

Abstract
Landmark discoveries made nearly two decades ago identified known transcriptional regulators as histone lysine methyltransferases; since then the field of lysine methylation signaling has been dominated by studies of how this small chemical posttranslational modification regulates gene expression and other chromatin-based processes. However, recent advances in mass spectrometry-based proteomics have revealed that histones are just a subset of the thousands of eukaryotic proteins marked by lysine methylation. As the writers, erasers, and readers of histone lysine methylation are emerging as a promising therapeutic target class for cancer and other diseases, a key challenge for the field is to define the full spectrum of activities for these proteins. Here we summarize recent discoveries implicating non-histone lysine methylation as a major regulator of diverse cellular processes. We further discuss recent technological innovations that are enabling the expanded study of lysine methylation signaling. Collectively, these findings are shaping our understanding of the fundamental mechanisms of non-histone protein regulation through this dynamic and multi-functional posttranslational modification.
eTOC Blurb
Lysine methylation is a key regulator of histone and non-histone protein function, cellular processes, and disease progression. Cornett et al. discuss recent progress in understanding molecular functions associated with lysine methylation signaling and highlight the need for continued study of this dynamic and widespread posttranslational modification.
Introduction
Covalent, reversible posttranslational modifications (PTMs) greatly expand the functional diversity of proteins beyond that provided by amino acid composition and tertiary structure. Methylation on the ε-amine of a lysine sidechain was first reported on a bacterial flagellar protein in 1959 (Ambler and Rees, 1959) and shortly thereafter on histone proteins (Murray, 1964). However, the functional implications of lysine methylation were not broadly appreciated until the turn of the century when Jenuwein and colleagues revealed the molecular activity of a known suppressor of position-effect variegation (PEV), Suppressor of Variegation 3–9 Homolog 1 S(UV39H1), as a histone lysine methyltransferase (Murn and Shi, 2017; Rea et al., 2000). Since this landmark discovery, substantial progress has been made characterizing proteins that write (lysine methyltransferases; KMTs), erase (lysine demethylases; KDMs), and read (effector proteins) lysine methylation (Figure 1) (Murn and Shi, 2017). The studies of lysine methylation signaling have focused largely on the modification of histone proteins, owing to their abundance, evolutionary conservation, biochemical tractability, and established phenotypic assays. This body of work has firmly established that histone lysine methylation is a major contributor to fundamental chromatin-templated biological processes including transcription, DNA replication, and DNA repair (Black et al., 2012; Rothbart and Strahl, 2014). However, as illustrated in Figure 1, the writers, erasers, and readers of lysine methylation have an increasing number of non-histone substrates and binding partners. Additionally, lysine methylation regulators are emerging as important targets for a variety of clinical implications, as evidenced by the number of molecules in various stages of preclinical and clinical development (Figure 1).
Figure 1. Lysine methylation writer, eraser, and reader families.

Wheel plots depicting current knowledge of the subcellular localization, substrates, and evaluation status of small molecular inhibitors for (A) lysine methyltransferases; KMTs, (B) lysine demethylases; KDMs, (C) proteins with UniProt-annotated PHD, BAH, CW, and SPIN domains, and (D) the Royal family of methyllysine readers with UniProt-annotated Tudor, MBT, PWWP, and Chromo domains. Data was curated from UniProt (2019) and ChromoHub (Liu et al., 2012).
Recent technical advances in mass spectrometry (MS)-based proteomics have revealed that the scope of lysine methylation extends far beyond histone proteins. According to the most recent datasets available on PhosphoSite (Hornbeck et al., 2015), nearly 3,000 non-histone proteins from human cells have been reported to be modified with lysine methylation at approximately 5,000 unique sites. The identification of new sites of lysine methylation has far outpaced functional annotation. In fact, only a small fraction of non-histone lysine methylation sites have been assigned a function and the majority of these studies have been limited to a few proteins (e.g., p53, ER, RB1), which have been extensively reviewed elsewhere (Biggar and Li, 2015; Biggar et al., 2017; Carlson and Gozani, 2016). Here, we discuss key findings in recent years that have expanded our understanding and appreciation of the functional role of lysine methylation in molecular processes by fine-tuning non-histone protein function (Figure 2). We also reflect on technological innovations that have enabled these discoveries. We then end with a perspective of the perceived challenges and opportunities that lie ahead for the blossoming field of lysine methylation research.
Figure 2. Molecular functions associated with lysine methylation.
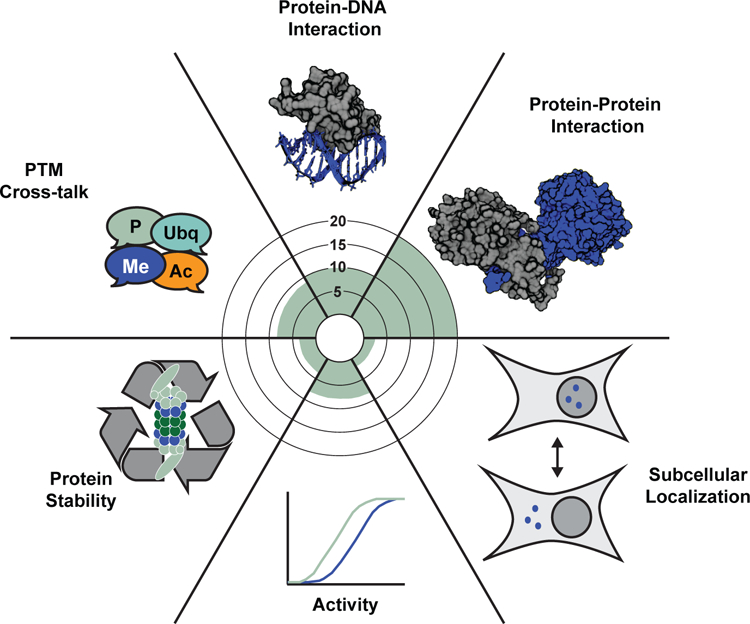
Radar plot counting the number of PubMed-archived papers assigning the indicated molecular function to lysine methylation.
Lysine Methylation Facilitates Protein-Protein Interactions
Lysine methylation, unlike acetylation, does little to the charge properties of the lysine side chain (Luo, 2018). In the context of histone proteins, it is appreciated that lysine methylation functions primarily as a signal for the selective recruitment of effector proteins (Musselman et al., 2012). On non-histone proteins as well, lysine methylation can facilitate protein-protein interactions (Figure 2). For example, a chemical proteomics screen for substrates of the G9a/GLP (EHMT2/EHMT1) dimer in mouse embryonic stem cells led to the identification of Activating Transcription Factor 7 Interacting Protein (ATF7IP) lysine 16 as a novel substrate (Tsusaka et al., 2018). In the same study, immunoprecipitation of ATF7IP followed by mass spectrometry revealed that the histone H3 lysine 9 (H3K9) methyl-effector protein M-Phase Phosphoprotein 8 (MPP8) selectively binds ATF7IP K16 tri-methylation. ATF7IP and MPP8 are both part of the HUman Silencing Hub (HUSH) complex which contains the H3K9 methyltransferase SET Domain Bifurcated 1 (SETDB1) (Figure 3A). The HUSH complex mediates gene silencing, and knockout of either ATF7IP or MPP8 impaired HUSH complex function. Interestingly, mutation or truncation of ATF7IP to prevent K16 methylation led to a delay in HUSH complex silencing of a virally introduced transgene, suggesting methylation of ATF7IP partially contributes to HUSH complex function (Tsusaka et al., 2018).
