

Abstract
Efavirenz (more specifically the S-enantiomer) is a cornerstone antiretroviral therapy for treatment of HIV infection. The major primary metabolite is S-8-hydroxyefavirenz, which does not have antiretroviral activity but is neurotoxic. Cytochrome P450 2B6 (CYP2B6) is the major enzyme catalyzing S-8-hydroxyefavirenz formation. CYP2B6 genetics and drug interactions are major determinants of clinical efavirenz disposition and dose adjustment. In addition, as a prototypic CYP2B6 substrate, S-efavirenz and analogs can inform on the structure, activity, catalytic mechanisms, and stereoselectivity of CYP2B6. Metabolism of R-efavirenz by CYP2B6 remains unexplored. This investigation assessed S-efavirenz metabolism by clinically relevant CYP2B6 genetic variants. This investigation also evaluated R-efavirenz hydroxylation by wild-type CYP2B6.1 and CYP2B6 variants. S-Efavirenz 8-hydroxylation by wild-type CYP2B6.1 and variants exhibited positive cooperativity and apparent cooperative substrate inhibition. On the basis of Clmax values, relative activities for S-efavirenz 8-hydroxylation were in the order CYP2B6.4 > CYP2B6.1 ≈ CYP2B6.5 ≈ CYP2B6.17 > CYP2B6.6 ≈ CYP2B6.7 ≈ CYP2B6.9 ≈ CYP2B6.19 ≈ CYP2B6.26; CYP2B6.16 and CYP2B6.18 showed minimal activity. Rates of R-efavirenz metabolism were approximately 1/10 those of S-efavirenz for wild-type CYP2B6.1 and variants. On the basis of Clmax values, there was 14-fold enantioselectivity (S > R-efavirenz) for wild-type CYP2B6.1, and 5- to 22-fold differences for other CYP2B6 variants. These results show that both CYP2B6 516G > T (CYP2B6*6 and CYP2B6*9) and 983T > C (CYP2B6*16 and CYP2B6*18) polymorphisms cause canonical diminishment or loss-of-function variants for S-efavirenz 8-hydroxylation, provide a mechanistic basis for known clinical pharmacogenetic differences in efavirenz disposition, and may predict additional clinically important variant alleles. Efavirenz is the most stereoselective CYP2B6 drug substrate yet identified and may be a useful probe for the CYP2B6 active site and catalytic mechanisms.
SIGNIFICANCE STATEMENT
Clinical disposition of the antiretroviral S-efavirenz is affected by CYP2B6 polymorphisms. Expressed CYP2B6 with 516G>T (CYP2B6*6 and CYP2B6*9), and 983T>C (CYP2B6*16 and CYP2B6*18) polymorphisms had a diminishment or loss of function for efavirenz 8-hydroxylation. This provides a mechanistic basis for efavirenz clinical pharmacogenetics and may predict additional clinically important variant alleles. Efavirenz metabolism showed both cooperativity and cooperative substrate inhibition. With greater than 10-fold enantioselectivity (S- vs. R- metabolism), efavirenz is the most stereoselective CYP2B6 drug substrate yet identified. These findings may provide mechanistic insights.
Introduction
Efavirenz [(S)-6-chloro-4-(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one] is a non-nucleoside reverse transcriptase inhibitor used as first-line therapy for human immunodeficiency virus (HIV) infection (Rakhmanina and van den Anker, 2010). One essential step in HIV replication is viral single-strand RNA conversion into double-strand DNA, catalyzed by viral reverse transcriptase, followed by viral DNA integration into the host genome. HIV reverse transcriptase has a catalytic p66 (66-kDa) subunit and a smaller p51 (55-kDa) subunit that functions mainly for structural support. The p66 subunit is further divided into N-terminal polymerase domain, which catalyzes complementary DNA polymerization from template RNA, and C-terminal RNase H domain, which digests viral RNA and removes RNA primers during DNA synthesis. Efavirenz binds to a hydrophobic pocket in the p66 polymerase domain about 10 Å from the active site and inhibits activity via an allosteric mechanism (Schauer et al., 2014). Shortly after synthesis of RS-6-chloro-4-(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one, it was identified that reverse transcriptase inhibition was highly stereospecific, as the R-enantiomer (R)-6-chloro-4-(cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one (henceforth referred to as R-efavirenz) was inactive (Young et al., 1995), and all further drug development proceeded with the single S-enantiomer (henceforth referred to as S-efavirenz).
S-Efavirenz is extensively metabolized by cytochrome P450 enzymes (Scheme 1). The major primary metabolite is S-8-hydroxyefavirenz, both in vitro and in vivo, and a minor primary metabolite is S-7-hydroxyefavirenz (Ward et al., 2003; Desta et al., 2007). Secondary metabolites include 8,14-dihydroxyefavirenz and 7,8-dihydroxyefavirenz (Ogburn et al., 2010; Avery et al., 2013). These metabolites are devoid of significant pharmacologic activity toward HIV-1 (Avery et al., 2013). Nevertheless, they are not inert, as S-8-hydroxyefavirenz has been associated with clinical neurotoxicity (Decloedt et al., 2015) and was at least an order of magnitude more neurotoxic than S-efavirenz or S-7-hydroxyefavirenz in vitro (Tovar-y-Romo et al., 2012). Cytochrome P450 2B6 (CYP2B6) is the major enzyme catalyzing S-8-hydroxyefavirenz and thence 8,14-dihydroxyefavirenz formation, whereas CYP2A6 is responsible for 7-hydroxylation (Ward et al., 2003; Damle et al., 2008). CYP2B6 is a major determinant of clinical efavirenz metabolism and elimination; drug interactions resulting from CYP2B6 inhibition increase efavirenz exposure (Damle et al., 2008; Desta et al., 2016); and diminished CYP2B6 activity unmasks the influence of CYP2A6 on efavirenz exposure (di Iulio et al., 2009).
Scheme 1.

Metabolism of S-efavirenz catalyzed by CYP2B6.
The CYP2B6 gene is highly polymorphic (Zanger and Klein, 2013), with at least 38 allelic variants described (https://www.pharmvar.org/gene/CYP2B6), of which 25 are considered important and eight are common in at least one racial/ethnic population (Zhou et al., 2017). CYP2B6 metabolizes a broad range of substrates, constituting nearly 8% of marketed drugs (Nolan et al., 2006), although the relative contribution of CYP2B6 to total hepatic P450 content is small. In addition to efavirenz, clinically important CYP2B6 substrates include methadone, bupropion, ketamine, cyclophosphamide, and artemisinin.
The pharmacogenetics of efavirenz disposition has been comprehensively reviewed (Čolić et al., 2015; Sinxadi et al., 2015; Russo et al., 2016). The CYP2B6 516G>T polymorphism, alone constituting CYP2B6*9 or together with 785A>G constituting CYP2B6*6, is a canonical loss-of-function variant that was the first and most studied and is consistently associated with increased efavirenz exposure and reduced clearance and metabolism (Haas et al., 2004; Tsuchiya et al., 2004; Rotger et al., 2005). Efavirenz clearance is approximately 25% and 50% lower in 516GT and 516TT carriers, respectively (Čolić et al., 2015; Robarge et al., 2016). The less common CYP2B6 983T>C polymorphism, alone constituting CYP2B6*18 or together with 785A>G constituting CYP2B6*16, is also associated with increased efavirenz exposure (Wyen et al., 2008; Dhoro et al., 2015; Röhrich et al., 2016). The 516G>T, 785A>G, and 983T>C polymorphisms are more common in African than Caucasian populations, and the lattermost is considered essentially Africa-specific (Čolić et al., 2015; Russo et al., 2016). In Africans or African-Americans, CYP2B6*6/*6 and CYP2B6*6/*18 genotypes had the highest single-dose (Haas et al., 2009) or steady-state efavirenz concentrations (3- to 4-fold higher than CYP2B6*1/*1) (Maimbo et al., 2012). CYP2B6*6, *9, *16, and *18 constitute a poor-metabolizer phenotype (Čolić et al., 2015; Russo et al., 2016). Efavirenz adverse effects in general and adverse neurologic and neuropsychiatric effects in particular (e.g., neurocognitive impairment, depression, suicidality) have been associated with higher plasma efavirenz exposure, slow efavirenz metabolizer phenotype, or 516G>T and/or 983T>C polymorphisms (Haas et al., 2004; Rotger et al., 2005; Apostolova et al., 2015; Vo and Varghese Gupta, 2016; Gallien et al., 2017; Mollan et al., 2017; Chang et al., 2018). In contrast, 785A>G alone (CYP2B6*4) codes for a protein with increased efavirenz hydroxylation in vitro (Bumpus et al., 2006), but the clinical significance is ambiguous (Russo et al., 2016). CYP2B6 genetically guided efavirenz dosing has been evaluated and recommended (Gatanaga et al., 2007; Mukonzo et al., 2014; Vo and Varghese Gupta, 2016). Other CYP2B6 variant activity and clinical implications for efavirenz disposition are less well characterized. Furthermore, and surprisingly, excepting CYP2B6.4 and CYP2B6.6 (Bumpus et al., 2006; Ariyoshi et al., 2011; Zhang et al., 2011; Xu et al., 2012; Radloff et al., 2013), comparatively less is known about S-efavirenz metabolism by CYP2B6 variants in vitro than in vivo. Therefore, the first purpose of this investigation was to assess S-efavirenz metabolism by clinically relevant CYP2B6 variants, coexpressed with P450 oxidoreductase and cytochrome b5 in a fully catalytically competent system.
CYP2B6 is pharmacologically and clinically relevant, and several CYP2B6 substrates are chiral, with varying degrees of enantioselective metabolism, and enantioselectivity may vary among different CYP2B6 variants (Wang et al., 2018). As a prototypic CYP2B6 substrate, S-efavirenz and analogs have been used to inform on the structure, activity, and catalytic mechanism of wild-type CYP2B6 (Bumpus and Hollenberg, 2008; Cox and Bumpus, 2014; Cox and Bumpus, 2016; Shah et al., 2018) and variants such as CYP2B6.4 (Bumpus et al., 2005). These compounds, together with molecular modeling, have provided insights into the active site configuration of CYP2B6. In this regard, the metabolism of R-efavirenz by CYP2B6, and by CYP2B6 variants, remains unexplored. Therefore, the second purpose of this investigation was to evaluate the metabolism of R-efavirenz by wild-type CYP2B6.1 and CYP2B6 variants, potentially to inform on CYP2B6 active-site character or activity.
Materials and Methods
Materials.
