

Abstract
Two-dimensional (2D) materials are a broad class of synthetic ultra-thin sheet-like solids whose rapid pace of development motivates systematic study of their biological effects and safe design. A challenge for this effort is the large number of new materials and their chemical diversity. Recent work suggests that many 2D materials will be thermodynamically unstable and thus non-persistent in biological environments. Such information could inform and accelerate safety assessment, but experimental data to confirm the thermodynamic predictions is lacking. Here we propose a framework for early hazard screening of nanosheet materials based on biodissolution studies in reactive media, specially chosen for each material to match chemically feasible degradation pathways. Simple dissolution and in vitro tests allow grouping of nanosheet materials into four classes: A, potentially biopersistent; B: slowly degradable (>24–48 hours); C, biosoluble with potentially hazardous degradation products; and D, biosoluble with low-hazard degradation products. The proposed framework is demonstrated through an experimental case study on MoO3 nanoribbons, which have a dual 2D / 1D morphology and have been reported to be stable in aqueous stock solutions. The nanoribbons are shown to undergo rapid dissolution in biological simulant fluids and in cell culture, where they elicit no adverse responses up to 100μg ml−1 dose. These results place MoO3 nanoribbons in Class D, and assigns them a low priority for further nanotoxicology testing. We anticipate use of this framework could accelerate the risk assessment for the large set of new powdered 2D nanosheet materials, and promote their safe design and commercialization.
Introduction
Two-dimensional (2D) materials are atomically-thin sheet-like solids, which are under active development in technologies that include catalysis,1 electronics,2 water treatment,3 and medical devices.4,5 As a class, 2D materials show enormous chemical diversity, and include, but are not limited to, metal chalcogenides, oxides, silicates and halides.6 The total number of materials prepared in the form of 2D nanosheets now exceeds 2506 and there is a pressing need for safe design strategies to specifically support this new branch of nanotechnology R&D. The most immediate need is for high throughput screening techniques to categorize hazard by class, and to prioritize a subset of the large 2D material family for more detailed study of biological responses. The approach to 2D materials may be informed by the large literature on the fate and toxicity of the more common 0D and 1D.7–13 Many types of nanomaterials chemically transform when introduced into complex environmental or biological systems14–17 leading to degradation, dissolution, or phase transformations such as oxidation or sulfidation.15 These transformations are typically media specific and can dramatically affect fate, uptake and toxicity.7,10–12 Incorporation of material transformation processes into biological uptake and toxicity models is ongoing,18 and will ultimately be required to adequately predict material fate.
Many of the emerging 2D materials have not been systematically studied in dilute aqueous suspensions relevant to environmental and biological exposures. A recent review6 used thermodynamic methods to estimate the intrinsic stability of 2D materials in such dilute aqueous suspensions, and proposed that most materials will be chemically unstable and ultimately non-persistent in the environment or in biological tissue.19 In order to reliably classify any of these new materials as biodegradable or persistent, it will be necessary to validate the thermodynamic predictions with experimental data and to characterize degradation kinetics and time-scales. It is particularly useful to identify materials that are rapidly degradable, as these will not induce biological responses through their characteristic 2D nanosheet geometry nor through other behaviors associated with the solid phase, such as surface redox activity. Instead, risks for such materials may be reasonably assessed on the basis of the chemical effects of their soluble dissolution or degradation products.
We therefore propose that dissolution screening be applied as a logical first step in the hazard assessment of emerging 2D materials. Such screening can identify and prioritize a smaller subset of high priority materials for full characterization of the solid properties relevant for biological responses, and thus reduce the total burden of nanotoxicity testing. Dissolution screening already plays an established and important role in hazard assessment for mineral fibers,20–22 which, in common with 2D materials, are a diverse set of 1D inorganic particles of unusual shape.
Recent literature has begun to address the stability of 2D materials under conditions relevant to environmental and biological effects.6,19,23–25 MoS2 has been experimentally confirmed to undergo oxidative dissolution,19,26 in agreement with thermodynamic predictions.6,19 Graphene-based materials are also thermodynamically unstable to oxidation, but like most other carbons show very slow oxidation kinetics, and have been reported to require reactive oxygen species or oxidative enzymes for degradation over laboratory time scales.27–30 General degradation mechanisms for 2D nanosheets may be oxidative, hydrolytic, or reductive,6,31 and careful consideration of this degradation chemistry will be important for selecting suitable simulant fluids and for classifying 2D materials based on abiotic stability.6 Most experimental studies to date have focused on oxidative degradation routes,6,19,27 but it is essential to also consider the other degradation modes.
A class of 2D materials resistant to oxidative degradation are metal oxides with high metallic oxidation states. An example is layered MoO3, which is being explored for applications that include antimicrobial agents in marine paints32,33 or medical devices6, lithium ion battery cathodes,34 chemotherapy agents,35 or smart chemochromic materials such as windows.36 Here we consider layered MoO3 nanoribbons as a case study in a dissolution screening framework for 2D materials. MoO3 is a layered material has a belt-like shape with approximate dimensions of 10μm (length) by 200 nm (width) by 10 nm (thickness).36 These dimensions indicate that MoO3 is a true 2D material, however the unusually large ratio (~50) between the two in-plane dimensions (10 um vs. 200 nm) also imparts a 1D or fibrous nature to the material. The dual 1D/2D structure gives the material properties characteristic of both nanosheet (2D) and fibrous (1D) materials. As a 2D material it is a few-layered nanoplate that can serve as host for intercalation of foreign guest molecules,36 or be potentially be exfoliated into monolayer sheets. As a 1D material, it has a high aspect ratio (>50) and a total length large enough to potentially challenge cells in phagocytic uptake or internal vesicular packaging.37,38 The Young’s modulus of MoO3 nanoribbons is estimated to be approximately 31 GPa while bulk MoO3 is reported as 540 GPa39 For comparison, crocidolite asbestos has an elastic modulus of ~160–170 GPa40. The bending rigidity for MoO3 nanoribbons is sufficient to maintain their confirmation as straight rods (no significant curvature) during handling, abiotic dissolution, or cell interactions as seen in Figs. 1,3, and 4.
Figure 1.
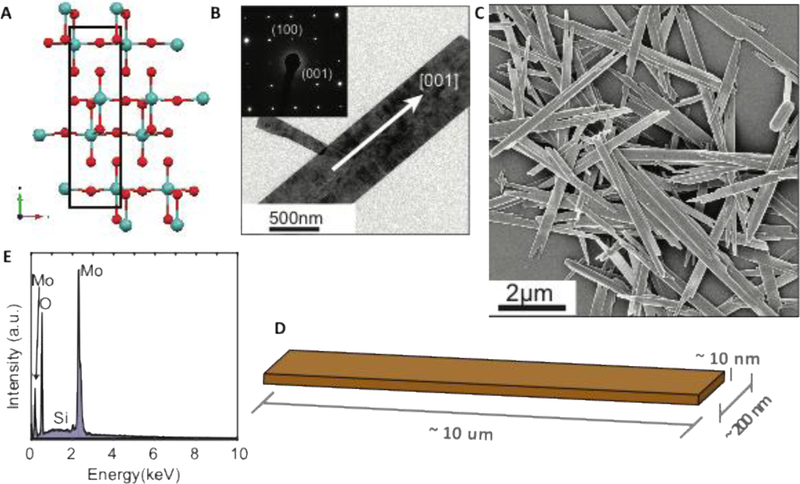
Morphology and composition of MoO3 nanoribbons. (A) atomic positions in three-layer MoO3 nanosheet structure (Mo in blue, O in red); (B) TEM images and SAED pattern of a MoO3 nanoribbon. Data were collected from JEOL2500 at 200keV (C) SEM image of nanoribbons deposited from stock suspension (Zeiss LEO 1530 5keV); (D) 3D sketch of typical nanoribbon dimensions; (E) EDS spectrum of nanoribbons on a Si substrate.
Figure 3.
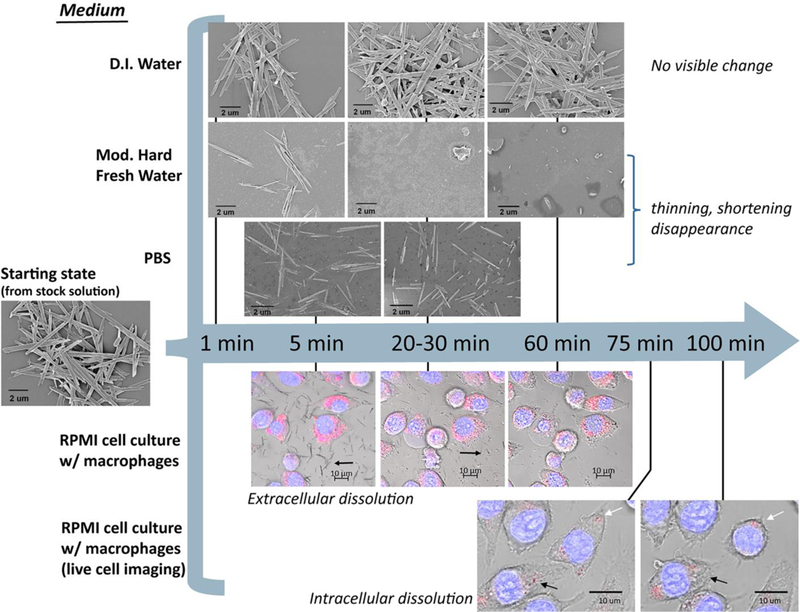
Evolution of nanoribbon morphology in various fluid media and in extracellular and intracellular environments during in vitro studies. Images above the blue arrow are from SEM analysis of dried samples after incubation in fluid simulants for defined times shown. Images below the blue arrow are confocal fluorescent images overlaid with brightfield DIC images. Black arrows in the images with macrophages (bottom two rows) show dissolution of the MoO3 nanoribbons while white arrows indicate MoO3 nanoribbons that persisted over 100 mins. Hoechst stain (blue) = DNA; LysoTracker Red stain (pink) = lysosomes.