Figure 3. Common phenotypes associated with histone and non-histone substrates of KMT activity.
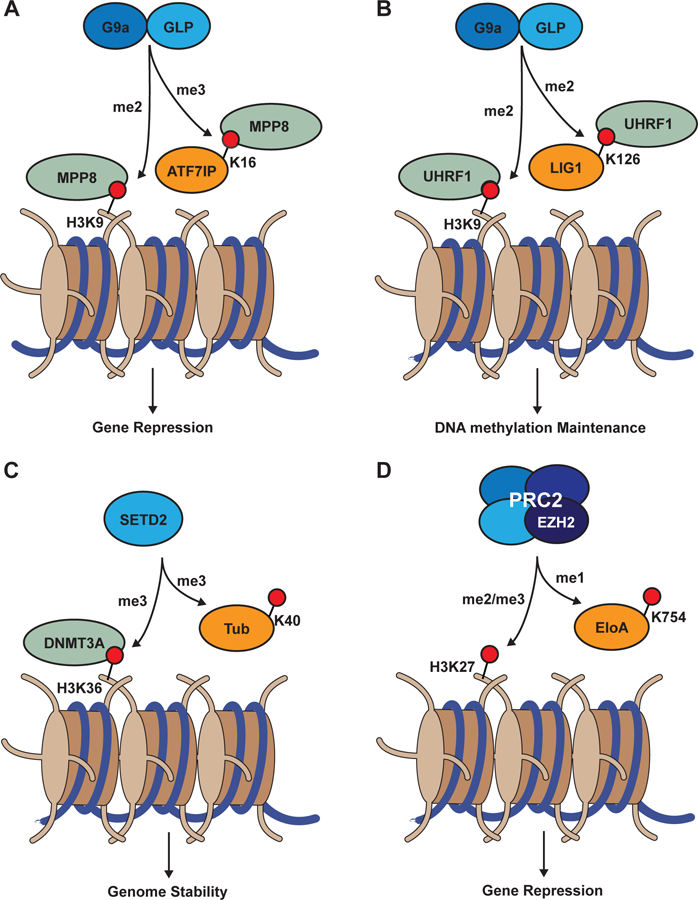
(A) MPP8 chromodomain recognition of G9a/GLP-dependent H3K9 and ATF7IPK16 methylation is associated with gene silencing. (B) UHRF1 tandem Tudor domain recognition of G9a/GLP-dependent H3K9 and LIG1 methylation is associated with DNA methylation inheritance. (C) SETD2-dependent H3K36 and αTubulinK40 methylation contributes to genomic stability. (D) The catalytic subunit of the PRC2 complex, EZH2, methylates H3K27 and ElonginAK754 to promote gene silencing.
Like MPP8, the DNA methylation regulatory protein Ubiquitin-like with PHD and Ring Finger Domains 1 (UHRF1) also reads H3K9 methylation (Figure 3B). An interaction between UHRF1 and DNA Ligase 1 (LIG1) was recently reported in a lysine methylation-dependent manner (Ferry et al., 2017; Kori et al., 2019). While loss of LIG1 had no detectable impact on DNA methylation, deletion of the region surrounding the methylation site on LIG1 showed a DNA methylation defect (Ferry et al., 2017).
As can be seen from these examples, KMTs can have both histone and non-histone substrates. Likewise, it is likely that most effector proteins, KMTs, and KDMs will target both histone and non-histone proteins. Interestingly, as illustrated by the above-described studies, histone and non-histone lysine methylation can converge on shared biological outcomes (Figure 3). Deconvoluting the contribution of specific lysine methylation events on histone and non-histone proteins will be a crucial line of future inquiry.
In part because high-throughput approaches to identify methyllysine-driven protein-protein interactions are limited, the progress connecting reader proteins to non-histone protein lysine methylation has been slow. For histone lysine methylation, the use of histone peptide microarrays has been instrumental in making connections between reader proteins and histone lysine methylation (Levy et al., 2011a; Rothbart et al., 2012; Wilkinson and Gozani, 2014). For non-histone proteins, most connections made thus far have been facilitated by low-throughput candidate-based approaches. For example, protein-domain microarrays displaying recombinant methyllysine reader domain libraries recently helped identify that the plant homeodomain (PHD) of PHD Finger Protein 20-Like protein (PHF20L1) reads methylation on DNA Methyltransferase 1 (DNMT1) (Estève et al., 2014) and Retinoblastoma Protein 1 (RB1) (Carr et al., 2017). Clearly, evidence is mounting that methyllysine reader domains recognize both methylated histone and non-histone proteins. Making additional connections between reader proteins and the vast non-histone lysine methylome is a major challenge and an important direction of future research.
Lysine Methylation Regulates Protein-DNA Interactions
Lysine methylation has been associated with the regulation of protein-DNA interactions since early studies characterizing non-histone lysine methylation on the tumor suppressor protein p53 (Chuikov et al., 2004), and this has been confirmed to be a major mechanism by which lysine methylation impacts protein function (Figure 2). Recently, the multifunctional transcription factors Yin Yang 1 (YY1) and Yin Yang 2 (YY2) were shown to be regulated by lysine methylation. Several KMTs interact with YY1, which prompted Zhang et al. to test whether YY1 might be directly methylated (Zhang et al., 2016). Indeed, YY1 is mono-methylated on lysines 173 and 411 by SET domain-containing protein 7/9 (SET7/9), resulting in increased YY1 DNA binding. YY2 was also shown to be mono-methylated by SET7/9. Mono-methylation of YY2 on lysine 247 (K247) enhanced YY2 binding to DNA, and knockout of SET7/9 phenocopied a K247 to arginine mutation of YY2. Furthermore, lysine-specific histone demethylase 1A (LSD1) removed YY2 K247me1 and knockout of LSD1 resulted in increased YY2 binding to DNA in cells. These recent examples and others (Ea and Baltimore, 2009; Liu et al., 2015; Xie et al., 2012) provide additional evidence in support of a role for lysine methylation in the regulation of protein-DNA interactions, particularly for transcription factors. Future structural and quantitative analyses of these interactions will be important to further understand this regulatory mechanism. Additionally, as evidence accumulates demonstrating transcription factor regulation by lysine methylation, so do questions about how upstream KMTs are themselves regulated. It is well appreciated that cascades of phosphorylation lead to transcriptional regulation, and emerging evidence suggests lysine methylation acts in a similar manner.
