

Abstract
The well accepted “free drug hypothesis” for small-molecule drugs assumes that only the free (unbound) drug concentration at the therapeutic target can elicit a pharmacologic effect. Unbound (free) drug concentrations in plasma are readily measurable and are often used as surrogates for the drug concentrations at the site of pharmacologic action in pharmacokinetic-pharmacodynamic analysis and clinical dose projection in drug discovery. Furthermore, for permeable compounds at pharmacokinetic steady state, the free drug concentration in tissue is likely a close approximation of that in plasma; however, several factors can create and maintain disequilibrium between the free drug concentration in plasma and tissue, leading to free drug concentration asymmetry. These factors include drug uptake and extrusion mechanisms involving the uptake and efflux drug transporters, intracellular biotransformation of prodrugs, membrane receptor–mediated uptake of antibody-drug conjugates, pH gradients, unique distribution properties (covalent binders, nanoparticles), and local drug delivery (e.g., inhalation). The impact of these factors on the free drug concentrations in tissues can be represented by Kp,uu, the ratio of free drug concentration between tissue and plasma at steady state. This review focuses on situations in which free drug concentrations in tissues may differ from those in plasma (e.g., Kp,uu > or <1) and discusses the limitations of the surrogate approach of using plasma-free drug concentration to predict free drug concentrations in tissue. This is an important consideration for novel therapeutic modalities since systemic exposure as a driver of pharmacologic effects may provide limited value in guiding compound optimization, selection, and advancement. Ultimately, a deeper understanding of the relationship between free drug concentrations in plasma and tissues is needed.
Introduction
One of the key questions for assessing new drug entities in the drug-discovery stage is whether drug concentration and duration at the site of action (target) are adequate to elicit a pharmacologic effect. The primary reasons highlighted for failure of drug candidates during phase 2 are lack of efficacy (48%) and safety (25%) (Harrison, 2016). The question of whether adequate concentration at the target has been achieved may underpin a low cumulative success rate in the clinic (estimated to be 11.6%) (Smietana et al., 2016). Since free drug concentration at the target sites ultimately drives pharmacodynamics (PD), including both the efficacy and, at times, the toxicity of a drug, the inability to measure or predict accurately the free drug concentrations at the target site is a possible factor in the failure in drug development (Smith et al., 2010; Rankovic, 2015).
The Free Drug Hypothesis.
Drug molecules bind to proteins and lipids in blood and tissues, and only the free drug is available for target engagement (Benet and Hoener, 2002; Smith et al., 2010; Liu et al., 2014; Di et al., 2017). This means that the driver for efficacy is the free drug concentration (Cu) at the site of action. The free drug hypothesis describes two key points: 1) the PD of a drug is driven by the Cu at the site of action. This is also the case for clearance and drug-drug interactions through interactions with metabolic enzymes and drug transporters. In addition, changes in free drug concentration will lead to PD changes. 2) According to the passive diffusion theory, the free drug concentration in tissue is equal to the free drug concentration in plasma at a pharmacokinetic (PK) steady state (Benet and Hoener, 2002; Smith et al., 2010). This means the ratio of tissue (Cu,t) to plasma (Cu,p) free drug concentration at steady state (Kp,uu) for a noneliminating tissue will equal 1 (Kp,uu = Cu,t/Cu,p = ∼1) when transporters are not involved in tissue distribution of the drug. Less well recognized is the fact that, even in the presence of passive diffusion, Kp,uu can be <1 if the drug is rapidly cleared (by metabolism or transport) from the tissue.
The free drug concentration in plasma has been commonly used as a surrogate for free drug concentrations in tissue for PKPD modeling and dose projection in supporting compound selection and advancement. Advanced bioanalytical technologies have made it easier to obtain the total plasma drug concentrations in vivo and to determine the fraction of unbound (fu) in vitro/ex vivo, thereby enabling determination of the plasma free drug concentration. The free drug hypothesis has been widely used in drug discovery, with greater success for small molecules. Several examples of Kp,uu = ∼1 are listed in Supplemental Table S1. As an example, the free plasma concentration (Cu,p) of fluconazole, a highly permeable compound, was similar to the free concentration in body fluids, such as vaginal secretions, breast milk, saliva, sputum, prostatic, cerebrospinal, and seminal vesicle fluid (Debruyne, 1997; Smith et al., 2010).
The plasma and tissue PK and Cu are determined by multiple factors, such as clearance, distribution/disposition characteristics, permeability and transporter effects, biotransformation, and drug-delivery methods. Cu,p is not driven by the free fraction (fu) after oral administration but is governed by unbound clearance CLu. This concept is exemplified by the case of D01-4582, in which plasma fu level was 17-fold higher in SD rats compared with CD rats (Supplemental Fig. S1) (Ito et al., 2007); however, the plasma free drug concentrations were similar between the two strains of rats, although the total plasma concentrations in Sprague-Dawley rats were approximately 19-fold lower than in CD rats after an oral dose. These and many other results reported in the literature support the notion that plasma protein binding need not be optimized in the drug-discovery process for oral drugs (Liu et al., 2014).
The presence of active transporters could create asymmetry of free drug concentrations between tissues and plasma at PK steady state. Consequently, in some situations, drug concentrations in tissues are different from that in plasma (i.e., Kp,uu
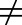 1). As new therapeutic modalities emerge and absorption, distribution, metabolism, and excretion sciences advance, it has become clear that some compounds do not appear to abide by the fundamental principle of equal free drug concentrations in plasma and tissues. Thus, when Kp,uu
1). As new therapeutic modalities emerge and absorption, distribution, metabolism, and excretion sciences advance, it has become clear that some compounds do not appear to abide by the fundamental principle of equal free drug concentrations in plasma and tissues. Thus, when Kp,uu
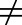 1, Cu,p may not serve as a good predictive surrogate of Cu,t; consequently, Cu,p may not provide a relevant framework for PKPD and dose prediction. This review focuses on a discussion of select cases highlighting scenarios in which free drug concentrations in tissues are likely different from those in plasma. We discuss total drug concentration in tissue-to-plasma ratio, and Kp is also discussed in cases where data for Kp,uu are limited; however, it is worth noting that Kp can be misleading if binding differs in the two compartments.
1, Cu,p may not serve as a good predictive surrogate of Cu,t; consequently, Cu,p may not provide a relevant framework for PKPD and dose prediction. This review focuses on a discussion of select cases highlighting scenarios in which free drug concentrations in tissues are likely different from those in plasma. We discuss total drug concentration in tissue-to-plasma ratio, and Kp is also discussed in cases where data for Kp,uu are limited; however, it is worth noting that Kp can be misleading if binding differs in the two compartments.
Methods for Determining Total Drug Concentrations in Plasma and Tissues
Drug concentrations in tissue can be readily measured in animals, although this obviously requires sacrificing the animals; so time-course data are often limited. The challenge is to measure tissue drug concentrations in humans. The only noninvasive and sensitive method for the latter is imaging [e.g., positron emission tomography (PET) imaging].
Liquid Chromatography-Tandem Mass Spectrometry.
Total drug concentrations in plasma, blood, and tissues can be measured in various ways. Because of selectivity, sensitivity, and ease of use, liquid chromatography-tandem mass spectrometry (LC-MS/MS) is the most common analytical choice. For LC-MS/MS, analytes are extracted from plasma/blood and tissue homogenates by organic solvents or other sample preparation techniques (e.g., solid-phase extraction) before analysis.
Autoradiography.
Examining tissue-to-plasma distribution of total drug (Kp) can be done by quantitative whole-body autoradiography (Stumpf, 2005; Solon et al., 2010) and microquantitative autoradiography (Stumpf, 2005; Solon et al., 2010; Drexler et al., 2011). These methods allow determination of local radioactivity in specific regions. Three limitations of the technique include not being able to distinguish parent drug from metabolites, providing only the total concentration of drug-related materials and the need for radiolabeled material, which is usually not available in discovery.
Positron Emission Tomography and Other Imaging Techniques.
Techniques such as PET and magnetic resonance imaging have been used to estimate drug concentrations in tissues (Sasongko et al., 2005; Eyal et al., 2009; He et al., 2014; Sundelin et al., 2017; Kaneko et al., 2018). Like the limitations of autoradiography, they cannot differentiate parent from metabolites and require a radiolabel (11C, 18F, etc.). Furthermore, for highly plasma protein drugs, corrections of tissue images for drug in the blood contained within the tissue should be performed to avoid overestimating total tissue drug concentration (Hsiao and Unadkat, 2014).
Matrix-Assisted Laser Desorption Ionization Mass Spectrometry Imaging.
More recently, matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) has become an emerging cutting-edge tool for determining the spatial localization of molecules of interest within tissues (Schwamborn and Caprioli, 2010; Caprioli, 2016). Unlike other imaging techniques, such as autoradiography, MALDI-MSI does not require molecular tags or labels. Further, this technique can generate distribution profiles (ion intensity maps) of many hundreds of compounds, including endogenous molecules that have different mass/charge (m/z) ratios simultaneously (Angel and Caprioli, 2013). The application of MALDI-MSI to visualize the spatial distribution of the antiretroviral drug tenofovir (TFV) and its diphosphorylated metabolite, TFV-diphosphate (the pharmacologically active form), in human colorectal tissue after TFV administration (as a rectal enema) demonstrated the heterogeneity in the distributions of these analytes (Seneviratne et al., 2018) (Supplemental Fig. S2). In this study, the potential ion suppression effects were determined by calculating tissue extinction coefficients for both TFV and TFV-DP.
MALDI-MSI technology can be further extended for the quantitation of drugs and their metabolites as well. Although this quantification can be achieved by incorporating an internal standard and creating a calibration curve, performing absolute quantitation has some challenges. For example, variation of signal (pixel-to-pixel), ion-suppression effects, and interference from matrix-related ions (or ion adducts) are confounding factors that must be addressed by applying appropriate normalization strategies and quantitative software (Källback et al., 2012; Schulz et al., 2019).
Modeling Approaches to Estimate Drug Concentrations.
Modeling approaches can be used to estimate drug concentrations in plasma and tissues (Watanabe et al., 2009; Tsamandouras et al., 2015; Li et al., 2018a,b); however, this is still an evolving area, and further validation is needed. One example of this is a proteomics-informed bottom-up approach to predict total drug concentrations in tissues where there is active transport and/or metabolism (Prasad and Unadkat, 2014). Specifically, metabolism- and transport-mediated clearance of a drug can be measured in vitro (in human liver microsomes, cytosol, S9 fraction, transporter expressing cell lines or primary cells), and protein expression-based scaling factors can then be used to extrapolate in vitro clearance to in vivo (IVIVE). Using quantitative proteomics, transporter tissue abundance has been established in the liver, kidney, and intestine (Drozdzik et al., 2014; Wang et al., 2015; Prasad et al., 2016). As a proof-of-concept, rosuvastatin hepatobiliary clearance in rats has been reasonably predicted using both rat organic-anion-transporting polypeptide (OATP)-expressing cell lines and sandwich-cultured rat hepatocytes (Ishida et al., 2018). Whereas IVIVE using transfected cell lines and quantitative proteomics is a promising technique for predicting intracellular drug concentrations, further validation of this approach is needed. In another example, global scaling factors were developed for physiology-based pharmacokinetic (PBPK) modeling involving transporter-mediated processes (Li et al., 2014). The modeling approach is intended for extrapolation or prediction of Kp or Kp,uu for drug candidates that have not been administered to humans.
Estimation of Free Drug Concentrations and Kp,uu
Measurement of fu and Total Drug Concentration.
Since direct measurement of free drug concentrations is challenging, it is typically obtained indirectly by measuring fu and total drug concentration. Measurement of total drug concentration was discussed earlier herein, and a number of methods are available to determine fu (Di et al., 2011, 2012); equilibrium dialysis is a commonly used method (Di et al., 2017). An extensive discussion of Kp,uu and experimental methods for measuring unbound drug concentrations in tissue can be found in the International Transporters Consortium white paper (Guo et al., 2018). Although it is reasonable to assume that the ex vivo binding is representative of the in vivo situation in liquid matrices (plasma), the same equivalence may not be as generalizable for solid matrices (tissues) where homogenization is required before separation of free and total drug. In addition, some of the measurements can be influenced by the presence of contaminants, such as plasticizers. Various in silico models have been developed to predict plasma protein binding based on molecular properties (Sun et al., 2018).
Prediction of In Vivo Kp,uu from In Vitro Kp,uu.
A new method has recently been developed to estimate in vivo liver-to-plasma Kp,uu from suspension hepatocytes in 4% bovine serum albumin (which is similar to albumin context in plasma) for OATP substrates (Supplemental Table S2). Good IVIVE has been demonstrated in both rats and humans. This approach has the potential to estimate in vivo free drug concentration in the liver from free plasma concentration (Riccardi et al., 2016, 2017). The method has been shown to be applicable to a number of solute carrier family, such as OATPs, OAT2, sodium-taurocholate cotransporting polypeptide, and SLC13A (Riccardi et al., 2016, 2017, 2019). Prospective predictions of Kp,uu have been successful based on human PD data or based on clearance data using the Kp,uu method (Riccardi et al., 2016, 2017, 2019). Several hypotheses about the mechanisms of the effect of albumin have been developed, and increasing numbers of examples of more accurate predictions in the presence of albumin have been reported (Bowman and Benet, 2018; Mao et al., 2018; Miyauchi et al., 2018; Bowman et al., 2019). The theoretical bases for a direct IVIV translation of Kp,uu have been discussed; in most cases, the differences between in vitro and in vivo Kp,uu values are minimal when the metabolism- or transporter-mediated clearance is not extensive (e.g., extraction ratios are not too high) (Li et al., 2019).
