional groups at multiple sites. Therefore, there is a premium on the ability to perform selective activation of these functional groups. We report here a supramolecular strategy where encapsulation of a hydrogenation catalyst enables selective olefin hydrogenation, even in the presence of multiple sites of unsaturation. While the reaction requires at least one sterically nondemanding alkene substituent, the rate of hydrogenation is not sensitive to the distance between the alkene and the functional group, including a carboxylate, on the other substituent. This observation indicates that only the double bond has to be encapsulated to effect hydrogenation. Going further, we demonstrate that this supramolecular strategy can overcome the inherent allylic alcohol selectivity of the free catalyst, achieving supra-molecular catalyst-directed regioselectivity as opposed to directing-group selectivity.
Supramolecular catalysis offers a unique means of achieving regioselectivity via three-dimensional control over steric and noncovalent interactions between substrate and the host cavity.1–6 This has been demonstrated in a number of instances ranging from organic to organometallic trans-formations.7–16 In one important example, supramolecular support of a metal-mediated hydroformylation reaction leads to a change in the selectivity of the transformation through the steric and secondary interactions achieved by a supramolecular scaffold (Figure 1a).17–21 Our group has also demonstrated the ability of a host-encapsulated rhodium (1) and ruthenium catalyst to perform isomerization of allylic alcohols to aldehydes (Figure 1b).22,23 Using rhodium, it was shown that the encapsulated organometallic complex exhibits divergent reactivity from that of the free species.
Figure 1.
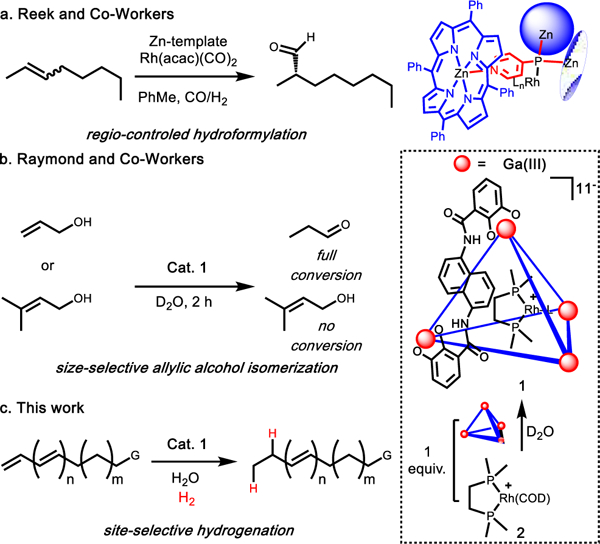
Selected applications of organometallic catalyzed reactions supported by supramolecular hosts. (a) Selective hydroformylation by a supramolecular supported rhodium catalysts selects for a single major regioisomer. (b) Selective metal (Ir or Rh) catalyzed allylic alcohol isomerization achieved by host induced size-selectivity. (c) Proposal for the selective hydrogenation based on size-selectivity where a single point of unsaturation could be selectivity activated over others in the same substrate.
These examples clearly demonstrate that the encapsulation of transition metal-catalysts in supramolecular assemblies can be leveraged to achieve unique selectivity; however, application of this strategy to more complex substrates, bearing multiple sites of reactivity, remains rare especially in catalytic processes.24 We hypothesized that the supramolecular strategies that enable selective transformations of subtly varied substrates might provide new avenues for site-selective reactions. Specifically, that size-selectivity could be harnessed to achieve exquisite catalyst-directed selectivity in a complex substrate, in contrast to size-discrimination between substrates.25–27 Herein we report the ability to hydrogenate a single point of unsaturation of a polyene in the absence of any substrate-driven selectivity to demonstrate a new approach for performing site- and size-selective hydrogenation (Figure 1c). We also provide evidence that complete encapsulation in the host cavity is not required to achieve selective alkene hydrogenation.
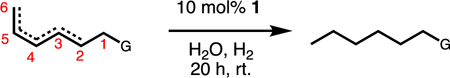
Initial experiments showed that supramolecular catalyst 1, which is prepared in situ by mixing rhodium complex 2 and the Raymond tetrahedron in a 1:1 ratio (Figure 1), hydrogenated a simple terminal olefin substrate (Table 1, entry 1). Free catalyst 2 also efficiently hydrogenated the same terminal olefin, but significantly faster (1 h versus 12 h) than supramolecular catalyst 1. To verify that this reactivity difference was due to host encapsulation of 2, a standard control reaction was performed: to in situ formed 1 was added strongly binding guest Et4N+ (as salt [Et4N][Cl]), resulting in ejection of 2 and Et4N-blocked host.28 With the cavity of the host blocked, complex 2 maintained the same levels of reactivity (>99% conversion, 1 h) since it was no longer encapsulated demonstrating that encapsulation does modify reactivity.
