

Abstract
The immunosuppressive agent mycophenolate is used extensively in kidney transplantation, yet dosing strategy applied varies markedly from fixed dosing (“one-dose-fits-all”), to mycophenolic acid (MPA) trough concentration monitoring, to dose optimization to an MPA exposure target (as area under the concentration-time curve [MPA AUC0-12]). This relates in part to inconsistent results in prospective trials of concentration-controlled dosing (CCD). In this review, the totality of evidence supporting mycophenolate CCD is examined: pharmacological characteristics, observational data linking exposure to efficacy and toxicities, and randomized controlled trials of CCD, with attention to dose optimization method and exposure achieved. Fixed dosing of mycophenolate consistently leads to underexposure associated with rejection, as well as overexposure associated with toxicities. When CCD is driven by pharmacokinetic calculation to a target concentration (target concentration intervention), MPA exposure is successfully controlled and clinical benefits are seen. There remains a need for consensus on practical aspects of mycophenolate target concentration intervention in contemporary tacrolimus-containing regimens and future research to define maintenance phase exposure targets. However, given ongoing consequences of both overimmunosuppression and underimmunosuppression in kidney transplantation, impacting short- and long-term outcomes, these should be a priority. The imprecise “one-dose-fits-all” approach should be replaced by the clinically proven MPA target concentration strategy.
INTRODUCTION
Graft Loss and Mortality
Outcomes from kidney transplantation remain suboptimal.1-3 Effective immunosuppressive drugs, along with attention to cardiovascular disease4 and prophylaxis against infection,5 have significantly reduced rates of acute rejection (15.4%), graft loss (3.6%), and death (2.8%) in the first posttransplant year for standard risk recipients.6 However, time to allograft failure remains substantially shorter than typical recipient life expectancy following transplantation, due largely to chronic antibody-mediated rejection.7-10 Approximately 20% of kidney allograft recipients have returned to dialysis 5 years after transplantation, increasing to around 50% after 15 years.11-13 At the same time, drug toxicities remain an important cause of morbidity and mortality from cardiovascular,14 infectious,15 and malignant16,17 diseases.
Immunosuppression and MPA
Immunosuppressant dosing aims for a sufficient biological drug effect to prevent rejection, while minimizing dose-dependent toxicities. Precision dosing requires an understanding of between-subject variability in both the pharmacokinetics (PK) and pharmacodynamics (PD) of the immunosuppressant agents.18-21
For all drugs, concentration at site of action (the “biophase”) is more directly linked to drug effect than dose.19,22 For certain drugs, concentrations vary widely between individuals on fixed dosing (FD), due primarily to differences in the extent of absorption (bioavailability) and rate of elimination (drug clearance). If FD leaves an unacceptable proportion of individuals outside the range of safe and effective concentrations,23 then dosing to a therapeutic range (therapeutic drug monitoring [TDM]) or a target concentration (target concentration intervention [TCI])24,25 has the potential to both maximize the beneficial effect and minimize toxicities (see Figure 1).
FIGURE 1.
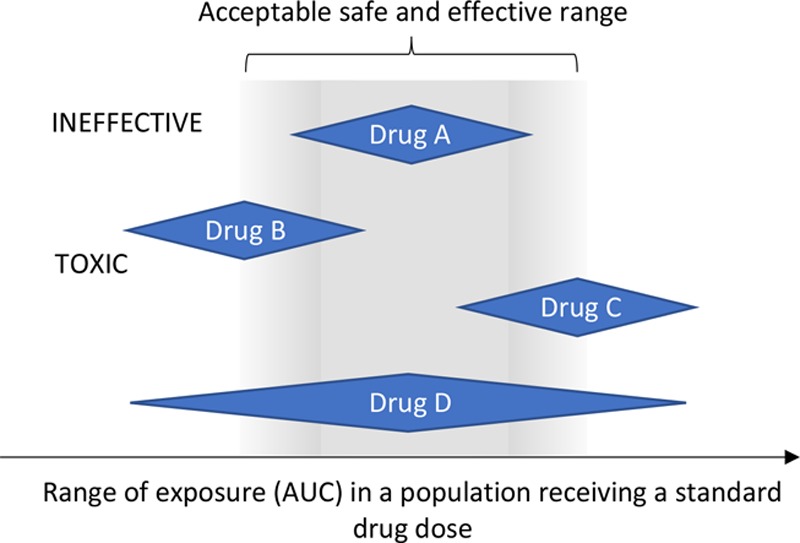
An explanation of how drug dosing decisions can be made by examining the relationship between drug exposure (AUC) at a fixed dose and the acceptable range for safe and effective exposure. Drug A can use fixed dosing, as this gives acceptable drug exposure in all. Drug B is being dosed too low—the population dose should be increased. Drug C is being dosed too high—the population dose should be decreased. Drug D shows both overexposure and underexposure on a fixed dose. Some form of dose optimization is required. AUC, area under the concentration-time curve.
Mycophenolate mofetil (MMF) was initially marketed as a “one-dose-suits-all” drug, despite evidence obtained during drug development supporting concentration-controlled dosing (CCD).26 It displays wide between-subject variability in PK,27,28 leading to an over 10-fold range in mycophenolic acid (MPA) exposure (area under the total MPA concentration-time curve from 0 to 12 h [AUCt0-12]) with mycophenolate FD. This ranges from <10 to >100 mg/L.h,29 well beyond the widely proposed therapeutic range of 30–60 mg/L.h.28-33
Two randomized controlled trials (RCTs) of CCD in kidney transplantation have demonstrated substantially reduced graft rejection when doses are individualized to a target MPA AUCt0-12.26,34,35 However, 2 decades and numerous publications later, the benefit of CCD over FD remains contentious.29,36-40 Critically, the 2 largest RCTs, “fixed-dose concentration-controlled trial (FDCC)” and “Opticept,” failed to significantly differentiate MPA exposure between treatment arms.31,41
To establish a role for CCD, it must first be shown that a measure of systemic exposure is associated with clinical outcomes. Biophase concentrations are rarely available in clinical practice; hence, easily accessible concentrations (eg, blood) are used as surrogate. Depending on the exposure metric (eg, trough or AUC), the matrices (eg, whole blood, plasma, or protein-free plasma for unbound concentrations), and the time-course of drug effect,19 measured concentrations may or may not predict outcomes. The pharmacokinetic-pharmacodynamic (PKPD) characteristics of MPA,30 including enterohepatic cycling (EHC),42 high protein binding,43,44 and presumed local gut toxicity,45 may have complicated assessment of the exposure-effect relationship.
For example, although trough concentrations are considered sufficiently well correlated with AUC for many therapeutic drugs,46,47 the relationship between trough and AUC for MPA is less precise.48,49 The use of MPA trough concentrations in clinical care is contentious.30,33,49-51 Despite this, reviews examining the MPA exposure-effect relationship have not distinguished exposure derived from trough concentrations versus AUC0-12. This has likely diluted the relationship between exposure and effect.
Drug concentrations are almost always measured as “total concentration,” the sum of unbound drug and drug bound to plasma proteins. However, it is the unbound concentration that is the “effective” concentration, as only an unbound drug can equilibrate across cellular membranes.43,44,52 While the relationship between unbound and total MPA concentrations is linear in normal physiological states, this is not the case in certain settings, including hypoalbuminemia or severe renal impairment.33,53
If an association between a measure of exposure and drug response is shown, the next question is whether using drug concentration to individualize dose, CCD, improves outcomes. Gold standard is the randomized concentration-controlled trial (RCCT), where participants are randomized to 2 or more treatment arms based on target concentration (or exposure) rather than dose size.24,54-56 This removes confounding influence between PK and PD characteristics55,56 and allows direct comparison of different exposure targets.
Attention should be drawn to the 2 different methods for CCD: TDM or TCI.24,25 The concentration-effect relationship is typically monotonic and continuous, approaching an asymptote of maximal effect (Figure 2, curve for beneficial effect).23,25 For a drug to be clinically useful, the beneficial effect needs to occur at lower concentrations than unacceptable toxicities (Figure 2, toxicity curves). The TDM approach uses a “therapeutic window,” a range of concentrations between ineffectiveness and toxicity. However, this entails a false categorization of a continuous covariate (drug concentration) into “subtherapeutic,” “therapeutic,” and “toxic.” Clear thresholds between these 3 categories do not exist,25 and drug response (both beneficial and toxic) is not the same at the bottom as at the top of such a window (Figure 2). In contrast, the TCI approach targets a specific concentration.25
FIGURE 2.
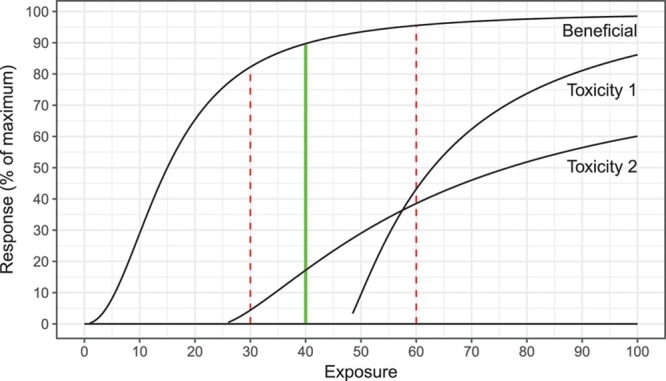
Schematic diagram of exposure-effect relationships for hypothetical “DRUG X,” with exposure-response curves for benefit (reduction in rejection from the baseline rate), toxicity 1 (infectious risk, including opportunistic), and toxicity 2 (suppression of hematopoeisis). Magnitude of response and likelihood and magnitude of toxicities increase with increasing exposure. From the bottom to the top of the therapeutic range (dashed red lines, 30–60 units), magnitude of beneficial response increases, as do toxicities. The optimal balance of efficacy and toxicities is seen at 40 units (optimal target exposure).
There are 2 distinct advantages to TCI.24,25,57 First, it promotes determination of the optimal point of balance between benefit and toxicity, a more precise goal consistent with the concentration-effect relationship. Second, the required dose can be calculated directly from the target concentration and clearance.24 This could be by proportional dose adjustment from an estimate of AUC or by maximum a posteriori Bayesian estimation (MAPBE).58-61 The latter involves estimation of an individual’s PK characteristics using a limited sample of concentrations and a population PK model (Bayesian prior).20,62
Given controversies regarding the benefit of CCD, and an ongoing need to improve immunosuppressant precision in kidney transplantation,2 a systematic literature review was performed. The aim was to provide an updated perspective on the MPA concentration-effect relationship and a critical analysis of exposure and effectiveness in the RCTs of CCD.
Literature Review Methodology
A systematic literature search was undertaken to identify studies in kidney transplant recipients:
assessing the relationship between MPA exposure and beneficial effects.
assessing the relationship between MPA exposure and toxicities.
assessing benefit of mycophenolate CCD by RCT.