Figure 4.
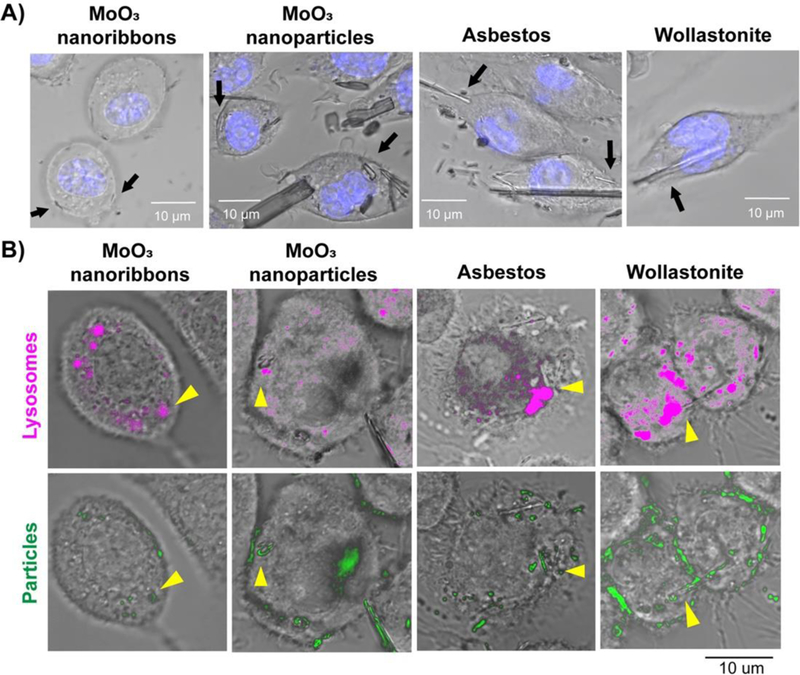
Internalization of high aspect ratio materials and particles by murine macrophages after 60min. A) Confocal and DIC images showing the internalization of 2D MoO3 nanoribbons, MoO3 nanoparticles, wollastonite and crocidolite asbestos. Nuclei were stained with Hoechst (blue fluorescence). B) Colocalization of MoO3 nanoribbons, MoO3 nanoparticles, asbestos and wollastonite with lysosomes. Lysosomes and particles were highlighted with magenta and green pseudocolors, respectively, using Image J software. Arrows indicate internalization of nanoribbons, particles or fibrous reference samples.
The 1D nature of MoO3 nanoribbons is of particular relevance for investigation of potential adverse human health impacts, and it will be useful to consider its properties in the context of the fiber pathogenicity paradigm. This paradigm, as described by Donaldson et al.41 identifies width, length and biopersistence of natural and man-made fibers as critical determinants of lung disease following inhalation. A fiber is considered biopersistent if the rate at which it dissolves is slow relative to the rate of physical removal from the lung.42 Both biopersistence and length of a fiber are critical factors in determining the removal of fibers from the lung. Dissolution can result in either complete disintegration or breakage of long fibers into several shorter fibers. Short fibers (<15um) are fully engulfed by macrophages and readily cleared from the lung. Longer fibers (>15–20um) are more difficult to clear from the lung because they are not easily taken up by macrophages, resulting in incomplete or frustrated phagocytosis.41 Thus, the persistence of long fibers in the lung determines the delivered dose to the lung tissue. Fibers that remain in the lung for an extended period of time have the potential to cause persistent inflammation, fibrosis and cancer.43,44
Experimental studies using silicate-based fibers, crocidolite asbestos and wollastonite, support the fiber pathogenesis paradigm. Wollastonite fibers undergo complete dissolution in phagolysosomal simulant fluid within 90 days.45 Elimination of wollastonite fibers from the lung is primarily attributed to dissolution, with short half-life of 15–21 days. Wollastonite exposure does not induce lung disease due to its rapid dissolution and lack of surface reactivity.46–48 In contrast, both in vitro and in vivo studies show that crocidolite asbestos fibers dissolve slowly, with a half-life of 240 days, while longer (>5um) fibers persist for over one year in rats.49,50 Long crocidolite asbestos fibers undergo frustrated phagocytosis and the retention of these fibers in the lungs and pleura results in extensive cytotoxicity due to the surface redox activity of iron (III) along the fiber surfaces leading to the formation of reactive oxygen species.43,51,52 Leakage of reactive oxygen species into adjacent tissues has been shown to induce DNA damage, inflammation, fibrosis and cancer.43,53,54 The fiber pathogenicity paradigm has recently been extended to include high-aspect-ratio nanomaterials (HARNs). Carbon nanotubes, titanium dioxide nanorods and silver or nickel nanowires have all been shown to be biologically durable and induce frustrated phagocytosis in macrophages.41,55,56 Several of these HARNs induce similar effects as crocidolite asbestos fibers, including cytotoxicity, inflammation and cancer in rodents.41,57,58
As the biological durability of fibrous nanomaterials is closely linked to adverse health impacts, this study focusing on the dissolution kinetics of MoO3 nanoribbons is particularly relevant for environmental and biological media. The cellular uptake and cytotoxicity of MoO3 nanoribbons in macrophages are compared to MoO3 particles, as well as to wollastonite and crocidolite asbestos as fibrous reference materials. Together, these chemical and in vitro biological studies provide insight into whether this novel HARN fits into the fiber pathogenicity paradigm. Finally, in the last section the results will be used as a case study to demonstrate how dissolution screening and in vitro biological studies can be incorporated in a tiered testing strategy for emerging 2D nanomaterials.
Materials and Methods
MoO3 Synthesis and Characterization
The MoO3 nanoribbons investigated herein were synthesized following the procedure described by Wang et al.6 Exactly 0.36g of molybdenum powder was mixed with 30ml distilled water in an Erlenmeyer flask. Drop-wise 2.5ml of 30% aqueous hydrogen peroxide solution was added followed by 20min of stirring. The solution was then transferred to a 50ml Parr acid digestion bomb with a Teflon liner and heated at 180°C for 12h. The cooled products were washed with ethanol three times using centrifugation and finally dispersed and stored in distilled water. The MoO3 stock suspension was characterized using a Scanning Electron Microscope (SEM, Zeiss LEO 1530 and FEI Scios Dual Beam FIB/SEM) with Energy Dispersive X-Ray (EDS), Transmission Electron Microscope (TEM, JEOL 2500), Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES, Thermo Scientific iCAPtm 7400), Raman Analysis (532 nm at 5mW) and X-ray diffraction (XRD, Bruker Eco Advance). The ICP-OES measured concentration of the final suspension was 4.7mg mL-1. The dimensions of these materials were confirmed to be 10μm by 200 nm by 10 nm (Fig. 1), and consist of ~7–9 monolayers separated by Van der Waals gaps, in which each fundamental MoO3 monolayer itself consists of three atomic layers of covalently bonded Mo and O atoms (Fig. 1A). The MoO3 nanoribbons were stable in the concentrated stock solution over the course of the study (>8 months). Note that all reagents used in this MoO3 synthesis were purchased from Sigma-Aldrich. Additional synthesis and characterization data can be found in Wang and Koski.36
Fluid media for accellular dissolution studies
MoO3 nanoribbons were studied in the following six fluid environments: nanopure water (NP H2O, pH 7.0), EPA moderately-hard water (Mod pH 7.8)59 phosphate buffered saline (PBS, pH 7.4), Roswell Park Memorial Institute Medium (RPMI, pH 7.4), simulated lung fluid (SLF, pH 7.5)60 and phagolysosomal simulant fluid (PSF pH 4.5).61 Complete formulations of each media are listed in SI (Suppl. Tables 1–5). These different media were chosen to mimic different exposure scenarios. EPA Mod mimics freshwater environments while PBS has the pH and ionic strength typical of biological fluids. RPMI is a commonly used cell culture medium and was supplemented with 10%, v/v Fetal Bovine Serum (FBS). SLF and PSF mimic two local environments relevant to inhalation exposure: the lung extracellular environment (SLF) and the phagolysosome after uptake by macrophages (PSF).33 All six fluid environments were never diluted below 90% of the initial solution composition when adding aliquots of the aqueous MoO3 stock for biopersistence testing. Reagents used to prepare Mod, SLF, and PSF were either ACS or reagent grade and purchased from Fisher Scientific (Fairlawn, NJ), Sigma Aldrich (St Louis, MO), Arcos Organics (NJ, USA) or Alpha Aesar (Ward Hill, MA, USA). PBS, RPMI and FBS were purchased from Fisher Scientific (Fairlawn, NJ).
Dissolution and aggregation screening studies by DLS
Screening level stability tests were performed using a Malvern Zetasizer DLS (Worcestershire, United Kingdom) using a 90o scattering angle. The MoO3 stock described above was vortexed for 1min and then bath sonicated for 15min prior to use. The stock was diluted 1:10 into plastic disposable polystyrene cuvettes using each test media described above, resulting in a nominal Mo concentration of 470mg L−1 and total volume of 1 ml. Each cuvette was capped mixed by inversion for 1 to 2s prior to placement into the DLS. No additional mixing was used during DLS measurements. Instrumental triplicate readings were taken every 20s resulting in 1 data point per minute. Z-average was the selected as the metric for this work due to its widespread use and minimization of reported variability. Although Z-average measures hydrodynamic diameter (HDD), it is employed here as a semi-quantitative indictor through which the evolution of the effective nanosheet size during dissolution or aggregation can be monitored over time.