Table 1.
Screening of General Hydrogenation Reactivity for Simple Olefinic Substrates by Catalyst 1
 | ||||
|---|---|---|---|---|
| Entry | G | Unsaturation | Conv. [%] | Yield [%] |
| 1 | −OH | 5–6 | >99 | 92a |
| 2 | −OH | 4–5 (E or Z) | >99 | 87 (E) 85(Z) |
| 3 | −OH | 3–4 (Z) | 15 | 10b |
| 4 | −OH | 3–4 (E) | <5 | <5b |
| 5 | −OH | 2–3 (E or Z) | <5 | <5 |
| 6 | −NAc | 5–6 | 88 | 80 |
| 7 | 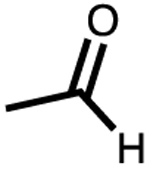 |
4–5 (Z) | >99 | 75 |
| 8 | 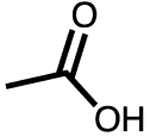 |
5–6 | >99 | 92 |
a 12h reaction time.
b Reactions performed at 50 °C.
Further experiments showed that catalyst 2 efficiently hydrogenated the compounds shown in entries 1–7 in Table 1 within 1 h. In contrast, 1 hydrogenates a methyl-substituted substrate (Table 1, entry 2), but ethyl-substituted substrates show only minor conversions with cis- giving slightly higher conversions than trans- (Table 1, entries 3 and 4). Performing the same reaction with Et4N-blocked host yields full conversion of these substrates, further demonstrating that the lack of reactivity is a result of the host prohibiting reactivity at the encapsulated organometallic catalyst.
This trend continued upon moving the double bond another carbon unit away from the terminal position, leading to no detectable conversion of the allylic alcohol (Table 1, entry 5). To demonstrate that alcohols were not the only substrates tolerated under these conditions, additional functional groups were tested (Table 1, entries 6–8). Each showed good conversion. Notably, carboxylic acid (Table 1, entry 8) remains reactive even under pH 8.0 buffered conditions. Encapsulation of a carboxylate anion with the anionic cage would be unfavorable, suggesting substrate encapsulation is not a requirement for hydrogenation.29 Moreover, catalyst 2 gave incomplete conversion in the presence of the carboxylic acid. This stark difference in reactivity between supramolecular supported and free catalyst offered promise for realizing selective olefin hydrogenation.
A competition experiment was then performed to determine if 1 could discriminate between methyl- and ethyl-substituted olefins (3 and 5, respectively). Good conversion of methyl-olefin (3) was observed while ethyl-olefin starting material (5) was recovered in high yield (Figure 2a). In the analogous control reaction without supramolecular host, full conversion of both substrates occurred rapidly to yield hexanol 4 (Figure 2b). Based on this selectivity, along with the lack of reactivity of allylic alcohol (Table 1, entry 5) it was proposed that catalyst control with 1 should be able to override inherent allylic alcohol directed rhodium hydrogenation. A competition between allylic alcohol 6 and methyl-olefin 3 with free catalyst 2 showed good selectivity for hydrogenation of 6 over 3 at an early time point (Figure 2c). In contrast, good conversion of 3 and recovery of 6 was observed in the presence of catalyst 1, demonstrating the ability of the supramolecular scaffold to overcome native catalyst selectivity (Figure 2d).
Figure 2.