Protein Stability is Regulated by Lysine Methylation
Lysine methylation has been shown to promote protein stability (Figure 2). For example, SET7/9 mediated lysine methylation stabilizes both p53 and Estrogen Receptor alpha (ERα), either by blocking polyubiquitination-dependent proteasomal degradation or by blocking the ubiquitin machinery (Chuikov et al., 2004; Subramanian et al., 2008). More recently, it was shown that a cystic fibrosis (CF)-associated mutation in the CFTR gene causes a shift from methylation to ubiquitination (Pankow et al., 2019). While functional studies are needed to determine whether the stability of this mutant CFTR ion channel is compromised, it is exciting to speculate that modulation of this lysine methylation site – perhaps through inhibition of a KDM – could offer a new treatment paradigm for CF by preventing degradation of the CFTR protein.
In addition to stabilizing proteins by blocking potential sites of ubiquitylation, lysine methylation can also play the opposite role as a signal for protein degradation; it accomplishes this by recruiting the ubiquitin ligase machinery, directly or indirectly, through adaptor proteins. Lysine methylation events that function in this way have been termed “methyl-degrons.” The KMT Enhancer of Zeste 2 (EZH2) was reported to generate a methyl-degron on the nuclear receptor Retinoid-related Orphan Receptor Alpha (RORalpha/NR1F1) that is recognized by a putative chromodomain of the DDB1-CUL4-Associated Factor 1 (DCAF1) (Lee et al., 2012). DCAF1 recognition of EZH2-directed mono-methylation on NR1F1 lysine 38 leads to ubiquitylation of NR1F1 by the DDB1/2/CUL4 E3 ligase complex. A similar methyl-degron mechanism was recently shown for SET7/9 directed mono-methylation of SMAD7 lysine 70, which promotes ubiquitylation by the E3-ligase Arkadia and degradation of SMAD7 (Elkouris et al., 2016).
These studies raise questions regarding how E3 ligases recognize methylated lysines. It is intriguing to speculate that some E3-ligases might themselves contain cryptic methyllysine effector domains. Methyllysine effectors have also been shown to recruit E3 ligases. For example, SET Domain Bifurcated 1 (SETDB1) mono-methylates RAC-alpha serine/threonine-protein kinase (AKT) on lysine 64, leading to ubiquitylation by the E3 ligase TNF Receptor Associated Factor 6 (TRAF6) or the Skp, Cullin, F-box containing (SCF) complex (Wang et al., 2019). The interaction between AKT and these E3 ligases is mediated through Lysine-specific Demethylase 4A (KDM4A), which recognizes methylated AKT through its Tandem Tudor domain. However, KDM4A facilitates ubiquitylation independent of its KDM activity. Surely, the role of lysine methylation in regulating protein stability will be an exciting area of research in the coming years.
Lysine Methylation can affect Protein Subcellular Localization
Lysine methylation is a major determinant of protein localization within cells. For instance, the nuclear localization of Catenin beta-1 (β-catenin) was shown to be positively regulated by SET and MYND domain containing 2 (SMYD2), a KMT that catalyzes mono-methylation of β-catenin on lysine 133 (Matsuo et al., 2017). SMYD2 is found in the nucleus (Figure 1), and methylation of β-catenin may promote its nuclear accumulation by facilitating an interaction with the import machinery. The nuclear-localizing KMT SET Domain Containing 1A (SETD1A) has also been shown to regulate the localization of several proteins. Di-methylation of HSP70 on lysine 561 by SETD1A leads to enhanced HSP70 nuclear localization (Cho et al., 2012). Similarly, SETD1A-dependent mono-methylation on lysine 342 of Yes-Associated Protein 1 (YAP1), a key transcriptional coactivator in the Hippo signaling pathway, promoted its nuclear localization. Interestingly, it was found that YAP1 K342me1 blocks its interaction with Chromosomal Maintenance 1 (CRM1), a nuclear export protein (Fang et al., 2018), thus offering a methylation-dependent mechanism to trap YAP1 in the nucleus. Interestingly, KMTs themselves can undergo dynamic localization changes. For example, Wnt signaling promotes SETDB1 shuttling from the nucleus to the cytoplasm during muscle differentiation (Beyer et al., 2016).
Crosstalk between Lysine Methylation and other PTMs
Histone PTMs regulate other chromatin modifications through dynamic crosstalk mechanisms. Lysine methylation is a major contributor to this signaling complexity on histone proteins (Rothbart and Strahl, 2014), and there is growing evidence it plays a similar role on non-histone proteins. Lysine methylation crosstalk with other PTMs occurs through a variety of mechanisms. One of the appreciated crosstalk mechanisms on histone proteins is the H3K9/S10 methyl-phospho switch that contributes to the ejection of methyllysine-reading Heterochromatin Protein 1 (HP1) from mitotic chromatin (Fischle et al., 2005; Hirota et al., 2005). Similar mechanisms for non-histone proteins have been described where methylation and phosphorylation at neighboring sites have opposing functions, and these have been reviewed elsewhere (Biggar and Li, 2015). Recent evidence suggests that additional mechanisms of crosstalk between lysine methylation and phosphorylation exist. The KMT SET Domain Bifurcated 2 (SETD2) was shown to mono-methylate Signal Transducer and Activator of Transcription 1 (STAT1) on lysine 525, leading to enhanced phosphorylation of STAT1 on Y701. Phosphorylation of STAT1 Y701 leads to STAT1 activation and ultimately downstream activation of the interferon-mediated antiviral response (Chen et al., 2017). How STAT1 K525me1 leads to increased phosphorylation remains unclear. Methylation may alter the conformation of the protein or the responsible kinase. Alternatively, an unidentified effector protein may recognize this methylation site to facilitate phosphorylation. It is important to note that SETD2 has been shown to tri-methylate other substrates, and it is not clear why SETD2 activity is restricted to mono-methylation on STAT1.
It is clear that crosstalk between phosphorylation and lysine methylation occurs at neighboring sites, but lysine methylation has also recently been shown to directly and indirectly affect the activity of kinases and phosphatases. On the one hand, cell cycle-associated Polo-like Kinase 1 (PLK1) was recently identified as a KMT substrate for SET Domain Containing 6 (SETD6). SETD6 methylates two lysine residues on PLK1, and loss of SETD6 and PLK1 methylation resulted in an increase in PLK1 kinase activity (Vershinin et al., 2019). On the other hand, Interferon Regulatory Factor 3 (IRF3) was shown to be methylated by Nuclear Receptor Binding SET Domain Protein 3 (NSD3) on K366 (Wang et al., 2017). IRF3 is activated through phosphorylation on S396 and is deactivated by removal of this phosphorylation event by the Protein Phosphatase 1 Catalytic Subunit Gamma (PP1cc). Notably, PP1cc is unable to recognize the methylated form of IRF3, leading to the stabilization of activated IRF3. It is interesting to speculate that a demethylase exists to remove this methylation and subsequently allow PP1cc to dephosphorylate IFR3 and disable its function.
