

Abstract
Although significant advances in experimental high throughput screening (HTS) have been made for drug lead identification, in silico virtual screening (VS) is indispensable owing to its unique advantage over experimental HTS, target-focused, cheap, and efficient, albeit its disadvantage of producing false positive hits. For both experimental HTS and VS, the quality of screening libraries is crucial and determines the outcome of those studies. In this paper, we first reviewed the recent progress on screening library construction. We realized the urgent need for compiling high-quality screening libraries in drug discovery. Then we compiled a set of screening libraries from about 20 million druglike ZINC molecules by running fingerprint-based similarity searches against known drug molecules. Lastly, the screening libraries were objectively evaluated using 5847 external actives covering more than 2000 drug targets. The result of the assessment is very encouraging. For example, with the Tanimoto coefficient being set to 0.75, 36% of external actives were retrieved and the enrichment factor was 13. Additionally, drug target family specific screening libraries were also constructed and evaluated. The druglike screening libraries are available for download from https://mulan.pharmacy.pitt.edu.
Graphical Abstract
1. INTRODUCTION
It is a challenging task to turn a new chemical compound into a real drug. First, a developable drug lead targeting the right protein or nucleic acid receptor must be discovered. A developable drug lead is then optimized to improve its activity of inhibition, selectivity against other targets, and its absorption, distribution, metabolism, excretion (ADME), and toxicity properties prior to the clinical trials.
Virtual Screening.
In modern drug discovery, high throughput screening is becoming an indispensable approach to identify drug leads. Compared to HTS, virtual screening is another technique which is complementary to experimental HTS. Although frequently produce false positives, VS is widely used because of its unique features, such as drug target-relevant, cheap, and efficient.
Virtual screening is a widely used technique to enrich developable drug leads in computer-aided drug design. A commonly used strategy in VS is the so-called hierarchical screenings,1,2 in the spirit of achieving both accuracy and efficiency. The basic idea of hierarchical screenings is to apply multiple filters to enrich the hits sequentially, with the efficient filters applied in the early stages and the more advanced and time-consuming filters applied in the later stages. Only the hits of the current screening enter the next round. The most efficient filters include those based on 2D-substructure, molecular fingerprints, and molecular shapes; less efficient filters include pharmacophore models and regular molecular docking; the most advanced filters include molecular docking using accurate scoring functions (such as the Glide extra precision docking scoring function3) and free energy-based methods (such as MM/GBSA and MM/PBSA4–8). We have successfully applied this strategy to identify drug leads for HIV-1 RT.1 An alternative screening strategy is parallel screening,2 where a set of docking screenings are performed in parallel using several complementary methods and the best hits ranked according to each individual method are combined for biological testing.
Virtual Screening Filters.
The VS filters are a key component of VS studies, which can loosely be classified into two categories depending on if the drug target information is used or not. The ligand-based screenings utilize known bioactives to define queries for VS; on the contrary, the structure-based screenings measure how tight a compound interacts with the drug target at the binding site. Molecular docking is the most widely used structure-based filter. Of course, structure-based pharmacophores9 and de novo design10 also rely on the structure information on protein or nucleic acid targets.2
The ligand-based filters, which are typically much cheaper compared to the structure-based ones, include substructures,11,12 topological indexes,13 molecular fingerprints,14 shape15 and electrostatic16 properties, and pharmacophore models.17,19 Li et al. developed a shape-based method called USR (ultrafast shape recognition) to identify potential drug leads. In about 2 s, 93.9 million 3D conformers expanded from 23.1 million purchasable molecules are screened.19 We recently constructed a set of molecular fingerprint-based artificial neural network (FANN) models for virtual screening of large diverse databases.20 Using 1361 ligands of cannabinoid receptor 2 as the training set, a FANN-QSAR model was derived and applied to screening a large NCI database to identify novel compounds bound to the CB2 receptor. The screening led to the discovery of several compounds which have CB2 binding affinities ranging from 6.70 nM to 3.75 μM.
Drug likeness is widely used to filter out those compounds unlikely to be drugs. Lipinski’s “Rule of 5” is the most famous drug likeness filter ever applied in virtual screening. Other popular molecular properties for characterizing drug likeness include polar surface area (PSA) and polar molecular volume (PMV). It was reported that 90% of orally bioavailable non-CNS drugs had a PSA below 120 Å2, and the criterion was dropped to 80 Å2 for CNS drugs.21 QSAR models can also serve as a filter in VS.22–25 A QSAR-based filter is widely applied in the drug lead optimization phase.
Screening Libraries.
Another key component of VS and experimental HTS is the screening library construction. There are two types of screening libraries to be applied in different phases of drug discovery. In the lead-identification phase, one wants the screening compounds structurally diverse so that novel drug leads can be identified; while in the sequential lead-optimization phase, one wants the screening compounds structurally akin to the drug lead to facilitate the construction of the structure–activity relationship. Ideally, the compounds in both kinds of libraries are druglike. Even though the current screening compounds only occupy a very tiny fraction of the entire chemical space which is estimated to be more than 1060 molecules, the registered compounds are more than 27 million.26,27 For example, the ZINC database28 collects more than 18 million commercially available compounds that satisfy Lipinski’s Rule of 529 at the time this paper was being written. It is not practical to screen all of them for a drug target both in the in vitro/in vivo HTS experiments and in the in silico virtual screenings. Therefore, screening compound libraries, which only collect fractions of the total available compounds, must be constructed for either HTS or VS studies. The outcome of an experimental HTS or a VS study strongly depends on the quality of the screening library.
There are a lot of compound libraries which are widely used in VS studies. The following are only some representatives: ZINC database,28 Pubchem,30,31 NCI open database,32 ChemDiv (www.chemdiv.com), Specs (www.specs.net), Chembridge (www.chembridge.com), ChemSpider, ChemNavigator (http://www.chemnavigator.com), etc. Most of the screening libraries collect a great number of compounds. Except for the most efficient filters, it is impractical to screen all the entries.
To construct diverse screening libraries, molecular diversity must be evaluated. A variety of molecular descriptors are applied to define a diversity metric, such as molecular fingerprints,14 topology index,13 and physicochemical properties.33 Recently, Koutsoukas et al. benchmarked a set of 13 molecular descriptors in assessing the diversity of chemical libraries.34 The descriptor performance was assessed by the coverage of bioactivity space. It was found that Bayes Affinity fingerprints35 and ECFP414 outperform others by retrieving more actives than random sampling.
Diverse screening libraries are then constructed by removing redundancy using a set of algorithms, including the dissimilarity-based method, cell-based method, cluster-based method, and optimization-based method.36,37 Principal component analysis was routinely applied to decrease the dimensionality of descriptor matrix and to extract the main variation of the descriptor data.38 We recently developed a Compound Library Acquisition and Prioritization (CAP) algorithm to construct diverse screening libraries.37 The CAP algorithm was established using the Euclidean distances of the BCUT39 chemical space. It was demonstrated that the CAP-selected subsets of an existing in-house screening library have their overall chemical diversities enhanced, as measured by using chemistry-space cell partition statistics and a similarity index.