ICP-AES measurements of dissolution rates
Nanoribbon dissolution rates were determined by time-resolved ICP-OES measurement of total soluble Mo in aqueous solution after filtration removal (0.02μm cutoff) of the remaining solid phases. A syringe pump was purchased from New Era Pump Systems, Inc. (Farmingdale, NY, USA) and was set to a flow rate of 1ml min−1 for a total time of 1 minute and 25 seconds. Reaction vessels were prepared in sterile centrifuge tubes and an aliquot of sample (~1.5ml) was removed for filtration using a 5ml BD disposable syringes (BD Medical, Franklin Lakes, NJ, USA) connected by luer-lok to 3.5in disposable hypodermic needles (Air-Tite, Virginia Beach, VA, USA). Syringe needles were removed after the subsample was collected and replaced with cartridge syringe filters syringe taking care to prevent introducing air onto the filter cartridge. The cartridge filters were 0.02μm Anatoptm 10+ and 25+ syringe filters (GE Healthcare Life Sciences, Buckinghamshire, UK). Both filters utilize the same membrane and only differ in the total sample volume each disposable filtration unit can accommodate. The total Mo concentration of all experimental replicates was 47mg L−1 nominally based on the measured concentration of the stock. Filtrates were collected at 1, 15, 30, 45 and 60min for all matrices. The time points listed indicate the start of the filtration procedure. PBS and PSF required additional time points between 1–15min to attempt capture of partial dissolution. Filtrates were analyzed using multiple ICP-OES/AES instruments, a Horiba Jobin Yvon 2000 Ultratrace ICP-AES (Kyoto, Japan) or a Thermo Scientific iCAPtm 7400 ICP-OES. Dissolution data was normalized to total Mo concentration measured from unfiltered aliquots of each sample.
Dissolution reaction stoichiometry
The evolution of pH during MoO3 nanoribbon dissolution was used to gain insight into the reaction stoichiometry. The initial pH of nanopure water was adjusted using 0.1M NaOH (Fisher Scientific, Lot 081317, Fair Lawn, NJ) and then measured as 9.89 using an Oriontm 8102 BNUWP ROSS Ultra pH electrode (Thermo Fisher Scientific, Waltham MA). A 45μL aliquot of the 4.7g L−1 stock was added to 45ml of nanopure water, and the change in pH was measured until the value stabilized for > 1h. The measured value was compared to the theoretical H+ produced using the stoichiometry shown in equation 1 and the modeled pH change using the chemical equilibrium model, visual MINTEQ 3.1.62 MINTEQ was used to mimic adjusting solution pH to 10, and subsequently the exact addition of MoO3. The pH predicted by adding MoO3 was compared to the experimental value. The modeled system was assumed to be open with respect to atmospheric CO2 as experiments were conducted in open atmospheric conditions.
Cellular maintenance and material treatments
Cellular studies were conducted using J774A.1 murine monocyte-derived macrophages (ATCC TIB-67, Snyderman, 1950)63 cultured in RPMI 1640 media supplemented with 10% fetal bovine serum and 0.1% penicillin/streptomycin. Cells were maintained in a 5% CO2 humidified environment at 37⁰C. Prior to exposure to test or reference samples, cells were seeded as adherent monolayers and incubated for 24 hours. 2D MoO3 nanoribbons36 and MoO3 nanoparticles (US Research Nanomaterials US3330) suspended in sterile water were sonicated in a bath sonicator for 1 hour before concentrations of 10–100μg ml−1 were prepared in supplemented RPMI media. Crocidolite asbestos and wollastonite fibers were used as positive and negative high aspect ratio reference materials, respectively. Crocidolite asbestos fibers originally prepared and characterized by the Union Internationale Contre le Cancer64 were purchased from Duke Scientific Corporation (Palo Alto, CA). NYAD 1250 wollastonite (Lot 11–28-94) was a gift from NYCO (Willsboro, NY). Crocidolite asbestos stocks were prepared as described in Sanchez et al. (2011).65 Wollastonite stocks were prepared in sterile water, autoclaved for 30min and stored at 4⁰C. Final concentrations of 10–100μg ml−1 were prepared in supplemented RPMI media. Images, dimensions and surface areas of all nanomaterials or fibers are included in SI (Suppl. Fig. S1, Table S6). Sodium molybdate (Sigma Aldrich, 737860) was dissolved in sterile water at 50mg ml−1 before dilutions were prepared in supplemented RPMI media at equivalent ionic Mo concentrations as in fully dissolved MoO3 nanoribbons (Suppl. Table S7).
Confocal Imaging
Nanoribbon internalization was assessed by staining murine macrophages with 500nM Lysotracker red DND (Invitrogen L7528) and Hoechst 33342 (1:3000, Invitrogen H3570) to visualize lysosomes and nuclei, respectively, in FBS-free media for 35min at 37⁰C. Cells were then exposed to MoO3 nanoribbons (470μg mL−1), MoO3 nanoparticles, crocidolite asbestos or wollastonite (100μg mL−1) in HEPES supplemented, complete RPMI media and incubated at 37⁰C in an environmental chamber on a Zeiss LSM 710 confocal microscope. Confocal images were taken at 63× every 10min over 2h with both fluorescent and DIC filters. Image J software (imagej.nih.gov/ij) was used to separate the color channels for each image, creating greyscale images. The look up table (LUT) was then used to assign artificial colors to the bright and dark regions of the image. This technique allowed us to artificially color the lysosomes magenta and the particles green in order to highlight their localization within the cell.
Cathepsin B localization was used as a marker of lysosomal integrity. After exposure to test and reference materials for 60min, macrophages were washed with PBS and incubated in Magic Red in vitro Cathepsin B reagent (1:10, ImmunoChemistry Technologies) and Hoechst 33342 (1:3000, Invitrogen H3570) for 15 min at 37⁰C. Cells were then washed with PBS and fixed in 4% paraformaldehyde for 10min before obtaining confocal fluorescent images of cathepsin B activity and localization at 63× on an Olympus IX81 microscope.
Cytotoxicity Assay
Cytotoxicity was determined using a LIVE/DEAD viability assay (Invitrogen L3224). Briefly, cells were seeded in 96 well Greiner μClear imaging plates (USA Scientific), allowed to attach for 24h, and exposed to 2D MoO3 nanoribbons, MoO3 nanoparticles, crocidolite asbestos, wollastonite or sodium molybdate for 1, 7 or 24h. Following exposure, cells were washed with PBS and incubated with 2μM calcein AM and 4μM ethidium homodimer 1 in phenol and FBS-free media for 10min at 37⁰C. This assay distinguishes live from dead cells by simultaneously staining the live cells with green fluorescence due to intracellular esterase activity and the dead cells with red nuclear fluorescence due to binding of ethidium homodimer to nuclear DNA following loss of plasma membrane integrity. Widefield, fluorescent images were obtained at 10× on an Olympus IX81 microscope. Three fields of view, each containing >500 cells, were taken of each condition to ensure accurate representation of the cell population. Each experiment was conducted with three biological replicates. Images were analyzed using CellProfiler software66 to obtain live (green fluorescence), dead (red fluorescence) and total cell counts. Statistical analysis was conducted as a two-way ANOVA with a Dunnett multiple comparison test in GraphPad Prism 7.
Results and Discussion
MoO3 nanoribbon behavior in fluid simulants
The MoO3 nanoribbons (Fig. 1) were added to various environmental and biological fluid media as aliquots from the concentrated stock suspension, and the ribbon behaviors were monitored by DLS, ICP-OES/AES (filtrates), and electron microscopies (dried solid residues) as shown in Fig. 2–3. The media dependence of MoO3 behavior is clearly visible to the naked eye. MoO3 dispersions were initially white colloidal suspensions and did not change significantly in nanopure water (NP) (Fig. 2A). In PBS, however, the suspensions cleared after 20min (Fig. 2B), and no settled material was observed in any test cuvettes, suggesting complete MoO3 dissolution.
Figure 2.
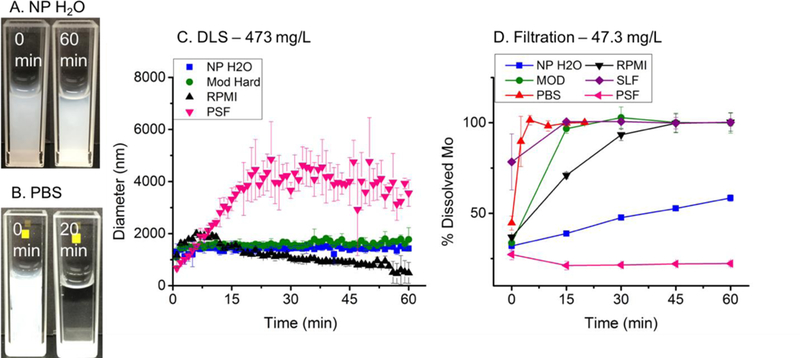
Aggregation and dissolution studies of MoO3 nanoribbons. A-B. Optical imaging in NP (A) and PBS (B) showing contrasting behavior. DLS monitoring in four fluid media at nanoribbon starting concentration of 473 mg L−1 (C). Note that dissolution in PBS and SLF were too fast to obtain good DLS data. ICP-AES measurements of soluble molybdenum after ultrafiltration in six media at starting nanoribbon loading of 47.3 mg L−1 (D). N=3 for all data points.