Estimation Based on PD Endpoints.
Assuming in vitro potency is the same as in vivo potency, the activity shift between in vivo and in vitro can be used to estimate Kp,uu. An example is illustrated using rosuvastatin and pravastatin (Riccardi et al., 2017). By comparing the in vivo and in vitro IC50 values of their inhibition potency for 3-hydroxyl-3-methylglutaryl coenzyme A reductase, liver-to-plasma Kp,uu values were derived. The key limitation for Kp,uu estimation is the underlying assumption that there is no potency difference between the in vitro and in vivo systems.
Microdialysis to Measure In Vivo Free Drug Concentration.
In vivo microdialysis can be used to measure free drug concentration. For example, cerebral microdialysis has been used to measure free drug concentration in brain (Tisdall and Smith, 2006). The limitations are that this method is technically challenging, it is low-throughput, and it suffers from high nonspecific binding to dialysis devices for lipophilic molecules.
Limitations of Current Approaches to Free Drug Estimation.
Most of the current methods to estimate free drug concentration use tissue homogenates to determine fu and then a total drug concentration measurement, which destroys all subcellular structures. The free drug concentration determined using this approach thus represents the average free drug concentration from all the subcellular compartments. As efficacy or toxicity depends on the drug’s interaction with specific targets within the subcellular compartment, it is important to understand whether the average free drug concentration represents the subcellular compartment of interest. For acidic compounds, the average free drug concentration is mostly the cytosolic concentration, whereas for basic compounds, it might be mostly lysosomal or mitochondrial and also depends on the cell type since different cell types can have different volumes of subcellular compartment. An example of heterogeneous subcellular drug distribution is metformin. Whereas the average Kp,uu of metformin from all the subcellular compartments in human embryonic kidney HEK293-OCT1 cells is 53.6, the model prediction of mitochondrial and endoplasmic reticulum Kp,uu is two to three times greater than the cytoplasm, lysosome, and nucleus (Chien et al., 2016). It is therefore important to convert fu from tissue homogenates to fu,cell of the subcellular compartment of interest to obtain the free drug concentration at the site of action. As Kp,uu is defined as a steady-state parameter, it is critical to ensure that both in vitro and in vivo measurements are made under steady-state conditions (e.g., by using intravenous infusion or intravenous area under the concentration curve (AUC) and using multiple time points to make sure steady state has been achieved). In some cases, contamination of blood or bile will need to be considered to estimate liver exposure. Typically, liver blood volume is set as the lower limit of liver Kp. For compounds with high biliary clearance, correction may need to be applied to obtain liver concentration.
Cases of Asymmetric Tissue Distribution
Substrates of Uptake and Efflux Transporters: Liver and Brain Examples.
Active transporters can create asymmetric drug distribution between tissues and plasma where Kp,uu
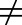 1. Different tissues can have different Kp,uu values for a particular compound. For example, an acidic compound can have a liver-to-plasma Kp,uu
1. Different tissues can have different Kp,uu values for a particular compound. For example, an acidic compound can have a liver-to-plasma Kp,uu
 1, whereas a brain-to-plasma Kp,uu
1, whereas a brain-to-plasma Kp,uu
 1. Tissue-to-plasma Kp,uu > 1 or <1 depends on a number of factors, including transporter expression difference. In a noneliminating tissue (e.g., brain), when active uptake is greater than efflux and other elimination mechanisms in the tissue (e.g., bulk flow), Kp,uu will be >1. In contrast, if active efflux from the tissue is predominant relative to passive diffusion, Kp,uu will be <1. For example, for drugs that are efflux transporter [polyglycoprotein (P-gp), breast cancer resistance protein (BCRP)] substrates, the brain-to-plasma Kp,uu is <1 (Feng et al., 2018). For an elimination organ (e.g., liver), Kp,uu is determined by multiple processes, including passive diffusion, active uptake and efflux, and metabolism based on the extended clearance concept (Yamazaki et al., 1996; Shitara et al., 2006; Watanabe et al., 2010; Patilea-Vrana and Unadkat, 2016). For instance, in vivo rat liver-to-plasma Kp,uu values of pravastatin, cerivastatin, fluvastatin, and rosuvastatin were 2.2, 29, 44, and 57, respectively (Riccardi et al., 2017). Of note, the rat liver samples collected in Riccardi et al. (2017) had all blood vessels removed to correct for blood but not bile contamination, and thus the Kp,uu values are likely reflective of true partitioning coefficients.
1. Tissue-to-plasma Kp,uu > 1 or <1 depends on a number of factors, including transporter expression difference. In a noneliminating tissue (e.g., brain), when active uptake is greater than efflux and other elimination mechanisms in the tissue (e.g., bulk flow), Kp,uu will be >1. In contrast, if active efflux from the tissue is predominant relative to passive diffusion, Kp,uu will be <1. For example, for drugs that are efflux transporter [polyglycoprotein (P-gp), breast cancer resistance protein (BCRP)] substrates, the brain-to-plasma Kp,uu is <1 (Feng et al., 2018). For an elimination organ (e.g., liver), Kp,uu is determined by multiple processes, including passive diffusion, active uptake and efflux, and metabolism based on the extended clearance concept (Yamazaki et al., 1996; Shitara et al., 2006; Watanabe et al., 2010; Patilea-Vrana and Unadkat, 2016). For instance, in vivo rat liver-to-plasma Kp,uu values of pravastatin, cerivastatin, fluvastatin, and rosuvastatin were 2.2, 29, 44, and 57, respectively (Riccardi et al., 2017). Of note, the rat liver samples collected in Riccardi et al. (2017) had all blood vessels removed to correct for blood but not bile contamination, and thus the Kp,uu values are likely reflective of true partitioning coefficients.
For compounds that have asymmetric tissue distribution owing to transporter involvement, free drug concentration in the liver should be used rather than plasma free concentration to understand drug-drug interaction (DDI) and the impact of genetic polymorphism owing to inhibition or induction of drug-metabolizing enzymes. The extended clearance model has been developed to understand the enzyme-transporter interplay on clearance, DDI, and genetic polymorphism (Yamazaki et al., 1996; Shitara et al., 2006; Watanabe et al., 2010; Patilea-Vrana and Unadkat, 2016). In the liver, when the metabolic plus biliary efflux clearances are much greater than sinusoidal efflux clearance, sinusoidal uptake clearance becomes the rate-determining step in the hepatic clearance of the drug. In other words, when the drug is eliminated out of the liver before it can return to plasma, the hepatic clearance is driven mainly by uptake clearance, even if there is significant metabolism and/or biliary efflux. In this case, inhibition of uptake transporters will increase plasma drug concentrations and exposure (AUC) but will not significantly impact the liver drug AUC, provided the drug is eliminated mainly by the liver. On the other hand, inhibition of metabolic enzyme or biliary transporters will increase the liver drug AUC while minimally impacting the plasma drug AUC (Watanabe et al., 2010). In both examples highlighted, the changes in the drug’s plasma concentrations will not reflect changes in the hepatic drug concentrations. This asymmetry may lead to misinterpretations of the impact of a DDI or genetic polymorphism on the drug’s PD effects when the target site of the drug is in the liver.
Atorvastatin, a permeable drug (logD7.4 = 1.53), is a substrate of OATP transporters and is eliminated from the body primarily by hepatic CYP3A metabolism. In a DDI study with atorvastatin, coadministration with rifampin, but not itraconazole, OATP, and CYP3A inhibitors, respectively, led to a significant increase in the plasma AUC of atorvastatin (Maeda et al., 2011). Based on these data, hepatic uptake clearance of atorvastatin via OATPs is the rate-determining step in the clearance of the drug. Compared with wild-type allele, patients with OATP1B1 polymorphism c.521T > C (conferring reduced function) have increased plasma atorvastatin AUC; however, no significant change in the PD response (i.e., lowering of low-density lipoprotein cholesterol) of atorvastatin is observed in patients with reduced OATP1B1 function (Shitara et al., 2013). In contrast, patients with homozygous CYP3A5*3 allele (conferring reduced function) have demonstrated an increased PD response to atorvastatin (Kivistö et al., 2004). PET imaging of rosuvastatin in the rat showed that inhibition of OATP uptake by rifampin led to a 2.3-fold increase in blood AUC, but it showed no significant change to the hepatic AUC (He et al., 2014). PBPK analysis of rosuvastatin in humans further illustrates that whereas OATP1B1 polymorphism impacts rosuvastatin plasma concentrations, the predicted rosuvastatin liver concentrations are not significantly impacted, and therefore neither is its PD response (Rose et al., 2014). These highlighted results demonstrate how the changes to a drug’s plasma exposure do not reflect changes to liver exposure and, therefore, PD. This is because the rate-determining step in the hepatic clearance of atorvastatin and rosuvastatin is uptake clearance via OATPs, even though, for the two drugs, there is metabolism via CYP3A enzymes and biliary efflux via MRP2/BCRP, respectively.
Metformin, an antidiabetic agent, is an interesting example. The pharmacologic site of action of metformin is in the liver, but it is eliminated primarily via the kidney as unchanged drug. In the liver, metformin is transported into the hepatocytes by OCT1 and perhaps is excreted in the bile by multidrug and toxin extrusion 1 (MATE1) as a minor route (Liang and Giacomini, 2017). Patients with OCT1 and MATE1 genetic polymorphism have an decreased and increased PD effect of metformin, respectively, but with no significant change in metformin plasma AUC (Shu et al., 2007; Stocker et al., 2013). PET imaging of [11C]metformin distribution into the liver demonstrated that patients with OCT1-reduced-function polymorphism had ∼30% lower metformin hepatic distribution versus those patients with wild-type OCT1 (Sundelin et al., 2017) without knowing whether this result is pharmacologically relevant. Likewise, in mice, coadministration with pyrimethamine (MATE inhibitor) results in a ∼2.5-fold increase in hepatic metformin AUC compared with controls (Jensen et al., 2016). Because most of metformin’s elimination is renal, hepatic transporters will impact its hepatic drug concentrations and therefore PD effect without impacting its plasma drug concentration.
The impact of efflux transporters on tissue-to-plasma Kp,uu is best demonstrated by interactions at the blood-brain barrier (BBB) (Supplemental Fig. S3). A critical functional component of the BBB that limits drug delivery is active efflux transport, mainly by P-gp and Bcrp, and brain drug delivery can be severely limited by this mechanism (Table 1) (Schinkel et al., 1996; Agarwal et al., 2011b; Gampa et al., 2017). For example, the brain distribution of [11C]verapamil increased 88% when coadministered with cyclosporine (i.e., P-gp and weak CYP3A inhibitor) without significantly affecting the systemic [11C]verapamil concentrations (Fig. 1) (Sasongko et al., 2005). In addition, reports indicate expression of efflux proteins in tumor cells, forming a secondary barrier to drug delivery to tumors in the brain (Demeule et al., 2001; Dean et al., 2005; Szakacs et al., 2006; Fattori et al., 2007; Luo et al., 2012). Even though the BBB/brain-tumor barrier may be relatively compromised in some regions of tumor, regions of brain that contain infiltrative tumor cells can have an intact and functional BBB. The development of systemic antitumor therapies (e.g., kinase inhibitors) that can adequately permeate an intact BBB and reach the target tumor cells has proven challenging (Table 1). On the other hand, restricted brain penetration owing to transporter-mediated drug efflux may be advantageous in some cases by reducing central nervous system toxicity.
TABLE 1.
