

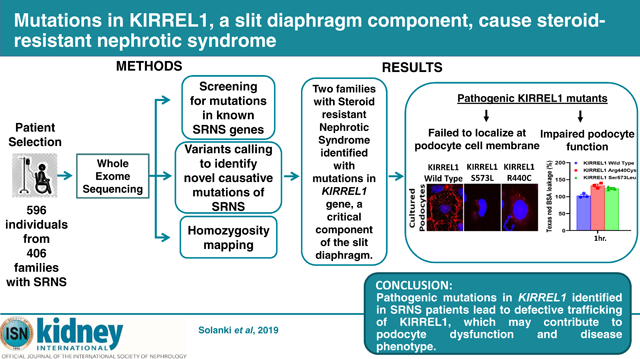
Abstract
Steroid-resistant nephrotic syndrome is a frequent cause of chronic kidney disease almost inevitably progressing to end-stage renal disease. More than 58 monogenic causes of steroid-resistant nephrotic syndrome have been discovered and majority of known steroid-resistant nephrotic syndrome-causing genes are predominantly expressed in glomerular podocytes, placing them at the center of disease pathogenesis. Herein, we describe two unrelated families with steroid-resistant nephrotic syndrome with homozygous mutations in the KIRREL1 gene. One mutation showed high frequency in the European population (minor allele frequency 0.0011) and this patient achieved complete remission following treatment, but later progressed to chronic kidney disease. We found that mutant KIRREL1 proteins failed to localize to the podocyte cell membrane, indicating defective trafficking and impaired podocytes function. Thus, the KIRREL1 gene product has an important role in modulating the integrity of the slit diaphragm and maintaining glomerular filtration function
Keywords: KIRREL1, FSGS, MCD, steroid-resistant nephrotic syndrome (SRNS)
Graphical Abstract
INTRODUCTION
Steroid-resistant nephrotic syndrome (SRNS) represents the second most frequent cause of CKD in the first 3 decades of life1. It is characterized by edema, nephrotic-range proteinuria, hyperlipidemia and its histological hallmarks, focal segmental glomerular sclerosis (FSGS). Although there have been advancements in treatment regimens no drug directly targets any of the primary causes of SRNS2. Currently, there are more than 58 genes that, if mutated, cause SRNS in a monogenic way3. Most of them are relevantly expressed in the glomerular visceral epithelial cell, the podocyte, placing it at the center of the pathogenesis of SRNS4. Podocytes are terminally differentiated cells that maintain glomerular filtration barrier. Foot processes of neighboring podocytes form a specialized intercellular junction, known as the slit diaphragm, which is essential for the filtration function of the kidney5. The pathophysiology of nephrotic syndrome is characterized by structural alteration of the cytoskeleton and molecular reorganization of slit diaphragm components leading to foot process effacement6. The first monogenic cause of SRNS in humans was discovered in NPHS1, an essential and major constituent of the slit diaphragm that is involved in forming glomerular filter via its homophilic or hetherophilic extracellular domain interaction7. Subsequently, several other monogenic causes in genes associated with slit diaphragm and actin cytoskeleton were discovered, emphasizing an essential role of this structure3. KIRREL1 has been described as a NPHS1-like Ig superfamily cell adhesion molecule8. Deletion of Kirrel in a mouse model results in podocyte foot processes effacement phenotype causing loss of glomerular filtration function9. A direct interaction between NPHS1 and KIRREL1 as well as their co-localization at slit diaphragm were described10. KIRREL1 is involved in rearrangement of actin cytoskeleton as a signaling mediator, emphasizing its essential role in organization and functional assembly of slit diaphragm11. Evidence suggest that in response to glomerular injury, NPHS1 and KIRREL1 trigger a signaling cascade that initiates downstream signaling and trafficking events, leading to morphological (effacement) and functional (proteinuria) changes in podocytes12. It has also been shown that injury to podocytes observed in various glomerular diseases is associated with mislocalization and displacement of KIRREL1 and NPHS1 from the podocyte cell membrane leading to podocyte damage13. The signaling events initiated by these proteins in response to injury govern the subsequent morphological and phenotypic changes in podocytes. Collectively, these observations suggest that a specific pathogenic link between SRNS development and alteration in KIRREL1 exists, indicating that similar to NPHS1, mutations in KIRREL1 may have pathological effects leading to renal dysfunction. Human mutations in KIRREL1 causing SRNS have been not reported to date.