Lysine methylation also directly competes with other PTMs directed toward lysine residues. As outlined above, lysine residues can be ubiquitylated or methylated to regulate protein stability. Similarly, lysine methylation and acetylation can occur at the same site: for instance SETD2 tri-methylates α-Tubulin on lysine 40, presumably preventing acetylation of this residue (Park et al., 2016) and contributing to cytoskeletal remodeling. As a result, loss of SETD2 leads to genomic instability, presumably through destabilization of microtubules. However, SETD2 also catalyzes tri-methylation of lysine 36 on histone H3 (H3K36me3), a binding site for the PWWP domain of the de novo DNA methyltransferase 3B (DNMT3B) (Baubec et al., 2015; Dhayalan et al., 2010). Mutations in the PWWP domain cause immunodeficiency, centromere instability, and facial abnormalities (ICF) syndrome (Ge et al., 2004), suggesting that the role of SETD2 in maintaining genomic stability may be through both histone and non-histone pathways (Figure 3C).
Evidence is mounting that lysine methylation on non-histone proteins functions as part of a dynamic network of PTMs to mediate signaling within cells. As shown exquisitely on histone proteins, lysine methylation can function to promote or prevent the addition of other PTMs. The examples outlined above demonstrate that similar mechanisms regulate PTMs on non-histone proteins. The concept that combinations of PTMs function together to mediate specific signaling events through histone proteins was coined the “histone code” hypothesis (Strahl and Allis, 2000). Inspired by this concept, a more broad “protein code” was hypothesized, encompassing both histone and non-histone proteins (Sims and Reinberg, 2008). Future study of the many ways by which lysine methylation works with and opposed to other PTMs will certainly expand our understanding of the diverse functions of lysine methylation, but also of PTM signaling in general.
Lysine Methylation Inside the Nucleus
Most lysine methylation regulators localize to the nucleus at least some of the time (Figure 1) (Carlson and Gozani, 2016). However, beyond chromatin function, few studies have revealed mechanisms of lysine methylation signaling in the nucleus. Growing evidence suggests lysine methylation regulates RNA splicing. A proteome-wide screen for methylated proteins enriched for many factors involved in splicing, yet at the time, little was known about the function of these lysine methylation events or the responsible enzymes (Moore et al., 2013). The KMT SET Domain and Mariner Transposase Fusion Gene-Containing Protein (SETMAR), disputed for its function as an H3K36 methyltransferase (Carlson et al., 2015; Fnu et al., 2011), was shown to mono-methylate the splicing factor Small Nuclear Ribonucleoprotein U1 Subunit 70 (snRNP70) on lysine 130 (Carlson et al., 2015). Notably, SETMAR was the first identified KMT to target a splicing factor, but the functional significance of snRNP70K130me1 is unclear. The splicing factor RNA Binding Motif Protein 25 (RBM25) was shown to be mono-methylated on lysine 77 (Carlson et al., 2017). While the responsible KMT is elusive, functional studies suggest RBM25K77me1 may regulate splicing by blocking the interaction between RBM25 and another splicing factor, Serine- and Arginine-rich Splicing Factor 2 (SRSF2). With the identification of a large number of splicing proteins that undergo lysine methylation, continued study of the role lysine methylation plays in regulating RNA splicing will be an important area of future study.
In addition to regulating splicing, studies suggest lysine methylation may regulate the transcriptional machinery itself. Posttranslational modification of the RNA polymerase II (RNAPII) C-terminal domain (CTD) is an important regulatory mechanism of RNAPII function. The CTD consist of heptad-repeats of a consensus sequence (Try-Ser-Pro-Thr-Ser-Pro-Ser), and phosphorylation of serines 2 and 5 are associated with transcription elongation and initiation, respectively (Buratowski, 2009). Notably, vertebrates also contain a variable number of non-canonical repeats that contain lysine substituted for serine in the seventh position. These non-canonical K7-containing RNAPII CTD repeats were shown to be mono- and di-methylated (Dias et al., 2015; Voss et al., 2015). RNAPII CTD-K7me1/me2 was found to be associated with the early stages of transcription, but the function of these modifications and the responsible regulators are unknown.
Determining whether biological effects associated with a lysine methylation regulator are due to a histone or non-histone protein remains a significant challenge, particularly for non-histone lysine methylation occurring on proteins in the nucleus. As discussed above for effector proteins, evidence is mounting that lysine methylation regulators modulate cellular processes through methylation and/or recognition of both histone and non-histone proteins (Figure 3). Recent studies found this is true even for proteins with appreciated biological functions associated with their histone targets. For example, Polycomb Repressive Complex 2 (PRC2) is well studied for its role in epigenetic regulation of facultative heterochromatin through KMT activity toward lysine 27 on histone H3 (H3K27) by the catalytic component of the complex, EZH2. Using a SPOT array to characterize the substrate selectivity of PRC2, it was discovered that the RNAPII transcription elongation factor Elongin A (EloA) is mono-methylated on lysine 754 by PRC2 (Ardehali et al., 2017). Mutation of the endogenous target lysine to methionine with CRISPR/Cas9 revealed that PRC2-directed methylation of EloAK754 helps reinforce suppression of a subset of PRC2 target genes. Details of this molecular mechanism involving EloAK754me1 are still unclear, but this recent study exemplifies the types of systematic approaches that will be necessary for delineating histone-versus non-histone-specific functions of nuclear KMTs. Notably, it was found that PRC2 activity toward H3K27 and EloAK754 both contribute to gene regulation by PRC2 (Figure 3D). PRC2 has other reported non-histone substrates (He et al., 2012), and EZH2 inhibitors are in clinical trials for hematologic malignancies. It is therefore fundamental to further study how the sum of all PRC2 substrates contributes to its chromatin regulatory and oncogenic functions.
Lysine Methylation Outside the Nucleus
Despite the majority of lysine methylation regulators being localized to the nucleus, several recent studies have begun to unveil an expansive role for lysine methylation in regulating processes in the cytoplasm. In particular, lysine methylation is emerging as a regulator of translation. Despite being first reported on the translation machinery nearly 50 years ago (Chang et al., 1976), it was not until recently that progress was made toward determining the function of this methylation or the responsible KMTs or KDMs. An example of the rapid progress that has recently been made are studies of Elongation Factor 1 Alpha (eEF1A), an essential GTPase associated with the translation machinery. Methylation of eEF1a was mapped on several lysine residues (Jakobsson et al., 2018a), and five unique KMTs were recently connected to methylation of eEF1a (Jakobsson et al., 2018b; Liu et al., 2019; Małecki et al., 2017). Two groups independently identified Methyltransferase-like Protein 13 (METTL13) as the KMT responsible for catalyzing di-methylation on lysine 55 of eEF1A (Jakobsson et al., 2018b; Liu et al., 2019). Notably, this methylation directly affects translation. It was mechanistically determined that METTL13-directed methylation of eEF1AK55me2 leads to an increased rate of eEF1A GTP hydrolysis and increased cellular protein synthesis (Liu et al., 2019). Indeed, investigating the role of lysine methylation in regulating translation may hold promise for new cancer treatments. Liu et al. showed the increased rate of translation mediated through METTL13 methylation of eEF1AK55 promotes tumorigenesis (Liu et al., 2019). Elevated METTL13 expression was also associated with poor survival in patients with pancreatic ductal adenocarcinoma (PDAC). METTL13 depletion reduced tumor size and extended life in a PDAC mouse model (Liu et al., 2019), supporting chemical inhibition of METTL13 as a rationale treatment approach.