S-Efavirenz was purchased from TCI America (Portland, OR). R-Efavirenz was purchased from Carbosynth US (San Diego, CA). The standards of rac 7-hydroxyefavirenz-d4, rac-8-hydroxyefavirenz, rac-7-hydroxyefavirenz, and rac-8,14-dihydroxyefavirenz were purchased from Toronto Research Chemicals (TRC, Toronto, ON, Canada). Spodoptera frugiperda (Sf9) cells and Sf-900 III SFM culture media were purchased from ThermoFisher (Waltham, MA). Trichoplusia ni cells and ESF AF culture media were from Expression Systems (Davis, CA). β-NADP, glucose-6-phosphate, and glucose-6-phosphate dehydrogenase were purchased from Sigma Aldrich (St. Louis, MO).
Generation of Baculovirus Constructs.
The production of recombinant proteins of CYP2B6 variants (Table 1), wild-type P450 reductase (POR), and cytochrome b5 were carried out as described previously (Wang et al., 2018). Briefly, the human genes of CYP2B6, POR, and b5 were amplified from the Human Liver Quick-Clone cDNA library (Clonetech, Mountain View, CA) and inserted individually into the transfer vector pVL1393 using the In-Fusion HD Cloning system (Clontech). The plasmid carrying the gene of wild-type CYP2B6 was used as the template, and the polymorphic variants of CYP2B6 were generated by site-directed mutagenesis using Quik-Change II XL Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA), as described (Wang et al., 2018). BestBac 2.0 Baculovirus Cotransfection Kit (Expression Systems) was used for production of recombinant baculovirus. Sf9 insect cells were cotransfected with BestBac linearized DNA and the plasmid DNA of transfer vector carrying the gene of interest on a six-well plate to produce p0 generation of recombinant baculovirus. Sf9 cells in suspension culture were infected with p0 to make subsequent viral generation. The maximal viral generation is limited to p3 to minimized undesirable variants owing to successive amplification. All viral titers were determined using the BacPAK Baculovirus Rapid Titer Kit (Clontech).
TABLE 1.
CYP2B6 variants
| CYP2B6 Allele | Variant | cDNA Sequence Mutation | Protein Sequence Mutationa | Allele Frequency (%) |
|---|---|---|---|---|
| CYP2B6*1 | Wild-type | Wild-type | ||
| CYP2B6*4 | rs2279343 | 785A>G | K262R | 2-4 Ca |
| CYP2B6*5 | rs3211371 | 1459C>T | R487C | 12 Ca |
| CYP2B6*6 | rs3745274, rs2279343 | 516G>T, 785A>G | Q172H/K262R | 33 Af, 28 Ca |
| CYP2B6*7 | rs3745274, rs2279343, rs3211371 | 515G>T, 785A>G, 1459C>T | Q172H/K262R/R487C | 3 Ca |
| CYP2B6*9 | rs3745274 | 516G>T | Q172H | |
| CYP2B6*16 | rs2279343, rs28399499 | 785A>G, 983T>C | K262R/I328T | 6.9 Af |
| CYP2B6*17 | rs33973337, rs33980385, rs33926104 | 76A>T, 83A>G, 85C>A, 86G>C | T26S/D28G/R29T | 6.3 Af |
| CYP2B6*18 | rs28399499 | 983T>C | I328T | 9.4 Af |
| CYP2B6*19 | rs34826503 | 516G>T, 785A>G, 1006C>T | Q172H/K262R/R336C | 1.6 Af |
| CYP2B6*26 | rs3826711, rs2279343, rs3745274 | 499C>G, 516G>T, 785A>G | P167A/Q172H/K262R | 1.3 As |
Af, African; As, Asian; Ca, Caucasian.
All CYP2B6 variants result in missense mutations.
Expression of Recombinant Proteins in Insect Cells.
Protein expression was performed as described previously (Wang et al., 2018). All CYP2B6 variants were coexpressed with redox partners P450 reductase (POR) and cytochrome b5 in insect cells by triple infection. Briefly, Trichoplusia ni cells in suspension culture in early log phase growth were infected with the recombinant baculoviruses carrying the genes of CYP2B6, POR and b5, with the multiplicities of infection at ratio of 4:2:1 (CYP2B6/POR/b5), in the presence of heme precursors 100 μM δ-aminolevulinic acid and 100 μM ferric citrate. After 48–72 hours growth post-infection, cells were harvested by centrifugation for 15 minutes at 3000g, and washed two times with phosphate-buffered saline followed by centrifugation in each wash step. The cell pellets were resuspended in 100 mM potassium phosphate buffer at pH 7.4, and stored frozen at −80°C. Frozen cells were thawed and lysed on ice in a Potter-Elvehjem tissue homogenizers. Full cell disruption was achieved by the combination of one freeze-thaw cycle and 10 strokes in the glass-Teflon Potter-Elvehjem pestle. Aliquots of 0.5 ml of homogenized cells were stored at −80°C.
P450 content, b5 content, and POR activity were measured as described previously (Wang et al., 2018). Total protein concentrations were determined using Bio-Rad Protein Assay Dye Regent Concentrate, the basis of which is the Bradford method. P450 concentration was determined by difference spectrum of ferrous-carbon monoxide complex in a CO binding assay using an extinction coefficient Δε450–490 nm of 91 mM−1cm−1. Cytochrome b5 content was determined by difference spectrum of NADH-reduced and oxidized b5 using an extinction coefficient Δε424–410 nm of 185 mM−1cm−1. POR activity was measured in an NADPH-cytochrome c reductase activity assay, and the reaction rate was calculated using an extinction coefficient of ε550 nm of 21 mM−1cm−1 for reduced cytochrome c. The POR activity was converted to POR content assuming that 3000 nmol of cytochrome c are reduced per minute per nanomoles POR at 23°C (Guengerich et al., 2009).
Efavirenz Metabolism.
Incubations were carried out in 96-well PCR plates with raised wells. The procedure used in this study was adapted with modifications from published protocols (Ward et al., 2003; Avery et al., 2013). Positive displacement pipettes were used in steps in which organic solvents were involved. Twenty-millimolar stock solutions of S- and R-efavirenz were prepared in 100% methanol. Substock solutions of efavirenz at 10 mM in 50% methanol, 2 mM in 25% methanol, and 200, 50, 10 μM in 10% methanol were prepared by dilution from the 20 mM stock. To a 96-well plate, efavirenz was added from the stock and substock solutions to 100 mM potassium phosphate buffer at pH7.4 containing CYP2B6/POR/b5 proteins. The final CYP2B6 concentration was 25 pmol/ml. S-efavirenz concentrations were 0, 0.25, 0.5, 1.25, 2.5, 5, 10, 20, 40, 70, and 100 μM and R-efavirenz concentrations were 0.11–45 μM, owing to limited solubility and availability. The total reaction volume was 200 μl and the methanol concentration was controlled at 0.5% for every incubation. After preincubation for 5 minutes at 37°C, the reaction was initiated by adding an NADPH-regenerating system (final concentrations: 10 mM glucose 6-phosphate, 1 mM β-NADP, 1 IU/ml glucose-6-phosphate dehydrogenase, and 5 mM magnesium chloride, preincubated at 37°C for 10 minutes). The reaction was allowed to proceed for 20 minutes at 37°C, then terminated by withdrawing 100 μl of reaction mixture and mixing with 200 μl of ice-cold acetonitrile containing 32 ng/ml internal standard rac-7-hydroxyefavirenz-d4 in glass tubes (16 × 125 mm). The metabolite products were extracted using a liquid/liquid extraction method as described previously (Avery et al., 2013) with modifications. Three hundred microliters of 50 mM ammonium formate was added to the quenched reaction mixture, followed by extraction with 1.0 ml of hexane/ethyl acetate (1:1). All samples were vortex-mixed for 30 seconds and centrifuged at 2500 rpm for 5 minutes. Six hundred, twenty-five microliters of organic layer was transferred to another clean glass tube (13 × 100 mm) and evaporated under nitrogen to dryness at 30°C using Turbo Vap LV Evaporator (Zymark, Hopkinton, MA). For high-performance liquid chromatography/mass spectrometry (HPLC/MS) analysis, the residues of the samples were reconstituted in 200 μl of 0.05% formic acid in 50% acetonitrile.
Analysis of Efavirenz Metabolites by HPLC/Tandem Mass Spectrometry.
Calibration samples were prepared using standards of rac-8-hydroxyefavirenz, rac-7-hydroxyefavirenz, and rac-8,14-dihydroxyefavirenz, with all three analytes at identical concentrations of 0, 0.5, 1, 2.5, 5, 10, 25, 50, 100, 250, and 500 ng/ml in 100 mM potassium phosphate buffer (pH 7.4) containing 33% methanol. Calibrators were processed identically to incubation samples.
LC-MS/MS analysis was performed on a Shimadzu HPLC system composed of two LC-20AD XR pumps, DGU20A5R degasser, CBM-20A system controller, CTO-20C column oven, FCV-11AL solvent selection valve, and a SIL-20AC XR temperature-regulated autosampler. The LC system was coupled to an API6500 triple quadrupole tandem mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA) operated with Analyst 1.6.2. software. MultiQuant 3.0.1 (AB Sciex) was used for peak integration, generation of calibration curves, and data analysis. Efavirenz metabolites were analyzed utilizing a Kinetex XB-C18 100A column (100 × 2.1 mm, 2.6 μm; Phenomenex) equipped with a Security Guard ULTRA Cartridge for C18 UHPLC (2 × 2.1 mm; Phenomenex). A 0.25-μm inline filter was additionally added prior to the sample entering the column. The column oven was at ambient temperature and the autosampler was at 4°C. The mobile phase A was 0.1% formic acid in Milli-Q water and mobile phase B was 0.1% formic acid in acetonitrile. Chromatographic separation was achieved using an isocratic condition of 50% mobile phase A and 50% mobile phase B and a flow rate of 0.15 ml/min. The injection volume was 5 μl and total run time is 12 minutes. Under these conditions, the approximate retention time was 6.5 minutes for 8-hydroxyefavirenz, 5.7 minutes for 7-hydroxyefavirenz, 5.6 minutes for 7-hydroxyefavirenz-d4, and 3.1 minutes for 7,8-dihydroxyefavirenz and 8,14-dihydroxyefavirenz. The mass spectrometer was operated with a turbo spray ion source in the negative mode with multiple reaction monitoring (MRM). Analytes were detected with the following MRM transitions: m/z 329.9 > 257.8 for 8-hydroxyefavirenz and 7-hydroxyefavirenz, m/z 346.0 > 274.0 for 7,8-dihydroxyefavirenz, and m/z 345.9 > 262.1 for 8,14-dihydroxyefavirenz.
Data Analysis.
Formation of 8-hydroxyefavirenz by enzyme variants at fixed concentrations was analyzed by analysis of variance with post-hoc Dunnett’s test (SigmaPlot 12.5; Systat). Results are the mean ± S.D.
