

Abstract
The three-dimensional organization of chromosomes can have a profound impact on their replication and expression. The chromosomes of higher eukaryotes possess discrete compartments that are characterized by differing transcriptional activities. Contrastingly, most bacterial chromosomes have simpler organization with local domains, the boundaries of which are influenced by gene expression. Numerous studies have revealed that the higher-order architectures of bacterial and eukaryotic chromosomes are dependent on the actions of Structural Maintenance of Chromosomes (SMC) superfamily protein complexes, in particular the near-universal condensin complex. Intriguingly, however, many archaea, including members of the genus Sulfolobus do not encode canonical condensin. We describe chromosome conformation capture experiments on Sulfolobus species. These reveal the presence of distinct domains along Sulfolobus chromosomes that undergo discrete and specific higher-order interactions, thus defining two compartment types. We observe causal linkages between compartment identity, gene expression and binding of a hitherto uncharacterized SMC superfamily protein that we term “coalescin”.
Graphical Abstract
In Brief
Insights into the chromosome architecture of archaea lacking canonical condensin reveal a two-domain compartmentalization resembling eukaryotic A/B compartments that influences gene expression and is maintained by a new class of SMC protein
Introduction
The interplay of the three-dimensional shape of chromosomes and DNA-based processes, including gene expression and DNA replication, is complex and can be causal and consequential in nature. Studies in bacteria and eukaryotes have revealed that chromosomes are organized into multiscale structural entities such as self-interacting domains (termed TADs in eukaryotes and CIDs in bacteria). In metazoans, an additional level of interactions leads to the definition of distinct A- and B-type compartments (Bonev and Cavalli, 2016; Lieberman-Aiden et al., 2009; Rowley and Corces, 2018). In both bacteria and eukaryotes. Structural Maintenance of Chromosomes (SMC) proteins play fundamental roles in mediating chromosome organization. In particular, the SMC complex, condensin, has been implicated in facilitating the higher-order organization of chromosomes (Hirano, 2016). Significantly, condensin is conserved in most bacteria and many archaea and contains two copies of a SMC subunit and two additional kleisen subunits, ScpA and ScpB. Recent studies in bacteria have revealed condensin-dependent higher-order organization of chromosomes. In Bacillus subtilis, condensin is loaded at ParB binding sites near the replication origin and is believed to facilitate “zipping” up of the two chromosome arms via a loop-extrusion process (Wang et al. 2015; Marbouty et al., 2015). In the γ-proteobacteria, condensin has been supplanted by an alternate SMC complex, comprised of the SMC protein MukB and two kleisen subunits MukE and MukF. Recent studies have implicated MukBEF in sculpting the higher-order organization of the E. coli chromosome (Lioy et al., 2018). Thus, in both bacteria and eukaryotes, members of the SMC family of ATPases play pivotal roles in shaping chromosome architecture (Jeppsson et al., 2014; Nolivos and Sherratt, 2014; van Ruiten and Rowland, 2018).
Phylogenetic studies indicated that the lineage that gave rise to Archaea and Eukarya separated from Bacteria prior to the emergence of archaeal and eukaryal domains. In agreement with this proposal, many aspects of the information processing machineries of Archaea and Eukarya are fundamentally related and distinct from their counterparts in Bacteria. Numerous studies in a range of archaeal model systems have confirmed the orthology of the archaeal and eukaryotic information processing machineries. The hyperthermophilic crenarchaeal genus Sulfolobus is of particular interest, as its chromosome dynamics exhibit both bacterial and eukaryotic features (Lindas and Bernander, 2013; Samson and Bell, 2011). Like many bacteria, Sulfolobus species have single circular chromosomes, ranging in size from 2.2 to 3 megabases. The chromosomes are replicated from three replication origins, each of which fires once per cell cycle (Duggin et al., 2008; Lundgren et al., 2004; Robinson and Bell, 2007; Robinson et al., 2004). Newborn Sulfolobus cells have a brief G1 period with a single copy of the chromosome. Upon completion of S phase, which is mediated by proteins homologous to components of the eukaryotic DNA replication machinery, cells spend half their cell cycle in a G2 phase during which sister chromatids exhibit cohesion (Robinson et al., 2007). Chromosomes segregation likely involves a system related to the ParAB system of bacteria (Kalliomaa-Sanford et al., 2012). Cells then divide using orthologs of the eukaryotic ESCRT apparatus (Lindas et al., 2008; Samson et al., 2008).
Although our knowledge of the biochemistry and structural biology of archaeal information processing machineries has expanded significantly, chromosome conformation in these organisms has only been described at the level of gross nucleoid morphology (Poplawski and Bernander, 1997). Essentially nothing is known about the nature, establishment and maintenance of archaeal chromosome conformation. Chromosome organization in Sulfolobus is especially enigmatic, because crenarchaea lack clear homologs of condensin-type SMC (Kamada and Barilla, 2018). In the following, we investigate the three-dimensional organization of the Sulfolobus chromosome. Our results reveal the presence of sub-megabase scale domains along the chromosome. These domains contribute to two compartment types. We uncover a role for a novel SMC protein, termed coalescin, in establishing domain identity. Further, we establish a causal relationship between transcription, ClsN occupancy and domain integrity. Our work thus establishes a paradigm for organization of chromosomes in the third domain of life and, given the evolutionary relationship between archaea and eukaryotes, yields insight into the evolution of eukaryotic chromosome conformation.
Results
Plaid pattern architecture of archaeal chromosomes
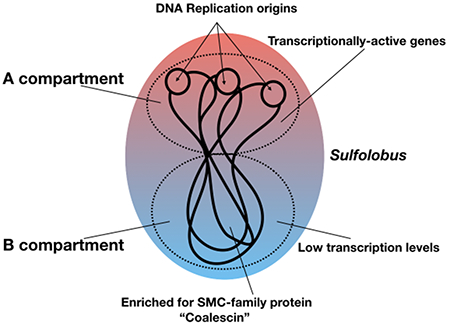
To investigate the three-dimensional (3D) organization of Sulfolobus chromosomes we first performed Hi-C experiments on Sulfolobus acidocaldarius and the distantly related Sulfolobus islandicus. Cells were crosslinked during asynchronous growth, in which most cells are in G2 phase (Bernander and Poplawski, 1997). The chromosomal DNA was digested with HindIII, end-labeled with biotin, and subjected to proximity ligation followed by biotin purification and paired-end sequencing (Figures 1A, S1 and S2). We were able to generate Hi-C contact matrices in a highly reproducible manner. Strikingly, the contact matrices we obtained exhibit plaid patterns formed by regional enrichment and depletion of long-range interactions. More specifically, we observe domains of interaction along the primary diagonal of the plot and substantial off-diagonal signals indicative of defined inter-domain interactions. Importantly, contact matrices of non-crosslinked DNA show uniform signal intensities, ruling out the possibility that the plaid patterns arose from sequencing artifacts (Figure S2).
Figure 1. Sulfolobus Chromosomes are Organized into Two Compartments.
(A) Hi-C results obtained from asynchronously growing S. acidocaldarius (left) and S. islandicus (right). The contact matrices represent interaction frequencies for pairs of 15-kb (S. acidocaldarius) and 30-kb (S. islandicus) bins. Three biological replicates were used for the analysis.
(B) The matrices in (A) were converted into Pearson correlation matrices (top) and subsequently used for principal component analysis. The first principal component is shown as a compartment index (bottom).
(C) 3D representations of the contact matrices in (A). Each bin in the matrices is represented as a bead. Beads corresponding to oriC1, oriC2, and oriC3 are shown in magenta, cyan, and green respectively. The other beads are colored based on the compartment to which they belong.
See also Figures S1, S2 and S3.
The exclusive and defined nature of inter-domain interactions that we observe in the plaid pattern suggest the existence of a compartment-like higher-order organization. To define the compartments in S. acidocaldarius and S. islandicus, we converted the contact matrices into Pearson correlation matrices and used them for principal component analysis as performed in the original Hi-C study (Figures 1B and S1B) (Lieberman-Aiden et al., 2009). The first principal component defined a compartment index. We found that all origins of replication (oriC1, oriC2, and oriC3) in each species fall into the same compartment as defined by positive compartment index values. As described later, the origin-containing and the inter-origin region compartments show higher and lower gene expression levels respectively. As this was reminiscent of the observation that eukaryotic A/B compartments exhibit higher and lower gene expression activities respectively, we shall therefore refer to the origin-containing compartment as the A compartment and the non-origin compartment as the B compartment.
We note that the domains we observe are significantly larger in scale than the CIDs observed in bacterial Hi-C experiments (Le et al., 2013; Lioy et al., 2018; Marbouty et al., 2015; Trussart et al., 2017; Wang et al., 2015). Indeed, the plaid patterns that we observe are reminiscent of the contact maps obtained in studies of eukaryotic chromosomes (Lieberman-Aiden et al., 2009). To test for the presence of smaller domains, we quantified the directional preference of Hi-C contacts, the metric used to identify bacterial CIDs (Le et al., 2013; Lioy et al., 2018; Marbouty et al., 2015; Wang et al., 2015). At the scale of 300 kb, this analysis identified domains that corresponded well with A/B domains. A major difference is that, in S. acidocaldarius, the directional preference plot divides a genomic segment containing oriC2 and oriC1 into three regions of similar sizes (Figure S3A). Smaller subdomains are not clearly observed at the scale of 300 kb or even of 90 kb, which is comparable with scales adopted to locate bacterial CIDs (Le et al., 2013; Lioy et al., 2018; Marbouty et al., 2015; Wang et al., 2015). We also calculated the expected contact probability of loci separated by genomic distance s to investigate the existence of self-interacting domains (Nuebler et al., 2018; Schwarzer et al., 2017). In the CID-harboring bacteria Caulobacter crescentus and B. subtilis (Le et al., 2013; Wang et al., 2015), contact probability declines gradually when s is smaller than ~100 kb and abruptly when s is larger (Figure S3B). The transition in the plots reflect the self-interacting nature of CIDs, whose average size is ~170 kb (Le et al., 2013). In contrast, there are no similar transitions in the contact probability plots of S. acidocaldarius and S. islandicus.
We generated 3D representations of the S. acidocaldarius and S. islandicus chromosomes by processing the Hi-C data with ShRec3D (Lesne et al., 2014). In the generated models, loci in the A and B compartments tend to segregate into distinct spaces (Figure 1C). As a result, the chromosomes are bent into arc-like shapes. Although the 3D representations are based on averages of DNA-DNA interactions in cell populations, we note that similar crescent structures have been reported in a light microscopy study that visualized the gross morphology of the chromosome of S. acidocaldarius (Poplawski and Bernander, 1997).
DNA replication and domain structure
The segregation of the replication origin-containing A-domains and inter-origin B-domains into distinct compartment classes suggested that DNA replication might be playing an active role in establishing chromosome architecture. The three origins of S. islandicus are each specified by a distinct initiator protein. Two origins are defined by proteins related to eukaryotic Orc1 and Cdc6; oriC1 by Orc1-1 and oriC2 by Orc1-3. The third origin, oriC3, is specified by WhiP, a distant relative of eukaryotic Cdt1 (Figure S4A). Accordingly, we exploited our previously described Δorc1-1 and Δorc-1,orc1-3 strains of S. islandicus that lack replication initiation at oriC1 or both oriC1 and oriC2 respectively (Samson et al., 2013). In addition, we created a new strain in which cis-acting oriC1 sequences were deleted. Marker frequency analysis confirmed that oriC1 is no longer active in this strain (Figure S4B). We emphasize that in Sulfolobus, replication termination is mediated by passive fork-collision rather than in a bacterial site-specific manner (Duggin et al., 2011; Samson et al., 2013). Thus, termination simply occurs half-way between the active replication origins. Strikingly, despite the reprogramming of the cells’ replication profiles, the Hi-C data that we generated for the wild-type and the three mutant strains are essentially indistinguishable (Figures S4C and S4D). Thus, the activity of origins of replication is not required for establishment or maintenance of either A- or B-type domain identity or compartment formation.
Coalescin, a novel Sulfolobus-encoded SMC protein
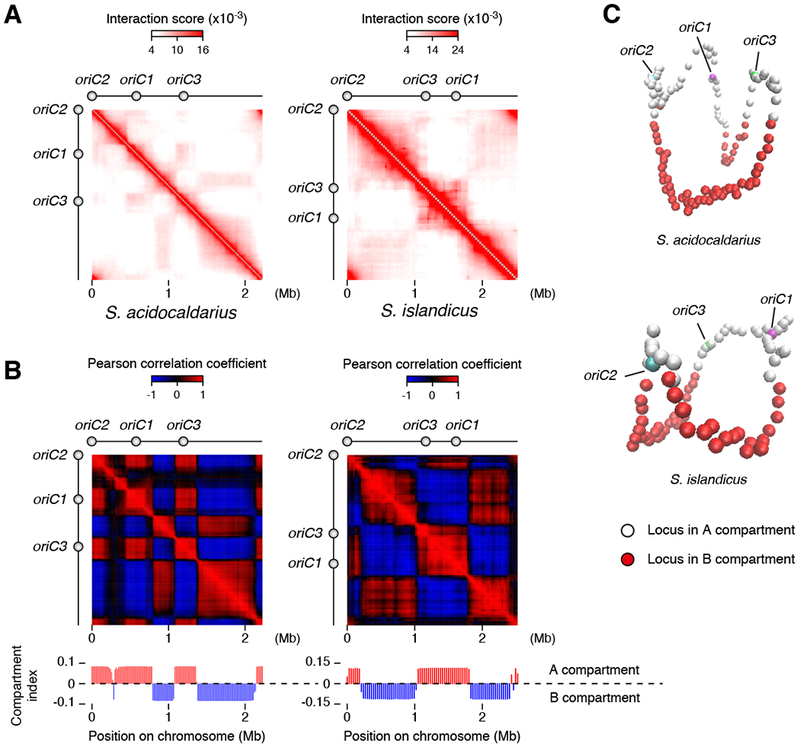
In both bacterial and eukaryotic species, SMC proteins play pivotal roles in sculpting local and higher-order chromosome architecture. In particular, the condensin complex is highly conserved across all three domains of life with the notable exception of the crenarchaea, the archaeal phylum to which Sulfolobus belongs. How then, in the absence of condensin, is the strikingly non-random organization of the Sulfolobus chromosome established? We speculated that Sulfolobus may have undergone a non-orthologous gene displacement event, substituting an unknown protein for condensin. Accordingly, we searched for genes encoding SMC superfamily members in the Sulfolobus genome. We identified the universally conserved gene for the Rad50 DNA repair protein and a second novel open reading frame, encoded by the saci_0046 gene. We focused on the saci_0046 gene; hereafter we will refer to it and its homologs as ClsN or “coalescin”. Significantly, the clsN gene is restricted to the Sulfolobales and is absent from other organisms that possess canonical condensin (Figure S5A). The protein encoded by clsN possesses the SMC signatures of a split ABC-type ATPase module separated by extensive regions of coiled-coil (Figure 2A). Structural modeling using the Phyre2 server predicted structural similarity between S. acidocaldarius ClsN and the Pyrococcus yayanosii condensin Smc head and adjacent coiled coil (Figure S5B) (Diebold-Durand et al., 2017; Kelley et al., 2015). 77% of the CIsN sequence was modeled with 100% confidence despite only 14% amino acid identity. Intriguingly, while the coiled-coils in condensin and cohesin are interrupted by a hinge domain, we note the presence of a pair of conserved cysteine residues in the middle of the ClsN coiled-coil, suggesting that, as in the Rad50 family of SMC-like proteins, ClsN may possess a zinc hook motif (Hopfner et al., 2002). Notably, ClsN and Rad50 are the only two SMC superfamily proteins encoded within the Sulfolobus chromosome.