Dynamic light scattering was then used as a simple in situ screening tool to monitor nanoribbon stability (Fig. 2C). DLS is a simple, common and rapid analysis technique67,68 that has been applied to measure changes in particle size distribution of nanomaterials in a range of complex media including river water, synthetic stomach fluid69 and various gastrointestinal simulant fluids.70 DLS measures hydrodynamic diameter (HDD), which in general is affected by aggregation, dissolution, or adsorption of macromolecules.71 For the complex nanoribbon geometry the reported diameters cannot be interpreted quantitatively, but the relative changes provide useful information here on dissolution and aggregation in situ. No change in HDD was observed in NP H2O or EPA Mod (Fig. 1C). In PSF, MoO3 HDD increased from 667nm to 3867nm in 22min, with no further change detected by 60min (Figure 1C). In RPMI augmented with FBS, HDD increased from 1167nm to 1915nm in 10min followed by a steady decrease in size to 487nm at 60min (Fig. 1C). The initial increase in HDD in RPMI was much smaller in magnitude than what was observed in PSF. Finally, DLS analysis on PBS and SLF yielded highly variable data (Suppl. Fig. S2), presumably due to rapid dissolution (see Fig. 2D) and thus very low light scattering signal throughout the experiment. The aqueous behavior of MoO3 nanoribbons is clearly complex and media dependent. Understanding this behavior requires quantitative measurements of dissolution products and pH effects.
Figure 2D shows ICP-OES/AES measurements of dissolved Mo fractions after ultrafiltration. Complete dissolution of the 2D MoO3 was achieved after one hour in MOD, PBS, RPMI and SLF, while the nanoribbons were stable in PSF and partially dissolved in NP (58%) at the time-scale. Note that in all experiments, 27%−37% of the total Mo is reported as soluble at the first-time point and likely represents dissolved Mo species coexisting with the nanoribbons in the stock suspension.
Dissolution was fastest in PBS and SLF, with more than 75% of Mo mass in filtrates at the 1-min time point. MoO3 dissolution kinetics were slower in RPMI, requiring 30–45min. Interestingly, filtration indicated that MoO3 was completely dissolved in Mod after 15-min, in apparent contradiction to the stability suggested by DLS screening (Fig. 2C). This contradiction is explained by the difference in total MoO3 concentration relative to the fixed components in Mod and is discussed further in the SI.
We are not aware of other solubility data on MoO3 nanoribbons, but data on bulk MoO3 forms may provide insight into the chemistry that underlies the complex media effects seen in Fig. 2. Bulk MoO3 has been observed to dissolve in both water or alkaline solutions.72–74 A very slow dissolution rate in pure water of 5.6 × 10−7 min−1 (characteristic dissolution time of 3.4 years) has been reported by Petrochenkov et al,72 for a powder form of MoO3. The particle size and crystalline form of this MoO3 powder were not reported. The proposed dissolution reaction72 is shown in Eq. 1:
| 1 |
with the molybdate anion as the main Mo containing species. The dissolution rate of MoO3 has been shown to increase with both pH and temperature72 and solubility increases with increasing pH.73 Dissolution of this MoO3 is reported to be favored at neutral or basic pH values, and dissolution-generated protons can slow the overall dissolution rate,75 becoming negligible at about pH 2.76 Evidence in support of this hypothetical reaction stoichiometry for the nanomaterial form can be obtained by monitoring pH evolution during dissolution and comparing it to predictions from Eq. 1. We adjusted the pH of NP water to ~10 with NaOH to promote rapid dissolution, and then added 4.7g L−1 MoO3 and took time-resolved measurements with a pH electrode. We observed decreases in pH over time, stabilizing at 5.5 after 24 hours. Assessing Eq. 1 requires a calculation of the moles of H+ produced per mole of Mo dissolved, for comparison to the 2:1 stoichiometry predicted by Eq. 1. MINTEQ modeling of the dissolution process, assuming molybdate as the Mo-containing product, predicted a final pH of 5.5 (see Table 1). The moles of protons generated are quite similar in the three cases (Based on Eq. 1 stoichiometry, measured pH, and modeled pH in an open system) providing support for this dissolution stoichiometry (Table 1). This agreement also supports the assumption that the dominant aqueous Mo species is MoO42- rather than any polymolybdate species.77
Table 1.
Effect of dissolution reaction on suspension pH following addition of 4.7mg L−1 MoO3 in NP water adjusted to pH 9.98 based on the hypothesized stoichiometry (Eq. 1), experimental data and MINTEQ modeled values. Measured pH data was obtained as the average of 3 triplicate experimental replicates. The agreement in estimated NH+ values between the three methods provides support for the product identification (molybdate) and reaction stoichiometry hypothesized in Eq. 1 (2 moles H+ per mole MoO3 dissolved).
| Method | pH change during dissolution |
Apparent moles of protons generated by dissolution reaction, NH+ (μmol/L) |
|---|---|---|
| Eq. 1 prediction (for 4.7mg L−1 MoO3) | n/a | 98.6 |
| Direct pH measurement | 9.98 → 5.5 | 98.7 |
| MINTEQ modelling of pH change in an open system | 9.98 → 5.4 | 99.4 |
This dissolution stoichiometry offers an explanation for many of the observed media behaviors. Our data show complete dissolution of MoO3 in RPMI, PBS, and SLF, all of which are buffered media (~pH 7), in which most of the dissolution-generated H+ are neutralized to prevent significant pH change. Removal of these protons shifts the Eq. 1 dissolution equilibrium to the right by Le Chatelier’s principle and favors dissolution. In contrast, there is little dissolution in the acidic buffer (PSF, pH 4.5) or in pure water, the latter being unbuffered and subject to rapid acidification by the dissolution process itself. The EPA moderately hard water is an intermediate case, in which the ~ 1M bicarbonate acts as a partial buffer to limit acidification (see SI for further discussion of MoO3 dissolution in these carbonate solutions). The dissolution stoichiometry of Eq. 1 could thus explain many of the observed media effects in Fig. 2.
Other features in the Fig. 2 data can be related to colloidal stability or aggregation of the nanoribbons. The rise in hydrodynamic diameter in PSF likely represents colloidal instability due to electrostatic screening in this high-salt (ionic strength = 0.14M) fluid. For comparison, the low ionic strength of NP and Mod hard water yields stable sizes over time. The cell culture medium (RPMI), despite being a high-salt environment, does not induce much aggregation, showing instead only a slight, early increase in apparent particle size compared to the aggregation observed in PSF. The relative lack of aggregation here is believed to be due to due to the dispersing effects of proteins adsorbed from medium. Indeed proteins such as BSA have been widely used to disperse nanomaterials for biological studies78.
Imaging of nanoribbon dissolution, cellular uptake and localization
It is noteworthy that the rate and extent of dissolution of the MoO3 nanoribbons is much higher in cell culture medium and simulated lung fluid (pH 7.5) than in phagolysosomal simulant fluid (pH 4.5). This pH dependence is consistent with the dissolution stoichiometry in Eq. 1, and this finding has potential implications for nanoribbon behavior in cell culture and in the human lung. First, the short dissolution time scales in RPMI/SLF (< 1hr) suggest that nanoribbon dissolution may occur partially or totally in the extracellular environments, in medium or lung lining fluid, prior to cell uptake, thus largely preventing direct cellular interaction with long, intact nanoribbons. Secondly, if some MoO3 nanoribbons do survive long enough for phagocytic uptake by macrophages and localization in phagolysosomes, the low pH of the phagolysosomal environment can be expected to slow the dissolution process and partially stabilize the ribbons as solids within the vesicle. Interestingly, this is the opposite behavior to that observed for many other metal-containing nanoparticles, which are more stable at extracellular (high) pH, and undergo acid-catalyzed dissolution with release of toxic ions in the lysosome or phagolysosome.14,31,79
To explore this interesting competition between cell uptake rates and dissolution rates in cell culture medium, and to monitor the shape and size of MoO3 nanoribbons during the degradation process, time-resolved imaging experiments were carried out in both cellular and cell-free environments (Fig. 3). In buffered cell-free environments, ex situ SEM imaging shows that the 1D/2D MoO3 nanoribbons undergo progressive shortening and thinning in all media buffered to prevent acidification by dissolution-generated protons (Fig. 3 upper images). The partially dissolved fibers continue to exist in straight, not curved or tangled states. While thinning decreases bending rigidity, shortening has the opposite effect (it inhibits fiber buckling), and shortening occurs here in parallel with thinning. Overall, we do not see a significant population of fibers that appear flexible in any stage of the process.
For the in vitro studies, macrophages were used because they are the initial cell to respond to inhaled fibers or particles deposited in the airways or alveoli. Macrophages phagocytize particles and fibers in order to clear them from the lungs.80 Since our acellular dissolution studies demonstrated that MoO3 nanoribbons dissolve over time in cell culture medium, we conducted confocal imaging to determine whether nanoribbons would be internalized by the macrophages prior to their dissolution. Nanoribbons suspended in RPMI media in the presence of macrophages underwent dissolution within one hour (Fig. 3), in agreement with the chemical dissolution studies in media without cells (Fig. 2D). Visual observation revealed that while most of the dissolution occurs in the extracellular media, some nanoribbons are internalized by macrophages prior to their complete dissolution (Fig. 3, Fig. 4A). The MoO3 nanoribbons observed intracellularly were shorter, compared to the initial MoO3 nanoribbons, providing morphological evidence of partial dissolution of the nanoribbons in the cell culture media prior to cellular uptake. Internalized MoO3 nanoribbons may become thinner during dissolution, however, no visual evidence of flexibility was observed as all MoO3 nanoribbons and fragments appeared rigid in confocal macrophage images. After 60 mins, confocal imaging overlaid with DIC images confirmed the presence of MoO3 nanoribbons within the cell membrane, while nanoribbons in the external media appeared to have dissolved (Fig. 3, Fig. 4A). MoO3 nanoribbons within the cells were localized in the same focal plane as the nucleus (Fig. 4A). The acellular dissolution study showed slower dissolution kinetics for the 2D MoO3 nanoribbons in phagolysosomal simulant fluid than in cell culture media, and therefore we used confocal imaging to assess whether nanoribbons were localized, and possibly stabilized within lysosomes. LysoTracker Red was used to visualize lysosomes and combined with confocal and DIC imaging to determine nanoribbon location within the cell. MoO3 nanoribbons are seen to co-localize with lysosomes (Fig. 4B), and live cell imaging revealed that some internalized 2D MoO3 nanoribbons appeared to dissolve completely within 2 hours, while others persisted longer (Fig. 3, bottom image row).