Some targeted agents implicated in the treatment of cancers, and their brain distribution
| Targeted Agent | Primary Indication | Primary Target | Substrate Status | Brain-to-Plasma Ratio (Kp), Mice | Reference | |
|---|---|---|---|---|---|---|
| P-gp (ABCB1) | BCRP (ABCG2) | |||||
| Vemurafeniba | Melanoma | Mutant BRAF | Yes | Yes | 0.01 | Durmus et al. (2012), Mittapalli et al. (2012) |
| Dabrafenibb | Melanoma | Mutant BRAF | Yes | Yes | 0.04 | Mittapalli et al. (2013) |
| Trametinibc | Melanoma | MEK | Yes | No | 0.15 0.15 (Kp,uu) | Vaidhyanathan et al. (2014) |
| Gampa et al. (2018) | ||||||
| Cobimetinibd | Melanoma | MEK | Yes | No | 0.32 0.027 (Kp,uu) | Choo et al. (2014) |
| Omipalisiba | Melanoma | PI3k/mTOR | Yes | Yes | 0.06 | Vaidhyanathan et al. (2016) |
| Palbociclib | Breast cancer | CDK4/6 | Yes | Yes | 0.2a 0.01 (Kp,uu)e | de Gooijer et al. (2015), Parrish et al. (2015) |
| Abemaciclibe | Breast cancer | CDK4/6 | Yes | Yes | 1.2 0.17 (Kp,uu)e | Raub et al. (2015) |
| Imatinibf | Leukemia | BCR-Abl TK | Yes | Yes | 0.2 | Soo et al. (2010) |
| Sorafenibc | Hepatocellular and renal carcinoma | CRAF, BRAF,KIT, FLT-3, VEGFR | Yes | Yes | 0.06 | Agarwal et al. (2011) |
| Erlotinibg | NSCLC | EGFR | Yes | Yes | 0.06 0.13 (Kp,uu) | Kim et al. (2019a) |
| Sunitinibh | Renal carcinoma | RTK | Yes | Yes | 2.3 | Chee et al. (2016) |
| Gefitinibg | NSCLC | EGFR | Yes | Yes | 0.36 (Kp,uu) 0.1 (Kp,uu)i | Kim et al. (2019a) |
| Lapatiniba | Breast cancer | EGFR/HER2 | Yes | Yes | 0.03 | Polli et al. (2009) |
| Dasatiniba | Leukemia | BCR-Abl TK | Yes | Yes | <0.12 | Chen et al. (2009) |
| Nilotinibj | Leukemia | BCR-Abl K | Yes | Yes | 0.002 | Lee et al. (2018) |
AUC, area under the curve; BCRP, breast cancer resistance protein; EGFR, epidermal growth factor receptor; Kp, Brain-to-plasma ratio of the total concentrations; Kp,uu, brain-to-plasma ratio of the unbound concentrations; MEK, mitogen-activated protein kinase enzyme; NSCLC, non–small cell lung cancer; P-gp, P-glycoprotein; RTK, rhabdoid tumor of the kidney; VEGFR, vascular endothelial growth factor receptor.
Steady-state brain-to-plasma concentration ratios.
Brain-to-plasma ratio of AUC0–4 h after oral dose.
Brain-to-plasma ratio of AUC0–tlast after intravenous dose.
Brain-to-plasma ratio of concentrations 6 hours after oral dose.
Brain-to-plasma ratios of concentrations 5 minutes after intravenous dose.
Brain-to-plasma ratios of concentrations Cmax after oral dose.
Brain-to-plasma ratio of AUC0–tlast after subcutaneous dose.
Brain-to-plasma ratio of AUC0–∞ after oral dose.
Brain-to-plasma ratio of AUC0–24 h after oral dose.
Fig. 1.

Impact of efflux transporter inhibition on tissue drug concentrations. Increased [11C]verapamil distribution in the human brain (increased red color) in the presence of P-gp inhibition by cyclosporine. Representative brain magnetic resonance (left panel) and PET (middle and right pane) images before (middle panel) and during (right panel) cyclosporine infusion (2.5 mg/kg per hour) are shown (Sasangko et al., 2005). Whereas the [11C]verapamil concentrations in the brain increased (∼2-fold increase in brain AUC) when cyclosporine was coadministered, the systemic radioactive content of verapamil changed minimally (Sasangko et al., 2005). Figure reprinted with permission from Sasangko et al. (2005).
Many examples in the literature support the utility of Kp (when binding factors cancelled out) and Kp,uu in characterizing the brain distribution (Table 1). The following practical example also demonstrates the utility of Kp and Kp,uu in characterizing the brain distribution of a novel MEK inhibitor E6201 (Gampa et al., 2018). E6201 demonstrates low nanomolar potency in several BRAF mutant melanoma cell lines (Byron et al., 2012; Narita et al., 2014; Babiker et al., 2018). In vitro accumulation experiments in Madin-Darby canine kidney cell II cells show that E6201 is not a substrate of P-gp or Bcrp. After a 40-mg/kg single intravenous bolus dose of E6201, the total concentrations were greater in brain compared with plasma in four genotypes of mice (wild-type or efflux transporter–deficient); the systemic drug concentrations were similar across the four genotypes. As a consequence, the brain partitioning represented by the brain-to-plasma AUC ratios (Kp) were >1: 2.66, 4.37, 3.72, and 5.40 in wild-type, P-gp-deficient (Mdr1a/b−/−), Bcrp-deficient (Bcrp1−/−), P-gp- and Bcrp-deficient (Mdr1a/b−/− Bcrp1−/−) mice, respectively. The brain Kp after single oral and intraperitoneal doses of 40 mg/kg E6201 were comparable to the Kp observed in intravenous dosing studies. The free fractions (fu) of E6201 were determined using in vitro rapid equilibrium dialysis experiments and were 0.14% and 3.4% in brain and plasma, respectively. The Kp,uu values after a single 40-mg/kg intravenous dose of E6201 in wild-type, Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/− Bcrp1−/− mice were 0.14, 0.24, 0.2, and 0.29, respectively. These findings demonstrate that the delivery of E6201 to the brain is minimally impacted by P-gp or Bcrp-mediated active efflux at the BBB. For E6201, the brain Kp in wild-type mice is considerably higher than that of trametinib and cobimetinib, and the Kp,uu is higher than that of cobimetinib, although similar to that of trametinib (Table 2).
TABLE 2.
Comparison of brain distribution of MEK inhibitors in wild-type mice (data are presented as means)
| MEK Inhibitor | Dose mg/kg | Kp | Kp,uu | Reference |
|---|---|---|---|---|
| Trametinib | 5 (i.v.) | 0.15 | 0.15 | Vaidhyanathan et al. (2014) |
| Gampa et al. (2018) | ||||
| Cobimetiniba | 10 (p.o.) | 0.32 | 0.027 | Choo et al. (2014) |
| E6201 | 40 (i.v.) | 2.66 | 0.14 | Gampa et al. (2018) |
MEK, mitogen-activated protein kinase enzyme; Kp (AUC ratio), the ratio of AUC(0-t,brain) to AUC(0-t,plasma) using total drug concentrations; Kp,uu (AUC ratio), the ratio of AUC(0-t,brain) to AUC(0-t,plasma) using free drug concentrations.
Kp and Kp,uu based on plasma and brain concentrations 6 hours postdose.
The following example demonstrates that the efflux transporters could affect drug distribution in tissues differently. Apixaban is a potent and highly selective Factor Xa inhibitor, which is widely used for treatment of arterial and venous thrombotic diseases (Wong et al., 2011). Apixaban is a substrate for P-gp and BCRP (Zhang et al., 2013b). In addition, BCRP appears to play a more important role for absorption and intestinal and renal elimination of apixaban than P-gp in transporter-KO rats after oral and intravenous dosing (Zhang et al., 2013a). The distribution of [14C]apixaban was investigated in rats after single oral doses in which tissue distribution of radioactivity was measured using quantitative whole-body autoradiography (Wang et al., 2011). After oral administration, radioactivity was distributed quickly in rats with Cmax at 1 hour for most tissues in lactating rats. The radioactivity concentration was lowest in brains, which was detectable only within the first 1 hour after dose (Kp < 0.1 by concentration, Kp < < 0.1 by AUC). In pregnant rats, low levels of radioactivity were present in fetal blood, liver, and kidney, which were much lower (up to 20-fold) than that in the respective maternal organs (Kp < 0.1). In lactating rats, apixaban showed extensive lacteal excretion; values were 15-fold higher for Cmax and 55-fold greater for AUC than those in blood (Kp > 15 by Cmax, Kp > 50 by AUC) (Wang et al., 2011). Apixaban showed Kp,uu < 0.1 in brain, Kp,uu > 50 in milk, Kp,uu ∼1 in blood in rats, assuming similar protein binding in plasma and brain or milk. In this case, there was a direct correlation between blood concentrations and apixaban anticoagulant activity (Kp = 1 at the target tissue) since Factor Xa is mostly present in blood, the target for pharmacologic activity of apixaban (He et al., 2011; Wong et al., 2011). This provides a good example for a small-molecule drug whose tissue-to-plasma ratios could be greater, equal to, or less than that in blood (Kp < 1 to >1), depending on the specific tissue considered.
Lysosomal Trapping and Albumin-Assisted Uptake Leading to Higher Free Drug Concentrations in Tissue.
The free drug hypothesis can be complicated by lysosomal trapping and protein-assisted drug uptake. These two scenarios can lead to higher tissue free drug concentrations than plasma and are described here.
In the first scenario, there can be pH differences between plasma, tissue, and cellular organelles (Berezhkovskiy, 2011). The pH in lysosomes is 4.5–5 and, therefore, much lower than plasma, having a significant impact on the cellular levels of basic compounds. Even though there is still equilibrium of the neutral form of a molecule, basic compounds (pKa > 5) will be ionized in the lysosomes, and their charged nature will greatly reduce diffusion out of the lysosome. This phenomenon, called lysosomal trapping, results in Kp,uu > 1 (Ufuk et al., 2017). Generally, the impact on efficacy is small if the molecular target is in the cytosol, endoplasmic reticulum, or nucleus because the pH of those organelles is similar to the pH of plasma; however, if the target is in the lysosome itself or on the luminal side of the lysosomal membrane, it can greatly enhance efficacy, and it may appear that efficacy occurs at lower unbound plasma or tissue concentrations than anticipated.
In the second scenario, compounds, usually acids, that are highly bound to albumin can be prone to “protein-facilitated uptake” and, in particular, “albumin-facilitated uptake” (Fig. 2). This phenomenon was observed as early 1966 (Baker and Bradley, 1966), and an elegant study described it in more detail in 1988 (Tsao et al., 1988). Subsequently, the phenomenon was forgotten while academia and the industry focused on drug-metabolizing enzymes and, thereafter, transporters. More recently, the hypothesis was applied (Poulin et al., 2012, 2016) to improve in vivo-in vitro correlation for highly plasma protein-bound compounds. Using traditional models, highly bound compounds tend to underestimate in vivo clearance based on in vitro microsomal or hepatocyte data. Other examples have also been presented and reviewed related to albumin-facilitated uptake (Bowman and Benet, 2018). For example, hepatic uptake experiments were performed with hepatocytes in the presence and absence of plasma or albumin (Kim et al., 2019b). Considering the free drug hypothesis, uptake should be identical in the presence and absence of plasma or albumin after correcting for plasma protein binding; however, the uptake was substantially greater in the presence of plasma after correcting for plasma protein binding, and this phenomenon was generally more pronounced for highly bound compounds (fu < 0.05, especially fu < 0.01); this results in Kp,uu > 1 in the presence of plasma or albumin. In another study, it was shown that increased hepatic uptake clearance (Vmax/unbound Km) was observed with increasing albumin concentrations, and this phenomenon was explained by albumin-facilitated uptake (Fukuchi et al., 2017). Although a substantial amount of data have been presented that seems to support this albumin-facilitated uptake hypothesis (Bowman and Benet, 2018), there is still a lot of speculation about the exact mechanism; several hypotheses have been proposed (Poulin et al., 2016; Bowman and Benet, 2018), including the following:
The presence of an albumin receptor (e.g., FcRn) on hepatocytes and enhanced drug cellular uptake from the receptor-albumin-drug complex (Christensen and Birn 2013)
Interaction between albumin and the hepatocyte cell surface accompanied by a conformational change or driven by ionic interaction between the albumin-drug complex and the negatively charged groups on the hepatocyte cell membrane surface
Rate-limiting dissociation of drug from the albumin-drug complex whereby the bound (instead of free) drug determines the overall rate of hepatic uptake
Rate-limiting diffusion of unbound drug through the unstirred water layer (i.e., the albumin-drug complex enhances diffusion through the unstirred water layer and increases uptake for highly lipophilic compounds with lower diffusional flux)
Involvement of a high-affinity uptake transporters that strip drug from the albumin-drug complex; this may explain why most drugs for which this effect has been invoked are both highly bound to albumin and uptake transporter substrates
In addition to the preceding hypotheses, allosterism is a possible mechanism as the presence of an endogenous compound in plasma can change the transport clearance of a drug, and many transporters have been shown to be allosteric, such as OATPs and MRP2 (Gerk et al., 2004; Kindla et al., 2011). Although albumin-facilitated uptake has been mainly invoked to explain higher than anticipated uptake of drug into the liver, it could also occur in other organs, especially if the phenomenon is general and does not involve an albumin receptor or transporters (e.g., ionic interaction between the albumin-drug complex and the negatively charged groups on any cell surface). Assuming the molecular target of the drug is not in the liver, what would be the net effect of albumin-facilitated uptake? If this is a general phenomenon happening across tissues, the net effect on the efficacious dose may well be quite small because 1) increased uptake at the site of action leads to enhanced local unbound concentrations and presumably efficacy (Kp,uu > 1), but 2) enhanced uptake in the liver leads to higher drug clearance (for low ER drugs) and a lower unbound plasma concentration. Thus, the difference between the efficacious dose for compounds with and without albumin-facilitated uptake may well be quite small.
Fig. 2.

Protein-facilitated drug uptake mechanism.
Targeted Covalent Inhibitors.