Figure 2. Coalescin is a Novel Sulfolobus-encoded SMC Protein.
(A) Diagram of the domain organization of ClsN showing the bipartite ATPase module separated by extensive predicted coiled-coil. The sequences of the Walker A, Walker B and candidate zinc-hook motifs are shown.
(B) Growth phase-dependent expression of ClsN. Protein samples collected from a culture of S. islandicus at the time points numbered on the growth curve (left) were run on a 10 % SDS-PAGE gel. ClsN and a loading control, PCNA3, were detected by western blotting (right).
(C) Chromatin fractionation of ClsN in exponential and stationary phase S. islandicus cells. After separating cell extract into chromatin (C) and soluble (S) fractions, total protein (T) and extract fractions were separated by SDS-PAGE. ClsN and soluble and chromatin markers, Vps4 and Reptin respectively, were detected by western blotting.
(D) ChIP-Seq analysis of ClsN enrichment on S. acidocaldarius (upper) and S. islandicus (lower) chromosomes. The B domains are shaded in gray. Two biological replicates were used for the analysis. All ChIP data is normalized to input DNA. Genome coordinates are shown on the X axes. Above the plots, the location of peaks of less than 5 kb width are shown in black. Peaks greater than 5 kb that are shared between the two species are in red and those that are species-specific are in blue. The positions of loci referred to in the text are indicated and insertion sequences (ISs) highlighted in pink. Insertion sequences were identified using ISsaga (Varani et al., 2011), and potential false-positive hits were omitted. Bar graphs to the right of the panels indicating chromosomal localization quantify the relative distribution of ClsN between A and B compartments. ClsN enrichment is calculated as ChIP read coverage normalized to input for the distinct compartments.
See also Figure S5.
Western blotting revealed that ClsN is present during exponential and stationary phases of cultures (Figure 2B). Semi-quantitative western blotting revealed it to be an abundant protein in S. islandicus cells, with approximately 70,000 molecules of monomer per cell. The protein is less abundant in S. acidocaldarius, with 4,400 monomers per cell (Figure S5C). Notably, fractionation revealed that a higher proportion of ClsN is chromatin-associated in S. acidocaldarius (Figures 2C and 5A). We performed ChIP-Seq analysis to determine the chromosomal enrichment sites of this protein in both S. acidocaldarius and S. islandicus (Figure 2D). On a global level, we observe a striking, non-uniform distribution of the protein across the chromosome, with broad (> 5kb) zones of enrichment within B-domains and regions with lower ClsN levels correlating with A-domains (Figure 2D and Figure S5). We observe conservation of binding loci between the two species with the flagellar operon, a heterodisulphide reductase gene cluster and fatty acid metabolism genes being sites of broad peaks of high ClsN occupancy. We also observed peaks of CIsN binding in the vicinity of the CRISPR-Cas arrays in both species and a significant enrichment of ClsN at insertion sequences (Figure S5).
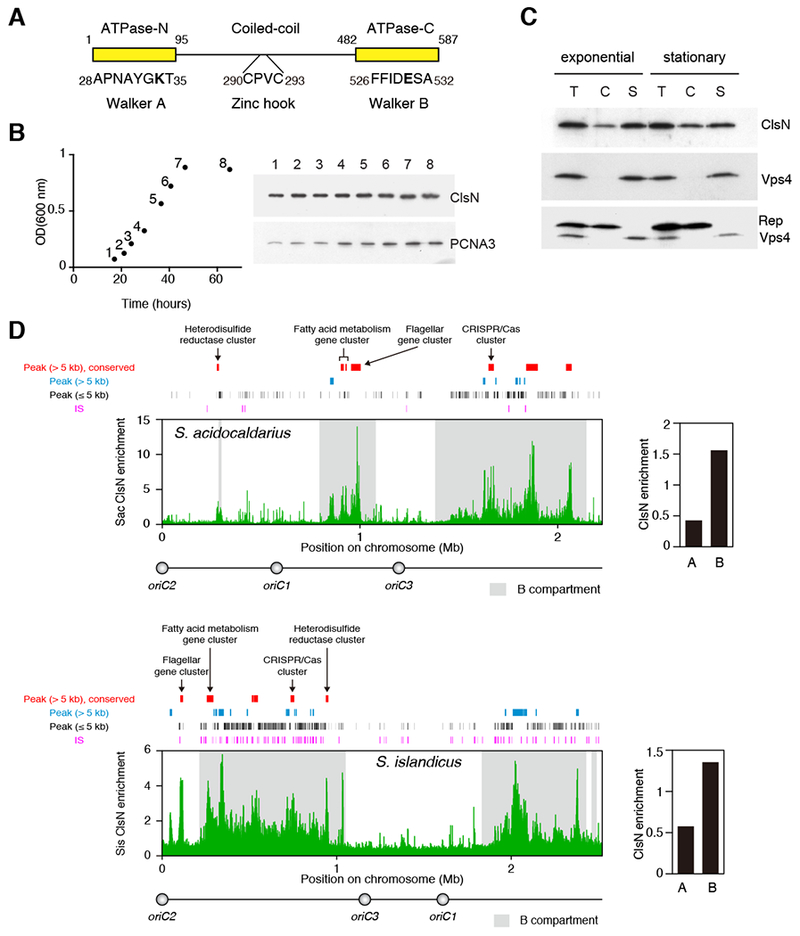
Figure 5. Perturbation of Transcriptional Landscape Alters the Chromosomal Distribution of ClsN and Chromosome Domain Organization.
(A) Chromatin fractionation of ClsN in S. acidocaldarius after treatment with actinomycin D. After separating cell extract into chromatin (C) and soluble (S) fractions, total protein (T) and extract fractions were run on SDS-PAGE. ClsN and soluble and chromatin markers, Vps4 and Reptin respectively, were detected by western blotting. Protein samples were collected before actinomycin D treatment and 15 and 30 minutes after treatment.
(B) Changes in chromosome conformation and ClsN distribution in S. acidocaldarius across a time course of actinomycin D treatment. Top panels: Hi-C contact matrices. Upper middle panels: compartment index plots, the A compartment is in red, the B compartment is in blue. Lower middle panels: ClsN ChIP-seq profiles at 1-kb resolution. All samples were normalized to the input DNA from the corresponding time point. Bottom panels: Enrichment of ClsN in the domains divided by the in the domains. ClsN enrichment is calculated as ChIP read coverage normalized to input for the domains. The location of each domain is based on the untreated condition.
(C) Distance-normalized frequency at the various timepoints of genomic contacts within the A compartment (A-A), within the B compartment (B-B) and between A and B compartments (A-B), based on the compartment identities in the untreated sample. See also Figure S7.
A recent genome-wide transposon mutagenesis screen in S. islandicus revealed cIsN to be an essential gene (Zhang et al., 2018). In agreement, our attempts to delete the gene were unsuccessful. We therefore cloned the clsN gene into a shuttle vector to direct arabinose-inducible expression in S. islandicus. In addition to the vector for overexpressing the wild-type gene, we created constructs to mediate arabinose-inducible overexpression of wild-type, Walker A, Walker B and zinc-hook mutants of ClsN. Significantly, we were only able to recover transformants with either empty vector or wild-type ClsN, suggesting that even basal expression of the mutated forms of ClsN was highly toxic to Sulfolobus. Indeed, flow cytometry revealed that even the modest, approximately 1.5-fold, levels of overexpression of ClsN prior to induction with arabinose (Figure 3A – lanes marked “T” and Figure S6A) resulted in an aberrant cell cycle distribution, with elevated populations of cells with less than 1C and greater than 2C chromosome content (Figure 3B). Induction by arabinose increased the ClsN level a further two-fold (Figure 3A and Figure S6A). Interestingly, elevating ClsN levels selectively augmented the chromatin-associated ClsN fraction (Figure 3A). ChIP-Seq revealed that ClsN overexpression leads to enhanced ClsN localization within the B-domains (Figure 3E and S6B).
Figure 3. Coalescin Overexpression from a Plasmid leads to Enhanced Compartmentalization and Intermolecular DNA Interactions at Coalescin-enriched Sites.
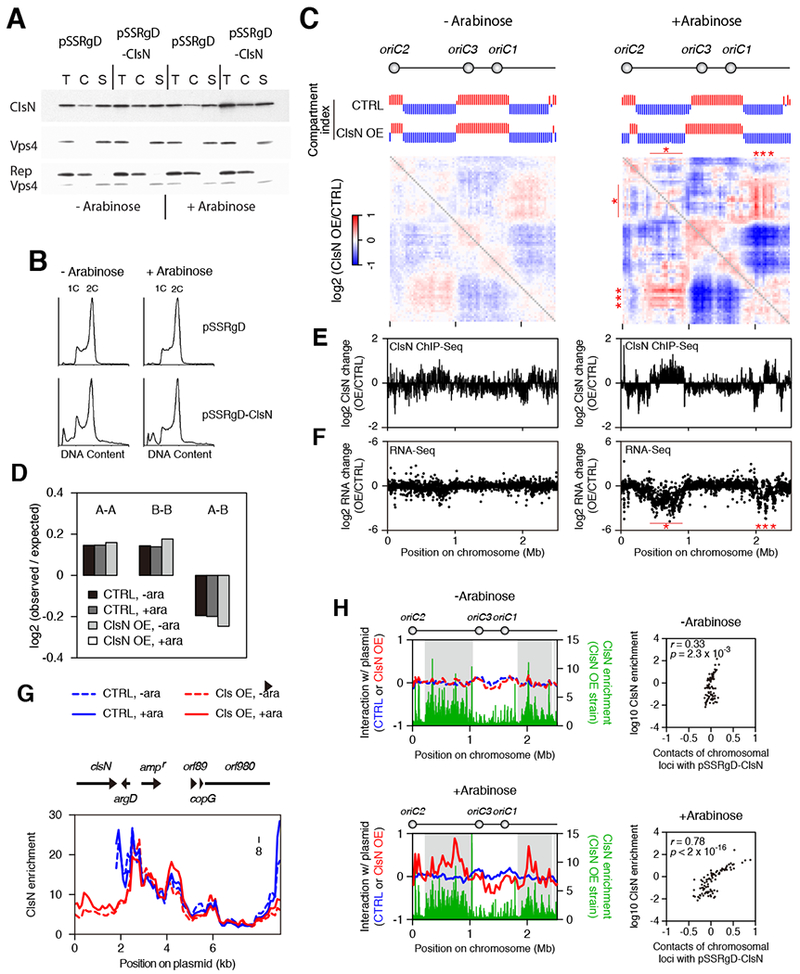
(A) Chromatin fractionation of ClsN in S. islandicus before and after four hours of induction of ClsN with arabinose. Cells containing the empty vector pSSRgD or the overexpression vector pSSRgD-ClsN were used. After separating cell extract into chromatin (C) and soluble (S) fractions, total protein (T) and extract fractions were run on SDS-PAGE. ClsN and soluble and chromatin markers, Vps4 and Reptin respectively, were detected by western blotting.
(B) Flow cytometry profiles of the ClsN overexpression strain and control strain before and after four hours of induction with arabinose. Cells containing one and two chromosomes (1C and 2C) are indicated.
(C) Ratios of Hi-C interaction scores between S. islandicus strains containing either of the pSSRgD-ClsN overexpression plasmid (ClsN OE) or pSSRgD empty control vector (CTRL). Cells were collected before and after arabinose induction (“−Arabinose” and “+Arabinose” respectively). Compartment index plots are also shown where the A and B compartments are indicated by red and blue bars respectively.
(D) Distance-normalized (observed/expected) frequency of genomic contacts is shown for interactions within the A compartment (A-A), within the B compartment (B-B), and between the different compartments (A-B). Frequencies were calculated for cells containing the pSSRgD empty vector or pSSRgD-ClsN overexpression vector before and after induction with arabinose (−ara and +ara respectively). See STAR Methods for more detail.
(E) ClsN ChIP-Seq. The log2 values of changes in ClsN occupancy (at 5-kb resolution) of the overexpression strain relative to the empty vector are plotted for cells grown in before (left panel) or after (right panel) the addition of arabinose. See also Figure S6B.
(F) RNA-Seq analysis on protein-coding genes. The log2 values of Transcripts Per Million (TPM) in the overexpression strain relative to the empty vector are plotted for cells grown in the absence (left panel) or presence of arabinose.
(G) ClsN ChIP-seq profiles on plasmids at 100-bp resolution. The locations and directions of the plasmid genes are shown on top. Profiles for the pSSRgD empty vector “CTRL” are shown in blue and pSSRgD-ClsN overexpression plasmid “ClsN OE” in red, with dashed lines indicating growth in the absence of arabinose and solid lines following 4 hours of induction with arabinose.