Schneider et al. applied a self-organizing map (SOM) to construct diverse screening libraries.40 According to the principle of SOM, “neuron vectors” are positioned in the data space such that the distribution of data points is represented by the distribution of neuron vectors. A training procedure, such as unsupervised learning, is needed to achieve this. SOMs cover diverse fields of drug discovery, such as scaffold-hopping, repurposing, and screening library design. Noeske et al. developed a SOM by using pharmacophore descriptors to map druglike chemical space.41 Naderi et al. recently presented a graph-based approach for constructing target-focused screening libraries.42 They developed an exhaustive graph-based search algorithm to conduct virtual new compound syntheses by reconnecting the building blocks according to their connectivity patterns. However, the acquisition of the virtual compounds may be an issue. Sukuru et al. presented an approach based on extended connectivity fingerprints to carry out diversity selection on a per plate basis.43 They found that a fingerprint-diverse subset of 250 K compounds selected from a 4-fold larger screening deck achieved significantly higher hit rates for most of the screenings. Recently, Horvath et al. described a method of constructing a general-purpose screening library.38 The work represented a collaborative effort to construct the general-purpose screening library of EU-OPENSCREEN. As this screening is not exclusively targeted at drug discovery, loose compliance to druglikeness criteria was applied during the library construction. Mok and Brenk established a workflow to generate a target-specific screening library.44 They mined the ChEMBL database to assemble an ion channel-focused screening library.
Application of VS in Drug Discovery.
VS is routinely applied in pharmaceutical industry to identify novel and optimize the known drug leads. Recently, applications of VS to a family of receptors have emerged. Perez-Regidor et al. reviewed the latest effort of applying VS to search novel chemical entities for a set of Toll-like receptor (TLR) modulators, which include TLR2, TLR3, TLR4, TLR7, and TLR8.45 The TLR family proteins are interesting drug targets as they can recognize a wide variety of pathogens. Senderowitz and Marantz conducted more than 10 docking VS to identify novel compounds targeting GPCRs using the ab initio derived GPCR models.46
Human histamine H4 receptor (H4R) is a key player in inflammatory responses. Istyastono et al. recently explored the possibilities and challenges of discovering compounds against H4R by performing structure-based virtual screening (SBVS). Of the 37 tested compounds, 9 fragments had affinities between 0.14 and 6.3 μM against the H4R target. They used the area under the ROC curves to evaluate a set of screening studies.47 Ballester et al. conducted hierarchical virtual screening to discover new molecular scaffolds for antibacterial drug leads.48 Using the combination of shape-based method for rapid screening and molecular docking for reliable screening, they were able to explore truly large and diverse databases. Fifty new active molecular scaffolds were identified from their VS studies.
Hierarchical VS has been routinely applied in our drug design projects. In the HIV-1 RT project, pharmacophore modeling, molecular docking, and binding free energy calculations using MM-PBSA were applied to identify novel drug leads targeting at the non-nucleoside binding site.1 In another project, we discovered the first small molecule that inhibits p18INK4C by using the computational chemical genomics screening approach and stem cells specific chemogenomics knowledgebase.49 p18INK4C, a potent negative regulator of human hematopoietic stem cell (HSC) self-renewal, is a member of the cyclin-dependent kinase inhibitory proteins. In the transient receptor potential vanilloid type 1 (TRPVl) project, we developed a five-point pharmacophore model using known antagonists and constructed a homology model for docking-based screening. The in silico screenings using both types of filters yielded a set of promising hits and some were confirmed as hTRPV1 antagonists in assay.50
Above we briefly reviewed the VS technique with an emphasis on screening library construction. More reviews on VS can be found in other recent publications.51–54 The reviews on the latest advances of molecular docking can be found elsewhere.55–58
As a summary, virtual screening has become a daily practice in modern drug discovery. The screening libraries which collect millions of compounds need to be condensed to be applied in VS studies. Most effort is focused on developing structurally diverse libraries and target-specific libraries. However, general-purpose druglike screening libraries, which have moderate sizes and are suitable for daily virtual screenings of arbitrary drug targets, are under construction. The objective of this work is to construct and evaluate this kind of screening libraries.
In the following, we first justify our effort of applying known drugs to enrich screening compounds by conduct a survey on drug–target interactions. Next, a set of generic and target-specific screening libraries have been compiled using known drugs. Last, the screening libraries have been critically evaluated using an external library of actives. Recommendations on how to use those screening libraries are also discussed.
2. METHODS
Drug–Target Interaction Analysis.
How should a VS be evaluated and how many hits should be selected to do a bioassay? Of course, the answer to this question is target-dependent. We hope to provide some hints to this question by performing a survey of drug–target interactions. A major challenge in drug repositioning is to identify interactions between known drugs and targets. In silico prediction of drug–target interaction (DTI) can speed up the process of identifying novel DTIs by providing the most promising DTI candidates to minimize the larger scale expensive and time-consuming experimental work.
In the following, we conducted a survey on DTIs collected by the Drug Bank Database.59,60 As a unique bioinformatics and cheminformatics resource, the DrugBank database combines detailed drug data with comprehensive drug–target information. The data used in the present study was released on April 1, 2017 (version 5.0.6). The drug–target interaction data were extracted from the “Drug Target Identifiers” table under the “Protein Identifiers” category on the DrugBank website (https://www.drugbank.ca/releases/latest#protein-identifiers). Other databases that collect DTIs include the TTD (Therapeutic Target Database).61
Special attention was paid to some drug target classes, including G-protein coupled receptors (GPCR), ion channels, kinase, and protease, which comprise a large fraction of the targets of the approved and investigational drugs.62 GPCRs play prominent roles in many physiological processes, which makes them ideal targets to be regulated by small molecule drugs.63 Owing to the key roles in many bioprocesses that involve rapid changes in cells, ion channels are another type of popular drug targets for pharmacological intervention. Those bioprocesses include cardiac, skeletal, and smooth muscle contraction, T-cell activation, epithelial transport of nutrients and ions, and pancreatic beta-cell insulin release.64 In many cellular processes, such as cell cycle progression and signal transduction, kinases play a key role.65 Proteases play an important role in many signaling pathways, and they are the prominent modulators of many diseases, such as cardiovascular disorders and cancer. Proteases are also drug targets for combating many parasites and viruses.66
The members of the four prominent target classes were determined using multiple UniProt sources. First, the <keyword> child elements of each protein entry in the UniProt XML file were identified. A protein was determined to be a GPCR if the “id” attribute of a <keyword> element was “KW-0297”. Similarly, the protein belonged to the ion channel family if the “id” attribute was either KW-0107, KW-0407, KW-0631, KW-0851, KW-0869, KW-0894, or KW-1071; the protein belonged to the kinase family if the “id” attribute was either KW-0418, KW-0723, or KW-0829; and the protein was a member of protease family if the “id” attribute was either KW-0031, KW-0064, KW-0121, KW-0224, KW-0482, KW-0645, KW-0720, KW-0788, or KW-0888.