Recent studies also support a role for KDM4A, a KDM appreciated for its activity toward H3K9 and H3K36 (Klose et al., 2006; Whetstine et al., 2006). KDM4A was found to associate with the translation machinery in the cytoplasm, and depletion or inhibition of its catalytic activity altered the distribution of translation factors in polysome fractions (Van Rechem et al., 2015). Intriguingly, this resulted in a net decrease in global protein synthesis. Substrates of KDM4A in the cytoplasm remain elusive, and their identification will be a decisive step toward mechanistic understanding of this protein’s translation regulatory function.
New Technologies Guiding the Study of Lysine Methylation
The progress made to date mapping lysine methylation and defining its functions is intimately linked to the development of innovative technologies. Perhaps the most critical advance has been the ability to detect lysine methylation by mass spectrometry (MS). A combination of developments in state-of-the-art high-sensitivity MS instrumentation and optimized sample preparation techniques have rapidly expanded the lysine methylome in recent years. In general, robust PTM analysis by MS requires enrichment for peptides containing the modification of interest. Lysine methylation presents a unique challenge in this regard, since the modification is relatively small and does not modify the charge properties of the amino acid side chain. Enrichment strategies for methylated peptides have relied heavily on the use of “pan” methyllysine antibody reagents (Bremang et al., 2013; Cao and Garcia, 2016; Guo et al., 2014). However, creating a reagent that is truly “pan” has proven difficult (Levy et al., 2011b). Essentially an antibody must be able to distinguish a single methyl group while also binding with high affinity to all possible sequences surrounding a target lysine – a tall task for an antibody whose epitope footprint is typically 5–8 residues. This enrichment step is a significant drawback of lysine methylation MS workflows, and as a result, several groups have proposed alternative approaches.
For example, as an alternative to antibody-based enrichment, effector protein domains with limited sequence selectivity have been employed. In one approach, the Malignant Brain Tumor (MBT) domain repeats from Lethal(3) MBT-like Protein 1 (L3MBTL1) were used to enrich for mono- and di-methylated lysine-containing peptides (Moore et al., 2013). The HP1β chromodomain was used in a similar approach that resulted in the identification of 184 methylated proteins (Wang et al., 2018). Other strategies follow in the footsteps of mapping phosphorylation, which was facilitated, in part, by chromatographic separation techniques to enrich for phospho-peptides (Roux and Thibault, 2013). A similar strategy for lysine methylation would be extremely useful, as it would bypass the sequence bias limitations stemming from the use of affinity reagents. Toward this goal, several chromatographic strategies to enrich for methyllysine peptides have recently been proposed (Ning et al., 2016; Wang et al., 2016; Wu et al., 2015). For example, a charge-suppression strategy has been implemented in which the charge on unmodified lysine and arginine residues after tryptic digestion is neutralized by a chemical reaction that has no effect on methylated residues (Ning et al., 2016). Coupling this approach to charge-based separation enriched for methylated peptides and identified 399 unique lysine methylation sites (Ning et al., 2016). Overall, the combination of these alternative affinity reagents and novel chromatographic enrichment strategies has helped expand cartography of the lysine methylome in recent years.
The functional annotation of lysine methylation sites has lagged significantly behind their identification. However, despite the relatively slow progress compared to mapping efforts, the molecular functions for lysine methylation in a variety of cellular processes is becoming clearer, as discussed above (Figure 2). These findings were facilitated, in large part, by approaches that connect the regulators of lysine methylation to their substrates. Knowledge of specific KMT or KDM substrate sites has greatly facilitated investigation of the function of specific lysine methylation events. This includes several MS-based approaches (Carlson et al., 2015; Olsen et al., 2016), but these approaches are limited since proteome-wide analysis of lysine methylation by MS is still not robust.
Beyond MS-based approaches, a variety of techniques have been developed that aim to identify substrates in vitro. One example uses an array fabricated with thousands of proteins immobilized on a functionalized glass surface. The microarray is subjected to an in vitro KMT reaction, facilitating protein-level identification of substrates. This approach was used to identify Mitogen-Activated Protein Kinase(3) 2 (MAP3K2) as a novel KMT substrate of SET and MYND domain containing Protein 3 (SMYD3), connecting lysine methylation signaling to RAS-driven cancers (Mazur et al., 2014). A key advantage to the use of protein arrays is identification of substrates at the protein level, removing the possibility of false positives due to the use of unstructured peptides. While it is appreciated that SET domain-containing KMTs recognize linear motifs, recent evidence suggests contacts outside of the active site impact substrate selectivity (Kublanovsky et al., 2018). A limitation to the use of protein arrays for substrate identification is that the common surface chemistry for protein immobilization relies on charge interactions, which prevents control over uniformity of protein display and may mask lysine side-chains. Control over protein folding is also a concern that increases the likelihood of false negatives with this approach.
Synthetic peptides on membrane support (SPOT) arrays have also found common use for KMT substrate identification (Rathert et al., 2008). In this approach, iterative on-membrane synthesis of peptides with differing amino acids in positions near the target lysine of a known substrate helps identify the positional dependence of a KMT motif. This approach was used to identify new substrates for several KMTs (Ardehali et al., 2017; Lanouette et al., 2015; Rathert et al., 2008; Rowe and Biggar, 2018). A limitation of this tolerance-based approach is the requirement of starting with a known substrate for derivation. As many KMT family members are still considered ‘orphan’ enzymes with no known substrates (Murn and Shi, 2017), this is considered a major drawback of this assay platform. In addition, since peptide substrates are synthesized directly on the array, precise quality control over peptide composition being queried is lacking.
An assay querying a lysine-oriented peptide library (K-OPL) was recently reported for KMT substrate identification (Cornett et al., 2018). Unlike SPOT peptide arrays, the approach required no prior knowledge of a substrate. The approach screened a large combinatorial peptide library to determine the sequence determinants of substrate selectivity plus and minus a defined number of residues from a target lysine. The resultant substrate selectivity profile was used in a variety of ways to guide downstream study of the lysine methylation regulator of interest. Novel substrates were revealed by ranking all the lysine residues in the proteome based on their propensity to be used as a substrate. The approach also has potential utility for evaluating how mutations in disease may rewire lysine methylation signaling networks, as was recently shown for phosphorylation (Creixell et al., 2015).
The demonstrated utility of these various techniques provides a comprehensive toolkit to map and annotate the function of lysine methylation. An important observation stemming from the use of these approaches is that the mapping of lysine methylation is likely incomplete. The use of substrate selectivity technologies (SPOT and K-OPL) reveal that the sequences most preferred by KMTs are difficult to detect by standard MS pipelines (Cornett et al., 2018). Likewise, as MS pipelines have improved with the use of newer affinity reagents, chromatographic strategies, and instrumentation, the lysine methylome has expanded. Overall, while the field has made progress developing new technologies to map lysine methylation and facilitate connections between lysine methylation sites and the enzymes responsible for regulating them, we predict there is still a lot of work ahead to comprehensively map and functionally annotate the lysine methylome. Future efforts integrating the use of these complementary technologies will help reveal the breadth of biological activities associated with lysine methylation and will be paramount for developing new strategies to exploit lysine methylation signaling for therapeutic benefit.