To assess the exposure-effect relationships, only studies using estimates of MPA AUC0-12 were included. This was to clarify the strength of association based upon the more reliable measure of drug exposure. MPA AUC0-12 is estimated by full PK profiling (numerous samples over the entire dosing interval), or from a more limited number of samples (limited sampling strategy [LSS]), using multilinear regression equation49 or MAPBE.60
For studies involving MMF, estimates of MPA AUCt0-12 were included whatever the method. In contrast, for studies involving mycophenolate sodium, only prolonged sampling profiles were included (to at least 8 h postdose), as shorter LSS’ have not been shown to adequately predict exposure63,64 due to slow absorption of mycophenolate sodium.
For outcomes, the relationship between MPA AUCt0-12 and rejection, hematological toxicity and infection were assessed. The relationship between MPA AUCt0-12 and gastrointestinal toxicities was not examined as the mechanism is thought due to direct toxicity from MPA metabolites in the gut via EHC,45,65,66 thus indirectly linked to plasma MPA concentrations.
Due to low patient numbers without prespecified power calculation in a significant number of studies, the likelihood of type II errors, particularly for toxicities,67 was considered high. Thus, in addition to reporting the number of articles where statistical significance was met (P < 0.05), the number showing an association or trend was reported. While it might be argued that these articles do not meet sufficient statistical standards, it would be erroneous to suggest that they support the null hypothesis of no association.
Studies were examined altogether, and after separation into concurrent calcineurin inhibitor (CNI) usage (if >75% use of specific agent by cohort or if separate data given). This is because concurrent CNI impacts MPA exposure.31 Cyclosporine inhibits the EHC of MPA, reducing dose-normalized MPA exposure, particularly in the initial posttransplant period with high cyclosporine concentrations.31,68,69 When tacrolimus is used, the initial reduction in dose-normalized exposure is less, while MPA AUCt0-12 above 60 mg/L.h is more common.31,70-72
Electronic databases were searched up to January 25, 2019. Medline (Ovid) and Embase (Ovid) databases were searched using the following thesaurus or keywords:
Population: “kidney transplantation”;
Intervention: “mycophenol*,” “pharmaco*,” “drug monitoring”;
Outcomes: “drug effects,” “rejection,” “survival,” “mortality” or “survival rate,” “severity of illness index,” “treatment outcome or treatment failure,” “infection,” “anemia,” “leucopenia,” “lymphopenia/lymphocytopenia/lymphocyte depletion,” “diarrhea,” “IMP dehydrogenase,” and “adverse outcome.”
In addition, PubMed was searched using keywords “mycophenol*” and “transplant*,” from 2013 onward, to identify e-pubs not yet indexed in Medline. Results were limited to the English language and merged with the references from Staatz and Tett.66,73 Finally, additional references were sourced through searching of reference lists of relevant retrieved articles.
Duplicate entries were identified and removed. Remaining articles were then screened for relevancy, first through perusing of their title and abstract, then if these appeared suitable, through a full-text examination.
RESULTS
A total of 6029 unique articles were identified through the literature search. This was reduced by title review to 476 articles and by abstract review to 104 articles. Following full-text review, a total of 36 publications were identified as appropriate and included in the systemic review.
Evidence for an Exposure-response Relationship for Reduction of Acute Rejection
Twenty-seven cohorts were identified that assessed the relationship between MPA AUCt0-12 and rejection, comprising 3794 individuals. Study features and findings are summarized in Table 1.
TABLE 1.
A summary of studies that have examined the relationship between MPA exposure and beneficial outcomes
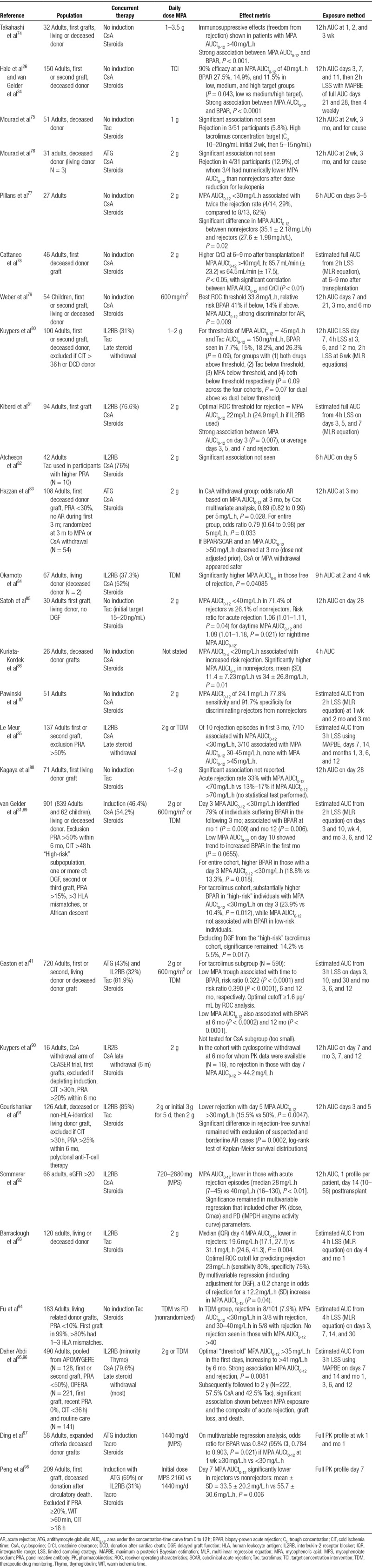
A statistically significant relationship between MPA AUCt0-12 and rejection was evident in 20 of the 27 cohorts (comprising 3382 of 3794 individuals, 89.1%).26,31,35,41,74,77-79,81,83-87,89,91-93,95-98 An additional 3 studies showed a trend in favor of this association (5.7% of individuals),80,90,94 leaving only 4 cohorts (5.1% of individuals) without association.75,76,82
For cyclosporine cotreated transplant recipients, 12 of 16 cohorts (comprising 1181 of 1518 individuals, 77.8%) reported a statistically significant association between MPA exposure and acute rejection.26,35,74,77-79,81,83,86,87,92,95,96 Of the remaining 4, 2 (18.1% of individuals) reported a trend between MPA exposure and acute rejection.89,90 Only 2 cohorts reported no relationship (4.2% of individuals).76,82 One of these negative cohorts involved 31 recipients receiving antithymocyte globulin, a lymphocyte-depleting agent with more potent immunosuppressive effects. Rejection occurred in 4 of 31 participants (12.9%), 3 of the 4 having a lower MPA AUCt0-12 than those without rejection (without application of a statistical test) following dose reduction for leukopenia.76
For tacrolimus cotreated transplant recipients, 7 of 11 cohorts (comprising 1373 of 1696 individuals, 81.0%) revealed a statistically significant association.41,85,89,91,93,97,98 Two further cohorts reported a trend (11.9% of individuals).80,94 This left 2 cohorts (7.2% of individuals).75,88 One reported twice the rate of AR with MPA AUCt0-12 below 70 mg/L.h, without application of a statistical test.88 The other involved 51 transplant recipients (2.7% of individuals) who received high target tacrolimus concentrations by today’s standards: 10–20 ng/mL in the initial 2 weeks and then 5–15 ng/mL thereafter.75 Rejection occurred in 3 of 51 recipients (5.8%), with no relationship to MPA exposure.
Evidence for an Exposure-response Relation for Reduction of Immunosuppressant Toxicity
Twenty-two cohorts involving 3225 kidney transplant recipients were identified that assessed the relationship between MPA AUCt0-12 and hematological or infectious toxicities. Study features and findings are summarized in Table 2.
TABLE 2.
A summary of studies that have examined the relationship between MPA exposure and toxicity
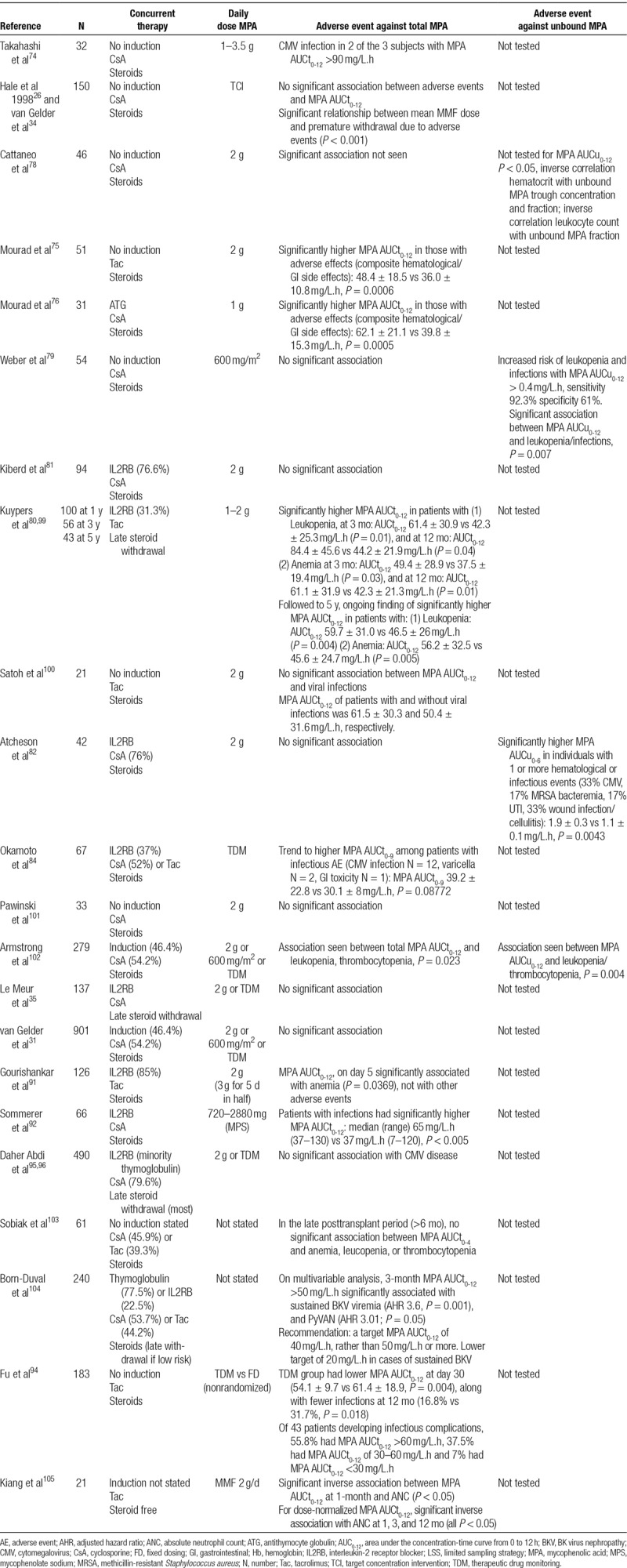
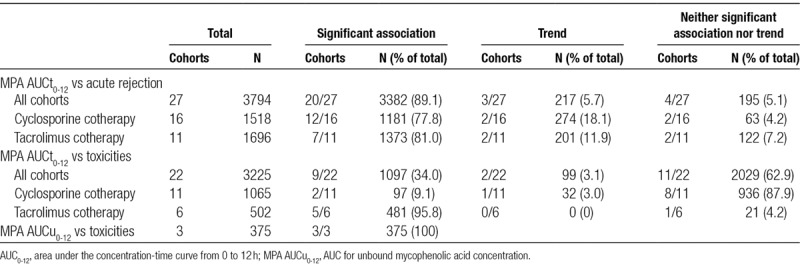
Only 9 of 22 cohorts reported a statistically significant association between MPA exposure and toxicities, comprising 1097 individuals (34.0% of the 3225 individuals).75,76,80,91,92,94,99,102,104,105 A further 2 cohorts (3.1% of individuals) supported a trend towards this association.74,84 Eleven of 22 cohorts (62.9% of individuals) reported no association.26,31,34,35,78,79,81,82,96,100,101,103
In cyclosporine cotreated cohorts, only 2 of 11 studies reported a statistically significant association between exposure and toxicities (comprising 9.1% of 1065 individuals),76,92 along with a trend in 1 study (3.0% of individuals).74
However, the association was far more consistent in cohorts cotreated with tacrolimus (6 relevant cohorts involving 502 individuals). A statistically significant association was reported in 5 of the 6 cohorts (comprising 481 of 502 individuals, 95.8%).75,80,91,94,99,105 There was just 1 cohort that did not report any relationship with toxicities (4.2% of individuals).100
There were 3 publications where unbound MPA concentrations were measured alongside total drug concentrations,79,82,102 comprising 375 individuals. All 3 reported a statistically significant association between unbound exposure (MPA AUCu0-12) and toxicities. Of these, 2 of the 3 studies concurrently failed to show an association between total concentrations (MPA AUCt0-12) and toxicities.79,82
Evidence for CCD and Improved Clinical Outcome
Five RCTs of mycophenolate CCD were identified. Study features and findings are summarized in Table 3.