The free drug hypothesis is more difficult to apply to targeted covalent inhibitor drugs because of PK and PD disconnect common with this mechanism of action. Since reactive moieties covalently bind to the target, target interaction is different from drugs that bind reversibly (Baillie, 2016). The following equation (eq. 1) describes the mechanism of how a covalent inhibitor (labeled C) modulates target enzyme or protein (labeled R):
| (1) |
The binding affinity of the covalent inhibitor to target proteins comes from both noncovalent (KI) and subsequent covalent (kinact) interactions. Permeation of free circulating covalent inhibitor through the cellular membrane and the reaction of free covalent inhibitors with either on-target or off-target enzymes are two different processes that are driven by concentration gradient (permeation) and reaction energy (covalent bonding). The differences in the reactive nucleophiles across the cellular membrane can alter free drug concentrations as a result of covalent bonding, assuming permeation is dominated by passive permeability. Depending on the molar ratio of intracellular free drug versus the target protein, the covalent interaction between inhibitor and target enzyme could play a role in the in vivo disposition of covalent inhibitors. With adequate inhibitor concentrations exceeding the target covalent binding threshold, an excess amount of free drug can re-establish a different free concentration gradient across cellular membranes. Several covalent inhibitors have demonstrated such capacity-limited nonlinear pharmacokinetic profile (Landersdorfer et al., 2012; Li et al., 2012).
Besides covalent interaction with targeted enzymes, it is common for covalent inhibitors to react covalently with off-target enzymes both in circulating blood and intracellular tissues, which also contributes to free drug disposition at steady state. The potential to form such covalent interaction is highly associated with the reactivity of different warhead moieties and their corresponding promiscuity toward other nucleophiles. Aspirin was shown to acetylate hemoglobin and serum albumin decades ago (Bridges et al., 1975; Burch and Blazer-Yost, 1981; Liyasova et al., 2010). Acrylamide-containing covalent inhibitors are intrinsically reactive to intracellular glutathione and free thiol groups in cysteine residues of proteins and enzymes. Numerous covalent inhibitors (e.g., neratinib, osimertinib, ibrutinib) show degrees of endogenous proteinaceous adduct formation. Neratinib formed covalent adducts on Lys-190 of human serum albumin (Wang et al., 2010). Similarly, [14C]osimertinib revealed covalent binding in incubations with hepatocyte, plasma, and serum albumin. In the case of ibrutinib, in vivo, more than 50% of total plasma radioactivity was attributed to covalently bound materials from 8 hours onward; as a result, covalent binding accounted for 38% and 51% of total radioactivity AUC0–24 h and AUC0–72 h, respectively (Scheers et al., 2015). The elimination of covalently bound radioactivity in human plasma was slower than the elimination of total radioactivity, indicating that ibrutinib likely has different reactivity toward protein targets in different tissues. Alternatively, the turnover rate of the proteins may differ.
In the free drug hypothesis, the interaction between drug and receptor is reversible. Within the time scale of receptor turnover half-life, covalent inhibitors can bind to receptors reversibly or irreversibly. The outcome of the covalent inhibition results in removal of receptor targets from the active naïve target pool. Thus, the pharmacologic activity from covalent inhibition is determined by both free drug concentration and the turnover of target protein or enzyme. If the time to resynthesize target protein is longer than covalent inhibitor in vivo half-life, a sustained pharmacology effect can occur even after the covalent inhibitor is eliminated from systemic circulation (Fig. 3A). After disappearance of drug, recovery to baseline level is dependent on resynthesis of the proteins, irrespective of drug concentration. In Fig. 3B, the animal model PKPD results for PRN1008, a reversible covalent inhibitor, showed a clear disconnect between PK exposure profile and BTK target occupancy. Clinically, covalent drug pharmacologic effect can last multiple days or even weeks after drug treatment discontinuation. Finasteride, a 5α-reductase inhibitor, prevents conversion of testosterone to dihydrotestosterone. Finasteride has a terminal pharmacokinetic half-life of 6–8 hours. After discontinuing daily finasteride administration, dihydrotestosterone concentrations do not return to pretreatment levels until approximately 2 weeks after withdrawal of therapy (https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020180s044lbl.pdf).
Fig. 3.

PKPD profiles of a covalent drug. (A) Modeled PKPD plot of a hypothetical covalent inhibitor alkylating a target with turnover half-life of 24 hours. (B) Exposure profile disconnect from BTK target occupancy for covalent BTK inhibitor PRN1008 in a rat collagen-induced arthritis model.
Small-Molecule Prodrugs.
For prodrugs that form the active drug in tissue, it is difficult to describe their pharmacologic activity using plasma free drug concentration. A prodrug is an administered molecular entity that is converted to the active form in the body (Testa, 2009). It is usually easier to measure the drug in plasma when the active drug is quickly formed from the prodrug by carboxylesterases in plasma, as is the case for drugs such as clofibrate and oseltamivir; however, for prodrugs like sofosbuvir, the active form is not permeable and is formed through biosynthesis or biotransformation intracellularly and may not result in a meaningful level in plasma (Kp,uu > > 1). The rationale for design of prodrugs has been extensively and well established in the literature; these concepts are beyond the scope of this review (Stella, 2010). The compound administered is chemically distinct from the compound driving the pharmacologic response for a prodrug. Consequently, both the mechanism of release and the physiologic distribution and expression of activating enzyme(s) are of great importance in the selection and development of prodrugs (Stella, 2010). This tissue drug concentration asymmetry is anticipated even under PK steady state.
Prodrugs represent approximately 12% of all new drugs approved in last decade (Rautio et al., 2018). Clofibrate, oseltamivir, tenofovir, and sofosbuvir are used in the treatment of dyslipidemia, influenza, HIV, and hepatitis C, respectively (Oberg, 2006; Menendez-Arias et al., 2014; Rivero-Juarez et al., 2018). Beyond the obvious commonality that each of these drugs is administered orally, there are significant differences, as illustrated in Fig. 4, in the activation and hydrolysis mechanism, enzyme kinetics of activation/hydrolysis, target tissue, and pharmacologic mechanism of target inhibition. Whereas the relatively simple ester hydrolysis of clofibrate and oseltamivir might suggest that an obvious relationship exists between the plasma exposure of the liberated acid and pharmacologic response, this would ignore the confounding effects of the site of hydrolysis and the endpoints used to assess pharmacologic activity (Chasseaud et al., 1974; Miller and Spence, 1998; Shi et al., 2006; Wattanagoon et al., 2009). These relationships between target loading, hydrolysis, and pharmacologic mechanism become even more complicated considering tenofovir and sofosbuvir (Oberg, 2006; Ray et al., 2016; Rivero-Juarez et al., 2018). Both these compounds undergo multistep activation to their ultimate pharmacologically active species (Fig. 4). Although it is likely that the prodrug is the chemical species entering the target cells, there are significant differences in the distribution of the target cells, lymphocytes (tenofovir), and hepatocytes (sofosbuvir), that would further complicate deconvolution of the relationship between systemic concentrations and pharmacologic effect (Oberg, 2006; Ray et al., 2016; Rivero-Juarez et al., 2018). Furthermore, it is conceivable that both compounds could load their target cells locally, in the gastrointestinal-associated lymph tissue in the case of tenofovir and the liver for sofosbuvir such that portal venous concentrations would be related more to cellular uptake than to systemic concentrations. Additionally, the pharmacologic mechanism of these compounds is DNA (tenofovir) or RNA (sofosbuvir) chain termination by the triphosphorylated metabolite of each compound that is impermeable (Oberg 2006). This process is irreversible and further obscures any relationship with the plasma concentrations of the administered compound. For prodrugs that form the active drug in tissue, dynamic modeling will need to be used to develop PK/PD relationships.
Fig. 4.

Biotransformation of prodrugs. A-E are examples.
Antibody-Drug Conjugates.
An antibody-drug conjugate (ADC), in general, is itself inactive and could be considered a prodrug that requires a transformation to expose or release the payload (active drug). ADCs consist of an antibody and a payload with a chemical linker and have proven an effective modality of selectively delivering a small-molecule payload to targeted cells. The ADC marketing approval of ADCETRIS, KADCYLA, and BESPONSA has drawn wide interest in expanding the applications of these powerful agents (Doronina et al., 2003; LoRusso et al., 2011; Ricart, 2011; Sliwkowski and Mellman, 2013; Chari et al., 2014; Shor et al., 2015; Beck et al., 2017). The payload exerts its biologic activities after ADC is internalized and the payload is released into cells at the site of action (e.g., in tumor cells). For example, different efficacy profiles were obtained for two ADCs (CD22 conjugates of cyclopropyl-disulfide-PBD and cyclobutyl-disulfide-PBD) after administration in xenograft mice (Ma et al., 2016; Zhang et al., 2016). The cyclobutyl-disulfide-PBD released the active PBD payload, but the former ADC released an inactive payload metabolite. For these two ADCs, the tumor and plasma exposures of total antibody (i.e., monoclonal antibodies) were similar in plasma and tumors; the drug-to-antibody ratios of the two conjugates were also similar for 7 days after dosing. The cyclobutyl-disulfide-PBD ADC showed Kp > 150 in tumors for its payload 96 hours after dose. Figure 5 illustrates hypothetical ADC payload concentration-time profiles in tumors, liver, and blood, in which tumor payload concentration is much greater than that in livers with very low concentration in plasma. The tumor growth inhibition correlated with intratumor PBD exposures, but not with systemic exposures of ADCs or the payload after administration of HER2-disulfide-PBD conjugates in xenograft mice (Zhang et al., 2018). These results indicate that analysis of ADC species in circulation is insufficient to explain or predict ADC efficacy and suggested that payload/catabolite identification and quantitation in the tumors of xenograft mice are important to understand the ADC efficacy. As a consequence of the cellular uptake, biotransformation, and pharmacologic mechanisms of ADCs, the concentration of the active molecule (payload) in tumors could be much higher than that in plasma.
Fig. 5.

Hypothetical ADC payload concentration-time profiles in tumor, liver, and plasma. The figure was depicted partially based on recently published data in xenograft mice after intravenous administration of the CD22-conjugates of cyclobutyl-disulfide-PBD (Ma et al., 2016; Zhang et al., 2016).
Local Drug Delivery (Inhalation).
Local delivery is a direct way to increase the free drug concentration at a given tissue relative to plasma (Kp,uu > 1). There are many modes of local drug delivery, including dermal delivery to the skin, oral ingestion of gastrointestinal-restricted agents, intranasal delivery to the nasal cavity, and oral inhalation to the lungs. With multiple blockbuster therapies having benefited millions of patients (Stein and Thiel, 2017), inhalation to the lungs will be used to highlight the principles of local drug delivery for this review.
In preclinical species, inhalation doses are typically administered by intranasal delivery, intratracheal instillation, or tower dosing of dry powders or nebulized solutions and suspensions (Turner et al., 2011; Price et al., 2019). Lung tissue can be routinely harvested from preclinical species, allowing total drug levels to be measured directly and relationships between lung and plasma concentrations to be established. In humans, inhaled drugs are commonly delivered as dry powders from breath-actuated devices or as solutions or suspensions administered via pressurized metered devices or nebulizers (Stein and Thiel, 2017; Kaplan and Price, 2018). It is typically not feasible to measure drug concentrations directly in human lung tissue because of the invasive nature of lung biopsy (Esmailpour et al., 1997). Instead, it is common to measure drug concentrations in plasma and then infer lung concentrations based on lung-to-plasma correlations established in preclinical species.
A key advantage of inhaled and lung-targeted drugs is that they are typically administered in lower doses than are oral systemic drugs. This is the case for two reasons. First, an inhaled drug is not subject to first-pass metabolism by the liver or gut wall before reaching the lung as would be the case with an oral drug. Second, an inhaled drug is immediately concentrated at the site of action (the lung), as opposed to an oral drug that reaches the lung only after significant dilution in blood and other tissues.
In addition to minimizing the total dose, inhalation can lead to a lung-biased drug distribution. Therapeutic index may thereby be increased by maximizing pharmacology in the lung while minimizing its exposure elsewhere in the body; however, many drugs, particularly those that are highly soluble or highly intrinsically permeable, pass rapidly through the lung after inhalation and enter the systemic circulation via the pulmonary vasculature. Inhaled and lung-targeted drugs must therefore be designed with specific properties to slow permeation and prolong retention in the lung. These properties generally include one or more of the following: 1) low solubility/slow dissolution rate, 2) high tissue affinity imparted by strongly basic center(s) or high lipophilicity, 3) low membrane permeability, or 4) slow off-rate from the target of interest. Specific examples of inhaled drugs using these lung-retention strategies are illustrated in Supplemental Fig. S4 and Table 3. High systemic clearance is typically another desirable feature for lung-targeted drugs since, even with inhalation dosing, a compound may be absorbed systemically (Millan et al., 2011; Jones et al., 2017). As discussed already herein, the lung-deposited portion of the dose is subject to pulmonary absorption into systemic circulation. Additionally, a large fraction of a human inhaled dose is typically swallowed, which may allow it to be absorbed from the gastrointestinal tract (Taburet and Schmit, 1994; Hochhaus et al., 2015). High systemic clearance will rapidly eliminate any systemically absorbed compound and will maximize the ratio of drug in the lung relative to the blood and peripheral organs.
TABLE 3.