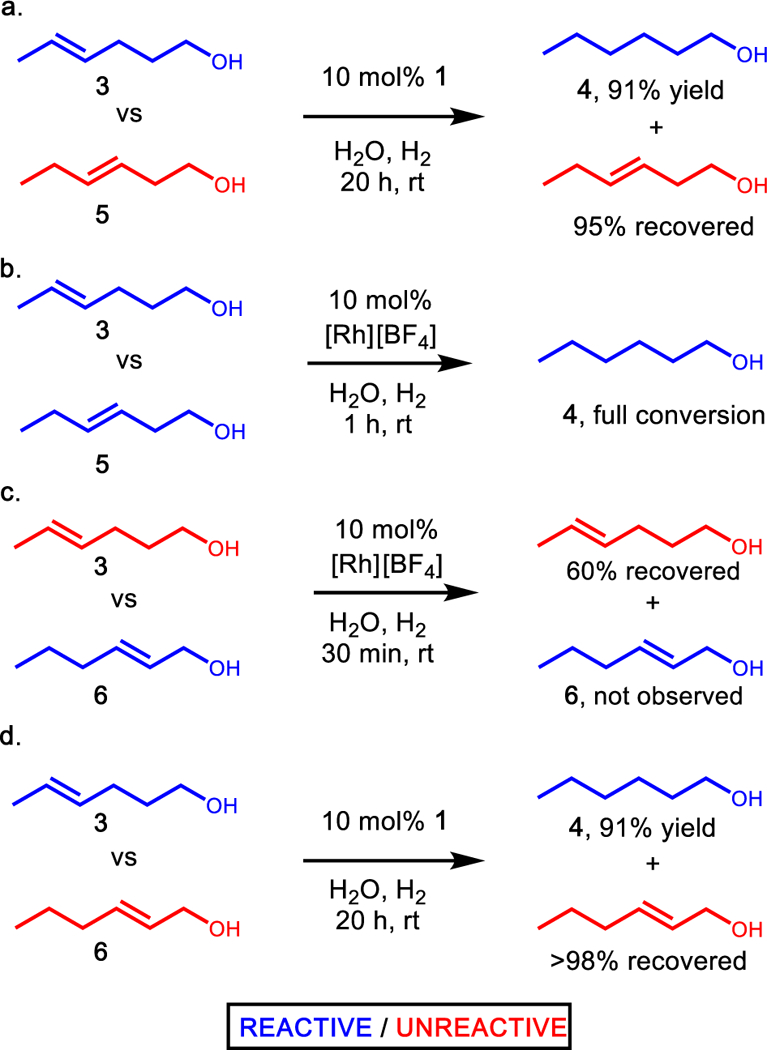
Competition experiments to demonstrate divergent selectivity of free catalyst 2 compared to the supramolecular supported catalyst 1. (a) Supramolecular supported size-selective hydrogenation of methyl- vs ethyl-olefin. (b) Catalyst 2 shows no selectivity between methyl- vs ethyl-olefin. (c) Allylic alcohol-directed hydrogenation results in selective hydrogenation of substrate 6 over 3 at early reaction times. (d) Supramolecular supported catalysis overrides native allylic alcohol directing effect and selectively hydrogenates 3.
To expand this strategy of selective hydrogenation, alkyne substrates were investigated. As with alkene hydrogenation, methyl substituted alkyne 7 was readily converted by catalyst 1. Under room temperature conditions, cis-alkene 8 was observed, and high conversion was realized upon warming the reaction mixture (Figure 3a, 9). In agreement with the results observed above, ethyl alkyne 10 was unreactive. A direct competition experiment between methyl- and ethyl-alkyne (7 and 10, respectively) showed selectivity for conversion of the methyl-alkyne (Figure 3b). Hydrogenation with 2 displays minimal selectivity and rapidly hydrogenates both 7 and 10.
Figure 3.
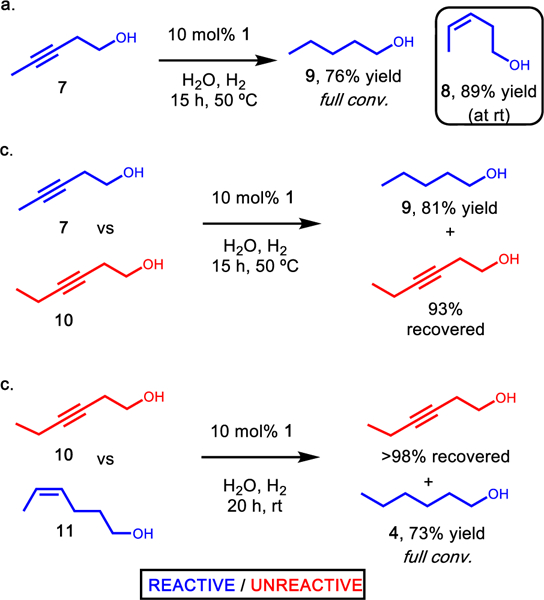
Supramolecular supported catalyst selectively hydrogenates alkynes based on size-exclusion. (a) Hydrogenation of methyl-alkyne 7 provided cis-alkene 8 at room temperature and heating afforded fully hydrogenated 9. (b) Competition between methyl- and ethyl-alkyne (7 and 10, respectively) provided size-selective hydrogenation of 7 and full recovery of 10. (c) Methyl-alkene 11 was selectively hydrogenated with full recovery of ethyl-alkyne 10.