TABLE 3.
RCTs of concentration-controlled dosing and clinical outcome
All used the MMF formulation. Three used a TCI strategy: the multitarget RCCT published in 1998,26,34 “APOMYGRE” published in 2007,35 and “OPERA” published in 2011.106 Two used a TDM strategy: the FDCC, published in 200831 and “Opticept” published in 2009.41
MPA Dose Individualization Using TCI
All 3 TCI trials optimized mycophenolate dose using MAPBE. Two showed a statistically significant and clinically important benefit. A third trial, with 2 distinct interventions in the treatment arm, neither supported nor refuted benefit of TCI.
Multitarget RCCT
The first trial26,34 was the only RCCT, with more than one target-exposure arm.55 One hundred and fifty recipients were randomized to 3 separate target MPA AUCt0-12 arms: 16.1 mg/L.h (low target), 32.2 mg/L.h (medium target), or 60.6 mg/L.h (high target). Though concentration targets were exceeded in later posttransplant periods (due to so-called “time-dependant clearance”),68,69 the trial was successful in separating treatment arms into 3 distinct MPA exposure groups (see Figure 1, trial publication).26 In each arm, within-group PK variability was reduced from 40%–50% to almost 30%.26
The primary end point, biopsy-proven acute rejection (BPAR) at 6 months, was less frequent with increasing exposure target: 27.5%, 14.9%, and 11.5% in low, medium, and high AUC target arms (P = 0.043, low versus medium/high target groups)34 The requirement for treatment with muromonab-CD3 or antithymocyte globulin (reflecting more severe rejection) also numerically decreased with increasing exposure targets—13.7%, 6.4%, and 3.9%, respectively—failing to reach statistical significance though in small numbers.34
By logistic regression analysis, the relationship between randomly assigned MPA AUCt0-12 and rejection was highly significant (P < 0.001).26 Increasing MPA AUCt0-12 was associated with a reduction in the probability of BPAR by 50%, 75%, and 90% at MPA AUCt0-12 values of 15, 25, and 40 mg/L.h, respectively.26 The association between rejection and trough MPA total concentration was also significant, though weaker (P < 0.01). With doses adjusted to randomly assigned exposure targets, the association between MMF dose and BPAR was not significant (P = 0.082).34
For toxicities, there was a significant relationship between serious adverse events or death and increased MMF dose (P < 0.001), but no significant relationship was found with total MPA AUC0-12, peak or trough concentration.34
APOMYGRE
The second RCT (“APOMYGRE”) randomized 137 renal transplant recipients to FD MMF (2 g/d) or TCI to a target MPA AUCt0-12 of 40 mg/L.h.35 The primary outcome, treatment failure, was a composite of acute rejection, death, graft loss, and MMF withdrawal at 12 months.
TCI improved MPA exposure. At day 14 (the first postadjustment MPA AUCt0-12), the proportion of patients above an MPA AUCt0-12 of 30 mg/L.h was 68.3% versus 30.2% in TCI versus FD groups, with no difference in proportion above 60 mg/L.h (1.6% in each). At the next MPA AUCt0-12 assessment (month 1), proportions were 90.8% versus 55.5%, respectively, with MPA AUCt0-12 above 60 mg/L.h in 13.8% versus 4.7%.
Treatment failure occurred in 47.7% versus 29.2% in the FD versus TCI arms, respectively (P = 0.03). This was entirely due to differences in rejection (BPAR in 24.6% versus 7.7%, P = 0.01). TCI led to early dose escalation in underexposed individuals, with 82% of recipients taking between 2.5 and 4 g/d MMF at month 1, as well as individualized dose reductions, with MMF dose below 2 g/d in 6% at 1 month, 26% at 3 months, and 48% at 6 months. These are low MMF doses with concomitant cyclosporine (some below 1 g/d), without apparent negative impact given overall superiority of the TCI arm.
Of acute rejection episodes in the first 3 months, 70% were associated with an MPA AUCt0-12 <30 mg/L/h, while the remaining 30% occurred in those with an MPA AUCt0-12 between 30 and 45 mg/L.h. Trial design dictated that dose adjustment was capped at 1g/d at a time; however, MAPBE predicted need for >1g/d dose increase for 70% of individuals based on day 7 AUC and 33% based on day 14 AUC. Thus, if larger dose increments had been allowed, the benefit of TCI may have been even greater.107
The TCI dosing in APOMYGRE cost <1% of total yearly costs (hospital and treatment) after a renal transplant. This can be compared with the marginal cost saving in preventing a single transplant failure of 8% of total yearly costs.108
OPERA
The third RCT, “OPERA,” was not a pure TCI trial. It involved 247 kidney transplant recipients considered to be at a low risk of rejection (primary allograft, panel reactive antibody at transplantation of 0%, cold ischemia time <36 h).106 Randomization was to either MMF 2 g/d (FD) or an MMF optimization arm with 2 aspects: an empiric increased dose of 3 g/d for 10 days following transplantation (“dose intensification”), followed by TCI to a target MPA AUCt0-12 of 40 mg/L.h. Steroids were withdrawn on day 7 in both arms.
The optimization arm received significantly higher dose and MPA exposure for the first 6 weeks after transplantation (P = 0.001 at week 2; P = 0.002 at week 6). MPA AUCt0-12 was >30 mg/L.h in 66% versus 38% of optimization versus FD patients at week 2 (due to “dose intensification”) and 81% versus 62% at week 6 (due to TCI). Doses ranged from 1 to 4 g/d in the TCI arm, with significantly reduced within-group AUC variability.106
The primary outcome, BPAR (including subclinical rejection) at 3 months, was lower than expected, with no significant difference between treatment arms.106 The optimization arm did not tolerate therapy as well, with significantly more dose reductions for adverse events (58.7% versus 42.2%, P = 0.009). Although lacking statistical significance, all toxicities associated with MPA were numerically higher in the optimization arm. Finally, there was a trend toward increased BPAR in the optimization arm (24.6% versus 14.9%, P = 0.06).
Given the initial substantive difference in dose between treatment arms, the independent impact of subsequent TCI cannot be objectively assessed in this low-risk steroid withdrawal protocol.
MPA Dose Individualization Using TDM
Fixed Dose Concentration-controlled Trial
“FDCC” was the largest of the RCTs, with 901 kidney transplant recipients randomized to either FD of 2 g/d or CCD.31 Although designed to achieve a target MPA AUCt0-12 (45 mg/L.h), actual implementation used a TDM approach.31 Exposure within 30–60 mg/L.h was considered acceptable. Clinicians could also choose a different target concentration for individual patients based on their assessment of immunological risk, as long as this fell within the 30–60-mg/L.h range.31 Finally, only MPA AUCt0-12 values were provided. The decision to adjust dose, and by how much, was left to the individual clinician.
The TDM approach in FDCC was unsuccessful in improving MPA exposure. There was “nonadherence to required early dose increments” by clinicians, with an overall lack of substantive dose changes. Consequently, “mean MPA AUC values, and the proportion of patients achieving AUC values within the therapeutic range,” were similar in the TDM and FD groups. Outcomes were also the same: treatment failure in 25.6% versus 25.7% (P = 0.81) and BPAR in 14.9% versus 15.5%, in the TDM and FD groups, respectively. However, with minimal difference in exposure between the 2 groups, differences in outcome “could not be expected, and were not observed.”31
As the CCD procedure was unsuccessful in differentiating MPA exposure between the 2 arms, a conclusion regarding method effectiveness of CCD cannot be drawn.56,109,110 This contrasts with the TCI trials, which clearly demonstrated that MPA exposure can be effectively controlled, leading to outcome benefits.26,34,35
Opticept
The second TDM trial, “Opticept,”41 was the only RCT of CCD using trough MPA concentrations. Seven hundred and twenty participants were randomized to 3 treatment arms with 2 intervention variables: MMF dosing strategy (TDM versus FD), and CNI therapeutic range (“standard” versus “reduced”). Group C was the control arm: FD mycophenolate and “standard” CNI. Group A was the primary intervention arm: MMF TDM and “reduced” CNI. Group B was halfway between: MMF TDM and “standard” CNI. The primary outcome was noninferiority of group A compared with C, based upon treatment failure at 12 months (a composite of BPAR, graft loss, loss to follow-up, or withdrawal).
MMF dose optimization was by TDM, to achieve MPA trough concentrations ≥1.3 or ≥1.9 µg/mL, alongside cyclosporine or tacrolimus, respectively. Dose individualization for MPA was according to clinician judgement rather than a centralized PK-guided calculation.
TDM led to significantly higher MMF dose in group A compared with groups B and C. The reason for dose difference between groups A and B—noting that both were TDM arms—was not made clear. Most importantly, however, as with FDCC, dose adjustments were insufficient to attain planned exposure, with “little differentiation among treatment groups in MPA exposure.” In tacrolimus-cotreated patients (81.9% of total participants), MPA trough concentrations were “identical at all time points with or without monitored dosing.”
The primary outcome end point was achieved: noninferiority of Group A (MMF TDM + “reduced” CNI) against Group C (MMF FD + “standard” CNI).41 In fact, there was numerically less rejection and treatment failure in the intervention arm, group A, despite lower CNI exposure, while outcomes in groups B and C were identical. Specifically, treatment failure occurred in 55 (22.6%), 67 (28.3%), and 67 (27.9%) subjects in groups A, B, and C, respectively (P = 0.13 for A versus B and P = 0.18 for A versus C). BPAR occurred in 15 (6.2%), 23 (9.7%), and 23 (9.6%), respectively (P = 0.17 for group A versus C). The occurrence of adverse events was similar across treatment groups.
As with FDCC, lack of differentiation in exposure to MPA between treatment arms means that method effectiveness of CCD was not tested.