Second, a protein was also recognized as a GPCR if it was listed as a 7- transmembrane G-linked receptor (http://www.uniprot.org/docs/7tmrlist). Similarly, a protein was determined to be a kinase if its name appears in http://www.uniprot.org/docs/pkinfam, or a protease, if its name shows in http://www.uniprot.org/docs/peptidas.
Screening Library Construction.
The screening libraries were compiled from the druglike data set of the ZINC database.28 In total, 18 855 206 entries were downloaded. All the ZINC entries obey the Lipinski’s Rule of 5 without any violation. Then the FP2 fingerprints were generated using the OpenBabel software.67 2D-similarity searches were performed using drug molecules as queries. A ZINC molecule was recognized as a hit when the Tanimoto coefficient between its FP2 fingerprint and any drug molecule’s FP2 fingerprint was equal to or larger than a threshold. A drug molecule belongs to one of the three categories: approved drug (AD, 1596), investigational drug (ID, 486), and experimental drug (ED, 4437). For each drug category, the number in the parentheses is the number of drugs for that category. The numbers of drugs belonging to the four major drug target classes are listed in Table 1. In this work, a series of structural similarity (SS) thresholds, 85, 80, 75, 70, 65, 60, 55, and 50%, were applied to prioritize the screening libraries.
Table 1.
Summary on the Total Entries of Drugs, Targets, and Drug–Target Pairs for All and Four Major Drug Target Classes
| all | AD | ID | ED | ||
|---|---|---|---|---|---|
| all | no. drugs | 6248 | 1596 | 486 | 4437 |
| no. targets | 4046 | 1858 | 774 | 2632 | |
| no. D-T pairs | 19057 | 10387 | 2492 | 7679 | |
| GPCR | no. drugs | 559 | 474 | 135 | 35 |
| no. targets | 117 | 111 | 83 | 25 | |
| no. D-T pairs | 1920 | 1759 | 426 | 53 | |
| ion channel | no. drugs | 407 | 277 | 55 | 103 |
| no. targets | 206 | 162 | 84 | 66 | |
| no. D-T pairs | 2075 | 1794 | 234 | 165 | |
| protease | no. drugs | 1091 | 125 | 36 | 947 |
| no. targets | 246 | 93 | 60 | 193 | |
| no. D-T pairs | 1388 | 237 | 78 | 1125 | |
| kinase | no. drugs | 758 | 99 | 43 | 644 |
| no. targets | 267 | 124 | 58 | 202 | |
| no. D-T pairs | 1143 | 275 | 101 | 846 |
Evaluation of VS Studies.
The success of VS is determined by the ability of VS filters to distinguish actives from inactives through ranking compounds.68 The ideal performance of VS is that all actives but not one inactive was selected, which is basically impractical. The performance of a VS is evaluated using a set of metrics and statistical parameters including hit rate (HR), enrichment factor (EF), true positive rate (TPR), false positive rate (FPR), false discovery rate (FDR), receiver operator characterization (ROC), and the resulting area under the curve. Other parameters, such as precision, recall, accuracy, F1 score, and Matthews correlation coefficient (MCC) are also applied to evaluate the performance of virtual screenings.69 The two most important metrics of evaluating the performance of a VS, HR, and EF, are defined below. Hit percentage (HP) is applied to measure how well a VS filter condense a screening library.
| (1) |
| (2) |
| (3) |
where M is known actives and m is known actives being selected as hits. N and n are total numbers of compounds in a screening library and number of hits, respectively. Unlike HP, HR and EF are calculated using known actives. HR measures the ratio of known actives being selected as hits. EF measures the ratio of the probability of selecting a true active from hits to the probability of selecting a true active randomly from the whole database. With HR and EF, one can calculate the hit rate for a random screening library of the same size: HRrandom = HR/EF.
Critical Evaluation of the Screening Libraries.
The developed screening libraries were assessed by a set of test data sets of bioactives. All the bioactives, which are more potent than 10 μM against at least one target, were extracted from the target subset of the ZINC database.28 The entries which are duplicated with any drug molecules were eliminated from the test data sets.
3. RESULTS
Drug–Target Interaction Analysis.
There are 7874 drug entries and 4704 drug targets in the Drug Bank as of May 2017. In total, 20 426 drug–target pairs were collected and 6854 drug entries and 4277 drug targets were involved. After the further elimination of drugs not having structures, 6248 drugs were left which cover 4046 drug targets. The total number of DTIs drops to 19 057. On average, each drug corresponds to 3.0 drug targets and each drug target corresponds to 4.7 drugs. The total numbers of drugs, targets, and drug–target pairs for the approved, investigational, and experimental drugs are summarized in Table 1.
The total number of approved drugs of the four major drug target classes is 975, which accounts for 61% of all the approved drugs. This percentile slightly drops to 55% in the investigational drugs, and it further drops to 39% in the experimental drugs. It is worthy to point out that the number of ED targeting proteases and kinases dramatically outnumber the numbers of AD and ID, reflecting the fact that proteases and kinases are becoming hot drug targets today. It is pointed out that there are some overlaps between the drugs of the four different classes. For the GPCR category, there are 85, 223, and 10 drugs overlapped with the ion channel, protease, and kinase categories, respectively. The numbers of overlapped drugs are 71, 16, and 18 between ion channel/protease, ion channel/kinase, and protease/kinase, respectively. When only approved drugs are considered, the numbers of overlapped drugs are much fewer: there are 77, 3, 7, 6, 7, and 4 drugs overlapped between GPCR/ion channel, GPCR/protease, GPCR/kinase, ion channel/protease, ion channel/kinase, and protease/kinase, correspondingly.
Useful information on the drug discovery trends can be revealed by studying Table 1 and Figure 1. On average every GPCR target has 16.4 drugs, and every ion channel target has 10.1 drugs, both are significantly more than the average of 4.7 for all drug target classes. Interestingly, the number of experimental GPCR drugs, 35, is much smaller than that of investigational GPCR drugs. On the contrary, there are much more experimental than investigational drugs for the protease and kinase drug targets.
Figure 1.

Summary of drug–target interaction. (A) Number of targets per drug. (B) Number of drugs per target.
The number of targets a drug molecule has is quite different for the approved, investigational, and experimental drugs, as demonstrated in Figure 1A. As to specific target classes, those numbers do not share common patterns at all. The protease and kinase drugs have fewer drug targets than the GPCR and ion channel drugs.
As suggested by Table 1 and Figure 1, a drug molecule is likely associated with many drug targets; therefore, it is a good idea to identify those drug molecules that may interact with the drug target in study. Moreover, more potential drug leads may be identified from screening libraries which collect structurally similar compounds to known drugs. Constructing a set of prioritized druglike screening libraries is the objective of this work.
Screening Library Construction.
It is a dogma that the properties of a molecule are determined by its structure, and as such, similar structures tend to bear similar properties.70 This similarity principle is the basis of the similarity searches. The higher the similarity between two compounds, the higher the chance the two compounds share the similar bioactivities for a drug target.