8-Hydroxyefavirenz formation versus substrate concentration data were analyzed by nonlinear regression analysis. Results are the parameter estimate ± S.E. of the estimate. Metabolism of both efavirenz enantiomers at low concentrations (up to 40–45 μM) exhibited homotropic positive cooperativity. At higher concentrations of S-efavirenz there was evidence of substrate inhibition. Two approaches were used, depending on the substrate concentrations modeled.
Metabolism over the substrate concentration range 0.25–40 μM S-efavirenz and 0.11–45 μM R-efavirenz was analyzed using an allosteric model of the Hill equation (eq. 1), where [S] is the substrate efavirenz concentration, n is the Hill coefficient, and S50 represents the substrate concentration at which the reaction reached half-maximal velocity.
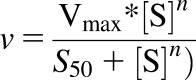 |
(1) |
R-efavirenz 8-hydroxylation by CYP2B6.19 showed weak substrate cooperativity. Therefore data were also analyzed using the Michaelis-Menten equation. R-efavirenz 8-hydroxylation by CYP2B6.19 also had high Km and 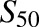 values relative to the substrate range. Thus it was also analyzed by linear regression. Specifically, when substrate concentrations are far below Km, the observed rate versus [S] approaches a linear function and the Michaelis-Menten equation can be simplified to v = (Vmax/Km)*[S], and the slope of the v versus [S] plot represents an estimate of Vmax/Km. R-Efavirenz 8-hydroxylation by CYP2B6.4 showed substrate inhibition. Therefore data were also analyzed using the LiCata model (vide infra).
values relative to the substrate range. Thus it was also analyzed by linear regression. Specifically, when substrate concentrations are far below Km, the observed rate versus [S] approaches a linear function and the Michaelis-Menten equation can be simplified to v = (Vmax/Km)*[S], and the slope of the v versus [S] plot represents an estimate of Vmax/Km. R-Efavirenz 8-hydroxylation by CYP2B6.4 showed substrate inhibition. Therefore data were also analyzed using the LiCata model (vide infra).
S-Efavirenz 8-hydroxylation over the substrate concentration range 0.25–100 μM exhibited both homotropic positive cooperativity and substrate inhibition for all active CYP2B6 variants. For R-efavirenz hydroxylation, only CYP2B6.4 showed substrate inhibition. Several models were evaluated for fitting positive cooperativity and substrate inhibition, including (i) a combination of the Hill equation with substrate inhibition (Müller et al., 2015), (ii) homotropic cooperativity with complete substrate inhibition and a single substrate molecule binding to an inhibitory site (a simplified version of the LiCata model, below) (Kapelyukh et al., 2008), (iii) cooperative catalysis and substrate inhibition (Pastra-Landis et al., 1978), (iv) substrate inhibition analogous to uncompetitive inhibition (Michaelis-Menten plus substrate inhibition) with facilitated sequential binding of two additional substrate (inhibitor) molecules (Bapiro et al., 2018), (v) uncompetitive inhibition (Michaelis-Menten plus substrate inhibition) assuming simultaneous binding of n molecules (determined from the data) of the substrate to the inhibitory site (Bapiro et al., 2018), and (vi) a modified Hill equation with cooperative substrate binding, substrate inhibition, and cooperative inhibitor binding (eq. 2) (LiCata and Allewell, 1997). In this model, K is the substrate dissociation constant, Ki is the inhibitor dissociation constant, Vi is the final velocity at infinite substrate concentration, and x represents a second Hill coefficient that allows for cooperativity of inhibitor substrate binding. To obtain convergence, the value of x must be fixed. The integer value for x that gave the best fit (x = 3) was determined empirically.
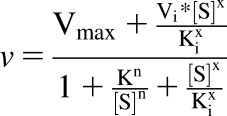 |
(2) |
Analysis using models i–iv did not produce acceptable fits or did not converge at all. Model v gave some acceptable fits and parameters but some required constraint on Km < Ki, and the model produced some unrealistic n and very high Km and Vmax values. Model vi achieved the best fits to the S-efavirenz metabolism data, on the basis of F values and parameter estimates and error variances, and was the final model chosen.
Because the in vitro intrinsic clearance parameter Vmax/Km is suitable only for reactions following Michaelis-Menten kinetics, data were further analyzed using Clmax, the maximal clearance (eq. 3) (Houston and Kenworthy, 2000):
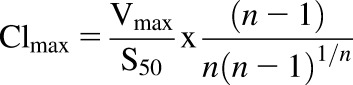 |
(3) |
Results
8-Hydroxyefavirenz was the predominant metabolite of wild-type CYP2B6-catalyzed S-efavirenz metabolism, as expected (Fig. 1A). Very low amounts of 8,14-dihydroxyefavirenz were formed. Neither S-7-hydroxyefavirenz nor 7,8-dihydroxyefavirenz were detected. Two aspects of S-efavirenz 8-hydroxylation by CYP2B6.1 are notable. First, S-efavirenz hydroxylation was maximal at substrate concentrations of 20–40 μM, and higher substrate concentrations resulted in substantially less 8-hydroxyefavirenz formation (Fig. 1B). The diminished formation at high substrate concentrations was not the result of facile secondary metabolism of 8-hydroxyefavirenz to 8,14-dihydroxyefavirenz. Indeed, rates of secondary 14-hydroxylation of 8-hydroxyefavirenz were generally low compared with primary metabolism (Fig. 1; Table 2). However at low S-efavirenz concentrations (0.25–1.25 μM), 8,14-dihydroxyefavirenz formation was predominant, representing 70%–100% of total substrate metabolism. In contrast, at 20 μM S-efavirenz, 8-hydroxyefavirenz was 95% of product formation and only 5% was converted to 8,14-dihydroxyefavirenz. Thus at low S-efavirenz concentrations, secondary metabolism of 8-hydroxyefavirenz to 8,14-dihydroxyeavirenz was facile but was inhibited at higher substrate concentrations. These observations are consistent with substrate inhibition of both primary and secondary CYP2B6.1-catalyzed hydroxylation. The second notable aspect of S-efavirenz hydroxylation was the atypical kinetics. Metabolism by CYP2B6.1 at low substrate concentrations deviated from standard Michaelis-Menten hyperbolic kinetics and instead showed a sigmoidal pattern suggesting cooperativity consistent with multiple substrate binding sites (Fig. 1A). The Eadie-Hofstee plot showed curvature indicative of such cooperativity (Fig. 1A, inset). At high substrate concentrations there was substrate inhibition (Fig. 1B). The Eadie-Hofstee plot showed a circular pattern, consistent with both cooperativity and substrate inhibition.
Fig. 1.

Primary and secondary metabolism of S-efavirenz catalyzed by coexpressed wild-type CYP2B6.1, P450 oxidoreductase, and cytochrome b5. Formation of 8-hydroxyefavirenz (8-OH-EFV) (●) and 8,14-dihydroxyefavirenz (8,14-diOH-EFV) (▲). Results are the mean ± S.D. of triplicate determinations. (A) Metabolism over the substrate range 0.25–40 μM. The solid line represents predicted concentrations on the basis of parameters from nonlinear regression using the Hill equation. The dotted line represents predicted concentrations on the basis of parameters from nonlinear regression using the Michaelis-Menten equation. The inset shows an Eadie-Hofstee plot for 8-hydroxyefavirenz formation. The solid line represents predicted values on the basis of parameters from nonlinear regression using the Hill equation. (B) Metabolism over the substrate range 0.25–100 μM, showing substrate inhibition. The solid line represents predicted concentrations on the basis of parameters from analysis using cooperative substrate binding and substrate inhibition with cooperativity in the inhibitory mode (LiCata model). The inset shows an Eadie-Hofstee plot for 8-hydroxyefavirenz formation. The solid line represents predicted concentrations on the basis of parameters from nonlinear regression using the LiCata model.
TABLE 2.
8-Hydroxyefavirenz and 8,14-dihydroxyefavirenz formation in low substrate concentration range
| S-Efavirenz | 8-Hydroxyefavirenz | 8,14-diOH-efavirenz | 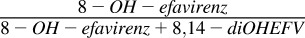 |
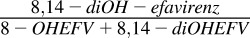 |
|---|---|---|---|---|
| µM | pmol/min per picomoles | pmol/min per picomoles | % | % |
| 0.25 | 0 | 0.014 ± 0.010 | 0 | 100 |
| 0.5 | 0.001 ± 0.001 | 0.055 ± 0.046 | 2 | 98 |
| 1.25 | 0.055 ± 0.009 | 0.209 ± 0.033 | 21 | 79 |
| 2.5 | 0.332 ± 0.048 | 0.460 ± 0.182 | 42 | 58 |
| 5.0 | 1.46 ± 0.29 | 0.405 ± 0.078 | 78 | 22 |
| 10 | 2.43 ± 0.10 | 0.505 ± 0.144 | 83 | 17 |
| 20 | 4.03 ± 0.28 | 0.201 ± 0.095 | 95 | 5 |
S-Efavirenz 8-hydroxylation at therapeutic (5–10 μM steady-state) substrate concentrations catalyzed by coexpressed CYP2B6 (wild-type and variants), wild-type POR, and cytochrome b5 is shown in Fig. 2. CYP2B6.6, CYP2B6.7, CYP2B6.9, CYP2B6.19, and CYP2B6.26 had diminished activity compared with CYP2B6.1, and CYP2B6.16 and CYP2B6.18 were essentially catalytically inactive. In contrast, CYP2B6.4 had higher activity than CYP2B6.1. Results at lower substrate concentrations showed comparatively little difference between variants, which may reflect differences in substrate cooperativity. At clinically relevant concentrations, hydroxylation rates were of the order CYP2B6.4 > CYP2B6.1 ≈ CYP2B6.5 ≈ CYP2B6.17 > CYP2B6.6 ≈ CYP2B6.7 ≈ CYP2B6.9 ≈ CYP2B6.19 ≈ CYP2B6.26 > > CYP2B6.16 and CYP2B6.18. For all CYP2B6 variants, 8,14-dihydroxefavirenz formation was less than by CYP2B6.1 (not shown).
Fig. 2.

Metabolism of S-efavirenz and R-efavirenz to 8-hydroxyefavirenz at therapeutic concentrations. Asterisks denote rates significantly different from wild-type (P < 0.05). Not shown are results for CYP2B6.16 and CYP2B6.18, which had negligible activity.