Performing whole exome sequencing and targeted exon sequencing, we here identify mutations in KIRREL1 gene from two unrelated families with a history of SRNS. Using different in vitro functional assays we demonstrate that these mutations result in altered trafficking and mislocalization of KIRREL1 protein as well as affected integrity of slit diaphragm.
RESULTS
Mutations in KIRREL1 in 2 patients with nephrotic syndrome
To identify novel monogenic causes of SRNS, we performed whole-exome sequencing14 (WES) and homozygosity mapping15 in a cohort of 596 individuals from 406 families with SRNS. We first evaluated this data set for mutations in 33 known SRNS causing genes16. We discovered two homozygous missense mutations (p.Arg440Cys and p.Ser573Leu) of the KIRREL1 gene in two individuals of unrelated consanguineous families (A666 and B742) with SRNS. So the frequency of this gene would be (2/406), which is 0.4 % emphasizing its rarity as a monogenetic cause in a highly preselected cohort with SRNS. The mutated alleles causing mutations p.Arg440Cys and p.Ser573Leu appear to be very rare based on the reference data base gnomAD with a minor allele frequency (MAF) of 3.97×10−6 and a MAF of 7.87×10−4 respectively (Fig.1, Suppl.Fig.1, Suppl.Table.1 and Suppl.Table.2). Notably, the p.Ser573Leu, identified in a child of Italian descent and found mutated at the homozygous state in one control subject in GnomAD, has an allele frequency of 0.0011 in European populations, which is very frequent. The respective amino acid residues are conserved to D.rerio for both mutations (Fig.1B and C, Suppl.Table.1 and Suppl.Figure.2), and mutations are located within a homozygosity peak (Fig.1D and E). Renal histology in both patients was characterized by low-grade mesangial hyper-cellularity (Fig.1F and G). Ultrastructural analysis yielded extensive foot process effacement, increased mesangial matrix and mesangial proliferation (Fig.1H).
Figure 1. Homozygosity mapping and whole exome sequencing identify recessive mutations of KIRREL1 in 2 families with steroid-resistant nephrotic syndrome.

(A) KIRREL1 (NM_018240.6/NP_060710.3). Exon structure of human cDNA with the corresponding protein including its functional domains obtained from SMART. Positions of start codons and of stop codon are indicated. Arrows indicate positions of pathogenic mutations detected in families with SRNS. H, homozygous. (B) Phylogram of KIRREL1 protein was generated using multiple sequence alignment in Clustal omega. Numbers are representing the percent identity graph data comparing evolutionary relationship among different organisms to human reference for KIRREL1 protein (0: 100% identical; 1: 0% identical). (C) Evolutionary conservation of amino acid residues that are altered in patients with SRNS. Altered amino acid residues of KIRREL1 (p.Arg440Cys, p.Ser573Leu). (D) Homozygosity mapping identifies regions of homozygosity with recessive candidate loci (red circles) in patient A666_21. Nonparametric LOD (NPL) scores and SNP positions (Affymetrix 250K StyI array) are plotted on human chromosomes concatenated from p-ter (left) to q-ter (right). Genetic distance is given in centimorgans (cM). Whole exome sequencing identifies a homozygous mutation of KIRREL1 (p.Arg440Cys) that is positioned within the maximum NPL peak on chromosome 1 (arrowhead). (E) Homozygosity mapping identifies regions of homozygosity with recessive candidate loci (red circles) in patient B742_21. Nonparametric LOD (log of the odds ratio) (NPL) score profile is plotted across the human genome. Within the maximum NPL peak on chromosome 1 (arrowhead), we identified a homozygous mutation in KIRREL1 (p.Ser573Leu). (F) Renal histology of individual A666_21 with KIRREL1 mutation. Presence of only minor findings of mesangial hypercellularity is consistent with minimal change disease on light microscopy (H&E staining). (Scale bars: upper panel 200 μm and lower panel 100 μm). (G) Renal histology of individual B742_21 with KIRREL1 mutation shows focal segmental glomerular sclerosis (arrows) on light microscopy (MPAS staining). (Scale bars: upper and lower panels 20 μm). (H) Renal histology of B742_21 with KIRREL1 mutation showing podocyte foot process effacement (arrowheads) on transmission electron microscopy (TEM). Glomerular basement membrane is highlighted by a dotted line. (Scale bars: upper and lower panels 2 μm).