The performance of fingerprint-based screenings is summarized in Table 2. The efficiency of a virtual screening, which is measured by HP, strongly depends on the SS threshold. With the SS threshold of 85%, only less than 1% of entries were selected; while the efficiency is dropped to 50% (only 50% of entries were filtered out) with the SS threshold of 50%. In the following text, we labeled the screening libraries using the threshold; for example, SS85 is the screening library retrieved using the SS threshold of 85%.
Table 2.
List of the Entry Numbers and Hit Percentages (%) of General-Purpose Druglike Data Sets
| drug | AD | AD_ID | ED | |||||
|---|---|---|---|---|---|---|---|---|
| SS | no. entries | HP | no. entries | HP | no. entries | HP | no. entries | HP |
| 85 | 123192 | 0.7 | 50220 | 0.3 | 51602 | 0.3 | 99042 | 0.5 |
| 80 | 255861 | 1.4 | 95044 | 0.5 | 99014 | 0.5 | 204480 | 1.1 |
| 75 | 529276 | 2.8 | 199205 | 1.1 | 210071 | 1.1 | 407909 | 2.2 |
| 70 | 1096003 | 5.8 | 448688 | 2.4 | 479925 | 2.5 | 833300 | 4.4 |
| 65 | 2165864 | 11.5 | 967861 | 5.1 | 1051010 | 5.6 | 1648630 | 8.7 |
| 60 | 3949711 | 20.9 | 1923581 | 10.2 | 2109277 | 11.2 | 3098326 | 16.4 |
| 55 | 6593073 | 34.9 | 3570446 | 18.9 | 3888542 | 20.6 | 5511034 | 29.2 |
| 50 | 9560618 | 50.7 | 5883362 | 31.2 | 6288146 | 33.3 | 8513983 | 45.1 |
With the continually increased computer power, it is practical to screen a database with 200 000 or more entries using regular docking filters. Taking the docking program of Glide as an example,3 Glide can screen 10 000 to 20 000 molecules for a typical drug target with one CPU core using the standard precision scoring function. As our screening libraries are prioritized, one should always start from SS85, followed by SS80, SS75, and so on.
Besides the general-purpose screening libraries, target class-specific screening libraries were also constructed. It is not a surprise that the screening efficiencies are much higher (smaller HP values) for all four drug target classes (Table 3).
Table 3.
List of the Entry Numbers and Hit Percentages (%) of Target-Specific Druglike Datasets
| GPCR | ion | kinase | protease | |||||
|---|---|---|---|---|---|---|---|---|
| SS | no. entries | HP | no. entries | HP | no. entries | HP | no. entries | HP |
| 85 | 25595 | 0.1 | 22000 | 0.1 | 9573 | 0.1 | 40101 | 0.2 |
| 80 | 44673 | 0.2 | 42126 | 0.2 | 17677 | 0.1 | 84264 | 0.4 |
| 75 | 96741 | 0.5 | 94513 | 0.5 | 36471 | 0.2 | 177168 | 0.9 |
| 70 | 231963 | 1.2 | 233155 | 1.2 | 83746 | 0.4 | 376400 | 2.0 |
| 65 | 557328 | 3.0 | 548882 | 2.9 | 197394 | 1.0 | 801259 | 4.2 |
| 60 | 1234291 | 6.5 | 1169750 | 6.2 | 467769 | 2.5 | 1661329 | 8.8 |
| 55 | 2505668 | 13.3 | 2363312 | 12.5 | 1161054 | 6.2 | 3293679 | 17.5 |
| 50 | 4293284 | 22.8 | 4177077 | 22.1 | 2676629 | 14.2 | 5733633 | 30.4 |
The average and maximum hit numbers of all the fingerprint-based screenings are listed in Tables S1–S4. Studying those tables helps us estimate how many hits we expect to retrieve for a given SS threshold. There are some orphan drug molecules which are not well represented in the ZINC screening library since zero hit was retrieved for them even using a very low SS threshold of 50%. The percentages of zero-hit drugs are listed in Table S5. Other drugs have hits from one to thousands. We allocated the drugs into 17 groups which have hits of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11–20, 21–50, 51–100, 101–200, 201–500, 501–1000, and 1001 and above. The distributions of drugs among the 17 groups are demonstrated in Figures 2–5 for different SS thresholds. Although the distribution patterns are different from one SS threshold to another and from one drug category to another, the most abundant groups are 11–20, 21–50, and 51–100 hits when SS thresholds are larger than 70%. The distributions of drugs among the 17 groups for SS thresholds of 65, 60, 55, and 50% are demonstrated in Figures S1–S4.
Figure 2.

Distributions of drug molecules (%) in 17 hit number groups. A molecule of the ZINC druglike data set is recognized as a hit if its 2D-similarity score against the query drug molecule is equal to or better than 0.85.
Figure 5.

Distributions of drug molecules (%) in 17 hit number groups. A molecule of the ZINC druglike data set is recognized as a hit if its 2D-similarity score against the query drug molecule is equal to or better than 0.70.
Critical Evaluation of the Screening Libraries.
In total, we collected 5847 bioactives and most of them were tested on human drug targets. Subsets of the bioactives targeting at GPCR, ion channel, kinase, and protease were compiled. Interestingly, only four compounds targeting proteases were left after the removal of the duplicated entries. Details on the composition of the test data sets are provided in Table S6.
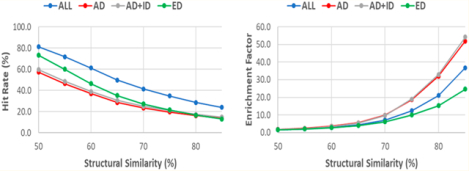
Table 4 lists the HR and EF for four drug sets using eight SS thresholds. The relationships between hit rates versus SS thresholds and enrichment factors versus SS thresholds are shown in Figure 6. The overall quality of the screening libraries is satisfactory. For example, the enrichment factors are 36.9, 53.5 and 55.5, 24.8 for SS85 of drug (all drugs), AD (approved drugs), AD_ID (approved and investigational drugs), and ED (experimental drugs), respectively. The corresponding hit rates are 24.1, 14.2, 15.2, and 13.0% for the four SS85 sets. For the drug SS75 set, although EF is dropped to 12.9, HR increases to 36.1%. To seek a balance between HR and EF, we would recommend drug SS75 and AD SS70 (HR = 25.4, EF = 10.7) for virtual screening studies. The HR and EF of the screening libraries against “Human”, a subset which collects 4078 actives of human drug targets, are listed in Table S7.
Table 4.