Properties of representative inhaled, lung targeted compounds
Lung retention mechanism(s) for each compound are denoted by checkmarks.
| Compound | Target | cLogP | cpKa | Lung Retention Mechanism(s) | ||||
|---|---|---|---|---|---|---|---|---|
| Dissolution-Limited | Tissue Affinity | Low Permeability | Slow Off-Rate | |||||
| Basic | Lipophilic | |||||||
| Fluticasone propionatea | Glucocorticoid receptor | 3.3 | <3 | ✓ | ✓ | |||
| Fluticasone furoateb | Glucocorticoid receptor | 3.7 | <3 | ✓ | ✓ | |||
| Salmeterol Xinafoatec | β2-adrenergic receptor | 4.1 | 9.4 | ✓ | ✓ | ✓ | ✓d | |
| Salbutamole | β2-adrenergic receptor | 0.6 | 9.4 | ✓ | ||||
| Tiotropium bromidef | Muscarinic receptors (M3, M1) | −2.3 | <3 | ✓ | ✓ | |||
| Compound 3.05g | Epithelial sodium channel | −2.0 | 8.5 | ✓ | ✓ | |||
| GSK2269557/Nemiralisibh | PI3Kδ | 4.8 | 8.7, 4.1 | ✓ | ✓ | |||
Apparent slow off-rate likely due to partitioning into lipid membrane bilayers.
Anderson et al. (1994), Dickson et al. (2016), and references therein.
Kley et al. (2016).
Figure 6A shows an example of a compound with high systemic clearance that is also poorly retained in the lungs after inhalation dosing. The compound diffuses quickly through the lung into systemic circulation, and there is negligible separation between lung and plasma concentrations. Lack of lung retention and high systemic clearance result in a short half-life in the lungs, and this profile is suboptimal for a lung-targeted drug. In contrast, Fig. 6B shows the profile of a compound with high systemic clearance that is well retained after inhalation dosing. The compound diffuses only slowly across the lung and, once eventually absorbed into blood, is metabolized rapidly. The result is a desirable profile for a lung-targeted inhaled therapy characterized by a long half-life in the lung and a marked separation between lung concentrations relative to plasma (Kp > > 1).
Fig. 6.

Typical concentration-time profiles after inhalation of (A) a poorly lung-retained drug with high clearance in plasma; (B) a significantly lung-retained drug with high clearance in plasma.
Although determining Kp between lung and plasma in preclinical species is straightforward, measuring Kp,uu is more challenging because of the uncertainty in quantifying “free” drug in the lung. Indeed, concentrations measured in the lung represent an aggregate of all states of the drug, and it is not typically possible to differentiate drug that is unbound and soluble from drug that is bound to lung tissue or insoluble. For that reason, a Kp,uu > 1 is typically inferred only indirectly from preclinical in vivo PKPD models. Typically, this is accomplished either by 1) demonstrating a potent PD response in the lung but a diminished response in other tissues with inhaled dosing (Hemmerling et al., 2017) or 2) demonstrating equivalent lung PD via both intravenous and inhaled dosing but at lower plasma concentrations with inhaled dosing (Shaw et al., 2016). Finally, in the clinical setting, a Kp,uu > 1 is inferred by observing the desired PD effect or disease improvement in the lung, with a corresponding lack of on-target pharmacology in the periphery.
Nanoparticles.
Nanoparticles (NPs) have applications in targeted drug delivery that can create asymmetric drug distribution between tissues and plasma. Several categories of NP have been developed and are depicted as cartoons in Supplemental Fig. S5. NPs have unique disposition properties. Most engineered NPs are in the size range from 30 to 400 nm and as such do not undergo renal clearance or suffer from extensive metabolism by liver enzymes that require 5.5 nm for these clearance mechanisms (Choi et al., 2007); however, when NPs are intravenously administered, serum proteins known as opsonins adhere to the surface of the NPs, thereby making them amenable to uptake from mononuclear phagocyte cells that are a part of the reticuloendothelial system. This results in selective uptake of NPs in the liver and spleen.
Tumor and other inflamed tissues have a leaky vasculature through which larger-sized molecules can either enter or leave. This principle is used for the extravasation of NPs from the systemic circulation to tumor tissues. The effect has been labeled as enhanced permeability and retention (EPR) and has been the bedrock for passive targeting using NPs. The EPR effect is accumulative and is therefore significantly enhanced by increasing systemic circulation of the NPs. From a drug-distribution standpoint, the drug concentration in the tumor clearly will differ from other normal tissues and plasma owing to the EPR effect.
For therapeutic benefit, the NP cargo (drug) should be readily released at the site of action (i.e., the tumor cell). For active targeting, the NP is decorated with a ligand that will bind with a receptor expressed on the cell surface of the target cell. Once the ligand binds to the receptor, it facilitates receptor-mediated endocytosis transporting the drug into the cell. For example, Lippard and coworkers showed that a lethal dose of cisplatin could be safely delivered to prostate cancer cells by using an aptamer-decorated poly(lactic-co-glycolic acid-b-PEG NP that encapsulates cisplatin (Dhar et al., 2008). Park et al. (2002) synthesized long-circulating immunoliposomes of doxorubicin that were decorated with PEG chains and anti-HER2 monoclonal antibody fragments. These immunoliposomes were administered intravenously for targeting tumor cells. Given the mechanism of action, when using NPs either for passive or targeted delivery, the tissue concentrations can be substantially different from the plasma concentrations.
Summary
The “free drug hypothesis” means that only free drug molecules can passively diffuse in and out of a cell, interact with their targets, and result in pharmacologic effects. Since the free drug concentration at the target site cannot be directly measured, the plasma free drug concentration is frequently used as a surrogate. The assumption of the free drug hypothesis in tissue has helped in the discovery of many small-molecule drugs. This typically occurs when Cu,p and Cu,t are equal (Kp,uu = 1); however, the assumption of Cu,p and Cu,t being equal does not always hold (Kp,uu
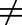 1) for new modalities. For some of these new modalities, tissue is an elimination organ as opposed to a distribution organ. Although some cases were discussed based on Kp instead of Kp,uu (because of limited Kp,uu data), free drug concentration asymmetry likely holds in these situations. When the free drug concentration in plasma does not equal that in the tissue, the tissue free drug concentration would need to be estimated to develop the PKPD relationship and predict dose and toxicity. The surrogate approach has limitations, and some of these situations are summarized in Table 4.
1) for new modalities. For some of these new modalities, tissue is an elimination organ as opposed to a distribution organ. Although some cases were discussed based on Kp instead of Kp,uu (because of limited Kp,uu data), free drug concentration asymmetry likely holds in these situations. When the free drug concentration in plasma does not equal that in the tissue, the tissue free drug concentration would need to be estimated to develop the PKPD relationship and predict dose and toxicity. The surrogate approach has limitations, and some of these situations are summarized in Table 4.
TABLE 4.
Applicability of surrogate approach of free drug equilibrium between plasma and tissues for different modalities
| Type of Modalities | Applicability | Examples | Kp or Kp, uu | Comments |
|---|---|---|---|---|
| Highly permeable compounds | Yes, in blood | Eliquis | =1 | As many other small molecules |
| Efflux transporter substrates | No, in brain or fetus | Eliquis | >1 or <1 | Low concentration in brain but high in milk |
| Uptake transporter substrates | No | Statins | >1 | Liver OATP uptake |
| Quick-forming prodrugs | Maybe | Oseltamivir | >1 | Hydrolysis removal of protecting groups |
| Lysosomal trapping | Could be | Garenoxacin | >1, =1 | pH gradient, lysosomal accumulation into macrophages |
| Target trapping | Not likely | Paclitaxel | >>1 | Microtubule binding, cancer cell accumulation |
| Covalent binders | Not likely | Afatinib | >1 | DNA alkylators, protein modifiers |
| Transforming prodrugs | No | Tenofovir | >>1 | Active drug formed through metabolism and biosynthesis in tissue |
| ADCs | No | Kadcyla | >>1 | Antigen-mediated uptake, catabolism, drug release |
| Nucleic acid | No | Fomivirsen Mipomersen | >1 | Unique distribution properties |
| Nanoparticles | No | >>1 | Unique distribution properties | |
| Inhalation | No | Fluticasone | >>1 | Designed to increase systemic clearance |
ADC, antibody drug conjugate; OATP, organic-anion-transporting polypeptide.
For drug modalities such as transporter substrates, prodrugs, antibody-drug conjugates, and drugs requiring tropical delivery, the surrogate approach does not apply (Kp,uu
 1). In addition, those new modalities, such as nucleic acids and protein degraders (Burslem and Crews, 2017) for hard-to-drug targets (Valeur et al., 2017), have unique distribution properties and may also exhibit higher drug concentrations in tissues than in plasma. These cases introduce difficulties for modeling scientists and regulatory agencies. When the free drug concentration in tissues cannot be measured or easily extrapolated from that in plasma, the plasma exposures may have limited utility in guiding candidate selection or compound advancement because plasma exposures cannot be related to effect for PKPD modeling and human dose projection. For these therapeutic modalities, IVIVE to predict tissue concentrations warrants further development. In addition, since drug discovery starts from developing a bioavailable and efficacious compound in animal models (e.g., xenograft mouse), bridging drug concentrations in plasma and target tissues is important and practical in preclinical models for understanding PKPD and in supporting late clinical development. Ultimately, a deeper understanding of the relationship between free drug concentrations in plasma and tissues is needed. Indeed, we believe this is the next frontier in absorption, distribution, metabolism, and excretion research.
1). In addition, those new modalities, such as nucleic acids and protein degraders (Burslem and Crews, 2017) for hard-to-drug targets (Valeur et al., 2017), have unique distribution properties and may also exhibit higher drug concentrations in tissues than in plasma. These cases introduce difficulties for modeling scientists and regulatory agencies. When the free drug concentration in tissues cannot be measured or easily extrapolated from that in plasma, the plasma exposures may have limited utility in guiding candidate selection or compound advancement because plasma exposures cannot be related to effect for PKPD modeling and human dose projection. For these therapeutic modalities, IVIVE to predict tissue concentrations warrants further development. In addition, since drug discovery starts from developing a bioavailable and efficacious compound in animal models (e.g., xenograft mouse), bridging drug concentrations in plasma and target tissues is important and practical in preclinical models for understanding PKPD and in supporting late clinical development. Ultimately, a deeper understanding of the relationship between free drug concentrations in plasma and tissues is needed. Indeed, we believe this is the next frontier in absorption, distribution, metabolism, and excretion research.
Acknowledgments
We thank the following scientists who contributed to discussion and ideas for this review: Dr. Dan Sutherlin (Genentech), Dr. Julie Lade (Amgen), Dr. Lijuan Jiang (Enanta), Dr. Matthew Durk (Genentech), Dr. Nina Isoherranen (University of Washington), Dr. Sheerin Shahidi-Latham (MyoKardia), and Dr. William Humphreys. D.Z. thanks the American Society for Pharmaceutical and Experimental Therapeutics (ASPET) for invitation to present a symposium on Tissue Free Drug Concentration at the 2018 annual meeting on April 25, 2018, in San Diego. D.Z. (Genentech), Lijuan Jiang (Enanta), Julie Lade (Amgen), Sheerin Shahidi-Latham (Genentech), Herana Seneviratne (Johns Hopkins University), and Gautham Gampa (University of Minnesota) presented related topics at the symposium chaired by D.Z. and Julie Lade.
Abbreviations
- ADC
antibody drug conjugate
- AUC
area under concentration time curve
- BBB
brain-blood barriers
- BCRP
breast-cancer resistance protein
- CLu
unbound drug clearance
- Cu
unbound drug concentration
- DDI
drug-drug interaction
- EPR
enhanced permeability and retention
- fu
unbound fraction
- IVIVE
in vitro in vivo extrapolation
- Kp
tissue-to-plasma drug concentration ratio
- Kp,uu
tissue-to-plasma free drug concentration ratio
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- MALDI-MSI
matrix-assisted laser desorption/ionization mass spectrometry imaging
- MATE
multidrug and toxin extrusion
- MEK
mitogen-activated protein kinase enzyme
- NP
nanoparticle
- OATP
organic-anion-transporting polypeptide
- OCT1
organic cation transport 1
- PBD
pyrrolo[2,1-c][1,4]benzodiazepine-dimer
- PBPK
physiology-based pharmacokinetic modeling
- PET
positron emission tomography
- P-gp
P-glycoprotein
- PK
pharmacokinetics
- PKPD
pharmacokinetics and pharmacodynamics
- TFV
tenofovir
Authorship Contributions
Participated in research design: Zhang, Hop, Patilea-Vrana, Gampa, Seneviratne, Unadkat, Kenny, Nagapudi, Di, Zhou, Zak, Wright, Bumpus, Zang, Liu, Lai, Khojasteh.
Wrote or contributed to the writing of the manuscript: Zhang, Hop, Patilea-Vrana, Gampa, Seneviratne, Unadkat, Kenny, Nagapudi, Di, Zhou, Zak, Wright, Bumpus, Zang, Liu, Lai, Khojasteh.