DISCUSSION
The consequences of both underimmunosuppression or overimmunosuppression, with potentially preventable morbidity and mortality, remain prevalent after kidney transplantation.1-3 For mycophenolate, the dosing strategy applied varies markedly, from “one-dose-suits-all” (FD),111 to trough concentration monitoring,40 to TCI to an estimated MPA AUCt0-12 target.112
This review demonstrates that mycophenolate FD consistently leaves a proportion of individuals with MPA underexposure associated with rejection (see Table 4). In addition, a link has been shown between MPA exposure and both hematological and infectious toxicities, more apparent with tacrolimus cotherapy or when unbound MPA is measured (see Table 4).
TABLE 4.
Summary table of observational exposure-effect data
The link between MPA AUCt0-12 and rejection is considered “definitive.”113 Five prospective RCTs of mycophenolate CCD have been performed. When critically analyzed, these trials show that CCD using TCI leads to effective control of MPA exposure and to improved clinical outcomes.
The 1998 multitarget RCCT randomly assigned participants into 1 of 3 exposure targets,26,34 the pharmacological gold standard for unbiased assessment of the exposure-response relationship.55,56,114 It was hailed at the time as a landmark demonstration of science-based drug development based on clinical trial simulation.114,115 Increasing exposure target significantly reduced BPAR.26 With random assignment of participants to exposure targets, the association between MPA exposure and BPAR was highly significant, while that between MPA dose and BPAR was not.26
In “APOMYGRE,”35 the TCI approach was superior to FD, with a 39% reduction in treatment failure. This involved initial individualized dose escalation followed by individualized dose reduction, with overall superiority and no increase in toxicities. In addition, TCI was cost neutral.108
In “OPERA,” TCI was effective in maintaining MPA exposure target and reducing within-group PK variability, beyond the initial “dose intensification” period. Notably, 3 other trials of MPA “dose intensification” (without subsequent TCI), in standard or higher-risk recipients, revealed either a significant reduction in rejection98,116 or strong trend,91 showing that this intervention alone can impact outcomes. In contrast, OPERA revealed no efficacy benefit at 3 months (and less tolerance). This suggests that intensified dose (3 g/d for 10 d) followed by TCI is not beneficial in a preselected low-risk early steroid withdrawal population. The trend to higher rejection at 12 months in the dose optimization arm is also of interest: perhaps more dose reductions secondary to toxicities might have contributed?106 Regardless, it is impossible to assess impact of increased precision in MPA exposure (by TCI) independent of the substantive dose difference in the initial phase.
Thus, 2 TCI trials (the multitarget RCCT26,34 and APOMYGRE35) reveal a statistically significant and clinically important benefit of TCI. This is not refuted by the subsequent OPERA trial.
The TCI trials, with effective control of MPA exposure, contrast with the 2 trials using TDM to individualize exposure. In FDCC31 and Opticept,41 TDM without consistent dosing advice did not reliably achieve target MPA exposure (nor even differentiate MPA exposure between treatment arms). As a result, both trials failed to show a clinical benefit of CCD.
A “dose optimization feedback loop” is recommended for RCCTs to maximize probability of target concentration attainment.55,56 A centralized system provides the clinician with a probability-based dose prediction that they can immediately use. Without this, CCD relies on the individual clinician having the time, and the experiential knowledge, to determine new doses themselves.
The clinical pharmacology community has long advocated active PK-guided dosing to a concentration target (TCI) in clinical care.24,25,117-119 TCI is more pharmacologically rational than TDM,23,117,120 although to the authors’ knowledge, the 2 have never been directly compared in terms of clinical outcomes. The RCTs of mycophenolate CCD, while not head-to-head, provide an indirect but noteworthy comparison.
The question arises as to why TCI and PK-guided dosing appear necessary to improve MPA exposure, contrary to other immunosuppressant drugs where TDM suffices. It may relate to clinician experience with CNIs, where doses are generally increased cautiously, perhaps reflecting the lesser precision of trough concentrations and desire to avoid overshoot. For MPA, however, concentration attainment may require greater than proportional dose adjustment.112,121 In addition, TDM leaves the dose unchanged if drug exposure lies anywhere within the broad therapeutic window set for MPA, contrasting the TCI trials showing benefit where active intervention to reach an optimal target was used, even if the measured value was within 30–60 mg/L.h.
Assessing the actual exposure achieved in CCD trials is critical. The Elite-Symphony trial6 reported superiority of low-dose tacrolimus over low- or standard-dose cyclosporine. However, while the target exposure in the tacrolimus arm was 3–7 ng/mL, the actual concentrations achieved were higher. Mean trough concentrations were above 7 ng/mL for the first 8 weeks (with almost 50% of individuals above the therapeutic range for this period).6,122 By 12 months, mean (SD) tacrolimus concentration was 6.4 ± 2.4 ng/mL, and at 3 years, 6.5 ± 2.3 ng/mL.6,123 This trial supports excellent outcomes out to 3 years posttransplant123 with the achieved tacrolimus trough concentrations. However, the results from the Elite-Symphony trail cannot be used to support the intended 3–7-ng/mL therapeutic range (outcomes achieving this have not been given). Equally, it is erroneous to assert that a trial of CCD with minimal difference in exposure between arms (because concentrations were not well controlled) proves the lack of benefit of CCD.
In 2013, Wang et al124 published a systematic review and meta-analysis of RCTs of mycophenolate CCD versus FD, concluding that the evidence was not supportive. However, whilst the result of this meta-analysis was technically correct, “an analysis is only as good as the data on which it was based.” Meta-analysis of the RCTs without careful consideration of the trial methodologies and exposures achieved has led to misinterpretation of the strength of the evidence.
First, Wang et al124 excluded the original multitarget RCCT34,125 due to lack of FD comparator.124 While this trial design does not test the degree of benefit of CCD over FD, it remains the most robust method for determining the causative relationship between exposure and effect.24,54-56,114,126 In this case, it revealed a highly significant relationship between MPA exposure and BPAR,26,34 with MPA exposure values spanning those seen in a population on FD. Second, Wang et al124 included RCTs that, as has been shown in this review, cannot be used to support or refute benefit of CCD. Not accounting for these critical differences in trial methodologies and exposures achieved led Wang et al124 to a conclusion that we reject in this review.
Only 2 RCTs have been able to independently test the benefit of CCD.26,34,35 Both reported clear benefit, using TCI to a target MPA AUCt0-12 of 40 mg/L.h. Together, they confirm MPA AUCt0-12 as a valid biomarker of drug exposure linked causally with drug effect.54,55 No subsequent RCTs have refuted this finding.
The link between exposure and toxicities has proven difficult to establish, particularly in cyclosporine cohorts. This is not because MPA has a “wide therapeutic index,” as dose-dependent toxicities remain prevalent with FD.26,35,127-130 Rather it relates to issues with the exposure metric in certain settings.
The use of total MPA concentration as surrogate for unbound concentration fails in certain pathophysiological states. Hypoalbuminemia (≤31 g/L) leads to a reduction in total MPA without changing unbound MPA concentration,33,131,132 potentially missing toxic unbound concentrations if only total MPA concentrations are measured (Figure 3).
FIGURE 3.
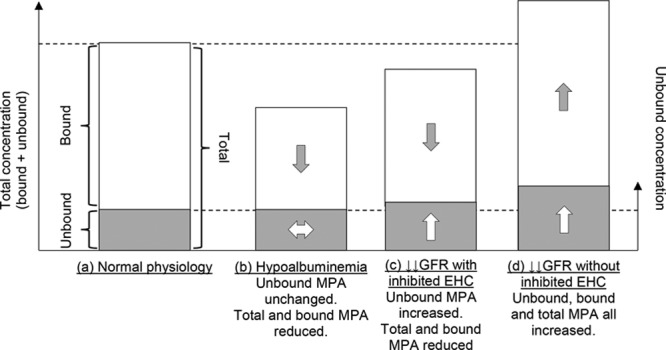
The relationship between the “effective” unbound MPA concentration and total concentration in normal physiological state and relative change (at steady state) in several pathophysiological states. (A) Normal physiology, (B) hypoalbuminemia, (C) severe renal impairment with inhibited EHC (eg, due to CsA), and (D) severe renal impairment without inhibited EHC (eg, Tac used instead of CsA). CsA, cyclosporine; EHC, enterohepatic cycling; GFR, glomerular filtration rate; MPA, mycophenolic acid.
Severe renal impairment (creatinine clearance <25 mL/min) leads to reduced excretion of the major MPA metabolite, MPA-glucuronide (MPAG). This leads to an increase in both total and unbound MPA concentrations, presumed due to EHC and reactivation of accumulating MPAG.53,133 However, with cyclosporine cotherapy, EHC is inhibited, significantly reducing reactivation of MPAG to MPA. Greater MPAG accumulation also increases displacement of MPA from albumin (hence a decrease in total MPA), with unbound concentration unchanged or elevated.53 Again, although for cyclosporine cohorts only, toxic unbound concentrations may be missed if only total MPA concentrations are measured (Figure 3).53
MPA exposure is higher overall alongside tacrolimus in the initial period,31,70-72 further explaining better correlation with toxicities in such cohorts (a greater prevalence of overexposure).
Finally, although not examined in this review, MPA-induced gastrointestinal side effects are thought related to local toxicity from metabolites undergoing EHC.45,65,66 This puts the biophase at a site distal to the plasma compartment, explaining greater difficulty correlating exposure with effect.
After the initial multitarget RCCT, it was noted that “the efficacy of MMF is primarily related to MPA AUC, whereas tolerability is more dependent on the dose of MMF. The apparent discrepancy between these findings cannot readily be explained.”26,34 We now have a plausible explanation: issues using total to predict unbound MPA concentrations in certain settings, and an indirect link between plasma concentration and amount of drug in the gut for GI toxicity.
To summarize, complicated PKPD characteristics and challenges in CCD have clouded understanding of the exposure-effect relationship of MPA in kidney transplantation. This has contributed to failure to recognize the better outcomes when dose optimization is based on PK-guided TCI25 compared with TDM and individual clinician-based dose adjustment. Only if MPA target exposure is effectively achieved are benefits seen.
It is of course noteworthy that the 2 RCTs that effectively tested mycophenolate CCD, showing benefit of TCI, were in cohorts concurrently receiving cyclosporine. Nowadays, tacrolimus use predominates in many centers, along with induction antibody therapy (“quadruple therapy”: induction, steroids, MPA, and CNI). In addition, rejection rates are low.6,111
However, the validity of MPA AUCt0-12 as a biomarker for drug exposure, causally linked to drug effect, will still apply with different drug combinations or populations, although exposure target may differ. Second, precision dosing aims to maximize benefits and minimize toxicities: in contemporary cohorts, there remains MPA underexposure associated with rejection,41,89,91,93,97,98 dose-dependent toxicities,129,130,134 and overexposure associated with toxicities,80,94,99,104,105 highlighting a potential value of TCI.