Concentration dependence of S-8-hydroxyefavirenz formation is shown in Fig. 3 for CYP2B6 variants, and kinetic parameters are provided in Table 3, for 0.25–40 μM S-efavirenz. Most variants had a sigmoidal curve indicating cooperativity, and data were analyzed by fitting the Hill equation. This was most apparent for CYP2B6.4 and CYP2B7.17, similar to wild-type CYP2B6.1. Hill coefficients (n), representing intensity of the cooperativity, varied from 1.4 to 2.5. Conversely, CYP2B6.9 data were hyperbolic, and regression analysis generated similar results from fitting either Hill or Michaelis-Menten equations, and n was close to 1, suggesting the absence of cooperative substrate binding. Differences in activity between CYP2B6 variants were the result of differences in Vmax, which varied approximately 3-fold, and 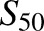 which varied approximately 2-fold. On the basis of the Clmax values, relative activities for S-efavirenz 8-hydroxylation were in the order CYP2B6.4 > CYP2B6.1 ≈ CYP2B6.5 ≈ CYP2B6.17 > CYP2B6.6 ≈ CYP2B6.7 ≈ CYP2B6.9 ≈ CYP2B6.19 ≈ CYP2B6.26 > > CYP2B6.16 and CYP2B6.18.
which varied approximately 2-fold. On the basis of the Clmax values, relative activities for S-efavirenz 8-hydroxylation were in the order CYP2B6.4 > CYP2B6.1 ≈ CYP2B6.5 ≈ CYP2B6.17 > CYP2B6.6 ≈ CYP2B6.7 ≈ CYP2B6.9 ≈ CYP2B6.19 ≈ CYP2B6.26 > > CYP2B6.16 and CYP2B6.18.
Fig. 3.

8-Hydroxyefavirenz formation from S-efavirenz (●) and R-efavirenz (▲) catalyzed by CYP2B6 variants, POR.1, and cytochrome b5. Solid lines represent predicted concentrations on the basis of parameters from nonlinear regression using the Hill equation. Parameter estimates are in Table 3.
TABLE 3.
Kinetic parameters for 8-hydroxyefavirenz formation from efavirenz enantiomers
Wild-type CYP2B6 and all variants were coexpressed with wild-type POR.1 and cytochrome b5. Results (Vmax and 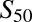 and n) are the parameter estimate and S.E. of the estimate, determined by nonlinear regression analysis of the Hill equation, over the substrate concentration range 0.25–40 μM S-efavirenz and 0.11–45 μM R-efavirenz.
and n) are the parameter estimate and S.E. of the estimate, determined by nonlinear regression analysis of the Hill equation, over the substrate concentration range 0.25–40 μM S-efavirenz and 0.11–45 μM R-efavirenz.
| CYP2B6 Variant | S-8-Hydroxyefavirenz Formation from S-Efavirenz | R-8-Hydroxyefavirenz Formation from R-Efavirenz | ||||||
|---|---|---|---|---|---|---|---|---|
| Vmax | 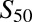 |
n | Clmax | Vmax | 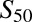 |
n | Clmax | |
| pmol/min per picomoles | mM | ml/min per nanomoles | pmol/min per picomoles | mM | ml/min per nanomoles | |||
| CYP2B6.1 | 4.2 ± 0.2 | 7.7 ± 0.6 | 2.1 ± 0.3 | 0.27 | 0.57 ± 0.09 | 16.1 ± 4.4 | 1.5 ± 0.3 | 0.019 |
| CYP2B6.4a | 4.5 ± 0.1 | 5.9 ± 0.3 | 2.5 ± 0.2 | 0.39 | 0.78 ± 0.06 | 7.0 ± 1.1 | 1.2 ± 0.1 | 0.071 |
| CYP2B6.5 | 3.3 ± 0.1 | 7.3 ± 0.7 | 1.4 ± 0.1 | 0.25 | 0.44 ± 0.02 | 6.5 ± 0.8 | 1.8 ± 0.3 | 0.034 |
| CYP2B6.6 | 3.4 ± 0.1 | 11.8 ± 0.5 | 1.8 ± 0.1 | 0.15 | 0.41 ± 0.05 | 14.5 ± 3.8 | 1.2 ± 0.2 | 0.018 |
| CYP2B6.7 | 1.5 ± 0.1 | 5.0 ± 0.3 | 1.5 ± 0.1 | 0.16 | 0.14 ± 0.01 | 6.3 ± 1.3 | 1.6 ± 0.4 | 0.011 |
| CYP2B6.9 | 1.7 ± 0.1 | 8.1 ± 1.0 | 1.2 ± 0.1 | 0.13 | 0.13 ± 0.03 | 13.5 ± 5.6 | 1.3 ± 0.3 | 0.006 |
| CYP2B6.16b | 0.19 ± 0.01 | 0.012 ± 0.002 | ||||||
| CYP2B6.17 | 4.4 ± 0.2 | 9.3 ± 0.8 | 1.7 ± 0.2 | 0.24 | 0.38 ± 0.07 | 9.2 ± 4.30 | 1.2 ± 0.3 | 0.055 |
| CYP2B6.18b | 0.20 ± 0.01 | 0.009 ± 0.001 | ||||||
| CYP2B6.19c | 3.4 ± 0.1 | 13.4 ± 0.9 | 1.5 ± 0.1 | 0.13 | 0.65 ± 0.33 | 31 ± 28 | 1.2 ± 0.4 | 0.013 |
| CYP2B6.26 | 1.5 ± 0.1 | 5.0 ± 0.3 | 1.8 ± 0.1 | 0.15 | 0.21 ± 0.01 | 4.2 ± 0.7 | 1.9 ± 0.4 | 0.025 |
CYP2B6.4 (only) showed substrate inhibition with R-efavirenz. Results in the table for CYP2B6.4and R-efavirenz were from the Hill equation and the substrate concentration range 0.11–18 μM. Data were also analyzed with the model for substrate inhibition (LiCata model, x = 3) over the substrate concentration range 0.11–45 μM, yielding Vmax = 0.90 ± 0.18, K = 9.0 ± 3.5, n = 1.1 ± 0.1, Clmax = 0.74, Vi = 0.09 ± 0.19, and Ki = 38 ± 11 μM.
CYP2B6.16 and CYP2B6.18 rates were measured at a fixed substrate concentration of 40 μM S- and R-efavirenz.
For R-efavirenz hydroxylation by CYP2B6.19, an alternative Michaelis-Menten model of linear regression analysis at low substrate concentrations generated a ratio of Vmax/Km = 0.019.
Concentration dependence of S-8-hydroxyefavirenz formation is shown in Fig. 4 for CYP2B6 variants, and kinetic parameters are provided in Table 4 for 0–100 μM S-efavirenz. In addition to cooperativity, substrate inhibition is apparent, and data were analyzed by fitting a modified Hill equation with cooperative substrate binding, substrate inhibition, and cooperative binding of three inhibitor molecules. For CYP2B6.7 and CYP2B6.9 minimal substrate binding cooperativity was suggested by n values of 1.0 and 0.9, and thus Clmax = Clint. Both Vmax and K varied approximately 2-fold. Ki values were 3- to 7-fold greater than K. CYP2B6.19 showed minimal substrate inhibition, and the model incorporating substrate inhibition did not fit the data well. On the basis of the Clmax values using the model for both cooperativity and substrate inhibition, the relative activities for S-efavirenz 8-hydroxylation were CYP2B6.4 > CYP2B6.1 ≈ CYP2B6.5 ≈ CYP2B6.17 > CYP2B6.6 ≈ CYP2B6.7 ≈ CYP2B6.9 ≈ CYP2B6.19 ≈ CYP2B6.26. This was similar to that using only limited substrate concentrations and cooperativity without inhibition.
Fig. 4.

S-Efavirenz hydroxylation by CYP2B6 variants, POR.1, and cytochrome b5 showing positive cooperativity and substrate inhibition. Solid lines represent predicted concentrations on the basis of parameters from nonlinear regression using model of cooperative substrate binding with substrate inhibition and cooperative inhibitor binding. Parameter estimates are in Table 4.
TABLE 4.
Kinetic parameters for 8-hydroxyefavirenz formation from S-efavirenz
Results (Vmax, K, n, Ki, and Vi) are the parameter estimates and S.E. of the estimate, determined by nonlinear regression analysis using a model of cooperative substrate binding with substrate inhibition and cooperative inhibitor binding over the substrate concentration range 0.25–100 μM.
| Vmax | K | n | Clmax | Ki | Vi | |
|---|---|---|---|---|---|---|
| pmol⋅min/pmol | µM | mM | pmol/min per picomoles | |||
| CYP2B6.1 | 7.4 ± 2.4 | 17 ± 8 | 1.4 ± 0.3 | 0.25 | 47 ± 10 | 0.0001 ± 0.42 |
| CYP2B6.4 | 6.2 ± 1.0 | 9 ± 2 | 1.7 ± 0.3 | 0.35 | 53 ± 8 | 0.0002 ± 0.44 |
| CYP2B6.5 | 4.1 ± 0.4 | 11 ± 2 | 1.2 ± 0.1 | 0.25 | 75 ± 10 | 5.8e−5 ± 0.42 |
| CYP2B6.6 | 3.6 ± 0.2 | 13 ± 1 | 1.7 ± 0.1 | 0.14 | 84 ± 16 | 1.46 ± 0.43 |
| CYP2B6.7 | 2.7 ± 1.0 | 16 ± 12 | 1.0 ± 0.2 | 0.17 | 46 ± 11 | 4.5e−5 ± 0.14 |
| CYP2B6.9 | 2.8 ± 0.7 | 21 ± 10 | 0.9 ± 0.1 | 0.13 | 57 ± 9 | 4.4e−5 ± 0.15 |
| CYP2B6.17 | 5.5 ± 0.5 | 13 ± 2 | 1.4 ± 0.1 | 0.23 | 71 ± 8 | 2.8e−5 ± 0.44 |
| CYP2B6.19 | 3.5 ± 0.2 | 14 ± 2 | 1.5 ± 0.1 | 0.13 | 173 ± 448 | 7.1e−5 ± 21 |
| CYP2B6.26 | 2.1 ± 0.4 | 9 ± 4 | 1.2 ± 0.3 | 0.15 | 56 ± 11 | 5.7e−5 ± 0.18 |
Evaluation of R-efavirenz metabolism by wild-type CYP2B6.1 showed that 8-hydroxyefavirenz was the only metabolite observed, and 7-hydroxyefavirenz, 8,14-dihydroxyefavirenz, and 7,8-dihydroxyefavirenz were not detected. Immediately apparent is that rates of R-efavirenz 8-hydroxylation were an order of magnitude less than those of S-efavirenz (Figs. 2 and 5). Metabolism of R-efavirenz by CYP2B6.1 showed a sigmoidal pattern suggesting cooperativity.
Fig. 5.

Stereoselectivity of efavirenz metabolism. Shown is formation of 8-hydroxyefavirenz from S-efavirenz (●) and R-efavirenz (▲) by CYP2B6.1. The solid line represents predicted concentrations on the basis of parameters from nonlinear regression using the Hill equation for 8-hydroxylation of S-efavirenz and R-efavirenz. The inset compares predicted concentrations for R-efavirenz hydroxylation on the basis of parameters from nonlinear regression using the Hill equation (solid line) and Michaelis-Menten equation (dotted line).