In vitro toxicity assessment of MoO3 nanoribbons and reference materials
Crocidolite asbestos and wollastonite are naturally-occurring silicate-based, fibrous minerals. 2D MoO3 nanoribbons, crocidolite asbestos and wollastonite all have a length many times greater than their width, making them high aspect ratio materials (Suppl. Fig. S1, Table S6). Wollastonite is reported to completely dissolve in phagolysosomal simulant fluid within 90 days, and to display little toxicity and no association with lung disease in humans.6,45,48 In contrast, crocidolite asbestos does not dissolve in phagolysosomal simulant fluid over 90 days and is biopersistent in lung tissue.45,46 Biopersistence of asbestos fibers leads to inflammation, fibrosis and cancer.43,53,81 Thus, crocidolite asbestos and wollastonite are used in this study as positive and negative reference materials, respectively, for the cytotoxicity experiments in this study. Long crocidolite asbestos fibers are not easily engulfed by macrophages, resulting in frustrated phagocytosis.41,81 Redox catalyzed generation of reactive oxygen species leads to disruption of the lysosomal membrane allowing the protease cathepsin B to leak into the cytoplasm of the cell and initiate cell death by apoptosis.82,83 Previous studies have also shown that other nanomaterials, such as multi-walled carbon nanotubes, also induce frustrated phagocytosis and cathepsin B release.84–86 As a fibrous, high aspect ratio nanomaterial, 2D MoO3 nanoribbons may also disrupt lysosomal membrane integrity leading to cell death.
To determine whether 2D MoO3 nanoribbons can disrupt the lysosomal membrane, we assessed intracellular release of cathepsin B after a 24-hour exposure (Fig. 5). We confirmed previous results by Palomaki et al84 showing that crocidolite asbestos fibers induce release of cathepsin B from the lysosomes, resulting in a diffuse fluorescent signal throughout the cytoplasm. In contrast, cells exposed to 2D MoO3 nanoribbons resembled untreated cells, with cathepsin B localized in punctate cytoplasmic foci. MoO3 nanoparticles and soluble molybdate ions also did not disrupt lysosomal integrity. Additionally, wollastonite also did not induce leakage of cathepsin B into the cytoplasm. Together these results show that the MoO3 nanoribbons can be internalized by macrophages prior to their dissolution and are localized within lysosomes but do not disrupt lysosomal membrane integrity. The possibility that the MoO3 nanoribbons or released soluble molybdate ions could induce cytotoxicity was assessed next.
Figure 5.
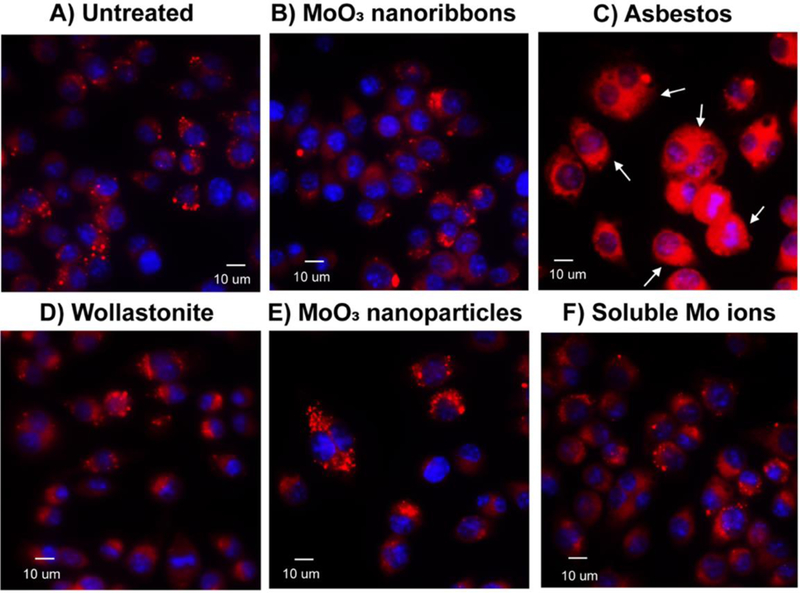
Fluorescent images of cathepsin B localization in murine macrophages. Confocal images of live cells stained for cathepsin B (red fluorescence) in untreated cells (A) and following 24h exposure to 10μg ml−1 2D MoO3 nanoribbons (B), crocidolite asbestos (C), wollastonite (D), MoO3 nanoribbons (E) or soluble molybdate ions (F). Cathepsin B release into the cytoplasm was only observed after asbestos exposure.
Cytotoxicity was determined after a 1h exposure, representing the time of initial internalization and extracellular dissolution, in addition to longer exposures, 7 and 24h, because some of the MoO3 nanoribbons appeared to persist intracellularly. MoO3 nanoribbons (up to 100ug ml−1) did not induce cytotoxicity after any timepoint (Fig. 6). Wollastonite also did not induce cytotoxicity, in agreement with previous reports.47,87 In contrast, our data show a significant increase in cytotoxicity 7 and 24h after exposure to crocidolite asbestos (≥ 50ug ml−1) (Fig. 6A,B,C). This is in agreement with previous reports of crocidolite asbestos toxicity.43,51 As the 2D MoO3 nanoribbons showed some evidence of intracellular dissolution, we also investigated the cytotoxicity of MoO3 nanoparticles and soluble molybdate ions. Soluble molybdate ions (up to 100ug ml−1 or 690uM) induced no cytotoxicity (Fig. 6. D,E,F, Suppl. Fig S3.). Previous studies report that soluble molybdate is non-cytotoxic at these concentrations. Extremely high doses, ≥3mM, are required for soluble molybdate to induce cytotoxicity after 24h.19 Cell survival decreased significantly to 83% after 24h exposure to 100μg ml−1 MoO3 reference nanoparticles (Fig. 6D,E,F). This may be due to the higher biodurability of the MoO3 nanoparticles, compared to the nanoribbons. DLS stability assays show that MoO3 nanoparticles (Suppl. Fig. S4) do not dissolve as rapidly as MoO3 nanoribbons (Figure 2C), especially in buffered media (pH ~7).
Figure 6.
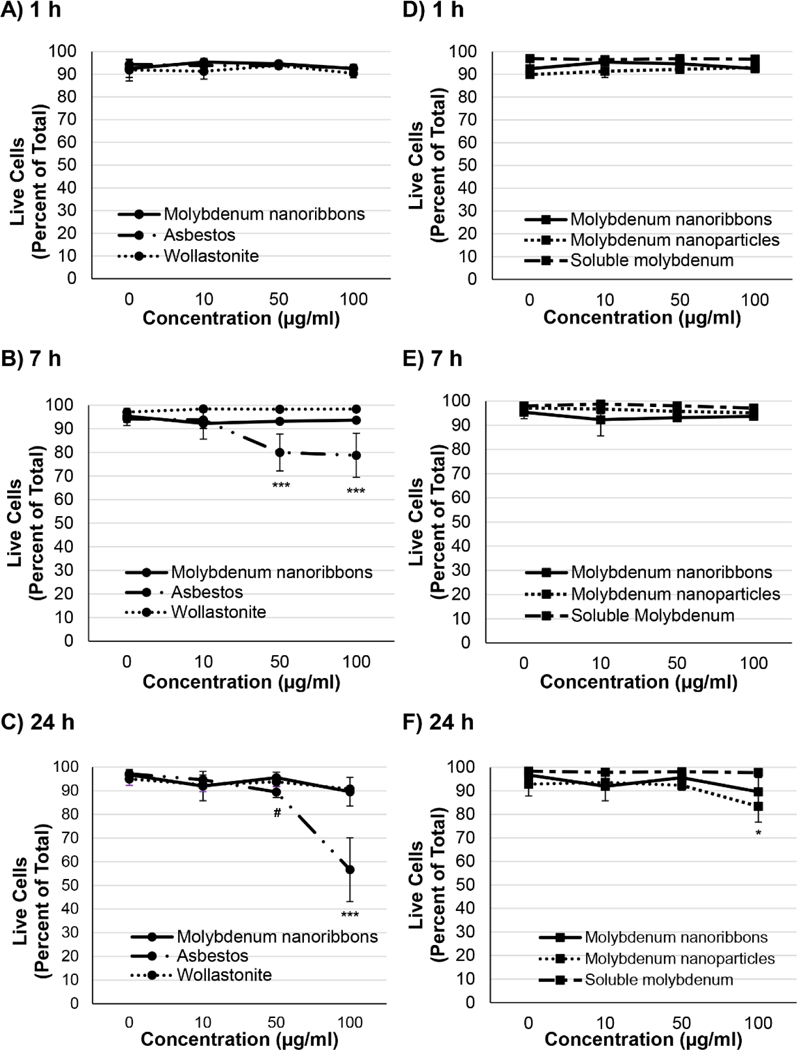
Cytotoxicity of 2D MoO3 nanoribbons compared against that of reference high aspect ratio materials, molybdenum nanoparticles and soluble molybdate ions. Assessment of murine macrophage viability after 1h (A, D), 7h (B, E) and 24h (C, F) exposures was determined using the Invitrogen LIVE/DEAD assay, with calcein AM staining live cells (green fluorescence) and ethidium homodimer 1 staining dead cells (red fluorescence). No decrease in viability was detected in cells exposed to 2D MoO3 nanoribbons. Only crocidolite asbestos, significantly decreased viability after both 7 and 24 h of exposure (A, B, C). A slight, but significant decrease in viability was induced by 24h exposure to MoO3 nanoparticles, while soluble molybdate ions did not adversely affect cell viability (F). #p<0.05; *p<0.01; ***p<0.0001.