The MPA AUCt0-12 target of 40 mg/L.h in the initial posttransplant period, based on the method effective RCTs, can reasonably be extrapolated to tacrolimus cohorts based on 2 lines of evidence. First, this approximates the typical (mean or median) MPA AUCt0-12 seen in the initial posttransplant week in tacrolimus cotreated cohorts on 2 g/d dosing (ie, this is the exposure the typical patient receives).31,91,135 Continuing 2 g/d, typical MPA AUCt0-12 then increases to over 50 mg/L.h by week 431,91,135 and to 60 mg/L.h by month 3,91,135 presumably due to higher serum albumin and glomerular filtration rate53,68 ± reduction in steroid dose.136
This target also aligns with the observational data that exist, at least for recipients at increased risk. A substantial increase in rejection rates has been reported with an initial MPA AUCt0-12 <30 mg/L.h and 1 of the following: >3 human leukocyte antigen mismatches, panel reactive antibody >15%, repeat transplant, delayed graft function, or African American descent;89 if concurrent underexposure to other immunosuppressants93 or for expanded criteria donation.97,98 Elsewhere, similar rejection “thresholds” have been reported in contemporary regimen.41,91 Rapid and effective target concentration attainment could ameliorate this risk in such individuals.
MPA underexposure in the immediate posttransplant week may not be detrimental in low-risk recipients.89,106 However, the authors would caution against concluding that early AUC estimation is unnecessary in this group. While a target of 40 mg/L.h may not be required in the immediate posttransplant period, identification of high exposure (ie, 60–100+ mg/L.h) provides an opportunity for early individualized dose reduction. Supporting benefit of such a strategy, reduced infection was reported in a nonrandomized MMF CCD trial alongside tacrolimus in the first posttransplant month.94
In the maintenance phase, “incomplete efficacy, patient intolerance, and side effects” to antiproliferatives remain an issue,2 although infrequency of hard outcomes makes it harder to show quantitative associations. Importantly, Daher Abdi et al95,96 used joint modeling to link longitudinal changes in MPA exposure with outcomes at 1 (490 subjects) and 2 years posttransplant (222 subjects), pooling cohorts from APOMYGRE, OPERA, and clinical care. Robust association was reported between MPA AUCt0-12 and hazard of rejection at 1 year (P = 0.0081), with suggestion to maintain exposure above a “threshold” of 37 mg/L.h at 1 month posttransplant, above 40 mg/L.h by month 3, and above 41 mg/L.h by month 6 and onward.95 Out to 2 years (excluding the OPERA cohort), all subjects having received induction therapy, MMF, CNI (42.5% tacrolimus), and steroid withdrawal after 3 months, a significant association was shown between MPA exposure and the composite of acute rejection, graft loss and death at 2 years (with each 1 mg/L.h increase in MPA AUCt0-12, there was a 4% hazard reduction).95
In contemporary “quadruple therapy” regimen with steroid continuation, equivalent data to support a maintenance phase MPA exposure target do not yet exist (although presumably a lower target than for steroid withdrawal cohorts would suffice). Furthermore, there has been a trend to empiric reduction of the population dose of mycophenolate in the first few months in such regimens (to 1.5 g/d, and eventually 1 g/d if low risk),111 due to an increase in toxicities including BK virus nephropathy.137,138 Nevertheless, an association has been reported between MPA dose reduction and rejection in steroid continuation cohorts.134,139 In addition, multivariable analysis of 240 kidney transplant recipients has revealed an association between an MPA AUCt0-12 >50 mg/L.h at 3 months posttransplantation and both sustained BK viremia (P < 0.0001) and polyomavirus-associated nephropathy (P = 0.013) over the subsequent 2 years.104 Just as targeted dose reductions occurred in the TCI arm of APOMYGRE, TCI in contemporary regimens has the potential to more effectively reduce BK virus disease and other toxicities than the current trend to empiric population dose reduction,111 while avoiding iatrogenic underexposure in those with already low MPA exposure on initial FD.
Finally, the impact of tacrolimus exposure on subclinical inflammation and de novo donor-specific antihuman leukocyte antigen antibody (dnDSA) formation has been reported in recent years.140-145 In contrast, while some studies have linked the use of mycophenolate to reduced dnDSA formation,146,147 the impact of MPA dose or exposure on dnDSA formation is largely absent. Torres et al141 linked tubulointerstitial inflammation in low-risk recipients with combination of low tacrolimus concentrations and reduced MMF dosing, while Filler et al148 reported a significant association between minimum MPA trough concentrations and dnDSA formation in pediatric renal transplant recipients.
This review provides strong evidence favoring MPA TCI in kidney transplantation. However, there is an urgent need to better define target concentration beyond the initial phase in steroid continuation regimens, and to correlate MPA exposure with dnDSA formation. This could first involve prospective collection of MPA exposure, both for total and unbound MPA, within contemporary steroid continuation drug regimen. PKPD time to event analyses could then be performed, like that by Daher Abdi et al,95,96 to link the time course of exposure with dnDSA formation and clinical outcomes. As a final definitive step, an RCT of FD versus TCI to an AUC target, with surrogate endpoints including dnDSA, would be of benefit.
There is in addition a need for consensus on practical aspects of MPA TCI. Frequent AUC estimation has been suggested in cyclosporine-cotreated cohorts: “in the first week after transplant, then each week for the first month, each month until month 3, and subsequently every 3 months up to 1 year.”33 This is due to a 30%–50% increase in dose-normalized exposure over the first 3 months, to avoid overshooting target. However, without the dose-dependent inhibitory effect of cyclosporine on EHC, the change in exposure over the first 3 months appears less substantial in tacrolimus-containing regimens,121,149 and thus a lesser frequency should suffice.
Access to methods for MPA TCI is also required, by broadening access to MAPBE,112,150 or using acceptably precise LSS methods for estimation of MPA AUCt0-12, for example, multilinear regression equation equation validated in an equivalent population49 or extended sampling for trapezoid estimation.63 To reduce practical burden of repeated blood sampling, validation of new technologies enabling precise dried blood spot testing is needed.151
Finally, more data are needed to determine optimal unbound MPA exposure in the initial posttransplant weeks to allow interpretation of MPA exposure in the setting of significant hypoalbuminemia or delayed graft function.33 In addition, the use of intracellular concentrations of MPA in peripheral lymphocytes152 or pharmacodynamic measurement of Inosine-5’-monophosphate dehydrogenase activity153,154 could in theory offer an alternative to systemic exposure estimation, though to date clinical value has not been shown.
The consequences of inefficacy and toxicities from current immunosuppressive agents remain significant, due to between-subject PKPD variability as well as individual patient susceptibilities. Expectations are for “slow, painstaking, stepwise improvements in outcomes from the techniques we have … and careful honing of new methods with better efficacy than old ones.”155 Increasing precision with MPA by individualizing dose to a target concentration (TCI) provides such an opportunity.
CONCLUSION
MPA AUCt0-12 is a valid biomarker of drug exposure, more directly linked to drug effect than mycophenolate dose. FD leads to both overexposure and underexposure and off-target toxicities. Along with the overwhelming observational evidence, 2 adequately designed and executed trials26,34,35 have tested the benefit of dosing to a target MPA exposure, revealing statistically significant and clinically important benefit. No subsequent evidence refutes these findings.
There remains a need for consensus on frequency of exposure estimation in the early phase; to increase access to estimation methods that balance precision and practicality; to better define exposure targets in the maintenance phase; and to better define the exposure-effect relationship for the unbound concentration. These should be seen as a priority, given ongoing prevalence of immune-mediated graft loss and life-limiting toxicities. The imprecise one-dose-suits-all approach with mycophenolate should come to end and be replaced by the scientifically based and evidence-proven TCI approach.
Footnotes
D.K.M. participated in the conception and design of the review, performance of the systematic literature search, analysis and interpretation of the data, writing of the paper, revising the manuscript critically, and approval of the version of the manuscript to be published. J.Y.K., A.W., and N.C. participated in the conception and design of the review, analysis and interpretation of the data, revising the manuscript critically, and approval of the version of the manuscript to be published. K.A.B. and C.E.S. participated in the analysis and interpretation of the data, revising the manuscript critically, and approval of the version of the manuscript to be published. F.I. and N.H. participated in the conception and design of the review, analysis and interpretation of the data, writing of the paper, revising the manuscript critically, and approval of the version of the manuscript to be published.
The authors declare no conflicts of interest.
D.K.M. undertook this work during his PhD candidature, which was supported by a Murdoch Children’s Research Institute Postgraduate Health Research Scholarship and a Royal Australasian College of Physicians Jacquot Research Entry Scholarship.