From these competition experiments, it was proposed that the supramolecular supported catalyst should be able to selectively hydrogenate a sterically accessible alkene in the presence of an inherently more reactive alkyne. This would provide support for the ability of this catalyst to perform site selective hydrogenation when cross reactivity is a concern. When subjecting a mixture of 10 and 11 to catalyst 2, alkyne hydrogenation was more rapid than that of the alkene. However, supramolecular catalyst 1 provides good conversion of alkene 11 and recovery of 10 (Figure 3c), providing another example of catalyst control that is different from reactivity of the free catalyst.
With the goal of performing site-selective hydrogenation on a substrate containing multiple sites of unsaturation, dieneol 12 was explored. Upon subjecting 12 to supramolecular hydro-genation conditions, the major product was cis-3-hexen-1-ol 13, which is challenging to produce under standard rhodium hydrogenation conditions.30–32 In contrast, free catalyst 2 provided a mixture of products that eventually converged to fully hydrogenated hexanol.
The selective formation of 13 was proposed to occur via a 1,4-hydride addition mechanism (Figure 4a). In this reaction sequence, the steric confinement provided by the host cavity is proposed to force the intermediate rhodium bound mono-alkene (I) into a cis-orientation to minimize steric clash with the walls, leading to host-selected product 13. To further support the 1,4-addition mechanism, the analogous reaction was performed with deuterium gas. From this reaction, the doubly deuterated cis-product (14) was exclusively observed. Interestingly, the product of this transformation was a single isomer indicating good regioselective and diastereoselective deuterium addition within the host (Figure 4b).33
Figure 4.
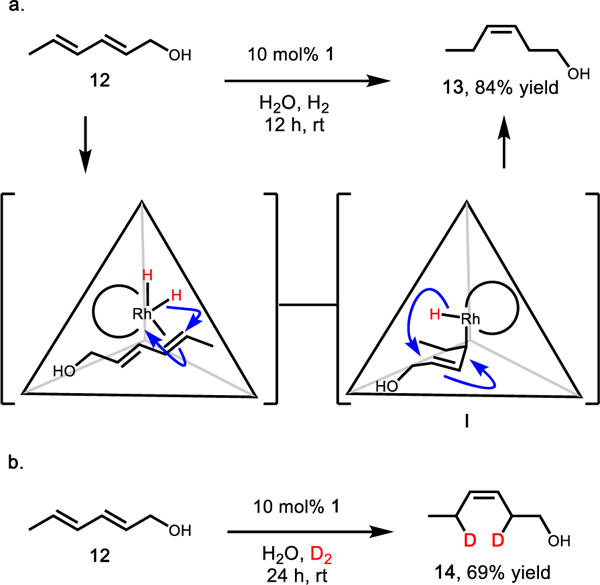
(a) Supramolecular host supported hydrogenation of trans,trans-2,4-hexadiene-1-ol (12) to cis-3-hexen-1-ol (13). (b) Under dideuterium conditions, selective 1,4-addition was observed to yield a single major isomer 14.
To further demonstrate that site-selective hydrogenation could be realized through catalyst control, polyenol 15, where the olefins are not in conjugation as above, was investigated (derived from lineolenic acid). Selective hydrogenation of any point of unsaturation on 15 would be challenging due to a lack of directing groups and similar reactivity. This proved to be the case upon subjecting 15 to hydrogenation with catalyst 2. Hydrogenation gives multiple intermediate products at early time points that rapidly converged to the fully hydrogenated alcohol. However, subjecting 15 to host-supported hydro-genation conditions gave minor conversion even with heating and extended reaction times (Figure 5). This lack of reactivity is in agreement with previous experiments indicating no reactivity at ethyl-substituted olefins.
Figure 5.