Treatment response and renal outcome in both families with KIRREL1 mutations
Both patients with detected homozygous mutations in KIRREL1 shared the phenotype of SRNS. There were no extrarenal manifestations or syndromic features in both affected children. Nevertheless, both affected individuals varied remarkably regarding the response to treatment course and the outcome of the renal function.
The patient A666_21 was diagnosed with NS at age of 5 years. He developed SRNS at his initial presentation and was resistant to different immunosuppressive drugs. He has never achieved a complete remission and remained having undulating proteinuria with partial response to treatment. Additional treatment with ACE-I and ARB was not beneficial for the patient. Nevertheless, his renal function remained normal all over the time, so he stopped his clinical visits after 12.5 years of follow up. Interestingly, this patient has a younger sister with reported periorbital edema and proteinuria as well as extended relatives with three children with ESRD and one child with nephrotic syndrome. However, neither the genotype of his sister nor the genotypes from his relatives are known because DNA samples for genetic analysis could not be obtained (Suppl. Table 1).
The patient B742_21 was diagnosed with NS at age of 14 years. He developed SRNS at his initial presentation. Throughout he was started on tacrolimus with weaning off of corticosteroids. He was additionally treated with CCB and ACE-I for his high blood pressure and proteinuria. During the course of treatment with Tacrolimus, CCB and gradual dose increase of ACE-I the patient succeeded a complete remission. However, over 4 years of follow up, he developed chronic kidney disease, which has continued to progress attaining CKD stage 2 at the time of this publication (Suppl.Table.1).
KIRREL1 mutations lead to mislocalization in cultured human podocytes
We have previously demonstrated, that KIRREL1 primarily localizes at the podocyte cell membrane and injury induces its mislocalization17. Since in silico analysis suggested, that the mutations may increase protein instability, we hypothesized, that this may induce mislocalization of these mutant proteins. To test this hypothesis, we generated cDNA constructs containing patient’s mutant alleles with inserted Flag tag at the N-terminus (Fig.2A). Stably transfected podocyte cell lines expressing these mutant proteins were generated, the expression was confirmed by western blot (Fig.2B). The localization of these mutant proteins and KIRREL1 wild type was analyzed by immunofluorescence microscopy using anti-Flag antibody. In comparison to the KIRREL1 wild type, a mislocalization of both KIRREL1 mutants (Arg440Cys and Ser573Leu) was noted (Fig.2C). The subcellular protein fractionation analysis of these cell lines further confirmed, that in comparison to KIRREL1 wild type protein, the mutant KIRREL1 proteins were present in the membrane fraction at a significant low level.(Suppl.Fig.3A and B). Further, the KIRREL1 mutations did not affect membrane localization of NPHS1 (Suppl.Fig.4A and B and Suppl.Fig.5A and B).
Figure 2. KIRREL1 mutants mislocalize in human podocytes.

(A) Schematic domain distribution, position of Flag tag and mutations in the KIRREL1 protein. (B) Western blot analysis showing the expression of the wild type control and KIRREL1 mutants plasmid constructs as indicated. (C) Unpermeabilized wild type human podocytes, human podocytes stably expressing flag tagged wild type KIRREL1 and flag tagged KIRREL1 mutants p.Ser573Leu and p.Arg440Cys were stained with Flag antibody, that labeled Flag-KIRREL1 extracellularly. Their membrane localization was evaluated by immunofluorescence microscopy using a confocal microscope. KIRREL1 mutants p.Ser573Leu and p.Arg440Cys show mislocalization compared to wild type control in cultured human podocytes cell lines. (Scale bars: 25 μm).