REFERENCES
- 1.Neuberger JM, Bechstein WO, Kuypers DR, et al. Practical recommendations for long-term management of modifiable risks in kidney and liver transplant recipients: A guidance report and clinical checklist by the consensus on managing modifiable risk in transplantation (COMMIT) group. Transplantation 20171014S Suppl 2S1–S56 [DOI] [PubMed] [Google Scholar]
- 2.O’Connell PJ, Kuypers DR, Mannon RB, et al. Clinical trials for immunosuppression in transplantation: the case for reform and change in direction. Transplantation 20171011527–1534 [DOI] [PubMed] [Google Scholar]
- 3.Wadström J, Ericzon BG, Halloran PF, et al. Advancing transplantation: new questions, new possibilities in kidney and liver transplantation. Transplantation 2017101Suppl 2SS1–S41 [DOI] [PubMed] [Google Scholar]
- 4.Pilmore H, Dent H, Chang S, et al. Reduction in cardiovascular death after kidney transplantation. Transplantation 201089851–857 [DOI] [PubMed] [Google Scholar]
- 5.Parasuraman R, Yee J, Karthikeyan V, et al. Infectious complications in renal transplant recipients. Adv Chronic Kidney Dis 200613280–294 [DOI] [PubMed] [Google Scholar]
- 6.Ekberg H, Tedesco-Silva H, Demirbas A, et al. ; ELITE-Symphony Study Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med 20073572562–2575 [DOI] [PubMed] [Google Scholar]
- 7.Einecke G, Sis B, Reeve J, et al. Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant 200992520–2531 [DOI] [PubMed] [Google Scholar]
- 8.Gaston RS, Cecka JM, Kasiske BL, et al. Evidence for antibody-mediated injury as a major determinant of late kidney allograft failure. Transplantation 20109068–74 [DOI] [PubMed] [Google Scholar]
- 9.Sellarés J, de Freitas DG, Mengel M, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant 201212388–399 [DOI] [PubMed] [Google Scholar]
- 10.Halloran PF, Chang J, Famulski K, et al. Disappearance of T cell-mediated rejection despite continued antibody-mediated rejection in late kidney transplant recipients. J Am Soc Nephrol 2015261711–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transplant 201111450–462 [DOI] [PubMed] [Google Scholar]
- 12.Gondos A, Döhler B, Brenner H, et al. Kidney graft survival in Europe and the United States: strikingly different long-term outcomes. Transplantation 201395267–274 [DOI] [PubMed] [Google Scholar]
- 13.Coemans M, Süsal C, Döhler B, et al. Analyses of the short- and long-term graft survival after kidney transplantation in Europe between 1986 and 2015. Kidney Int 201894964–973 [DOI] [PubMed] [Google Scholar]
- 14.Bamgbola O. Metabolic consequences of modern immunosuppressive agents in solid organ transplantation. Ther Adv Endocrinol Metab 20167110–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karuthu S, Blumberg EA. Common infections in kidney transplant recipients. Clin J Am Soc Nephrol 201272058–2070 [DOI] [PubMed] [Google Scholar]
- 16.Chapman JR, Webster AC, Wong G. Cancer in the transplant recipient. Cold Spring Harb Perspect Med 201336–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Au E, Wong G, Chapman JR. Cancer in kidney transplant recipients. Nat Rev Nephrol 201814508–520 [DOI] [PubMed] [Google Scholar]
- 18.Holford NH, Sheiner LB. Understanding the dose-effect relationship: clinical application of pharmacokinetic-pharmacodynamic models. Clin Pharmacokinet 19816429–453 [DOI] [PubMed] [Google Scholar]
- 19.Wright DF, Winter HR, Duffull SB. Understanding the time course of pharmacological effect: a PKPD approach. Br J Clin Pharmacol 201171815–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duffull SB, Wright DF, Winter HR. Interpreting population pharmacokinetic-pharmacodynamic analyses–a clinical viewpoint. Br J Clin Pharmacol 201171807–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Standing JF. Understanding and applying pharmacometric modelling and simulation in clinical practice and research. Br J Clin Pharmacol 201783247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holford N. Concentration controlled therapy. In: Breckenridge A, editor. In: Esteve Foundation Workshop. Amsterdam, The Netherlands: Elsevier Science; 2001. [Google Scholar]
- 23.Holford NH, Buclin T. Safe and effective variability–a criterion for dose individualization. Ther Drug Monit 201234565–568 [DOI] [PubMed] [Google Scholar]
- 24.Morris RG. Target concentration strategy for cyclosporin monitoring. Clin Pharmacokinet 199732175–179 [DOI] [PubMed] [Google Scholar]
- 25.Holford NH. Target concentration intervention: beyond Y2K. Br J Clin Pharmacol 1999489–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hale MD, Nicholls AJ, Bullingham RE, et al. The pharmacokinetic-pharmacodynamic relationship for mycophenolate mofetil in renal transplantation. Clin Pharmacol Ther 199864672–683 [DOI] [PubMed] [Google Scholar]
- 27.Shaw LM, Kaplan B, DeNofrio D, et al. Pharmacokinetics and concentration-control investigations of mycophenolic acid in adults after transplantation. Ther Drug Monit 20002214–19 [DOI] [PubMed] [Google Scholar]
- 28.Shaw LM, Holt DW, Oellerich M, et al. Current issues in therapeutic drug monitoring of mycophenolic acid: report of a roundtable discussion. Ther Drug Monit 200123305–315 [DOI] [PubMed] [Google Scholar]
- 29.Kuypers DR, Le Meur Y, Cantarovich M, et al. ; Transplantation Society (TTS) Consensus Group on TDM of MPA Consensus report on therapeutic drug monitoring of mycophenolic acid in solid organ transplantation. Clin J Am Soc Nephrol 20105341–358 [DOI] [PubMed] [Google Scholar]
- 30.Jeong H, Kaplan B. Therapeutic monitoring of mycophenolate mofetil. Clin J Am Soc Nephrol 20072184–191 [DOI] [PubMed] [Google Scholar]
- 31.van Gelder T, Silva HT, de Fijter JW, et al. Comparing mycophenolate mofetil regimens for de novo renal transplant recipients: the fixed-dose concentration-controlled trial. Transplantation 2008861043–1051 [DOI] [PubMed] [Google Scholar]
- 32.Tönshoff B, David-Neto E, Ettenger R, et al. Pediatric aspects of therapeutic drug monitoring of mycophenolic acid in renal transplantation. Transplant Rev (Orlando) 20112578–89 [DOI] [PubMed] [Google Scholar]
- 33.Tett SE, Saint-Marcoux F, Staatz CE, et al. Mycophenolate, clinical pharmacokinetics, formulations, and methods for assessing drug exposure. Transplant Rev (Orlando) 20112547–57 [DOI] [PubMed] [Google Scholar]
- 34.van Gelder T, Hilbrands LB, Vanrenterghem Y, et al. A randomized double-blind, multicenter plasma concentration controlled study of the safety and efficacy of oral mycophenolate mofetil for the prevention of acute rejection after kidney transplantation. Transplantation 199968261–266 [DOI] [PubMed] [Google Scholar]
- 35.Le Meur Y, Büchler M, Thierry A, et al. Individualized mycophenolate mofetil dosing based on drug exposure significantly improves patient outcomes after renal transplantation. Am J Transplant 200772496–2503 [DOI] [PubMed] [Google Scholar]
- 36.Knight SR, Morris PJ. Does the evidence support the use of mycophenolate mofetil therapeutic drug monitoring in clinical practice? A systematic review. Transplantation 2008851675–1685 [DOI] [PubMed] [Google Scholar]
- 37.Byrne R, Yost SE, Kaplan B. Mycophenolate mofetil monitoring: is there evidence that it can improve outcomes? Clin Pharmacol Ther 201190204–206 [DOI] [PubMed] [Google Scholar]
- 38.van Gelder T. Therapeutic drug monitoring for mycophenolic acid is value for (little) money. Clin Pharmacol Ther 201190203–204 [DOI] [PubMed] [Google Scholar]
- 39.Kiang TK, Ensom MH. Therapeutic drug monitoring of mycophenolate in adult solid organ transplant patients: an update. Expert Opin Drug Metab Toxicol 201612545–553 [DOI] [PubMed] [Google Scholar]
- 40.Filler G, Alvarez-Elías AC, McIntyre C, et al. The compelling case for therapeutic drug monitoring of mycophenolate mofetil therapy. Pediatr Nephrol 20173221–29 [DOI] [PubMed] [Google Scholar]
- 41.Gaston RS, Kaplan B, Shah T, et al. Fixed- or controlled-dose mycophenolate mofetil with standard- or reduced-dose calcineurin inhibitors: the Opticept trial. Am J Transplant 200991607–1619 [DOI] [PubMed] [Google Scholar]
- 42.Roberts MS, Magnusson BM, Burczynski FJ, et al. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet 200241751–790 [DOI] [PubMed] [Google Scholar]
- 43.Benet LZ, Hoener BA. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther 200271115–121 [DOI] [PubMed] [Google Scholar]
- 44.Dasgupta A. Therapeutic drug monitoring of mycophenolic acid. Adv Clin Chem 201676165–184 [DOI] [PubMed] [Google Scholar]
- 45.Arns W. Noninfectious gastrointestinal (GI) complications of mycophenolic acid therapy: a consequence of local GI toxicity? Transplant Proc 20073988–93 [DOI] [PubMed] [Google Scholar]
- 46.van Rossum HH, Press RR, den Hartigh J, et al. Point: a call for advanced pharmacokinetic and pharmacodynamic monitoring to guide calcineurin inhibitor dosing in renal transplant recipients. Clin Chem 201056732–735 [DOI] [PubMed] [Google Scholar]
- 47.Marquet P. Counterpoint: is pharmacokinetic or pharmacodynamic monitoring of calcineurin inhibition therapy necessary? Clin Chem 201056736–739 [DOI] [PubMed] [Google Scholar]
- 48.Prémaud A, Debord J, Rousseau A, et al. A double absorption-phase model adequately describes mycophenolic acid plasma profiles in de novo renal transplant recipients given oral mycophenolate mofetil. Clin Pharmacokinet 200544837–847 [DOI] [PubMed] [Google Scholar]
- 49.Barraclough KA, Isbel NM, Franklin ME, et al. Evaluation of limited sampling strategies for mycophenolic acid after mycophenolate mofetil intake in adult kidney transplant recipients. Ther Drug Monit 201032723–733 [DOI] [PubMed] [Google Scholar]
- 50.Kaplan B. Mycophenolic acid trough level monitoring in solid organ transplant recipients treated with mycophenolate mofetil: association with clinical outcome. Curr Med Res Opin 2006222355–2364 [DOI] [PubMed] [Google Scholar]
- 51.Miura M, Niioka T, Kato S, et al. Monitoring of mycophenolic acid predose concentrations in the maintenance phase more than one year after renal transplantation. Ther Drug Monit 201133295–302 [DOI] [PubMed] [Google Scholar]
- 52.Smith DA, Di L, Kerns EH. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov 20109929–939 [DOI] [PubMed] [Google Scholar]
- 53.de Winter BC, van Gelder T, Sombogaard F, et al. Pharmacokinetic role of protein binding of mycophenolic acid and its glucuronide metabolite in renal transplant recipients. J Pharmacokinet Pharmacodyn 200936541–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanathanan LP, Peck CC. The randomized concentration-controlled trial: an evaluation of its sample size efficiency. Control Clin Trials 199112780–794 [DOI] [PubMed] [Google Scholar]
- 55.Sanathanan LP, Peck C, Temple R, et al. Randomization, Pk-controlled dosing, and titration: an integrated approach for designing clinical trials. Drug Info J 199125425–431 [Google Scholar]
- 56.Kraiczi H, Jang T, Ludden T, et al. Randomized concentration-controlled trials: motivations, use, and limitations. Clin Pharmacol Ther 200374203–214 [DOI] [PubMed] [Google Scholar]
- 57.Jelliffe R. Goal-oriented, model-based drug regimens: setting individualized goals for each patient. Ther Drug Monit 200022325–329 [DOI] [PubMed] [Google Scholar]
- 58.Sheiner LB, Beal S, Rosenberg B, et al. Forecasting individual pharmacokinetics. Clin Pharmacol Ther 197926294–305 [DOI] [PubMed] [Google Scholar]
- 59.Jelliffe RW, Schumitzky A, Van Guilder M, et al. Individualizing drug dosage regimens: roles of population pharmacokinetic and dynamic models, bayesian fitting, and adaptive control. Ther Drug Monit 199315380–393 [PubMed] [Google Scholar]