(H) Left panels: Hi-C interaction scores between plasmids (color coding as in panel 4F) and the chromosome at 30-kb resolution. The upper panel shows the results from cells grown without arabinose, the lower panel from cells following four hours of induction with arabinose. In each dataset, values are shown as log2 ratios relative to the median. ChIP-Seq profiles of ClsN enrichment (1-kb resolution) in the over-expression strain plots are superimposed in green. Grey shading indicates the position of the B domains in the parental S. islandicus strain. Right panels: Hi-C interaction score between pSSRgD-ClsN and the chromosome are compared with ClsN enrichment for each 30-kb region of the chromosome.
See also Figure S6.
Hi-C experiments showed a complex pattern of changes upon ClsN overexpression, with a general trend of enhanced intra-domain interactions (Figure 3C and 3D and Figure S6C). Additionally, enhancement of B-compartment formation occurs upon overexpression of ClsN. Interestingly, principle component analysis revealed that the region spanning genome coordinates 2430 – 120 kb is identified as a B-type domain in the overexpression strain, rather than part of an A-domain as it is in the empty-vector-containing and parental strains. Furthermore, this switch in domain identity correlates with a strongly enhanced peak of ClsN binding and reduction of transcription at that locus (Figure 3E and 3F). In addition, overexpression of ClsN leads to enhanced binding of the protein within its pre-existing zones of enrichment within the B domains. Importantly, the elevation of ClsN within B-domains correlates with strengthened compartmental organization. Finally, we examined the consequence of ClsN expression on gene expression and observed a reduction in levels of transcripts arising from the B domains (Figure 3F). Notably, the loci showing the greatest reductions in RNA level on ClsN overexpression are coincident with regions that show enhanced inter-domain interaction in the Hi-C maps (marked with asterisks in Figures 3C and 3F).
Coalescin overexpression modulates plasmid localization
Our ChIP-Seq analysis additionally revealed that both empty vector and overexpression plasmids were contacted by ClsN and ClsN was most highly associated with the bacterial plasmid-derived regions of the vectors (Figure 3G).
Given the above results and the frequent interactions between ClsN-enriched sites in the chromosome (Figures 1A and 3D), we wished to determine whether the ClsN overexpression plasmid might associate with specific regions on the S. islandicus chromosome. Fortuitously, the overexpression plasmid and the empty vector contain a HindIII site, which allowed us to use Hi-C to investigate their physical contacts with the chromosome. Whereas the empty vector and overexpression vector in non-inducing conditions interacted with all chromosomal regions uniformly, the overexpression plasmid interacted preferentially with ClsN-bound regions in the chromosome when ClsN was maximally overexpressed (Figure 3H, lower panels). Thus, in addition to the elevated levels of chromosomally associated ClsN facilitating strengthening of compartmentalization, they also promote a trans-association of the plasmid with those sites of chromosomal ClsN enrichment.
The influence of the transcription program on chromosome conformation
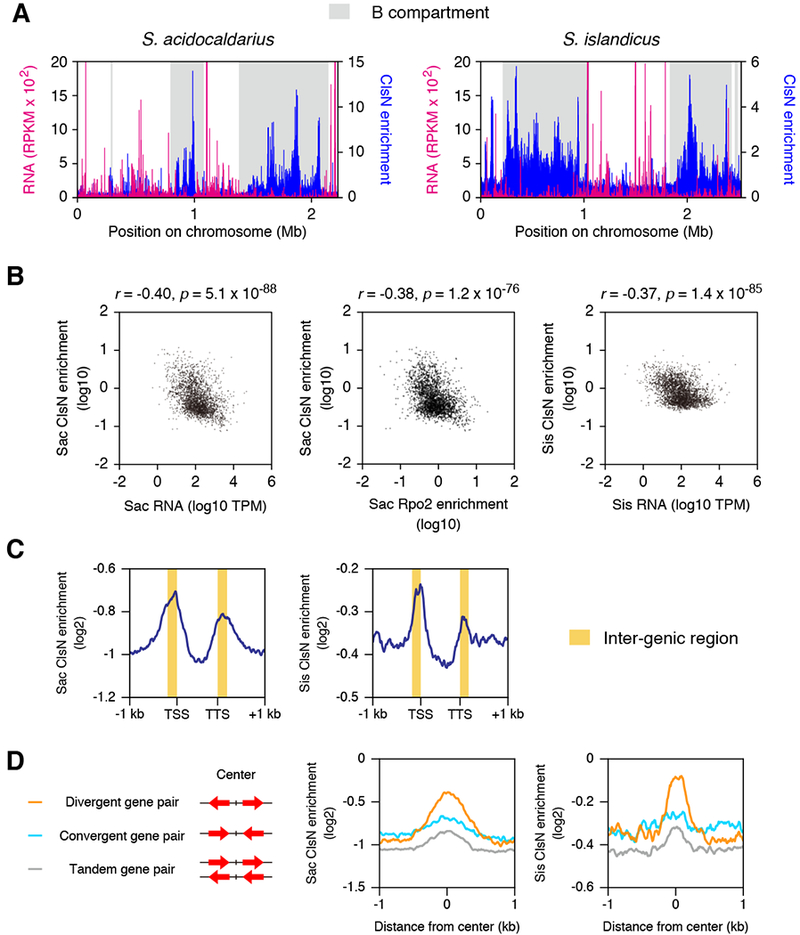
Overexpression of ClsN led to extensive reduction in transcription at sites with enhanced ClsN occupancy (Figure 3E and 3F). We wished to test whether the reciprocal was also true – could transcription influence ClsN localization and chromosome conformation? A previous study showed that essential, highly-expressed genes are more enriched in origin-proximal regions of Sulfolobus genomes than in origin-distal regions (Andersson et al., 2010). Thus, we speculated that there might be a relationship between ClsN binding and transcription. Comparison of ChIP-seq and RNA-seq profiles in wild-type cells revealed that ClsN was enriched in weakly expressed loci in both S. acidocaldarius and S. islandicus (Figures 4A), and this tendency was statistically significant (Figure 4B). A similar negative correlation was observed between ClsN and RNA polymerase occupancies in S. acidocaldarius (Figure 4B), indicating that ClsN binding anticorrelates with transcriptional activity rather than just RNA levels. Examination of the distribution of ClsN with respect to open reading frames across the chromosome reveals clsN to preferentially binds to inter-genic regions (Figure 4C). Further metagene analyses parsing gene pairs into convergent, divergent or tandem pairs revealed a preference for ClsN to be associated with promoter regions, with maximal enrichment at intergenic regions between divergent genes.
Figure 4. Coalescin Binding Anticorrelates with Gene Expression.
Two biological replicates per sample were used for RNA-seq and ChIP-seq analyses.
(A) RNA-seq and ChIP-seq profiles from asynchronously growing S. acidocaldarius and S. islandicus. RNA transcript abundance is plotted in magenta for 1-kb bins as Reads Per Kilobase per Million mapped reads (RPKM). Enrichment of ClsN is plotted for each 1 -kb bin in blue. Shaded regions indicate the B compartment. Genome coordinates are shown on the x-axis.
(B) Scatter plots comparing RNA level (TPM) and ClsN enrichment for protein-coding genes in S. acidocaldarius (left panel) and S. islandicus (right panel). Additionally, ClsN enrichment versus analysis of RNAP enrichment as adjudged by anti-Rpo2 ChIP-Seq are plotted (middle panel). Spearman rank correlation coefficients (r) and corresponding p-values are shown.
(C) Metagene analysis of ClsN distribution over open reading frames and intergenic regions are plotted for S. acidocaldarius (left) and S. islandicus (right). TTS = transcription termination site, TSS = transcription start site. The yellow shading indicates the mean intergenic spacing in the genomes.
(D) Metagene analysis of ClsN distribution over intergenic regions. The intergenic regions were categorized into three groups according to the orientations of two adjacent genes.
To determine whether transcription affects the genomic distribution of ClsN, we perturbed transcription in S. acidocaldarius cells by treating them with actinomycin D. This drug has previously been exploited in studies of mRNA stability in Sulfolobus (Andersson et al., 2006). Importantly, no detectable alterations to DNA integrity over the 30-minute timescale of the experiment were imparted by actinomycin D administration (Figures S7A and S7B). Also, the levels of chromatin-associated ClsN, as adjudged by fractionation, were unaffected by the treatment (Figure 5A). However, ChIP-Seq revealed ClsN distribution to be altered, in particular with decreased ClsN occupancy in the B1 and B2 domains (Figures 5B). In correlation with the observed de-localization of ClsN, Hi-C studies show that actinomycin D treatment almost completely eliminated the plaid pattern in the contact matrix (Figure 5B). The loss of the plaid pattern was manifested as a decrease in intra-compartment interactions and an increase in intercompartment interactions (Figure 5C). These results indicate that transcriptional state defines chromosomal distribution of ClsN and plays a role in maintaining the domain and compartment organization.
Restructuring of the transcription profile and chromosome conformation in stationary phase
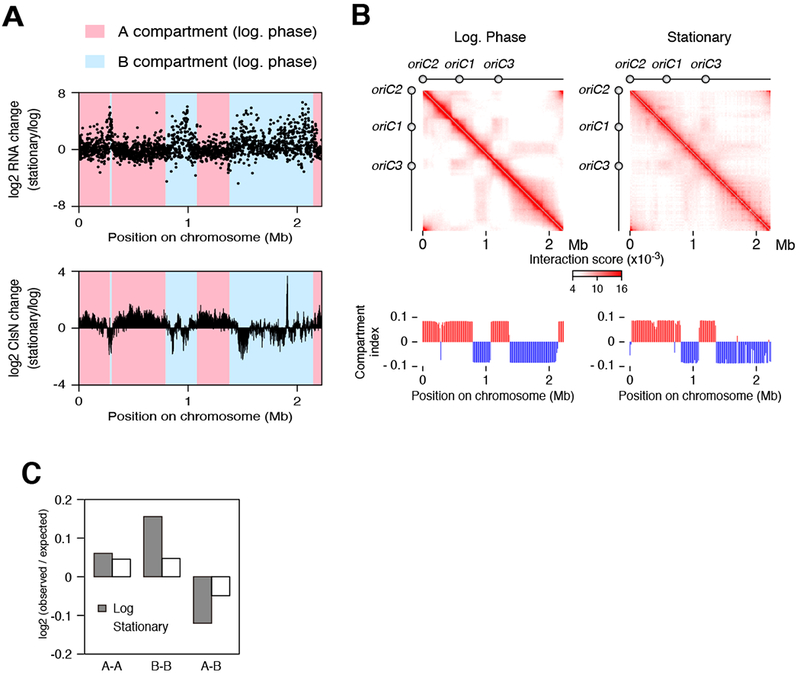
To address the interplay of transcription and chromosome conformation in unperturbed cells, we compared chromosome conformation in S. acidocaldarius cells growing in exponential phase and cells in stationary phase. Transcriptome profiling by RNA-Seq showed a significant remodeling of the gene expression program, with a clear increase in expression of genes localized in the B compartment (Figure 6A, upper panel). This biased distribution of stationary-phase induced genes mirrors the enrichment of essential, constitutively expressed genes in origin-proximal regions (Andersson et al., 2010). As described above, fractionation experiments demonstrated that ClsN remains chromatin-associated in stationary phase (Figure 2C). Indeed, there is a modest but reproducible increase in the chromatin-associated ClsN. Notably, however, ChIP-Seq revealed that the relative increase in expression of B compartment genes was accompanied by decreased occupancy of those domains by ClsN (Figure 6A, lower panel). Hi-C of the stationary-phase cells revealed that the extent of compartmentalization was weakened in stationary phase, manifested by a diffuse plaid pattern of the contact matrix (Figure 6B), a decrease in intra-compartment interactions, and an increase in inter-compartment interactions (Figure 6C). In agreement with our observations, previous light microscopy studies of exponential and stationary phase S. acidocaldarius cells revealed that nucleoids in stationary phase appear more diffuse and less structured than those in logarithmically growing cells (Poplawski and Bernander, 1997). Taken together, our data indicate that transcriptional activation of B-compartment-associated genes in stationary phase is accompanied by migration of ClsN from that compartment and consequent alterations in chromosome conformation.
Figure 6. Transcriptome Changes in Stationary Phase are Associated With Alterations of ClsN Localization and Chromosomal Interactions.
Two biological replicates per sample were used for RNA-seq and ChIP-seq analyses.
(A) Upper panel: changes in RNA expression after the entry into stationary phase. log2fold change in RNA level (TPM) is plotted for S. acidocaldarius protein-coding genes. Lower panel: log2-fold change in ClsN enrichment is plotted for each 1-kb bin.
(B) Hi-C contact matrices and compartment index profiles of S. acidocaldarius cells in log phase (left panel) and stationary phase (right panel).
(C) Distance-normalized frequency at the various timepoints of genomic contacts within the A compartment (A-A), within the B compartment (B-B) and between A and B compartments (A-B).
Alterations to gene expression could also result in local changes to DNA topology and clsN localization could be modulated by this rather than transcription per se. The genomes of Sulfolobus and other hyperthermophiles are believed to be maintained in a positively supercoiled state by the action of reverse gyrase. Sulfolobus islandicus responds to cold shock by rapidly introducing negative supercoiling, as adjudged by the topology of plasmids isolated from treated cells (Lopez-Garcia and Forterre, 1997, 1999). Specific linking differences (σ) were approximately +0.010 at the physiological 80 °C and shifted to as low as −0.033 after one hour of treatment at 65 °C. In vitro studies revealed that negative supercoiling facilitates gene transcription at reduced temperatures by overcoming barriers to promoter melting (Bell et al., 1998). We incubated cells at 65 °C for one hour and compared them to cells maintained at 78 °C. Flow cytometry revealed a modest decrease in 1C cells and a corresponding increase in S-phase cells upon cold-shock (Figure S8A). ClsN protein levels and bulk chromatin-association were unaltered by cold-shock (Figure S8B). ChIP-Seq revealed modest (<2-fold) changes in occupancy of loci distributed around the genome. (Figure S8C). Hi-C demonstrated that the location and boundaries of domains were unaltered as a consequence of cold-shock, with the exception of a small region of enhanced interaction emanating from a locus at genome coordinate ~1.5 Mb. However, while domain boundaries were preserved in general, short-range interactions were reduced and longer-range interactions were enhanced by cold-shock (Figures S8D and S8E). RNA-Seq reveals extensive and widely distributed reductions in gene expression that display a very modest (r = −0.19) but significant (p = 4 e−22) anti-correlation with changes in ClsN occupancy (Figure S8G).