Footnotes
G.P.-V. was supported by the Rene Levy Fellowship. G.P.-V. and J.D.U. were supported in part by the National Institutes of Health National Institute of Drug Abuse [Grant P01DA032507]. G.G. was supported by the Ronald J. Sawchuk Fellowship in Pharmacokinetics and University of Minnesota Doctoral Dissertation Fellowship. His work was supported by the NIH [Grants R01-NS077921, R01-NS073610, U54-CA210180] and Strategia Therapeutics Inc. H.K.S. and N.N.B. were funded by the NIH [Grants U19AI11327, UM1 AI068613, R01AI128781].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. (2011a) Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med 13:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Sane R, Ohlfest JR, Elmquist WF. (2011b) The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther 336:223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GP, Lindén A, Rabe KF. (1994) Why are long-acting beta-adrenoceptor agonists long-acting? Eur Respir J 7:569–578. [DOI] [PubMed] [Google Scholar]
- Angel PM, Caprioli RM. (2013) Matrix-assisted laser desorption ionization imaging mass spectrometry: in situ molecular mapping. Biochemistry 52:3818–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiker HM, Byron SA, Hendricks WPD, Elmquist WF, Gampa G, Vondrak J, Aldrich J, Cuyugan L, Adkins J, De Luca V, et al. (2018) E6201, an intravenous MEK1 inhibitor, achieves an exceptional response in BRAF V600E-mutated metastatic malignant melanoma with brain metastases. Invest New Drugs 10.1007/s10637-018-0668-8 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie TA. (2016) Targeted covalent inhibitors for drug design. Angew Chem Int Ed Engl 55:13408–13421. [DOI] [PubMed] [Google Scholar]
- Baker KJ, Bradley SE. (1966) Binding of sulfobromophthalein (BSP) sodium by plasma albumin. Its role in hepatic BSP extraction. J Clin Invest 45:281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A, Goetsch L, Dumontet C, Corvaïa N. (2017) Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov 16:315–337. [DOI] [PubMed] [Google Scholar]
- Benet LZ, Hoener BA. (2002) Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther 71:115–121. [DOI] [PubMed] [Google Scholar]
- Berezhkovskiy LM. (2011) The corrected traditional equations for calculation of hepatic clearance that account for the difference in drug ionization in extracellular and intracellular tissue water and the corresponding corrected PBPK equation. J Pharm Sci 100:1167–1183. [DOI] [PubMed] [Google Scholar]
- Bowman CM, Benet LZ. (2018) An examination of protein binding and protein-facilitated uptake relating to in vitro-in vivo extrapolation. Eur J Pharm Sci 123:502–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CM, Okochi H, Benet LZ. (2019) The presence of a transporter-induced protein binding shift: a new explanation for protein-facilitated uptake and improvement for in vitro-in vivo extrapolation. Drug Metab Dispos 47:358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges KR, Schmidt GJ, Jensen M, Cerami A, Bunn HF. (1975) The acetylation of hemoglobin by aspirin. In vitro and in vivo. J Clin Invest 56:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch JW, Blazer-Yost B. (1981) Acetylation of albumin by low doses of aspirin. Thromb Res 23:447–452. [DOI] [PubMed] [Google Scholar]
- Burslem GM, Crews CM. (2017) Small-molecule modulation of protein homeostasis. Chem Rev 117:11269–11301. [DOI] [PubMed] [Google Scholar]
- Byron SA, Loch DC, Wellens CL, Wortmann A, Wu J, Wang J, Nomoto K, Pollock PM. (2012) Sensitivity to the MEK inhibitor E6201 in melanoma cells is associated with mutant BRAF and wildtype PTEN status. Mol Cancer 11:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprioli RM. (2016) Imaging mass spectrometry: molecular microscopy for the new age of biology and medicine. Proteomics 16:1607–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari RV, Miller ML, Widdison WC. (2014) Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl 53:3796–3827. [DOI] [PubMed] [Google Scholar]
- Chasseaud LF, Cooper AJ, Saggers VH. (1974) Plasma concentrations and bioavailability of clofibrate after administration to human subjects. J Clin Pharmacol 14:382–386. [DOI] [PubMed] [Google Scholar]
- Chee EL, Lim AY, Modamio P, Fernandez-Lastra C, Segarra I. (2016) Sunitinib tissue distribution changes after coadministration with ketoconazole in mice. Eur J Drug Metab Pharmacokinet 41:309–319. [DOI] [PubMed] [Google Scholar]
- Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. (2009) P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther 330:956–963. [DOI] [PubMed] [Google Scholar]
- Chien HC, Zur AA, Maurer TS, Yee SW, Tolsma J, Jasper P, Scott DO, Giacomini KM. (2016) Rapid method to determine intracellular drug concentrations in cellular uptake assays: application to metformin in organic cation transporter 1-transfected human embryonic kidney 293 cells. Drug Metab Dispos 44:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV. (2007) Renal clearance of quantum dots. Nat Biotechnol 25:1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo EF, Ly J, Chan J, Shahidi-Latham SK, Messick K, Plise E, Quiason CM, Yang L. (2014) Role of P-glycoprotein on the brain penetration and brain pharmacodynamic activity of the MEK inhibitor cobimetinib. Mol Pharm 11:4199–4207. [DOI] [PubMed] [Google Scholar]
- Christensen EI, Birn H. (2013) Proteinuria: tubular handling of albumin-degradation or salvation? Nat Rev Nephrol 9:700–702. [DOI] [PubMed] [Google Scholar]
- Crim C, Pierre LN, Daley-Yates PT. (2001) A review of the pharmacology and pharmacokinetics of inhaled fluticasone propionate and mometasone furoate. Clin Ther 23:1339–1354. [DOI] [PubMed] [Google Scholar]
- Daley-Yates PT. (2015) Inhaled corticosteroids: potency, dose equivalence and therapeutic index. Br J Clin Pharmacol 80:372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, Fojo T, Bates S. (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5:275–284. [DOI] [PubMed] [Google Scholar]
- Debruyne D. (1997) Clinical pharmacokinetics of fluconazole in superficial and systemic mycoses. Clin Pharmacokinet 33:52–77. [DOI] [PubMed] [Google Scholar]
- de Gooijer MC, Zhang P, Thota N, Mayayo-Peralta I, Buil LC, Beijnen JH, van Tellingen O. (2015) P-glycoprotein and breast cancer resistance protein restrict the brain penetration of the CDK4/6 inhibitor palbociclib. Invest New Drugs 33:1012–1019. [DOI] [PubMed] [Google Scholar]
- Demeule M, Shedid D, Beaulieu E, Del Maestro RF, Moghrabi A, Ghosn PB, Moumdjian R, Berthelet F, Béliveau R. (2001) Expression of multidrug-resistance P-glycoprotein (MDR1) in human brain tumors. Int J Cancer 93:62–66. [DOI] [PubMed] [Google Scholar]
- Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. (2008) Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci USA 105:17356–17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L, Breen C, Chambers R, Eckley ST, Fricke R, Ghosh A, Harradine P, Kalvass JC, Ho S, Lee CA, et al. (2017) Industry perspective on contemporary protein-binding methodologies: considerations for regulatory drug-drug interaction and related guidelines on highly bound drugs. J Pharm Sci 106:3442–3452. [DOI] [PubMed] [Google Scholar]
- Di L, Umland JP, Trapa PE, Maurer TS. (2012) Impact of recovery on fraction unbound using equilibrium dialysis. J Pharm Sci 101:1327–1335. [DOI] [PubMed] [Google Scholar]
- Dickson CJ, Hornak V, Velez-Vega C, McKay DJ, Reilly J, Sandham DA, Shaw D, Fairhurst RA, Charlton SJ, Sykes DA, et al. (2016) Uncoupling the structure-activity relationships of β2 adrenergic receptor ligands from membrane binding. J Med Chem 59:5780–5789. [DOI] [PubMed] [Google Scholar]
- Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, et al. (2003) Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol 21:778–784. [DOI] [PubMed] [Google Scholar]
- Down K, Amour A, Baldwin IR, Cooper AW, Deakin AM, Felton LM, Guntrip SB, Hardy C, Harrison ZA, Jones KL, et al. (2015) Optimization of novel indazoles as highly potent and selective inhibitors of phosphoinositide 3-kinase δ for the treatment of respiratory disease. J Med Chem 58:7381–7399. [DOI] [PubMed] [Google Scholar]
- Drexler DM, Tannehill-Gregg SH, Wang L, Brock BJ. (2011) Utility of quantitative whole-body autoradiography (QWBA) and imaging mass spectrometry (IMS) by matrix-assisted laser desorption/ionization (MALDI) in the assessment of ocular distribution of drugs. J Pharmacol Toxicol Methods 63:205–208. [DOI] [PubMed] [Google Scholar]
- Drozdzik M, Gröer C, Penski J, Lapczuk J, Ostrowski M, Lai Y, Prasad B, Unadkat JD, Siegmund W, Oswald S. (2014) Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine. Mol Pharm 11:3547–3555. [DOI] [PubMed] [Google Scholar]
- Durmus S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. (2012) Oral availability and brain penetration of the B-RAFV600E inhibitor vemurafenib can be enhanced by the P-GLYCOprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Mol Pharm 9:3236–3245. [DOI] [PubMed] [Google Scholar]
- Eriksson J, Sjögren E, Thörn H, Rubin K, Bäckman P, Lennernäs H. (2018) Pulmonary absorption–estimation of effective pulmonary permeability and tissue retention of ten drugs using an ex vivo rat model and computational analysis. Eur J Pharm Biopharm 124:1–12. [DOI] [PubMed] [Google Scholar]
- Esmailpour N, Högger P, Rabe KF, Heitmann U, Nakashima M, Rohdewald P. (1997) Distribution of inhaled fluticasone propionate between human lung tissue and serum in vivo. Eur Respir J 10:1496–1499. [DOI] [PubMed] [Google Scholar]
- Eyal S, Chung FS, Muzi M, Link JM, Mankoff DA, Kaddoumi A, O’Sullivan F, Hebert MF, Unadkat JD. (2009) Simultaneous PET imaging of P-glycoprotein inhibition in multiple tissues in the pregnant nonhuman primate. J Nucl Med 50:798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattori S, Becherini F, Cianfriglia M, Parenti G, Romanini A, Castagna M. (2007) Human brain tumors: multidrug-resistance P-glycoprotein expression in tumor cells and intratumoral capillary endothelial cells. Virchows Arch 451:81–87. [DOI] [PubMed] [Google Scholar]
- Feng B, Doran AC, Di L, West MA, Osgood SM, Mancuso JY, Shaffer CL, Tremaine L, Liras J. (2018) Prediction of human brain penetration of P-glycoprotein and breast cancer resistance protein substrates using in vitro transporter studies and animal models. J Pharm Sci 107:2225–2235. [DOI] [PubMed] [Google Scholar]
- Fukuchi Y, Toshimoto K, Mori T, Kakimoto K, Tobe Y, Sawada T, Asaumi R, Iwata T, Hashimoto Y, Nunoya KI, et al. (2017) Analysis of nonlinear pharmacokinetics of a highly albumin-bound compound: contribution of albumin-mediated hepatic uptake mechanism. J Pharm Sci 106:2704–2714. [DOI] [PubMed] [Google Scholar]
- Gampa G, Kim M, Cook-Rostie N, Laramy JK, Sarkaria JN, Paradiso L, DePalatis L, Elmquist WF. (2018) Brain distribution of a novel MEK inhibitor E6201: implications in the treatment of melanoma brain metastases. Drug Metab Dispos 46:658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gampa G, Vaidhyanathan S, Sarkaria JN, Elmquist WF. (2017) Drug delivery to melanoma brain metastases: can current challenges lead to new opportunities? Pharmacol Res 123:10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerk PM, Li W, Vore M. (2004) Estradiol 3-glucuronide is transported by the multidrug resistance-associated protein 2 but does not activate the allosteric site bound by estradiol 17-glucuronide. Drug Metab Dispos 32:1139–1145. [DOI] [PubMed] [Google Scholar]
- Guo Y, Chu X, Parrott NJ, Brouwer KLR, Hsu V, Nagar S, Matsson P, Sharma P, Snoeys J, Sugiyama Y, et al. (2018) Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and physiologically based pharmacokinetic modeling approaches. Clin Pharmacol Ther 104:865–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RK. (2016) Phase II and phase III failures: 2013-2015. Nat Rev Drug Discov 15:817–818. [DOI] [PubMed] [Google Scholar]
- He J, Yu Y, Prasad B, Link J, Miyaoka RS, Chen X, Unadkat JD. (2014) PET imaging of Oatp-mediated hepatobiliary transport of [(11)C] rosuvastatin in the rat. Mol Pharm 11:2745–2754. [DOI] [PubMed] [Google Scholar]