- 60.Marquet P. Clinical application of population pharmacokinetic methods developed for immunosuppressive drugs. Ther Drug Monit 200527727–732 [DOI] [PubMed] [Google Scholar]
- 61.Keizer RJ, Ter Heine R, Frymoyer A, et al. Model-informed precision dosing at the bedside: scientific challenges and opportunities. CPT Pharmacometrics Syst Pharmacol 20187785–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beal SL, Sheiner LB. Estimating population kinetics. Crit Rev Biomed Eng 19828195–222 [PubMed] [Google Scholar]
- 63.Hougardy JM, Maufort L, Cotton F, et al. Therapeutic drug monitoring of enteric-coated mycophenolate sodium by limited sampling strategies is associated with a high rate of failure. Clin Kidney J 20169319–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brooks EK, Tett SE, Isbel NM, et al. Evaluation of multiple linear regression-based limited sampling strategies for enteric-coated mycophenolate sodium in adult kidney transplant recipients. Ther Drug Monit 201840195–201 [DOI] [PubMed] [Google Scholar]
- 65.Shipkova M, Armstrong VW, Oellerich M, et al. Acyl glucuronide drug metabolites: toxicological and analytical implications. Ther Drug Monit 2003251–16 [DOI] [PubMed] [Google Scholar]
- 66.Staatz CE, Tett SE. Pharmacology and toxicology of mycophenolate in organ transplant recipients: an update. Arch Toxicol 2014881351–1389 [DOI] [PubMed] [Google Scholar]
- 67.Rawlins M. De testimonio: on the evidence for decisions about the use of therapeutic interventions. Lancet 20083722152–2161 [DOI] [PubMed] [Google Scholar]
- 68.van Hest RM, Mathot RA, Pescovitz MD, et al. Explaining variability in mycophenolic acid exposure to optimize mycophenolate mofetil dosing: a population pharmacokinetic meta-analysis of mycophenolic acid in renal transplant recipients. J Am Soc Nephrol 200617871–880 [DOI] [PubMed] [Google Scholar]
- 69.van Hest RM, van Gelder T, Bouw R, et al. Time-dependent clearance of mycophenolic acid in renal transplant recipients. Br J Clin Pharmacol 200763741–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Gelder T, Klupp J, Barten MJ, et al. Comparison of the effects of tacrolimus and cyclosporine on the pharmacokinetics of mycophenolic acid. Ther Drug Monit 200123119–128 [DOI] [PubMed] [Google Scholar]
- 71.van Gelder T, Bouamar R, Shuker N, et al. The optimal MMF dose in tacrolimus treated patients. Am J Transplant. 2014;14:1221. doi: 10.1111/ajt.12682. [DOI] [PubMed] [Google Scholar]
- 72.van Gelder T, Hesselink DA. Mycophenolate revisited. Transpl Int 201528508–515 [DOI] [PubMed] [Google Scholar]
- 73.Staatz CE, Tett SE. Clinical pharmacokinetics and pharmacodynamics of mycophenolate in solid organ transplant recipients. Clin Pharmacokinet 20074613–58 [DOI] [PubMed] [Google Scholar]
- 74.Takahashi K, Ochiai T, Uchida K, et al. Pilot study of mycophenolate mofetil (RS-61443) in the prevention of acute rejection following renal transplantation in Japanese patients. RS-61443 investigation committee–Japan. Transplant Proc 1995271421–1424 [PubMed] [Google Scholar]
- 75.Mourad M, Malaise J, Chaib Eddour D, et al. Pharmacokinetic basis for the efficient and safe use of low-dose mycophenolate mofetil in combination with tacrolimus in kidney transplantation. Clin Chem 2001471241–1248 [PubMed] [Google Scholar]
- 76.Mourad M, Malaise J, Chaib Eddour D, et al. Correlation of mycophenolic acid pharmacokinetic parameters with side effects in kidney transplant patients treated with mycophenolate mofetil. Clin Chem 20014788–94 [PubMed] [Google Scholar]
- 77.Pillans PI, Rigby RJ, Kubler P, et al. A retrospective analysis of mycophenolic acid and cyclosporin concentrations with acute rejection in renal transplant recipients. Clin Biochem 20013477–81 [DOI] [PubMed] [Google Scholar]
- 78.Cattaneo D, Gaspari F, Ferrari S, et al. Pharmacokinetics help optimizing mycophenolate mofetil dosing in kidney transplant patients. Clin Transplant 200115402–409 [DOI] [PubMed] [Google Scholar]
- 79.Weber LT, Shipkova M, Armstrong VW, et al. The pharmacokinetic-pharmacodynamic relationship for total and free mycophenolic acid in pediatric renal transplant recipients: a report of the german study group on mycophenolate mofetil therapy. J Am Soc Nephrol 200213759–768 [DOI] [PubMed] [Google Scholar]
- 80.Kuypers DR, Claes K, Evenepoel P, et al. Clinical efficacy and toxicity profile of tacrolimus and mycophenolic acid in relation to combined long-term pharmacokinetics in de novo renal allograft recipients. Clin Pharmacol Ther 200475434–447 [DOI] [PubMed] [Google Scholar]
- 81.Kiberd BA, Lawen J, Fraser AD, et al. Early adequate mycophenolic acid exposure is associated with less rejection in kidney transplantation. Am J Transplant 200441079–1083 [DOI] [PubMed] [Google Scholar]
- 82.Atcheson BA, Taylor PJ, Mudge DW, et al. Mycophenolic acid pharmacokinetics and related outcomes early after renal transplant. Br J Clin Pharmacol 200559271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hazzan M, Labalette M, Copin MC, et al. Predictive factors of acute rejection after early cyclosporine withdrawal in renal transplant recipients who receive mycophenolate mofetil: results from a prospective, randomized trial. J Am Soc Nephrol 2005162509–2516 [DOI] [PubMed] [Google Scholar]
- 84.Okamoto M, Wakabayashi Y, Higuchi A, et al. Therapeutic drug monitoring of mycophenolic acid in renal transplant recipients. Transplant Proc 200537859–860 [DOI] [PubMed] [Google Scholar]
- 85.Satoh S, Tada H, Murakami M, et al. Circadian pharmacokinetics of mycophenolic acid and implication of genetic polymorphisms for early clinical events in renal transplant recipients. Transplantation 200682486–493 [DOI] [PubMed] [Google Scholar]
- 86.Kuriata-Kordek M, Boratyńska M, Urbaniak J, et al. Mycophenolic acid concentration profiles may select recipients with high-risk of acute rejection in renal transplant recipients. Pol Merkur Lekarski 200621161–3discussion 164 [PubMed] [Google Scholar]
- 87.Pawinski T, Durlik M, Szlaska I, et al. The weight of pharmacokinetic parameters for mycophenolic acid in prediction of rejection outcome: the receiver operating characteristic curve analysis. Transplant Proc 20063886–89 [DOI] [PubMed] [Google Scholar]
- 88.Kagaya H, Miura M, Satoh S, et al. No pharmacokinetic interactions between mycophenolic acid and tacrolimus in renal transplant recipients. J Clin Pharm Ther 200833193–201 [DOI] [PubMed] [Google Scholar]
- 89.van Gelder T, Tedesco Silva H, de Fijter JW, et al. Renal transplant patients at high risk of acute rejection benefit from adequate exposure to mycophenolic acid. Transplantation 201089595–599 [DOI] [PubMed] [Google Scholar]
- 90.Kuypers DR, Ekberg H, Grinyó J, et al. Mycophenolic acid exposure after administration of mycophenolate mofetil in the presence and absence of cyclosporin in renal transplant recipients. Clin Pharmacokinet 200948329–341 [DOI] [PubMed] [Google Scholar]
- 91.Gourishankar S, Houde I, Keown PA, et al. The CLEAR study: a 5-day, 3-g loading dose of mycophenolate mofetil versus standard 2-g dosing in renal transplantation. Clin J Am Soc Nephrol 201051282–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sommerer C, Müller-Krebs S, Schaier M, et al. Pharmacokinetic and pharmacodynamic analysis of enteric-coated mycophenolate sodium: limited sampling strategies and clinical outcome in renal transplant patients. Br J Clin Pharmacol 201069346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barraclough KA, Staatz CE, Johnson DW, et al. Kidney transplant outcomes are related to tacrolimus, mycophenolic acid and prednisolone exposure in the first week. Transpl Int 2012251182–1193 [DOI] [PubMed] [Google Scholar]
- 94.Fu L, Huang Z, Song T, et al. Short-term therapeutic drug monitoring of mycophenolic acid reduces infection: a prospective, single-center cohort study in Chinese living-related kidney transplantation. Transpl Infect Dis 201416760–766 [DOI] [PubMed] [Google Scholar]
- 95.Daher Abdi Z, Essig M, Rizopoulos D, et al. Impact of longitudinal exposure to mycophenolic acid on acute rejection in renal-transplant recipients using a joint modeling approach. Pharmacol Res 20137252–60 [DOI] [PubMed] [Google Scholar]
- 96.Daher Abdi Z, Prémaud A, Essig M, et al. Exposure to mycophenolic acid better predicts immunosuppressive efficacy than exposure to calcineurin inhibitors in renal transplant patients. Clin Pharmacol Ther 201496508–515 [DOI] [PubMed] [Google Scholar]
- 97.Ding CG, Jiao LZ, Han F, et al. Early immunosuppressive exposure of enteric-coated-mycophenolate sodium plus tacrolimus associated with acute rejection in expanded criteria donor kidney transplantation. Chin Med J (Engl) 20181311302–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Peng W, Liu G, Huang H, et al. Short-term intensified dosage regimen of mycophenolic acid is associated with less acute rejection in kidney transplantation from donation after circulatory death. Urol Int 2018101443–449 [DOI] [PubMed] [Google Scholar]
- 99.Kuypers DR, de Jonge H, Naesens M, et al. Current target ranges of mycophenolic acid exposure and drug-related adverse events: a 5-year, open-label, prospective, clinical follow-up study in renal allograft recipients. Clin Ther 200830673–683 [DOI] [PubMed] [Google Scholar]
- 100.Satoh S, Tada H, Murakami M, et al. The influence of mycophenolate mofetil versus azathioprine and mycophenolic acid pharmacokinetics on the incidence of acute rejection and infectious complications after renal transplantation. Transplant Proc 2005371751–1753 [DOI] [PubMed] [Google Scholar]
- 101.Pawinski T, Durlik M, Szlaska I, et al. Comparison of mycophenolic acid pharmacokinetic parameters in kidney transplant patients within the first 3 months post-transplant. J Clin Pharm Ther 20063127–34 [DOI] [PubMed] [Google Scholar]
- 102.Armstrong V, Heller T, Brandhorst G, et al. Relationship between free mycophenolic acid and hematologic side effects: interim results from the FDCC study. Transplantation. 2006;82(Suppl 2):344. [Google Scholar]
- 103.Sobiak J, Kamińska J, Głyda M, et al. Effect of mycophenolate mofetil on hematological side effects incidence in renal transplant recipients. Clin Transplant 201327E407–E414 [DOI] [PubMed] [Google Scholar]
- 104.Borni-Duval C, Caillard S, Olagne J, et al. Risk factors for BK virus infection in the era of therapeutic drug monitoring. Transplantation 2013951498–1505 [DOI] [PubMed] [Google Scholar]
- 105.Kiang TKL, Partovi N, Shapiro RJ, et al. Regression and genomic analyses on the association between dose-normalized mycophenolic acid exposure and absolute neutrophil count in steroid-free, de novo kidney transplant recipients. Clin Drug Investig 2018381011–1022 [DOI] [PubMed] [Google Scholar]
- 106.Le Meur Y, Thierry A, Glowacki F, et al. Early steroid withdrawal and optimization of mycophenolic acid exposure in kidney transplant recipients receiving mycophenolate mofetil. Transplantation 2011921244–1251 [DOI] [PubMed] [Google Scholar]
- 107.Prémaud A, Rousseau A, Le Meur Y, et al. Feasibility of, and critical paths for mycophenolate mofetil bayesian dose adjustment: pharmacological re-appraisal of a concentration-controlled versus fixed-dose trial in renal transplant recipients. Pharmacol Res 201061167–174 [DOI] [PubMed] [Google Scholar]