Validation of Hi-C results by DNA fluorescence in situ hybridization
To independently test the genome compartmentalization observed in Hi-C, we performed DNA fluorescence in situ hybridization (DNA FISH) for S. acidocaldarius. In each experiment, probes were designed for two loci in the same compartment and a third locus in the opposing compartment (Figure S9A). In exponentially growing cells, despite similar genomic distances, loci in the same compartment were closer in three dimensions than loci in the different compartments (Figures S9B–D). These results corroborate our Hi-C results showing genome compartmentalization in exponentially growing cells. In contrast, upon entry into stationary phase, the differences in the distances of intra-compartment and inter-compartment probe pairs were diminished. Indeed, in stationary phase, loci found in distinct compartments in exponential phase were closer to one another than those previously found within a given exponential phase compartment type (Figure S9B–D).
Locus-specific effects of transcription on ClsN occupancy
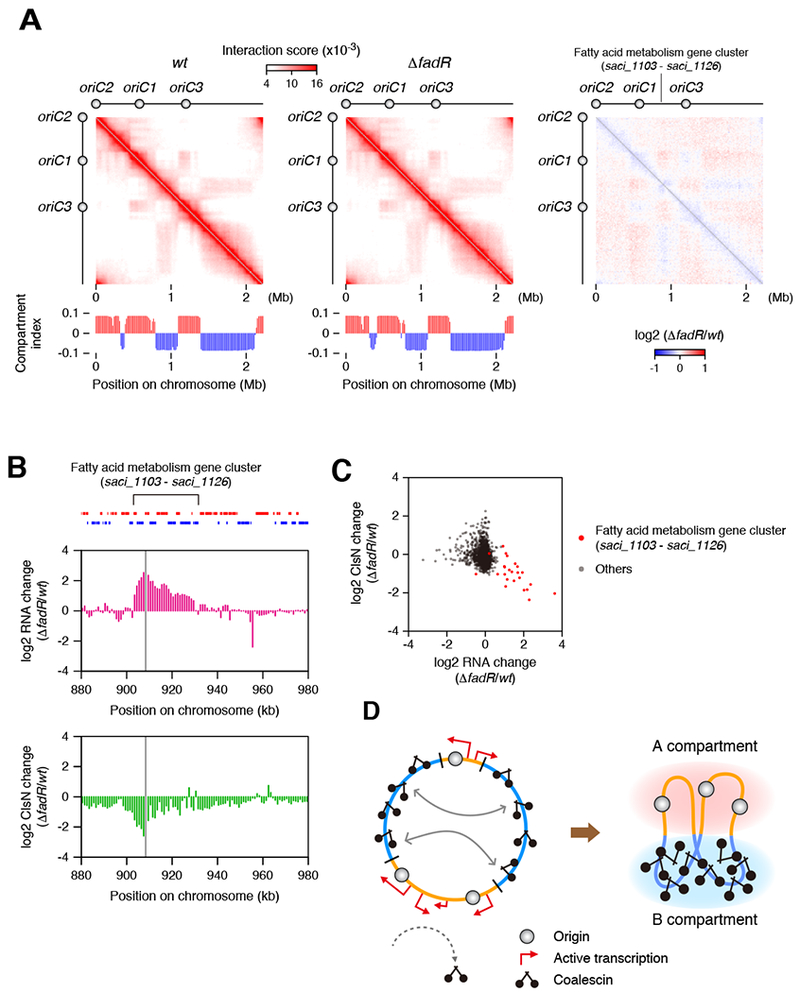
As described above, in both S. islandicus and S. acidocaldarius, strong peaks of ClsN occupancy are observed at a conserved fatty acid metabolism gene cluster (Figure 2D) that is transcriptionally repressed under standard growth conditions. Recent work in S. acidocaldarius has demonstrated that this 30 kb region containing 23 genes in at least 17 distinct transcription units is a regulon under the control of a TetR-family transcriptional repressor, FadR (Wang et al., 2019). Deletion of FadR results in constitutive expression of the locus. We obtained the fadR deletion strain (a kind gift from Dr. Eveline Peeters) and performed Hi-C, RNA-Seq and clsN ChIP-Seq on it and the parental strain MW001. Hi-C did not reveal any gross global differences in chromosome conformations of the two strains. Careful examination reveals some subtle alterations in the contact profile emanating from the vicinity of the fatty acid metabolism regulon (Figure 7A). RNA-Seq confirms the constitutive expression of the regulon in the deletion strain and ChIP-Seq reveals strong depletion of ClsN from this locus (Figure 7B). A genome-wide analysis of changes in RNA and ClsN binding reveals the changes to be specific to this locus (Figure 7C).
Figure 7. Locus Specific Effects on ClsN Occupancy Upon Constitutive Expression of a Regulon.
Two biological replicates per sample were used for analyses.
(A) Hi-C analysis in the wild-type and ΔfadR deletion strains of S. acidocaldarius.
(B) RNA-seq and ClsN ChIP-seq profiles in the vicinity of the fatty acid metabolism regulon. Log2-fold changes in RNA level (RPKM) and ClsN enrichment in ΔfadR were quantified for each 1-kb bin. The bin containing the fadR gene was omitted from the plots and shaded in gray. Locations of genes are shown on top (gene orientations are indicated by different colors).
(C) Log2-fold changes in RNA level (TPM) and ClsN enrichment in ΔfadR were quantified for protein-coding genes and shown as a scatter plot.
(D) Model for the organization of Sulfolobus chromosomes. Transcription excludes ClsN from the origin-containing A-domains. ClsN binding to the B-domains promotes coalescence of these regions. Coalescence could be promoted either by protein-protein interactions or via a loop-extrusion/catenation activity.
Discussion
We have shown that the chromosomes of S. acidocaldarius and S. islandicus are organized into two types of spatial compartments, the A/B compartments. The B compartment is enriched for coalescin, a hitherto uncharacterized SMC-like protein. Based on the lack of condensin in Sulfolobus and the data presented here, we propose that Sulfolobus species have evolved a unique condensin-independent mechanism of chromosome organization in which coalescin binds to less active genes and promotes coalescence of those loci to establish the B compartment (Figure 7D). Whether the Sulfolobus A compartment is actively structured by currently unidentified protein(s) or if it is simply defined by exclusion from the B-compartment is currently unknown.
In Sulfolobus, we observe that domain identity is largely governed by the transcription landscape. Upon cold-shock, Sulfolobus species induce topological changes in their DNA (Lopez-Garcia and Forterre, 1997, 1999). It is notable that, while we observe a general increase in long-range contacts and corresponding decrease in short-range contacts following cold-shock, the domain and compartment boundaries are largely preserved. Similarly, the global transcription and ClsN occupancy profiles show only modest local alterations. Thus, changes in topology do not grossly perturb chromosome conformation and, rather, presumably act to allow maintenance of the transcription program at reduced temperatures (Bell et al., 1998).
In contrast, ClsN relocalizes concomitant with cessation or alteration of the transcription program and this correlates with a genome-wide remodeling of chromosome structure. Furthermore, even modest elevation of ClsN levels alters the level of gene expression within the B-compartment. This suggests that ClsN acts as an effector to modulate chromosome conformation in response to transcriptional cues. Notably, ClsN facilitates tethering of plasmid sequences to ClsN-rich regions of the chromosome. Taken together, these observations suggest that, while the gross chromosomal architecture of sub-megabase domains and compartmentalization is morphologically reminiscent of eukaryotic chromosomes, the mechanistic basis of their formation may be quite distinct. ClsN (587 amino acids) is significantly smaller than the SMC subunits of condensin and possesses a candidate zinc hook domain - a feature found in the Rad50 family of SMC proteins, rather than a hinge. The Sulfolobus Rad50 is 864 amino acids and shares 19% sequence identity with ClsN. Rad50 has been implicated in tethering sister chromatids and juxtaposing DNA ends during DNA repair (Oh and Symington, 2018; Seeber et al., 2016). Another SMC superfamily member, RecN, is also believed to mediate tethering of sister chromatids (Vickridge et al., 2017). We speculate that ClsN may function in a similar manner to these proteins in facilitating both intra-domain and inter-domain interactions. One possibility is that clsN may help impose or constrain repressive loop-structures in the DNA. It is notable in this regard that the FadR repressor has been proposed to effect the coordinate control of its 30 kb regulon via long-range interactions between its four distinct binding sites (Wang et al., 2019).
The Sulfolobus chromosome conformation described here is fundamentally distinct from that of bacteria. More specifically, higher-order interactions of domains into genome compartments have not been reported for bacteria. Instead, most bacterial genomes adopt the colinear ori-ter configuration of chromosome arms formed by directional condensin-mediated loop extrusion from the origin towards the terminus (Le et al., 2013; Marbouty et al., 2015; Tran et al., 2017; Wang et al., 2017; Wang et al., 2015). Bacteria have presumably evolved this process to enable efficient chromosome segregation concomitant with DNA replication (Gruber and Errington, 2009; Sullivan et al., 2009). It may be significant that euryarchaea have cell cycle parameters reminiscent of those of bacteria, with multiple copies of their chromosomes and no discernable gap phases in the cell cycle (Samson and Bell, 2011). Euryarchaea also possess clear orthologs of the SMC and kleisen subunits of bacterial condensin (Figure S5A). In contrast, chromosome replication and segregation occur at temporally distinct cell cycle stages in eukaryotes and in members of the crenarchaeal phylum (Bernander and Poplawski, 1997; Robinson et al., 2007). It is possible that the shared cell cycle logic of crenarchaea and eukaryotes, with gap phases separating replication and segregation, may have loosened the constraints on chromosome conformation, thereby facilitating the evolution of domain and compartment-based chromosome architectures. In this regard, it is notable that eukaryotes may share a more recent ancestor with crenarchaea than with euryarchaea (Williams et al., 2013).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen D. Bell (stedbell@indiana.edu).The materials generated in this study are available from the Lead Contact with no restrictions.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strains, Media, and Growth Conditions
The S. acidocaldarius wild type strain DSM639 was grown in Brock’s medium (Brock et al., 1972) containing 0.2 % sucrose and 0.1 % tryptone, pH 3.2 at 78°C. For the MW001 and ΔfadR strains, uracil was added to 10 μg/ml (Wang et al., 2019). The S. islandicus Δorc1-1 and Δorc1-1/Δorc1-3 strains were constructed previously from S. islandicus REY15A (E233S) (Deng et al., 2009; Samson et al., 2013) and were grown in TSVY medium (containing mineral salts, 0.2% sucrose, 0.1% tryptone, 0.05% yeast extract, and 1X vitamin solution) supplemented with 20 μg/ml of uracil. The S. islandicus agmatine auxotroph, E235, was grown with 50 μg/ml of agmatine (Zhang and Whitaker, 2018). Except for stationary-phase experiments, cells were collected during log phase (OD600 ≈ 0.2). Stationary-phase cells of were collected after OD600 reached ~1.0. To block transcription, 1 mg/ml of actinomycin D (dissolved in DMSO) was added to a log-phase culture of DSM639 to a final concentration of 15 μg/ml. For the temperature shift experiment, S. islandicus E233S was cultured in a 78 °C water bath and shifte d to a 65 °C water bath for one hour. E. coli strains were grown in L-Broth (1% tryptone, 0.5% yeast estract 0.05%NaCl) supplemented with the appropriate antibiotics.
Genetic Manipulation of Sulfolobus
S. islandicus REY15A (E235) was grown to early log phase and pelleted at room temperature. The cells were washed three times with 20 mM sucrose at room temperature and were then resuspended in 20 mM sucrose to an OD600 = 10-20. 300-700 ng of pSSRgD-based plasmid or 1 μg of the linearized oriC1 knockout construct were added to 50 μl of cells. The cells were then electroporated at 1.2 kV, 25 μF, 600 Ω. 900 μl of pre-warmed high salt buffer (22.7 mM ammonium sulfate, 2.9 mM potassium sulfate, 1.3 mM potassium chloride, 9.3 mM glycine) were added and the cells were transferred to sterile tubes and incubated at 78 °C for 1 hour before plating on TSVY plates + uracil [20 μg/ml]. After incubation at 78 °C for 7-14 days, isolated colonies were used to inoculate 1.5-ml TSVY + uracil [20 μg/ml]. pSSRgD transformants were confirmed by colony PCR and western blot detection of the overexpressed ClsN protein. ΔoriC1 transformants were confirmed by colony PCR and sequencing of the oriC1 locus.
clsN overexpression
Strains containing pSSRgD (empty vector) and pSSRgD-clsN were grown to approximately OD600= 0.2 in TSVY + uracil [20 μg/ml]. D-(-)-arabinose was added to cultures to a final concentration of 0.2 %. All induced samples were collected at four hours post-induction. Overexpression of ClsN was confirmed by western analysis.
METHODS
Plasmid construction
The gene encoding ClsN was amplified from S. acidocaldarius genomic DNA with the SacSMC_F and SacSMC_noSTOP_R primers and cloned into pET30 via NdeI and XhoI restriction sites to generate pET30-SacSMC. The gene encoding ClsN was amplified from S. islandicus genomic DNA with the SMC_F and SMCnoSTOP_R primers and cloned into pET33 via NcoI and XhoI restriction sites to generate pET33-SisSMC.
pSSRgD was generated by PCR amplification of the Sulfolobus tokodaii argD gene (ST1348) using ArgD_BstXI and ArgD_XmaI. Following digestion with BstXI and XmaI, the fragment was ligated into the XmaI and SacI sites of pSSR to replace the simvastatin resistance cassette in pSSR (Zheng et al., 2012). The gene encoding ClsN was amplified from S. islandicus REY15A, digested with FauI and SalI, and cloned in to the NdeI and SalI sited of pSSRgD.
pCR-ArgD was generated by PCR amplification of the Sulfolobus tokodaii argD gene (ST1348) using StoArgD_FT7 StoArgD_RT3. The amplimer was assembled using the HiFi DNA Assembly kit (New England Biolabs) with the pCR-Script (Stratagene) backbone that had been amplified with primers reverse complementary to the T3 and T7 sequencing primers.
oriC1 Deletion Construct
ORBs 2 and 3 (Origin Recognition Box) and intervening DNA were deleted from oriC1 in S. islandicus REY15A (E235) by gene deletion. The S. tokadaii argD gene was amplified from pCR-ArgD with OriC1-UP and oriC1-DO forward and reverse primers containing flanks that were homologous to 75 bp of sequence upstream and downstream of the deletion target.