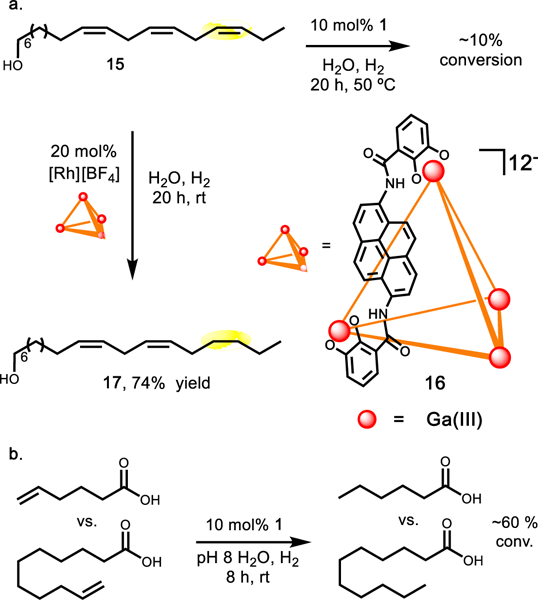
(a) Minor conversion of 15 was observed using catalyst 1. Catalyst 16 provides good conversion of 15 to the singly hydrogenated product 17. (b) Competition between two acids with different alkane lengths show similar conversion, indicating remote group does not affect reactivity.
To overcome this lack of reactivity, the larger pyrene-walled supramolecular host 16 was investigated.34 In analogy to our observations with the naphthalene host, 2 is a good guest within 16. Performing hydrogenation of 15 with this larger supramolecular host provided good conversion of the starting material to yield the monohydrogenated product 17. This product was verified by direct analogy to authentic material (see Supporting Information for details). Increasing the catalyst loading gave complete conversion to provide the singly hydrogenated product 17 in 74% yield (Figure 5a).
Interpreting the larger size of host 16 to be the reason for increased reactivity is reasonable. However, the increased size does not mean that 16 fully encapsulates the substrate during the reaction, whereas the smaller host cannot. Instead, it is proposed that 16 is able to allow enough of the guest to enter and undergo hydrogenation at the metal center while some portion of the substrate remains outside of the host. This is in agreement with similar conversions observed with two carboxylic acid substrates of different alkyl lengths (Figure 5b), which are proposed to undergo conversion in this way due to unfavorable coulombic interactions.
In conclusion, we have demonstrated the ability of a supramolecular-supported hydrogenation catalyst to perform site-selective hydrogenation. With this catalyst, hydrogenation of alkenes is dictated by the steric profile of the substrate (as well as the microenvironment of the metal catalyst), enabling selective reactivity that is not observed with the parent hydrogenation catalyst. Moreover, selective olefin hydro-genation of inherently more reactive alkynes can be realized when these substrates differ by only a methyl-substituent. This site-selectivity allowed for the monohydrogenation of the fatty alcohol of lineolenic acid which contains three points of unsaturation. This high size- and site-selectivity, as well as the lack of requirement for full encapsulation of the substrate, provide a promising tool for performing precise transformation on complex substrates.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Director, Office of Science, Office of Basic Energy Sciences, and the Division of Chemical Sciences, Geosciences, and Bioscience of the U.S. Department of Energy at Lawrence Berkeley National Laboratory (Grant No. DE-AC02-05CH11231) and a NIH Postdoctoral Fellowship to T.A.B. (Grant No. 1F32GM129933-01).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b05604.