Metabolism of R-efavirenz by the various CYP2B6 variants was evaluated. Secondary metabolism to 8,14-dihydroxyefavirenz was not observed for any 2B6 variant. R-Efavirenz 8-hydroxylation at 2–9 μM substrate concentrations catalyzed by coexpressed CYP2B6 (wild-type and variants), wild-type POR, and cytochrome b5 are shown in Fig. 2. Rates of R-efavirenz metabolism were approximately 1/10 those of S-efavirenz. Compared with CYP2B6.1, CYP2B6.7, and CYP2B6.9 had diminished activity, and CYP2B6.16 and CYP2B6.18 were essentially inactive, and CYP2B6.4, CYP2B6.5, and CYP2B7.17 had higher activity.
Concentration dependence of R-8-hydroxyefavirenz formation is shown in Fig. 3 for CYP2B6 variants, and kinetic parameters are provided in Table 3. The Hill equation was used to model the data. CYP2B6.4 had the highest Clmax
R-efavirenz 8-hydroxylation, similar to S-efavirenz. CYP2B6.4 was the only variant showing substrate inhibition. CYP2B6.4 data were analyzed using both the Hill equation over a limited concentration range, and the LiCata model over the broader concentration range. Both analyses afforded similar Vmax, K, or 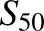 , and Clmax values. CYP2B6.19 fitting showed a high
, and Clmax values. CYP2B6.19 fitting showed a high 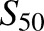 value relative to the substrate concentrations, and high S.E., suggesting uncertainty in the model parameters. Since cooperativity was minor (n = 1.2), data were also modeled using the Michaelis Menten equation, which yielded parameters of Vmax = 0.88 pmol/min per picomoles, Km = 54 ± 26 μM, and Clint (Vmax/Km) = 0.016 (not shown). Linear regression analysis was also performed in the linear range of 0.23–4.5 μM R-efavirenz and yielded Vmax/Km = slope = 0.019 (not shown). This is similar to the value obtained using the Hill (0.021) and Michaelis-Menten (0.016) equations. CYP2B6.19 was the only isoform with a high K or
value relative to the substrate concentrations, and high S.E., suggesting uncertainty in the model parameters. Since cooperativity was minor (n = 1.2), data were also modeled using the Michaelis Menten equation, which yielded parameters of Vmax = 0.88 pmol/min per picomoles, Km = 54 ± 26 μM, and Clint (Vmax/Km) = 0.016 (not shown). Linear regression analysis was also performed in the linear range of 0.23–4.5 μM R-efavirenz and yielded Vmax/Km = slope = 0.019 (not shown). This is similar to the value obtained using the Hill (0.021) and Michaelis-Menten (0.016) equations. CYP2B6.19 was the only isoform with a high K or 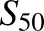 value. Differences in activity between CYP2B6 variants reflected differences in both Vmax and K values. On the basis of Clmax values, relative activities for R-efavirenz 8-hydroxylation were in the order CYP2B6.4 > CYP2B6.17 > CYP2B6.5 > CYP2B6.1 ≈ CYP2B6.6 ≈ CYP2B6.7 ≈ CYP2B6.19 ≈ CYP2B6.26 > CYP2B6.9 > > CYP2B6.16 and CYP2B6.18. Parameter estimates and reaction order should be interpreted cautiously, however, because of the low rates of metabolism.
value. Differences in activity between CYP2B6 variants reflected differences in both Vmax and K values. On the basis of Clmax values, relative activities for R-efavirenz 8-hydroxylation were in the order CYP2B6.4 > CYP2B6.17 > CYP2B6.5 > CYP2B6.1 ≈ CYP2B6.6 ≈ CYP2B6.7 ≈ CYP2B6.19 ≈ CYP2B6.26 > CYP2B6.9 > > CYP2B6.16 and CYP2B6.18. Parameter estimates and reaction order should be interpreted cautiously, however, because of the low rates of metabolism.
Efavirenz 8-hydroxylation was stereoselective (S > R), and stereoselectivity was similar across all CYP2B6 variants. On the basis of Clmax values, there was 14-fold enantioselectivity (Clmax, S-efavirenz/Clmax, R-efavirenz = 14) for wild-type CYP2B6.1 and 5- to 25-fold differences for other CYP2B6 variants (Table 3).
Discussion
CYP2B6-catalyzed 8-hydroxylation accounts for approximately 90% of S-efavirenz oxidative metabolic clearance (Ward et al., 2003). This investigation provides novel insight into the roles of CYP2B6 genetic polymorphisms in the metabolism of S-efavirenz, additional CYP2B6 variants probably of clinical significance, mechanisms of CYP2B6-catalyzed efavirenz metabolism, and expression system influences on CYP2B6 variants’ catalytic activity. In addition, results demonstrate the remarkable stereoselectivity of efavirenz metabolism by CYP2B6, and an unusual combination of cooperative metabolism and substrate inhibition, which may provide additional insights about this important P450 isoform.
The first major observation was that S-efavirenz 8-hydroxylation by CYP2B6 exhibited positive homotropic cooperativity, and that cooperativity was generally preserved across CYP2B6 variants. Cooperativity, rather than Michaelis-Menten kinetics, was evidenced by nonlinear Eadie-Hofstee plots. Cooperativity (n) was greatest at higher rates of S-efavirenz metabolism (CYPs 2B6.1 and 2B6.4) but occurred with all genetic variants. Previous studies reported that S-efavirenz 8-hydroxylation by human liver microsomes was cooperative (Ward et al., 2003) or followed single-site hyperbolic Michaelis-Menten kinetics (Ogburn et al., 2010) and was hyperbolic with baculovirus-expressed (Ward et al., 2003; Ogburn et al., 2010; Xu et al., 2012) and Escherichia coli-expressed wild-type CYP2B6 (Bumpus et al., 2006; Zhang et al., 2011). It is well known that some P450s exhibit allosteric regulation and cooperative behaviors (Denisov et al., 2009). For example, CYP3A4 has a large and flexible substrate binding pocket that allows simultaneous binding of multiple ligands, leading to cooperativity, but ligand binding to nearby allosteric sites could also be involved. CYP2B6 has a smaller substrate binding pocket that is only about 50% of the CYP3A4 active site volume (Gay et al., 2010). Nonetheless, CYP2B6 is still spacious relative to small molecules (Ekins et al., 1999; Lewis et al., 1999; Ekins et al., 2008; Wang et al., 2018) and can accommodate ligands of various geometries by movement of residues in the active site (Shah et al., 2018). Cooperativity among CYP2B6 substrates is relatively uncommon, having been described for CYP2B6.1 and 7-hydroxy-4-trifluoromethylcoumarin (n = 1.4) (Ekins et al., 1997), testosterone (n = 1.3) (Ekins et al., 1998), methadone (Totah et al., 2007), and S-efavirenz (n = 1.5) (Ward et al., 2003) but not several other substrates (Ekins et al., 1998; Liu et al., 2016), and also for CYP2B6.4 and 7-ethoxycoumarin (n = 1.7) (Ariyoshi et al., 2001). Interestingly, heteroactivation by efavirenz was recently reported, with enhanced midazolam hydroxylation by CYP3A4 via interaction at an allosteric site (Ichikawa et al., 2018).
In addition to positive cooperativity, S-efavirenz 8-hydroxylation showed apparent substrate inhibition. S-8-Hydroxyefavirenz formation by CYP2B6.1 was highest at 40 μM S-efavirenz and declined at higher concentrations. At 100 μM S-efavirenz, 8-hydroxyefavirenz formation was reduced to 4% of Vmax for CYP2B6.1 and also for CYP2B6.4. Substrate inhibition was influenced by CYP2B6 polymorphism. Less inhibition was observed with CYP2B6.6. Efavirenz is a known mechanism-based CYP2B6.1 inhibitor (as is S-8-hydroxyefavirenz) (Bumpus et al., 2006). Substrate inhibition was observed previously with expressed CYP2B6.1 and efavirenz (Ward et al., 2003) and efavirenz analogs (Cox and Bumpus, 2016), but not always reported (Bumpus et al., 2006; Zhang et al., 2011; Xu et al., 2012), and not observed with human liver microsomes (Ward et al., 2003; Ogburn et al., 2010). It is interesting that CYP2B6.1 and CYP2B6.4 had the greatest intrinsic clearances, cooperativity (n > 2), and substrate inhibition. Further investigation is necessary to better understand the interactions of CYP2B6 with efavirenz, substrate binding cooperativity, and the influence on metabolism.
S-Efavirenz 8-hydroxylation data were best fit to a model with positive homotropic cooperativity of both metabolism and inhibitory substrate binding, and a second Hill coefficient of 3 for inhibitory substrate binding. Although homotropic cooperativity, multiple substrate binding to the active site, and substrate inhibition have often been reported with P450s (Denisov et al., 2009), multiple inhibitor binding (Bapiro et al., 2018) and concomitant catalytic and inhibitory cooperativity are relatively uncommon (Müller et al., 2015). Comprehensive modeling of both catalytic and inhibitory cooperativity resulted in many parameters relative to the number of experimental observations, with a concern for an over-parameterized model. Thus we included both this analysis and the analysis of the noninhibited data using the Hill equation alone over the uninhibited substrate concentrations. Both models afforded similar conclusions with respect to the relative activities of the CYP2B6 variants.
The second major observation was that CYP2B6 genetic variants had altered activity toward S-efavirenz. At the recommended adult efavirenz dose of 600 mg, therapeutic plasma concentrations are 1–4 μg/ml (3–13 μM) (Bednasz et al., 2017). At 10 μM S-efavirenz, relative activities were CYP2B6.4 > CYP2B6.1 > CYP2B6.5, CYP2B6.17 > CYP2B6.6, CYP2B6.7, CYP2B6.9, CYP2B6.19, and CYP2B6.26; CYP2B6.16 and CYP2B6.18 were relatively inactive. Rank order was different at lower substrate concentrations, owing in part to differing cooperativity for the variants. Clmax values were CYP2B6.4 > CYP2B6.1 ≈ CYP2B6.5 ≈ CYP2B6.17 > CYP2B6.6 ≈ CYP2B6.7 ≈ CYP2B6.9 ≈ CYP2B6.19 ≈ CYP2B6.26 > > CYP2B6.16 and CYP2B6.18. Kinetic parameters for S-efavirenz 8-hydroxylation by CYP2B6 variants, mainly CYP2B6.1, CYP2B6.4, CYP2B6.6, and CYP2B6.9, have been reported (Table 5) (Bumpus et al., 2006; Ariyoshi et al., 2011; Zhang et al., 2011; Xu et al., 2012; Radloff et al., 2013; Watanabe et al., 2018). CYP2B6.4 (785G>T, K262R) activity was greater than wild-type when expressed in T. ni (144%, this investigation), Sf9 cells (142%) (Ariyoshi et al., 2011), and E. coli (170%) (Bumpus et al., 2006), or similar to wild-type in E. coli (96%) (Zhang et al., 2011). Greater CYP2B6.4 activity toward S-efavirenz in vitro is thus a relatively consistent observation.