Hi-C
To crosslink DNA-DNA contacts, 20 ml of cell culture were mixed with (80-X) ml of ambient PBS buffer and X ml of 37% formaldehyde (Fisher Scientific). X was 5.4 for S. acidocaldarius (final formaldehyde concentration of 2%) and 10.8 for S. islandicus (final formaldehyde concentration of 4%). The reaction was incubated for 30 min at 25°C before quenching with glycine (at final concentrations of 125 mM for S. acidocaldarius and of 250 mM for S. islandicus) for 10 min at room temperature. The fixed cells were collected by centrifugation and washed twice with PBS buffer. The cell pellet was stored at −80°C until use.
A frozen pellet of S. acidocaldarius was resuspended and diluted to an OD600 of 4 in resuspension buffer (PBS buffer containing 1 mM EDTA), and 600 μl of the cell suspension were treated with proteinase K (at a final concentration of 24 μg/ml) for 15 min at 37°C to partially disrupt cell walls. The cells were collected by centrifugation and washed four times with resuspension buffer and once with 1 x NEBuffer 2. Centrifugation was done at 21,000 x g, 4°C for 5 min. A frozen pellet of S. islandicus was resuspended and diluted to an OD600 of 4 in 1 x NEBuffer 2, and 600 μl of the cell suspension were centrifuged. Each cell pellet was resuspended in 75 μl of 1 x NEBuffer 2. 75 μl of the cell suspension were mixed with 8.33 μl of 10% SDS and heated at 65°C for 15 min. After cooling on ice for 90 sec, 25 μl of cell lysate (corresponding to ~4 × 108 cells) were mixed with 41.8 μl of 1 x NEBuffer 2, 3.2 μl of 10 x NEBuffer 2, 20 μl of 10% Triton X-100. Chromosomal DNA was digested by adding 10 μl of 100 U/μl HindIII (NEB) to the mixture and incubating the reaction for 4 h at 37°C. After cooling on ice, the reaction was mixed with 12 μl of dNTP mixture (0.67 mM dATP, 0.67 mM dGTP, and 0.67 mM dTTP in TE buffer), 20 μl of 0.4 mM biotin-14-dCTP (Thermo Fisher Scientific), 3.6 μl of 10 x NEBuffer 2, and 1 μl of 5 U/μl DNA Polymerase i, Large (Klenow) Fragment (NEB). After incubation for 30 min at 20°C, the reaction was quenched by adding 15.4 μl of 10% SDS and 2.8 μl of 0.5 M EDTA and incubating for 5 min at room temperature. For ligation, the reaction was mixed with 1426 μl of nuclease-free water, 200 μl of 10 x T4 DNA Ligase Reaction Buffer (NEB), 200 μl of 10% Triton X-100, and 20 μl of 400 U/μl T4 DNA Ngase (NEB). The Ngation reaction was incubated for 4 h at 16°C before adding 200 μl of 10% SDS, 100 μl of 0.5 M EDTA, and 10 μl of 20 mg/ml proteinase K. To reverse crosslinks, the reaction was incubated for 6 h at 65°C and then for at least 6 h at 37°C. The DNA was extracted twice with phenol:chloroform:isoamyl alcohol and isopropanol-precipitated together with 40 μg of glycogen. The purified DNA was dissolved in 40 μl of 1 x NEBuffer 2 containing 0.1 mg/ml RNase A and incubated for 30 min at 37°C. To remove biotin from unligated DNA, the solution was mixed with 48 μl of nudease-free water, 6 μl of 10 x NEBuffer 2, 0.5 μl of 20 mg/ml BSA (Thermo Fisher Scientific, 5 μl of 2mM dGTP (in TE buffer), and 0.5 μl of 3 U/μl T4 DNA polymerase (NEB). After incubating for 4 h at 20°C, the DNA was extracted with phenol:chloroform:isoamyl alcohol and ethanol-precipitated. The DNA was dissolved in 100 μl of Buffer EB (QIAGEN) and sheared with a Bioruptor (Diagenode) at low power for 60 cycles (30-sec on, 30-sec off).
55.5 μl of the sheared DNA was used for library construction with NEBNext Ultra DNA Library Prep Kit for Illumina and NEBNex Multiplex Oligos for Illumina (NEB). First, end repair, adapter ligation, and size selection were performed according to the manufacturer’s instructions. The purified DNA was subjected to biotin purification with Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific). Before use, 10 μl/sample of beads were washed four times with B&W Buffer (5 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1M NaCl) and resuspended in 135 μl/sample of B&W Buffer. 135 μl of the bead suspension was added to each DNA solution and rotated for 30 min at room temperature. The beads were washed three times with B&W Buffer followed by one wash with 1 x NEBuffer 2 without DTT. Each sample was resuspended in 15 μl of 0.1 x TE (pH 8.0) and used for PCR in a 50-μl reaction for 14 cycles. The PCR products were purified with AMPure XP Beads (Beckman Coulter) and paired-end sequenced (43 bp x 2) on the Illumina NextSeq platform at the Center for Genomics and Bioinformatics at Indiana University.
Hi-C of Non-crosslinked DNA
Cells were harvested and resuspended in 600 μl of lysis buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 0.42% SDS, 0.1 mg/ml RNase A). After incubation for 20 min at room temperature, the cell suspension was treated with 0.2 mg/ml of proteinase K for 1.5 h at 37°C. DNA was then extracted four times with phenol:chloroform:isoamyl alcohol, ethanol-precipitated, and dissolved in 1 x NEBuffer 2. 55 μl of the solution (containing approximately 25 μg of DNA) were mixed with 42 μl of 1 x NEBuffer 2 and 3 μl of 100 U/ml of HindIII (NEB). After incubation for 2 h at 37°C, the reaction was mixed with 6 μl of dNTP mixture (0.67 mM dATP, 0.67 mM dGTP, and 0.67 mM dTTP in TE buffer), 10 μl of 0.4 mM biotin-14-dCTP (Thermo Fisher Scientific), 2 μl of 10 x NEBuffer 2, and 1 μl of 5 U/μl DNA Polymerase I, Large (Klenow) Fragment (NEB). After incubation for 30 min at 20°C, the DNA was extracted with phenol:chloroform:isoamyl alcohol, ethanol-precipitated, and dissolved in 25 μl of 1 x T4 DNA Ligase Reaction Buffer (NEB). 20 μl of the solution was treated with 1 μl of 400 U/μl T4 DNA ligase (NEB) for 3h at 16°C. After heating for 20 min at 65°C, the reaction was mixed with 23 μl of nuclease-free water, 10 x T4 DNA Ligase Reaction Buffer (NEB), 20 mg/ml BSA (Thermo Fisher Scientific), 2.5 μl of 2 mM dGTP (in TE buffer), and 0.25 μl of 3 U/μl T4 DNA polymerase (NEB). After incubation for 4 h at 20°C, the DNA was extracted with phenol:chloroform:isoamyl alcohol, ethanol-precipitated, and dissolved in 22 μl of Buffer EB (NEB). 15 μl of the solution were diluted in 87 μl of Buffer EB and sheared with a Bioruptor (Diagnogen) at low power for 40 cycles (30-sec on, 30-sec off). After treatment with 0.5 μl of 10 mg/ml RNase A for 30 min at 37°C, the DNA was used for library preparation as described for the Hi-C experiment with crosslinking.
Generating Hi-C Contact Maps
We mapped and processed raw Hi-C reads using HiC-Pro version 2.9.0 (Servant et al., 2015). Reads were mapped to the genome of S. acidocaldarius DSM639 (GenBank ID: CP000077.1) or S. islandicus REY15A (GenBank ID: CP002425.1) with default parameters. The start positions of the genomes were modified as explained in the next paragraph. For the overexpression experiment of clsN, reads were also mapped to the sequence of pSSRgD-ClsN. Invalid reads pairs including dangling ends, self-circle ligation, and duplicates were discarded. Remaining valid read were used to generate raw Hi-C matrices (see Table S1). The genome was binned at 15-kb for S. acidocaldarius and 30-kb for S. islandicus. When combining biological replicates, valid read pairs from them were pooled before generating raw Hi-C matrices. The intra-bin ligations were discarded by setting the values on the corresponding diagonal to zero. The data were iteratively corrected with the MAX_ITER parameter of 500. The obtained matrices were normalized so that the sum of interaction scores is equal to 1 for each row and column.
In our preliminary analysis of S. acidocaldarius, we noticed that interaction score was extraordinarily high between the first bin and the last bin. These bins are adjacent on the circular chromosome of S. acidocaldarius but not are neighbors based on the genomic coordinates. HiC-Pro failed to discard self-ligation junctions of the restriction fragment spanning the boundary of the two bins, as it was originally developed to analyze Hi-C data from eukaryotic linear chromosomes. To solve this problem, we redefined the genomic coordinates of each Sulfolobus species so that they started from the first base of the first restriction site in the original definition (S. acidocaldarius DSM639: 1,011 bp from the start, S. islandicus REY15A: 966 bp from the start). These modified genomes were used to map reads and generate Hi-C matrices. The obtained data were visualized and analyzed with R software (http://www.R-project.org).
Identification of Compartments
Compartment index was calculated according to previous work (Lieberman-Aiden et al., 2009). First, distance-normalized Hi-C matrices were generated by calculating the log2 ratio of interaction score versus expected value (genome-wide average of interaction scores for the corresponding genomic distance) for each pair of bins. Obtained matrices were converted into Pearson correlation matrices, in which the entry ai,j represents the Pearson correlation coefficient between the ith row and the jth column of the corresponding observed/expected matrices. Obtained Pearson correlation matrices were then used for principal component analysis. We defined the sign of the first principal component such that the value is positively correlated with RNA expression level. The first principal component value was used as compartment index. Generation of Pearson correlation matrices and principal component analysis were done with HiTC (Servant et al., 2012).
3D Model of Chromosome Structure
We converted Hi-C contact matrices into 3D models of chromosome structure using ShRec3D (Lesne et al., 2014). This algorithm assumes that the spatial distance between two loci are inversely proportional to the interaction score. All the 3D structures were rendered with VMD (Humphrey et al., 1996).
Quantification of Compartment Strength
To quantify compartment strength, every genomic bin was assigned to one of the two compartments according to compartment index. Based on this assignment, every bin pairs were classified as either of “A-A,” “B-B,” and “A-B.” For each of the three groups, the sum of observed interaction scores and the sum of expected interaction scores was calculated. Before this calculation, DNA contacts within 45 kb (S. acidocaldarius) or 60 kb (S. islandicus) were discarded, because such short-range interactions constitute a large part of genomic contacts in cells but reflect compartment strength very little. The log2 ratio of the observed sum versus the expected sum was used to estimate compartment strength.
Directional Preference of Hi-C contacts
Directional preference was calculated according to a previous study (Le et al. 2013). To calculate the directional preference for a given bin, we collected Hi-C interaction scores between the bin and bins located either downstream or upstream within the distance of 90 kb or 300 kb. We then compared log2 values of the upstream and downstream vectors by paired t-test. Directional preference was defined as t-value of the test.
Purification of Recombinant ClsN
S. acidocaldarius and S. islandicus clsN proteins were expressed with C-terminal His6-tags in E. coli Rosetta (DE3) cells. Cultures were grown in LB at 37 °C to an OD 600 = 0.6-0.8 and induced with 1 mM IPTG for 3 hours at 37 °C. Cells were lysed by French press in buffer (50 mM sodium phosphate, 300 mM NaCl, 5 mM imidazole, 10 % glycerol, 5 mM β-mercaptoethanol, pH 7.5) containing Roche Mini-Complete Protease Inhibitors. The soluble lysate was heat treated for 20 minutes at 65 °C, and the heat-stable fraction was purified first over Ni-NTA agarose (Qiagen) and then over a HiTrap Heparin column (GE Healthcare). Proteins were further purified over a HiLoad 26/600 Superdex 200 column (GE Healthcare) in 50 mM sodium phosphate, 150 mM NaCl, 10 % glycerol, 5 mM β-mercaptoethanol, pH 7.5.
Affinity Purification of Antibodies
1 mg of purified, recombinant S. acidocaldarius ClsN protein was coupled to a 1-ml HiTrap NHS-activated HP column (GE Healthcare) in standard coupling buffer (200 mM sodium carbonate, 500 mM NaCl, pH 8.3) according to the manufacturer’s protocol (GE Healthcare).
Rabbit anti-ClsN polyclonal serum was diluted 1:10 in 1X TBS, passed through a 0.45 μm filter, and recirculated through the affinity column at <1 ml/minute for 45 minutes at room temperature. The column was washed with 1X TBS, TBS-T, and then eluted with 100 mM glycine, pH 2.5 into Tris, pH 8.5, 100 mM final molarity. Fractions were spotted onto Immobilon-P membrane (Millipore) and those containing antibody were detected with anti-rabbit HRP secondary antibody.