Performance of Druglike Datasets against an External Active Set with 5847 Entries
| drag | AD | AD_ID | ED | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SS | no. hits | HR | EF | no. hits | HR | EF | no. hits | HR | EF | no. hits | HR | EF |
| 85 | 1409 | 24.1 | 36.9 | 832 | 14.2 | 53.5 | 886 | 15.2 | 55.4 | 761 | 13.0 | 24.8 |
| 80 | 1712 | 29.3 | 21.6 | 996 | 17.0 | 33.8 | 1066 | 18.2 | 34.7 | 982 | 16.8 | 15.5 |
| 75 | 2109 | 36.1 | 12.9 | 1229 | 21.0 | 19.9 | 1329 | 22.7 | 20.4 | 1289 | 22.0 | 10.2 |
| 70 | 2508 | 42.9 | 7.4 | 1486 | 25.4 | 10.7 | 1586 | 27.1 | 10.7 | 1624 | 27.8 | 6.3 |
| 65 | 3059 | 52.3 | 4.6 | 1867 | 31.9 | 6.2 | 1975 | 33.8 | 6.1 | 2153 | 36.8 | 4.2 |
| 60 | 3715 | 63.5 | 3.0 | 2350 | 40.2 | 3.9 | 2462 | 42.1 | 3.8 | 2843 | 48.6 | 3.0 |
| 55 | 4346 | 74.3 | 2.1 | 2926 | 50.0 | 2.6 | 3051 | 52.2 | 2.5 | 3652 | 62.5 | 2.1 |
| 50 | 4865 | 83.2 | 1.6 | 3540 | 60.5 | 1.9 | 3671 | 62.8 | 1.9 | 4398 | 75.2 | 1.7 |
Figure 6.

Performance of virtual screenings for external actives (5847 entries) against screening libraries. (A) Hit rates. (B) Enrichment factors.
The screening performance for specific drug target classes was also investigated (Tables S8–S11). The HR versus SS threshold plots are shown in Figure 7, and the EF vs SS threshold plots are shown in Figure 8. Although the target class-specific screening libraries usually have larger enrichment factors (Figure 8), the hit rates are lower than the general-purpose screening libraries (Figure 7), such as Drug SS75 and AD SS70. The striking differences between the general-purpose and target-class-specific screening libraries occur when SS thresholds are larger than 0.8. It is also noted that the patterns of HR ~ SS threshold and EF ~ threshold plots may not be representative for the protease class, as there are only four actives of protease targets left after those overlapping with the known drugs are excluded.
Figure 7.

Hit rates of virtual screenings for external target-specific active sets against general-purpose and target-class-specific screening libraries. (A) GPCR. (B) Ion channel. (C) Kinase. (D) Protease.
Figure 8.

Enrichment factors of virtual screenings for external target-specific active sets against general-purpose and target-class-specific screening libraries. (A) GPCR. (B) Ion channel. (C) Kinase. (D) Protease.
4. DISCUSSION
Drug–Target Interaction Prediction.
The above drug–target interaction analysis was based on the experimentally confirmed drug–target interactions. It is of great interest to identify unknown drug-target interactions. In silico prediction of DTI promotes drug repurposing and facilitates us to explore the potential side effects of a drug due to multiple drug–target interactions. DTIs also provide insights about potential drug–drug interactions. Recently, Wen et al. developed a deep-learning-based method to predict drug-target interactions.71 Using both the extended connectivity fingerprints of drugs and the protein sequence composition (the protein sequence compositions consist of amino acid composition, dipeptide composition, and tripeptide composition) of drug targets as descriptors, they built up a deep-belief-network (DBN) model which outperforms the other state-of-the-art methods. The DBN model can be further applied to predict whether a new target interacts with some existing drugs or whether a drug molecule can bind to some existing targets.
We developed an online tool, TargetHunter, to identify potential drug targets for a chemical compound.72 The basic idea is to predict ligand–target interactions by comparing a query compound with bioactives for which their drug targets are well documented. The bioactive database TargetHunter explored is ChEMBL,73 which collects millions of bioactivity data covering more than 8000 drug targets.72 An algorithm, called TAMOSIC (targets associated with its most similar counterparts) was developed by us to assign the targets which are associated with the compounds most similar to a query compound as the potential drug targets of the query compound.72 The case studies demonstrated that Target-Hunter was a promising technique for new target identification or repurposing drugs.
Further Development on Screening Library Construction.
Screening library construction plays a critical role in both HTS experiments and virtual screenings. There are two types of screening libraries, diverse screening libraries for drug lead identification and focused libraries for drug lead optimization. The former may not be druglike as they are compiled to maximize the structure diversity or molecular property diversity. The target-focused library using a limit number of known actives might not be able to cover other potential hits. On the contrary, the druglike data sets constructed in this work have the following advantages. First of all, those screening libraries are druglike as they are enriched by known drugs; second, those screening libraries, although belong to focused libraries, have considerable diversity as they are enriched using all kinds of known drugs. Therefore, the screening libraries developed in this work are general-purpose screening libraries which can be applied in both drug lead identification and optimization for arbitrary drug targets. With our screening libraries, one may identify more actives from VS for the target in study, as the molecules enriched by the drugs of the other drug targets may be potential hits of the drug target in study, as revealed by the complicate DTIs between drugs and drug targets. The advantage of our general-purpose screening libraries is even more obvious for new drug targets for which there are no or only few known actives.
Although the screening libraries constructed in this work are mainly for VS studies, they may provide useful hints on compound library construction for experimental HTS because of the following reasons. First, all the entries of the screening libraries are commercially available; second, the compounds of screening libraries (especially Drug SS80 and the AD SS75) are druglike; last but not least, a compound library for experimental HTS is usually constructed for multiple drug discovery projects.
Certainly, it is worthwhile to continue to improve the enrichment factors of the screening data sets. We plan to further condense the screening data sets by removing those that are least druglike. The drug likeness score of a compound is a function of both molecular properties (aqueous solubility, human oral bio availability, human intestinal absorption, plasma protein binding, and other ADME and pharmacokinetic properties) and druglike fingerprints described by the building blocks of drugs and bioactives. Deng et al. proposed to use biological relevance to assemble screening libraries.69 The biological relevance score will also become a component of our drug likeness score. We are in the process of developing such a kind of druglike function.
The opposite trend is to include more compounds into the screening libraries. Natural products could be applied as queries to recruit compounds in the ZINC database. Gu et al. found that there was a large portion of overlap in chemical space between FDA-approved drugs and natural products.74 Therefore, we are planning to compile natural product-like screening libraries.