- Hemmerling M, Nilsson S, Edman K, Eirefelt S, Russell W, Hendrickx R, Johnsson E, Kärrman Mårdh C, Berger M, Rehwinkel H, et al. (2017) Selective nonsteroidal glucocorticoid receptor modulators for the inhaled treatment of pulmonary diseases. J Med Chem 60:8591–8605. [DOI] [PubMed] [Google Scholar]
- Hochhaus G, Horhota S, Hendeles L, Suarez S, Rebello J. (2015) Pharmacokinetics of orally inhaled drug products. AAPS J 17:769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao P, Unadkat JD. (2014) Predicting the outer boundaries of P-glycoprotein (P-gp)-based drug interactions at the human blood-brain barrier based on rat studies. Mol Pharm 11:436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida K, Ullah M, Tóth B, Juhasz V, Unadkat JD. (2018) Successful prediction of in vivo hepatobiliary clearances and hepatic concentrations of rosuvastatin using sandwich-cultured rat hepatocytes, transporter-expressing cell lines, and quantitative proteomics. Drug Metab Dispos 46:66–74. [DOI] [PubMed] [Google Scholar]
- Ito T, Takahashi M, Sudo K, Sugiyama Y. (2007) Marked strain differences in the pharmacokinetics of an alpha4beta1 integrin antagonist, 4-[1-[3-Chloro-4-[N-(2-methylphenyl)-ureido]phenylacetyl]-(4S)-fluoro-(2S)-pyrrolidine-2-yl]-methoxybenzoic Acid (D01-4582), in Sprague-Dawley rats are associated with albumin genetic polymorphism. J Pharmacol Exp Ther 320:124–132. [DOI] [PubMed] [Google Scholar]
- Jensen JB, Sundelin EI, Jakobsen S, Gormsen LC, Munk OL, Frøkiær J, Jessen N. (2016) [11C]-Labeled metformin distribution in the liver and small intestine using dynamic positron emission tomography in mice demonstrates tissue-specific transporter dependency. Diabetes 65:1724–1730. [DOI] [PubMed] [Google Scholar]
- Jones P, Storer RI, Sabnis YA, Wakenhut FM, Whitlock GA, England KS, Mukaiyama T, Dehnhardt CM, Coe JW, Kortum SW, et al. (2017) Design and synthesis of a pan-janus kinase inhibitor clinical candidate (PF-06263276) suitable for inhaled and topical delivery for the treatment of inflammatory diseases of the lungs and skin. J Med Chem 60:767–786. [DOI] [PubMed] [Google Scholar]
- Kaneko K, Tanaka M, Ishii A, Katayama Y, Nakaoka T, Irie S, Kawahata H, Yamanaga T, Wada Y, Miyake T, et al. (2018) A clinical quantitative evaluation of hepatobiliary transport of [11C]dehydropravastatin in humans using positron emission tomography. Drug Metab Dispos 46:719–728. [DOI] [PubMed] [Google Scholar]
- Källback P, Shariatgorji M, Nilsson A, Andrén PE. (2012) Novel mass spectrometry imaging software assisting labeled normalization and quantitation of drugs and neuropeptides directly in tissue sections. J Proteomics 75:4941–4951. [DOI] [PubMed] [Google Scholar]
- Kaplan A, Price D. (2018) Matching inhaler devices with patients: the role of the primary care physician. Can Respir J 2018:9473051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Laramy JK, Mohammad AS, Talele S, Fisher J, Sarkaria JN, Elmquist WF. (2019a) Brain distribution of a panel of epidermal growth factor receptor inhibitors using cassette dosing in wild-type and Abcb1/Abcg2-deficient mice. Drug Metab Dispos 47:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Lee KR, Miyauchi S, Sugiyama Y. (2019b) Extrapolation of in vivo hepatic clearance from in vitro uptake clearance by suspended human hepatocytes for anionic drugs with high binding to human albumin: improvement of in vitro-to-in vivo extrapolation by considering the “albumin-mediated” hepatic uptake mechanism on the basis of the “facilitated-dissociation model”. Drug Metab Dispos 47:94–103. [DOI] [PubMed] [Google Scholar]
- Kindla J, Müller F, Mieth M, Fromm MF, König J. (2011) Influence of non-steroidal anti-inflammatory drugs on organic anion transporting polypeptide (OATP) 1B1- and OATP1B3-mediated drug transport. Drug Metab Dispos 39:1047–1053. [DOI] [PubMed] [Google Scholar]
- Kivistö KT, Niemi M, Schaeffeler E, Pitkälä K, Tilvis R, Fromm MF, Schwab M, Eichelbaum M, Strandberg T. (2004) Lipid-lowering response to statins is affected by CYP3A5 polymorphism. Pharmacogenetics 14:523–525. [DOI] [PubMed] [Google Scholar]
- Landersdorfer CB, He Y-L, Jusko WJ. (2012) Mechanism-based population pharmacokinetic modelling in diabetes: vildagliptin as a tight binding inhibitor and substrate of dipeptidyl peptidase IV. Br J Clin Pharmacol 73:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GL, Winter H, Arends R, Jay GW, Le V, Young T, Huggins JP. (2012) Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br J Clin Pharmacol 73:706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Barton HA, Yates PD, Ghosh A, Wolford AC, Riccardi KA, Maurer TS. (2014) A “middle-out” approach to human pharmacokinetic predictions for OATP substrates using physiologically-based pharmacokinetic modeling. J Pharmacokinet Pharmacodyn 41:197–209. [DOI] [PubMed] [Google Scholar]
- Li R, Kimoto E, Niosi M, Tess DA, Lin J, Tremaine LM, Di L. (2018a) A study on pharmacokinetics of bosentan with systems modeling, part 2: prospectively predicting systemic and liver exposure in healthy subjects [published correction appears in Drug Metab Dispos (2018) 46:484]. Drug Metab Dispos 46:357–366 DOI: 10.1124/dmd.117.078808. [DOI] [PubMed] [Google Scholar]
- Li R, Niosi M, Johnson N, Tess DA, Kimoto E, Lin J, Yang X, Riccardi KA, Ryu S, El-Kattan AF, et al. (2018b) A study on pharmacokinetics of bosentan with systems modeling, part 1: translating systemic plasma concentration to liver exposure in healthy subjects [published correction appears in Drug Metab Dispos (2018) 46:483; Drug Metab Dispos (2019) 47:269]. Drug Metab Dispos 46:346–356 DOI: 10.1124/dmd.117.078790. [DOI] [PubMed] [Google Scholar]
- Li Z, Di L, Maurer TS. (2019) Theoretical considerations for direct translation of unbound liver-to-plasma partition coefficient from in vitro to in vivo. AAPS J 21:43. [DOI] [PubMed] [Google Scholar]
- Liang X, Giacomini KM. (2017) Transporters involved in metformin pharmacokinetics and treatment response. J Pharm Sci 106:2245–2250. [DOI] [PubMed] [Google Scholar]
- Liu X, Wright M, Hop CE. (2014) Rational use of plasma protein and tissue binding data in drug design. J Med Chem 57:8238–8248. [DOI] [PubMed] [Google Scholar]
- Liyasova MS, Schopfer LM, Lockridge O. (2010) Reaction of human albumin with aspirin in vitro: mass spectrometric identification of acetylated lysines 199, 402, 519, and 545. Biochem Pharmacol 79:784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. (2011) Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res 17:6437–6447. [DOI] [PubMed] [Google Scholar]
- Luo Y, Ellis LZ, Dallaglio K, Takeda M, Robinson WA, Robinson SE, Liu W, Lewis KD, McCarter MD, Gonzalez R, et al. (2012) Side population cells from human melanoma tumors reveal diverse mechanisms for chemoresistance. J Invest Dermatol 132:2440–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Khojasteh SC, Hop CE, Erickson HK, Polson A, Pillow TH, Yu SF, Wang H, Dragovich PS, Zhang D. (2016) Antibody drug conjugates differentiate uptake and DNA alkylation of pyrrolobenzodiazepines in tumors from organs of xenograft mice. Drug Metab Dispos 44:1958–1962. [DOI] [PubMed] [Google Scholar]
- Maeda K, Ikeda Y, Fujita T, Yoshida K, Azuma Y, Haruyama Y, Yamane N, Kumagai Y, Sugiyama Y. (2011) Identification of the rate-determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin Pharmacol Ther 90:575–581. [DOI] [PubMed] [Google Scholar]
- Mao J, Doshi U, Wright M, Hop CECA, Li AP, Chen Y. (2018) Prediction of the pharmacokinetics of pravastatin as an OATP substrate using plateable human hepatocytes with human plasma data and PBPK modeling. CPT Pharmacometrics Syst Pharmacol 7:251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menéndez-Arias L, Álvarez M, Pacheco B. (2014) Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: mechanism of action and resistance. Curr Opin Virol 8:1–9. [DOI] [PubMed] [Google Scholar]
- Millan DS, Bunnage ME, Burrows JL, Butcher KJ, Dodd PG, Evans TJ, Fairman DA, Hughes SJ, Kilty IC, Lemaitre A, et al. (2011) Design and synthesis of inhaled p38 inhibitors for the treatment of chronic obstructive pulmonary disease. J Med Chem 54:7797–7814. [DOI] [PubMed] [Google Scholar]
- Miller DB, Spence JD. (1998) Clinical pharmacokinetics of fibric acid derivatives (fibrates). Clin Pharmacokinet 34:155–162. [DOI] [PubMed] [Google Scholar]
- Mittapalli RK, Vaidhyanathan S, Dudek AZ, Elmquist WF. (2013) Mechanisms limiting distribution of the threonine-protein kinase B-RaF(V600E) inhibitor dabrafenib to the brain: implications for the treatment of melanoma brain metastases. J Pharmacol Exp Ther 344:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittapalli RK, Vaidhyanathan S, Sane R, Elmquist WF. (2012) Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on the brain distribution of a novel BRAF inhibitor: vemurafenib (PLX4032). J Pharmacol Exp Ther 342:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi S, Masuda M, Kim SJ, Tanaka Y, Lee KR, Iwakado S, Nemoto M, Sasaki S, Shimono K, Tanaka Y, et al. (2018) The phenomenon of albumin-mediated hepatic uptake of organic anion transport polypeptide substrates: prediction of the in vivo uptake clearance from the in vitro uptake by isolated hepatocytes using a facilitated-dissociation model. Drug Metab Dispos 46:259–267. [DOI] [PubMed] [Google Scholar]
- Mundy C, Kirkpatrick P. (2004) Tiotropium bromide. Nat Rev Drug Discov 3:643–644. [DOI] [PubMed] [Google Scholar]
- Narita Y, Okamoto K, Kawada MI, Takase K, Minoshima Y, Kodama K, Iwata M, Miyamoto N, Sawada K. (2014) Novel ATP-competitive MEK inhibitor E6201 is effective against vemurafenib-resistant melanoma harboring the MEK1-C121S mutation in a preclinical model. Mol Cancer Ther 13:823–832. [DOI] [PubMed] [Google Scholar]
- Oberg B. (2006) Rational design of polymerase inhibitors as antiviral drugs. Antiviral Res 71:90–95. [DOI] [PubMed] [Google Scholar]
- Park JW, Hong K, Kirpotin DB, Colbern G, Shalaby R, Baselga J, Shao Y, Nielsen UB, Marks JD, Moore D, et al. (2002) Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin Cancer Res 8:1172–1181. [PubMed] [Google Scholar]
- Parrish KE, Pokorny J, Mittapalli RK, Bakken K, Sarkaria JN, Elmquist WF. (2015) Efflux transporters at the blood-brain barrier limit delivery and efficacy of cyclin-dependent kinase 4/6 inhibitor palbociclib (PD-0332991) in an orthotopic brain tumor model. J Pharmacol Exp Ther 355:264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patilea-Vrana G, Unadkat JD. (2016) Transport vs. metabolism: what determines the pharmacokinetics and pharmacodynamics of drugs? Insights from the extended clearance model. Clin Pharmacol Ther 100:413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, Otto V, Castellino S, Demby VE. (2009) An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab Dispos 37:439–442. [DOI] [PubMed] [Google Scholar]
- Poulin P, Burczynski FJ, Haddad S. (2016) The role of extracellular binding proteins in the cellular uptake of drugs: impact on quantitative in vitro-to-in vivo extrapolations of toxicity and efficacy in physiologically based pharmacokinetic-pharmacodynamic research. J Pharm Sci 105:497–508. [DOI] [PubMed] [Google Scholar]
- Poulin P, Kenny JR, Hop CE, Haddad S. (2012) In vitro-in vivo extrapolation of clearance: modeling hepatic metabolic clearance of highly bound drugs and comparative assessment with existing calculation methods. J Pharm Sci 101:838–851. [DOI] [PubMed] [Google Scholar]
- Prasad B, Johnson K, Billington S, Lee C, Chung GW, Brown CD, Kelly EJ, Himmelfarb J, Unadkat JD. (2016) Abundance of drug transporters in the human kidney cortex as quantified by quantitative targeted proteomics. Drug Metab Dispos 44:1920–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B, Unadkat JD. (2014) Optimized approaches for quantification of drug transporters in tissues and cells by MRM proteomics. AAPS J 16:634–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DN, Kunda NK, Muttil P. (2019) Challenges associated with the pulmonary delivery of therapeutic dry powders for preclinical testing. Kona Powder Particle J 36:129–144. [Google Scholar]