To narrow down the subcellular localization of mutant KIRREL1 proteins we investigated their co-localization with the markers of early endosomes (Rab5a), late endosomes (Rab7a), Golgi complex (N-acetylgalactosaminyltransferase), and lysosomes (Lamp1). Both KIRREL1 mutants showed increased localization at Golgi apparatus, early endosomes and lysosomes (Suppl.Fig.6A, left panels 1, 2, 3 and 6B). In comparison, KIRREL1 wild type protein was predominantly localized at the cell membrane (Suppl.Fig.3A). No significant differences were noted in the localization of KIRREL wild type and mutant proteins in late endosomes (Suppl.Fig.6A, left panel 4). These findings suggest that the KIRREL1 mutations induce trafficking defects, which allows the retention of mutant proteins in subcellular organelles.
KIRREL1 Mutations induce loss of transepithelial permeability in cultured human podocytes
Since KIRREL1 mutants failed to localize at cell membrane, we hypothesized that this may affect integrity of cell-cell adhesions and transcellular permeability. To assess these changes, we performed a transepithelial permeability assay, where permeability of Texas Red-labeled albumin was measured across the stable podocyte monolayer expressing either KIRREL1 wild type or KIRREL1 mutant proteins. The respective stable podocytes were grown on transwell filters and analyzed for Albumin influx across the podocytes monolayer. In comparison to cells expressing KIRREL1 wild type protein, a significant increase in albumin flux (an indicator of increase in transepithelial permeability) was noted within 1 hour (p <0.01), with a further increase at 4 hours (p< 0.001) (Suppl.Fig.7). Our results conclude that KIRREL1 mutations adversely affect the podocyte cell junctions and intercellular integrity.
DISCUSSION
In this report, we have identified and characterized two novel recessive mutations in KIRREL1 gene in two patients with SRNS of 2 unrelated consanguineous families in well conserved amino acid residues indicating the physiological significance of these regions. It was interesting to note that the p.Ser573Leu mutation appeared very frequent in European population with MAF 0.0011 and was found mutated at the homozygous state in one control subject in GnomAD being even slightly more common than the mutation of most common gene (NPHS2; p.Arg138Gln)18. However it is to be noted that a pathogenic variant is by definition a variant, which contributes mechanistically to disease, but is not necessarily fully penetrant (i.e., may not be sufficient in isolation to cause disease) and thus, there is a possibility of variable expressivity or incomplete penetrance for this variant (MacArthur et al., Nature 2014; 469). Further, as per our knowledge, the gnomAD data base is a reference of healthy individuals; although there is still not a 100% guarantee for it, since those individuals haven’t been screened on a standard procedure, leaving a possibility of including false negative subjects. However, the in silico predicting scores, good conservation and the functional data for p.Ser573Leu variant, are conclusive with its functional impact. Interestingly, although the patient with this mutation achieved complete remission upon treatments with Tacrolimus, CCB and ACE-I, however a later follow up showed progression to CKD. We further, demonstrated that stable expression of KIRREL1 protein containing human mutations in human podocyte cell lines leads to its mislocalization and accumulation in early endosomes, Golgi apparatus, and lysosomes indicating impaired protein trafficking. Additionally, our findings conclude that mislocalization of KIRREL1 alters podocyte cell junctions and leads to impaired intercellular integrity.
It is likely that the differences in the pathophysiology of these mutations are due to the functional differences of these regions. While the extracellular domain of KIRREL1 is involved in interaction with slit membrane protein NPHS1 ensuring structural integrity and filtering function of slit diaphragm19, the intracellular domain has been shown to be actively involved in signaling events that define actin cytoskeleton organization and modulation11. The redistribution of NPHS1 and KIRREL1 from the podocyte cell membrane to the subcellular compartments is strongly associated with the initiation of podocyte dysfunction and eventual loss of glomerular filtration function20,21. It was interesting to note that both mutations induced protein mislocalization, where its ability to localize at the podocyte cell membrane was severely diminished indicating affected protein trafficking. It is likely that these mutant proteins are transported to the podocyte cell membrane but due to their increased instability, they are removed and remain trapped in early endosomes which serve as sorting protein recycling station enabling their restoring back to plasma membrane22. We have previously shown that mislocalization of KIRREL1 is associated with increased cellular permeability and loss of intercellular junctions13. Since these mutant proteins phenocopied that effect, it is likely that this mislocalization may contribute to the disease phenotype. It is difficult to predict which cellular pathway is exactly affected due to these mutations and will need further detailed investigation. Although the follow up studies are required to investigate detailed signaling and trafficking events affected by these mutations. Nevertheless, these novel mutations highlight the biological importance of KIRREL1 in podocytes homeostasis.