Experimental details, characterization data, and methods (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Hong CM; Bergman RG; Raymond KN; Toste FD Self-Assembled Tetrahedral Hosts as Supramolecular Catalysts. Acc. Chem. Res. 2018, 51 (10), 2447–2455. [DOI] [PubMed] [Google Scholar]
- (2).Jongkind LJ; Caumes X; Hartendorp APT; Reek JNH Ligand Template Strategies for Catalyst Encapsulation. Acc. Chem. Res. 2018, 51 (9), 2115–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhang Q; Catti L; Tiefenbacher K Catalysis inside the Hexameric Resorcinarene Capsule. Acc. Chem. Res. 2018, 51 (9), 2107–2114. [DOI] [PubMed] [Google Scholar]
- (4).Yu Y; Rebek J Reactions of Folded Molecules in Water. Acc. Chem. Res. 2018, 51 (12), 3031–3040. [DOI] [PubMed] [Google Scholar]
- (5).Raynal M; Ballester P; Vidal-Ferran A; van Leeuwen PWNM Supramolecular Catalysis. Part 2: Artificial Enzyme Mimics. Chem. Soc. Rev. 2014, 43 (5), 1734–1787. [DOI] [PubMed] [Google Scholar]
- (6).Ward MD; Hunter CA; Williams NH Coordination Cages Based on Bis(Pyrazolylpyridine) Ligands: Structures, Dynamic Behavior, Guest Binding, and Catalysis. Acc. Chem. Res. 2018, 51 (9), 2073–2082. [DOI] [PubMed] [Google Scholar]
- (7).Lee SJ; Cho S-H; Mulfort KL; Tiede DM; Hupp JT; Nguyen ST Cavity-Tailored, Self-Sorting Supramolecular Catalytic Boxes for Selective Oxidation. J. Am. Chem. Soc. 2008, 130 (50), 16828–16829. [DOI] [PubMed] [Google Scholar]
- (8).Catti L; Tiefenbacher K Intramolecular Hydroalkoxylation Catalyzed inside a Self-Assembled Cavity of an Enzyme-like Host Structure. Chem. Commun. 2015, 51 (5), 892–894. [DOI] [PubMed] [Google Scholar]
- (9).Zhang Q; Tiefenbacher K Terpene Cyclization Catalysed inside a Self-Assembled Cavity. Nat. Chem. 2015, 7, 197. [DOI] [PubMed] [Google Scholar]
- (10).Kaphan DM; Toste FD; Bergman RG; Raymond KN Enabling New Modes of Reactivity via Constrictive Binding in a Supramolecular-Assembly-Catalyzed Aza-Prins Cyclization. J. Am. Chem. Soc. 2015, 137 (29), 9202–9205. [DOI] [PubMed] [Google Scholar]
- (11).Brown CJ; Toste FD; Bergman RG; Raymond KN Supramolecular Catalysis in Metal–Ligand Cluster Hosts. Chem. Rev. 2015, 115 (9), 3012–3035. [DOI] [PubMed] [Google Scholar]
- (12).Zhang Q; Catti L; Pleiss J; Tiefenbacher K Terpene Cyclizations inside a Supramolecular Catalyst: Leaving-Group- Controlled Product Selectivity and Mechanistic Studies. J. Am. Chem. Soc. 2017, 139 (33), 11482–11492. [DOI] [PubMed] [Google Scholar]
- (13).Wu N-W; Rebek J Cavitands as Chaperones for Monofunctional and Ring-Forming Reactions in Water. J. Am. Chem. Soc. 2016, 138 (24), 7512–7515. [DOI] [PubMed] [Google Scholar]
- (14).Shi Q; Masseroni D; Rebek J Macrocyclization of Folded Diamines in Cavitands. J. Am. Chem. Soc. 2016, 138 (34), 10846–10848. [DOI] [PubMed] [Google Scholar]
- (15).Wu N-W; Petsalakis ID; Theodorakopoulos G; Yu Y; Rebek J Jr. Cavitands as Containers for α,ω-Dienes and Chaperones for Olefin Metathesis. Angew. Chem., Int. Ed. 2018, 57 (46), 15091–15095. [DOI] [PubMed] [Google Scholar]
- (16).Bender TA; Morimoto M; Bergman RG; Raymond KN; Toste FD Supramolecular Host-Selective Activation of Iodoarenes by Encapsulated Organometallics. J. Am. Chem. Soc. 2019, 141 (4), 1701–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jiang X-B; Lefort L; Goudriaan PE; de Vries AHM; van Leeuwen PWNM; de Vries JG; Reek JNH Screening of a Supramolecular Catalyst Library in the Search for Selective Catalysts for the Asymmetric Hydrogenation of a Difficult Enamide Substrate. Angew. Chem., Int. Ed. 2006, 45 (8), 1223–1227. [DOI] [PubMed] [Google Scholar]