- Rankovic Z. (2015) CNS drug design: balancing physicochemical properties for optimal brain exposure. J Med Chem 58:2584–2608. [DOI] [PubMed] [Google Scholar]
- Raub TJ, Wishart GN, Kulanthaivel P, Staton BA, Ajamie RT, Sawada GA, Gelbert LM, Shannon HE, Sanchez-Martinez C, De Dios A. (2015) Brain exposure of two selective dual CDK4 and CDK6 inhibitors and the antitumor activity of CDK4 and CDK6 inhibition in combination with temozolomide in an intracranial glioblastoma xenograft. Drug Metab Dispos 43:1360–1371. [DOI] [PubMed] [Google Scholar]
- Rautio J, Meanwell NA, Di L, Hageman MJ. (2018) The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Discov 17:559–587. [DOI] [PubMed] [Google Scholar]
- Ray AS, Fordyce MW, Hitchcock MJ. (2016) Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of Human Immunodeficiency Virus. Antiviral Res 125:63–70. [DOI] [PubMed] [Google Scholar]
- Ricart AD. (2011) Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin Cancer Res 17:6417–6427. [DOI] [PubMed] [Google Scholar]
- Riccardi K, Li Z, Brown JA, Gorgoglione MF, Niosi M, Gosset J, Huard K, Erion DM, Di L. (2016) Determination of unbound partition coefficient and in vitro-in vivo extrapolation for SLC13A transporter-mediated uptake. Drug Metab Dispos 44:1633–1642. [DOI] [PubMed] [Google Scholar]
- Riccardi K, Lin J, Li Z, Niosi M, Ryu S, Hua W, Atkinson K, Kosa RE, Litchfield J, Di L. (2017) Novel method to predict in vivo liver-to-plasma Kpuu for OATP substrates using suspension hepatocytes. Drug Metab Dispos 45:576–580. [DOI] [PubMed] [Google Scholar]
- Riccardi KA, Tess DA, Lin J, Patel R, Ryu S, Atkinson K, Di L, Li R. (2019) A novel unified approach to predict human hepatic clearance for both enzyme- and transporter-mediated mechanisms using suspended human hepatocytes. Drug Metab Dispos 47:484–492. [DOI] [PubMed] [Google Scholar]
- Rivero-Juarez A, Brieva T, Frias M, Rivero A. (2018) Pharmacodynamic and pharmacokinetic evaluation of the combination of daclatasvir/sofosbuvir/ribavirin in the treatment of chronic hepatitis C. Expert Opin Drug Metab Toxicol 14:901–910. [DOI] [PubMed] [Google Scholar]
- Rose RH, Neuhoff S, Abduljalil K, Chetty M, Rostami-Hodjegan A, Jamei M. (2014) Application of a physiologically based pharmacokinetic model to predict OATP1B1-related variability in pharmacodynamics of rosuvastatin. CPT Pharmacometrics Syst Pharmacol 3:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasongko L, Link JM, Muzi M, Mankoff DA, Yang X, Collier AC, Shoner SC, Unadkat JD. (2005) Imaging P-glycoprotein transport activity at the human blood-brain barrier with positron emission tomography. Clin Pharmacol Ther 77:503–514. [DOI] [PubMed] [Google Scholar]
- Scheers E, Leclercq L, de Jong J, Bode N, Bockx M, Laenen A, Cuyckens F, Skee D, Murphy J, Sukbuntherng J, et al. (2015) Absorption, metabolism, and excretion of oral 14C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos 43:289–297. [DOI] [PubMed] [Google Scholar]
- Schinkel AH, Wagenaar E, Mol CA, van Deemter L. (1996) P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest 97:2517–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwamborn K, Caprioli RM. (2010) Molecular imaging by mass spectrometry--looking beyond classical histology. Nat Rev Cancer 10:639–646. [DOI] [PubMed] [Google Scholar]
- Schulz S, Becker M, Groseclose MR, Schadt S, Hopf C. (2019) Advanced MALDI mass spectrometry imaging in pharmaceutical research and drug development. Curr Opin Biotechnol 55:51–59. [DOI] [PubMed] [Google Scholar]
- Seneviratne HK, Hendrix CW, Fuchs EJ, Bumpus NN. (2018) MALDI mass spectrometry imaging reveals heterogeneous distribution of tenofovir and tenofovir diphosphate in colorectal tissue of subjects receiving a tenofovir-containing enema. J Pharmacol Exp Ther 367:40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw DE, Baig F, Bruce I, Chamoin S, Collingwood SP, Cross S, Dayal S, Drückes P, Furet P, Furminger V, et al. (2016) Optimization of platelet-derived growth factor receptor (PDGFR) inhibitors for duration of action, as an inhaled therapy for lung remodeling in pulmonary arterial hypertension. J Med Chem 59:7901–7914. [DOI] [PubMed] [Google Scholar]
- Shi D, Yang J, Yang D, LeCluyse EL, Black C, You L, Akhlaghi F, Yan B. (2006) Anti-influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J Pharmacol Exp Ther 319:1477–1484. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Horie T, Sugiyama Y. (2006) Transporters as a determinant of drug clearance and tissue distribution. Eur J Pharm Sci 27:425–446. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Maeda K, Ikejiri K, Yoshida K, Horie T, Sugiyama Y. (2013) Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm Drug Dispos 34:45–78. [DOI] [PubMed] [Google Scholar]
- Shor B, Gerber HP, Sapra P. (2015) Preclinical and clinical development of inotuzumab-ozogamicin in hematological malignancies. Mol Immunol 67 (2 Pt A):107–116. [DOI] [PubMed] [Google Scholar]
- Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG, et al. (2007) Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest 117:1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwkowski MX, Mellman I. (2013) Antibody therapeutics in cancer. Science 341:1192–1198. [DOI] [PubMed] [Google Scholar]
- Smietana K, Siatkowski M, Møller M. (2016) Trends in clinical success rates. Nat Rev Drug Discov 15:379–380. [DOI] [PubMed] [Google Scholar]
- Smith DA, Di L, Kerns EH. (2010) The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov 9:929–939. [DOI] [PubMed] [Google Scholar]
- Solon EG, Schweitzer A, Stoeckli M, Prideaux B. (2010) Autoradiography, MALDI-MS, and SIMS-MS imaging in pharmaceutical discovery and development. AAPS J 12:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo GW, Law JH, Kan E, Tan SY, Lim WY, Chay G, Bukhari NI, Segarra I. (2010) Differential effects of ketoconazole and primaquine on the pharmacokinetics and tissue distribution of imatinib in mice. Anticancer Drugs 21:695–703. [PubMed] [Google Scholar]
- Stein SW, Thiel CG. (2017) The history of therapeutic aerosols: a chronological review. J Aerosol Med Pulm Drug Deliv 30:20–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella VJ. (2010) Prodrugs: some thoughts and current issues. J Pharm Sci 99:4755–4765. [DOI] [PubMed] [Google Scholar]
- Stocker SL, Morrissey KM, Yee SW, Castro RA, Xu L, Dahlin A, Ramirez AH, Roden DM, Wilke RA, McCarty CA, et al. (2013) The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther 93:186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf WE. (2005) Drug localization and targeting with receptor microscopic autoradiography. J Pharmacol Toxicol Methods 51:25–40. [DOI] [PubMed] [Google Scholar]
- Sun L, Yang H, Li J, Wang T, Li W, Liu G, Tang Y. (2018) In silico prediction of compounds binding to human plasma proteins by QSAR models. ChemMedChem 13:572–581. [DOI] [PubMed] [Google Scholar]
- Sundelin E, Gormsen LC, Jensen JB, Vendelbo MH, Jakobsen S, Munk OL, Christensen M, Brøsen K, Frøkiaer J, Jessen N. (2017) Genetic polymorphisms in organic cation transporter 1 attenuates hepatic metformin exposure in humans. Clin Pharmacol Ther 102:841–848. [DOI] [PubMed] [Google Scholar]
- Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234. [DOI] [PubMed] [Google Scholar]
- Taburet AM, Schmit B. (1994) Pharmacokinetic optimisation of asthma treatment. Clin Pharmacokinet 26:396–418. [DOI] [PubMed] [Google Scholar]
- Testa B. (2009) Prodrugs: bridging pharmacodynamic/pharmacokinetic gaps. Curr Opin Chem Biol 13:338–344. [DOI] [PubMed] [Google Scholar]
- Tisdall MM, Smith M. (2006) Cerebral microdialysis: research technique or clinical tool. Br J Anaesth 97:18–25. [DOI] [PubMed] [Google Scholar]
- Tsamandouras N, Dickinson G, Guo Y, Hall S, Rostami-Hodjegan A, Galetin A, Aarons L. (2015) Development and application of a mechanistic pharmacokinetic model for simvastatin and its active metabolite simvastatin acid using an integrated population PBPK approach. Pharm Res 32:1864–1883. [DOI] [PubMed] [Google Scholar]
- Tsao SC, Sugiyama Y, Shinmura K, Sawada Y, Nagase S, Iga T, Hanano M. (1988) Protein-mediated hepatic uptake of rose bengal in analbuminemic mutant rats (NAR). Albumin is not indispensable to the protein-mediated transport of rose bengal. Drug Metab Dispos 16:482–489. [PubMed] [Google Scholar]
- Turner PV, Pekow C, Vasbinder MA, Brabb T. (2011) Administration of substances to laboratory animals: equipment considerations, vehicle selection, and solute preparation. J Am Assoc Lab Anim Sci 50:614–627. [PMC free article] [PubMed] [Google Scholar]
- Ufuk A, Assmus F, Francis L, Plumb J, Damian V, Gertz M, Houston JB, Galetin A. (2017) In vitro and in silico tools to assess extent of cellular uptake and lysosomal sequestration of respiratory drugs in human alveolar macrophages. Mol Pharm 14:1033–1046. [DOI] [PubMed] [Google Scholar]
- Valeur E, Guéret SM, Adihou H, Gopalakrishnan R, Lemurell M, Waldmann H, Grossmann TN, Plowright AT. (2017) New modalities for challenging targets in drug discovery. Angew Chem Int Ed Engl 56:10294–10323. [DOI] [PubMed] [Google Scholar]
- Wang J, Li-Chan XX, Atherton J, Deng L, Espina R, Yu L, Horwatt P, Ross S, Lockhead S, Ahmad S, et al. (2010) Characterization of HKI-272 covalent binding to human serum albumin. Drug Metab Dispos 38:1083–1093. [DOI] [PubMed] [Google Scholar]
- Wang L, He K, Maxwell B, Grossman SJ, Tremaine LM, Humphreys WG, Zhang D. (2011) Tissue distribution and elimination of [14C]apixaban in rats. Drug Metab Dispos 39:256–264. [DOI] [PubMed] [Google Scholar]
- Wang L, Prasad B, Salphati L, Chu X, Gupta A, Hop CE, Evers R, Unadkat JD. (2015) Interspecies variability in expression of hepatobiliary transporters across human, dog, monkey, and rat as determined by quantitative proteomics. Drug Metab Dispos 43:367–374. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y. (2009) Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther 328:652–662. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kusuhara H, Sugiyama Y. (2010) Application of physiologically based pharmacokinetic modeling and clearance concept to drugs showing transporter-mediated distribution and clearance in humans. J Pharmacokinet Pharmacodyn 37:575–590. [DOI] [PubMed] [Google Scholar]
- Wattanagoon Y, Stepniewska K, Lindegårdh N, Pukrittayakamee S, Silachamroon U, Piyaphanee W, Singtoroj T, Hanpithakpong W, Davies G, Tarning J, et al. (2009) Pharmacokinetics of high-dose oseltamivir in healthy volunteers. Antimicrob Agents Chemother 53:945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Pinto DJ, Zhang D. (2011) Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis 31:478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki M, Suzuki H, Sugiyama Y. (1996) Recent advances in carrier-mediated hepatic uptake and biliary excretion of xenobiotics. Pharm Res 13:497–513. [DOI] [PubMed] [Google Scholar]
- Zhang D, Frost CE, He K, Rodrigues AD, Wang X, Wang L, Goosen TC, Humphreys WG. (2013a) Investigating the enteroenteric recirculation of apixaban, a factor Xa inhibitor: administration of activated charcoal to bile duct-cannulated rats and dogs receiving an intravenous dose and use of drug transporter knockout rats. Drug Metab Dispos 41:906–915. [DOI] [PubMed] [Google Scholar]
- Zhang D, He K, Herbst JJ, Kolb J, Shou W, Wang L, Balimane PV, Han YH, Gan J, Frost CE, et al. (2013b) Characterization of efflux transporters involved in distribution and disposition of apixaban. Drug Metab Dispos 41:827–835. [DOI] [PubMed] [Google Scholar]
- Zhang D, Yu SF, Khojasteh SC, Ma Y, Pillow TH, Sadowsky JD, Su D, Kozak KR, Xu K, Polson AG, et al. (2018) Intratumoral payload concentration correlates with the activity of antibody-drug conjugates. Mol Cancer Ther 17:677–685. [DOI] [PubMed] [Google Scholar]
- Zhang D, Yu SF, Ma Y, Xu K, Dragovich PS, Pillow TH, Liu L, Del Rosario G, He J, Pei Z, et al. (2016) Chemical structure and concentration of intratumor catabolites determine efficacy of antibody drug conjugates. Drug Metab Dispos 44:1517–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]