In conclusion, this is the first report describing mutations in human KIRREL1 that results in SRNS emphasizing its important role in modulating the integrity of slit diaphragm and maintaining glomerular filtration function. The fact that one patient has responded to Tacrolimus, CCB and ACE-I treatment cannot be generalized at this point with regards to other patients with KIRREL1 mutations that may be discovered in the future, because we feel that it is not justified to draw conclusions on genotype-treatment relationships based on a single case. To study the detailed pathophysiology of these mutations, further studies, especially construction of Knock-in mouse model, will be needed.
Short Methods:
Study Approval and participants
Approval for human subject research was obtained from the University of Michigan and the Boston Children’s Hospital Institutional Review Boards. Patients were enrolled between 1998 and 2016. We performed whole exome sequencing in 596 individuals from 406 families. Patients were recruited in collaboratingon with centers worldwide and were ethnically diverse. Individuals of consanguineous kindred were preferentially sent for whole exome sequencing. All participants or their guardians provided written informed consent. Following informed consent, we obtained pedigree information, clinical data, and blood samples from individuals with proteinuria or nephrotic syndrome (NS) (onset < 25 years of age). Clinical data and blood samples were obtained from individuals with NS or their guardians. Clinical data were obtained using an established questionnaire (http://www.renalgenes.org). The diagnosis of NS was made by (pediatric) nephrologists, on the basis of standardized clinical and renal histologic criteria. Renal biopsy samples were evaluated by renal pathologists. Treatment response to immunosuppressive therapy in NS was defined as follows:
Complete response: remission of proteinuria, with a urine protein to creatinine ratio of <0.2 mg/mg, normalization of albumin.
Partial response: decrease in proteinuria by 50%.
Resistance: persistent, fixed proteinuria.
In cases with complete response, the absence of proteinuria has been confirmed in laboratory values of negative proteinuria either in 24-hours urine specimen or in the first morning urine specimen.
Whole exome sequencing
Whole exome sequencing was performed on genomic DNA isolated from blood lymphocytes or saliva and subjected to library preparation and exome capture using Agilent SureSelect™ human exome capture arrays ( Life technologies™) followed by next-generation sequencing on a HiSeq Illumina sequencing platform. Sequence reads were mapped to the human reference genome (NCBI build 37/hg19) using CLC Genomics Workbench™ (version 6.5.2; CLC bio, Aarhus, Denmark). Subsequent bioinformatics analysis excluded all variants with minor allele frequencies greater than 1% in the dbSNP database (version 142). Non-coding variants outside canonical splice side regions as well as synonymous variants were excluded, and variants were annotated using the ExAC database. Variants that were reported more than 10 times in the homozygous state in the ExAC database were excluded as likely non-pathogenic polymorphisms. Remaining variants were ranked based on established criteria3,23,24 and were evaluated for potential pathogenicity under consideration of phenotypic as well as pedigree information.
Cell culture and generation of podocytes overexpressing KIRREL1 and mutants
The immortalized human podocyte cell line was obtained from Dr. Moin Saleem25 and the cells were cultured in RPMI 1640-based medium supplemented with 10% FBS (Invitrogen), 2 g/liter of sodium bicarbonate (NaHCO3), insulin-transferrin-selenium supplement (Sigma-Aldrich), and 200 U/ml of penicillin and streptomycin (Invitrogen). Retroviruses overexpressing KIRREL1 and its mutants were generated by the transfection of the respective plasmids, cloned into the pBABE vector into Phoenix cells according to the manufacturer’s instructions.