ChIP-Seq
Chromatin immunoprecipitation (ChIP) was performed as described in (Samson et al., 2013). Cultures of Sulfolobus were crosslinked with 1 % formaldehyde for 20 minutes. After quenching with 125 mM glycine, the cells were pelleted and washed with 1X PBS. The pellets were resuspended in TBS-TT (20 mM Tris, 150 mM NaCl, 0.1 % Tween-20, 0.1 % Triton X-100, pH 7.5) and sonicated using a Diagenode Bioruptor to generate DNA fragments ranging from 200-1000 bp. Extract was then clarified by centrifugation. 10 μg of extract (based on protein concentration) were used in each 100 μl ChIP reaction. Samples were rotated for 2-3 hours with 3 μl of antiserum or 75 μl of purified anti-S. acidocaldarius ClsN antibodies at 4 °C. 25 μl of a 50 % slurry of protein A sepharose were then added and the samples were rotated for another hour at 4 °C. Each ChIP reaction was then washed five times at room temperature with TBS-TT, once with TBS-TT containing 500 mM NaCl, and once with TBS-TT containing 0.5 % Tween-20 and 0.5 % Triton X-100. Protein-DNA complexes were eluted from the protein A sepharose in 20 mM Tris, 10 mM EDTA, 0.5 % SDS, pH 7.8 at 65 °C for 30 minutes. Crosslinking was reversed and protein was digested by incubating the samples with 10 ng/μl Proteinase K for 6 hours at 65 °C followed by 10 hours at 37 °C. The samples were extracted with phenol/chloroform/isoamyl alcohol first, then chloroform alone and the DNA was precipitated in 100 % ethanol containing 20 μg of glycogen. After washing with 70 % ethanol and air drying, the DNA was resuspended in 50 μl TE buffer.
ChIP reactions were performed in triplicate and pooled. 50 μl of ChIP reactions and 100 ng of input DNA were used to construct DNA libraries using Illumina TruSeq ChIP Library Preparation Kits. The manufacturer’s instructions were followed, except DNA size selection was performed using a 0.6 X ratio of AMPure XP beads rather than purification by agarose gel. DNA quality, size and quantity were verified using a High Sensitivity DNA chip on an Agilent Technologies 2100 Bioanalyzer and with a Qubit dsDNA High Sensitivity Assay kit on a Qubit 2.0 Fluorometer (Invitrogen). Libraries were sequenced using the Illumina HiSeq platform at the Wellcome Trust Center for Human Genetics, Oxford, UK or the Illumina NextSeq platform at the Center for Genomics and Bioinformatics at Indiana University.
Reads from single or multiple biological replicates were mapped to the genome of S. acidocaldarius DSM639 or S. islandicus REY15A using Bowtie 2 version 2.3.2 with default parameters (Langmead and Salzberg, 2012). Multi-mapping reads were retained for downstream analyses. For a plasmid-containing strain of S. islandicus, reads were separately mapped to the genome or the corresponding plasmid. To calculate protein enrichment on the chromosome, ChIP-seq coverage of the region of interest was divided by input coverage of the region after normalizing the total number of reads mapped to the chromosome. To calculate protein enrichment on the plasmid, numbers of reads mapped to the chromosome and the plasmid were summed and used for read number normalization. Protein enrichment in protein-coding genes was calculated according to RefSeq annotation downloaded from UCSC Archaeal Genome Browser (http://archaea.ucsc.edu/index.html).
Metagene analyses on RefSeq protein-coding genes were performed using functions implemented in deepTools version 3.0.0 (Ramirez et al., 2016). After mapping, ChIP reads and input reads were compared using bamCompare (parameters: -bs 10, --scaleFactorsMethod readCount). The output file was processed using computeMatrix and plotProfile.
Pooled paired-end reads were used for peak calling with MACS2 (Zhang et al., 2008) using the option -m 1 50. Overlap of broad ClsN peaks with conserved gene cluster was examined using SynMap2 (Haug-Baltzell et al., 2017).
Marker Frequency Analysis (MFA-Seq)
MFA was performed by NGS sequencing on the Illumina NextSeq at the Center for genome Biology at Indiana University. DNA was isolated from exponentially growing (OD600 nm = 0.2) S. islandicus ΔoriC1 cells and, as reference, from a stationary phase population confirmed by flow cytometry to be exclusively G2 cells. ~28 million reads were obtained for both samples. Read counts were grouped in 1 kilobase bins and the count for exponentially growing cells divided by the stationary phase values. The midpoint of the lowest trough was set arbitrarily to a value of 1.
RNA-seq
RNA was extracted from S. acidocaldarius and S. islandicus using the method described previously (Samson et al., 2008). After treatment with DNase, the RNA was ethanol precipitated and dissolved in DEPC-treated water. Without ribosomal RNA depletion, the RNA samples were used for preparing strand-specific libraries with NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB) according to the protocol for use with purified mRNA or rRNA-depleted RNA in the manufacturer’s manual. RNA fragmentation was carried out based on RIN values determined by TapeStation (Agilent) at the Center for Genomics and Bioinformatics at Indiana University. The libraries were paired-end sequenced (43 bp x 2) on the Illumina NextSeq at the Center for Genomics and Bioinformatics at Indiana University.
Reads from biological replicates were pooled and mapped to both strands using Bowtie 2 version 2.3.2 (parameter: --maxins 1000) (Langmead and Salzberg, 2012). After removal of multi-mapping reads, the remaining reads were processed using bamCoverage implemented in deepTools version 3.0.0 (Ramirez et al., 2016) to calculate Reads Per Kilobase region per Million mapped reads (RPKM) for genomic bins of fixed size. To calculate Transcripts Per Million (TPM), reads from each biological replicate were mapped to RefSeq protein-coding genes in a strand-specific manner using Salmon version 0.8.2 (Patro et al., 2017). The average of the TPM values from biological replicates was used for downstream analyses.
Probe Preparation for DNA FISH
DNA fragments of 6.1-6.3 kb were amplified by PCR from S. acidocaldarius genomic DNA and cloned into pCR-XL-2-TOPO using TOPO XL-2 Complete PCR Cloning Kit (Thermo Fisher Scientific). The primers used for amplification are as follows. P1F and P1R, P2F and P2R, P3F and P3R, P4F and P4R and P5F and P5R. Fluorescently-labeled FISH probes were generated from these plasmids using FISH Tag DNA Multicolor Kit (Thermo Fisher Scientific). For each probe, 1 μg of plasmid DNA was used and the final product was dissolved in 12 μl of nuclease-free water.
DNA FISH
DNA FISH was performed as described previously (Robinson et al., 2007) with modifications. Approximately 5 × 108 cells were collected by centrifugation at room temperature. The cell pellet was resuspended in PBS containing 1.5 % paraformaldehyde and incubated at 25°C for 30 min. After centrifugation, the cell pellet was permeabilized by resuspension in 0.2% Triton X-100 and 0.04% SDS at 25°C for 20 min. After washing once with PBS, the cells were resuspended in PBS containing 0.1 mg/ml of RNase A and incubated at 25°C for 30 min. The cells were washed once with PBS and then once with pre-hybridization buffer (2 x SSC, 36.4 mM phosphate buffer [pH 6.9], 0.02% Ficoll, 0.02% polyvinylpyrrolidone, 60% formamide, 50 μg/ml BSA). The cells were resuspended in 40 μl of hybridization buffer (1.15 μl of a probe labeled with Alexa Fluor 488, 1.15 μl of a probe labeled with Alexa Fluor 647, 2 x SSC, 36.4 mM phosphate buffer [pH 6.9], 0.02% Ficoll, 0.02% polyvinylpyrrolidone, 60% formamide, 50 μg/ml BSA). The suspension was heated at 60°C for 10 min and then incubated at 30°C overnight. After adding 200 μl of wash buffer (2 x SSC, 60% formamide, prewarmed to 30°C), the cells were pelleted and washed twice with wash buffer, once with 2 x SSC, once with 1 x SSC, and then once with PBS. The cell pellet was resuspended in 120-200 μl of PBS, and 10 μl of the suspension were spread onto a polyD-lysine-coated coverslip. After partially air-dried, the coverslip was rinsed twice with PBS and inverted onto a slide spotted with 13 μl of VECTASHIELD (Vector Laboratories) containing 1.5 ng/μl of DAPI. The coverslip was sealed with nail polish.
Microscopy
DNA FISH samples were imaged using a Metemorph-driven Olympus BX-61, with a Z stack of 21 planes (0.1 μm step size) collected for each channel. 3D distances between fluorescent foci were quantified using the ImageJ plugin SpotDistance (Schober et al., 2008) with a visual inspection. Between 161 and 333 cells were analyzed per probe pair, as indicated in Figure 2C.
Chromatin Fractionation
Cells were harvested from Sulfolobus cultures at OD600 = 0.2 for exponential samples and OD600 = 1.0 for stationary samples. Pelleted cells were resuspended in 100 μl/0.1 OD600unit/ml of chromatin extraction buffer (25 mM HEPES, 15 mM MgCl2, 100 mM NaCl, 400 mM sorbitol, 0.5 % Triton-X100, pH 7.5) and incubated on ice for 10 minutes. Extracts were centrifuged for 20 minutes at 14,000 g, 4 °C. The soluble fraction was transferred to a new tube and the pelleted chromatin fraction was resuspended in a volume of chromatin extraction buffer equivalent to the soluble fraction. The chromatin fraction was then sonicated three times for 30 seconds each.
Flow Cytometry
Sulfolobus cells were fixed with 72% ethanol, stained with SYTOX Green (Invitrogen) and DNA contents were analyzed with an LSRII flow cytometer in the Flow Cytometry Facility at Indiana University.
Sequence Homology Search
To search for archaeal homologs of condensin-type SMC, blastp search was carried out on the NCBI website against each archaeal group shown in Figure S3. The sequence of Pyrococcus furiosus SMC (accession number Q8TZY2.2) was used as a query and the E-value cutoff was set to 10−5 We found that even hits of very high E-values and query coverages showed low sequence identities (~30%). This diversity is due to less stringent conservation of coiled-coil regions at the primary sequence level. Based on this variability, we defined hits of > 30% sequence identity and > 90% query coverage as bona fide homologs of condensin-type SMC. The blastp hits from Korarchaeota do not fulfil these criteria, but some of the hits resemble SMC in that they have Walker A motifs in N-termini, Walker B motifs in C-termini, and predicted coiled coils in the middle. These SMC-like sequences are composed of ~750 amino acids and quite shorter than the query sequence (1,177 amino acids). We defined these SMC-like proteins as distant homologs of condensin-type SMC. The distant homologs lack the zinc-hook motif (Cys-X-X-Cys) and therefore do not seem to be homologs of ClsN.
To search for archaeal homologs of ClsN, blastp search was carried out on the NCBI website against all archaea. The sequence of S. islandicus ClsN (accession numbers ADX84150.1) was used as a query and the the E-value cutoff was set to 10−5 The retrieved sequences form three groups: (i) sequences that are from the order Sulfolobales and exhibit relatively high sequence identities (36-100%) and high query coverages (> 90%); (ii) sequences that are from diverse orders and exhibit low sequence identities (21-26%) and high query coverages (> 90%); (iii) sequences whose sequences identities and query coverages are both low. We defined the first group as bona fide homologs of ClsN and the second as distant homologs. We obtained a few distant homologs of ClsN from each of the orders Heimdalarchaeota, Methanosarcinales, and Archaeoblobales. There were also three distant homologs in Bathyarchaeota, but they lack the zinc-hook motif and thus were discarded. The number of distant homologs obtained was larger (17 sequences) in Halobacteria, and blastp search using one of the hits as a query identified many homologous proteins in Halobacteria. Therefore, we conclude that distant homologs of ClsN are widely present in Halobacteria.
QUANTIFICATION AND STATISTICAL ANALYSIS
Hi-C data were analyzed using HiC-Pro (Servant et al., 2015), HiTC (Servant et al., 2012), and ShRec3D (Lesne et al., 2014). ChIP-seq data were analyzed using deepTools (Ramirez et al., 2016) and MACS2 (Zhang et al. 2008). RNA-seq data were analyzed using deepTools (Ramirez et al., 2016) and Salmon (Patro et al., 2017). DNA FISH data were analyzed using SpotDistance (Schober et al., 2008). If needed, additional quantification and statistical analyses were performed using R software (http://www.R-project.org). See Figure Legends and Methods for more details.
All experiments were repeated using independent cultures. The number of replicates was described in Figure Legends. When not stated otherwise, results from a single replicate were presented.
DATA AND CODE AVAIALBILITY
Data generated in the study are available at Gene Expression Omnibus (GEO) databank with accession code GSE128063.
Supplementary Material
Figure S1. Reproducibility of Hi-C Data. Related to Figure 1
(A) Scatter plots comparing Hi-C interaction scores between biological replicates. r indicates the Pearson correlation coefficient.
(B) Heat maps comparing Hi-C interaction scores between biological replicates.
(C) Comparison of the compartment index between biological replicates.
Figure S8. Effects of Cold Shock on the Chromosome Conformation of S. islandicus. Related to Figures 1, 5 and 6.
Two biological replicates were used for ChIP-seq, Hi-C, and RNA-seq analyses.
(A) Flow cytometry profiles of S. islandicus cells cultured at 78°C and 65°C.
(B) Chromatin fractionation. After separating cell extract into chromatin (C) and soluble (S) fractions, total protein (T) and extract fractions were run on SDS-PAGE. ClsN and soluble and chromatin markers, Vps4 and Reptin respectively, were detected by western blotting.
(C) log2-fold change in ClsN enrichment is plotted for each 1-kb bin.
(D) Effects of the cold shock were analyzed by Hi-C.
(E) Distance-normalized frequency of genomic contacts within the A compartment (A-A), within the B compartment (B-B) and between A and B compartments (A-B).
(F) log2 fold change in RNA level (TPM) was plotted for protein-coding genes in S. islandicus.
Figure S9. Correlation of Physical Proximities Measured by Hi-C and DNA FISH. Related to Figure 1 and Figure 6.
(A) Schematic showing the locations of FISH probes in the S. acidocaldarius chromosome.
(B) Representative images of S. acidocaldarius cells. Chromosomal DNA was stained with DAPI (blue) and either of the FISH probe pairs shown in (A). The scale bar indicates 1 μm.
(C) Violin plots of 3D distances between FISH foci. The black box indicates the interquartile range. The white circle in the box indicates the median. The whisker denotes all values of 1.5 x interquartile range. p-values were calculated by Wilcoxon rank sum test.
Figure S2. Sequence Properties of Sulfolobus Genomes in Relation to Hi-C. Related to Figure 1
(A) Length distributions of the HindIII fragments in the genomes of S. acidocaldarius and S. islandicus.
(B) GC % of genomic regions segmented by compartment index plots. The compartment index plots were generated using the datasets in Figure 1.