TABLE 5.
Summary of reported 8-hydroxylation of S-efavirenz
Relative activities are shown as percentage of CYP2B6 variants Clint (Clmax for this study) compared with the wild type, on the basis of data in Table 3.
| Bumpus et al., 2006 | Ariyoshi et al., 2011 | Zhang et al., 2011 | Xu et al., 2012 | Radloff et al., 2013 | Watanabe et al., 2018 | This Study (Clmax) | |
|---|---|---|---|---|---|---|---|
| Expression system | E. coli | Sf9 | E. coli | SF9 | COS-1 | HEK | T. ni |
| CYP2B6.1 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| CYP2B6.4 | 170 | 142 | 96 | 122 | 144 | ||
| CYP2B6.5 | 138 | 83 | 109 | 92 | |||
| CYP2B6.6 | 50 | 20 | 49 | 183 | 266 | 56 | |
| CYP2B6.7 | 156 | 166 | 59 | ||||
| CYP2B6.9 | 33 | 172 | 48 | ||||
| CYP2B6.17 | 85 | 89 | |||||
| CYP2B6.19 | 37 | 48 | |||||
| CYP2B6.26 | 183 | 56 |
More generally, CYP2B6 variant catalytic activity is variant-, substrate-, and expression system-dependent. With other substrates, CYP2B6.4 was more active than CYP2B6.1 toward methadone (Gadel et al., 2013, 2015) and artmether (Honda et al., 2011) but less active toward cyclophosphamide (Ariyoshi et al., 2011), ifosfamide (Calinski et al., 2015), bupropion (Zhang et al., 2011), and ketamine (Wang et al., 2018). CYP2B6.6 (516G>T, 785A>G, Q172H/K262R) had lesser activity toward S-efavirenz (53% of wild-type), consistent with most (20%–50%) (Ariyoshi et al., 2011; Zhang et al., 2011; Xu et al., 2012) but not all (Radloff et al., 2013; Watanabe et al., 2018) reports. CYP2B6.6 was also less active than CYP2B6.1 toward methadone (Gadel et al., 2013, 2015), ketamine (Wang et al., 2018), and bupropion (Zhang et al., 2011) but more active toward artmether and cyclophosphamide (Ariyoshi et al., 2011; Honda et al., 2011). CYP2B6.9 (516G>T, Q172H) had even lower activity toward S-efavirenz (38% of wild-type) than CYP2B6.6 in the current investigation. Likewise, CYP2B6.9 also had lower 8-hydroxylation activity (33%) in one investigation (Zhang et al., 2011) but not another (Watanabe et al., 2018), and lower activity than wild-type in metabolizing methadone (Gadel et al., 2013, 2015), bupropion (Zhang et al., 2011), and ketamine (Wang et al., 2018) but greater with ifosfamide (Calinski et al., 2015). Thus, the CYP2B6 516G>T polymorphism (coding for both CYP2B6*6 and CYP2B6*9) is a canonical loss-of-function polymorphism for efavirenz.
Some structure-activity information is available on the CYP2B6 variants. CYP2B6.6 and CYP2B6.9 have the common Q172H mutation. Several in vitro studies have evaluated these variants, yet it is not clear how Q172H remotely (Q172 is about 15 Å away from the heme) affects the reaction in the active site. Moreover, effects of Q172H are moderated by K262R, and effects can be substrate-dependent (Ariyoshi et al., 2011). CYP2B6.16 and 2B6.18 share the I328T mutation in the J-helix, causing structural changes in the C and I helices that disrupt heme binding, alter ligand recognition, and reduce the ligand-binding pocket volume from 78 (CYP2B6.1) to 14 Å3 (CYP2B6.18) (Kobayashi et al., 2014; Wang et al., 2018).
While this report was in preparation, another investigation of efavirenz metabolism by CYP2B6 variants was published (Watanabe et al., 2018). Variants were expressed in human embryonic kidney (HEK)293 cells, without coexpression of P450 oxidoreductase or cytochrome b5. As described previously (Wang et al., 2018), expression systems can influence P450 activity. Mammalian systems (e.g., monkey kidney COS, HEK cells) allow easy P450 expression and use native reductase and b5, but P450 expression levels and protein integrity can vary widely. Some HEK results (Watanabe et al., 2018) differed substantially from previous reports (Table 5). Comparing kinetic parameters for efavirenz 8-hydroxylation using HEK versus our insect cell-expressed CYP2B6 shows that Vmax with insect cell expression was higher than with HEK expression, for example, 12-fold for CYP2B6.1 (4.2 vs. 0.35 pmol/min per picomoles). Such activity differences may influence reported Clint values for the variants.
There is potential clinical significance to the genetic variability in S-efavirenz metabolism in vitro. CYPB6*6 (516G>T, 785A>G), CYPB6*9 (516G>T), CYPB6*16 (785A>G, 983T>C), and CYPB6*18 (983T>C) constitute a poor efavirenz metabolizer clinical phenotype, associated with reduced clearance and increased plasma concentrations (Čolić et al., 2015; Russo et al., 2016; Robarge et al., 2016), and with common general and neuropsychiatric side effects (Haas et al., 2004; Rotger et al., 2005; Apostolova et al., 2015; Vo and Varghese Gupta, 2016; Gallien et al., 2017; Mollan et al., 2017; Chang et al., 2018). The present results, showing moderately (CYP2B6.6, CYP2B6.9) or markedly (CYP2B6.16, CYP2B6.18) lower metabolism, provide a mechanistic explanation for the clinical observations that the CYP2B6 516G>T polymorphism (Haas et al., 2004; Tsuchiya et al., 2004; Rotger et al., 2005; Čolić et al., 2015; Dhoro et al., 2015; Robarge et al., 2016) and the 983T>C polymorphism (Haas et al., 2009; Maimbo et al., 2012; Dhoro et al., 2015; Röhrich et al., 2016) are associated with increased efavirenz exposure and reduced clearance and metabolism. Other CYP2B6 variants we tested with these two polymorphisms (CYP2B6.19, CYP2B6.26) also had diminished activity. Thus, both CYP2B6 516G>T and 983T>C are canonical loss-of-function variants for S-efavirenz 8-hydroxylation. Other variants with these polymorphisms (CYP2B6.13, CYP2B6.20, CYP2B6.29, CYP2B6.34, CYP2B6.36, CYP2B6.37, CYP2B6.38) would be predicted to also have diminished activity. With the consistent association between CYP2B6 516G>T or 983T>C and increased efavirenz exposure, these other alleles would also be expected to be phenotypic poor metabolizers. This has been reported for CYP2B6*20 (Čolić et al., 2015). Although the clinical significance of 785A>G alone (CYP2B6*4) for efavirenz disposition is ambiguous, (Russo et al., 2016) the above in vitro-in vivo correlations, together with increased efavirenz hydroxylation in vitro by CYP2B6.4, would predict lower plasma efavirenz exposures and suggest further clinical investigation. These findings further strengthen the rationale for patient genotyping (specifically for CYP2B6 516G>T, 785A>G, and 983T>C polymorphisms) and for CYP2B6 genetically-guided efavirenz dosing (Mukonzo et al., 2014; Vo and Varghese Gupta, 2016).
The third major result was the surprising observation that efavirenz 8-hydroxylation was highly stereoselective. In this novel evaluation of R-efavirenz, metabolism at specific concentration and Clmax was generally at least 10-fold greater for S- versus R-efavirenz, for wild-type CYP2B6.1 and the active CYP2B6 variants. Differences in 8-hydroxylation were primarily owing to lower Vmax, as substrate affinity (K) was not substantially different between enantiomers. In addition, whereas both the primary metabolite 8-hydroxyefavirenz and very low amounts of the secondary metabolite 8,14-dihydroxyefavirenz was observed with S-efavirenz, only 8-hydroxyefavirenz was detected from R-efavirenz. This may well relate, however, to the lower R-efavirenz turnover and assay sensitivity. The considerable stereoselectivity of efavirenz 8-hydroxylation is a novel observation and contrasts with other CYP2B6 substrates. For example, N-demethylation of individual enantiomers by CYP2B6.1 was 2-fold greater for R- versus S-methadone (Totah et al., 2007) and S- versus R-ketamine (Wang et al., 2018), and hydroxylation of S-bupropion was 3-fold greater than R-bupropion (Coles and Kharasch, 2008). Similar enantioselectivities occurred with methadone and ketamine with several CYP2B6 variants (Gadel et al., 2015; Wang et al., 2018). N-Dechloroethylation by CYP2B6.1 was approximately 1.5- to 2-fold greater for S- versus R-ifosfamide, although the difference between S- and R-ifosfamide was substantially greater for 4-hydroxylation (Roy et al., 1999). Although these other CYP2B6 substrates follow Michaelis-Menten kinetics and are characterized by Clint, and efavirenz was characterized by Clmax, Clmax from nonhyperbolic metabolism can be used as a substitute for Clint when assessing metabolism (Houston and Kenworthy, 2000). Thus, efavirenz appears to be the CYP2B6 substrate with the greatest metabolic enantioselectivity yet observed.
A crystal structure of CYP2B6 in complex with an efavirenz analog, with a methyl group replacing the carbonyl oxygen, has been reported (Shah et al., 2018). Docking was described as consistent with the major and minor efavirenz metabolites. The chlorine of the efavirenz analog formed a Cl-π bond with the aromatic side chain of the F108 phenylalanine residue. Regarding the chiral carbon, the cyclopropyl group on the 4-cyclopropylethynyl substituent was between the side chains of F206 phenylalanine and T302 threonine, whereas the trifluoromethyl substituent was near the I101 isoleucine and F115 phenylalanine side chains. The considerable influence of efavirenz chirality on 8-hydroxylation demonstrates the importance of these residues in the active site. In addition, the protein for crystallization contains engineered mutations of K262R (as in CYP2B6.4) and Y226H, for stability and solubility. The structure shows the side chain of arginine (similar to R262 of CYP2B6.4) in close contact with the side chains of residues threonine T255 and aspartic acid D266 to form hydrogen bonding. In wild-type CYP2B6.1, the side chain of lysine K262 is not able to form similar hydrogen bonds with the neighboring residues. This difference between CYP2B6.4 and CYP2B6.1 may influence their structures and functions and account for differences in the metabolism of efavirenz and other substrates by these variants. The highly substrate-specific effect of the K262R substitution further informs on CYP2B6. Catalytic data for S- and R-efavirenz metabolism by wild-type CYP2B6 and variants may be useful in future computational studies to better understand mechanisms of metabolism by this clinically important isoform.