Supplementary Material
Figure S1. Two homozygous mutations in KIRREL1 gene in two unrelated consanguineous families with SRNS. (A and C) Pedigrees: squares indicate males and circles females family members. A666–22 is reported to have edema and proteinuria. However, there is no DNA and no objective laboratory data available. (B and D) Segregation analysis and sequencing traces of family members as indicated. The red boxes highlight mutated alleles.
{kind=link}
Supplementary Table 2. Filtering process for variants following whole exome sequencing (WES) in families A666 and B742
Figure S2. A scheme of the domain architecture of human and mouse KIRREL1. Localization of the two mutations in human and mouse are highlighted in red.
{kind=link}
Figure S3. (A) Immunoblot of the membrane fraction and total cell lysates of human podocyte cell lines stably expressing wild type and mutant KIRREL1 proteins. Compared to wild type control mutant KIRREL1 proteins are not present in the membrane fraction. Na-K ATPase has been used as marker for membrane fraction. GAPDH has been used to probe any cytoplasmic contribution. (B) Quantitative analysis of the western blot respectively. Data represent mean ± SEM (n=3). *p < 0.01, **p < 0.001, ***p < 0.0001 (p-values were calculated using paired t-test).
{kind=link}
Figure S4. Cloning, expression and localization of NPHS1 in human podocytes (A) Schematic of Flag-NPHS1 construct and position of Flag tag in the NPHS1 protein. (B) Upper panel: Western blot analysis of NPHS1 expression. Lower panel: NPHS1 expressing cells were fixed and immunofluorescence analysis was performed on unpermeabilized cells using NPHS1 antibody that labeled NPHS1 extracellularly. The surface expression of Flag-NPHS1 was visualized using confocal microscopy (Scale bars: 25 μm).
{kind=link}
Figure S5. Localization of NPHS1 in human podocytes expressing KIRREL1 mutants (A) Human podocyte cell lines stably expressing wild type and mutant KIRREL1 proteins were transduced with NPHS1 viral construct and the membrane fractions from these cells were prepared. Immunoblotting analysis showed comparable amounts of NPHS1 in all cell lines. Na-K ATPase and GAPDH were used as membrane and cytoplasmic markers respectively. (B) Immunofluorescence analysis using anti NPHS1 antibody also showed comparable amounts of NPHS1 on cell surface in wild type and mutant KIRREL1 expressing cell lines. (C) Quantification of the fluorescent intensity (data represent mean, error bars represents SEM (n=10), p-values were calculated using paired t-test; ns: non-significant).
{kind=link}
Figure S6. (A)To analyze subcellular localization of KIRREL1, podocytes stably expressing either KIRREL1 wild type or mutant KIRREL1-Arg440Cys and KIRREL1-Ser573Leu proteins were labelled with markers of the Golgi complex (N-acetylgalactosaminyltransferase) (panel 1), lysosomes (Lamp1) (panel 2), early endosomes (Rab5a) (designated as E. Endosome, panel 3), and late endosomes designated as L. Endosome (Rab7a) (designated as L. Endosome, panel 4) using Cell Light reagents. Cell imaging was performed using confocal microscopy, Scale bars represent 10 μm. (B) Single-plane images were analyzed for co-localization of KIRREL1 wild type and KIRREL1 mutants with indicated subcellular compartments marker using Image J software. Pearson's correlation coefficients are presented as mean, error bar represents SEM (n=10). *p <0.01, **p< 0.001 (p values were calculated using paired t-test).
{kind=link}
Figure S7. Bovine serum albumin (BSA) permeability assay was performed with wild type and mutant KIRREL1 stably expressing human podocyte cell lines, which were cultured as a monolayer on Transwell filters. The passage of albumin across the podocytes monolayer was assessed by a paracellular albumin flux assay using Texas red-labeled albumin. The cells stably expressing KIRREL1 mutants showed increased albumin leakage as compared with cells stably expression wild type protein (data represent mean, error bars represents SEM (n=3), p-values were calculated using paired t-test; *p< 0.01, **p< 0.001).