Wenlock et al. identified the trends in physio chemical properties that likely lead to a drug successful passes through clinical development and go to the market.75 Those physio chemical properties include aqueous solubility, logP (logarithm of octanol–water partition coefficient), molecular weight, numbers of hydrogen donors and acceptors, and so on. The drug likeness of a compound can be further measured by its ADME properties. Among the many ADME/PK properties, oral bio availability is particularly important for the orally administered drugs26,76–78
Besides the molecular properties, molecular scaffolds are widely used to measure the druglike score of a compound. We have developed a brutal-force algorithm to enumerate all the possible building blocks of a given molecule. Druglike scaffolds and fragments were then identified and assigned drug likeness scores.26 Bemis and Murcko identified the molecular frameworks that are frequently found in the drug molecules of the Comprehensive Medicinal Chemistry (CMC) database.79 It is possible to build a drug likeness fingerprint using the results of building block analysis to facilitate screening library construction. We recently performed a large-scale analysis on the interaction of ligand fragments with 20 ammonia acids.80 First, the small molecule binders of protein targets were extracted and dissected into fragments. Then, LigFrag-RPM, an algorithm matching the preference of ligand fragments and amino acid residues, was developed by comparing the profiles of the interactions between ligand fragments and the 20 proteinogenic amino acid residues. The LigFrag-RMP algorithm could serve as a filter to remove ineligible compounds in the screening libraries for a given protein target. In addition, the recent construction of an allosteric ligand fragment library by us provides promising venue to design functional allosteric modulators for GPCRs.81
5. CONCLUSION
In this work, a set of druglike data sets for virtual screenings have been constructed. These data sets have been prioritized and critically evaluated using a large data set of bioactives. Some data sets, particularly drug SS80 and AD SS75 are highly recommended to serve as general purpose screening libraries. These data sets can be used for both drug lead identification and drug lead optimization.
Supplementary Material
Figure 3.

Distributions of drug molecules (%) in 17 hit number groups. A molecule of the ZINC druglike data set is recognized as a hit if its 2D-similarity score against the query drug molecule is equal to or better than 0.80.
Figure 4.

Distributions of drug molecules (%) in 17 hit number groups. A molecule of the ZINC druglike data set is recognized as a hit if its 2D-similarity score against the query drug molecule is equal to or better than 0.75.
ACKNOWLEDGMENTS
We thank the NIH (R01GM79383, J.W.; 1R21GM097617–01, J.W.; P30 DA035778–01A1, X.-Q.X.; DOD W81XWH-16-1-0490:412288, X.-Q.X.) for research support.
ABBREVIATIONS USED
- AD
approved drugs
- AD_ID
approved and investigational drugs
- ADME
absorption, distribution, metabolism, excretion
- CAP
compound library acquisition and prioritization
- DTI
drug–target interaction
- ED
experimental drugs
- EF
enrichment factor
- FANN
fingerprint-based artificial neural network
- FDR
false discovery rate
- FPR
false positive rate
- HR
hit rate
- HTS
high throughput screening
- HP
hit percentage
- GPCR
G-protein coupled receptors
- ID
investigational drugs
- MCC
Matthews correlation coefficient
- MM/GBSA
molecular mechanics/generalized Born surface area
- MM/PBSA
molecular mechanics/Poisson–Boltzmann surface area
- PMV
polar molecular volume
- PSA
polar surface area
- QSAR
quantitative structure–activity relationship
- ROC
receiver operator characterization
- SOM
self-organizing map
- SS
structural similarity
- TPR
true positive rate
- TTD
therapeutic target database
- USR
ultrafast shape recognition
- VS
virtual screening
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.8b00537.
Tables S1–S11 and Figures S1–S4 mentioned in the text (PDF)
The authors declare no competing financial interest.
The screening libraries developed in this work are available to download from https://mulan.pharmacy.pitt.edu.
REFERENCES
- (1).Wang J; Kang X; Kuntz ID; Kollman PA Hierarchical database screenings for HIV-1 reverse transcriptase using a pharmacophore model, rigid docking, solvation docking, and MM-PB/SA. J. Med. Chem 2005, 48, 2432–2444. [DOI] [PubMed] [Google Scholar]
- (2).Kumar A; Zhang KYJ Hierarchical virtual screening approaches in small molecule drug discovery. Methods 2015, 71, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (4).Wang J; Hou T; Xu X Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Curr. Comput.-Aided Drug Des. 2006, 2, 287–306. [Google Scholar]
- (5).Kollman PA; Massova I; Reyes C; Kuhn B; Huo S; Chong L; Lee M; Lee T; Duan Y; Wang W; Donini O; Cieplak P; Srinivasan J; Case DA; Cheatham TE 3rd Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res 2000, 33, 889–897. [DOI] [PubMed] [Google Scholar]
- (6).Hou T; Wang J; Li Y; Wang W Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model 2011, 51, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kuhn B; Gerber P; Schulz-Gasch T; Stahl M Validation and use of the MM-PBSA approach for drug discovery. J. Med. Chem 2005, 48, 4040–4048. [DOI] [PubMed] [Google Scholar]
- (8).Page CS; Bates PA Can MM-PBSA calculations predict the specificities of protein kinase inhibitors? J. Comput. Chem 2006, 27, 1990–2007. [DOI] [PubMed] [Google Scholar]
- (9).Pirhadi S; Shiri F; Ghasemi JB Methods and Applications of Structure Based Pharmacophores in Drug Discovery. Curr. Top. Med. Chem 2013, 13, 1036–1047. [DOI] [PubMed] [Google Scholar]
- (10).Kutchukian PS; Shakhnovich EI De novo design: balancing novelty and confined chemical space. Expert Opin. Drug Discovery 2010, 5, 789–812. [DOI] [PubMed] [Google Scholar]
- (11).Maggiora G; Vogt M; Stumpfe D; Bajorath J Molecular Similarity in Medicinal Chemistry. J. Med. Chem 2014, 57, 3186–3204. [DOI] [PubMed] [Google Scholar]
- (12).Ehrlich HC; Henzler AM; Rarey M Searching for Recursively Defined Generic Chemical Patterns in Nonenumerated Fragment Spaces. J. Chem. Inf. Model 2013, 53, 1676–1688. [DOI] [PubMed] [Google Scholar]
- (13).Ivanciuc O Chemical Graphs, Molecular Matrices and Topological Indices in Chemoinformatics and Quantitative Structure-Activity Relationships. Curr. Comput.-Aided Drug Des 2013, 9, 153–163. [DOI] [PubMed] [Google Scholar]
- (14).Rogers D; Hahn M Extended-Connectivity Fingerprints. J. Chem. Inf. Model 2010, 50, 742–754. [DOI] [PubMed] [Google Scholar]
- (15).Hawkins PCD; Skillman AG; Nicholls A Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem 2007, 50, 74–82. [DOI] [PubMed] [Google Scholar]