- (18).García-Simón C; Gramage-Doria R; Raoufmoghaddam S; Parella T; Costas M; Ribas X; Reek JNH Enantioselective Hydroformylation by a Rh-Catalyst Entrapped in a Supramolecular Metallocage. J. Am. Chem. Soc. 2015, 137 (7), 2680–2687. [DOI] [PubMed] [Google Scholar]
- (19).Nowicki A; Zhang Y; Léger B; Rolland J-P; Bricout H; Monflier E; Roucoux A Supramolecular Shuttle and Protective Agent: A Multiple Role of Methylated Cyclodextrins in the Chemoselective Hydrogenation of Benzene Derivatives with Ruthenium Nanoparticles. Chem. Commun. 2006, 3, 296–298. [DOI] [PubMed] [Google Scholar]
- (20).Nurttila SS; Brenner W; Mosquera J; van Vliet KM; Nitschke JR; Reek JNH Size-Selective Hydroformylation by a Rhodium Catalyst Confined in a Supramolecular Cage. Chem. - Eur. J. 2019, 25 (2), 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nurttila SS; Linnebank PR; Krachko T; Reek JNH Supramolecular Approaches To Control Activity and Selectivity in Hydroformylation Catalysis. ACS Catal. 2018, 8 (4), 3469–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Leung DH; Bergman RG; Raymond KN Highly Selective Supramolecular Catalyzed Allylic Alcohol Isomerization. J. Am. Chem. Soc. 2007, 129 (10), 2746–2747. [DOI] [PubMed] [Google Scholar]
- (23).Brown CJ; Miller GM; Johnson MW; Bergman RG; Raymond KN High-Turnover Supramolecular Catalysis by a Protected Ruthenium(II) Complex in Aqueous Solution. J. Am. Chem. Soc. 2011, 133 (31), 11964–11966. [DOI] [PubMed] [Google Scholar]
- (24).Takezawa H; Kanda T; Nanjo H; Fujita M Site-Selective Functionalization of Linear Diterpenoids through U-Shaped Folding in a Confined Artificial Cavity. J. Am. Chem. Soc. 2019, 141 (13), 5112–5115. [DOI] [PubMed] [Google Scholar]
- (25).Huang Z; Dong G Site-Selectivity Control in Organic Reactions: A Quest To Differentiate Reactivity among the Same Kind of Functional Groups. Acc. Chem. Res. 2017, 50 (3), 465–471. [DOI] [PubMed] [Google Scholar]
- (26).Hartwig JF Catalyst-Controlled Site-Selective Bond Activation. Acc. Chem. Res. 2017, 50 (3), 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Toste FD; Sigman MS; Miller SJ Pursuit of Noncovalent Interactions for Strategic Site-Selective Catalysis. Acc. Chem. Res. 2017, 50 (3), 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
(28).Upon addition of [Et4N][Cl] salt to 1, rhodium complex 2 is ejected to give Et4N-blocked Raymond tetrahedron.
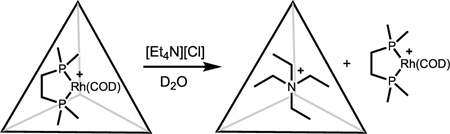
- (29).Tiedemann BEF; Raymond KN Dangling Arms: A Tetrahedral Supramolecular Host with Partially Encapsulated Guests. Angew. Chem., Int. Ed. 2006, 45 (1), 83–86. [DOI] [PubMed] [Google Scholar]
- (30).Dada R; Wei Z; Gui R; Lundgren RJ. Chemoselective Synthesis of Z-Olefins through Rh-Catalyzed Formate-Mediated 1,6- Reduction. Angew. Chem. Int. Ed. 2018, 57 (15), 3981–3984. [DOI] [PubMed] [Google Scholar]
- (31).Sodeoka M; Shibasaki M Arene Chromium Tricarbonyl Catalyzed Reactions in Organic Synthesis. Synthesis 1993, 1993 (7), 643–658. [Google Scholar]
- (32).Vasil’ev AA; Vlasyuk AL; Gamalevich GD; Serebryakov EP A Versatile and Convenient Protocol for the Stereocontrolled Synthesis of Olefinic Insect Pheromones. Bioorg. Med. Chem. 1996, 4 (3), 389–400. [DOI] [PubMed] [Google Scholar]
- (33).The only product formed in this reaction is the didueterated cis- olefin. Out of the potential diastereomers of deuterium addition, only one product is formed; however, the relative configuration was not determined.
- (34).Hart-Cooper WM; Zhao C; Triano RM; Yaghoubi P; Ozores HL; Burford KN; Toste FD; Bergman RG; Raymond KN. The Effect of Host Structure on the Selectivity and Mechanism of Supramolecular Catalysis of Prins Cyclizations. Chem. Sci 2015, 6 (2), 1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.