{kind=link}
Supplementary Table 1. Genetic and clinical data of 2 families with SRNS and mutations in KIRREL1
Acknowledgments
We are grateful to the families and study individuals for their contribution. We thank the Yale Center for Mendelian Genomics for whole-exome sequencing analysis. F.H. is the William E. Harmon Professor.
This research was supported by grants from the National Institutes of Health to F.H. DK076683, to D.N. RO1 2R01DK087956-06A1, and to the Yale Center for Mendelian Genomics U54HG006504 and N.M T32-DK007726–33 grant at Boston Children’s Hospital; Ben J. Lipps Research Fellowship from ASN to A.K.S. TBH was supported for this study by the DFG (CRC1140), by the BMBF (01GM1518C) and by the European Research Council-ERC grant 616891. E.W. was supported by the German National Academy of Sciences Leopoldina (LPDS-2015–07). A.J.M. was supported by an NIH Training Grant (T32DK-007726), by the 2017 Post-doctoral Fellowship Grant from the Harvard Stem Cell Institute, and by the American Society of Nephrology Lipps Research Program 2018 Polycystic Kidney Disease Foundation Jared J. Grantham Research Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
F.H. is a cofounder of Goldfinch-Bio. No other authors have competing financial interests. C.B. is an employee of Bioscientia.
Accession Number
KIRREL1/KIRREL1 (NM_018240.6/NP_060710.3)
References:
- 1.Smith JM, Stablein DM, Munoz R, Hebert D & McDonald RA Contributions of the Transplant Registry: The 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS). Pediatr Transplant 11, 366–73 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Greenbaum LA, Benndorf R & Smoyer WE Childhood nephrotic syndrome--current and future therapies. Nat Rev Nephrol 8, 445–58 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Lovric S, Ashraf S, Tan W & Hildebrandt F Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant 31, 1802–1813 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Machuca E, Benoit G & Antignac C Genetics of nephrotic syndrome: connecting molecular genetics to podocyte physiology. Hum Mol Genet 18, R185–94 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Tryggvason K, Patrakka J & Wartiovaara J Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med 354, 1387–401 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Asanuma K & Mundel P The role of podocytes in glomerular pathobiology. Clin Exp Nephrol 7, 255–9 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Ruotsalainen V et al. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci U S A 96, 7962–7 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sellin L et al. NEPH1 defines a novel family of podocin interacting proteins. FASEB J 17, 115–7 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Donoviel DB et al. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol 21, 4829–36 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barletta GM, Kovari IA, Verma RK, Kerjaschki D & Holzman LB Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J Biol Chem 278, 19266–71 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Harita Y et al. Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J Biol Chem 283, 9177–86 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garg P, Verma R, Nihalani D, Johnstone DB & Holzman LB Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol Cell Biol 27, 8698–712 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arif E et al. Slit diaphragm protein Neph1 and its signaling: a novel therapeutic target for protection of podocytes against glomerular injury. J Biol Chem 289, 9502–18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou W et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet 44, 910–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hildebrandt F et al. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet 5, e1000353 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warejko JK et al. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin J Am Soc Nephrol 13, 53–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sagar A et al. Targeting Neph1 and ZO-1 protein-protein interaction in podocytes prevents podocyte injury and preserves glomerular filtration function. Sci Rep 7, 12047 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boute N et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24, 349–54 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Gerke P, Huber TB, Sellin L, Benzing T & Walz G Homodimerization and heterodimerization of the glomerular podocyte proteins nephrin and NEPH1. J Am Soc Nephrol 14, 918–26 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Doublier S et al. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes 52, 1023–30 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Hulkko J et al. Neph1 is reduced in primary focal segmental glomerulosclerosis, minimal change nephrotic syndrome, and corresponding experimental animal models of adriamycin-induced nephropathy and puromycin aminonucleoside nephrosis. Nephron Extra 4, 146–54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komada M & Soriano P Hrs, a FYVE finger protein localized to early endosomes, is implicated in vesicular traffic and required for ventral folding morphogenesis. Genes Dev 13, 1475–85 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vivante A & Hildebrandt F Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 12, 133–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacArthur DG et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saleem MA et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 13, 630–8 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Two homozygous mutations in KIRREL1 gene in two unrelated consanguineous families with SRNS. (A and C) Pedigrees: squares indicate males and circles females family members. A666–22 is reported to have edema and proteinuria. However, there is no DNA and no objective laboratory data available. (B and D) Segregation analysis and sequencing traces of family members as indicated. The red boxes highlight mutated alleles.