(C) Contact matrices arising from analyses of proximity ligation performed using non-crosslinked DNA purified from S. acidocaldarius (left panel) and S. islandicus (right panel). The ligation products were sequenced and analyzed as performed in Hi-C of crosslinked DNA.
(D) Hi-C reads were mapped to the genomes of S. acidocaldarius and S. islandicus. Read coverage is shown for each 1-kb bin.
Figure S3. Analyses of Domains. Related to Figure 1
(A) Directional preference of genomic contacts was calculated at 90-kb (top panels) and 300-kb scales (bottom panels) from the contact matrices of S. acidocaldarius and S. islandicus in Figure 1A. Contact biases towards downstream and upstream regions are indicated by positive (red) and negative (green) values respectively. Black horizontal lines indicate threshold values (p = 0.05, paired t-test). Arrowheads indicate potential domain boundaries identified by the directional preference analysis but not by the principal component analysis in Figure 1B.
(B) Contact probability plots of asynchronously growing S. acidocaldarius and S. islandicus are compared with those of C. crescentus and B. subtilis (Le et al., 2013; Wang et al., 2015). Contact probability P(s) was defined as the average of Hi-C interaction scores of bin pairs at a distance of s. P(s) was calculated for each of three biological replicates, and the average of them was further normalized to its maximum value Pmax(s). To calculate contact probabilities for C. crescentus and B. subtilis, normalized Hi-C data from previous studies were retrieved from Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo). The accession numbers of the data are C. crescentus: GSM1120445 and GSM1120446, B. subtilis: GSM1671399 and GSM1671400.
Figure S4. Inactivation of Replication Origin(s) Does Not Affect Chromosome Conformation in S. islandicus. Related to Figure 1
(A) Organization of replication origins, initiator protein genes and replication termination zones on the chromosome of S. islandicus.
(B) Marker frequency analysis of the ΔoriC1 strain.
(C) Hi-C analysis of wild-type and mutant strains of S. islandicus. Top panels: Hi-C contact matrices. Middle panels: compartment index plots. A-domains are shown in red, B-domains in blue. Bottom panels: Pearson correlation matrices.
(D) Ratios of Hi-C interaction scores between mutants and wild type.
Figure S5. Coalescin is an Abundant and Novel Sulfolobus-Encoded SMC superfamily Protein. Related to Figure 2
(A) Phyletic distribution of ClsN. The schematic phylogenetic tree is depicted according to Spang et al. (Spang et al., 2017). See STAR Methods for details.
(B) Model for ClsN structure generated using the Phyre 2 server.
(C) Western blot analyses correlating molar amounts of ClsN protein from specific numbers of cells to defined amounts of recombinant proteins. Analyses were performed with both S. islandicus extract and recombinant protein and S. acidocaldarius extract and recombinant protein. Calibration curves appear in the right-hand panels. Quantitation was performed using ImageJ.
(D) Analyses on ClsN peaks in S. acidocaldarius (Sac). Left panel: Width distribution of ClsN peaks. Middle panel: Scatter plot comparing peak width and enrichment of ClsN. Right panel: Box plots comparing ClsN enrichment in protein-coding genes and insertion sequences (ISs). All data points were additionally plotted for the ISs because of their small number (yellow diamonds). p-values were calculated by Wilcoxon rank sum test.
(E) The same analyses as in panel (D) were conducted on ClsN peaks in S. islandicus (Sis).
Figure S6. Effects of Coalescin Overexpression. Related to Figure 3
(A) Western analysis of ClsN levels in strains containing an empty vector control, pSSRgD, or the ClsN overexpression vector, pSSRgD-ClsN, before and after four hours of induction with arabinose. Vps4 was detected as a loading control (lower panel).
(B) ChIP-Seq analyses of ClsN enrichment using the overexpression strain (ClsN OE) and the empty vector control strain (CTRL). ChIP-seq was performed before (left panels) and after (right panels) four hours of induction with arabinose. All ChIP data is normalized to input DNA. Genome coordinates are shown on the X axes. Grey shading indicates the position of the B domains in the parental S. islandicus strain.
(C) Hi-C contact matrices of the overexpression strain and the empty vector strain before and after incubation with arabinose.
Figure S7. DNA Integrity is Unaffected by a 30-Minute Treatment with Actinomycin D. Related to Figure 5
(A) 3 μg of genomic DNA purified from S. acidocaldarius cells treated with actinomycin D for 0, 15, and 30 minutes were run on a 0.7 % agarose gel, stained with ethidium bromide, and detected by UV illumination. The genome of S. acidocaldarius is 2.2 Mbp.
(B) Flow cytometry profiles of S. acidocaldarius 0, 15, and 30 minutes after treatment with actinomycin D. Cells containing one and two chromosomes (1C and 2C) are indicated.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit αclsN (custom) | Eurogentec | N/A |
| Rabbit αPCNA3 (custom) | CovalAb | N/A |
| Rabbit αVps4 (custom) | CovalAb | N/A |
| Rabbit αReptin (custom) | Covance | N/A |
| Rabbit αRpo2 | Qureshi et al., 1997 | N/A |
| Goat αRabbit HRP | Pierce | Cat #65-6120 |
| Archaeal Strains | ||
| S. acidocaldarius DSM639 | Lab collection | N/A |
| S. islandicus E233S (ΔpyrEF ΔlacS) | Deng et al., 2009 | N/A |
| S. islandicus Δorc1-1 | Samson et al., 2013 | N/A |
| S. islandicus Δorc1-1 Δorc1-3 | Samson et al., 2013 | N/A |
| S. islandicus E235 (ΔargD) | Zhang and Whitaker, 2018 | N/A |
| S. acidocaldarius MW001 | Wang et al., 2019 | N/A |
| S. acidocaldarius ΔfadR | Wang et al., 2019 | N/A |
| S. islandicus ΔoriC1 | This study | N/A |
| Bacterial Strains | ||
| E. coli NEB 10 beta was used for cloning | New England Biolabs | Cat # C3019I |
| E. coli Rosetta (DE3) was used for protein expression | Millipore Sigma | Cat # 70954 |
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Actinomycin D | Sigma Aldrich | Cat# A1410-2MG |
| Restriction enzymes | New England Biolabs | N/A |
| Biotin-14-dCTP | Thermo Fisher Scientific | Cat# 19518018 |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | Cat# M0210L |
| T4 DNA ligase | New England Biolabs | Cat# M0202L |
| Phenol:chloroform:isoamyl alcohol | Sigma Aldrich | Cat# 77618-100ML |
| BSA | Thermo Fisher Scientific | Cat# B14 |
| T4 DNA polymerase | New England Biolabs | Cat# M0203S |
| Dynabeads MyOne Streptavidin C1 | Thermo Fisher Scientific | Cat# 65001 |
| AMPure XP Beads | Beckman Coulter | Cat# A63880 |
| 37 % Formaldehyde | Thermo Fisher Scientific | Cat# BP531-500 |
| nProteinA Sepharose fast Flow | GE Healthcare | Cat# 17-5280-01 |
| HiTrap NHS-activated HP | GE Healthcare | Cat# 17-0716-01 |
| Proteinase K | Thermo Fisher Scientific | Cat# 25530015 |
| Glycogen | Invitrogen | Cat# 10814-010 |
| Critical Commercial Assays | ||
| NEBNext Ultra DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7370S |
| NEBNex Multiplex Oligos for Illumina | New England Biolabs | Cat# E7335S, E7500S, E7710S |
| NEBNext Ultra II Directional RNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7760S |
| NEBNext Ultra II Directional RNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7760S |
| NEBuilder HiFI DNA Assembly Master Mix | New England Biolabs | Cat# E2621S |
| TruSeq ChIP Library Preparation Kit | Illumina | IP-202-1012, IP-202-1024 |
| FISH Tag DNA Multicolor Kit | Thermo Fisher Scientific | Cat# F32951 |
| Deposited Data | ||
| The NGS datasets generated in this study are available at GEO | https://www.ncbi.nlm.nih.gov/geo/ | GSE128063 |
| The ChIP Seq data for fadR were obtained from GEO | https://www.ncbi.nlm.nih.gov/geo/ | GSE108039 |
| Experimental Models: Cell Lines | ||
| N/A | ||
| Experimental Models: Organisms/Strains | ||
| Information shown in “Archaeal Strains” | ||
| Oligonucleotides | ||
| See Table S2 | ||
| Recombinant DNA | ||
| pCR-XL-2-TOPO: DNA FISH P1 probe | This study | pNTB3 |
| pCR-XL-2-TOPO: DNA FISH P2 probe | This study | pNTB4 |
| pCR-XL-2-TOPO: DNA FISH P3 probe | This study | pNTB5 |
| pCR-XL-2-TOPO: DNA FISH P4 probe | This study | pNTB6 |
| pCR-XL-2-TOPO: DNA FISH P5 probe | This study | pNTB7 |
| pET30-SacSMC | This study | pET30-SacSMC |
| pET33-SisSMC | This study | pET33-SisSMC |
| pCR-ArgD | This study | pCR-ArgD |
| pSSRgD | This study | pSSRgD |
| pSSRgD-ClsN | This study | pSSRgD-ClsN |
| Software and Algorithms | ||
| HiC-Pro | Servant et al., 2015 | https://github.com/nservant/HiC-Pro |
| HiTC | Servant et al., 2012 | https://bioconductor.org/packages/release/bioc/html/HiTC.html |
| ShRec3D | Lesne et al., 2014 | https://sites.google.com/site/julienmozziconacci/home/softwares |
| VMD | Humphrey et al., 1996 | https://www.ks.uiuc.edu/Research/vmd/ |
| Bowtie 2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| deepTools | Ramirez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| Salmon | Patro et al., 2017 | https://combine-lab.github.io/salmon/ |
| SpotDistance | Schober et al., 2008 | http://bigwww.epfl.ch/sage/soft/spotdistance/ |
| Other | ||
Highlights.
Chromosomes of the archaea Sulfolobus have domain and compartment level organization
Domain specification and identity is dependent on transcriptional status
A previously uncharacterized SMC protein, ClsN, marks repressive domains
Acknowledgements
NT is funded by a JSPS Overseas Research Fellowship (Japan Society for the Promotion of Science). RYS, and SDB’s laboratory, are funded by the College of Arts and Science, Indiana University and NIH grant R01GM125579. We are indebted to Qunxin She (University of Copenhagen) and Norio Kurosawa (Soka University) for agmatine auxotroph strains and Eveline Peeters (Vrije Universiteit Brussel) for her kind gift of the fadR deletion strain. We thank Dave Miller (Center for Genomics and Bioinformatics, Indianas University) for help and advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Andersson AF, Lundgren M, Eriksson S, Rosenlund M, Bernander R, and Nilsson P (2006). Global analysis of mRNA stability in the archaeon Sulfolobus. Genome Biol 7, R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson AF, Pelve EA, Lindeberg S, Lundgren M, Nilsson P, and Bernander R (2010). Replication-biased genome organisation in the crenarchaeon Sulfolobus. BMC Genomics 11, 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SD, Jaxel C, Nadal M, Kosa PF, and Jackson SP (1998). Temperature, template topology, and factor requirements of archaeal transcription. Proc Natl Acad Sci U S A 95, 15218–15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernander R, and Poplawski A (1997). Cell cycle characteristics of thermophilic archaea. J Bacteriol 179, 4963–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, and Cavalli G (2016). Organization and function of the 3D genome. Nat Rev Genet 17, 661–678. [DOI] [PubMed] [Google Scholar]
- Brock TD, Brock KM, Belly RT, and Weiss RL (1972). Sulfolobus: a new genus of sulfur-oxidizing bacteria living at low pH and high temperature. Arch Mikrobiol 84, 54–68. [DOI] [PubMed] [Google Scholar]
- Deng L, Zhu H, Chen Z, Liang YX, and She Q (2009). Unmarked gene deletion and host-vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles 13, 735–746. [DOI] [PubMed] [Google Scholar]
- Diebold-Durand ML, Lee H, Ruiz Avila LB, Noh H, Shin HC, Im H, Bock FP, Burmann F, Durand A, Basfeld A, et al. (2017). Structure of Full-Length SMC and Rearrangements Required for Chromosome Organization. Mol Cell 67, 334–347 e335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggin IG, Dubarry N, and Bell SD (2011). Replication termination and chromosome dimer resolution in the archaeon Sulfolobus solfataricus. EMBO J 30, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggin IG, McCallum SA, and Bell SD (2008). Chromosome replication dynamics in the archaeon Sulfolobus acidocaldarius. Proc Natl Acad Sci U S A 105, 16737–16742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber S, and Errington J (2009). Recruitment of condensin to replication origin regions by ParB/SpoOJ promotes chromosome segregation in B. subtilis. Cell 137, 685–696. [DOI] [PubMed] [Google Scholar]
- Haug-Baltzell A, Stephens SA, Davey S, Scheidegger CE, and Lyons E (2017). SynMap2 and SynMap3D: web-based whole-genome synteny browsers. Bioinformatics 33, 2197–2198. [DOI] [PubMed] [Google Scholar]
- Hirano T (2016). Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell 164, 847–857. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, Karcher A, Henderson B, Bodmer JL, McMurray CT, et al. (2002). The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 418, 562–566. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- Jeppsson K, Kanno T, Shirahige K, and Sjogren C (2014). The maintenance of chromosome structure: positioning and functioning of SMC complexes. Nat Rev Mol Cell Biol 15, 601–614. [DOI] [PubMed] [Google Scholar]
- Kalliomaa-Sanford AK, Rodriguez-Castaneda FA, McLeod BN, Latorre-Rosello V, Smith JH, Reimann J, Albers SV, and Barilla D (2012). Chromosome segregation in Archaea mediated by a hybrid DNA partition machine. Proc Natl Acad Sci U S A 109, 3754–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada K, and Barilla D (2018). Combing Chromosomal DNA Mediated by the SMC Complex: Structure and Mechanisms. Bioessays 40. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TB, Imakaev MV, Mirny LA, and Laub MT (2013). High-resolution mapping of the spatial organization of a bacterial chromosome. Science 342, 731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne A, Riposo J, Roger P, Cournac A, and Mozziconacci J (2014). 3D genome reconstruction from chromosomal contacts. Nat Methods 11, 1141–1143. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindas AC, and Bernander R (2013). The cell cycle of archaea. Nat Rev Microbiol 11, 627–638. [DOI] [PubMed] [Google Scholar]
- Lindas AC, Karlsson EA, Lindgren MT, Ettema TJ, and Bernander R (2008). A unique cell division machinery in the Archaea. Proc Natl Acad Sci U S A 105, 18942–18946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy VS, Cournac A, Marbouty M, Duigou S, Mozziconacci J, Espeli O, Boccard F, and Koszul R (2018). Multiscale Structuring of the E. coli Chromosome by Nucleoid-Associated and Condensin Proteins. Cell 172, 771–783 e718. [DOI] [PubMed] [Google Scholar]