Concluding Remarks
Remarkable progress has been made in the last decade to reveal how lysine methylation fine-tunes the function of proteins in a variety of ways. These findings were fueled by new tools and technologies focused on mapping lysine methylation and connecting sites of lysine methylation to their regulatory proteins. The field of lysine methylation is now entering a new era that will be dominated by studies detailing the functional significance of lysine methylation at the mechanistic and biological levels. As evidenced by recent studies highlighted in this review, we suggest the field is just beginning to appreciate the expansive role of lysine methylation in regulating protein function, cellular processes, and disease progression.
Figure 4. Technologies developed to reveal KMT substrates.
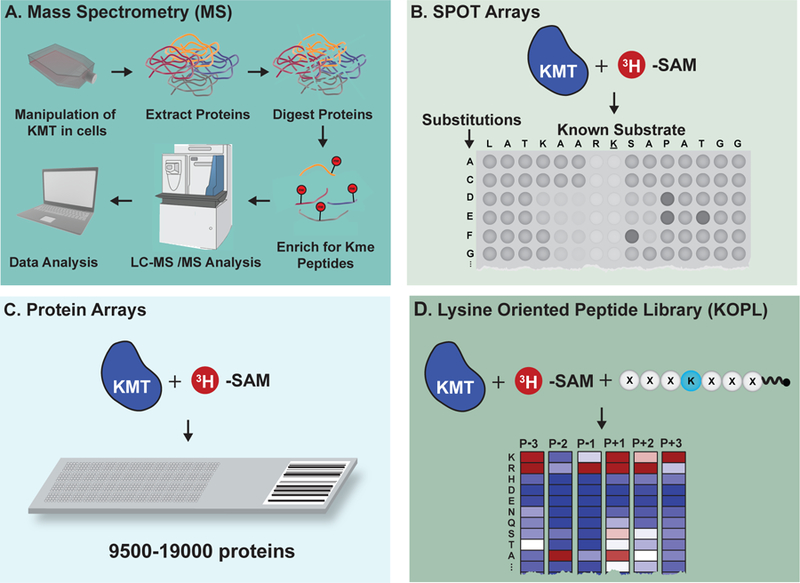
(A) Typical mass spectrometry (MS)-based proteomics pipeline for the identification of lysine methylation sites from cultured cells. (B) SPOT arrays evaluate the sequence tolerance of known KMT peptide substrates. (C) Protein arrays facilitate de novo identification of KMT substrates. (D) Lysine-oriented peptide libraries (K-OPL) reveal the sequence determinants of KMT substrate selectivity.
Acknowledgements
We thank members of the Rothbart laboratory for their insightful discussions and critical reading of this review. Work in the Rothbart laboratory is supported by grants from the National Institutes of Health (R35GM124736) and Van Andel Research Institute. Work in the Defossez lab is supported by Association pour la Recherche contre le Cancer (PGA1 RF20180206807), Agence Nationale de la Recherche (ANR-15-CE12–0012-01 and ANR-11-LABX-0071 under ANR-11-IDEX-0005–01), and Institut National du Cancer (INCa PLBio 2015–1-PLBio-01-DR A-1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ambler RP, and Rees MW (1959). Epsilon-N-Methyl-lysine in bacterial flagellar protein. Nature 184, 56–57. [DOI] [PubMed] [Google Scholar]
- Ardehali MB, Anselmo A, Cochrane JC, Kundu S, Sadreyev RI, and Kingston RE (2017). Polycomb Repressive Complex 2 Methylates Elongin A to Regulate Transcription. Mol. Cell 68, 872–884.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, Akalin A, and Schübeler D (2015). Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247. [DOI] [PubMed] [Google Scholar]
- Beyer S, Pontis J, Schirwis E, Battisti V, Rudolf A, Le Grand F, and Ait-Si-Ali S (2016). Canonical Wnt signalling regulates nuclear export of Setdb1 during skeletal muscle terminal differentiation. Cell Discov 2, 16037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggar KK, and Li SSC (2015). Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol 16, 5–17. [DOI] [PubMed] [Google Scholar]
- Biggar KK, Wang Z, and Li SSC (2017). SnapShot: Lysine Methylation beyond Histones. Mol. Cell 68, 1016–1016.e1. [DOI] [PubMed] [Google Scholar]
- Black JC, Van Rechem C, and Whetstine JR (2012). Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 48, 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremang M, Cuomo A, Agresta AM, Stugiewicz M, Spadotto V, and Bonaldi T (2013). Mass spectrometry-based identification and characterisation of lysine and arginine methylation in the human proteome. Mol. Biosyst 9, 2231. [DOI] [PubMed] [Google Scholar]
- Buratowski S (2009). Progression through the RNA Polymerase II CTD Cycle. Mol. Cell 36, 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X-J, and Garcia BA (2016). Global Proteomics Analysis of Protein Lysine Methylation. Curr. Protoc. Protein Sci 86, 24.8.1–24.8.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SM, and Gozani O (2016). Nonhistone Lysine Methylation in the Regulation of Cancer Pathways. Cold Spring Harb. Perspect. Med 6, a026435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SM, Moore KE, Sankaran SM, Reynoird N, Elias JE, and Gozani O (2015). A Proteomic Strategy Identifies Lysine Methylation of Splicing Factor snRNP70 by the SETMAR Enzyme. J. Biol. Chem 290, 12040–12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SM, Soulette CM, Yang Z, Elias JE, Brooks AN, and Gozani O (2017). RBM25 is a global splicing factor promoting inclusion of alternatively spliced exons and is itself regulated by lysine mono-methylation. J. Biol. Chem 292, 13381–13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr SM, Munro S, Sagum CA, Fedorov O, Bedford MT, and La Thangue NB (2017). Tudor-domain protein PHF20L1 reads lysine methylated retinoblastoma tumour suppressor protein. Cell Death Differ 24, 2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang FN, Navickas IJ, Chang CN, and Dancis BM (1976). Methylation of ribosomal proteins in HeLa cells. Arch. Biochem. Biophys 172, 627–633. [DOI] [PubMed] [Google Scholar]
- Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, Jiang Y, Wang C, and Cao X (2017). Methyltransferase SETD2-Mediated Methylation of STAT1 Is Critical for Interferon Antiviral Activity. Cell 170, 492–506.e14. [DOI] [PubMed] [Google Scholar]
- Cho H-S, Shimazu T, Toyokawa G, Daigo Y, Maehara Y, Hayami S, Ito A, Masuda K, Ikawa N, Field HI, et al. (2012). Enhanced HSP70 lysine methylation promotes proliferation of cancer cells through activation of Aurora kinase B. Nat. Commun 3, 1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, et al. (2004). Regulation of p53 activity through lysine methylation. Nature 432, 353–360. [DOI] [PubMed] [Google Scholar]
- Cornett EM, Dickson BM, Krajewski K, Spellmon N, Umstead A, Vaughan RM, Shaw KM, Versluis PP, Cowles MW, Brunzelle J, et al. (2018). A functional proteomics platform to reveal the sequence determinants of lysine methyltransferase substrate selectivity. Sci. Adv 4, eaav2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creixell P, Schoof EM, Simpson CD, Longden J, Miller CJ, Lou HJ, Perryman L, Cox TR, Zivanovic N, Palmeri A, et al. (2015). Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell 163, 202–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, and Jeltsch A (2010). The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem 285, 26114–26120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias JD, Rito T, Torlai Triglia E, Kukalev A, Ferrai C, Chotalia M, Brookes E, Kimura H, and Pombo A (2015). Methylation of RNA polymerase II non-consensus Lysine residues marks early transcription in mammalian cells. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea C-K, and Baltimore D (2009). Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc. Natl. Acad. Sci. U. S. A 106, 18972–18977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkouris M, Kontaki H, Stavropoulos A, Antonoglou A, Nikolaou KC, Samiotaki M, Szantai E, Saviolaki D, Brown PJ, Sideras P, et al. (2016). SET9-Mediated Regulation of TGF-beta Signaling Links Protein Methylation to Pulmonary Fibrosis. Cell Rep 15, 2733–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estève P-O, Terragni J, Deepti K, Chin HG, Dai N, Espejo A, Corrêa IR, Bedford MT, and Pradhan S (2014). Methyllysine Reader Plant Homeodomain (PHD) Finger Protein 20-like 1 (PHF20L1) Antagonizes DNA (Cytosine-5) Methyltransferase 1 (DNMT1) Proteasomal Degradation. J. Biol. Chem 289, 8277–8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Teng H, Wang Y, Liao G, Weng L, Li Y, Wang X, Jin J, Jiao C, Chen L, et al. (2018). SET1A-Mediated Mono-Methylation at K342 Regulates YAP Activation by Blocking Its Nuclear Export and Promotes Tumorigenesis. Cancer Cell 34, 103–118.e9. [DOI] [PubMed] [Google Scholar]
- Ferry L, Fournier A, Tsusaka T, Adelmant G, Shimazu T, Matano S, Kirsh O, Amouroux R, Dohmae N, Suzuki T, et al. (2017). Methylation of DNA Ligase 1 by G9a/GLP Recruits UHRF1 to Replicating DNA and Regulates DNA Methylation. Mol. Cell 67, 550–565.e5. [DOI] [PubMed] [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, and Allis CD (2005). Regulation of HP1–chromatin binding by histone H3 methylation and phosphorylation. Nature 438, 1116–1122. [DOI] [PubMed] [Google Scholar]
- Fnu S, Williamson EA, De Haro LP, Brenneman M, Wray J, Shaheen M, Radhakrishnan K, Lee S-H, Nickoloff JA, and Hromas R (2011). Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci 108, 540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y-Z, Pu M-T, Gowher H, Wu H-P, Ding J-P, Jeltsch A, and Xu G-L (2004). Chromatin targeting of de novo DNA methyltransferases by the PWWP domain. J. Biol. Chem 279, 25447–25454. [DOI] [PubMed] [Google Scholar]
- Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee KA, Yang V, Aguiar M, Kornhauser J, Jia X, et al. (2014). Immunoaffinity Enrichment and Mass Spectrometry Analysis of Protein Methylation. Mol. Cell. Proteomics [DOI] [PMC free article] [PubMed]
- He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, Wang G, Marquez VE, Orkin SH, and Pu WT (2012). PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev 26, 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Lipp JJ, Toh B-H, and Peters J-M (2005). Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 438, 1176–1180. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res [DOI] [PMC free article] [PubMed]
- Jakobsson ME, Małecki J, and Falnes PØ (2018a). Regulation of eukaryotic elongation factor 1 alpha (eEF1A) by dynamic lysine methylation. RNA Biol 15, 314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson ME, Małecki JM, Halabelian L, Nilges BS, Pinto R, Kudithipudi S, Munk S, Davydova E, Zuhairi FR, Arrowsmith CH, et al. (2018b). The dual methyltransferase METTL13 targets N terminus and Lys55 of eEF1A and modulates codon-specific translation rates. Nat. Commun 9, 3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, and Zhang Y (2006). The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 442, 312–316. [DOI] [PubMed] [Google Scholar]
- Kori S, Ferry L, Matano S, Jimenji T, Kodera N, Tsusaka T, Matsumura R, Oda T, Sato M, Dohmae N, et al. (2019). Structure of the UHRF1 Tandem Tudor Domain Bound to a Methylated Non-histone Protein, LIG1, Reveals Rules for Binding and Regulation. Structure 27, 485–496.e7. [DOI] [PubMed] [Google Scholar]
- Kublanovsky M, Aharoni A, and Levy D (2018). Enhanced PKMT-substrate recognition through non active-site interactions. Biochem. Biophys. Res. Commun 501, 1029–1033. [DOI] [PubMed] [Google Scholar]
- Lanouette S, Davey JA, Elisma F, Ning Z, Figeys D, Chica RA, and Couture J-F (2015). Discovery of substrates for a SET domain lysine methyltransferase predicted by multistate computational protein design. Structure 23, 206–215. [DOI] [PubMed] [Google Scholar]
- Lee JM, Lee JS, Kim H, Kim K, Park H, Kim J-Y, Lee SH, Kim IS, Kim J, Lee M, et al. (2012). EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell 48, 572–586. [DOI] [PubMed] [Google Scholar]
- Levy D, Kuo AJ, Chang Y, Schaefer U, Kitson C, Cheung P, Espejo A, Zee BM, Liu CL, Tangsombatvisit S, et al. (2011a). Lysine methylation of the NF-κB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-κB signaling. Nat. Immunol 12, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Liu CL, Yang Z, Newman AM, Alizadeh AA, Utz PJ, and Gozani O (2011b). A proteomic approach for the identification of novel lysine methyltransferase substrates. Epigenetics Chromatin 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhen XT, Denton E, Marsden BD, and Schapira M (2012). ChromoHub: a data hub for navigators of chromatin-mediated signalling. Bioinformatics 28, 2205–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Hausmann S, Carlson SM, Fuentes ME, Francis JW, Pillai R, Lofgren SM, Hulea L, Tandoc K, Lu J, et al. (2019). METTL13 Methylation of eEF1A Increases Translational Output to Promote Tumorigenesis. Cell 176, 491–504.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Chen Z, Xu C, Leng X, Cao H, Ouyang G, and Xiao W (2015). Repression of hypoxia-inducible factor α signaling by Set7-mediated methylation. Nucleic Acids Res 43, 5081–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M (2018). Chemical and Biochemical Perspectives of Protein Lysine Methylation. Chem. Rev 118, 6656–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Małecki J, Aileni VK, Ho AYY, Schwarz J, Moen A, Sørensen V, Nilges BS, Jakobsson ME, Leidel SA, and Falnes PØ (2017). The novel lysine specific methyltransferase METTL21B affects mRNA translation through inducible and dynamic methylation of Lys-165 in human eukaryotic elongation factor 1 alpha (eEF1A). Nucleic Acids Res 45, gkx002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Suzuki T, Vougiouklakis T, Nakamura Y, Yoshioka Y, Dohmae N, Hamamoto R, Deng X, Wang R, and Park J-H (2017). Critical roles of SMYD2-mediated β-catenin methylation for nuclear translocation and activation of Wnt signaling. Oncotarget 8, 55837–55847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur PK, Reynoird N, Khatri P, Jansen PWTC, Wilkinson AW, Liu S, Barbash O, Van Aller GS, Huddleston M, Dhanak D, et al. (2014). SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 510, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KE, Carlson SM, Camp ND, Cheung P, James RG, Chua KF, Wolf-Yadlin A, and Gozani O (2013). A general molecular affinity strategy for global detection and proteomic analysis of lysine methylation. Mol. Cell 50, 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murn J, and Shi Y (2017). The winding path of protein methylation research: Milestones and new frontiers. Nat. Rev. Mol. Cell Biol 18, 517–527. [DOI] [PubMed] [Google Scholar]