Supplementary Table 2. Filtering process for variants following whole exome sequencing (WES) in families A666 and B742
Figure S2. A scheme of the domain architecture of human and mouse KIRREL1. Localization of the two mutations in human and mouse are highlighted in red.
Figure S3. (A) Immunoblot of the membrane fraction and total cell lysates of human podocyte cell lines stably expressing wild type and mutant KIRREL1 proteins. Compared to wild type control mutant KIRREL1 proteins are not present in the membrane fraction. Na-K ATPase has been used as marker for membrane fraction. GAPDH has been used to probe any cytoplasmic contribution. (B) Quantitative analysis of the western blot respectively. Data represent mean ± SEM (n=3). *p < 0.01, **p < 0.001, ***p < 0.0001 (p-values were calculated using paired t-test).
Figure S4. Cloning, expression and localization of NPHS1 in human podocytes (A) Schematic of Flag-NPHS1 construct and position of Flag tag in the NPHS1 protein. (B) Upper panel: Western blot analysis of NPHS1 expression. Lower panel: NPHS1 expressing cells were fixed and immunofluorescence analysis was performed on unpermeabilized cells using NPHS1 antibody that labeled NPHS1 extracellularly. The surface expression of Flag-NPHS1 was visualized using confocal microscopy (Scale bars: 25 μm).
Figure S5. Localization of NPHS1 in human podocytes expressing KIRREL1 mutants (A) Human podocyte cell lines stably expressing wild type and mutant KIRREL1 proteins were transduced with NPHS1 viral construct and the membrane fractions from these cells were prepared. Immunoblotting analysis showed comparable amounts of NPHS1 in all cell lines. Na-K ATPase and GAPDH were used as membrane and cytoplasmic markers respectively. (B) Immunofluorescence analysis using anti NPHS1 antibody also showed comparable amounts of NPHS1 on cell surface in wild type and mutant KIRREL1 expressing cell lines. (C) Quantification of the fluorescent intensity (data represent mean, error bars represents SEM (n=10), p-values were calculated using paired t-test; ns: non-significant).
Figure S6. (A)To analyze subcellular localization of KIRREL1, podocytes stably expressing either KIRREL1 wild type or mutant KIRREL1-Arg440Cys and KIRREL1-Ser573Leu proteins were labelled with markers of the Golgi complex (N-acetylgalactosaminyltransferase) (panel 1), lysosomes (Lamp1) (panel 2), early endosomes (Rab5a) (designated as E. Endosome, panel 3), and late endosomes designated as L. Endosome (Rab7a) (designated as L. Endosome, panel 4) using Cell Light reagents. Cell imaging was performed using confocal microscopy, Scale bars represent 10 μm. (B) Single-plane images were analyzed for co-localization of KIRREL1 wild type and KIRREL1 mutants with indicated subcellular compartments marker using Image J software. Pearson's correlation coefficients are presented as mean, error bar represents SEM (n=10). *p <0.01, **p< 0.001 (p values were calculated using paired t-test).
Figure S7. Bovine serum albumin (BSA) permeability assay was performed with wild type and mutant KIRREL1 stably expressing human podocyte cell lines, which were cultured as a monolayer on Transwell filters. The passage of albumin across the podocytes monolayer was assessed by a paracellular albumin flux assay using Texas red-labeled albumin. The cells stably expressing KIRREL1 mutants showed increased albumin leakage as compared with cells stably expression wild type protein (data represent mean, error bars represents SEM (n=3), p-values were calculated using paired t-test; *p< 0.01, **p< 0.001).
Supplementary Table 1. Genetic and clinical data of 2 families with SRNS and mutations in KIRREL1