- 108.Rousseau A, Laroche ML, Venisse N, et al. Cost-effectiveness analysis of individualized mycophenolate mofetil dosing in kidney transplant patients in the APOMYGRE trial. Transplantation 2010891255–1262 [DOI] [PubMed] [Google Scholar]
- 109.Johnston A, Holt DW. Concentration-controlled trials. What does the future hold? Clin Pharmacokinet 19952893–99 [DOI] [PubMed] [Google Scholar]
- 110.Sheiner LB. Is intent-to-treat analysis always (ever) enough? Br J Clin Pharmacol 200254203–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pascual J, Berger SP, Witzke O, et al. ; TRANSFORM Investigators Everolimus with reduced calcineurin inhibitor exposure in renal transplantation. J Am Soc Nephrol 2018291979–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saint-Marcoux F, Vandierdonck S, Prémaud A, et al. Large scale analysis of routine dose adjustments of mycophenolate mofetil based on global exposure in renal transplant patients. Ther Drug Monit 201133285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Le Meur Y, Borrows R, Pescovitz MD, et al. Therapeutic drug monitoring of mycophenolates in kidney transplantation: report of the transplantation society consensus meeting. Transplant Rev (Orlando) 20112558–64 [DOI] [PubMed] [Google Scholar]
- 114.Sheiner LB, Steimer JL. Pharmacokinetic/pharmacodynamic modeling in drug development. Annu Rev Pharmacol Toxicol 20004067–95 [DOI] [PubMed] [Google Scholar]
- 115.Holford NH, Kimko HC, Monteleone JP, et al. Simulation of clinical trials. Annu Rev Pharmacol Toxicol 200040209–234 [DOI] [PubMed] [Google Scholar]
- 116.Sommerer C, Glander P, Arns W, et al. Safety and efficacy of intensified versus standard dosing regimens of enteric-coated mycophenolate sodium in de novo renal transplant patients. Transplantation 201191779–785 [DOI] [PubMed] [Google Scholar]
- 117.Matthews I, Kirkpatrick C, Holford N. Quantitative justification for target concentration intervention–parameter variability and predictive performance using population pharmacokinetic models for aminoglycosides. Br J Clin Pharmacol 2004588–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Neely M, Jelliffe R. Practical, individualized dosing: 21st century therapeutics and the clinical pharmacometrician. J Clin Pharmacol 201050842–847 [DOI] [PubMed] [Google Scholar]
- 119.Darwich AS, Ogungbenro K, Vinks AA, et al. Why has model-informed precision dosing not yet become common clinical reality? Lessons from the past and a roadmap for the future. Clin Pharmacol Ther 2017101646–656 [DOI] [PubMed] [Google Scholar]
- 120.McCune JS, Bemer MJ, Barrett JS, et al. Busulfan in infant to adult hematopoietic cell transplant recipients: a population pharmacokinetic model for initial and bayesian dose personalization. Clin Cancer Res 201420754–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.de Winter BC, Mathot RA, Sombogaard F, et al. Nonlinear relationship between mycophenolate mofetil dose and mycophenolic acid exposure: implications for therapeutic drug monitoring. Clin J Am Soc Nephrol 20116656–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ekberg H, Mamelok RD, Pearson TC, et al. The challenge of achieving target drug concentrations in clinical trials: experience from the symphony study. Transplantation 2009871360–1366 [DOI] [PubMed] [Google Scholar]
- 123.Ekberg H, Bernasconi C, Tedesco-Silva H, et al. Calcineurin inhibitor minimization in the symphony study: observational results 3 years after transplantation. Am J Transplant 200991876–1885 [DOI] [PubMed] [Google Scholar]
- 124.Wang X, Qin X, Wang Y, et al. Controlled-dose versus fixed-dose mycophenolate mofetil for kidney transplant recipients: a systematic review and meta-analysis of randomized controlled trials. Transplantation 201396361–367 [DOI] [PubMed] [Google Scholar]
- 125.Hale MD, Nicholls AJ, Bullingham RE, et al. The pharmacokinetic-pharmacodynamic relationship for mycophenolate mofetil in renal transplantation. Clin Pharmacol Ther 199864672–683 [DOI] [PubMed] [Google Scholar]
- 126.Peck CC, Barr WH, Benet LZ, et al. Opportunities for integration of pharmacokinetics, pharmacodynamics, and toxicokinetics in rational drug development. J Clin Pharmacol 199434111–119 [DOI] [PubMed] [Google Scholar]
- 127.Halloran P, Mathew T, Tomlanovich S, et al. Mycophenolate mofetil in renal allograft recipients: a pooled efficacy analysis of three randomized, double-blind, clinical studies in prevention of rejection. The international mycophenolate mofetil renal transplant study groups. Transplantation 19976339–47 [DOI] [PubMed] [Google Scholar]
- 128.Laftavi MR, Hai F, Laftavi H, et al. Mycophenolic acid dose reductions result in poor long-term renal allograft survival: comparison between mycophenolate sodium and mycophenolate mofetil. Transplant Proc 201143478–481 [DOI] [PubMed] [Google Scholar]
- 129.Doria C, Greenstein S, Narayanan M, et al. Association of mycophenolic acid dose with efficacy and safety events in kidney transplant patients receiving tacrolimus: an analysis of the mycophenolic acid observational renal transplant registry. Clin Transplant 201226E602–E611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vanhove T, Kuypers D, Claes KJ, et al. Reasons for dose reduction of mycophenolate mofetil during the first year after renal transplantation and its impact on graft outcome. Transpl Int 201326813–821 [DOI] [PubMed] [Google Scholar]
- 131.Dasgupta A. Usefulness of monitoring free (unbound) concentrations of therapeutic drugs in patient management. Clin Chim Acta 20073771–13 [DOI] [PubMed] [Google Scholar]
- 132.Bohnert T, Gan LS. Plasma protein binding: from discovery to development. J Pharm Sci 20131022953–2994 [DOI] [PubMed] [Google Scholar]
- 133.Kuypers DR, Claes K, Evenepoel P, et al. Long-term changes in mycophenolic acid exposure in combination with tacrolimus and corticosteroids are dose dependent and not reflected by trough plasma concentration: a prospective study in 100 de novo renal allograft recipients. J Clin Pharmacol 200343866–880 [DOI] [PubMed] [Google Scholar]
- 134.Langone A, Doria C, Greenstein S, et al. Does reduction in mycophenolic acid dose compromise efficacy regardless of tacrolimus exposure level? An analysis of prospective data from the mycophenolic renal transplant (MORE) registry. Clin Transplant 20132715–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kuypers DR, Vanrenterghem Y, Squifflet JP, et al. Twelve-month evaluation of the clinical pharmacokinetics of total and free mycophenolic acid and its glucuronide metabolites in renal allograft recipients on low dose tacrolimus in combination with mycophenolate mofetil. Ther Drug Monit 200325609–622 [DOI] [PubMed] [Google Scholar]
- 136.Cattaneo D, Perico N, Gaspari F, et al. Glucocorticoids interfere with mycophenolate mofetil bioavailability in kidney transplantation. Kidney Int 2002621060–1067 [DOI] [PubMed] [Google Scholar]
- 137.Hirsch HH, Randhawa P; AST Infectious Diseases Community of Practice BK polyomavirus in solid organ transplantation. Am J Transplant 201313Suppl 4179–188 [DOI] [PubMed] [Google Scholar]
- 138.Barraclough KA, Isbel NM, Staatz CE, et al. BK virus in kidney transplant recipients: the influence of immunosuppression. J Transplant. 2011;2011:750836. doi: 10.1155/2011/750836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Su V, Greanya ED, Ensom MH. Impact of mycophenolate mofetil dose reduction on allograft outcomes in kidney transplant recipients on tacrolimus-based regimens: a systematic review. Ann Pharmacother 201145248–257 [DOI] [PubMed] [Google Scholar]
- 140.Israni AK, Riad SM, Leduc R, et al. ; DeKAF Genomics Investigators Tacrolimus trough levels after month 3 as a predictor of acute rejection following kidney transplantation: a lesson learned from dekaf genomics. Transpl Int 201326982–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Torres IB, Reisaeter AV, Moreso F, et al. Tacrolimus and mycophenolate regimen and subclinical tubulo-interstitial inflammation in low immunological risk renal transplants. Transpl Int 2017301119–1131 [DOI] [PubMed] [Google Scholar]
- 142.Wiebe C, Rush DN, Nevins TE, et al. Class II eplet mismatch modulates tacrolimus trough levels required to prevent donor-specific antibody development. J Am Soc Nephrol 2017283353–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Girerd S, Schikowski J, Girerd N, et al. Impact of reduced exposure to calcineurin inhibitors on the development of de novo DSA: a cohort of non-immunized first kidney graft recipients between 2007 and 2014. BMC Nephrol. 2018;19:232. doi: 10.1186/s12882-018-1014-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Béland MA, Lapointe I, Noël R, et al. Higher calcineurin inhibitor levels predict better kidney graft survival in patients with de novo donor-specific anti-HLA antibodies: a cohort study. Transpl Int 201730502–509 [DOI] [PubMed] [Google Scholar]
- 145.Davis S, Gralla J, Klem P, et al. Lower tacrolimus exposure and time in therapeutic range increase the risk of de novo donor-specific antibodies in the first year of kidney transplantation. Am J Transplant 201818907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.O’Leary JG, Samaniego M, Barrio MC, et al. The influence of immunosuppressive agents on the risk of de novo donor-specific HLA antibody production in solid organ transplant recipients. Transplantation 201610039–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lederer SR, Friedrich N, Banas B, Welser G, Albert ED, Sitter T. Effects of mycophenolate mofetil on donor-specific antibody formation in renal transplantation. Clin Transplant 200519168–174 [DOI] [PubMed] [Google Scholar]
- 148.Filler G, Todorova EK, Bax K, et al. Minimum mycophenolic acid levels are associated with donor-specific antibody formation. Pediatr Transplant 20162034–38 [DOI] [PubMed] [Google Scholar]
- 149.Zhang HX, Sheng CC, Liu LS, et al. Systematic external evaluation of published population pharmacokinetic models of mycophenolate mofetil in adult kidney transplant recipients co-administered with tacrolimus. Br J Clin Pharmacol 201985746–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dong M, Fukuda T, Vinks AA. Optimization of mycophenolic acid therapy using clinical pharmacometrics. Drug Metab Pharmacokinet 2014294–11 [DOI] [PubMed] [Google Scholar]
- 151.Zwart TC, Gokoel SRM, van der Boog PJM, et al. Therapeutic drug monitoring of tacrolimus and mycophenolic acid in outpatient renal transplant recipients using a volumetric dried blood spot sampling device. Br J Clin Pharmacol 2018842889–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Md Dom ZI, Coller JK, Carroll RP, et al. Mycophenolic acid concentrations in peripheral blood mononuclear cells are associated with the incidence of rejection in renal transplant recipients. Br J Clin Pharmacol 2018842433–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Raggi MC, Siebert SB, Steimer W, et al. Customized mycophenolate dosing based on measuring inosine-monophosphate dehydrogenase activity significantly improves patients” outcomes after renal transplantation. Transplantation 2010901536–1541 [DOI] [PubMed] [Google Scholar]
- 154.Thi MT, Mourad M, Capron A, et al. Plasma and intracellular pharmacokinetic-pharmacodynamic analysis of mycophenolic acid in de novo kidney transplant patients. Clin Biochem 201548401–405 [DOI] [PubMed] [Google Scholar]
- 155.Chapman JR. The consequences of successful transplantation. Lancet 20113781357–1359 [DOI] [PubMed] [Google Scholar]