- Lopez-Garcia P, and Forterre P (1997). DNA topology in hyperthermophilic archaea: reference states and their variation with growth phase, growth temperature, and temperature stresses. Mol Microbiol 23, 1267–1279. [DOI] [PubMed] [Google Scholar]
- Lopez-Garcia P, and Forterre P (1999). Control of DNA topology during thermal stress in hyperthermophilic archaea: DNA topoisomerase levels, activities and induced thermotolerance during heat and cold shock in Sulfolobus. Mol Microbiol 33, 766–777. [DOI] [PubMed] [Google Scholar]
- Lundgren M, Andersson A, Chen L, Nilsson P, and Bernander R (2004). Three replication origins in Sulfolobus species: synchronous initiation of chromosome replication and asynchronous termination. Proc Natl Acad Sci U S A 101, 7046–7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbouty M, Le Gall A, Cattoni DI, Cournac A, Koh A, Fiche JB, Mozziconacci J, Murray H, Koszul R, and Nollmann M (2015). Condensin- and Replication-Mediated Bacterial Chromosome Folding and Origin Condensation Revealed by Hi-C and Super-resolution Imaging. Mol Cell 59, 588–602. [DOI] [PubMed] [Google Scholar]
- Nolivos S, and Sherratt D (2014). The bacterial chromosome: architecture and action of bacterial SMC and SMC-like complexes. FEMS Microbiol Rev 38, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, and Symington LS (2018). Role of the Mre11 Complex in Preserving Genome Integrity. Genes (Basel) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplawski A, and Bernander R (1997). Nucleoid structure and distribution in thermophilic Archaea. J Bacteriol 179, 7625–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi SA, Bell SD, and Jackson SP (1997). Factor requirements for transcription in the Archaeon Sulfolobus shibatae. EMBO J 16, 2927–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NP, and Bell SD (2007). Extrachromosomal element capture and the evolution of multiple replication origins in archaeal chromosomes. Proc Natl Acad Sci U S A 104, 5806–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NP, Blood KA, McCallum SA, Edwards PA, and Bell SD (2007). Sister chromatid junctions in the hyperthermophilic archaeon Sulfolobus solfataricus. EMBO J 26, 816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NP, Dionne I, Lundgren M, Marsh VL, Bernander R, and Bell SD (2004). Identification of two origins of replication in the single chromosome of the archaeon Sulfolobus solfataricus. Cell 116, 25–38. [DOI] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nat Rev Genet 19, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson RY, and Bell SD (2011). Cell cycles and cell division in the archaea. Curr Opin Microbiol 14, 350–356. [DOI] [PubMed] [Google Scholar]
- Samson RY, Obita T, Freund SM, Williams RL, and Bell SD (2008). A role for the ESCRT system in cell division in archaea. Science 322, 1710–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson RY, Xu Y, Gadelha C, Stone TA, Faqiri JN, Li D, Qin N, Pu F, Liang YX, She Q, et al. (2013). Specificity and function of archaeal DNA replication initiator proteins. Cell Rep 3, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober H, Kalck V, Vega-Palas MA, Van Houwe G, Sage D, Unser M, Gartenberg MR, and Gasser SM (2008). Controlled exchange of chromosomal arms reveals principles driving telomere interactions in yeast. Genome Res 18, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeber A, Hegnauer AM, Hustedt N, Deshpande I, Poli J, Eglinger J, Pasero P, Gut H, Shinohara M, Hopfner KP, et al. (2016). RPA Mediates Recruitment of MRX to Forks and Double-Strand Breaks to Hold Sister Chromatids Together. Mol Cell 64, 951–966. [DOI] [PubMed] [Google Scholar]
- Servant N, Lajoie BR, Nora EP, Giorgetti L, Chen CJ, Heard E, Dekker J, and Barillot E (2012). HiTC: exploration of high-throughput ‘C’ experiments. Bioinformatics 28, 2843–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A, Caceres EF, and Ettema TJG (2017). Genomic exploration of the diversity, ecology, and evolution of the archaeal domain of life. Science 357. [DOI] [PubMed] [Google Scholar]
- Sullivan NL, Marquis KA, and Rudner DZ (2009). Recruitment of SMC by ParB-parS organizes the origin region and promotes efficient chromosome segregation. Cell 137, 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NT, Laub MT, and Le TBK (2017). SMC Progressively Aligns Chromosomal Arms in Caulobacter crescentus but Is Antagonized by Convergent Transcription. Cell Rep 20, 2057–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussart M, Yus E, Martinez S, Bau D, Tahara YO, Pengo T, Widjaja M, Kretschmer S, Swoger J, Djordjevic S, et al. (2017). Defined chromosome structure in the genome-reduced bacterium Mycoplasma pneumoniae. Nat Commun 8, 14665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ruiten MS, and Rowland BD (2018). SMC Complexes: Universal DNA Looping Machines with Distinct Regulators. Trends Genet 34, 477–487. [DOI] [PubMed] [Google Scholar]
- Varani AM, Siguier P, Gourbeyre E, Charneau V, and Chandler M (2011). ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol 12, R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickridge E, Planchenault C, Cockram C, Junceda IG, and Espeli O (2017). Management of E. coli sister chromatid cohesion in response to genotoxic stress. Nat Commun 8, 14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Sybers D, Maklad HR, Lemmens L, Lewyllie C, Zhou X, Schult F, Brasen C, Siebers B, Valegard K, et al. (2019). A TetR-family transcription factor regulates fatty acid metabolism in the archaeal model organism Sulfolobus acidocaldarius. Nat Commun 10, 1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Brandao HB, Le TB, Laub MT, and Rudner DZ (2017). Bacillus subtilis SMC complexes juxtapose chromosome arms as they travel from origin to terminus. Science 355, 524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Le TB, Lajoie BR, Dekker J, Laub MT, and Rudner DZ (2015). Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev 29, 1661–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TA, Foster PG, Cox CJ, and Embley TM (2013). An archaeal origin of eukaryotes supports only two primary domains of life. Nature 504, 231–236. [DOI] [PubMed] [Google Scholar]
- Zhang C, Phillips APR, Wipfler RL, Olsen GJ, and Whitaker RJ (2018). The essential genome of the crenarchaeal model Sulfolobus islandicus. Nat Commun 9, 4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, and Whitaker RJ (2018). Microhomology-Mediated High-Throughput Gene Inactivation Strategy for the Hyperthermophilic Crenarchaeon Sulfolobus islandicus. Appl Environ Microbiol 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng T, Huang Q, Zhang C, Ni J, She Q, and Shen Y (2012). Development of a Simvastatin Selection Marker for a Hyperthermophilic Acidophile, Sulfolobus islandicus. Applied and Environmental Microbiology 78, 568–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Reproducibility of Hi-C Data. Related to Figure 1
(A) Scatter plots comparing Hi-C interaction scores between biological replicates. r indicates the Pearson correlation coefficient.
(B) Heat maps comparing Hi-C interaction scores between biological replicates.
(C) Comparison of the compartment index between biological replicates.
Figure S8. Effects of Cold Shock on the Chromosome Conformation of S. islandicus. Related to Figures 1, 5 and 6.
Two biological replicates were used for ChIP-seq, Hi-C, and RNA-seq analyses.
(A) Flow cytometry profiles of S. islandicus cells cultured at 78°C and 65°C.
(B) Chromatin fractionation. After separating cell extract into chromatin (C) and soluble (S) fractions, total protein (T) and extract fractions were run on SDS-PAGE. ClsN and soluble and chromatin markers, Vps4 and Reptin respectively, were detected by western blotting.
(C) log2-fold change in ClsN enrichment is plotted for each 1-kb bin.
(D) Effects of the cold shock were analyzed by Hi-C.
(E) Distance-normalized frequency of genomic contacts within the A compartment (A-A), within the B compartment (B-B) and between A and B compartments (A-B).
(F) log2 fold change in RNA level (TPM) was plotted for protein-coding genes in S. islandicus.
Figure S9. Correlation of Physical Proximities Measured by Hi-C and DNA FISH. Related to Figure 1 and Figure 6.
(A) Schematic showing the locations of FISH probes in the S. acidocaldarius chromosome.
(B) Representative images of S. acidocaldarius cells. Chromosomal DNA was stained with DAPI (blue) and either of the FISH probe pairs shown in (A). The scale bar indicates 1 μm.
(C) Violin plots of 3D distances between FISH foci. The black box indicates the interquartile range. The white circle in the box indicates the median. The whisker denotes all values of 1.5 x interquartile range. p-values were calculated by Wilcoxon rank sum test.
Figure S2. Sequence Properties of Sulfolobus Genomes in Relation to Hi-C. Related to Figure 1
(A) Length distributions of the HindIII fragments in the genomes of S. acidocaldarius and S. islandicus.
(B) GC % of genomic regions segmented by compartment index plots. The compartment index plots were generated using the datasets in Figure 1.
(C) Contact matrices arising from analyses of proximity ligation performed using non-crosslinked DNA purified from S. acidocaldarius (left panel) and S. islandicus (right panel). The ligation products were sequenced and analyzed as performed in Hi-C of crosslinked DNA.
(D) Hi-C reads were mapped to the genomes of S. acidocaldarius and S. islandicus. Read coverage is shown for each 1-kb bin.
Figure S3. Analyses of Domains. Related to Figure 1
(A) Directional preference of genomic contacts was calculated at 90-kb (top panels) and 300-kb scales (bottom panels) from the contact matrices of S. acidocaldarius and S. islandicus in Figure 1A. Contact biases towards downstream and upstream regions are indicated by positive (red) and negative (green) values respectively. Black horizontal lines indicate threshold values (p = 0.05, paired t-test). Arrowheads indicate potential domain boundaries identified by the directional preference analysis but not by the principal component analysis in Figure 1B.
(B) Contact probability plots of asynchronously growing S. acidocaldarius and S. islandicus are compared with those of C. crescentus and B. subtilis (Le et al., 2013; Wang et al., 2015). Contact probability P(s) was defined as the average of Hi-C interaction scores of bin pairs at a distance of s. P(s) was calculated for each of three biological replicates, and the average of them was further normalized to its maximum value Pmax(s). To calculate contact probabilities for C. crescentus and B. subtilis, normalized Hi-C data from previous studies were retrieved from Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo). The accession numbers of the data are C. crescentus: GSM1120445 and GSM1120446, B. subtilis: GSM1671399 and GSM1671400.
Figure S4. Inactivation of Replication Origin(s) Does Not Affect Chromosome Conformation in S. islandicus. Related to Figure 1
(A) Organization of replication origins, initiator protein genes and replication termination zones on the chromosome of S. islandicus.
(B) Marker frequency analysis of the ΔoriC1 strain.
(C) Hi-C analysis of wild-type and mutant strains of S. islandicus. Top panels: Hi-C contact matrices. Middle panels: compartment index plots. A-domains are shown in red, B-domains in blue. Bottom panels: Pearson correlation matrices.
(D) Ratios of Hi-C interaction scores between mutants and wild type.
Figure S5. Coalescin is an Abundant and Novel Sulfolobus-Encoded SMC superfamily Protein. Related to Figure 2
(A) Phyletic distribution of ClsN. The schematic phylogenetic tree is depicted according to Spang et al. (Spang et al., 2017). See STAR Methods for details.
(B) Model for ClsN structure generated using the Phyre 2 server.
(C) Western blot analyses correlating molar amounts of ClsN protein from specific numbers of cells to defined amounts of recombinant proteins. Analyses were performed with both S. islandicus extract and recombinant protein and S. acidocaldarius extract and recombinant protein. Calibration curves appear in the right-hand panels. Quantitation was performed using ImageJ.
(D) Analyses on ClsN peaks in S. acidocaldarius (Sac). Left panel: Width distribution of ClsN peaks. Middle panel: Scatter plot comparing peak width and enrichment of ClsN. Right panel: Box plots comparing ClsN enrichment in protein-coding genes and insertion sequences (ISs). All data points were additionally plotted for the ISs because of their small number (yellow diamonds). p-values were calculated by Wilcoxon rank sum test.
(E) The same analyses as in panel (D) were conducted on ClsN peaks in S. islandicus (Sis).
Figure S6. Effects of Coalescin Overexpression. Related to Figure 3
(A) Western analysis of ClsN levels in strains containing an empty vector control, pSSRgD, or the ClsN overexpression vector, pSSRgD-ClsN, before and after four hours of induction with arabinose. Vps4 was detected as a loading control (lower panel).
(B) ChIP-Seq analyses of ClsN enrichment using the overexpression strain (ClsN OE) and the empty vector control strain (CTRL). ChIP-seq was performed before (left panels) and after (right panels) four hours of induction with arabinose. All ChIP data is normalized to input DNA. Genome coordinates are shown on the X axes. Grey shading indicates the position of the B domains in the parental S. islandicus strain.
(C) Hi-C contact matrices of the overexpression strain and the empty vector strain before and after incubation with arabinose.
Figure S7. DNA Integrity is Unaffected by a 30-Minute Treatment with Actinomycin D. Related to Figure 5
(A) 3 μg of genomic DNA purified from S. acidocaldarius cells treated with actinomycin D for 0, 15, and 30 minutes were run on a 0.7 % agarose gel, stained with ethidium bromide, and detected by UV illumination. The genome of S. acidocaldarius is 2.2 Mbp.
(B) Flow cytometry profiles of S. acidocaldarius 0, 15, and 30 minutes after treatment with actinomycin D. Cells containing one and two chromosomes (1C and 2C) are indicated.