- Murray K (1964). The occurence of epsilon-N-methyl lysine in histones. Biochemistry 3, 10–15. [DOI] [PubMed] [Google Scholar]
- Musselman CA, Lalonde M-E, Côté J, and Kutateladze TG (2012). Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol 19, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Z, Star AT, Mierzwa A, Lanouette S, Mayne J, Couture J-F, and Figeys D (2016). A charge-suppressing strategy for probing protein methylation. Chem. Commun. (Camb) 52, 5474–5477. [DOI] [PubMed] [Google Scholar]
- Olsen JB, Cao X-J, Han B, Chen LH, Horvath A, Richardson TI, Campbell RM, Garcia BA, and Nguyen H (2016). Quantitative Profiling of the Activity of Protein Lysine Methyltransferase SMYD2 Using SILAC-Based Proteomics. Mol. Cell. Proteomics 15, 892–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankow S, Bamberger C, and Yates JR (2019). A posttranslational modification code for CFTR maturation is altered in cystic fibrosis. Sci. Signal 12, eaan7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IY, Powell RT, Tripathi DN, Dere R, Ho TH, Blasius TL, Chiang Y-C, Davis IJ, Fahey CC, Hacker KE, et al. (2016). Dual Chromatin and Cytoskeletal Remodeling by SETD2. Cell 166, 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, Komatsu Y, Shinkai Y, Cheng X, and Jeltsch A (2008). Protein lysine methyltransferase G9a acts on non-histone targets. Nat. Chem. Biol 4, 344–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599. [DOI] [PubMed] [Google Scholar]
- Van Rechem C, Black JC, Boukhali M, Aryee MJ, Graslund S, Haas W, Benes CH, and Whetstine JR (2015). Lysine Demethylase KDM4A Associates with Translation Machinery and Regulates Protein Synthesis. Cancer Discov 5, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, and Strahl BD (2014). Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta - Gene Regul. Mech 1839, 627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, Krajewski K, Strahl BD, and Fuchs SM (2012). Peptide Microarrays to Interrogate the “Histone Code.” In Methods in Enzymology, pp. 107–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, and Thibault P (2013). The coming of age of phosphoproteomics--from large data sets to inference of protein functions. Mol. Cell. Proteomics 12, 3453–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe EM, and Biggar KK (2018). An optimized method using peptide arrays for the identification of in vitro substrates of lysine methyltransferase enzymes. MethodsX 5, 118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, and Reinberg D (2008). Is there a code embedded in proteins that is based on post-translational modifications? Nat. Rev. Mol. Cell Biol 9, 815–820. [DOI] [PubMed] [Google Scholar]
- Strahl BD, and Allis CD (2000). The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Subramanian K, Jia D, Kapoor-Vazirani P, Powell DR, Collins RE, Sharma D, Peng J, Cheng X, and Vertino PM (2008). Regulation of Estrogen Receptor α by the SET7 Lysine Methyltransferase. Mol. Cell 30, 336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsusaka T, Kikuchi M, Shimazu T, Suzuki T, Sohtome Y, Akakabe M, Sodeoka M, Dohmae N, Umehara T, and Shinkai Y (2018). Tri-methylation of ATF7IP by G9a/GLP recruits the chromodomain protein MPP8. Epigenetics Chromatin 11, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vershinin Z, Feldman M, Levy D, Goliand I, and Elia N (2019). The methyltransferase SETD6 regulates Mitotic progression through PLK1 methylation. Proc. Natl. Acad. Sci 116, 1235–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss K, Forne I, Descostes N, Hintermair C, Schuller R, Maqbool MA, Heidemann M, Flatley A, Imhof A, Gut M, et al. (2015). Site-specific methylation and acetylation of lysine residues in the C-terminal domain (CTD) of RNA polymerase II. Transcription 6, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wang Q, Xu X, Xie B, Zhao Y, Li N, and Cao X (2017). The methyltransferase NSD3 promotes antiviral innate immunity via direct lysine methylation of IRF3. J. Exp. Med 214, 3597–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Long J, Gao Y, Zhang W, Han F, Xu C, Sun L, Yang S-C, Lan J, Hou Z, et al. (2019). SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat. Cell Biol 21, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Dong M, Mao J, Wang Y, Jin Y, Ye M, and Zou H (2016). Antibody-Free Approach for the Global Analysis of Protein Methylation. Anal. Chem 88, 11319–11327. [DOI] [PubMed] [Google Scholar]
- Wang R, Huang M, Li L, Kaneko T, Voss C, Zhang L, Xia J, and Li SSC (2018). Affinity Purification of Methyllysine Proteome by Site-Specific Covalent Conjugation. Anal. Chem 90, 13876–13881. [DOI] [PubMed] [Google Scholar]
- Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, et al. (2006). Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125, 467–481. [DOI] [PubMed] [Google Scholar]
- Wilkinson A, and Gozani O (2014). Histone-Binding Domains: Strategies for Discovery and Characterization. Biochim. Biophys. Acta 1839, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Cheng Z, Sun M, Wan X, Liu P, He T, Tan M, and Zhao Y (2015). A chemical proteomics approach for global analysis of lysine monomethylome profiling. Mol. Cell. Proteomics 14, 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Hao Y, Tao L, Peng S, Rao C, Chen H, You H, Dong M, and Yuan Z (2012). Lysine methylation of FOXO3 regulates oxidative stress-induced neuronal cell death. EMBO Rep 13, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wu X, Shi T, Xu H, Yi J, Shen H, Huang M, Shu X, Wang F, Peng B, et al. (2016). Regulation of Transcription Factor Yin Yang 1 by SET7/9-mediated Lysine Methylation. Sci. Rep 6, 21718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]