- (16).Berenger F; Voet A; Lee XY; Zhang KY J. A rotation-translation invariant molecular descriptor of partial charges and its use in ligand-based virtual screening. J. Cheminf 2014, DOI: 10.1186/1758-2946-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Leelananda SP; Lindert S Computational methods in drug discovery. Beilstein J. Org. Chem 2016, 12, 2694–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Caporuscio F; Tafi A Pharmacophore Modelling: A Forty Year Old Approach and its Modern Synergies. Curr. Med. Chem 2011, 18, 2543–2553. [DOI] [PubMed] [Google Scholar]
- (19).Li HJ; Leung KS; Wong MH; Ballester PJ USR-VS: a web server for large-scale prospective virtual screening using ultrafast shape recognition techniques. Nucleic Acids Res 2016, 44, W436–W441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Myint KZ; Xie XQ Ligand biological activity predictions using fingerprint-based artificial neural networks (FANN-QSAR). Methods Mol. Biol 2015, 1260, 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kelder J; Grootenhuis PDJ; Bayada DM; Delbressine LPC; Ploemen JP Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res 1999, 16, 1514–1519. [DOI] [PubMed] [Google Scholar]
- (22).Zhang L; Fourches D; Sedykh A; Zhu H; Golbraikh A; Ekins S; Clark J; Connelly MC; Sigal M; Hodges D; Guiguemde A; Guy RK; Tropsha A Discovery of novel antimalarial compounds enabled by QSAR-based virtual screening. J. Chem. Inf. Model 2013, 53, 475–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lu P; Wang Y; Ouyang P; She J; He M 3D-QSAR Based Pharmacophore Modeling and Virtual Screening for Identification of Novel G Protein-Coupled Receptor40 Agonists. Curr. Comput.-Aided Drug Des 2015, 11, 51–56. [DOI] [PubMed] [Google Scholar]
- (24).Liu Y; Huang L; Ye H; Lv X Combined QSAR-based virtual screening and fluorescence binding assay to identify natural product mediators of Interferon Regulatory Factor 7 (IRF-7) in pulmonary infection. SAR and QSAR in environmental research 2016, 27, 939. [DOI] [PubMed] [Google Scholar]
- (25).Neves BJ; Melo-Filho CC; Moreira Filho JT; Muratov EN; Andrade CH; Braga RC QSAR-based Virtual Screening: Advances and Applications in Drug Discovery Front. Front. Pharmacol 2018, 9, 1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang J; Hou T Drug and drug candidate building block analysis. J. Chem. Inf. Model 2010, 50, 55–67. [DOI] [PubMed] [Google Scholar]
- (27).Dobson CM Chemical space and biology. Nature 2004, 432, 824–828. [DOI] [PubMed] [Google Scholar]
- (28).Irwin JJ; Sterling T; Mysinger MM; Bolstad ES; Coleman RG ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- (30).Wang Y; Cheng T; Bryant SH PubChem BioAssay: A Decade’s Development toward Open High-Throughput Screening Data Sharing. SLAS Discov 2017, 22, 655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Xie XQS Exploiting PubChem for virtual screening. Expert Opin. Drug Discovery 2010, 5, 1205–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Voigt JH; Bienfait B; Wang S; Nicklaus MC Comparison of the NCI open database with seven large chemical structural databases. J. Chem. Inf. Comput Sci 2001, 41, 702–712. [DOI] [PubMed] [Google Scholar]
- (33).Gillet VJ New directions in library design and analysis. Curr. Opin. Chem. Biol 2008, 12, 372–378. [DOI] [PubMed] [Google Scholar]
- (34).Koutsoukas A; Paricharak S; Galloway WRJD; Spring DR; IJzerman AP; Glen RC; Marcus D; Bender A How Diverse Are Diversity Assessment Methods? A Comparative Analysis and Benchmarking of Molecular Descriptor Space. J. Chem. Inf. Model 2014, 54, 230–242. [DOI] [PubMed] [Google Scholar]
- (35).Bender A; Jenkins JL; Glick M; Deng Z; Nettles JH; Davies JW Bayes affinity fingerprints″ improve retrieval rates in virtual screening and define orthogonal bioactivity space: When are multitarget drugs a feasible concept? J. Chem. Inf. Model 2006, 46, 2445–2456. [DOI] [PubMed] [Google Scholar]
- (36).Gorse AD Diversity in medicinal chemistry space. Curr. Top. Med. Chem 2006, 6, 3–18. [DOI] [PubMed] [Google Scholar]
- (37).Ma C; Lazo JS; Xie XQ Compound acquisition and prioritization algorithm for constructing structurally diverse compound libraries. ACS Comb. Sci 2011, 13, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Horvath D; Lisurek M; Rupp B; Kuhne R; Specker E; von Kries J; Rognan D; Andersson CD; Almqvist F; Elofsson M; Enqvist PA; Gustavsson AL; Remez N; Mestres J; Marcou G; Varnek A; Hibert M; Quintana J; Frank R Design of a General-Purpose European Compound Screening Library for EU-OPENSCREEN. ChemMedChem 2014, 9, 2309–2326. [DOI] [PubMed] [Google Scholar]
- (39).Pirard B; Pickett SD Classification of kinase inhibitors using BCUT descriptors. J. Chem. Inf. Comput. Sci 2000, 40, 1431–1440. [DOI] [PubMed] [Google Scholar]
- (40).Schneider P; Tanrikulu Y; Schneider G Self-Organizing Maps in Drug Discovery: Compound Library Design, Scaffold-Hopping, Repurposing. Curr. Med. Chem 2009, 16, 258–266. [DOI] [PubMed] [Google Scholar]
- (41).Noeske T; Sasse BC; Stark H; Parsons CG; Weil T; Schneider G Predicting compound selectivity by self-organizing maps: Cross-activities of metabotropic glutamate receptor antagonists. ChemMedChem 2006, 1, 1066–1068. [DOI] [PubMed] [Google Scholar]
- (42).Naderi M; Alvin C; Ding Y; Mukhopadhyay S; Brylinski M A graph-based approach to construct target-focused libraries for virtual screening. J. Cheminf 2016, 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sukuru SCK; Jenkins JL; Beckwith REJ; Scheiber J; Bender A; Mikhailov D; Davies JW; Glick M Plate-Based Diversity Selection Based on Empirical HTS Data to Enhance the Number of Hits and Their Chemical Diversity. J. Biomol. Screening 2009, 14, 690–699. [DOI] [PubMed] [Google Scholar]
- (44).Mok NY; Brenk R Mining the ChEMBL Database: An Efficient Chemoinformatics Workflow for Assembling an Ion Channel-Focused Screening Library. J. Chem. Inf. Model 2011, 51, 2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Perez-Regidor L; Zarioh M; Ortega L; Martin-Santamaria S Virtual Screening Approaches towards the Discovery of Toll-Like Receptor Modulators. Int. J. Mol. Sci 2016, 17, 1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Senderowitz H; Marantz YG Protein-Coupled Receptors: Target-Based In Silico Screening. Curr. Pharm. Des 2009, 15, 4049–4068. [DOI] [PubMed] [Google Scholar]
- (47).Istyastono EP; Kooistra AJ; Vischer HF; Kuijer M; Roumen L; Nijmeijer S; Smits RA; de Esch IJP; Leurs R; de Graaf C Structure-based virtual screening for fragment-like ligands of the G protein-coupled histamine H-4 receptor. MedChemComm 2015, 6, 1003–1017. [Google Scholar]