Together, our data show that the fibrous remnants of the original MoO3 nanoribbons are localized within lysosomes after they are engulfed by macrophages. Once internalized, the nanoribbons do not disrupt lysosomal membrane integrity or induce cytotoxicity. Since the pH of extracellular lung lining fluid is 7.244, our data suggest that most of the MoO3 nanoribbons would dissolve extracellularly following inhalation and any MoO3 nanoribbons that persist could be engulfed and cleared by macrophages, without any resulting cytotoxicity.
Proposed Framework for Early Hazard Screening of Emerging 2D Materials
The present experiments on MoO3 nanoribbons were conducted as a case study in the context of a larger effort to develop tools for managing risks associated with emerging 2D materials as a general class. The large number of 2D materials and the rapid pace of ongoing discovery in this area provide strong motivation to devise simple screening strategies for grouping these sheet-like materials into rational sub-categories to guide and prioritize toxicity testing. A number of tiered testing strategies have been proposed in the general nanotoxicology literature,88–91 often beginning with characterization of nanomaterial physicochemical properties, or high-throughput in vitro toxicology screening. Many 2D materials are predicted to be unstable in biological media, however, and MoO3 is both predicted and observed to undergo dissolution at time scales equal to or shorter than those for cell uptake and processing. In such cases, the detailed physicochemical properties of the nanosheet solid (size, shape, surface charge, surface redox activity) become less important than the chemical properties of the dissolution products, which can be expected to be the drivers of most potential adverse outcome pathways. An alternative approach is therefore needed for efficient and systematic hazard screening of emerging 2D materials.
Figure 7 outlines our proposal for a biodissolution-based inhalation hazard screening method tailored for 2D material nanosheet powders. For a given test material, the screening method begins with a literature search on its chemistry, stability, and solubility. Because the current data available on nanosheet forms are limited, in many cases this first step in the framework must rely on data available for bulk forms, and should focus not on dissolution kinetics, but rather on the likely chemical pathways of biodegradation. Plausible pathways are identified from the set of (i) oxidative, (ii) reductive, or (iii) hydrolytic. The identified transformation pathway is then used to select one or more fluid simulant phases for dissolution testing (Fig. 7, central rectangle).
Figure. 7.
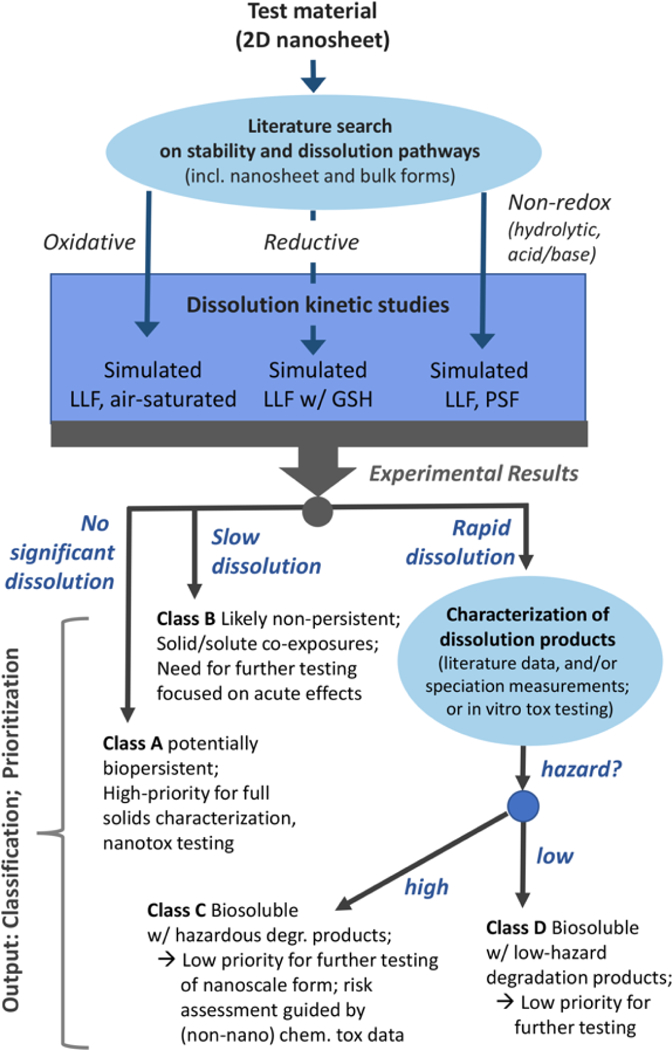
Proposed framework for 2D material hazard screening.
For materials susceptible to oxidation, experiments are recommended in simulated lung lining fluid with continuous or periodic exposure to atmospheric air to maintain oxic conditions. For materials susceptible to reductive degradation, simulated lung fluid (SLF) supplemented with glutathione at physiological concentrations (0.4–0.5mM)92 and refreshed as needed to ensure stoichiometric excess is recommended. For materials likely to undergo hydrolytic or acid/base dissolution, experiments are recommended in both lung lining fluid as a model for the extracellular space, and phagolysosomal fluid as a model for macrophage uptake. Analytical methods used for screening or quantitative evaluation of 2D material dissolution or biopersistence should be selected based on the 2D material elemental composition and suspension characteristics. The present case study used DLS and ICP-OES soluble metals analysis as simple techniques to monitor degradation. Materials that are poorly dispersible or have broad size distributions may be less suitable for DLS screening, and materials with insoluble or complex degradation products may require other methods. A large number of different analytical techniques have been explored and reviewed for characterizing nanomaterial mass, number, or size distributions.93–98
The results of simple dissolution kinetic studies are used to provisionally place 2D materials in one of four classification/prioritization groups, A to D (Fig. 7). Materials showing no measureable degradation in laboratory assays are referred to as Class A substances. Classes C or D materials exhibit rapid dissolution and are distinguished based on the hazard level of the dissolution products: high for Class C; low for class D. Class B materials exhibit an intermediate behavior in the form of slow degradation, where the effects of both the solid material and dissolved products must be considered.
Class A substances must be regarded as potentially biopersistent and become high-priority candidates for full solid-phase characterization and nanotoxicity testing using conventional approaches. For example, the dissolution rate constant (kdis = ng/cm2/h) for crocidolite asbestos in simulated lung fluid (pH 7.4) is extremely low (0.3 ± 0.1) and shows <1% dissolution in phagolysosomal simulant fluid (pH 4.6) after 56 days.99 Thus, crocidolite asbestos would be classified as a Class A substance. Class B materials exhibit an intermediate behavior in the form of slow degradation, as has reported recently for 2D MoS2.19 Their hazard assessment is complicated by the simultaneous co-exposure to solid particles and soluble ions, but their finite degradation rates suggest no long-term biopersistence The practical application of this classification framework will require a definition of “slow” dissolution (Class B) and its distinction from “fast” dissolution (Classes C,D). For this inhalation-based framework, we propose to distinguish between the two by using the time scale associated with macrophage clearance in the lung. Inhaled particles that dissolve slowly (>24–48 hours) in simulated lung fluid (pH 7.4) are most likely be taken up by macrophages as solid particles and cleared from the lungs by mucociliary transport towards the larynx where they would be expectorated or swallowed. In this case (Class B), additional studies would be warranted to assess dissolution in simulated saliva and gastric fluid31,101, which become secondary targets following the initial lung target. In contrast, we define Class C,D materials as those undergoing majority dissolution in < 24–48 hrs. These “rapidly” dissolving materials would likely be converted to molecular species in situ in the lung following inhalation, thus evading macrophage clearance and the degradation products would follow a different biokinetic pathway from those in Class B materials.
Overall, Class B materials are lower priority than recalcitrant Class A materials as solids, but hazards may be present and need to be assessed,. An example of a Class B material outside the world of 2D materials is the non-pathogenic synthetic vitreous fiber MMVF22. This material undergoes dissolution, with a dissolution rate constant of 119 ± 41 ng/cm2/hr in simulated lung fluid (pH 7.4) and 51% dissolution after 56 days in phagolysosomal simulant fluid.99 This places these fibers at a lower priority that Class A materials, but hazards may be present and need to be assessed, especially with respect to acute effects or repeated occupational exposures.
Class C and D materials both exhibit rapid dissolution. Materials in these classes are unlikely to present hazards as intact solid materials but may release toxic soluble material in their dissolution products, and this release may be the primary factor driving adverse biological responses. Other rapidly dissolving materials may have soluble dissolution products of relatively low hazard (such as MoO3). Being chemicals rather than nanoscale materials, the hazards associated with soluble dissolution products may reasonably be assessed in many cases from existing literature if the degradation products are (a) already known, (b) can be determined by chemical speciation of the products of the dissolution testing, or (c) can be tested in vitro by exposing cells to the full degradation product mixture. Based on one or more of these three approaches, a rapidly dissolving material may be placed into Class C (biosoluble with hazardous degradation products) or D (biosoluble with low hazard degradation products). Examples of nanomaterials with potentially hazardous dissolution products include some forms of nano-Cu or nano-Ag, which undergo oxidative dissolution (and fall within Class B or C) and for which biological activity is most commonly associated with the release of the copper or silver ion.79,102 Finally, the MoO3 nanoribbons used here as an experimental case study are a clear example of a class D material, and would thus be a low priority for further, more detailed nanotoxicity testing. We anticipate the use of this framework, based on simple dissolution testing in appropriately selected chemically reactive fluid simulants, will accelerate and simplify the hazard assessment process and enable the safe design of emerging 2D materials. In future work, the framework may be extended to cover exposure routes other than inhalation, or to other classes of emerging materials, where stability and persistence in biological compartments is poorly understood.