Abbreviations
- HEK
human embryonic kidney
- HIV
human immunodeficiency virus
- HPLC
high-performance liquid chromatography
- MS
mass spectrometry
- POR
NADPH cytochrome P450 oxidoreductase
- P450
cytochrome P450
Authorship Contributions
Participated in research design: Wang, Kharasch.
Conducted experiments: Wang, Neiner.
Performed data analysis: Wang, Neiner.
Wrote or contributed to the writing of the manuscript: Wang, Kharasch.
Footnotes
This work was supported by the National Institutes of Health [Grant R01-DA14211] and by the Washington University in St. Louis Department of Anesthesiology Russell B. and Mary D. Shelden fund. No author has any competing or conflict of interest.
References
- Apostolova N, Fumes HA, Blas-Garcia A, Galindo MJ, Alvarez A, Esplugues JV. (2015) Efavirenz and the CNS: what we already know and questions that need to be answered. J Antimicrob Chemother 70:2693–2708. [DOI] [PubMed] [Google Scholar]
- Ariyoshi N, Miyazaki M, Toide K, Sawamura Yi, Kamataki T. (2001) A single nucleotide polymorphism of CYP2b6 found in Japanese enhances catalytic activity by autoactivation. Biochem Biophys Res Commun 281:1256–1260. [DOI] [PubMed] [Google Scholar]
- Ariyoshi N, Ohara M, Kaneko M, Afuso S, Kumamoto T, Nakamura H, Ishii I, Ishikawa T, Kitada M. (2011) Q172H replacement overcomes effects on the metabolism of cyclophosphamide and efavirenz caused by CYP2B6 variant with Arg262. Drug Metab Dispos 39:2045–2048. [DOI] [PubMed] [Google Scholar]
- Avery LB, VanAusdall JL, Hendrix CW, Bumpus NN. (2013) Compartmentalization and antiviral effect of efavirenz metabolites in blood plasma, seminal plasma, and cerebrospinal fluid. Drug Metab Dispos 41:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapiro TE, Sykes A, Martin S, Davies M, Yates JWT, Hoch M, Rollison HE, Jones B. (2018) Complete substrate inhibition of cytochrome P450 2C8 by AZD9496, an oral selective estrogen receptor degrader. Drug Metab Dispos 46:1268–1276. [DOI] [PubMed] [Google Scholar]
- Bednasz CJ, Venuto CS, Ma Q, Daar ES, Sax PE, Fischl MA, Collier AC, Smith KY, Tierney C, Yang Y, et al. (2017) Efavirenz therapeutic range in HIV-1 treatment-naive participants. Ther Drug Monit 39:596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumpus NN, Hollenberg PF. (2008) Investigation of the mechanisms underlying the differential effects of the K262R mutation of P450 2B6 on catalytic activity. Mol Pharmacol 74:990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumpus NN, Kent UM, Hollenberg PF. (2006) Metabolism of efavirenz and 8-hydroxyefavirenz by P450 2B6 leads to inactivation by two distinct mechanisms. J Pharmacol Exp Ther 318:345–351. [DOI] [PubMed] [Google Scholar]
- Bumpus NN, Sridar C, Kent UM, Hollenberg PF. (2005) The naturally occurring cytochrome P450 (P450) 2B6 K262R mutant of P450 2B6 exhibits alterations in substrate metabolism and inactivation. Drug Metab Dispos 33:795–802. [DOI] [PubMed] [Google Scholar]
- Calinski DM, Zhang H, Ludeman S, Dolan ME, Hollenberg PF. (2015) Hydroxylation and N-dechloroethylation of Ifosfamide and deuterated Ifosfamide by the human cytochrome p450s and their commonly occurring polymorphisms. Drug Metab Dispos 43:1084–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JL, Lee SA, Tsai AC, Musinguzi N, Muzoora C, Bwana B, Boum Y, II, Haberer JE, Hunt PW, Martin J, et al. (2018) CYP2B6 genetic polymorphisms, depression, and viral suppression in adults living with HIV initiating efavirenz-containing antiretroviral therapy regimens in Uganda: pooled analysis of two prospective studies. AIDS Res Hum Retroviruses 34:982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles R, Kharasch ED. (2008) Stereoselective metabolism of bupropion by cytochrome P4502B6 (CYP2B6) and human liver microsomes. Pharm Res 25:1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čolić A, Alessandrini M, Pepper MS. (2015) Pharmacogenetics of CYP2B6, CYP2A6 and UGT2B7 in HIV treatment in African populations: focus on efavirenz and nevirapine. Drug Metab Rev 47:111–123. [DOI] [PubMed] [Google Scholar]
- Cox PM, Bumpus NN. (2014) Structure-activity studies reveal the oxazinone ring Is a determinant of cytochrome P450 2B6 activity toward efavirenz. ACS Med Chem Lett 5:1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox PM, Bumpus NN. (2016) Single heteroatom substitutions in the efavirenz oxazinone ring impact metabolism by CYP2B6. ChemMedChem 11:2630–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damle B, LaBadie R, Crownover P, Glue P. (2008) Pharmacokinetic interactions of efavirenz and voriconazole in healthy volunteers. Br J Clin Pharmacol 65:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decloedt EH, Rosenkranz B, Maartens G, Joska J. (2015) Central nervous system penetration of antiretroviral drugs: pharmacokinetic, pharmacodynamic and pharmacogenomic considerations. Clin Pharmacokinet 54:581–598. [DOI] [PubMed] [Google Scholar]
- Denisov IG, Frank DJ, Sligar SG. (2009) Cooperative properties of cytochromes P450. Pharmacol Ther 124:151–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desta Z, Metzger IF, Thong N, Lu JB, Callaghan JT, Skaar TC, Flockhart DA, Galinsky RE. (2016) Inhibition of cytochrome P450 2B6 activity by voriconazole profiled using efavirenz disposition in healthy volunteers. Antimicrob Agents Chemother 60:6813–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desta Z, Saussele T, Ward B, Blievernicht J, Li L, Klein K, Flockhart DA, Zanger UM. (2007) Impact of CYP2B6 polymorphism on hepatic efavirenz metabolism in vitro. Pharmacogenomics 8:547–558. [DOI] [PubMed] [Google Scholar]
- Dhoro M, Zvada S, Ngara B, Nhachi C, Kadzirange G, Chonzi P, Masimirembwa C. (2015) CYP2B6*6, CYP2B6*18, Body weight and sex are predictors of efavirenz pharmacokinetics and treatment response: population pharmacokinetic modeling in an HIV/AIDS and TB cohort in Zimbabwe. BMC Pharmacol Toxicol 16:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Iulio J Fayet A Arab-Alameddine M Rotger M Lubomirov R Cavassini M Furrer H Günthard HF Colombo S Csajka C et al. ; Swiss HIV Cohort Study (2009) In vivo analysis of efavirenz metabolism in individuals with impaired CYP2A6 function. Pharmacogenet Genomics 19:300–309. [DOI] [PubMed] [Google Scholar]
- Ekins S, Bravi G, Ring BJ, Gillespie TA, Gillespie JS, Vandenbranden M, Wrighton SA, Wikel JH. (1999) Three-dimensional quantitative structure activity relationship analyses of substrates for CYP2B6. J Pharmacol Exp Ther 288:21–29. [PubMed] [Google Scholar]
- Ekins S, Iyer M, Krasowski MD, Kharasch ED. (2008) Molecular characterization of CYP2B6 substrates. Curr Drug Metab 9:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S, Vandenbranden M, Ring BJ, Gillespie JS, Yang TJ, Gelboin HV, Wrighton SA. (1998) Further characterization of the expression in liver and catalytic activity of CYP2B6. J Pharmacol Exp Ther 286:1253–1259. [PubMed] [Google Scholar]
- Ekins S, VandenBranden M, Ring BJ, Wrighton SA. (1997) Examination of purported probes of human CYP2B6. Pharmacogenetics 7:165–179. [DOI] [PubMed] [Google Scholar]
- Gadel S, Crafford A, Regina K, Kharasch ED. (2013) Methadone N-demethylation by the common CYP2B6 allelic variant CYP2B6.6. Drug Metab Dispos 41:709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadel S, Friedel C, Kharasch ED. (2015) Differences in methadone metabolism by CYP2B6 variants. Drug Metab Dispos 43:994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallien S Journot V Loriot MA Sauvageon H Morlat P Reynes J Reliquet V Chêne G Molina JM; ANRS 099 ALIZE trial study group (2017) Cytochrome 2B6 polymorphism and efavirenz-induced central nervous system symptoms : a substudy of the ANRS ALIZE trial. HIV Med 18:537–545. [DOI] [PubMed] [Google Scholar]
- Gatanaga H, Hayashida T, Tsuchiya K, Yoshino M, Kuwahara T, Tsukada H, Fujimoto K, Sato I, Ueda M, Horiba M, et al. (2007) Successful efavirenz dose reduction in HIV type 1-infected individuals with cytochrome P450 2B6 *6 and *26. Clin Infect Dis 45:1230–1237. [DOI] [PubMed] [Google Scholar]
- Gay SC, Roberts AG, Halpert JR. (2010) Structural features of cytochromes P450 and ligands that affect drug metabolism as revealed by X-ray crystallography and NMR. Future Med Chem 2:1451–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP, Martin MV, Sohl CD, Cheng Q. (2009) Measurement of cytochrome P450 and NADPH-cytochrome P450 reductase. Nat Protoc 4:1245–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas DW, Gebretsadik T, Mayo G, Menon UN, Acosta EP, Shintani A, Floyd M, Stein CM, Wilkinson GR. (2009) Associations between CYP2B6 polymorphisms and pharmacokinetics after a single dose of nevirapine or efavirenz in African americans. J Infect Dis 199:872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas DW, Ribaudo HJ, Kim RB, Tierney C, Wilkinson GR, Gulick RM, Clifford DB, Hulgan T, Marzolini C, Acosta EP. (2004) Pharmacogenetics of efavirenz and central nervous system side effects: an Adult AIDS Clinical Trials Group study. AIDS 18:2391–2400. [PubMed] [Google Scholar]
- Honda M, Muroi Y, Tamaki Y, Saigusa D, Suzuki N, Tomioka Y, Matsubara Y, Oda A, Hirasawa N, Hiratsuka M. (2011) Functional characterization of CYP2B6 allelic variants in demethylation of antimalarial artemether. Drug Metab Dispos 39:1860–1865. [DOI] [PubMed] [Google Scholar]
- Houston JB, Kenworthy KE. (2000) In vitro-in vivo scaling of CYP kinetic data not consistent with the classical Michaelis-Menten model. Drug Metab Dispos 28:246–254. [PubMed] [Google Scholar]
- Ichikawa T, Tsujino H, Miki T, Kobayashi M, Matsubara C, Miyata S, Yamashita T, Takeshita K, Yonezawa Y, Uno T. (2018) Allosteric activation of cytochrome P450 3A4 by efavirenz facilitates midazolam binding. Xenobiotica 48:1227–1236. [DOI] [PubMed] [Google Scholar]