- (48).Ballester PJ; Mangold M; Howard NI; Robinson RLM; Abell C; Blumberger J; Mitchell JBO Hierarchical virtual screening for the discovery of new molecular scaffolds in antibacterial hit identification. J. R. Soc., Interface 2012, 9, 3196–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gao Y; Yang P; Shen H; Yu H; Song X; Zhang L; Zhang P; Cheng H; Xie Z; Hao S; Dong F; Ma S; Ji Q; Bartlow P; Ding Y; Wang L; Liu H; Li Y; Cheng H; Miao W; Yuan W; Yuan Y; Cheng T; Xie XQ Small-molecule inhibitors targeting INK4 protein p18(INK4C) enhance ex vivo expansion of haematopoietic stem cells. Nat. Commun 2015, 6, 6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Feng ZW; Pearce LV; Xu XM; Yang XL; Yang P; Blumberg PM; Xie XQ Structural Insight into Tetrameric hTRPV1 from Homology Modeling, Molecular Docking, Molecular Dynamics Simulation, Virtual Screening, and Bioassay Validations. J. Chem. Inf. Model 2015, 55, 572–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Xu X; Zhang W; Huang C; Li Y; Yu H; Wang Y; Duan J; Ling Y A novel chemometric method for the prediction of human oral bioavailability. Int. J. Mol. Sci 2012, 13, 6964–6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Glaab E Building a virtual ligand screening pipeline using free software: a survey. Briefings Bioinf. 2016, 17, 352–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Rognan D Development and virtual screening of target libraries. J. Physiol 2006, 99, 232–244. [DOI] [PubMed] [Google Scholar]
- (54).Gilad Y; Nadassy K; Senderowitz H A reliable computational workflow for the selection of optimal screening libraries. J. Cheminf 2015, DOI: 10.1186/s13321-015-0108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kitchen DB; Decornez H; Furr JR; Bajorath J Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discovery 2004, 3, 935–949. [DOI] [PubMed] [Google Scholar]
- (56).Bissantz C; Folkers G; Rognan D Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J. Med. Chem 2000, 43, 4759–4767. [DOI] [PubMed] [Google Scholar]
- (57).Chaput L; Martinez-Sanz J; Saettel N; Mouawad L Benchmark of four popular virtual screening programs: construction of the active/decoy dataset remains a major determinant of measured performance. J. Cheminf 2016, 8, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wallach I; Jaitly N; Nguyen K; Schapira M; Lilien R Normalizing Molecular Docking Rankings using Virtually Generated Decoys. J. Chem. Inf. Model 2011, 51, 1817–1830. [DOI] [PubMed] [Google Scholar]
- (59).Law V; Knox C; Djoumbou Y; Jewison T; Guo AC; Liu Y; Maciejewski A; Arndt D; Wilson M; Neveu V; Tang A; Gabriel G; Ly C; Adamjee S; Dame ZT; Han B; Zhou Y; Wishart DS DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Wishart DS; Knox C; Guo AC; Cheng D; Shrivastava S; Tzur D; Gautam B; Hassanali M DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Yang H; Qin C; Li YH; Tao L; Zhou J; Yu CY; Xu F; Chen Z; Zhu F; Chen YZ Therapeutic target database update 2016: enriched resource for bench to clinical drug target and targeted pathway information. Nucleic Acids Res. 2016, 44, D1069–D1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Bull SC; Doig AJ Properties of Protein Drug Target Classes. PLoS One 2015, 10, 0117955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Lane JR; Abdul-Ridha A; Canals M Regulation of G Protein-Coupled Receptors by Allosteric Ligands. ACS Chem. Neurosci 2013, 4, 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Bagal S; Brown AD; Cox PJ; Omoto K; Owen R; Pryde DC; Sidders B; Skerratt SE; Stevens EB; Storer RI; Swain NA. Ion Channels as Therapeutic Targets: A Drug Discovery Perspective. J. Med. Chem 2013, 56, 593–624. [DOI] [PubMed] [Google Scholar]
- (65).Manning G; Whyte DB; Martinez R; Hunter T; Sudarsanam S The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- (66).Drag M; Salvesen GS Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discovery 2010, 9, 690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Melville JL; Hirst JD TMACC: Interpretable correlation descriptors for quantitative structure-activity relationships. J. Chem. Inf. Model 2007, 47, 626–634. [DOI] [PubMed] [Google Scholar]
- (68).Wildman SA Approaches to Virtual Screening and Screening Library Selection. Curr. Pharm. Des 2013, 19, 4787–4796. [DOI] [PubMed] [Google Scholar]
- (69).Deng ZL; Du CX; Li X; Hu B; Kuang ZK; Wang R; Feng SY; Zhang HY; Kong DX Exploring the Biologically Relevant Chemical Space for Drug Discovery. J. Chem. Inf. Model 2013, 53, 2820–2828. [DOI] [PubMed] [Google Scholar]
- (70).Hendrickson JB Concepts and Applications of Molecular Similarity - Johnson,Ma, Maggiora,Gm. Science 1991, 252, 1189. [Google Scholar]
- (71).Wen M; Zhang ZM; Niu SY; Sha HZ; Yang RH; Yun YH; Lu HM Deep-Learning-Based Drug-Target Interaction Prediction. J. Proteome. Res 2017, 16, 1401–1409. [DOI] [PubMed] [Google Scholar]
- (72).Wang L; Ma C; Wipf P; Liu H; Su W; Xie XQ TargetHunter: an in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. 2013, 15, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Gaulton A; Bellis LJ; Bento AP; Chambers J; Davies M; Hersey A; Light Y; McGlinchey S; Michalovich D; Al-Lazikani B; Overington JP ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Gu JY; Gui YS; Chen LR; Yuan G; Lu HZ; Xu XJ Use of Natural Products as Chemical Library for Drug Discovery and Network Pharmacology. PLoS One 2013, 8, 0062839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Wenlock MC; Austin RP; Barton P; Davis AM; Leeson PD A comparison of physiochemical property profiles of development and marketed oral drugs. J. Med. Chem 2003, 46, 1250–1256. [DOI] [PubMed] [Google Scholar]
- (76).Hou T; Wang J; Zhang W; Wang W; Xu X Recent advances in computational prediction of drug absorption and permeability in drug discovery. Curr. Med. Chem 2006, 13, 2653–2667. [DOI] [PubMed] [Google Scholar]
- (77).Hou T; Wang J; Zhang W; Xu X ADME evaluation in drug discovery. 6. Can oral bio availability in humans be effectively predicted by simple molecular property-based rules? J. Chem. Inf. Model 2007, 47, 460–463. [DOI] [PubMed] [Google Scholar]
- (78).Hou T; Wang J; Zhang W; Xu X ADME evaluation in drug discovery. 7. Prediction of oral absorption by correlation and classification. J. Chem. Inf. Model 2007, 47, 208–218. [DOI] [PubMed] [Google Scholar]
- (79).Bemis GW; Murcko MA The properties of known drugs 0.1. Molecular frameworks. J. Med. Chem 1996, 39, 2887–2893. [DOI] [PubMed] [Google Scholar]
- (80).Wang L; Xie Z; Wipf P; Xie XQ Residue preference mapping of ligand fragments in the Protein Data Bank. J. Chem. Inf. Model 2011, 51, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Bian YM; Feng ZW; Yang P; Xie XQ Integrated In Silico Fragment-Based Drug Design: Case Study with Allosteric Modulators on Metabotropic Glutamate Receptor 5. AAPS J. 2017, 19, 1235–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.