Supplementary Material
Acknowledgments
The authors acknowledge financial support from the Superfund Research Program of the National Institute of Environmental Health Sciences, Grant 2P42 ES013660 and the NIEHS T32 Training Grant (ES00727225).
References
- 1.Deng D, Novoselov KS, Fu Q, Zheng N, Tian Z and Bao X, Nat. Nanotechnol, 2016, 11, 218–230. [DOI] [PubMed] [Google Scholar]
- 2.Nicolosi V, Chhowalla M, Kanatzidis MG, Strano MS and Coleman JN, Science, 2013, 340, 1226419–1226419. [Google Scholar]
- 3.Perreault F, Fonseca de Faria A and Elimelech M, Chem Soc Rev, 2015, 44, 5861–5896. [DOI] [PubMed] [Google Scholar]
- 4.Holzinger M, Le Goff A and Cosnier S, Front. Chem, , DOI: 10.3389/fchem.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mao HY, Laurent S, Chen W, Akhavan O, Imani M, Ashkarran AA and Mahmoudi M, Chem. Rev, 2013, 113, 3407–3424. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Zhu W, Qiu Y, Yi X, von dem Bussche A, Kane A, Gao H, Koski K and Hurt R, Chem Soc Rev, 2016, 45, 1750–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klaine SJ, Alvarez PJJ, Batley GE, Fernandes TF, Handy RD, Lyon DY, Mahendra S, McLaughlin MJ and Lead JR, Environ. Toxicol. Chem, 2008, 27, 1825–1851. [DOI] [PubMed] [Google Scholar]
- 8.Handy RD, Cornelis G, Fernandes T, Tsyusko O, Decho A, Sabo-Attwood T, Metcalfe C, Steevens JA, Klaine SJ, Koelmans AA and Horne N, Environ. Toxicol. Chem, 2012, 31, 15–31. [DOI] [PubMed] [Google Scholar]
- 9.Gottschalk F and Nowack B, J. Environ. Monit, 2011, 13, 1145–1155. [DOI] [PubMed] [Google Scholar]
- 10.Tourinho PS, van Gestel CAM, Lofts S, Svendsen C, Soares AMVM and Loureiro S, Environ. Toxicol. Chem, 2012, 31, 1679–1692. [DOI] [PubMed] [Google Scholar]
- 11.Hou W-C, Westerhoff P and Posner JD, Environ. Sci. Process. Impacts, 2013, 15, 103–122. [DOI] [PubMed] [Google Scholar]
- 12.Mitrano DM, Motellier S, Clavaguera S and Nowack B, Environ. Int, 2015, 77, 132–147. [DOI] [PubMed] [Google Scholar]
- 13.Stone V, Johnston H and Schins RPF, Crit. Rev. Toxicol, 2009, 39, 613–626. [DOI] [PubMed] [Google Scholar]
- 14.Liu J and Hurt RH, Env. Sci Technol, 2010, 44, 2169–2175. [DOI] [PubMed] [Google Scholar]
- 15.Lowry GV, Hotze EM, Bernhardt ES, Dionysiou DD, Pedersen JA, Wiesner MR and Xing BS, J. Environ. Qual, 2010, 39, 1867–1874. [DOI] [PubMed] [Google Scholar]
- 16.Reed RB, Ladner DA, Higgins CP, Westerhoff P and Ranville JF, Environ. Toxicol. Chem, 2012, 31, 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furtado LM, Hoque ME, Mitrano DM, Ranville JF, Cheever B, Frost PC, Xenopoulos MA, Hintelmann H and Metcalfe CD, Environ. Chem, 2014, 11, 419–430. [Google Scholar]
- 18.Thomas DG, Smith JN, Thrall BD, Baer DR, Jolley H, Munusamy P, Kodali V, Demokritou P, Cohen J and Teeguarden JG, Part. Fibre Toxicol, , DOI: 10.1186/s12989-018-0243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z, von dem Bussche A, Qiu Y, Valentin TM, Gion K, Kane AB and Hurt RH, Environ. Sci. Technol, 2016, 50, 7208–7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arts JHE, Hadi M, Keene AM, Kreiling R, Lyon D, Maier M, Michel K, Petry T, Sauer UG, Warheit D, Wiench K and Landsiedel R, Regul. Toxicol. Pharmacol, 2014, 70, 492–506. [DOI] [PubMed] [Google Scholar]
- 21.Arts JHE, Hadi M, Irfan M-A, Keene AM, Kreiling R, Lyon D, Maier M, Michel K, Petry T, Sauer UG, Warheit D, Wiench K, Wohlleben W and Landsiedel R, Regul. Toxicol. Pharmacol, 2015, 71, S1–S27. [DOI] [PubMed] [Google Scholar]
- 22.Utembe W, Potgieter K, Stefaniak AB and Gulumian M, Part. Fibre Toxicol, , DOI: 10.1186/s12989-015-0088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chowdhury I, Duch MC, Mansukhani ND, Hersam MC and Bouchard D, Environ. Sci. Technol, 2013, 47, 6288–6296. [DOI] [PubMed] [Google Scholar]
- 24.Kurapati R, Muzi L, de Garibay APR, Russier J, Voiry D, Vacchi IA, Chhowalla M and Bianco A, Adv. Funct. Mater, 2017, 27, 1605176. [Google Scholar]
- 25.Yang S, Cai H, Chen B, Ko C, Özçelik VO, Ogletree DF, White CE, Shen Y and Tongay S, Nanoscale, 2017, 9, 12288–12294. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, Zhang Y-J, Liu M, Peterson A and Hurt RH, Nanoscale, 2017, 9, 5398–5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotchey GP, Allen BL, Vedala H, Yanamala N, Kapralov AA, Tyurina YY, Klein-Seetharaman J, Kagan VE and Star A, ACS Nano, 2011, 5, 2098–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xing W, Lalwani G, Rusakova I and Sitharaman B, Part. Part. Syst. Charact, 2014, 31, 745–750. [Google Scholar]
- 29.Lalwani G, Xing W and Sitharaman B, J Mater Chem B, 2014, 2, 6354–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurapati R, Russier J, Squillaci MA, Treossi E, Ménard-Moyon C, Del Rio-Castillo AE, Vazquez E, Samorì P, Palermo V and Bianco A, Small, 2015, 11, 3985–3994. [DOI] [PubMed] [Google Scholar]
- 31.Liu JY, Wang ZY, Liu FD, Kane AB and Hurt RH, Acs Nano, 2012, 6, 9887–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krishnamoorthy K, Premanathan M, Veerapandian M and Kim SJ, NANOTECHNOLOGY, , DOI: 10.1088/0957-4484/25/31/315101. [DOI] [PubMed] [Google Scholar]
- 33.Maze E, Mocquery C, Millet B and Espinosa AFI, Use of MoO3 as corrosion inhibitor, and coating composition containing such an inhibitor, Google Patents, 2006.