- Kapelyukh Y, Paine MJ, Maréchal JD, Sutcliffe MJ, Wolf CR, Roberts GC. (2008) Multiple substrate binding by cytochrome P450 3A4: estimation of the number of bound substrate molecules. Drug Metab Dispos 36:2136–2144. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Takahashi O, Hiratsuka M, Yamaotsu N, Hirono S, Watanabe Y, Oda A. (2014) Evaluation of influence of single nucleotide polymorphisms in cytochrome P450 2B6 on substrate recognition using computational docking and molecular dynamics simulation. PLoS One 9:e96789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DF, Lake BG, Dickins M, Eddershaw PJ, Tarbit MH, Goldfarb PS. (1999) Molecular modelling of CYP2B6, the human CYP2B isoform, by homology with the substrate-bound CYP102 crystal structure: evaluation of CYP2B6 substrate characteristics, the cytochrome b5 binding site and comparisons with CYP2B1 and CYP2B4. Xenobiotica 29:361–393. [DOI] [PubMed] [Google Scholar]
- LiCata VJ, Allewell NM. (1997) Is substrate inhibition a consequence of allostery in aspartate transcarbamylase? Biophys Chem 64:225–234. [DOI] [PubMed] [Google Scholar]
- Liu J, Shah MB, Zhang Q, Stout CD, Halpert JR, Wilderman PR. (2016) Coumarin derivatives as substrate probes of mammalian cytochromes P450 2B4 and 2B6: assessing the importance of 7-alkoxy chain length, halogen substitution, and non-active site mutations. Biochemistry 55:1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maimbo M, Kiyotani K, Mushiroda T, Masimirembwa C, Nakamura Y. (2012) CYP2B6 genotype is a strong predictor of systemic exposure to efavirenz in HIV-infected Zimbabweans. Eur J Clin Pharmacol 68:267–271. [DOI] [PubMed] [Google Scholar]
- Mollan KR Tierney C Hellwege JN Eron JJ Hudgens MG Gulick RM Haubrich R Sax PE Campbell TB Daar ES et al. ; AIDS Clinical Trials Group (2017) Race/ethnicity and the pharmacogenetics of reported suicidality with efavirenz among clinical trials participants. J Infect Dis 216:554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukonzo JK, Owen JS, Ogwal-Okeng J, Kuteesa RB, Nanzigu S, Sewankambo N, Thabane L, Gustafsson LL, Ross C, Aklillu E. (2014) Pharmacogenetic-based efavirenz dose modification: suggestions for an African population and the different CYP2B6 genotypes. PLoS One 9:e86919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller CS, Knehans T, Davydov DR, Bounds PL, von Mandach U, Halpert JR, Caflisch A, Koppenol WH. (2015) Concurrent cooperativity and substrate inhibition in the epoxidation of carbamazepine by cytochrome P450 3A4 active site mutants inspired by molecular dynamics simulations. Biochemistry 54:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan D, Phillips E, Mallal S. (2006) Efavirenz and CYP2B6 polymorphism: implications for drug toxicity and resistance. Clin Infect Dis 42:408–410. [DOI] [PubMed] [Google Scholar]
- Ogburn ET, Jones DR, Masters AR, Xu C, Guo Y, Desta Z. (2010) Efavirenz primary and secondary metabolism in vitro and in vivo: identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab Dispos 38:1218–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastra-Landis SC, Evans DR, Lipscomb WN. (1978) The effect of pH on the cooperative behavior of aspartate transcarbamylase from Escherichia coli. J Biol Chem 253:4624–4630. [PubMed] [Google Scholar]
- Radloff R, Gras A, Zanger UM, Masquelier C, Arumugam K, Karasi JC, Arendt V, Seguin-Devaux C, Klein K. (2013) Novel CYP2B6 enzyme variants in a Rwandese population: functional characterization and assessment of in silico prediction tools. Hum Mutat 34:725–734. [DOI] [PubMed] [Google Scholar]
- Rakhmanina NY, van den Anker JN. (2010) Efavirenz in the therapy of HIV infection. Expert Opin Drug Metab Toxicol 6:95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robarge JD, Metzger IF, Lu J, Thong N, Skaar TC, Desta Z, Bies RR. (2016) Population pharmacokinetic modeling to estimate the contributions of genetic and nongenetic factors to efavirenz disposition. Antimicrob Agents Chemother 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röhrich CR, Drögemöller BI, Ikediobi O, van der Merwe L, Grobbelaar N, Wright GE, McGregor N, Warnich L. (2016) CYP2B6*6 and CYP2B6*18 predict long-term efavirenz exposure measured in hair samples in HIV-positive South African women. AIDS Res Hum Retroviruses 32:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotger M Colombo S Furrer H Bleiber G Buclin T Lee BL Keiser O Biollaz J Décosterd L Telenti A; Swiss HIV Cohort Study (2005) Influence of CYP2B6 polymorphism on plasma and intracellular concentrations and toxicity of efavirenz and nevirapine in HIV-infected patients. Pharmacogenet Genomics 15:1–5. [DOI] [PubMed] [Google Scholar]
- Roy P, Tretyakov O, Wright J, Waxman DJ. (1999) Stereoselective metabolism of ifosfamide by human P-450s 3A4 and 2B6. Favorable metabolic properties of R-enantiomer. Drug Metab Dispos 27:1309–1318. [PubMed] [Google Scholar]
- Russo G, Paganotti GM, Soeria-Atmadja S, Haverkamp M, Ramogola-Masire D, Vullo V, Gustafsson LL. (2016) Pharmacogenetics of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in resource-limited settings: influence on antiretroviral therapy response and concomitant anti-tubercular, antimalarial and contraceptive treatments. Infect Genet Evol 37:192–207. [DOI] [PubMed] [Google Scholar]
- Schauer GD, Huber KD, Leuba SH, Sluis-Cremer N. (2014) Mechanism of allosteric inhibition of HIV-1 reverse transcriptase revealed by single-molecule and ensemble fluorescence. Nucleic Acids Res 42:11687–11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MB, Zhang Q, Halpert JR. (2018) Crystal structure of CYP2B6 in complex with an efavirenz analog. Int J Mol Sci 19:E1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinxadi PZ, Leger PD, McIlleron HM, Smith PJ, Dave JA, Levitt NS, Maartens G, Haas DW. (2015) Pharmacogenetics of plasma efavirenz exposure in HIV-infected adults and children in South Africa. Br J Clin Pharmacol 80:146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totah RA, Allen KE, Sheffels P, Whittington D, Kharasch ED. (2007) Enantiomeric metabolic interactions and stereoselective human methadone metabolism. J Pharmacol Exp Ther 321:389–399. [DOI] [PubMed] [Google Scholar]
- Tovar-y-Romo LB, Bumpus NN, Pomerantz D, Avery LB, Sacktor N, McArthur JC, Haughey NJ. (2012) Dendritic spine injury induced by the 8-hydroxy metabolite of efavirenz. J Pharmacol Exp Ther 343:696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya K, Gatanaga H, Tachikawa N, Teruya K, Kikuchi Y, Yoshino M, Kuwahara T, Shirasaka T, Kimura S, Oka S. (2004) Homozygous CYP2B6 *6 (Q172H and K262R) correlates with high plasma efavirenz concentrations in HIV-1 patients treated with standard efavirenz-containing regimens. Biochem Biophys Res Commun 319:1322–1326. [DOI] [PubMed] [Google Scholar]
- Vo TT, Varghese Gupta S. (2016) Role of cytochrome P450 2B6 pharmacogenomics in determining efavirenz-mediated central nervous system toxicity, treatment outcomes, and dosage adjustments in patients with human immunodeficiency virus infection. Pharmacotherapy 36:1245–1254. [DOI] [PubMed] [Google Scholar]
- Wang PF, Neiner A, Kharasch ED. (2018) Stereoselective ketamine metabolism by genetic variants of cytochrome P450 CYP2B6 and cytochrome P450 oxidoreductase. Anesthesiology 129:756–768. [DOI] [PubMed] [Google Scholar]
- Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z. (2003) The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J Pharmacol Exp Ther 306:287–300. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Saito T, Rico EMG, Hishinuma E, Kumondai M, Maekawa M, Oda A, Saigusa D, Saito S, Yasuda J, et al. (2018) Functional characterization of 40 CYP2B6 allelic variants by assessing efavirenz 8-hydroxylation. Biochem Pharmacol 156:420–430. [DOI] [PubMed] [Google Scholar]
- Wyen C Hendra H Vogel M Hoffmann C Knechten H Brockmeyer NH Bogner JR Rockstroh J Esser S Jaeger H et al. ; German Competence Network for HIV/AIDS (2008) Impact of CYP2B6 983T>C polymorphism on non-nucleoside reverse transcriptase inhibitor plasma concentrations in HIV-infected patients. J Antimicrob Chemother 61:914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Ogburn ET, Guo Y, Desta Z. (2012) Effects of the CYP2B6*6 allele on catalytic properties and inhibition of CYP2B6 in vitro: implication for the mechanism of reduced efavirenz metabolism and other CYP2B6 substrates in vivo. Drug Metab Dispos 40:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SD, Britcher SF, Tran LO, Payne LS, Lumma WC, Lyle TA, Huff JR, Anderson PS, Olsen DB, Carroll SS, et al. (1995) L-743, 726 (DMP-266): a novel, highly potent nonnucleoside inhibitor of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother 39:2602–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanger UM, Klein K. (2013) Pharmacogenetics of cytochrome P450 2B6 (CYP2B6): advances on polymorphisms, mechanisms, and clinical relevance. Front Genet 4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sridar C, Kenaan C, Amunugama H, Ballou DP, Hollenberg PF. (2011) Polymorphic variants of cytochrome P450 2B6 (CYP2B6.4-CYP2B6.9) exhibit altered rates of metabolism for bupropion and efavirenz: a charge-reversal mutation in the K139E variant (CYP2B6.8) impairs formation of a functional cytochrome p450-reductase complex. J Pharmacol Exp Ther 338:803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Ingelman-Sundberg M, Lauschke VM. (2017) Worldwide distribution of cytochrome P450 alleles: a meta-analysis of population-scale sequencing projects. Clin Pharmacol Ther 102:688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]