- 34.Zhou L, Yang L, Yuan P, Zou J, Wu Y and Yu C, J. Phys. Chem. C, 2010, 114, 21868–21872. [Google Scholar]
- 35.Tran TA, Krishnamoorthy K, Song YW, Cho SK and Kim SJ, ACS Appl. Mater. INTERFACES, 2014, 6, 2980–2986. [DOI] [PubMed] [Google Scholar]
- 36.Wang M and Koski KJ, ACS Nano, 2015, 9, 3226–3233. [DOI] [PubMed] [Google Scholar]
- 37.Nagai H and Toyokuni S, Cancer Sci, 2012, 103, 1378–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu W, von dem Bussche A, Yi X, Qiu Y, Wang Z, Weston P, Hurt RH, Kane AB and Gao H, Proc. Natl. Acad. Sci, 2016, 113, 12374–12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mai L, Yang F, Zhao Y, Xu X, Xu L, Hu B, Luo Y and Liu H, Mater. Today, 2011, 14, 346–353. [Google Scholar]
- 40.Aveston J, J. Mater. Sci, 1969, 4, 625–633. [Google Scholar]
- 41.Donaldson K, Murphy FA, Duffin R and Poland CA, Part. Fibre Toxicol, 2010, 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stefaniak AB, Part. Fibre Toxicol, 2010, 7, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shukla A, Gulumian M, Hei TK, Kamp D, Rahman Q and Mossman BT, Free Radic. Biol. Med, 2003, 34, 1117–1129. [DOI] [PubMed] [Google Scholar]
- 44.Sanchez VC, Pietruska JR, Miselis NR, Hurt RH and Kane AB, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol, 2009, 1, 511–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X, Hurt RH and Kane AB, Carbon, 2010, 48, 1961–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Macdonald JL and Kane AB, Toxicol. Sci, 1997, 38, 173–183. [DOI] [PubMed] [Google Scholar]
- 47.Maxim LD and McConnell EE, Inhal. Toxicol, 2005, 17, 451–466. [DOI] [PubMed] [Google Scholar]
- 48.Maxim LD, Niebo R, Utell MJ, McConnell EE, Larosa S and Segrave AM, Inhal. Toxicol, 2014, 26, 95–112. [DOI] [PubMed] [Google Scholar]
- 49.Muhle H, Bellmann B and Pott F, Environ. Health Perspect, 1994, 102, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lippmann M, Crit. Rev. Toxicol, 2014, 44, 643–695. [DOI] [PubMed] [Google Scholar]
- 51.Goodglick LA and Kane AB, Cancer Res, 1986, 46, 5558–5566. [PubMed] [Google Scholar]
- 52.Donaldson K, Beswick PH and Gilmour PS, Toxicol. Lett, 1996, 88, 293–298. [DOI] [PubMed] [Google Scholar]
- 53.Toyokuni S, Nagoya J Med. Sci, 2009, 71, 1–10. [PMC free article] [PubMed] [Google Scholar]
- 54.Nagai H and Toyokuni S, Arch. Biochem. Biophys, 2010, 502, 1–7. [DOI] [PubMed] [Google Scholar]
- 55.Hamilton RF, Wu N, Porter D, Buford M, Wolfarth M and Holian A, Part. Fibre Toxicol, 2009, 6, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schinwald A, Murphy FA, Prina-Mello A, Poland CA, Byrne F, Movia D, Glass JR, Dickerson JC, Schultz DA, Jeffree CE, Macnee W and Donaldson K, Toxicol. Sci. Off. J. Soc. Toxicol, 2012, 128, 461–470. [DOI] [PubMed] [Google Scholar]
- 57.Donaldson K, Poland CA, Murphy FA, MacFarlane M, Chernova T and Schinwald A, Adv. Drug Deliv. Rev, 2013, 65, 2078–2086. [DOI] [PubMed] [Google Scholar]
- 58.Luanpitpong S, Wang L, Davidson DC, Riedel H and Rojanasakul Y, Environ. Sci. Nano, 2016, 3, 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.EPA, Methods for Measureing the Acute Toxicity of Effluents and recieveing Waters to Freshwater and Marine Organisms, 2002. [Google Scholar]
- 60.Gray JE, Plumlee GS, Morman SA, Higueras PL, Crock JG, Lowers HA and Witten ML, Environ. Sci. Technol, 2010, 44, 4782–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stefaniak AB, Guilmette RA, Day GA, Hoover MD, Breysse PN and Scripsick RC, Toxicol. In Vitro, 2005, 19, 123–134. [DOI] [PubMed] [Google Scholar]
- 62.J. P. Gustafsson, Visual MINTEQ, Stockholm, Sweden, 2013.
- 63.Snyderman R, Pike MC, Fischer DG and Koren HS, J. Immunol. Baltim. Md 1950, 1977, 119, 2060–2066. [PubMed] [Google Scholar]
- 64.Timbrell V and Rendall REG, Powder Technol, 1972, 5, 279–287. [Google Scholar]
- 65.Sanchez VC, Weston P, Yan A, Hurt RH and Kane AB, Part. Fibre Toxicol, 2011, 8, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P and Sabatini DM, Genome Biol, 2006, 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nobbmann U and Morfesis A, Mater. Today, 2009, 12, 52–54. [Google Scholar]
- 68.Brar SK and Verma M, TrAC Trends Anal. Chem, 2011, 30, 4–17. [Google Scholar]
- 69.Mwilu SK, El Badawy AM, Bradham K, Nelson C, Thomas D, Scheckel KG, Tolaymat T, Ma L and Rogers KR, Sci. Total Environ, 2013, 447, 90–98. [DOI] [PubMed] [Google Scholar]
- 70.Walczak AP, Fokkink R, Peters R, Tromp P, Herrera Rivera ZE, Rietjens IMCM, Hendriksen PJM and Bouwmeester H, Nanotoxicology, 2012, 7, 1198–1210. [DOI] [PubMed] [Google Scholar]
- 71.del Pino P, Pelaz B, Zhang Q, Maffre P, Nienhaus GU and Parak WJ, Mater Horiz, 2014, 1, 301–313. [Google Scholar]
- 72.Petrochenkov VA, Gorichev IG, Batrakov VV, Izotov AD and Kutepov AM, Theor. Found. Chem. Eng, 2004, 38, 386–393. [Google Scholar]
- 73.Angelidis TN, Tourasanidis E, Marinou E and Stalidis GA, Resour. Conserv. Recycl, 1995, 13, 269–282. [Google Scholar]
- 74.Yao JN, Loo BH and Fujishima A, Berichte Bunsenges. Für Phys. Chem, 1990, 94, 13–17. [Google Scholar]
- 75.Liu B and Zeng HC, J. Phys. Chem. B, 2004, 108, 5867–5874. [Google Scholar]
- 76.Irmawati R and Shafizah M, Int. J. Basic Appl. Sci, 2009, 9, 241–244. [Google Scholar]
- 77.Oyerinde OF, Weeks CL, Anbar AD and Spiro TG, Inorganica Chim. Acta, 2008, 361, 1000–1007. [Google Scholar]
- 78.Vippola M, Falck GCM, Lindberg HK, Suhonen S, Vanhala E, Norppa H, Savolainen K, Tossavainen A and Tuomi T, Hum. Exp. Toxicol, 2009, 28, 377–385. [DOI] [PubMed] [Google Scholar]
- 79.Wang Z, von dem Bussche A, Kabadi PK, Kane AB and Hurt RH, ACS Nano, 2013, 7, 8715–8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blank F, Fytianos K, Seydoux E, Rodriguez-Lorenzo L, Petri-Fink A, von Garnier C and Rothen-Rutishauser B, J. Nanobiotechnology, , DOI: 10.1186/s12951-016-0242-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mossman BT, Lippmann M, Hesterberg TW, Kelsey KT, Barchowsky A and Bonner JC, J. Toxicol. Environ. Health B Crit. Rev, 2011, 14, 76–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Castro M, Bunt G and Wouters F, Cell Death Discov, , DOI: 10.1038/cddiscovery.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT and Tschopp J, Science, 2008, 320, 674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Palomäki J, Välimäki E, Sund J, Vippola M, Clausen PA, Jensen KA, Savolainen K, Matikainen S and Alenius H, ACS Nano, 2011, 5, 6861–6870. [DOI] [PubMed] [Google Scholar]
- 85.Shi X, von dem Bussche A, Hurt RH, Kane AB and Gao H, Nat. Nanotechnol, 2011, 6, 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang H, Dunphy DR, Jiang X, Meng H, Sun B, Tarn D, Xue M, Wang X, Lin S, Ji Z, Li R, Garcia FL, Yang J, Kirk ML, Xia T, Zink JI, Nel A and Brinker CJ, J. Am. Chem. Soc, 2012, 134, 15790–15804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pailes WH, Judy DJ, Resnick H and Castranova V, J. Toxicol. Environ. Health, 1984, 14, 497–510. [DOI] [PubMed] [Google Scholar]
- 88.Thomas CR, George S, Horst AM, Ji Z, Miller RJ, Peralta-Videa JR, Xia T, Pokhrel S, Mädler L, Gardea-Torresdey JL, Holden PA, Keller AA, Lenihan HS, Nel AE and Zink JI, ACS Nano, 2011, 5, 13–20. [DOI] [PubMed] [Google Scholar]
- 89.George S, Xia T, Rallo R, Zhao Y, Ji Z, Lin S, Wang X, Zhang H, France B, Schoenfeld D, Damoiseaux R, Liu R, Lin S, Bradley KA, Cohen Y and Nel AE, ACS Nano, 2011, 5, 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nel A, Xia T, Meng H, Wang X, Lin S, Ji Z and Zhang H, Acc. Chem. Res, 2013, 46, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hendren CO, Lowry M, Grieger KD, Money ES, Johnston JM, Wiesner MR and Beaulieu SM, Environ. Sci. Technol, 2013, 47, 1190–1205. [DOI] [PubMed] [Google Scholar]
- 92.Cantin AM, North SL, Hubbard RC and Crystal RG, J. Appl. Physiol. Bethesda Md 1985, 1987, 63, 152–157. [DOI] [PubMed] [Google Scholar]
- 93.Helfrich A and Bettmer J, Int. J. Mass Spectrom, 2011, 307, 92–98. [Google Scholar]
- 94.von der Kammer F, Ferguson PL, Holden PA, Masion A, Rogers KR, Klaine SJ, Koelmans AA, Horne N and Unrine JM, Environ. Toxicol. Chem, 2012, 31, 32–49. [DOI] [PubMed] [Google Scholar]
- 95.Gallego-Urrea JA, Tuoriniemi J and Hassellov M, TrAC Trends Anal Chem, 2011, 30, 473–483. [Google Scholar]
- 96.Laborda F, Bolea E, Cepriá G, Gómez MT, Jiménez MS, Pérez-Arantegui J and Castillo JR, Anal. Chim. Acta, 2016, 904, 10–32. [DOI] [PubMed] [Google Scholar]
- 97.Laborda F, Jimenez-Lamana J, Bolea E and Castillo JR, J Anal Spectrom, 2013, 28, 1220–1232. [Google Scholar]
- 98.Montaño MD, Lowry GV, von der Kammer F, Blue J and Ranville JF, Environ. Chem, 2014, 11, 351–366. [Google Scholar]
- 99.IARC, World Health Organization, International Agency for Research on Cancer, Lyon, France, 2002, vol. 81. [Google Scholar]
- 100.Geiser M and Kreyling WG, Part. Fibre Toxicol, 2010, 7, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sohal IS, Cho YK, O’Fallon KS, Gaines P, Demokritou P and Bello D, ACS Nano, , DOI: 10.1021/acsnano.8b02978. [DOI] [PubMed] [Google Scholar]
- 102.Xiu Z, Zhang Q, Puppala HL, Colvin VL and Alvarez PJJ, Nano Lett, 2012, 12, 4271–4275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.